

Abstract
The intersection of protein and lipid biology is of growing importance for understanding how cells address structural challenges during adhesion and migration. While protein complexes engaged with the cytoskeleton play a vital role, support from the phospholipid membrane is crucial for directing localization and assembly of key protein complexes. During angiogenesis, dramatic cellular remodeling is necessary for endothelial cells to shift from a stable monolayer to invasive structures. However, the molecular dynamics between lipids and proteins during endothelial invasion are not defined. Here, we utilized cell culture, immunofluorescence, and lipidomic analyses to identify a novel role for the membrane binding protein Annexin A2 (ANXA2) in modulating the composition of specific membrane lipids necessary for cortical F-actin organization and adherens junction stabilization. In the absence of ANXA2, there is disorganized cortical F-actin, reduced junctional Arp2, excess sprout initiation, and ultimately failed sprout maturation. Furthermore, we observed reduced filipin III labeling of membrane cholesterol in cells with reduced ANXA2, suggesting there is an alteration in phospholipid membrane dynamics. Lipidomic analyses revealed that 42 lipid species were altered with loss of ANXA2, including an accumulation of phosphatidylcholine (16:0_16:0). We found that supplementation of phosphatidylcholine (16:0_16:0) in wild-type endothelial cells mimicked the ANXA2 knock-down phenotype, indicating that ANXA2 regulated the phospholipid membrane upstream of Arp2 recruitment and organization of cortical F-actin. Altogether, these data indicate a novel role for ANXA2 in coordinating events at endothelial junctions needed to initiate sprouting and show that proper lipid modulation is a critical component of these events.
Keywords: actin-related protein 2, adherens junctions, Annexin A2, cholesterol, cytoskeleton, endothelial cells, lipidomics, membrane lipids, phosphatidylcholines, phospholipids
1 |. INTRODUCTION
The formation of new blood vessels from pre-existing structures creates an exceptional problem for endothelial cells, which are responsible for maintaining the vascular barrier. During angiogenesis, cell–cell junctions maintain the endothelial barrier and remodel to allow endothelial cells to move collectively during the outgrowth of new sprouts.1,2 To accomplish this balance, specialized intra-cellular machinery must coordinate events at the junction that allow endothelial cells to both initiate sprouting and maintain cell–cell contacts.3–7 However, the mechanisms responsible for this regulation are not fully understood.
Angiogenesis is driven by a variety of signals, which can be generalized as enhancing or opposing barrier formation. An example is vascular endothelial growth factor (VEGF), which loosens the barrier between endothelial cells, leading to increased vessel permeability and vascular development.8,9 Another potent growth factor, basic fibroblast growth factor (bFGF), stabilizes the endothelial barrier at low levels, yet increases vessel permeability in a dose-dependent manner at higher concentrations.10–14 Lastly, sphingosine 1-phosphate (S1P) is an abundant plasma lysosphingolipid secreted by activated platelets,15 which greatly enhances barrier stability. As plasma levels of S1P are maintained through incorporation into red blood cells and high-density lipoproteins,16,17 endothelial cells consistently preserve a stable vascular barrier.18–20 Despite opposing effects on barrier formation between growth factors (VEGF and bFGF) and S1P, these agonists synergize to enhance endothelial sprouting in multiple models.11,21,22 One explanation for this observation is that the dynamic process of angiogenesis is optimized by a careful balance of signals that loosen junctions to initiate sprouting, then stabilize junctions as new multicellular structures form into a vessel with sufficient barrier integrity.4
Transitioning from a stable monolayer to an invasive sprout requires coordination between transmembrane protein complexes and the actin cytoskeleton. During this transition, the actin cytoskeleton must actively remodel to minimize stress on the cell.23,24 Adaptor proteins such as Annexin A2 (ANXA2) are ideal candidates to minimize stress at the cytoskeletal-membrane interface, through binding to both F-actin and phospholipids. ANXA2 heavily localizes wherever dynamic cytoskeletal-membrane structures form,25–29 which correlates with its increased expression in cell types that are motile, complex in shape, and/or stress bearing, such as macrophages, fibroblasts, epithelial cells, and endothelial cells.30,31 While ANXA2 is known to support endothelial junctional maintenance, barrier integrity, and sprout formation, the precise mechanisms by which ANXA2 supports these processes are unclear.32–34
It has long been argued that a critical function of ANXA2 is regulation of the phospholipid membrane, which directs protein localization. Traditionally, membrane lipid biology has broadly focused on lipid class ratios or levels of saturation; however, it is now appreciated that particular lipid species influence cellular functions,35,36 and minor lipid species can drive protein localization to shape cell behaviors.37 Lipid-dependent signaling events involving lipoproteins, lysophospholipids, and phosphatidylinositol (PI) phosphates have been studied during angiogenesis,38,39 but how membrane lipid composition is altered when endothelial cells transition from a stable monolayer to an invasive structure is not understood, as the dynamic nature of angiogenesis likely requires equally dynamic shifts in the membrane itself.
