

Abstract
Hereditary hemorrhagic telangiectasia (HHT) is a rare autosomal dominant hereditary disorder characterized by recurrent spontaneous epistaxis, mucocutaneous telangiectasias, and solid organ arteriovenous malformations (AVMs). Pulmonary hypertension (PH) is an increasingly recognized complication in patients with HHT, most often precipitated by high‐output heart failure in the presence of hepatic AVMs as well as pulmonary arterial hypertension in the form of a proliferative vasculopathy. The presence of PH in patients with HHT is associated with significant elevations in rates of morbidity and mortality. Additionally, there is growing recognition of a thromboembolic propensity in this population that increases the risk of chronic thromboembolic PH, posing unique clinical considerations regarding the use of anticoagulation. Patients with HHT are also at risk of PH due to disorders commonly seen in the general population, including left‐sided heart and lung disease. The etiology of PH in HHT is multifaceted and complex; the diagnostic approach and treatment strategies must consider the underlying pathophysiology of HHT. This comprehensive review summarizes current knowledge of PH in HHT, detailing the pathogenesis of known etiologies, diagnostic evaluation, and suggested treatment modalities as well as emerging therapies that may be of future interest.
Keywords: chronic thromboembolic pulmonary hypertension, hereditary hemorrhagic telangiectasia, high‐output heart failure, pulmonary arterial hypertension, pulmonary hypertension
Abbreviations
- AVM
arteriovenous malformation
- BMP
bone morphogenetic protein
- CI
cardiac index
- CO
cardiac output
- CSA
cross‐sectional area
- CTEPH
chronic thromboembolic pulmonary hypertension
- GDF
growth differentiation factor
- HHT
hereditary hemorrhagic telangiectasia
- HOHF
high‐output heart failure
- LVOT
left ventricular outflow tract
- mPAP
mean pulmonary arterial pressure
- PAH
pulmonary arterial hypertension
- PAWP
pulmonary arterial wedge pressure
- PH
pulmonary hypertension
- PVR
pulmonary vascular resistance
- RHC
right heart catheterization
- VTI
velocity time integral
HEREDITARY HEMORRHAGIC TELANGIECTASIA
Hereditary hemorrhagic telangiectasia (HHT) is a rare autosomal dominant hereditary disorder with an estimated incidence of 1–2 cases per 10,000 individuals that results in systemic vascular dysplasia. 1 , 2 The genetic disorder leads to mucocutaneous telangiectasias and visceral arteriovenous malformations (AVMs), producing a phenotypically diverse clinical syndrome that ranges from recurrent epistaxis to critical solid organ bleeds. Pulmonary hypertension (PH) is a chronic, progressive condition that was initially described in patients with HHT in 1969 and is increasingly recognized as a clinical complication of the disorder, with an estimated prevalence of 8%–40%. 3 , 4 , 5 , 6 , 7 Although several etiologies of HHT‐related PH have been characterized, appropriate clinical evaluation and management remain incompletely defined. This comprehensive review summarizes current knowledge of PH in HHT, detailing mechanisms of pathogenesis for known etiologies, diagnostic evaluation, and suggested therapies for this clinical entity.
Pathogenesis
HHT is a genetically heterogenous disorder in which all known mutations occur in genes encoding proteins of the transforming growth factor beta (TGF‐β) signaling pathway. 8 Ligand‐binding induces complexing of a membrane glycoprotein, endoglin, with multiple receptor serine/threonine kinases such as activin receptor‐like kinase 1 (ALK1). This leads to phosphorylation of a group of receptor‐regulated SMAD proteins—critical downstream effectors. The SMAD proteins complex with co‐factor SMAD4 and migrate to the cell nucleus to function as transcription factors. Signal transduction also occurs via interactions with other classes of proteins within the TGF‐β superfamily, including the bone morphogenetic protein (BMP) and growth differentiation factor (GDF) classes. Among the former, BMP9 and BMP10 are especially involved in endoglin/ALK1 molecular interactions, with pathogenic mutations linked to vascular anomalies. 9 There is a high degree of expression of the proteins of the TGF‐β signaling pathway in the endothelium, promoting vascular integrity. Pathologic defects disrupt angiogenetic mechanisms and impair endothelial remodeling, resulting in fragile, chronically dilated vessels in the form of telangiectasias and AVMs.
HHT type 1 (HHT1) is due to mutations in endoglin (ENG, chromosome 9q34), while HHT2 is caused by mutations in ALK1 (ACVRL1, chromosome 12q13), a receptor serine/threonine kinase. 10 , 11 Mutations in SMAD4 (MADH4, chromosome 18q21) result in combined Juvenile Polyposis/HHT. 12 Up to 85% of causative mutations of HHT are found in ENG and ACVRL1, with the frequency exceeding 95% when restricting observations to patients with definite HHT based on established diagnostic criteria. 13 Causative mutations in this group of patients with HHT are predominantly observed in ENG (50%–60%) followed by ACVRL1 (30%–40%) and MADH4 (2%–5%). Hundreds of pathogenic mutations have been identified; in cases of HHT1, HHT2, and combined Juvenile Polyposis/HHT, underlying mutations primarily lead to haploinsufficiency.
The genetic basis of the remaining cases of HHT is not well characterized. Linkage analysis has demonstrated HHT loci at chromosome 5q31 (HHT3) and chromosome 7p14 (HHT4), although the specific genes remain unknown. 14 , 15 , 16 , 17 Mutations in BMP9 (GDF2, chromosome 10q11) have been associated with HHT‐like symptoms (HHT5). 18 The identified causative mutations of HHT express variable penetrance depending on site and type; some mutations in the same gene may not be linked to the development of HHT at all. For instance, different GDF2 mutations result in varying measurable plasma BMP9 and BMP10 levels with or without clinical evidence of HHT. 19 This highlights the role of epigenetic, environmental, and other factors in mechanisms of HHT development. The current gene HHT panel tests for pathologic mutations in ACVRL1, ENG, SMAD4, and GDF2 as well as RASA1 and EPHB4. Genetic alterations in the RASA1 gene, which encodes a protein in the RAS‐MAPK (mitogen‐activated protein kinase) signal transduction pathway, have been implicated in the development of HHT, although stronger evidence suggests associations with the HHT‐mimic capillary malformation‐arteriovenous malformation syndrome. 20 Mutations in EPHB4, a transmembrane tyrosine kinase receptor, also result in capillary malformation‐arteriovenous malformation syndrome type II. 21 Novel mutations in the bone morphogenetic protein receptor type II (BMPR2, chromosome 2q33), which is an important pathogenic genetic factor in the development of heritable PH, have been linked to HHT. 22 Mutations in the ADAM17 and PTPN14 genes have been identified as genetic modifiers of angiogenetic processes in patients with HHT, with pathogenic variants resulting in diversity of phenotype and clinical severity. 13
Clinical characteristics
Clinical manifestations of HHT exhibit age‐related penetrance. 10 , 11 , 12 Patients gradually develop recurrent spontaneous epistaxis, mucocutaneous telangiectasias, and solid organ AVMs. Visceral AVMs are most commonly present in the liver, lungs, gastrointestinal tract, and brain. The sequelae of these malformed, dilated vessels include critical hemorrhage, symptomatic iron deficiency anemia, and hemorrhagic and/or ischemic stroke. Although onset and presentation can vary, clinical diagnosis can be made in an adult patient based on the presence of four Curaçao criteria: (1) spontaneous, recurrent epistaxis, (2) a first‐degree relative with HHT, (3) multiple cutaneous or mucosal telangiectasias, and (4) visceral AVMs. 23 , 24 , 25 The presence of three or more criteria confers a definite diagnosis, two criteria elicit a possible diagnosis, and one or less criteria suggest an unlikely diagnosis of HHT.
Although a negative result is not exclusionary, as approximately 10‐15% of patients with HHT have no identified pathogenic mutation, genetic testing should be conducted in all suspected cases of HHT. 13 Not only does genetic testing guide screening and management (e.g., colonoscopy in combined Juvenile Polyposis/HHT), but there is substantial evidence of phenotypic variation by molecular subtype, most notably in regard to the relative frequency and distribution of visceral AVMs. In patients with HHT, up to 75% develop hepatic AVMs, up to 45% have pulmonary AVMs, and up to 10% demonstrate cerebral AVMs. 26 , 27 Pulmonary and cerebral AVMs are more common in patients with HHT1, while hepatic manifestations are more often observed in HHT2. In the case of pulmonary and hepatic AVMs, clinical features may also be a consequence of arteriovenous shunting, including cerebral stroke, portal hypertension, and ischemic cholangiopathy.
PULMONARY HYPERTENSION
PH is a heterogeneous group of chronic, progressive hemodynamic disorders of different etiologies characterized by increased blood pressure in the pulmonary arteries that over time may lead to right heart dysfunction. In general, PH is defined on right heart catheterization (RHC) by a mean pulmonary arterial pressure (mPAP) > 20 mmHg at rest. 28 , 29
Diagnostic evaluation
Patients with PH will often present with nonspecific progressive exertional dyspnea, peripheral edema, chest pain, or fatigue. Multiple diagnostic and radiologic findings may suggest an underlying diagnosis of PH. Electrocardiogram may demonstrate evidence of right atrial enlargement (e.g., P pulmonale) and right ventricular stress (e.g., right axis deviation and right ventricular strain). Radiographic imaging can show right heart and pulmonary artery enlargement and etiology‐specific findings, such as signs of left‐sided heart disease, chronic pulmonary emboli, or parenchymal lung disease. Echocardiogram is a vital component of evaluation, allowing the assignment of an initial level of probability of PH using a comprehensive sum of parameters. Findings may include an elevated peak tricuspid regurgitation velocity which, when combined with noninvasive estimates of right atrial pressure, can also demonstrate increased right ventricular systolic pressure. Additional morphologic and functional variables that may aid in the evaluation of PH include an increased right atrial area, right ventricular dilatation, interventricular dependence, decreased tricuspid annular plane systolic excursion, and decreased tricuspid annular plane systolic excursion‐systolic pulmonary arterial pressure ratio.
