

Abstract
Sporadic findings in humans suggest that reinduction of heat acclimation (AC) after its loss occurs markedly faster than that during the initial AC session. Animal studies substantiated that the underlying acclimatory processes are molecular. Here we test the hypothesis that faster reinduction of AC (ReAC) implicates “molecular memory.” In vivo measurements of colonic temperature profiles during heat stress and ex vivo assessment of cross-tolerance to ischemia-reperfusion or anoxia insults in the heart demonstrated that ReAC only needs 2 days vs. the 30 days required for the initial development of AC. Stress gene profiling in the experimental groups highlighted clusters of transcriptionally activated genes (37%), which included heat shock protein (HSP) genes, anti-apoptotic genes, and chromatin remodeling genes. Despite a return of the physiological phenotype to its preacclimation state, after a 1 mo deacclimation (DeAC) period, the gene transcripts did not resume their preacclimation levels, suggesting a dichotomy between genotype and phenotype in this system. Individual detection of hsp70 and hsf1 transcripts agreed with these findings. HSP72, HSF1/P-HSF1, and Bcl-xL protein profiles followed the observed dichotomized genomic response. In contrast, HSP90, an essential cytoprotective component mismatched transcriptional activation upon DeAC. The uniform activation of the similarly responding gene clusters upon De-/ReAC implies that reacclimatory phenotypic plasticity is associated with upstream denominators. During AC, DeAC, and ReAC, the maintenance of elevated/phosphorylated HSF1 protein levels and transcriptionally active chromatin remodeling genes implies that chromatin remodeling plays a pivotal role in the transcriptome profile and in preconditioning to rapid cytoprotective acclimatory memory.
Keywords: cellular memory, cytoprotection, stress proteins, deacclimation, reacclimation
heat acclimation (AC) is a reversible “within lifetime” phenotypic adaptation to long-term elevations in environmental temperature that evolves via a continuum of temporally varying processes (18, 20, 38). Successful AC is characterized by enhanced thermal tolerance manifesting as improved endurance and resistance to temperature extremes, collectively delaying the onset of heat injury. AC can reinforce or interfere with the ability to combat novel acute stressors. In this way, AC was found to confer protection to a variety of conditions with impaired oxygen supply/oxygen demand ratios. Among these, cross-tolerance between ischemia-reperfusion insult in the heart (7, 20) and hyperoxia in the brain (1) have been extensively studied. The available evidence substantiates that a reprogramming of gene expression and translational processes are essential events in the pathway to AC. The important hallmarks of AC and AC-mediated cross-tolerance identified to date include enhanced reserves of heat shock protein 72 (HSP72) and hypoxia-inducible factor-1 alpha (HIF-1α), anti-apoptotic and antioxidative pathways, accelerated (vs. nonacclimated) stress-mediated transcriptional activation of the hsp70 gene and in turn, the heat shock response (HSR). Transcriptional activation of HIF-1α targets such as erythropoietin, erythropoietin receptor and glycolytic pathways have also been documented (7, 20, 26, 28). The complexity of the pathways essential to generate the mammalian acclimated phenotype include a continuum of processes that involve the interconnected activity of a large battery of constitutive and inducible genes encompassing metabolic, transporting/trafficking, and cytoprotective functions (18).
Although multiple studies have disclosed the physiological and molecular processes underlying AC in mammals, those dealing with the decay and loss of AC are sparse, and even fewer have explored the processes of its reinduction (31, 32). The early pioneering studies with this respect were confined to characterizing the physiological acclimated phenotype. It was shown then, for the first time, that the AC phenotype, when defined by consensus acclimatory criteria such as decreased heart rate and rectal temperature, persists during a long period of 7–20 days of deacclimation (DeAC), depending on whether humid or dry heat acclimation was applied (reviewed by Pandolf, Ref. 31). It was hypothesized that the physiological AC phenotype decays at a rate of a 1 day loss of acclimatized status for every 2 days spent without heat exposure (31). Reinduction of AC after the DeAC period occurs markedly faster than the initial/original AC session (31). Interestingly, a similar phenomenon was documented for cold temperature (16) and altitude reacclimation (ReAC) 25), implying that slow DeAC/rapid ReAC is not an AC-specific feature and may be a conserved adaptive response.
Memory in response to a stimulus is considered a reversible regulatory system in which a self-propagating “on” state is generated from common signaling molecules, such that once a system has been “sensitized” by a strong initiating stimulus, a second exposure elicits a more rapid response. Generally, memory processes are attributed to molecular signaling (2). Given that the underlying long-term acclimatory responses are molecular, we hypothesize that the faster reinduction of AC after a DeAC period implicates molecular memory. To test this hypothesis, we undertook a multidisciplinary approach to 1) characterize the DeAC/ReAC physiological phenotype, 2) profile the transcriptional activation of stress-associated genes to identify cues predisposing toward a rapid reinduction of the acclimated state, and 3) assess gene activation by measuring the expression level of their encoded proteins. Our results indicate that reinduction of the physiological AC phenotype requires only 2 days rather than the 30 days required for achieving initial acclimatory homeostasis, thus confirming that AC has a memory. The analyses presented here point to a divergence between the physiologic and genotypic responses. We found that despite the return of the AC phenotype to its preacclimated physiological state, certain gene clusters maintained an active transcriptional state throughout the entire DeAC/ReAC period. This dichotomy led us to conclude that acclimation induces persistent, selective transcriptional changes, predisposing individuals who have undergone an initial heat acclimation session to reacclimate to subsequent periods of elevated temperatures within a shorter time period. This phenomenon was evident both in thermoregulatory responses and in AC cross-tolerance. Given the enhanced transcriptional activation of chromatin remodeling gene clusters, our data support the hypothesis that “acclimation memory” may be linked to epigenetic information and cellular/molecular memory.
MATERIALS AND METHODS
Male Rattus norvegicus (Sabra strain), initially weighing 80–90 g (3-wk-old), fed Ambar laboratory chow with water ad libitum, and held under light-dark cycled conditions (12 h:12 h), were randomly assigned to the following groups (see Fig. 1): heat acclimated for 2 and 30 days (AC2d, AC) (17); deacclimated (DeAC) for 1 mo [DeAC(1)] and 2 mo [DeAC(2)]; reacclimated for 2d (ReAC) after deacclimation of 1 mo [ReAC(1)] and 2 mo [ReAC(2)]; and control, normothermic (C). Each of these groups was randomly subdivided into animals undergoing no further treatments and those subjected to an additional superimposed stress described below. This allowed us to test the acclimation status effect both on basal acclimatory properties and on the superimposed (novel) stress response. Controls and deacclimated rats were held at an ambient temperature of 24 ± 1°C, whereas AC was attained by a continuous exposure to 34 ± 1°C and 30%–40% relative humidity as previously described (17). To assess whether acclimation has a memory, we initially studied the loss or induction of AC using established physiological experimental paradigms characterizing the acclimated phenotype. These included 1) physiological, in vivo measurements of colonic temperature (Tc) profile during exposure to heat stress (HS) (17, 38) and 2) ex vivo assessment of the development of cross-tolerance to ischemia-reperfusion or anoxia in the isolated heart (measurement of infarct size) and in isolated cardiomyocytes (time to development of rigor contracture) (7, 26). To investigate acclimation memory, we studied 1) temporal global genomic responses under basal conditions and in response to acute HS in the AC-DeAC-ReAC groups using a stress-associated cDNA Atlas Array, 2) the expression of selected proteins, and 3) the transcriptional dynamics of selected genes.
Fig. 1.