In this study, we identify a new mechanism of function for ANXA2 in angiogenesis. In the absence of ANXA2, cortical actin and adherens junctions are disorganized and there is failed recruitment of Arp2, resulting in immediate excess sprout initiation, but ultimately failed sprout maturation. These events are explained in part by reduced labeling of cholesterol by filipin III, which suggests cholesterol presentation within the membrane is affected by loss of ANXA2. Lipidomic analyses show ANXA2 is required for proper levels of 42 lipid species. Notably, loss of ANXA2 results in accumulation of phosphatidylcholine (PC) (16:0_16:0), also known as dipalmitoylphosphatidylcholine (DPPC). Treatment of endothelial cells with DPPC mimics the shANXA2 phenotype, leading to disruption of Arp2 recruitment, enhanced sprout initiation, and inhibition of sprout maturation. Altogether, the data reveal a novel role for ANXA2 in S1P-mediated junctional stabilization and lipid homeostasis; ANXA2 maintains composition of the plasma membrane, which limits DPPC accumulation and excessive sprout initiation, while allowing for Arp2 recruitment. This ultimately enables maturation into a multicellular sprouting structure, which is crucial for angiogenesis.
2 |. MATERIALS AND METHODS
2.1 |. Cell culture
Authenticated human umbilical vein endothelial cells (HUVEC) obtained from Lonza (cat. C2517A, CC-2517) were cultured on 1 mg/ml gelatin-coated flasks prior to use for experiments at passage 3–6. Growth medium previously described40 consisted of M199 (Gibco, NY) supplemented with 0.4 mg/ml bovine hypothalamic extract (Pel-Freeze Biologicals, AR), 100 μg/ml heparin, 10% fetal bovine serum (FBS) (Gibco, NY), penicillin, and streptomycin. Human embryonic kidney 293 cells (HEK 293FT) obtained from Lonza were cultured on flasks coated with 20 μg/ml collagen type I. Growth medium previously described34 consisted of Dulbecco’s modified eagle medium (Gibco, NY) supplemented with 10% FBS, and antibiotics. HEK 293 used were within passages 3–14.
2.2 |. Making lentivirus
For each shRNA construct, 1250 ng plasmid DNA was transfected into 1 × 106 HEK 293FT cells with 1250 ng pLP1, pLP2, and pLP/VSVG packaging plasmids in a 25 cm2 flask. Lentiviral backbone vectors for shβ2M, shANXA2, and shArp2 were purchased from Millipore Sigma (TRCN057254, TRCN0000056145, and TRCN0000113861, respectively). To enhance packaging of all constructs, they were incubated for 20 min in polyethylenimine (3:1 ratio to DNA used) in lactate-buffered saline (PH 4.0). After 72 h, the lentivirus was collected and concentrated using Lenti-X™ concentrator per manufacturer instructions (Clontech, CA). Following concentration, lentivirus was suspended in M199 at 1/20th of the original volume and stored at −80°C.