Echocardiogram can also provide a noninvasive estimation of cardiac output (CO) with good correlation to thermodilution‐derived CO measurements, which may be particularly helpful in patients in a high‐flow state. 30 CO is defined as , where SV is stroke volume and HR is heart rate. SV is approximated in echocardiogram as a cylinder of blood volume leaving the left ventricle via the left ventricular outflow tract (LVOT), such that , where CSA is the cross‐sectional area and VTI is the velocity time integral. CSA is clinically obtained by measurement of the diameter of the LVOT: . The diameter of the LVOT is measured in the parasternal long‐axis view of the aortic valve at mid‐systole, and VTI is obtained in the apical five‐chamber view through pulsed wave doppler interrogation of the LVOT.
RHC is the gold standard for diagnosis and classification of PH. Important hemodynamic variables include pulmonary vascular resistance (PVR), pulmonary arterial wedge pressure (PAWP), and CO and cardiac index (CI). PH can be divided into precapillary and postcapillary PH. Precapillary PH is a result of pulmonary vascular remodeling leading to increased vascular resistance and is hemodynamically defined by mPAP > 20 mmHg, PAWP ≤ 15 mmHg, and PVR > 2 Wood units (WUs). Postcapillary PH is caused by increased pulmonary venous pressure, such as in left‐sided heart disease, and is defined as mPAP > 20 mmHg, PAWP > 15 mmHg, and PVR ≤ 2 WUs, and combined pre‐ and postcapillary PH is defined as an mPAP > 20 mmHg, PAWP > 15 mmHg, and PVR > 2 WUs. 28 PH is also defined by etiology, being divided into one of five clinical classifications (groups 1–5) based on underlying pathophysiology and characteristics of the condition. 29 Therefore, additional studies such as pulmonary function tests, imaging studies of the liver and lungs, and ventilation/perfusion lung scans are part of a complete diagnostic workup of PH. Precapillary PH is generally compromised of clinical group 1 (pulmonary arterial hypertension [PAH]) and group 3 (associated with lung diseases and/or hypoxia), while postcapillary PH is typically a result of clinical group 2 (associated with left‐sided heart disease and high CO states). Clinical group 4 (associated with pulmonary artery obstructions) and group 5 (unclear and/or multifactorial mechanisms) may exhibit both hemodynamic classifications.
Hereditary hemorrhagic telangiectasia
PH is an increasingly recognized clinical phenomenon in patients with HHT, although there are currently no recommendations for PH screening in this population. 25 A sizeable portion of studies rely on transthoracic echocardiographic data with no subsequent RHC to diagnose PH. There is also a reported lack of systematic echocardiographic examinations in this patient population. Investigations may also be subject to referral bias given the advanced care required to manage patients with this rare disease. Therefore, true prevalence is currently unknown. Nonetheless, the number off hospitalizations of patients with both HHT and PH exhibited a relative increase of over 200% between 2000 and 2014. 31 The age at presentation is highly etiology‐specific. Patients with HHT and PH are disproportionately more likely to be women, even when compared to the baseline female sex predilection observed in studies of HHT. 31 Concomitant HHT and PH are associated with worse overall age‐adjusted survival when compared to patients with HHT and no PH. 3 , 5 Inpatient mortality is also higher with more comorbidity burden, namely congestive heart failure, liver disease, and blood loss or iron deficiency anemia. 31 While elevated mortality in patients with HHT and PH is multi‐factorial, the underlying mechanism seems to be related to advanced PH and right‐sided heart failure.
In patients with HHT‐related PH, the two most common etiologies are, in order of prevalence, postcapillary PH secondary to high‐output heart failure (HOHF) in the presence of hepatic AVMs and anemia followed by PAH in the form of a proliferative, obliterative plexiform vasculopathy of the small pulmonary arteries as a result of pathogenic gene mutations. 4 Both clinical phenotypes are more common in HHT2; in the case of HHT‐related PAH, patients almost exclusively exhibit ACVLR1 mutations. HHT‐mediated states of hypercoagulability resulting in chronic thromboembolic pulmonary hypertension (CTEPH) have been rarely observed. 32 Moreover, other types of PH—not necessarily related to HHT—may exist in these patients as well. In patients with HHT, up to 70% can exhibit clinical signs of left‐sided heart disease and up to 20% may demonstrate lung disease. 33 Multiple clinical etiologies of PH per individual have been observed in studies of patients with HHT. 34 Thus, a complete hemodynamic profiling of patients with HHT presenting with signs and symptoms suggestive of PH is required. An algorithm to assess PH in patients with HHT is presented to augment established diagnostic evaluations (Figure 1).
Figure 1.
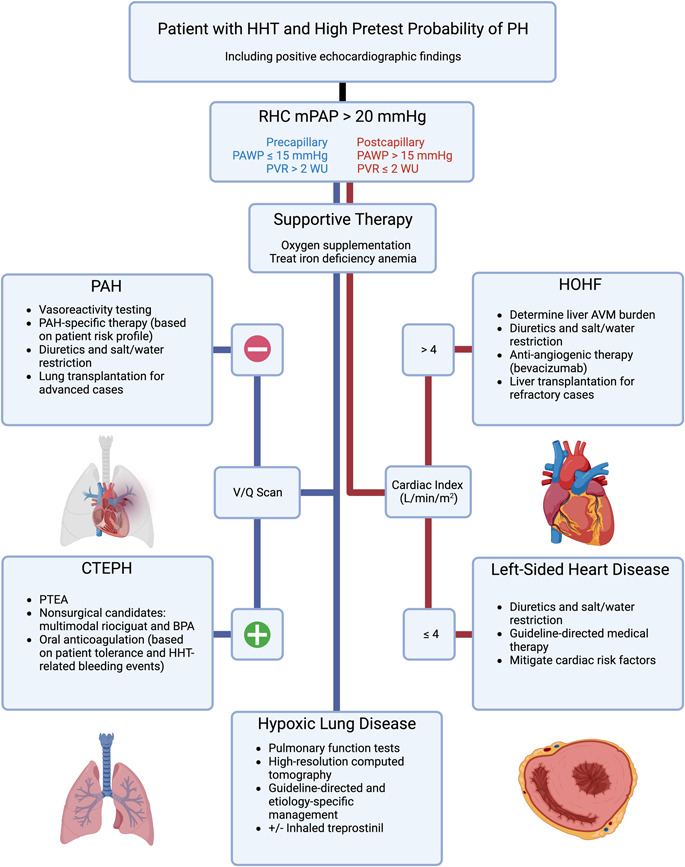
A practical algorithm to assist in the detection of and treatment for pulmonary hypertension (PH) in patients with hereditary hemorrhagic telangiectasia (HHT). Etiologies of precapillary PH in patients with HHT are in blue text and/or on the left‐hand flowchart line, while etiologies of postcapillary PH are in red text and/or on the right‐hand flowchart line. The latter is divided by cardiac index, in which a value greater than 4 L/min/m2 indicates a high output state. 35 , 36 Recommendations are on the basis of the most recently established guidelines for the treatment of PH in the general population as well as for the management of complications of HHT. 25 , 28 In PH due to hypoxic lung disease, inhaled treprostinil may be indicated in cases of pathologic interstitial subtypes. 37 AVM, arteriovenous malformation; BPA, balloon pulmonary angioplasty; CTEPH, chronic thromboembolic pulmonary hypertension; HOHF, high‐output heart failure; mPAP, mean pulmonary artery pressure; PAH, pulmonary arterial hypertension; PAWP, pulmonary arterial wedge pressure; PTEA, pulmonary thromboendarterectomy; PVR, pulmonary vascular resistance; RHC, right heart catheterization; V/Q, ventilation/perfusion lung scan; WU, Wood unit.
Right heart catheterization in hereditary hemorrhagic telangiectasia
There are considerations to be appraised when performing and evaluating the results of RHC in patients with HHT. All patients undergoing RHC should receive a thorough evaluation to assess the type of PH, including determination of hemodynamic profile as well as evaluation for right‐to‐left shunting. Measurements of right atrial pressure, pulmonary arterial pressures and mPAP, PAWP, and CO/CI should be performed. For evaluation of PH, the thermodilution method of CO/CI measurement is recommended. 38 In patients with evidence of a high CO state and/or a central mixed venous saturation greater than 70%, an oxygen saturation run study should be conducted in which oxygen saturations are assessed at the level of the pulmonary artery, right ventricle, right atrium, superior vena cava, and inferior vena cava. In patients with HOHF due to liver AVMs, a step‐up in oxygen saturation within the inferior vena cava at the level of the liver would be present. Additional complications to be considered when performing RHC in patients with HHT include the difficulty in laying some patients flat or in raising the legs due to exacerbation of epistaxis. In addition, in patients with suspected HOHF from a liver or abdominal AVM, a femoral approach may be required to obtain a detailed saturation study at varying levels in the inferior vena cava, if indicated.
HIGH‐OUTPUT HEART FAILURE
HOHF is a rare clinical phenotype of heart failure associated with decreased systemic vascular resistance and elevated CI on RHC (>4 L/min/m2), resulting in signs and symptoms of diffuse venous congestion. 35 , 36 Etiologies can be broadly classified into either a pathologic state of increased metabolic demand (e.g., hyperthyroidism and myeloproliferative disorders) or a disorder of arteriocapillary bypass resulting in increased flow into venous circulation (e.g., liver disease and arteriovenous shunts). Postcapillary PH in HOHF is due to high pulmonary flow; although PH in HOHF technically falls within PH clinical group 2 due to elevated PAWP pressures, the disease state is not inherently a disorder of the left ventricle and evaluation as well as management for this phenotype of heart failure can significantly differ from the standard approach.
Pathophysiology and presentation
Liver AVMs can be present in up to 75% of patients with HHT but manifest with clinical signs and symptoms in less than 10% of patients. 39 AVMs of the body and extremities can be classified based on angioarchitecture, which morphologically separates AVMs into the feeding arteries, the network of arteriovenous shunts (referred to as the nidus), and the draining veins. AVMs bypass high‐resistance arteriocapillary beds and lead to increased flow of blood to the right side of the heart. In the case of the liver, AVMs can result in three patterns of shunting: hepatic arteries to hepatic veins, hepatic arteries to portal veins, and portal veins to hepatic veins.