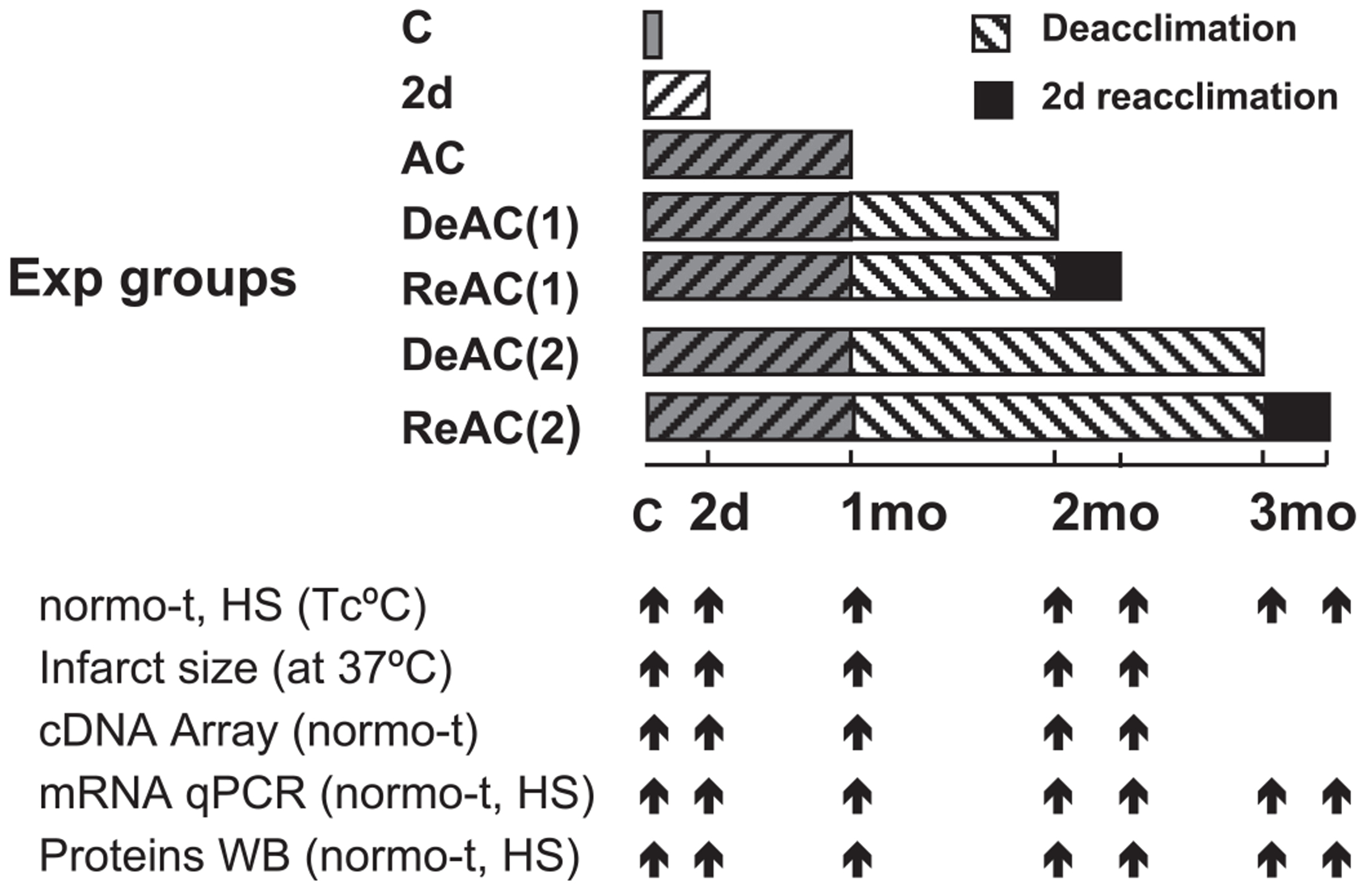
Protocol bar illustrating experimental plan. Transcripts and proteins were measured in heart (left ventricle) tissues. For details, see text. C, controls (at 24°C); AC, long-term heat acclimation (30 days at 34°C); HS, heat stress (at 41°C); normo-t, basal ambient temperature of the specific experimental (Exp) group; DeAC(1), DeAC(2), deacclimation for 1 and 2 mo at 24°C; ReAC, reacclimation for 2d at 34°C; Tc, colonic temperature; qPCR, real-time PCR; WB, Western immunoblot.
All experimental protocols were approved by the Ethics Committee for Animal Experimentation of The Hebrew University (Jerusalem, Israel).
Physiological Assessments of Acclimation Memory
Tc profile.
Conscious rats were exposed to HS for 2 h at 41°C. Tc was measured for 2 h at 20 min intervals, using a YL 402 thermistor inserted 6 cm deep beyond the anal sphincter (17, 38).
Ischemia-reperfusion injury (infarct size) in isolated hearts.
Rats were euthanized by ketamine-xylazine (8.5 mg/100 g body wt ketamine in 0.5% xylazine ip) anesthesia followed by cervical dislocation, and the heart was removed and mounted on a Langendorff perfusion system, retrogradely perfused with Krebs-Henseleit buffer (KHB) containing (in mM) 120 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 1.25 CaCl2, 25 NaHCO3, and 11 glucose, at pH 7.4, and aerated with a mixture of 95% O2-5% CO2 at 37°C and perfusion pressure of 100 cmH2O. After 10 min of equilibration the heart underwent global ischemia for 30 min, followed by 40 min of reperfusion. Then immediately, a 10% 2,3,5-triphenyltetrazolium chloride solution was infused until the coronary vasculature stained dark red. The hearts were removed, dried, and kept at −80°C. We fixed 1-mm-wide slices in 10% paraformaldehyde for 72 h and then photographed them. Total slice vs. infarct area was calculated using Adobe Photoshop (26).
Rigor contracture in cardiomyocytes.
Animals were euthanized as above, and the hearts were rapidly removed and placed in a physiological solution at 4°C. Cardiomyocytes were isolated using a modification of the technique described by Silverman’s group (11). In brief, the hearts were retrogradely perfused for 3 min with KHB, followed by perfusion for 5 min with calcium-free KHB, and then for 20–30 min with calcium-free KHB containing 0.2 mg/ml collagenase (type II, Sigma). The left ventricle was dissociated, and the myocytes were resuspended twice in calcium-free KHB. The myocytes were then suspended in KHB containing 100 mg/ml BSA and 50 μM CaCl2 and placed on the bottom of an anoxic chamber on an inverted microscope stage attached to a video-motion unit and Felix data acquisition software (PTI) (detailed in Ref. 4). During anoxia, an upward laminar flow of ultrapure argon was maintained to exclude virtually all oxygen (PO2 <0.02 Torr) (40). Time from the onset of anoxia to rigor contracture development for individual myocytes was measured.
Genomic Responses and Protein Analyses
For the various experimental series, the animals were under basal conditions or subjected to HS and allowed to recover at 24 ± 1°C for 0, 20, 40, or 60 min. The hearts were then removed, mounted on a Langendorff perfusion system as above, then perfused for 2 min with KHB to wash out the blood. The left ventricle was then excised and stored at −70°C until analysis (20).
Stress-gene expression using Clontech cDNA Atlas array.
Clontech Atlas rat stress array (cat. no. 7735-1, BD Biosciences Clontech) was used. Total RNA was extracted using TRI Reagent (Molecular Research Center) according to the manufacturer’s protocol (20). Individual mRNA samples were pooled into mRNA preparations of three hearts each. Probes were labeled for 1 h by the reverse transcription of total RNA (3 μg) at 42°C in a primer mix (Clontech) containing [32P]dATP (Amersham Biosciences, Buckinghamshire, UK). The reaction was terminated by 0.1 M EDTA and 1 mg/ml glucogen (Sigma). The unincorporated 32P-labeled nucleotides were removed by Nucleo-Spin extraction columns (Clontech). The membranes were prehybridized for 1 h at 68°C in a hybridization solution (Clontech) with 0.1 mg/ml sheared salmon testes DNA (Sigma) to block nonspecific binding. The synthesized radiolabeled cDNA probe (5 to 15 × 106 cpm) was hybridized overnight at 68°C. Cot-1 DNA (Clontech) (1 μg/ml) was added to block nonspecific binding. For analyses, the membranes were exposed to a phosphor screen for 15 and 24 h and detected using a Bio-imaging Analyzer BAS2000. We used Atlas Image 2.01 software (Clontech) to grid the phosphor image, record pixel density of each spot, and perform background subtraction.
For analysis, in brief, we standardized different recordings (radioactivity level at three time points) of the same hybridization experiment via a linear regression of the separate time points for each probe and averaged their regressed values to the recorded intensity at the middle time point. We then rescaled each measured expression value by the average expression of all probes in that hybridization, thereby normalizing all hybridizations to the same radioactive intensity, and transformed these values to make the control conditions a “baseline” for estimating the relative changes in expression. A base-2 log was used, so that a relative expression of 1 or −1 would indicate a twofold change over the average control conditions. Up-/downregulation beyond logbase 2 treat/C >0.8 has been considered physiologically meaningful (20). The data were then clustered using an agglomerative probabilistic method that groups genes together based on similar expression profiles (ScoreGenes package, http://compbio.cs.huji.ac.il/scoregenes/). For further details see Ref. 20. For an overall functional interpretation of the common responses, gene ontology (GO) database categories for biological processes (GO: http://www.geneontology.org) was used. The data discussed in this publication have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE8329.
Protein and mRNA Detection
Western blot analysis.
Total protein (50 μg per lane) was fractionated by electrophoresis on 9% polyacrylamide gels under denaturing conditions (23), transferred onto nitrocellulose membranes, blocked, then probed overnight at 4°C with the primary antibody followed by 1 h incubation at room temperature with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Jackson) diluted 1:10,000.