2.3 |. Lentiviral transduction
HUVECs were transduced with lentiviral shRNA and polybrene prior to being seeded onto gelatin-coated flasks at 50% confluency, as previously described.34 After 5 h, growth medium was replaced, and cells were incubated for 4 days.41 Knock-down was confirmed via Western blot.
2.4 |. Western blotting
Cell lysates were collected in preheated Laemmli sample buffer with 2% β-mercaptoethanol. After heating for 5 min at 95°C, samples were briefly vortexed and spun down before separating via SDS-PAGE. Polyacrylamide concentration varied from 8% to 12% depending on the molecular weight of target proteins. Electrophoresis was performed at 150 V, 400 amp, for 1 h. Samples were then transferred onto nitrocellulose membrane (GE Healthcare, IL) at 140 V, 400 amp, for 90 min. After transfer, blots were trimmed and placed in Western blocking solution (5% milk, Tris-buffered saline with 0.1% Tween 20) for 30 min, then primary antibodies were added for 3 h at room temperature or overnight at 4°C. Blots were rinsed three times for 5 min in Tris-buffered saline with 0.1% Tween 20, then put in fresh Western blocking solution with secondary antibodies tagged with horseradish protein (HRP) for 1 h at room temperature. Blots were washed again three times for 5 min, then HRP tags were activated with enhanced chemiluminescence solution (Millipore, MA). Signal was detected and imaged via ChemiDoc™ MP Imaging System (Bio-Rad, CA).
2.5 |. Collagen I preparation
Collagen type I was isolated from rat tail tendons as previously described.40 After collection, suspended collagen was lyophilized, resuspended in 0.1% acetic acid at 7.1 mg/ml, and stored at 4°C.
2.6 |. Seeding coverslips
Glass coverslips were placed in 24-well plates and coated with 50 μl collagen I (20 μg/ml) for 1 h. HUVECs were then seeded (25 000 cells/50 μl M199) and allowed to adhere for 15 min in 5% CO2 at 37°C. After 15 min, cells on coverslips were gently mixed with a pipette for even distribution and allowed to adhere an additional 15 min. The cells were then given 500 μl of a 1:1 mix of M199 and growth medium and allowed to grow overnight. The next day, cells were serum starved in M199 with Reduced Serum II42 for at least 1 h and treated according to experimental design. After treatment, the coverslips were fixed in 4% paraformaldehyde in PBS for 20 min. Following two 15-min washes in Tris-glycine, cells were permeabilized in 0.5% Triton X-100 in PBS for 20 min, then blocked with 1% bovine serum albumin, 1% goat serum, 0.2% sodium azide, and 0.1% Triton X-100 in PBS overnight at 4°C.
2.7 |. Invasion assay
To observe the ability of HUVEC to invade, 2.5 mg/ml collagen matrices with 1 μM S1P were prepared in 96-well A/2 plates (Corning, NY) or customized silicone plates as previously described.40,43 After 30 min of equilibration at 37°C in 5% CO2, HUVECs were seeded at 30 000 cells per 100 μl of M199 containing 50 μg/ml ascorbic acid, 40 ng/ml VEGF, 40 ng/ml bFGF, and Reduced Serum II, as described.40,42 Cells were allowed to invade for the allotted time, then fixed in 3% glutaraldehyde or 4% paraformaldehyde in PBS. If fixed with glutaraldehyde, collagen matrices were rinsed with water twice prior to toluidine blue staining. Samples fixed with paraformaldehyde were rinsed with Tris-glycine twice for 15 min, permeabilized with 0.5% Triton X-100 for 30 min, and incubated in immunofluorescence blocking solution (1% bovine serum albumin, 1% goat serum, 0.2% sodium azide, and 0.1% Triton X-100) overnight and stored at 4°C.