In instances of shunting of oxygenated blood from the hepatic artery to the hepatic veins, there is decreased effective hepatic perfusion. Increased oxygen demand and decreased systemic vascular resistance along with reduced hepatic and renal perfusion results in the activation of neurohormonal cascades, including the renin‐angiotensin‐aldosterone system and the adrenergic nervous system. The consequent elevated CO leads to enhanced venous return and cardiac filling pressures which may, over time, manifest in cardiac failure once the pump capacity of the left ventricle is exceeded. This hyperdynamic circulation also facilitates backward transmission of blood flow and pressure that can supersede the distensibility of the pulmonary vasculature to cause postcapillary PH via endothelial shear stress and pulmonary venous congestion. Chronic progression of left‐sided heart dysfunction and PH can contribute to the onset of atrial fibrillation as well as right heart failure. Of note, the volume of blood involved in arteriovenous shunting typically exceeds 20% of the CO to produce such hemodynamic disorders. 40
While the majority of hemodynamic profiles of patients with HHT and PH secondary to HOHF demonstrate isolated postcapillary PH, a considerable number of studies reports combined pre‐ and postcapillary PH. 41 , 42 , 43 This may be due to mild pulmonary arterial remodeling in the setting of chronic exposure to increased blood flow. However, investigations have also highlighted an unmasking of heritable pulmonary arteriopathy following successful treatment of initial HOHF symptoms as well as cases of portopulmonary hypertension concomitant with HOHF‐mediated PH. 42 , 44 In the case of the latter, patients have presented with symptoms of HOHF as well as hepatic encephalopathy. 45 Furthermore, one study has shown that 40% of patients with HHT have elevated ammonia levels. 46 Elevated ammonia levels were significantly associated with the presence of hepatic AVMs as well as elevated right ventricular systolic pressure; elevated serum ammonia provided an 80% sensitivity and 71% specificity in the prediction of PH in this cohort. These findings affirm that portopulmonary hypertension may serve as a contributory component in cases of HOHF‐mediated PH with combined pre‐ and postcapillary PH.
Clinically, patients with HOHF will present with signs and symptoms of volume overload (e.g., orthopnea, exertional dyspnea, and ascites). In patients with HHT, this is often also accompanied by worsening epistaxis and increased transfusion requirements. Patients will usually be female and in the fifth to seventh decades of life. 47 Therefore, HOHF and HOHF‐mediated PH are unlikely to be the initial clinical manifestation of HHT. In the event of a patient with no relevant medical history presenting with HOHF and evidence of liver shunts, the clinical provider should firstly consider a diagnosis of HHT; the presence of a congenital extrahepatic portosystemic shunt, though rare, would be an alternative diagnosis. As noted, liver AVMs are far more common in HHT2. Thus, patients with HHT presenting with HOHF will most often possess mutations in ACVRL1, although some reports have demonstrated HOHF‐mediated PH in patients with pathogenic ENG variants. 48 , 49
Evaluation, treatment, and outcomes
Clinical findings of heart failure combined with evidence of a high CO state, most often via echocardiogram, yield the diagnosis of HOHF. However, determination of underlying etiology is paramount. In the case of patients with HHT, almost all individuals will have a confirmed history of the disorder due to prior phenotypic manifestations. Thus, both Doppler ultrasound and abdominal computed tomography can be subsequently employed to confirm the presence of liver AVMs. RHC will show elevated CO/CI and, most often, postcapillary PH. Central venous oxygen saturation is often elevated and an oxygen saturation step‐up at the level of the inferior vena cava will confirm shunting at the level of hepatic AVM. The presence of combined pre‐ and postcapillary PH can indicate chronic HOHF or concomitant etiologies of PAH, which may need further evaluation.
Moreover, the presence of underlying hyperthyroidism must also be considered in any patient with high CO. Hyperthyroidism induces systemic vasodilation and increases metabolic demand. This disease state has been linked with the onset of precapillary as well as postcapillary PH. 35 Because thyroid dysfunction is relatively common in the hospital setting, elucidating its exact etiology in patients with high CO is paramount. This may be complicated by the effects of medical therapies with known alterations to thyroid function, such as intravenous diuretics or prostanoid therapies. 50 , 51 , 52 Dysregulated levels of thyroid‐stimulating hormone, T4 and T3 can obscure the identification of the pathogenic mechanism of high CO. It is suggested that the clinician guide suspicions based on the overt presence of signs and symptoms of thyrotoxicosis. While there may be a benefit to maintaining a low threshold to treat hyperthyroidism, a high CO state in patients with HHT should primarily prompt further evaluation for hepatic AVMs, which are a more likely etiology and have effective therapies for management.
Initial management consists of diuretic therapy with salt and water restriction as well as oxygen supplementation as needed. There is no systematic appraisal of the efficacy of antihypertensive or antiarrhythmic therapies. Vasodilators and inotropes may exacerbate the hyperdynamic state. All patients should be evaluated for iron deficiency anemia as the subsequent low blood viscosity and increased metabolic demand can exacerbate HOHF. If present, the patient can be treated with oral iron supplementation. Intravenous iron replacement may be considered for patients in whom oral repletion is not effective or tolerated. Intravenous bevacizumab is an antiangiogenic inhibitor of vascular endothelial growth factor A. Currently, the use of bevacizumab is indicated for various primary and metastatic malignancies as per guidelines of the Food and Drug Administration. However, bevacizumab has also successfully been employed for epistaxis and gastrointestinal bleeding in patients with HHT. 53 , 54 , 55 , 56 , 57 Additionally, the 2020 International HHT Guidelines recommend the use of bevacizumab for patients with HHT and symptomatic HOHF due to liver AVMs who have failed to respond to initial management. 25
A single‐center, phase II trial of 25 patients with confirmed HHT, severe liver involvement, and a high CI investigated response to intravenous bevacizumab. Results indicated normalization of CI in five and partial response in 15 patients at 6 months of follow‐up. Rates of epistaxis also markedly improved. 58 In a survey of 31 centers treating HHT, encompassing 150 patients, bevacizumab was reported to result in significant improvement in CI and HOHF symptoms in 55% of cases; however, sustained normalization of cardiac parameters was not common. 59 Adverse events secondary to therapy occurred in less than 10% of cases. Relapse following bevacizumab discontinuation remains a concern, but there is a paucity of data regarding its incidence. Per recommendations, the induction regimen for bevacizumab therapy involves administration of six doses every 2 weeks at a dose of 5 mg/kg. 25 The maintenance regimen may vary between patients, but generally involves one dose every 4–6 months at a dose of 5 mg/kg.
The only known curative treatment of HOHF secondary to liver AVMs is orthotopic liver transplantation. Consideration for liver transplantation is recommended for patients with HHT who have refractory HOHF despite bevacizumab therapy, biliary ischemia, or complicated portal hypertension. 25 Following transplantation, 5‐ and 10‐year survival rates are estimated to be greater than 80%. 60 , 61 Although recurrence of hepatic involvement was considered to be rare, a recent study of 14 patients demonstrated a cumulative 47.9% recurrence risk at 15 years after a median of 127 months. 62 However, these recurrent hepatic vascular lesions remained largely asymptomatic. It is recommended that a model for end‐stage liver disease exception score of 22 be supplied for patients with HHT and intractable heart failure. 63 Currently, it is also recommended that liver transplantation can be undertaken even in the presence of PH if the PVR is less than 3 WUs. In a systematic review of 22 patients with HHT and HHT‐related HOHF who underwent orthotopic liver transplantation, hemodynamic parameters, and symptoms improved or normalized in all but two patients. 64 Furthermore, one patient with combined pre‐ and postcapillary PH (PVR > 3 WUs) demonstrated normalization of parameters following transplantation. The suspected etiology of the patient's presentation was HOHF and portopulmonary hypertension. This suggests cases of PH with combined pre‐ and postcapillary components in this population may require additional appraisal regarding the utility of liver transplantation. Other invasive treatments, including hepatic artery ligation or embolization, are not recommended as they are temporizing measures with significant morbidity and mortality. 25
PULMONARY ARTERIAL HYPERTENSION
PAH is a rare hemodynamic disorder typified by elevated pulmonary arterial pressures and vascular resistance as a result of proliferative vasculopathy (clinical group 1). PAH can be idiopathic, heritable, or associated with certain drugs and toxins as well as various disease states (e.g., connective tissue diseases, portal hypertension, and congenital heart disease). 28 Rare etiologies of PAH include pulmonary veno‐occlusive disease, characterized by chronic, progressive obstruction of small pulmonary veins, and pulmonary capillary hemangiomatosis, distinguished by abnormal pulmonary capillary proliferation. PAH is a precapillary PH that can result in right‐sided cardiac enlargement, dysfunction, and heart failure.