Specific antibody binding was detected using enhanced chemiluminescence (Amersham) and visualized by exposing X-ray film to the membrane (26–28, 38). Band density of the scanned film was calculated using Tina software (Raytest, Straubenhardt, Germany). The primary antibodies used were polyclonal anti-rabbit anti-HSP72 (1:10,000), monoclonal anti-mouse anti-HSP90 (1:100),polyclonal anti-Bcl-xL (1:1,000) (Stressgen, Victoria, BC, and Delta Biolabs Ca, Canada, respectively), and anti-mouse anti-HSF1 (1:1,000; NeoMarkers, Fremont, CA). Polyclonal anti-rabbit β-actin (1:10,000; Santa Cruz Biotechnology, Santa Cruz, CA) was used for normalization.
Quantitative real-time reverse-transcription PCR.
For array hybridization confirmatory analysis and transcriptional dynamics, hsp70 and hsf1 mRNA were chosen. Previous studies (20, 28) confirmed the altered acclimatory dynamics of hsp70 gene. mRNA was measured using quantitative real-time reverse-transcription PCR (qRT-PCR; ABI Prism 7000 Sequence Detection System, Applied Biosystems). Reaction volumes of 20 μl contained 10 μl of SYBR Green Master Mix (Applied Biosystems), 500 nM each of the forward and reverse primer, and 5 μl of diluted cDNA. The appropriate cDNA dilution was determined using calibration curves established for each primer pair. The thermal profile for SYBR Green real-time RT-PCR was 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. The primers for real-time RT-PCR were designed using Primer Express software (Applied Biosystems). The primers, sense and antisense, respectively, were: HSP70: 5′-caagaatgcgctcgagtccta-3’, 5’-ctctttctcagccagcgtgtta-3, HSF1:5′-tcggtgaccatgcccgacatgag-3′, 5′-cagttcactgctccctgtgtccac-3′, β-Actin: 5′-tgtggcatccatgaaactac-3′, 5′-atttgcggtgcacgatggag-3′.
Statistics
For statistical analyses, we used commercially available computer software (SigmaStat 2.03). The treatment time points (TTP: C, AC2d, AC, DeAC, ReAC) and the acute stressors (HS, ischemia-reperfusion, or anoxia) were taken as the independent categorical variables, and individual animals or hearts were considered a random sample from the population. The following analyses were conducted. To test the effects of TTP as the only factor on the basal values or on the superimposed HS of the dependent expressed variables (for variables see Fig. 1) one-way ANOVA was used. For comparing continuous on line temperature measurements, we used repeated-measures one-way ANOVA. For post hoc pairwise comparisons between the control (nonacclimated) and the various treatment time point groups, Dunnett’s test was applied unless otherwise specified. To test whether TTP affect the response to superimposed HS, two-way ANOVA was used. If significant interaction (with HS and TTP as the two factors) was indicated, a multiple-comparisons test to detect if origin of significance stems from HS vs. basal (Tukey) was conducted. Comparisons between HS and basal groups within matched TTP groups were also conducted using two-sample Student’s t-test. Additional details are specified in the figure legends. Data are expressed as means ± SE. Values of P < 0.05 were considered statistically significant.
RESULTS
Acclimation Memory: Physiological Evidence
We previously showed that in rats, AC is attained via exposure to temperatures at the upper limit of the thermoneutral zone for ~1 mo (17). Loss of AC was detected after a return to normothermic conditions for 2–3 wk (M. Eynan and M. Horowitz, unpublished observation and Ref. 1). To assess whether acclimation has a memory, we initially studied the loss or induction of AC at integrative, organ, and cellular levels.
Tc and heating rate during HS.
During HS (Fig. 2), Tc rises to a hyperthermic plateau (Tc-pl). The normal thermoregulatory response of the rat thus provides a reliable criterion for the acclimation status (17, 38). Basal Tc and Tc-pl records (Table 1, Fig. 2) demonstrate that Tc-pl of rats heat acclimated for 30 days (AC) was significantly higher than controls (P < 0.05). As evident from Tc-pl (Table 1, Fig. 2), after a DeAC period of 1 mo, a loss of acclimation and a return to the preacclimated state was observed. Two days of re-exposure of the rats to the AC protocol, both after 1 and 2 mo of DeAC, restored the AC phenotype (P < 0.05). In contrast, the Tc-pl of short-term (2 day) acclimated rats resembled the Tc-pl of the control group. These differences among the groups are also reflected by changes in their heating rates (to Tc-pl) at the onset of HS (see Table 1 for details). The heating rates of the AC, ReAC (1), and ReAC (2) groups were significantly faster than those of the control, AC2d, or DeAC groups (P < 0.02).
Fig. 2.

Tc before and during heat stress (HS) at 41°C. C, controls; AC2d, heat acclimation (at 34°C) for 2 days; AC, heat acclimation for 30 days; DeAC, deacclimation at normothermic conditions for 1 or 2 mo. ReAC (1) and ReAC (2), reacclimation for 2 days following 1 or 2 mo of DeAC. Each symbol represents the average value for the particular time point. The repeated-measures 1-way ANOVA model results in significant differences between groups (P < 0.001). *Significant difference of AC, ReAC(1), and ReAC(2) vs. C P < 0.05, Dunnett’s; n = 5–10 rats per group.
Table 1.
Basal and hyperthermic plateau temperatures and heating rates of rats undergoing heat acclimation (AC2d, AC), deacclimation of 1 and 2 mo [DeAC (1), DeAC(2)], and reacclimated for 2 days [ReAC(1), ReAC(2)]
| C | AC2d | AC | DeAC(1) | ReAC(1) | DeAC(2) | ReAC(2) | |
|---|---|---|---|---|---|---|---|
| Basal Tc | 37.68±0.08 | 37.88±0.03 | 37.96±0.05 | 37.64±0.09 | 38.04±0.02 | 37.92±0.03 | 38.06±0.04 |
| Tc Plateau | 40.01±0.009 | 39.92±0.017 | 40.78±0.035† | 40.19±0.09 | 40.68±0.10† | 40.06±0.03 | 40.51±0.07† |
| Heating Rate, °C/min | 0.037±0.001 | 0.030±0.001 | 0.045±0.001* | 0.032±0.001 | 0.041±0.003* | 0.034±0.0006 | 0.04±0.001* |
Values are means ± SE and were derived from representative groups of 6–12 animals in each treatment group. Control (C) and deacclimated (DeAC) animals were maintained at 24 ± 1°C. Heat-acclimated (AC) and reacclimated (ReAC) rats were maintained at 34°C and 35% relative humidity. Tc, basal colonic temperature before heat stress; Tc plateau, average Tc at time points 80, 100, and 120 min exposure to heat stress at 41°C. For significance test, 2-way ANOVA followed by Dunnett was conducted. Significant difference from the C group:
P < 0.02;
P < 0.001.
Ischemic and anoxic cross-tolerance (7, 26) upon ReAC was demonstrated at both organ and cellular levels. Figure 3 shows infarct area measurements and the infarct-risk area ratio in heart slices from the experimental groups. The area of infarction was 3 and 2.4 times smaller in the AC and ReAC groups than in the control group (P < 0.05), respectively. In contrast, the infarct sizes of hearts from AC2d rats and DeAC groups were not significantly different from the control group average.
Fig. 3.
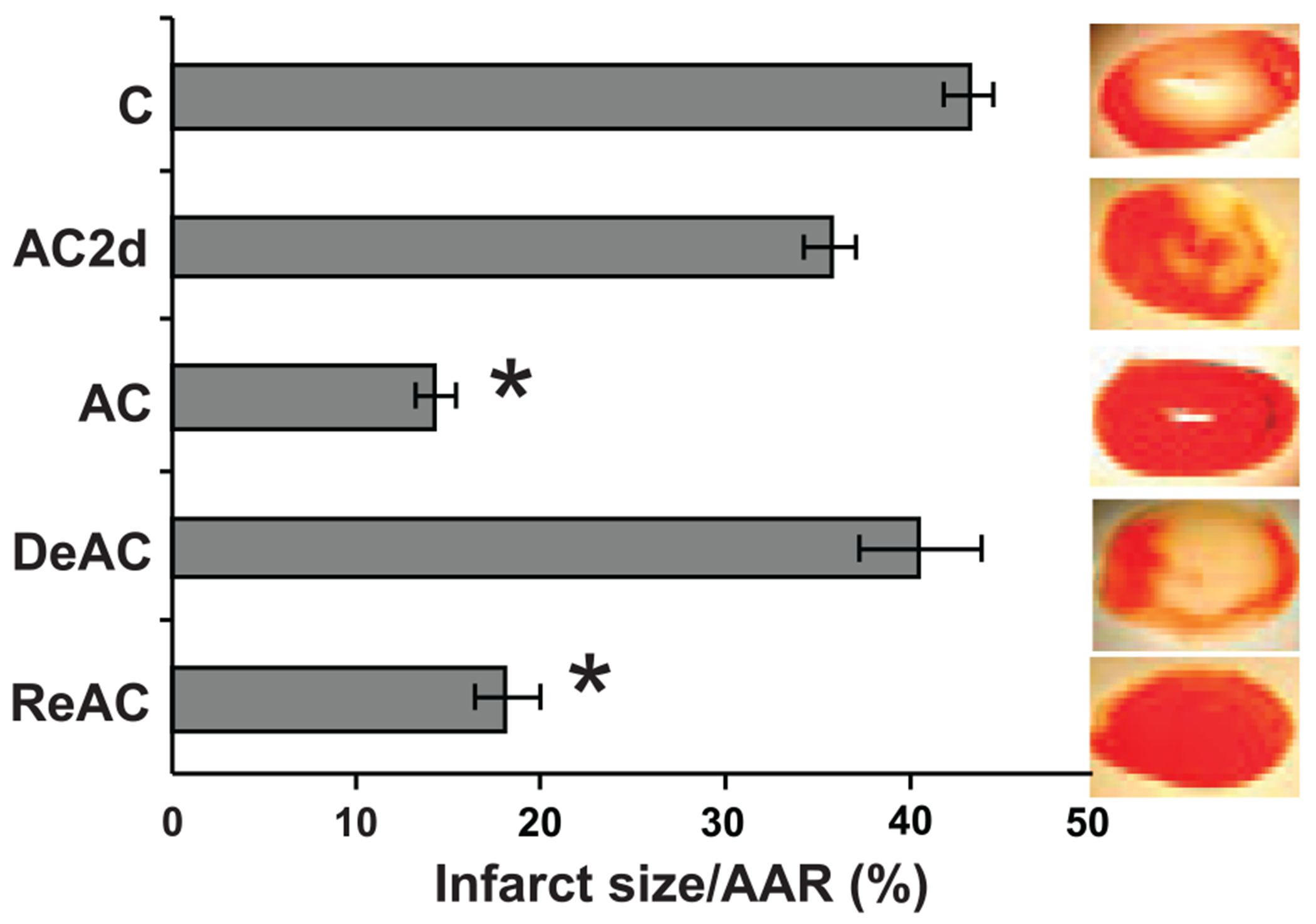
Infarct size, expressed as the percentage of infarct size/area at risk (AAR) in isolated hearts subjected to global ischemia (30 min) followed by reperfusion (40 min). The effects of treatment time point (TTP) on AAR was significant (1-way ANOVA, P < 0.001), *Significant difference of AC and ReAC from C (P < 0.05, Dunnett’s); n = 5–10 rats per groups. For abbreviations see legend Fig. 2.
In the single isolated myocyte, an imbalance between glycogen stores and the rate of anaerobic glycolytic energy consumption, leading to ATP depletion, results in a sudden rigor (40). This discrete event enabled us to measure the time to onset of ATP-depletion rigor and use the result as an additional (metabolic) criterion for acclimation status and cross-tolerance. Previously we demonstrated that AC hearts acquire higher glycogen stores and a slower rate of glycolysis, thus improving ATP supplementation to the ischemic heart (7). Figure 4 shows that the average time to contracture of the AC myocytes population was profoundly longer than in control myocytes (28.35 ± 0.17 vs. 14.6 ± 0.11 min, P < 0.05). The ReAC cells resembled AC myocytes, whereas AC2d and DeAC myocyte populations did not differ from controls. The enhanced anoxic endurance was maintained in cardiomyocytes of rats deacclimating for 2–2.5 wk.
Fig. 4.
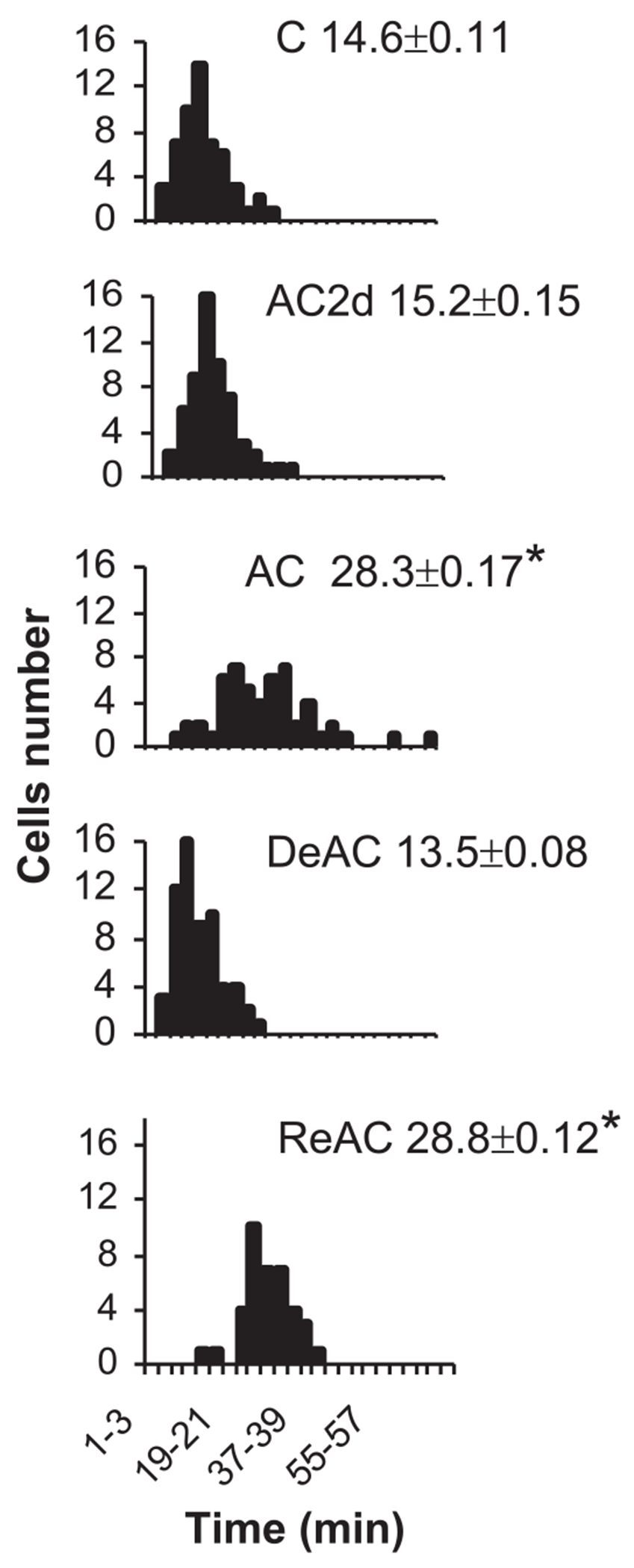
The time elapses from the onset of anoxia to rigor contraction in individual cardiomyocytes. Each panel depicts the distribution curve of time to rigor in cardiomyocytes population from rats subjected to one of the assigned treatments and the average onset time to rigor contraction (means ± SE, min); n = 6 rats per group. The effects of TTP on the time to rigor was significant (1-way ANOVA, P < 0.001). *Significant difference of the time to rigor of AC and ReAC from the C group. P < 0.05, Dunnett’s. For abbreviations see legend Fig. 2.