Pathophysiology and presentation
Idiopathic and heritable PAH are clinically and histologically indistinguishable; the latter entity is designated when heritable genetic alterations implicated in the onset of PAH are identified in an individual. The pathophysiology of PAH involves uncontrolled endothelial and smooth muscle cell proliferation with dysregulated programmed cell death, vessel fibrosis, and microthrombi formation. These processes result in vessel narrowing and elevation of pulmonary arterial pressures that impart a burden on the right ventricle, ultimately exceeding the compensatory mechanisms of the chamber to lead to clinical heart failure. Pathologic findings in PAH correlate to disease severity, starting with medial hypertrophy and intimal hyperplasia of pulmonary arterioles, transitioning to collagenous replacement and fibrosis of the intimal cells, and concluding with the gradual development of plexiform lesions. 65
Up to 80% of cases of heritable PAH are due to pathogenic mutations in BMPR2, which encodes for the BMPR2 protein that is a member of the TGF‐β family and regulates endothelial cell proliferation. 9 , 66 Of the remaining cases, genetic alterations in ACVLR1, ENG, SMAD4, and GDF2 have been increasingly identified and, in many cases, are associated with patients with HHT. 28 , 67
Histologically, PAH in patients with HHT is indistinguishable from those with heritable PAH due to alterations in BMPR2, including the presence of intimal hyperplasia and end‐stage plexogenic lesions. 68 Observations of PAH in patients with HHT have underlined the pleiotropic nature of TGF‐β signaling proteins, which may promote divergent intimal growth and apoptotic processes that explain the presence of both occlusive and dilated vascular phenotypes in patients with HHT. 69 Overall, the penetrance of these heritable mutations in the development of PAH is 20%–30%. 66 , 70
Almost all cases of PAH in patients with HHT have been associated with pathogenic mutations in ACVLR1. 4 , 71 , 72 Reports of HHT‐related PAH associated with ENG mutations have typically lacked patient‐specific data, including results from RHC studies, and have often involved other precipitating factors, such as the concomitant use of dexfenfluramine or extensive stimulant abuse. 73 , 74 Therefore, patients with HHT1 exhibiting precapillary PH should be thoroughly evaluated to exclude other etiologies (e.g., CTEPH and congenital heart disease). Rarely, PAH has been identified in patients with HHT and mutations in GDF2 and BMPR2. 22 , 75 Alternative causes of PAH, including pulmonary capillary hemangiomatosis and portal hypertension, have also been observed in patients with HHT. 76 , 77
Clinically, patients with PAH will present with progressive exertional dyspnea and decreased exercise tolerance. This may be accompanied by orthopnea and edema in cases of right heart failure. Patients will usually be female and younger than patients with HHT who present with HOHF (typically in the second to fourth decades of life). 71 Some reports present PAH as the initial manifestation of HHT in pediatric patients. 72 , 78 , 79
Evaluation, treatment, and outcomes
Because PAH involves a chronic, nonspecific clinical course, a complete diagnostic workup is indicated. This includes imaging of the chest (demonstrating pruning of peripheral vasculature), pulmonary function tests, echocardiography, ventilation/perfusion lung scan, and RHC (indicating precapillary PH). The presence of liver AVMs or high CO/CI should be investigated. Vasoreactivity testing should be performed in heritable PAH to identify potential responders to calcium channel blocker therapy. Although understudied, patients with HHT and PAH have largely been found to have minimal pulmonary vasoreactivity. 80
Beyond supportive therapy (e.g., oxygen supplementation, diuretics, and iron replacement), the treatment of PAH in patients with HHT currently involves the use of standard PAH vasodilator therapies, including endothelin receptor antagonists, phosphodiesterase inhibitors, soluble guanylate cyclase stimulators, and prostanoids. Individual studies have reported success, either in normalization of hemodynamic parameters or improvement of functional status, with the use of bosentan, sildenafil, selexipag, epoprostenol, treprostinil, and iloprost. 81 , 82 , 83 , 84 , 85 , 86 , 87 One investigation included a cohort of five patients as well as an analysis of 32 subjects across 21 studies, all of whom were patients with HHT and PAH treated with oral or intravenous prostanoids. 87 Results showed significant improvement in mPAP (65 ± 19 pretreatment vs. 51 ± 16 mmHg posttreatment; p = 0.04) and PVR (12 ± 6 pretreatment vs. 8 ± 4 WUs posttreatment; p = 0.01) following therapy, with no significant difference in survival between those who received oral or intravenous medication.
The collection of studies assessing the efficacy of pulmonary vasodilator therapy in patients with HHT and PAH has highlighted the known side effects of such therapies that pose unique concerns in patients with HHT. Endothelin receptor antagonists (bosentan and macitentan, but not ambrisentan) and soluble guanylate cyclase stimulators increase the risk of anemia. 88 , 89 Phosphodiesterase inhibitors are associated with increased rates of epistaxis and prostanoids possess antiplatelet properties. 90 The frequency of such adverse events in this patient population is not yet characterized and should remain an important consideration when selecting therapy for PAH. Patients with HHT and PAH should have their therapeutic regimen tailored to their clinical needs and individual side effect profiles. Escalation of therapy is generally based on assessment of risk status (low, intermediate, or high), which is comprised of a combination of hemodynamic, clinical, exercise, and morphologic parameters to estimate 1‐year mortality. 91 Ideally, patients should be managed with oral pulmonary vasodilator therapy with the addition of one or more agents if they do not achieve a low‐risk PAH profile. Of note, parenteral prostacyclin therapy is usually reserved for patients with high‐risk features and progressive disease.
When the clinician is initiating pulmonary vasodilator therapy for PAH in patients with HHT, they should identify and document the baseline epistaxis in the patient. The Epistaxis Severity Score, developed from a prospective, survey‐based study of nine hundred participants from over 20 countries, is a six‐question evaluation tool, scored from 0 to 10, that aids in determining the current severity of the patient's nosebleeds. 92 More recently, the Nasal Outcome Score for Epistaxis has been published as a 29‐item instrument to describe the biopsychosocial components of patient epistaxis burden more adequately. 93 Both tools may assist in discerning the evolution of epistaxis severity during treatment for PAH in patients with HHT. Of note, clinicians should consider that epistaxis may be transiently exacerbated during periods of up‐titration of vasodilator therapy.
Lung transplantation is sought in patients with severe PAH refractory to standard therapy. One study has described successful double lung transplant in a patient with HHT and severe PAH, resulting in resolution of symptoms and normalization of hemodynamic parameters. 83 Another investigation reviewed the clinical outcomes of three female patients with HHT who received a double lung transplant for PAH. 94 Patients were 36, 19, and 14 years of age at the time of transplant, and were alive four, 196, and 224 months later, respectively. Mild epistaxis was the only reported bleeding complication post‐transplant.
Patients with HHT who develop PAH have elevated mortality rates, typically succumbing to cardiac disease, major bleeding events, and sepsis. 31 , 43 One‐year and 3‐year survival rates of these patients are 78% and 53%, respectively—lower than matched patients with idiopathic PAH. 95 Clinical outcomes in patients with mutations in the ACVRL1 gene may be even worse. Patients with HHT2 develop PAH at a significantly younger age than patients with heritable PAH due to BMPR2 mutation or with idiopathic PAH and exhibit a lower overall survival. 80
There are several emerging therapies with potential utility in the treatment of PAH in patients with HHT. Bevacizumab may be employed as adjunct therapy as it is hypothesized to reduce adverse bleeding events in patients receiving standard pulmonary vasodilators. Thalidomide is an oral medication employed in the treatment of several cancers. 96 Its mechanism of action is multifactorial, including enhancing expression of platelet‐derived growth factor‐B and reducing activity of vascular endothelial growth factor, resulting in normalization of vessel maturation. The administration of thalidomide has successfully mitigated significant adverse bleeding events associated with tadalafil and ambrisentan use in a patient with HHT and PAH. 97 However, adverse effects such as neuropathy limit its therapeutic use.
Sotatercept is a novel fusion protein therapy consisting of the extracellular domain of the human activin receptor type IIA fused to the Fc domain of human IgG1. It targets members of the TGF‐β superfamily, particularly pro‐proliferative activin ligands (e.g., activin A, GDF8, GDF11), by serving as a “ligand trap” to restore regulation within the pathway. 98 , 99 A phase 2 placebo‐controlled trial of 106 adults receiving background medical therapy for PAH showed significant reduction in the primary endpoint of PVR. 98 A subsequent phase 3 trial of 163 patients receiving sotatercept compared to 160 placebo‐controlled patients demonstrated improvement in the primary endpoint of exercise capacity (i.e., 6‐min walk distance) as well as multiple secondary endpoints, including PVR, N‐terminal pro–B‐type natriuretic peptide levels, and functional class. 99 Although generally well tolerated, an elevated risk of epistaxis, thrombocytopenia, and, notably, telangiectasias was noted. Therefore, the role of this agent in the treatment of HHT‐related PAH remains unclear until its long‐term safety profile is better characterized.
CHRONIC THROMBOEMBOLIC PULMONARY HYPERTENSION
CTEPH is a progressive hemodynamic disorder characterized by chronic obstruction of vessels of the pulmonary arterial tree by organizing thromboembolisms, resulting in increased resistance to flow and development of PH (clinical group 4). 28 Risk factors for this recurrent, inflammatory thrombotic state include malignancy, inflammatory bowel disease, the presence of antiphospholipid antibodies, hypothyroidism, or a history of splenectomy. 100 Signs and symptoms are nonspecific, but may include progressive exertional dyspnea, edema, and hemoptysis. Features more specific to a thromboembolic event (e.g., unilateral leg swelling, and pleuritic chest pain) may raise clinical suspicion. Chronically elevated pulmonary arterial pressures can result in right‐sided heart failure.
Pathophysiology and presentation
Although HHT is characterized by its bleeding complications, the vascular disorder also imparts a prothrombic state and is associated with increased clotting events. In a study of 309 patients with HHT, factor VIII and von Willebrand factor antigen concentrations were found to be significantly elevated when compared to patients without HHT. 101 Levels of factor VIII antigen were also significantly associated with increasing age, with the presence of pulmonary AVMs, and with the incidence of venous thromboembolism. A subsequent prospective study recruited 609 patients with HHT to identify potential biomarkers associated with high factor VIII in this population. 102 Investigators discovered a notable, significant association between elevated factor VIII levels and low serum iron levels in the absence of concurrent, confounding inflammation. Low serum iron levels were also significantly associated with the incidence of venous thromboembolisms, depending on concomitant factor VIII levels. It was postulated that low intracellular iron promotes interactions between iron reactive proteins and iron response elements, eventually leading to elevated levels of factor VIII transcripts. 103 Another study demonstrating significant association between iron deficiency and the rate of ischemic stroke in patients with HHT presented platelet studies indicating iron deficiency amplifies platelet aggregation in response to serotonin. 104 Considering the significant degree of iron deficiency anemia secondary to bleeding events observed in patients with HHT, these findings revealed the propensity for clotting events in this population. Importantly, elevated factor VIII has been identified as a prothrombotic risk factor for CTEPH, with higher serum levels compared to patients with PAH as well as to healthy patients. 105 , 106
Patients with HHT and CTEPH have only been detailed in a few case studies. One study presented data from RHC in 31 patients with HHT; among the 23 patients with an mPAP > 20 mmHg, four patients had significant perfusion defects on ventilation/perfusion lung scans and evidence of thromboembolic patterns on computed tomography, suggestive of CTEPH. 34 Another report detailed a 54‐year‐old female patient with HHT and recurrent venous thromboembolisms despite active warfarin therapy who presented with over 6 months of exertional dyspnea. 32 Chest computed tomography demonstrated bilateral pulmonary emboli with filling defects. Multiple large AVMs were noted in the upper and lower lobes. Echocardiogram followed by RHC yielded an mPAP of 53.7 mmHg, PAWP of 16 mmHg, PVR of 7.6 WUs, and CI of 2.38 L/min/m2. The patient was diagnosed with CTEPH. After unsuccessful treatment with tadalafil and treprostinil, the patient underwent pulmonary thromboendarterectomy. Postoperative mPAP was 19.3 mmHg, and 6‐month follow‐up demonstrated resolution of cardiopulmonary symptoms.