Dichotomy Between the Genotypic and Phenotypic Responses
Gene profiling analysis during the course of AC displays a continuum of changes in the expression of stress-inducible genes, with an initial transient activation of genes coding for DNA integrity, replaced (if challenge continues) by the activation of genes encoding proteins linked with cytoprotective networks (20). Many cytoprotective genes are also targeted by chronic exposure to low ambient temperatures (10), suggesting that such genes contribute to thermal plasticity in general. Taken together, we hypothesized that stress-associated gene profiling in the De-/ReAC state could facilitate understanding of the phenomenon of rapid ReAC. Stress-cDNA atlas array analysis of altered gene expression (before and after HS) revealed the enhanced activation of several cytoprotective programs in hearts from both DeAC and ReAC groups (Fig. 5). The gene profiling of these groups (92 visible genes) differed markedly from that of control and AC groups but partially resembled the profile of non-HS 2d acclimated hearts. The cluster analysis highlights a very important finding, namely that ~37% of the visible genes, the majority of which were upregulated, did not resume their preacclimation levels after 1 mo of DeAC, despite the return of the physiological phenotype to its preacclimation state. Furthermore, the genes assigned to these clusters (no. 5–9 out of 10 clusters; Fig. 5, A–F) were significantly upregulated (4 clusters) or downregulated (1 cluster) (log2Treat/C ratio =>1) in the DeAC, ReAC, and the superimposed heat-stressed in the ReAC groups. Genes assigned to cluster no. 8 were also upregulated following short acclimation (AC2d) and genes assigned to cluster 7 (comprising members of the HSP superfamily, see below), in addition to their basal upregulation in response to AC per se, were further upregulated in response to the superimposed acute HS (log2Treat/C ratio =>2 vs. 1 in the basal, Fig. 5B). The annotation for biological processes of genes assigned to clusters representing the dichotomy from the physiological phenotype, comprised genes involved in cytoprotective pathways, such as e.g., the HSP species (HSP70.1, HSC73, HSP90) and DNAJ, nuclear responses, and transcriptional regulation linked with chromatin organization e.g., HMGB2 (high mobility groups protein) and SSRP1 (structural specific recognition protein 1), perhaps underpinning the regulated cytoprotection (discussed below). Additional GO categories of interest were CREBP1 (cAMP responsive binding protein 1) and SAPK (stress-activated protein kinase) associated with cellular memory and transcription processes in both central and peripheral organs (33). Detailed gene profiling of this experimental series is provided in GEO accession no. GSE8329.
Fig. 5.
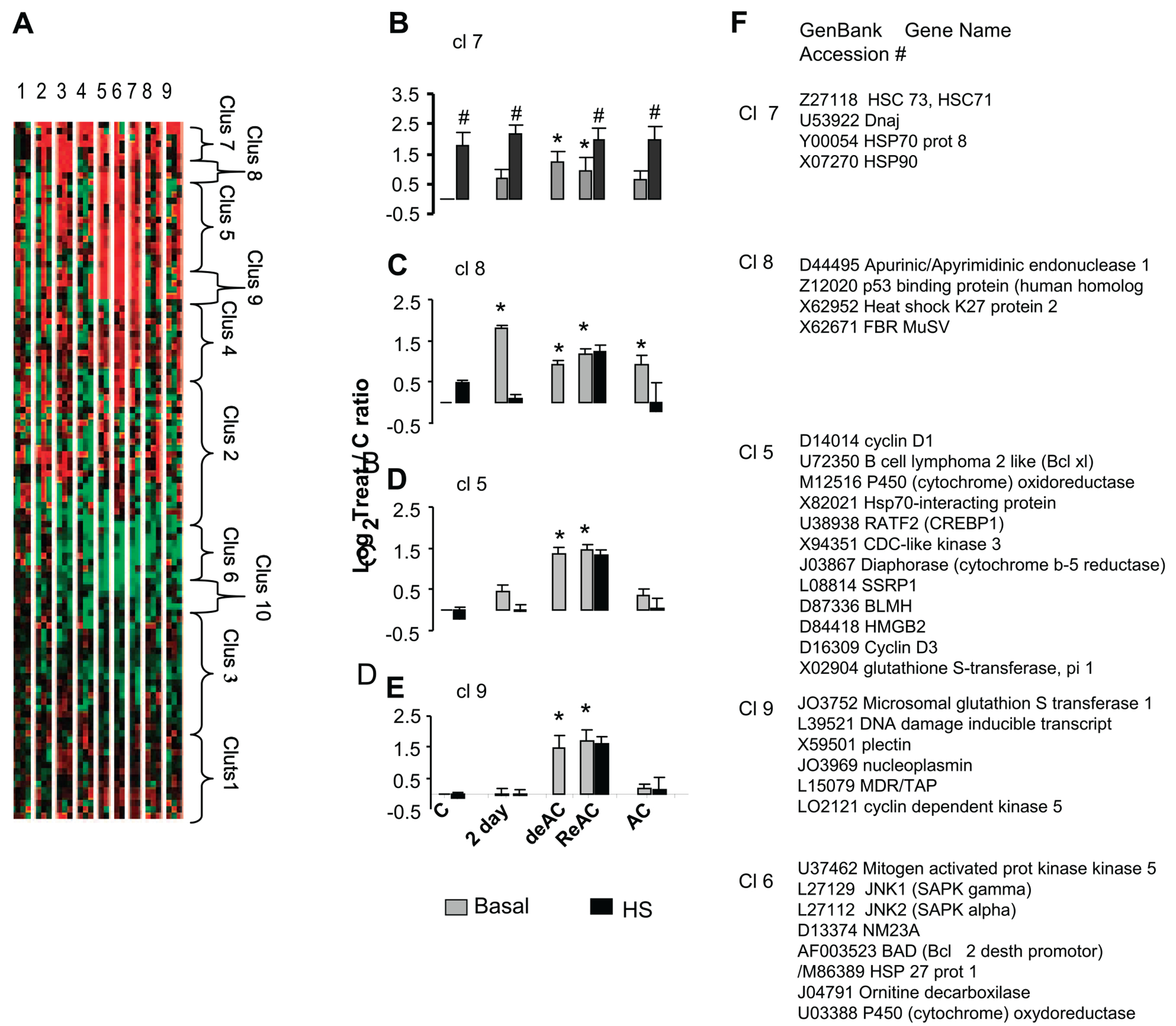
Analysis of stress-associated genes in (heart) left ventricle. A: tree view of all visible genes included in cluster analysis (Clontech stress array). Red, upregulation; green, downregulation; black, no change. Each row represents 1 gene; columns represent treatment groups (lanes 1–9: C, C-HS, AC2d, AC2d+HS, DeAC, ReAC, ReAC+HS, AC, AC+HS, respectively). B–E: bar graphs of representative clusters (left). F: names of genes assigned to clusters 5–9. Each bar presents average expression (mean ± SE), calculated as logbase 2 to control. Each cluster shown represents groups of genes that did not resume their preacclimation levels after 1 mo of DeAC despite the return of the preacclimation physiological phenotype; n = 9 rats (3 pools) per group. The effect of TTP on gene expression within basal levels was significant in all clusters (1-way ANOVA, P < 0.001). *Significant difference (P < 0.05, Dunnett’s) of TTP vs. C. To confirm significant HS vs. basal effect on gene expression at each TTP (observed in Cl 7 only) t-test was conducted. #Significant difference from the matched TTP basal value (P < 0.001, t-test). Cl, cluster; for all other abbreviations and experimental conditions see legend Fig. 1.
In the present investigation, we focused on representative cytoprotective genes and products that retained their alerted state following DeAC. To validate the results obtained in the array, we initially measured hsp70 transcript levels in the RNA samples using qRT PCR. When measured individually, the temporal changes in hsp70 levels (Fig. 6 inset) along the entire experimental protocol (see Fig. 1) were similar to that detected in the array. Furthermore, faster transcriptional activation in response to stress is a hallmark of acclimation (compared with nonacclimated) (20, 28). Hence, in an additional experimental series, the kinetics of hsp70 transcription following HS was studied (Fig. 6). Both TTP and HS contributed to hsp70 dynamics with statistically significant interaction between the two (Fig. 6, two-way ANOVA P < 0.001). The hsp70 transcript kinetics of DeAC, ReAC, and the acclimated (AC) phenotype were similar, with peak mRNA levels at 40 min post-HS, whereas the control phenotype demonstrated peak levels at 60 min post-HS. In contrast, HS blunted transcription in the AC2d group as was previously demonstrated (28). Basal transcript levels differed significantly according to the TTP groups (one-way ANOVA P < 0.006) with AC2d, DeAC, and ReAC significantly upregulated and AC downregulated compared with the nonacclimated (C) group (Fig. 6 inset). Figure 7 presents the protein profiles of HSP72, HSP90, and Bcl-xL, representative products of genes assigned to clusters 5 and 7, all part of interrelated cytoprotective networks. The effects of TTP on basal protein levels was significant (one-way ANOVA, P < 0.002, for HSP70 and P < 0.001 for HSP90 and Bcl-xL). Following AC, the level of these three proteins was significantly higher than in C (P < 0.05, Dunnett’s). Both HSP72 and Bcl-xL maintained this elevated level and did not return to their preacclimation levels throughout DeAC and ReAC (Fig. 7). In contrast, HSP90, (an additional chaperone of the HSR assemblage), returned to preacclimation levels in the DeAC groups (1 and 2 mo) but was upregulated significantly (P < 0.05, Dunnett’s) following ReAc. Notably, AC2d did not induce significant elevation in HSP90 reserves (Fig. 7). HS induced further elevation in the protein levels. The increase varied among TTPs and for each protein. However, values (for each protein) at the various TTPs, even when demonstrating significant difference within each TTP, did not differ significantly from the heat-stressed C value. In view of the upregulation of the hsp gene transcripts in the DeAC and ReAC groups, we studied heat shock factor 1 (HSF1), the consensus transcription factor of the hsp genes. Both the array analysis and the individual transcript measurements revealed hsf1 upregulation in AC, DeAC, and ReAC groups (Fig. 8 inset, one-way ANOVA P < 0.001). The effect of TTP on both basal and HS level of the proteins was significant. HSF1 protein level (total as well as the phosphorylated, Fig. 8, bottom and middle panels) was significantly upregulated (one-way ANOVA, P < 0.001, P < 0.02, respectively). All groups except for AC2d differed significantly from the C group (P < 0.05, Dunnett’s). The differences in basal vs. HS levels of the HSF1 vs. C were only observed in the control and AC2d groups (Fig. 8).