Evaluation, treatment, and outcomes
In patients with CTEPH, echocardiogram and RHC will generally demonstrate PH with precapillary hemodynamics; however, some patients may exhibit combined pre‐ and postcapillary components. Ventilation/perfusion lung scan will demonstrate one or more mismatched perfusion defects, and subsequent computed pulmonary arteriography will confirm the presence of organized thromboembolic material with associated filling defects. Patients diagnosed with CTEPH typically receive indefinite anticoagulant therapy. Pulmonary thromboendarterectomy is the only definitive therapy. In patients with HHT, the presence of pulmonary AVMs does not prohibit the need for pulmonary thromboendarterectomy, but consideration should be given to embolize any known large pulmonary AVMs before planned pulmonary thromboendarterectomy surgery. In cases of inoperable or recurrent CTEPH, riociguat, a soluble guanylate cyclase stimulator, is the only approved medical therapy as it has shown sustained improvement of functional capacity at 1‐year evaluations. 107 Patients with CTEPH who are not surgical candidates may be eligible for multimodal therapy in the form of targeted medical therapy with or without interventional balloon pulmonary angioplasty. 28
The use of anticoagulant and antiplatelet therapy in patients with HHT requires additional appraisal. Despite the potential for exacerbation of bleeding events in patients with HHT, antithrombotic therapy is not absolutely contraindicated and may be employed under careful observation. 25 Based on early studies, the 2020 International HHT Guidelines recommended that if anticoagulant therapy is deemed necessary in patients with HHT, the use of unfractionated heparin, low‐molecular‐weight heparin, and vitamin K antagonists is preferred over the administration of direct‐acting oral anticoagulants. 25 Furthermore, avoidance of combination anticoagulant/antiplatelet therapy or dual antiplatelet therapy was recommended.
Recently, a review of over 500 episodes of antithrombic therapy use in over 400 patients with HHT across over 70 unique studies was conducted. 108 Treatment regimens primarily consisted of anticoagulant therapy only, followed by antiplatelet therapy only. Anticoagulant therapy was largely in the form of warfarin or low‐molecular‐weight heparin. Exacerbation of HHT‐related bleeding events (e.g., epistaxis and gastrointestinal bleeding) occurred in almost 40% of cases, and therapy discontinuation was observed in almost 30% of cases. Approximately 15% of cases required additional local ablative or systemic therapies to target adverse bleeding events. Findings demonstrated significant variability between studies in the frequency and severity of observed adverse events following antithrombotic therapy use in patients with HHT, emphasizing the need for additional investigations to determine optimal therapy in this patient population.
A study of patients with HHT and suspected precapillary PH notably demonstrated that less than half of patients received ventilation/perfusion lung scans, reinforcing the need for a complete diagnostic approach in this setting. 43 Finally, in light of increasing recognition of the association between iron deficiency and thromboembolic events in patients with HHT, a more proactive approach in the screening and treatment of iron deficiency regardless of the presence of clinical signs and symptoms of anemia may be warranted.
ADDITIONAL ETIOLOGIES OF PULMONARY HYPERTENSION IN HEREDITARY HEMORRHAGIC TELANGIECTASIA
Left‐sided heart disease (clinical group 2) or lung diseases and/or hypoxia (clinical group 3) are the most common etiologies of PH in the general population; while likely under‐reported, signs and symptoms of these pathologies are also prevalent in a significant portion of patients with HHT. 33 , 109 , 110 Left‐sided heart disease as well as restrictive and obstructive lung disease have been observed in patients with HHT and PH. 34 Patients with HHT are at an elevated risk of myocardial infarction due to paradoxical embolism through pulmonary AVMs, leading to subsequent cardiomyopathy. Mutations in ENG and ACVRL1 may also promote a chronic maladaptive response to hypoxia. 8 , 9
In patients with HHT and evidence of left‐sided heart or lung disease, established guideline‐directed and etiology‐specific management is recommended along with mitigation of risk factors. The mechanism of action and known side effects of medical therapies, and their potential to exacerbate pre‐existing complications in this population, should be considered. For instance, use of vasodilator therapy in left‐sided heart failure may unmask or encourage a hyperkinetic state secondary to known or unknown liver AVMs. Additionally, the use of inhaled prostanoid treprostinil, approved for group 3 PH associated with interstitial lung disease, is indicated even in patients with HHT but may worsen bleeding events. Clinicians should remain aware of the unknown effects of more specialized therapy in the context of HHT pathophysiology; use of tyrosine kinase inhibitor nintedanib for idiopathic pulmonary fibrosis in a patient with HHT was correlated with an unanticipated improvement in the frequency of epistaxis and skin telangiectasias. 111 As the understanding of the pathogenesis of HHT continues to be improved, such therapeutic agents may garner additional interest.
SPECIAL CONSIDERATIONS
Pulmonary AVMs
Pulmonary AVMs are abnormal capillary‐free vascular structures and anatomic right‐to‐left shunts with low‐resistance, high‐flow properties. As a significant clinical concern in patients with HHT, who most often demonstrate asymptomatic hypoxemia followed by exertional dyspnea, the 2020 International HHT Guidelines recommend screening for pulmonary AVMs via bubble echocardiogram or low‐dose non‐contrast chest computed tomography. 25 Due to an elevated risk of neurological and hemorrhagic complications, it is recommended that pulmonary AVMs are treated, if the size is amenable, with transcatheter embolization using metallic coils or vascular plugs. There is a theoretical concern that closure of the low‐resistance vessel may result in a consequent rise in pulmonary vasculature pressures; however, studies have demonstrated successful improvement in arterial oxygen saturation without significant acute or sustained increase in mPAP following embolization. 112 Due to the high burden of pulmonary AVMs in patients with HHT as well as the overall small diameter of feeding arteries and potential for recanalization, treatment most often results in significant shunt reduction rather than complete eradication of AVMs. 113 , 114 Therefore, continuing medical follow‐up is necessary. Patients’ iron status should be optimized before the procedure. In patients with concomitant HHT‐related PAH and significantly elevated PVR, a course of medical vasodilator therapy before embolization may be warranted. Risks of the procedure include pulmonary edema, paradoxical emboli, and hemorrhagic events (e.g., hemoptysis, hemothorax secondary to rupture of pulmonary AVMs). In addition, the presence of pulmonary AVMs, especially when large, should be taken into consideration when initiating parenteral prostacyclin therapy in patients with HHT and PAH given the possible risk of paradoxical emboli.
Pregnancy and hereditary hemorrhagic telangiectasia
In patients with HHT, the physiologic adaptations of pregnancy, including markedly decreased peripheral vascular resistance and increased CO, can deleteriously affect the course and severity of PH. The degree of HOHF can worsen, particularly in the third trimester. Bevacizumab is classified as category C by the Food and Drug Administration due to evidence of teratogenicity and is therefore contraindicated in pregnancy. In women with PAH, mortality during pregnancy remains high, including within the post‐partum period, and there is an elevated risk of fetal complications such as prematurity. It is currently recommended that patients with PAH avoid pregnancy. 28 Hypercoagulability and an associated elevated thromboembolic potential during pregnancy are well documented, which could further increase the risk of CTEPH in patients with HHT. Finally, the burden of shunting lesions, including both pulmonary and liver AVMs, increases during pregnancy due to elevated blood volume, increasing the risk of fatal hemorrhagic complications as well as thromboembolic events. 78
CONCLUSION
PH is an increasingly recognized complication in patients with HHT with a multifaceted pathophysiology and varying etiologies, requiring specialized management. The overall prevalence of PH in this population remains unknown. The most common clinical phenotypes are PH secondary to HOHF followed by PAH. Moreover, there is a recent growing understanding of the thromboembolic propensity in HHT, raising the risk for CTEPH in this population. Therefore, a complete diagnostic approach in evaluating PH in patients with HHT is encouraged to ensure proper diagnosis of the underlying etiology. Due to disease complexity, patients should be referred for treatment at a center with expertise in management of both HHT and PH.
AUTHOR CONTRIBUTIONS
Akash Mathavan and Akshay Mathavan performed literature review and drafted the manuscript. Renuka Reddy, Kirk Jones, and Christina Eagan assisted in literature review and drafted portions of the manuscript. Ali Ataya supervised the manuscript, provided subject matter expertise, revised the manuscript, and was responsible for the final content. All authors have agreed on the journal to which the article will be submitted, the final version for publication, and accountability for all aspects of the work.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
This review paper was performed in accordance with the ethical standards of the Helsinki Declaration of 1975.
Mathavan A, Mathavan A, Reddy R, Jones K, Eagan C, Alnuaimat H, Ataya A. Pulmonary hypertension in hereditary hemorrhagic telangiectasia: a clinical review. Pulm Circ. 2023;13:e12301. 10.1002/pul2.12301
Akash Mathavan and Akshay Mathavan contributed equally to this study and shared the first authorship.