Fig. 6.
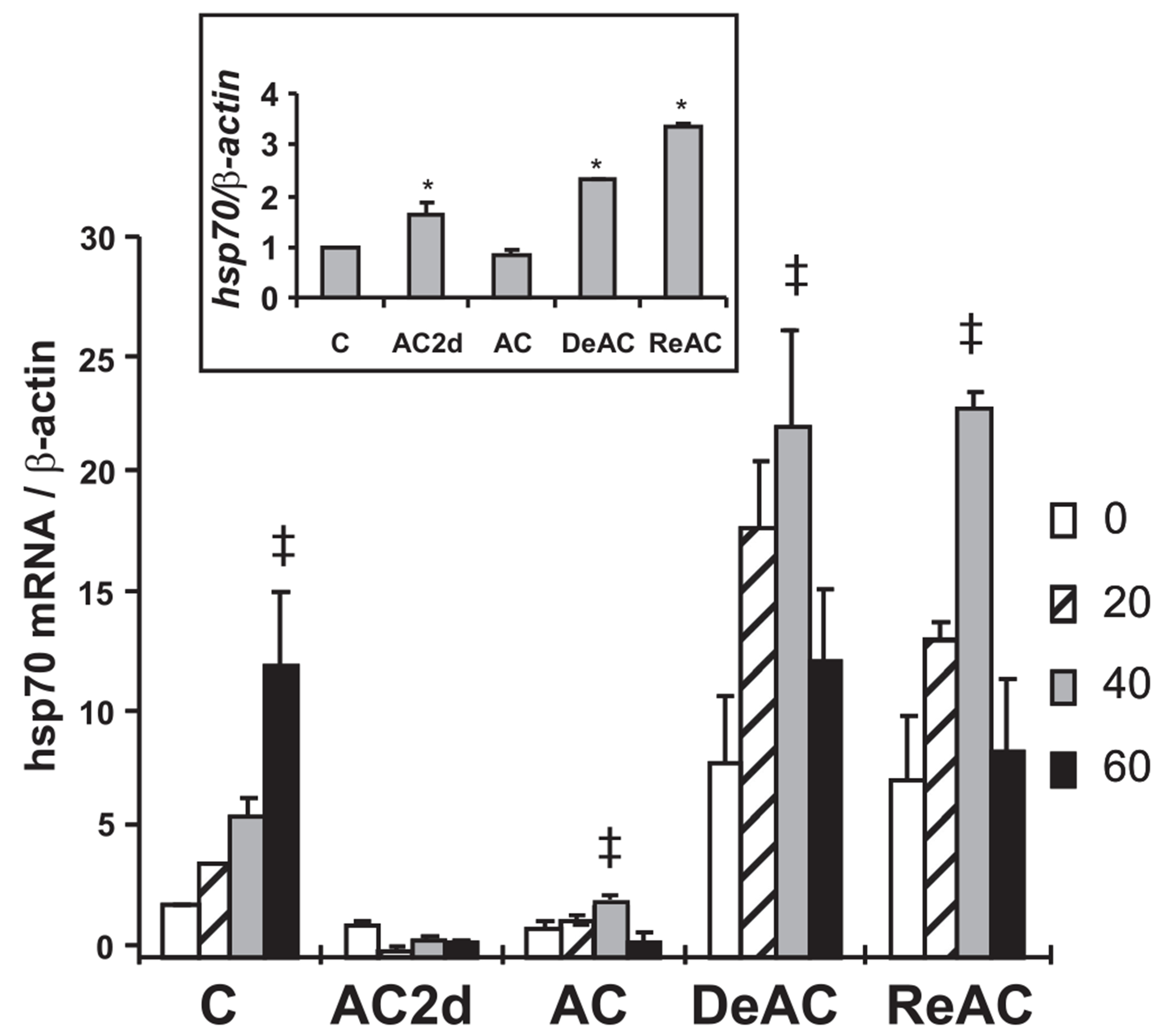
hsp70 mRNA 0, 20, 40, and 60 min post-HS at 41°C in left ventricle of the heart and hsp70 basal mRNA levels at each acclimation time point relative to basal C value (inset). Each bar represents mean ± SE, n = 4 rats per group. TTP showed significant effects on hsp dynamics post-HS (2-way ANOVA P < 0.001). There was statistically significant interaction between the 2 factors (P ≤ 0.001). To explore interaction in mRNA transcription dynamics among the groups, 1-way ANOVA followed by multiple pairwise comparisons (Tukey procedure) within each individual acclimation time point was conducted. ‡Significance (P < 0.05) of peak transcript level within the group. To test for significant effects of TTP within basal mRNA levels (inset), 1-way ANOVA was applied (P < 0.001). *Significant difference vs. C (P < 0.05, Dunnett’s).
Fig. 7.

HSP72, HSP90, and Bcl-xL protein profiles under basal and HS conditions. HSP72 and Bcl-xL profiles follow the observed dichotomized genomic vs. physiological phenotypic response. In contrast, HSP90, an essential cytoprotective component shows mismatched transcriptional activation upon DeAC. Protein levels are presented as protein/β-actin ratio. Each bar represents mean ± SE, n = 5–10 rats per group. The effect of TTP on basal levels of all proteins was statistically significant (1-way ANOVA, 0.001). *Significant difference (P < 0.05, Dunnett’s) of the protein level in the TTP vs. basal C level; to confirm significant HS vs. basal effect on gene expression at each TTP t-test was conducted. ##P < 0.001–0.009 and #P < 0.01–0.03 denote significant difference from the matched TTP basal values. For abbreviations and experimental conditions see legend Fig. 2.
Fig. 8.

Total HSF1 and phosphorylated (P) HSF1/HSF1 ratio as well as hsf1 transcript (inset) in the experimental groups. n = 5–10 rats per group. For abbreviations and experimental conditions see legend Fig. 2. The effect of TTP on basal levels of the proteins was statistically significant (1-way ANOVA, 0.001). *Significant difference from the C group (P < 0.05 Dunnett’s). †Significant difference from the HS value in the C group (Dunnett’s). To confirm significant HS vs. basal effect on gene expression at each TTP t-test was conducted. #Significant difference from the time point matched basal value (P < 0.001).
DISCUSSION
The physiological criteria used here provide substantial evidence that AC involves memory because reinduction of the AC phenotype to a second bout of acclimation occurred in a remarkably shorter period than required to achieve the initial AC phenotype. In this investigation acclimation memory was retained for 2 mo. The expression profile of a battery of stress-associated genes, as well as the set of genes and proteins individually studied, point to common responding, up-/down-regulated genes. These genes retain their AC altered levels upon DeAC, despite a return of the physiological phenotype to its preacclimated state. We propose that the rapid ReAC is associated with a predisposition of these continuously transcriptionally alerted genes/downstream pathways (AC throughout the DeAC phase) toward a prompt response when ReAC is applied.
The results of this study provide evidence for acclimation memory in adult rats at three levels of body organization: in vivo, in intact animals, and ex vivo, in isolated organs and cells. The in vivo confirmation for a memory in central thermoregulatory structures was demonstrated by a faster heating rate and higher Tc-pl, which is well matched with the temperature threshold for salivation (19). Collectively, the elevated Tc-pl leads to improved physical pathways for heat dissipation (17, 39).
Temperature thresholds for activating thermoregulatory effectors play a pivotal role in the process of acclimation. AC is characterized by elevated Tc-pl (28) and associated with 1) a considerable decrease in the number of warm-sensitive neurons, 2) an ability to convert numerous insensitive to warm sensitive neurons by mediators (34), and (3) an elevated angiotensin AT2/AT1 receptor ratio (38), all demarcating neuronal plasticity. Physiological evidence for neuronal plasticity was documented by Maruyama et al. (29) with respect to circadian temperature profiles, but the extent to which such changes are maintained has never been examined. Our present experiments demonstrate restoration of the AC phenotype following DeAC of 2 mo. Both adjustability to chronic environmental sensory inputs and neuronal plasticity are generally attributed to long-term cellular processes and transcriptional plasticity, leading to functional neuronal remodeling and lasting changes in behavior and physiological responses. Such processes, involving signaling molecules, are evident during environmental enrichment (8), hypoxia (45), and learning and memory (13, 36). This aspect of central memory concerning thermoregulatory functions was beyond the scope of the present investigation.