REFERENCES
- 1. Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333(14):918–924. [DOI] [PubMed] [Google Scholar]
- 2. Bideau A, Plauchu H, Brunet G, Robert J. Epidemiological investigation of Rendu‐Osler disease in France: its geographical distribution and prevalence. Population. 1989;44(1):3–22. [PubMed] [Google Scholar]
- 3. Chizinga M, Rudkovskaia AA, Henderson K, Pollak J, Garcia‐Tsao G, Young LH, Fares WH. Pulmonary hypertension prevalence and prognosis in a cohort of patients with hereditary hemorrhagic telangiectasia undergoing embolization of pulmonary arteriovenous malformations. Am J Respir Crit Care Med. 2017;196(10):1353–1356. [DOI] [PubMed] [Google Scholar]
- 4. Vorselaars V, Velthuis S, van Gent M, Westermann C, Snijder R, Mager J, Post M. Pulmonary hypertension in a large cohort with hereditary hemorrhagic telangiectasia. Respiration. 2017;94(3):242–250. [DOI] [PubMed] [Google Scholar]
- 5. Lyle MA, Fenstad ER, McGoon MD, Frantz RP, Krowka MJ, Kane GC, Swanson KL. Pulmonary hypertension in hereditary hemorrhagic telangiectasia. Chest. 2016;149(2):362–371. [DOI] [PubMed] [Google Scholar]
- 6. Sopeña B, Pérez‐Rodríguez MT, Portela D, Rivera A, Freire M, Martínez‐Vázquez C. High prevalence of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. Eur J Intern Med. 2013;24(3):e30–e34. [DOI] [PubMed] [Google Scholar]
- 7. Sapru RP, Hutchison DC, Hall JI. Pulmonary hypertension in patients with pulmonary arteriovenous fistulae. Heart. 1969;31(5):559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernández‐L A, Sanz‐Rodriguez F, Blanco FJ, Bernabéu C, Botella LM. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF‐beta signaling pathway. Clin Med Res. 2006;4(1):66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tillet E, Bailly S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front Genet. 2015;5:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrel J, McCormick MK, Pericak‐Vance MA, Heutink P, Oostra BA, Haitjema T, Westerman CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA. Endoglin, a TGF‐β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nature Genet. 1994;8(4):345–351. [DOI] [PubMed] [Google Scholar]
- 11. Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak‐Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous MEM, Marchuk DA. Mutations in the activin receptor–like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nature Genet. 1996;13(2):189–195. [DOI] [PubMed] [Google Scholar]
- 12. Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin É, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363(9412):852–859. [DOI] [PubMed] [Google Scholar]
- 13. McDonald J, Wooderchak‐Donahue W, VanSant Webb C, Whitehead K, Stevenson DA, Bayrak‐Toydemir P. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet. 2015;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cole SG. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42(7):577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Piantanida M, Buscarini E, Dellavecchia C, Minelli A, Rossi A, Buscarini L, Danesino C. Hereditary haemorrhagic telangiectasia with extensive liver involvement is not caused by either HHT1 or HHT2. J Med Genet. 1996;33(6):441–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wallace GMF. A hereditary haemorrhagic telangiectasia family with pulmonary involvement is unlinked to the known HHT genes, endoglin and ALK‐1. Thorax. 2000;55(8):685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bayrak‐Toydemir P, McDonald J, Akarsu N, Toydemir RM, Calderon F, Tuncali T, Tang W, Miller F, Mao R. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet Part A. 2006;140(20):2155–2162. [DOI] [PubMed] [Google Scholar]
- 18. Wooderchak‐Donahue WL, McDonald J, O'Fallon B, Upton PD, Li W, Roman BL, Young S, Plant P, Fülöp GT, Langa C, Morrell NW, Botella LM, Bernabeu C, Stevenson DA, Runo JR, Bayrak‐Toydemir P. BMP9 mutations cause a vascular‐anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet. 2013;93(3):530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Upton P, Richards S, Bates A, Niederhoffer KY, Morrell NW, Christian S. A rare homozygous missense GDF2 (BMP9) mutation causing PAH in siblings: does BMP10 status contribute? Am J Med Genet Part A. 2023;191(1):228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hernandez F, Huether R, Carter L, Johnston T, Thompson J, Gossage JR, Chao E, Elliott AM. Mutations in RASA1 and GDF2 identified in patients with clinical features of hereditary hemorrhagic telangiectasia. Hum Genome Var. 2015;2(1):15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu J, Streicher JL, Medne L, Krantz ID, Yan AC. EPHB4 mutation implicated in capillary malformation‐arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34(5):e227–e230. [DOI] [PubMed] [Google Scholar]
- 22. Ye F, Jiang W, Lin W, Wang Y, Chen H, Zou H, Huang S, Zhu N, Han S. A novel BMPR2 mutation in a patient with heritable pulmonary arterial hypertension and suspected hereditary hemorrhagic telangiectasia. Medicine. 2020;99(31):e21342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJJ, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu‐Osler‐Weber syndrome). Am J Med Genet. 2000;91(1):66–67. [DOI] [PubMed] [Google Scholar]
- 24. Faughnan ME, Palda VA, Garcia‐Tsao G, Geisthoff UW, McDonald J, Proctor DD, Spears J, Brown DH, Buscarini E, Chesnutt MS, Cottin V, Ganguly A, Gossage JR, Guttmacher AE, Hyland RH, Kennedy SJ, Korzenik J, Mager JJ, Ozanne AP, Piccirillo JF, Picus D, Plauchu H, Porteous MEM, Pyeritz RE, Ross DA, Sabba C, Swanson K, Terry P, Wallace MC, Westermann CJJ, White RI, Young LH, Zarrabeitia R. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48(2):73–87. [DOI] [PubMed] [Google Scholar]
- 25. Faughnan ME, Mager JJ, Hetts SW, Palda VA, Lang‐Robertson K, Buscarini E, Deslandres E, Kasthuri RS, Lausman A, Poetker D, Ratjen F, Chesnutt MS, Clancy M, Whitehead KJ, Al‐Samkari H, Chakinala M, Conrad M, Cortes D, Crocione C, Darling J, de Gussem E, Derksen C, Dupuis‐Girod S, Foy P, Geisthoff U, Gossage JR, Hammill A, Heimdal K, Henderson K, Iyer VN, Kjeldsen AD, Komiyama M, Korenblatt K, McDonald J, McMahon J, McWilliams J, Meek ME, Mei‐Zahav M, Olitsky S, Palmer S, Pantalone R, Piccirillo JF, Plahn B, Porteous MEM, Post MC, Radovanovic I, Rochon PJ, Rodriguez‐Lopez J, Sabba C, Serra M, Shovlin C, Sprecher D, White AJ, Winship I, Zarrabeitia R. Second international guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia. Ann Intern Med. 2020;173(12):989–1001. [DOI] [PubMed] [Google Scholar]
- 26. Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Human Genet. 2009;17(7):860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cottin V, Plauchu H, Bayle JY, Barthelet M, Revel D, Cordier JF. Pulmonary arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. Am J Respir Crit Care Med. 2004;169(9):994–1000. [DOI] [PubMed] [Google Scholar]
- 28. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano‐Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke‐Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S, Schwerzmann M, Dinh‐Xuan AT, Bush A, Abdelhamid M, Aboyans V, Arbustini E, Asteggiano R, Barberà JA, Beghetti M, Čelutkienė J, Cikes M, Condliffe R, de Man F, Falk V, Fauchier L, Gaine S, Galié N, Gin‐Sing W, Granton J, Grünig E, Hassoun PM, Hellemons M, Jaarsma T, Kjellström B, Klok FA, Konradi A, Koskinas KC, Kotecha D, Lang I, Lewis BS, Linhart A, Lip GYH, Løchen ML, Mathioudakis AG, Mindham R, Moledina S, Naeije R, Nielsen JC, Olschewski H, Opitz I, Petersen SE, Prescott E, Rakisheva A, Reis A, Ristić AD, Roche N, Rodrigues R, Selton‐Suty C, Souza R, Swift AJ, Touyz RM, Ulrich S, Wilkins MR, Wort SJ. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618–3731. [DOI] [PubMed] [Google Scholar]
- 29. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y, Wang Y, Shi J, Hua Z, Xu J. Cardiac output measurements via echocardiography versus thermodilution: a systematic review and meta‐analysis. PLoS One. 2019;14(10):e0222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harder EM, Fares WH. Hospitalizations with hereditary hemorrhagic telangiectasia and pulmonary hypertension in the United States from 2000 to 2014. Respir Med. 2019;147:26–30. [DOI] [PubMed] [Google Scholar]
- 32. Wille KM, Bellot SC, Acharya D, Madani MM, Singh SP, McGiffin DC. Thromboendarterectomy for chronic thromboembolic pulmonary hypertension in hereditary hemorrhagic telangiectasia. Am J Respir Crit Care Med. 2014;189(1):112–114. [DOI] [PubMed] [Google Scholar]
- 33. Droege F, Thangavelu K, Stuck BA, Stang A, Lang S, Geisthoff U. Life expectancy and comorbidities in patients with hereditary hemorrhagic telangiectasia. Vasc Med. 2018;23(4):377–383. [DOI] [PubMed] [Google Scholar]
- 34. Margelidon‐Cozzolino V, Cottin V, Dupuis‐Girod S, Traclet J, Ahmad K, Mornex JF, Turquier S. Pulmonary hypertension in hereditary haemorrhagic telangiectasia is associated with multiple clinical conditions. ERJ Open Res. 2021;7(1):00078‐2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qaiser KN, Sahay S, Tonelli AR. Pulmonary hypertension due to high cardiac output. Respir Med. 2023;206:107034. [DOI] [PubMed] [Google Scholar]
- 36. Mehta PA, Dubrey SW. High output heart failure. QJM. 2009;102(4):235–241. [DOI] [PubMed] [Google Scholar]
- 37. Waxman A, Restrepo‐Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, Allen R, Feldman J, Argula R, Smith P, Rollins K, Deng C, Peterson L, Bell H, Tapson V, Nathan SD. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. 2021;384(4):325–334. [DOI] [PubMed] [Google Scholar]
- 38. Alkhodair A, Tsang MYC, Cairns JA, Swiston JR, Levy RD, Lee L, Huckell VF, Brunner NW. Comparison of thermodilution and indirect Fick cardiac outputs in pulmonary hypertension. Int J Cardiol. 2018;258:228–231. [DOI] [PubMed] [Google Scholar]
- 39. Memeo M, Stabile Ianora AA, Scardapane A, Buonamico P, Sabbà C, Angelelli G. Hepatic involvement in hereditary hemorrhagic telangiectasia: CT findings. Abdom Imaging. 2004;29(2):211–220. [DOI] [PubMed] [Google Scholar]
- 40. Ariza Ordoñez N, Pino Marín A, Bonilla Crespo JS, Navarro Navajas A, Oliver GA, Medina HM, Forero JF. An unusual cause of right heart dysfunction and high output heart failure in a young woman. J Cardiovasc Dev Dis. 2022;9(12):418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamaguchi K, Ogihara Y, Fujimoto N, Nomura Y, Dohi K. Histopathological findings of pulmonary hypertension with elevated pulmonary arterial wedge pressure in hereditary hemorrhagic telangiectasia. Circ Rep. 2022;4(6):287–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aramalla S, Bhyravavajhala S, Vanaparty B, Narayanan R, Yerram S. Severe pulmonary arterial hypertension in a patient with hereditary hemorrhagic telangiectasia and multiple pulmonary and hepatic arteriovenous malformations. Ann Pediatr Cardiol. 2022;15(1):73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Revuz S, Decullier E, Ginon I, Lamblin N, Hatron PY, Kaminsky P, Carette MF, Lacombe P, Simon AC, Rivière S, Harlé JR, Fraisse A, Lavigne C, Leguy‐Seguin V, Chaouat A, Khouatra C, Dupuis‐Girod S, Hachulla E. Pulmonary hypertension subtypes associated with hereditary haemorrhagic telangiectasia: haemodynamic profiles and survival probability. PLoS One. 2017;12(10):e0184227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greco A, Plumitallo S, Scelsi L, Maggi G, Sobrero M, Turco A, Raineri C, Arseni N, Cappelletti D, Visconti LO, Pagella F, Spinozzi G, Ghio S, Olivieri C, Danesino C. Different forms of pulmonary hypertension in a family with clinical and genetic evidence for hereditary hemorrhagic teleangectasia type 2. Pulm Circ. 2018;8(4):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ford T, Wong M, Cheah B, Alexopolous C. Pulmonary hypertension and hepatic encephalopathy: lethal complications of Rendu‐Osler‐Weber disease. J R Coll Physicians Edinb. 2014;44(2):126–129. [DOI] [PubMed] [Google Scholar]
- 46. Bloom PP, Rodriguez‐Lopez J, Witkin AS, Al‐Samkari H, Kuter DJ, Mojtahed A, Luther J. Ammonia predicts hepatic involvement and pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. Clin Transl Gastroenterol. 2020;11(1):e00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Faughnan ME, Granton JT, Young LH. The pulmonary vascular complications of hereditary haemorrhagic telangiectasia. Eur Respir J. 2009;33(5):1186–1194. [DOI] [PubMed] [Google Scholar]
- 48. Harrison RE. Molecular and functional analysis identifies ALK‐1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40(12):865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yamamoto H, Itamoto C, Yamaguchi T, Koshyo T. Severe hypoxemia caused by high‐output heart failure and pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia. JACC Case Rep. 2021;3(17):1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Srimatkandada P. Severe autoimmune hyperthyroidism in two patients with pulmonary arterial hypertension after treatment with epoprostenol. J Thyroid Disord Ther. 2015;4:1. [Google Scholar]
- 51. Chadha C, Pritzker M, Mariash CN. Effect of epoprostenol on the thyroid gland: enlargement and secretion of thyroid hormone. Endocr Pract. 2009;15(2):116–121. [DOI] [PubMed] [Google Scholar]
- 52. Stockigt JR, Lim CF, Barlow JW, Wynne KN, Mohr VS, Topliss DJ, Hamblin PS, Sabto J. Interaction of furosemide with serum thyroxine binding sites: in vivo and in vitro studies and comparison with other inhibitors. J Clin Endocrinol Metab. 1985;60(5):1025–1031. [DOI] [PubMed] [Google Scholar]
- 53. Thompson AB, Ross DA, Berard P, et al. Very low dose bevacizumab for the treatment of epistaxis in patients with hereditary hemorrhagic telangiectasia. Allergy Rhinol. 2014;5(2):ar.2014.5.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Iyer VN, Apala DR, Pannu BS, Kotecha A, Brinjikji W, Leise MD, Kamath PS, Misra S, Begna KH, Cartin‐Ceba R, DuBrock HM, Krowka MJ, O'Brien EK, Pruthi RK, Schroeder DR, Swanson KL. Intravenous bevacizumab for refractory hereditary hemorrhagic telangiectasia–related epistaxis and gastrointestinal bleeding. Mayo Clin Proc. 2018;93(2):155–166. [DOI] [PubMed] [Google Scholar]
- 55. Epperla N, Kapke JT, Karafin M, Friedman KD, Foy P. Effect of systemic bevacizumab in severe hereditary hemorrhagic telangiectasia associated with bleeding. Am J Hematol. 2016;91(6):E313–E314. [DOI] [PubMed] [Google Scholar]
- 56. Al‐Samkari H, Kritharis A, Rodriguez‐Lopez JM, Kuter DJ. Systemic bevacizumab for the treatment of chronic bleeding in hereditary haemorrhagic telangiectasia. J Intern Med. 2019;285(2):223–231. [DOI] [PubMed] [Google Scholar]
- 57. Rosenberg T, Fialla AD, Kjeldsen J, Kjeldsen AD. Does severe bleeding in HHT patients respond to intravenous bevacizumab? Review of the literature and case series. Rhinology. 2019;57(4):242–251. [DOI] [PubMed] [Google Scholar]
- 58. Dupuis‐Girod S, Ginon I, Saurin JC, Marion D, Guillot E, Decullier E, Roux A, Carette MF, Gilbert‐Dussardier B, Hatron PY, Lacombe P, Lorcerie B, Rivière S, Corre R, Giraud S, Bailly S, Paintaud G, Ternant D, Valette PJ, Plauchu H, Faure F. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307(9):948–955. [DOI] [PubMed] [Google Scholar]
- 59. Al‐Samkari H, Albitar HA, Olitsky SE, Clancy MS, Iyer VN. Systemic bevacizumab for high‐output cardiac failure in hereditary hemorrhagic telangiectasia: an international survey of HHT centers. Orphanet J Rare Dis. 2019;14(1):256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lerut J, Orlando G, Adam R, Sabbà C, Pfitzmann R, Klempnauer J, Belghiti J, Pirenne J, Thevenot T, Hillert C, Brown CM, Gonze D, Karam V, Boillot O. Liver transplantation for hereditary hemorrhagic telangiectasia: report of the European liver transplant registry. Ann Surg. 2006;244(6):854–864 [DOI] [PMC free article] [PubMed] [Google Scholar]; discussion 862–864.
- 61. Dupuis‐Girod S, Chesnais AL, Ginon I, Dumortier J, Saurin JC, Finet G, Decullier E, Marion D, Plauchu H, Boillot O. Long‐term outcome of patients with hereditary hemorrhagic telangiectasia and severe hepatic involvement after orthotopic liver transplantation: a single‐center study. Liver Transpl. 2010;16(3):340–347. [DOI] [PubMed] [Google Scholar]
- 62. Dumortier J, Dupuis‐Girod S, Valette PJ, Valent A, Guillaud O, Saurin JC, Hervieu V, Robinson P, Plauchu H, Paliard P, Boillot O, Scoazec JY. Recurrence of hereditary hemorrhagic telangiectasia after liver transplantation: clinical implications and physiopathological insights. Hepatology. 2019;69(5):2232–2240. [DOI] [PubMed] [Google Scholar]
- 63. Garcia‐Tsao G, Gish RG, Punch J. Model for end‐stage liver disease (MELD) exception for hereditary hemorrhagic telangiectasia. Liver Transpl. 2006;12(S3):S108–S109. [DOI] [PubMed] [Google Scholar]
- 64. Perrodin SF, Vogt AP, Berzigotti A, Kim‐Fuchs C, Luedi MM, Candinas D, Banz VM. Resolution of precapillary pulmonary hypertension after liver transplantation for hereditary hemorrhagic telangiectasia: systematic review and case report. Transplant Proc. 2022;54(1):135–143. [DOI] [PubMed] [Google Scholar]
- 65. Heath D, Edwards JE. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation. 1958;18(4 Part 1):533–547. [DOI] [PubMed] [Google Scholar]
- 66. Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, Trembath RC, Loyd JE. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chida A, Shintani M, Yagi H, Fujiwara M, Kojima Y, Sato H, Imamura S, Yokozawa M, Onodera N, Horigome H, Kobayashi T, Hatai Y, Nakayama T, Fukushima H, Nishiyama M, Doi S, Ono Y, Yasukouchi S, Ichida F, Fujimoto K, Ohtsuki S, Teshima H, Kawano T, Nomura Y, Gu H, Ishiwata T, Furutani Y, Inai K, Saji T, Matsuoka R, Nonoyama S, Nakanishi T. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol. 2012;110(4):586–593. [DOI] [PubMed] [Google Scholar]
- 68. Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, Simonneau G, Galie N, Loyd JE, Humbert M, Nichols WC, Berg J, Manes A, McGaughran J, Pauciulo M, Wheeler L, Morrell NW. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345(5):325–334. [DOI] [PubMed] [Google Scholar]
- 69. Schulick AH, Taylor AJ, Zuo W, Qiu C, Dong G, Woodward RN, Agah R, Roberts AB, Virmani R, Dichek DA. Overexpression of transforming growth factor β1 in arterial endothelium causes hyperplasia, apoptosis, and cartilaginous metaplasia. Proc Natl Acad Sci U S A. 1998;95(12):6983–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yuan SM. Pulmonary artery hypertension in childhood: the transforming growth factor‐β superfamily‐related genes. Pediatr Neonatol. 2018;59(2):112–119. [DOI] [PubMed] [Google Scholar]
- 71. Vorselaars V, Hosman A, Westermann C, Snijder R, Mager J, Goumans MJ, Post M. Pulmonary arterial hypertension and hereditary haemorrhagic telangiectasia. Int J Mol Sci. 2018;19(10):3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Farhan A, Latif MA, Minhas A, Weiss CR. Cardiac and hemodynamic manifestations of hereditary hemorrhagic telangiectasia. Int J Angiol. 2022;31(2):75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chaouat A. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax. 2004;59(5):446–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ayala E, Kudelko KT, Haddad F, Zamanian RT, de Jesus Perez V. The intersection of genes and environment. Chest. 2012;141(6):1598–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gallego N, Cruz‐Utrilla A, Guillén I, Bonora AM, Ochoa N, Arias P, Lapunzina P, Escribano‐Subias P, Nevado J, Tenorio‐Castaño J. Expanding the evidence of a semi‐dominant inheritance in GDF2 associated with pulmonary arterial hypertension. Cells. 2021;10(11):3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Varnholt H, Kradin R. Pulmonary capillary hemangiomatosis arising in hereditary hemorrhagic telangiectasia. Hum Pathol. 2004;35(2):266–268. [DOI] [PubMed] [Google Scholar]