We examined cellular processes in the context of acclimatory memory in a peripheral organ, namely the denervated isolated heart (marked decrease in infarct size) and isolated cardiomyocytes (delayed rigor contracture due to larger ATP stores). The results support the view that the cross-tolerance occurring in the ReAC groups is a purely cellular phenomenon. The data observed suggest that both cytoprotective networks and energy metabolism pathways have a memory.
The stress-associated gene profiling delineated in this investigation showed a marked difference between nonacclimated HS rats and DeAC, ReAC, and ReAC-HS animals. Thirty-seven percent of the genes maintained an up-/downregulated state upon DeAC/ReAC, despite the return of the preacclimated physiological phenotype. Notably, a large number of the similarly responding transcripts retaining their alerted state upon DeAC/ReAC are assigned to the HSR assemblage e.g., hsp70, hsp90, HSPB2, and DNAJ. The antiapoptotic bcl-xl gene is jointly regulated. The promoter region of this gene contains sequences resembling the classical heat shock element (HSE) on hsp genes, explaining its behavior during thermal stimuli (37). In the experiments described here as well as in our previous reports (20, 28), faster hsp70 transcriptional dynamics (Fig. 6) together with augmented HSP72 protein reserves (Fig. 7) were detected in AC, supporting the functional contribution of this gene to cytoprotective memory (2, 7). Notably, the latter phenomenon was also reported (Ref. 20 and unpublished observations, M. Assayag and M. Horowitz) for Bcl-xL. Our findings here, demonstrating that both DeAC and ReAC demonstrated similar (to AC) transcript dynamics, suggest that faster transcript dynamics is part of the memory repertory. The DeAC phenotype, however, is characterized by a mismatch between the greater HSP72 reserves and the loss of cardioprotection. This finding agrees with the results of Xi et al. (44), who demonstrated that heat shock and HSP72 induction fail to protect the heart against ischemic injury. Based on our data at hand regarding HSP72 and HSP90, two essential players in the HSR, one explanation for the dichotomy between the phenotypic and the genotypic response in DeAC could stem from posttranslational changes in HSP90. In contrast to the elevated HSP72 protein levels and despite the upregulation of the hsp90 gene transcripts in both DeAC and ReAC groups, the DeAC hearts did not display augmented HSP90 protein levels. HSP90 is an essential component in the HSR and the duration of its upregulation is critical to cellular integrity (6, 43). Furthermore, its role in cardioprotection is also attributed to enhanced NO signaling (22). Hence, the dichotomy between the transcription and posttranslational activation of this gene is, perhaps, one reason for the failure of cytoprotection even when there are large HSP72 reserves in the DeAC state. Environmental effects result in global genomic/proteomic responses (9). Therefore, a change in HSP90 is likely to be only one of many factors involved in shaping the DeAC phenotype.
The marked upregulation of the hsf1 transcript (array and qRT-PCR) and its encoded protein, as well as the enhanced phosphorylated HSF1 fraction in the DeAC and ReAC hearts, provides one mechanism for rapid ReAC and cytoprotection. The binding of HSF1 to the HSE initiates the transcriptional activation of the HSP chaperone family. Although heat shock is a consensus trigger for this stress-response pathway, other stressors that result in protein damage, e.g., oxygen deprivation, can also initiate the HSR. Sorger et al. (41), Høj and Jakobsen (15), and Hashikawa and Sakurai (12) demonstrated that Hsf1 binding causes a conformational change required to overcome promoter and chromatin repression in a stress-induced and promoter-specific manner. Concomitantly, Duncan (5) proposed that posttranscriptional changes in hsf1 in yeasts, due to hyperphosphorylation, may also confer chromatin changes. Here, the significant upregulation of the hsf1 transcript, as well as HSF1 and the phosphorylated HSF1 protein levels in the DeAC and ReAC groups (and in AC) imply chromatin remodeling. Interestingly, a preliminary finding in our laboratory provided evidence for a constitutive HSF1 binding to the HSE following an acclimation period of 30 days (Maloyan and Horowitz, unpublished. For figure see supplement file).1 A similar phenomenon was detected for HIF-1α (26). The HIF-1α finding, together with the occurrence of constitutive HSE-HSF1 binding may imply a preconditioned chromatin state, allowing rapid chromatin changes and enhanced transcription and posttranslational changes (including that of HSP90) upon re-exposure to heat. This conclusion is strengthened by the finding that chronic exposure to mild hypoxia significantly increased HIF-1 signaling, as well as “memory” of HIF-1 and HIF target gene induction upon reoxygenation for 1 wk (21). The findings in this investigation of delayed time threshold for rigor contracture in the AC and ReAC myocytes, reflecting changes in anaerobic ATP production are in agreement with these findings. Our previous findings (4, 7, 20, 26) link between HIF-1-targeted transcriptional activation and altered glycolysis in AC hearts. HSF1 binding to the HSE is responsible for the transcriptional activation of the HSPs species linked to the heat shock response, including that of HSP90. Given that the chaperoning function of HSP90 traffics Hif-1α to the nucleus, our data provide the rationale for hypothesizing a linkage between HSPs regulation and HIF-1α, with HSF1 as an upstream component. Evidence presented by Zhou et al. (46) that HSP70 and HSP90 promote HIF-1 accumulation and stabilization, with their expression provoked by PI3K/AKT, supports this hypothesis.
An interesting finding emerging from this investigation is the marked upregulation of the HMGB2 and SSRP1 transcripts, both involved in chromatin remodeling and transcriptional regulation. While HMG boxes can bend DNA, causing allosteric transitions, thereby allowing other proteins to bind or alternatively, loosen the wrapped DNA, and thus enhance accessibility to chromatin remodeling complexes and transcription factors (3), SSRP1, having an HMG-DNA binding domain, alters the repressive nature of chromatin by affecting the transcriptional elongation of specific sets of genes, either independently or as a subunit of FACT (facilitating chromatin transcription) (24). The altered CREB detected among the alerted genes are additional implications of chromatin change in the De/ReAC groups. Hirota et al.(14) reported that particular environmental stresses alter chromatin structure via phosphorylation around CRE-like (cAMP-response element, found in the promoters of many inducible genes) sequences (30). The phosphorylation causes selective chromatin openings that activate the SAPK signaling cascade in response to distinct environmental stresses. Interestingly, CRE and the CREB transcription factor are associated with cellular memory and chromatin-mediated synaptic plasticity in the brain (35), as well as functioning as memory-storage molecules in the heart (33).
In summary, in this investigation we substantiated the phenomenon of acclimation memory at the integrative physiological level and provided cues for the underlying mechanisms. The uniform expression profile of many genes and their products implies that the phenotypic plasticity seen in the ReAC state is associated with upstream information. If one takes into account the significant changes in chromatin structure that occur as genes switch from quiescent to transcriptionally active states (42) along with the finding of upregulated chromatin remodeling gene transcripts, one can argue that the constitutive chromatin remodeling emerging during AC is an upstream (epigenetic?) regulator of the transcription state upon DeAC that leads to cytoprotective cellular acclimatory memory.
Supplementary Material
GRANTS
This study was supported by the USA-Israel Binational Fund BSF Grant 2003-298, Israel Science Foundation ISF Grant 321/06, and (in part) by the Intramural Research Program of the NIH National Institute on Aging. The PTI and the Video motion detector systems were granted by Israel Science Foundation and the Hebrew University.
Footnotes
The online version of this article contains supplemental material.