- 77. Pousada G, Baloira A, Valverde D. Pulmonary arterial hypertension and portal hypertension in a patient with hereditary hemorrhagic telangiectasia. Med Clin. 2015;144(6):261–264. [DOI] [PubMed] [Google Scholar]
- 78. Dupuis‐Girod S, Cottin V, Shovlin CL. The lung in hereditary hemorrhagic telangiectasia. Respiration. 2017;94(4):315–330. [DOI] [PubMed] [Google Scholar]
- 79. Ishiwata T, Terada J, Tanabe N, Abe M, Sugiura T, Tsushima K, Tada Y, Sakao S, Kasahara Y, Nakanishi N, Morisaki H, Tatsumi K. Pulmonary arterial hypertension as the first manifestation in a patient with hereditary hemorrhagic telangiectasia. Intern Med. 2014;53(20):2359–2363. [DOI] [PubMed] [Google Scholar]
- 80. Girerd B, Montani D, Coulet F, Sztrymf B, Yaici A, Jaïs X, Tregouet D, Reis A, Drouin‐Garraud V, Fraisse A, Sitbon O, O'Callaghan DS, Simonneau G, Soubrier F, Humbert M. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181(8):851–861. [DOI] [PubMed] [Google Scholar]
- 81. Chang SA, Jang SY, Ki CS, Kang IS, Kim DK. Successful bosentan therapy for pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia. Heart Vessels. 2011;26(2):231–234. [DOI] [PubMed] [Google Scholar]
- 82. Bonderman D, Nowotny R, Skoro‐Sajer N, Adlbrecht C, Lang IM. Bosentan therapy for pulmonary arterial hypertension associated with hereditary haemorrhagic telangiectasia. Eur J Clin Invest. 2006;36(Suppl 3):71–72. [DOI] [PubMed] [Google Scholar]
- 83. Smoot LB, Obler D, McElhinney DB, Boardman K, Wu BL, Lip V, Mullen MP. Clinical features of pulmonary arterial hypertension in young people with an ALK1 mutation and hereditary haemorrhagic telangiectasia. Arch Dis Child. 2009;94(7):506–511. [DOI] [PubMed] [Google Scholar]
- 84. Miyake R, Fujino T, Abe K, Hosokawa K, Ohtani K, Morisaki H, Yamada O, Higo T, Ide T. Pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia successfully treated with sildenafil. Int J Cardiol. 2016;214:275–276. [DOI] [PubMed] [Google Scholar]
- 85. Walsh LJ, Collins C, Ibrahim H, Kerins DM, Brady AP, O Connor TM. Pulmonary arterial hypertension in hereditary hemorrhagic telangiectasia associated with ACVRL1 mutation: a case report. J Med Case Rep. 2022;16(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Minai OA, Rigelsky C, Eng C, Arroliga AC, Stoller JK. Long‐term outcome in a patient with pulmonary hypertension and hereditary hemorrhagic telangiectasia. Chest. 2007;131(4):984–987. [DOI] [PubMed] [Google Scholar]
- 87. Abston E, Hon S, Rodriguez‐Lopez J, Moll M, Lanuti M, Farber HW, Wilson KC. Treatment of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia—a case series and systematic review. Pulm Pharmacol Ther. 2021;68:102033. [DOI] [PubMed] [Google Scholar]
- 88. Wei A, Gu Z, Li J, Liu X, Wu X, Han Y, Pu J. Clinical adverse effects of endothelin receptor antagonists: insights from the meta‐analysis of 4894 patients from 24 randomized double‐blind placebo‐controlled clinical trials. J Am Heart Assoc. 2016;5(11):e003896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kingman M, Archer‐Chicko C, Bartlett M, Beckmann J, Hohsfield R, Lombardi S. Management of prostacyclin side effects in adult patients with pulmonary arterial hypertension. Pulm Circ. 2017;7(3):598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Schwartz BG, Kloner RA. Drug interactions with phosphodiesterase‐5 inhibitors used for the treatment of erectile dysfunction or pulmonary hypertension. Circulation. 2010;122(1):88–95. [DOI] [PubMed] [Google Scholar]
- 91. Galiè N, Channick RN, Frantz RP, Grünig E, Jing ZC, Moiseeva O, Preston IR, Pulido T, Safdar Z, Tamura Y, McLaughlin VV. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hoag JB, Terry P, Mitchell S, Reh D, Merlo CA. An epistaxis severity score for hereditary hemorrhagic telangiectasia. Laryngoscope. 2010;120(4):838–843. [DOI] [PubMed] [Google Scholar]
- 93. Peterson AM, Kallogjeri D, Spitznagel E, Chakinala MM, Schneider JS, Piccirillo JF. Development and validation of the nasal outcome score for epistaxis in hereditary hemorrhagic telangiectasia (NOSE HHT). JAMA Otolaryngol Head Neck Surg. 2020;146(11):999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kim J, Studer SM, Crespo MM, Pilewski J, Toyoda Y, Johnson BA, Oggionni T, Vitulo P. 62: Outcomes following lung transplantation for pulmonary hypertension secondary to hereditary hemorrhagic telangiectasia. J Heart Lung Transplant. 2008;27(2):S82. [Google Scholar]
- 95. Li W, Xiong C, Gu Q, Wang X, Cheng X, Huang L, Yang T, Luo Q, Zhao Z, Ni X, Liu Z, He J. The clinical characteristics and long‐term prognosis of pulmonary arterial hypertension associated with hereditary hemorrhagic telangiectasia. Pulm Circ. 2018;8(2):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rehman W, Arfons LM, Lazarus HM. The rise, fall and subsequent triumph of thalidomide: lessons learned in drug development. Ther Adv Hematol. 2011;2(5):291–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nakamura T, Ogo T, Tahara N, Fukui S, Tsuji A, Ueda J, Fukumoto Y, Nakanishi N, Ogawa H, Yasuda S. Thalidomide for hereditary hemorrhagic telangiectasia with pulmonary arterial hypertension. Circ J. 2018;82(4):1205–1207. [DOI] [PubMed] [Google Scholar]
- 98. Humbert M, McLaughlin V, Gibbs JSR, Gomberg‐Maitland M, Hoeper MM, Preston IR, Souza R, Waxman A, Escribano Subias P, Feldman J, Meyer G, Montani D, Olsson KM, Manimaran S, Barnes J, Linde PG, de Oliveira Pena J, Badesch DB. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med. 2021;384(13):1204–1215. [DOI] [PubMed] [Google Scholar]
- 99. Hoeper MM, Badesch DB, Ghofrani HA, Gibbs JSR, Gomberg‐Maitland M, McLaughlin VV, Preston IR, Souza R, Waxman AB, Grünig E, Kopeć G, Meyer G, Olsson KM, Rosenkranz S, Xu Y, Miller B, Fowler M, Butler J, Koglin J, de Oliveira Pena J, Humbert M. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N Engl J Med. 2023;388(16):1478–1490. [DOI] [PubMed] [Google Scholar]
- 100. Al Abri Q, Lu AJ, Ramchandani MK. Chronic thromboembolic pulmonary hypertension: a comprehensive review and multidisciplinary approach to surgical treatment. Methodist Debakey Cardiovasc J. 2021;17(2):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shovlin CL, Sulaiman NL, Govani FS, Jackson JE, Begbie ME. Elevated factor VIII in hereditary haemorrhagic telangiectasia (HHT): association with venous thromboembolism. Thromb Haemostasis. 2007;98(5):1031–1039. [PubMed] [Google Scholar]
- 102. Livesey JA, Manning RA, Meek JH, Jackson JE, Kulinskaya E, Laffan MA, Shovlin CL. Low serum iron levels are associated with elevated plasma levels of coagulation factor VIII and pulmonary emboli/deep venous thromboses in replicate cohorts of patients with hereditary haemorrhagic telangiectasia. Thorax. 2012;67(4):328–333. [DOI] [PubMed] [Google Scholar]
- 103. Shovlin CL, Angus G, Manning RA, Okoli GN, Govani FS, Elderfield K, Birdsey GM, Mollet IG, Laffan MA, Mauri FA. Endothelial cell processing and alternatively spliced transcripts of factor VIII: potential implications for coagulation cascades and pulmonary hypertension. PLoS One. 2010;5(2):e9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shovlin CL, Chamali B, Santhirapala V, Livesey JA, Angus G, Manning R, Laffan MA, Meek J, Tighe HC, Jackson JE. Ischaemic strokes in patients with pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia: associations with iron deficiency and platelets. PLoS One. 2014;9(2):e88812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bonderman D, Turecek P, Jakowitsch J, Weltermann A, Adlbrecht C, Schneider B, Kneussl M, Rubin L, Kyrle P, Klepetko W, Maurer G, Lang I. High prevalence of elevated clotting factor VIII in chronic thromboembolic pulmonary hypertension. Thromb Haemostasis. 2003;90(09):372–376. [DOI] [PubMed] [Google Scholar]
- 106. Wong CL, Szydlo R, Gibbs S, Laffan M. Hereditary and acquired thrombotic risk factors for chronic thromboembolic pulmonary hypertension. Blood Coagul Fibrinolysis. 2010;21(3):201–206. [DOI] [PubMed] [Google Scholar]
- 107. Simonneau G, D'Armini AM, Ghofrani HA, Grimminger F, Hoeper MM, Jansa P, Kim NH, Wang C, Wilkins MR, Fritsch A, Davie N, Colorado P, Mayer E. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: a long‐term extension study (CHEST‐2). Eur Respir J. 2015;45(5):1293–1302. [DOI] [PubMed] [Google Scholar]
- 108. Zhang E, Virk ZM, Rodriguez‐Lopez J, Al‐Samkari H. Anticoagulation and antiplatelet therapy in hereditary hemorrhagic telangiectasia: a scoping review. Thromb Res. 2023;226:150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bofarid S, Hosman AE, Mager JJ, Snijder RJ, Post MC. Pulmonary vascular complications in hereditary hemorrhagic telangiectasia and the underlying pathophysiology. Int J Mol Sci. 2021;22(7):3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Farooqui M, Suriya S, Qeadan F, Vigil C, Wegele A, Ikram A, Quadri SA, Robinson M, Rodriguez‐Lopez J, Ortega‐Gutierrez S, Zafar A. Cerebrovascular and cardiovascular disease burden in patients with hereditary hemorrhagic telangiectasia. Neurol Sci. 2021;42(12):5117–5122. [DOI] [PubMed] [Google Scholar]
- 111. Kovacs‐Sipos E, Holzmann D, Scherer T, Soyka MB. Nintedanib as a novel treatment option in hereditary haemorrhagic telangiectasia. BMJ Case Rep. 2017;2017:bcr2017219393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Shovlin CL, Gibbs JSR, Jackson JE. Management of pulmonary arteriovenous malformations in pulmonary hypertensive patients: a pressure to embolise? Eur Respir Rev. 2009;18(111):4–6. [DOI] [PubMed] [Google Scholar]
- 113. Shovlin CL, Condliffe R, Donaldson JW, Kiely DG, Wort SJ. British Thoracic Society Clinical statement on pulmonary arteriovenous malformations. Thorax. 2017;72(12):1154–1163. [DOI] [PubMed] [Google Scholar]
- 114. Shovlin CL. Pulmonary arteriovenous malformations. Am J Respir Crit Care Med. 2014;190(11):1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]