REFERENCES
- 1.Arieli Y, Eynan M, Gancz H, Arieli R, Kashi Y. Heat acclimation prolongs the time to central nervous system oxygen toxicity in the rat. Possible involvement of HSP72. Brain Res 962: 15–20, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Barco A, Bailey CH, Kandel ER. Common molecular mechanisms in explicit and implicit memory. J Neurochem 97: 1520–1533, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev 15: 496–506, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Cohen O, Kanana H, Zoizner R, Gross C, Meiri U, Stern MD, Gerstenblith G, Horowitz M. Altered Ca2+ handling and myofilaments desensitization underlie cardiomyocyte performance in normothermic and hyperthermic heat acclimated rat hearts. J Appl Physiol 103: 266–275, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Duncan RF. Inhibition of Hsp90 function delays and impairs recovery from heat shock. FEBS J 272: 5244–5256, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Eynan M, Knubuvetz T, Meiri U, Navon G, Gerstenblith G, Bromberg Z, Hasin Y, Horowitz M. Heat acclimation-induced elevated glycogen, glycolysis, and low thyroxine improve heart ischemic tolerance. J Appl Physiol 93: 2095–2104, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Gagne J, Gelinas S, Martinoli M, Foster T, Ohayon M, Thompson R, Baudry M, Massicotte G. AMPA receptor properties in adult rat hippocampus following environmental enrichment. Brain Res Mol Brain Res 799: 16–25, 1998. [DOI] [PubMed] [Google Scholar]
- 9.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gracey AY, Fraser EJ, Li W, Fang Y, Taylor RR, Rogers J, Brass A, Cossins AR. Coping with cold: an integrative, multitissue analysis of the transcriptome of a poikilothermic vertebrate. Proc Natl Acad Sci USA 101: 16970–16975, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffiths EJ, Stern MD, Silverman HS. Measurement of mitochondrial calcium in single living cardiomyocytes by selective removal of cytosolic indo 1. Am J Physiol Cell Physiol 273: C37–C44, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Hashikawa N, Sakurai H. Phosphorylation of the yeast heat shock transcription factor is implicated in gene-specific activation dependent on the architecture of the heat shock element. Mol Cell Biol 24: 3648–3659, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hess U, Gall C, Granger R, Lynch G. Differential patterns of c-fos mRNA expression in amygdale during successive stages of odor discrimination learning. Learn Mem 4: 262–283, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Hirota K, Hasemi T, Yamada T, Mizuno KI, Hoffman CS, Shibata T, Ohta K. Fission yeast global repressors regulate the specificity of chromatin alteration in response to distinct environmental stresses. Nucleic Acids Res 32: 855–862, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Høj A, Jakobsen BK. A short element required for tuning off heat shock transcription factor: evidence that phosphorylation enhances deactivation. EMBO J 13: 2617–2624, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hori K, Ishigaki T, Koyama K, Kaya M, Tsujita J, Hori S. Adaptive changes in the thermogenesis of rats by cold acclimation and deacclimation. Jpn J Physiol 48: 505–508, 1998. [DOI] [PubMed] [Google Scholar]
- 17.Horowitz M Acclimatization of rats to moderate heat: body water distribution and adaptability of the submaxillary salivary gland. Pflügers Arch 366: 173–176, 1976. [DOI] [PubMed] [Google Scholar]
- 18.Horowitz M Heat acclimation and cross-tolerance against novel stressors: genomic-physiological linkage. In: The Neurobiology of Hyperthermia (Progress in Brain Res, vol. 162), edited by Sharma HS. Elsevier Science. Chapt. 18, p. 373–393, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Horowitz M, Argov D, Mizrahi R. Interrelationships between heat acclimation and salivary cooling mechanism in conscious rats. Comp Biochem Physiol A 74: 945–949, 1983. [DOI] [PubMed] [Google Scholar]
- 20.Horowitz M, Eli-Berchoer L, Wapinski I, Friedman N, Kodesh E. Stress-related genomic responses during the course of heat acclimation and its association with ischemic-reperfusion cross-tolerance. J Appl Physiol 97: 1496–1507, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Kamat CD, Thorpe JE, Shenoy SS, Ceriello A, Green DE, Warnke LA, Ihnat MA. A long-term “memory” of HIF induction in response to chronic mild decreased oxygen after oxygen normalization. BMC Cardiovasc Disord 7: 4, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kupatt C, Dessy C, Hinkel R, Raake P, Daneau G, Bouzin C, Boekstegers P, Feron O. Heat shock protein 90 transfection reduces ischemia-reperfusion-induced myocardial dysfunction via reciprocal endothelial NO synthase serine 1177 phosphorylation and threonine 495 dephosphorylation. Arterioscler Thromb Vasc Biol 24: 1435–1441, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685, 1970. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Zeng SX, Landais I, Lu H. Human SSRP1 has Spt16-dependent and -independent roles in gene transcription. J Biol Chem 282: 6936–6945, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Lyons TP, Muza SR, Rock PB, Cymerman A. The effect of altitude pre-acclimatization on acute mountain sickness during reexposure. Aviat Space Environ Med 66: 957–962, 1995. [PubMed] [Google Scholar]
- 26.Maloyan A, Eli-Berchoer L, Semenza GL, Gerstenblith G, Stern MD, Horowitz M. HIF-1alpha-targeted pathways are activated by heat acclimation and contribute to acclimation-ischemic cross-tolerance in the heart. Physiol Genomics 23: 79–88, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Maloyan A, Horowitz M. β-Adrenergic signaling and thyroid hormones affect HSP72 expression during heat acclimation. J Appl Physiol 93: 107–115, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Maloyan A, Palmon A, Horowitz M. Heat acclimation increases the basal HSP72 level and alters its production dynamics during heat stress. Am J Physiol Regul Integr Comp Physiol 276: R1506–R1515, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Maruyama M, Hara T, Katakura M, Hashimoto M, Haque A, Li G, Shido O. Contribution of the suprachiasmatic nucleus to the formation of a time memory for heat exposure in rats. J Physiol Sci 57: 107–114, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Montminy MR, Sevarino KA, Wagner JA, Mandel G, Goodman RH. Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc Natl Acad Sci USA 83: 6682–6686, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandolf KB. Adaptation to exercise in the heat: time course of heat acclimation and its decay. Intl J Sport Med 19, Suppl 2: S157–S160, 1998. [DOI] [PubMed] [Google Scholar]
- 32.Pandolf KB, Burse RL, Goldman RF. Role of physical fitness in heat acclimatization decay and reinduction. Ergonomics 20: 399–408, 1977. [DOI] [PubMed] [Google Scholar]
- 33.Patberg KW, Rosen MR. Molecular determinants of cardiac memory and their regulation. J Mol Cell Cardiol 36: 195–204, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Pierau F, Sann H, Yakimova K, P H. Plasticity of hypothalamic temperature-sensitive neurons. Prog Brain Res 115: 63–84, 1998. [DOI] [PubMed] [Google Scholar]
- 35.Pollak DD, Scharl T, Leisch F, Herkner K, Villar SR, Hoeger H, Lubec G. Strain-dependent regulation of plasticity-related proteins in the mouse hippocampus. Behav Brain Res 165: 240–246, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Rattiner L, Davis M, Ressler K. Differential regulation of brain-derived neurotrophic factor transcripts during the consolidation of fear learning. Learn Mem 11: 727–731. [DOI] [PubMed] [Google Scholar]
- 37.Samali A, Orrenius S. Heat shock proteins: regulators of stress response and apoptosis. Cell Stress Chaperones 3: 228–236, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwimmer H, Gerstberger R, Horowitz M. Heat acclimation affects the neuromodulatory role of AngII and nitric oxide during combined heat and hypohydration stress. Brain Res Mol Brain Res 130: 95–108, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Schwimmer H, Gerstberger R, Horowitz M. Nitric oxide and angiotensin II: neuromodulation of thermoregulation during combined heat and hypohydration stress. Brain Res 1006: 177–189, 2004. [DOI] [PubMed] [Google Scholar]
- 40.Silverman HS, Wei S, Haigney MC, Ocampo CJ, Stern MD. Myocyte adaptation to chronic hypoxia and development of tolerance to subsequent acute severe hypoxia. Circ Res 80: 699–707, 1997. [DOI] [PubMed] [Google Scholar]
- 41.Sorger PK, Lewis MJ, HRP. Heat shock factor is regulated differently in yrast and HeLa cells. Nature (London) 329: 81–84, 1987. [DOI] [PubMed] [Google Scholar]
- 42.Turner BM. Chromatin and Gene Regulation: Molecular Mechanisms in Epigenetics. Blackwell Science, Chapt. 6, p. 101–125, 2001. [Google Scholar]
- 43.Uffenbeck SR, Krebs JK. The role of chromatin structure in regulating stress-induced transcription in Saccharomyces cerevisiae. Biochem Cell Biol: 5244–5256, 2005. [DOI] [PubMed] [Google Scholar]
- 44.Xi L, Chelliah J, Nayeem MA, Levasseur JE, Hess ML, Kukreja RC. Whole body heat shock fails to protect mouse heart against ischemia/reperfusion injury: role of 72 kDa heat shock protein and antioxidant enzymes. J Mol Cell Cardiol 30: 2213–2227, 1998. [DOI] [PubMed] [Google Scholar]
- 45.Xia Y, Haddad G. Effect of prolonged O2 deprivation on Na+ channels: differential regulation in adult versus fetal rat brain. Neuroscience 94: 1231–1241, 1999. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J, Schmid T, Frank R, Brune B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1alpha from pVHL-independent degradation. J Biol Chem 279: 13506–13513, 2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.