2.8 |. 2D immunofluorescence
Coverslips were probed with primary antibodies (final concentration 1–4 μg/ml) in immunofluorescence blocking solution (see above) at 4°C overnight, then washed for 1 h in 0.1% Triton X-100 in PBS, changing the wash buffer every 20 min. Secondary AlexaFluor 488 or 594 antibodies (Invitrogen, MA) of the appropriate species were then added at room temperature for a 1 h incubation. After a final 1 h wash, 100 μM 4′,6-diamidino-2-phenylindole (DAPI) was added for 10 min, then coverslips were mounted onto a slide with Fluoro Gel (Electron Microscopy Sciences, PA) for confocal imaging.
2.9 |. 3D immunofluorescence
After incubation in immunofluorescence blocking solution, collagen matrices were placed with primary antibodies (final concentration 1–4 μg/ml) in fresh immunofluorescence blocking solution for 3 h at room temperature. Samples were then washed in 0.1% Triton X-100 in PBS for 2 h, changing the wash buffer every 20 min. Collagen matrices were incubated with secondary antibodies (AlexaFluor 488, 594, and/or 647; 1:600 in immunofluorescence blocking solution) for 1 h at room temperature. They were then washed in 0.1% Triton X-100 in PBS for 2 h, changing the buffer every 20 min. Samples underwent an additional wash overnight in fresh 0.1% Triton X-100 in PBS before labeling with DAPI and imagining via confocal microscope.
2.10 |. Antibodies
ANXA2, Santa Cruz sc28385, http://antibodyregistry.org/AB_626677; Arp2, Abcam ab47654, http://antibodyregistry.org/AB_1139848; α-actinin 1, Santa Cruz sc17829, http://antibodyregistry.org/AB_626633; β2M, Santa Cruz sc46697, http://antibodyregistry.org/AB_626749; FAK, Cell Signaling 3285, http://antibodyregistry.org/AB_2269034; Paxillin, Santa Cruz sc365059, http://antibodyregistry.org/AB_10844193; Phalloidin-FITC, Life Technologies A12379; VE-cadherin, Santa Cruz sc9989, http://antibodyregistry.org/AB_2077957; VE-cadherin, Abcam ab33168, http://antibodyregistry.org/AB_870662; Vinculin, Sigma V9131, http://antibodyregistry.org/AB_477629; Zyxin, Sigma ABC1387-25UL, https://antibodyregistry.org/AB_2905521.
2.11 |. Filipin III labeling
Filipin III stocks were made in ethanol and stored in aliquots at −80°C according to kit instructions (Abcam, MA, ab133116). When ready for use, a working solution was made at 1:100 in PBS. Coverslips were placed cell-side down on parafilm with 20 μl of the working solution for 1 h at room temperature, followed by two 10 min washes in PBS. Coverslips were immediately transferred to a PBS-filled well in a glass-bottom plate and imaged.
2.12 |. Imaging
Phase contrast images of toluidine blue stained sprouts were obtained using an Olympus CKX41 microscope, Olympus QColor3 camera, and QCapture Pro acquisition software (2048 × 1536 resolution). Density counts were captured with a 10× objective, while invasion distance was obtained from sliced matrices imaged at 20×. Confocal images of fluorescently labeled samples were obtained using a Nikon TE 2000 confocal microscope, Nikon A1 camera, and NIS-Elements acquisition software. All confocal images were obtained at 1024 resolution, with 1.4 numerical aperture. Whole field images of cell monolayers were obtained using a 60× oil immersion objective, except for images for FiberFit Software™ analysis which used a 100× oil immersion objective. For imaging cells invading into collagen matrices, a 40× water immersion objective was used. To image filipin III labeled cells, monolayers were imaged on a Nikon Eclipse TE2000-U microscope with a 60× oil immersion objective, using an ANDOR Zyla 4 sCMOS camera. Excitation was provided by an X-cite 120 Fluorescence Illumination System, which was managed by a Prior ProScan II controller and Nikon Elements.
2.13 |. Image analysis
With the Fiji package in ImageJ, 16-bit Nikon image files (*.nd2) were opened using the Bio-Formats plugin, then maximum intensity projections (MIPs) or 3D projections were created from the Z-stack. For colocalization measurements, MIPs were split into individual color channels, then analyzed using the Coloc2 plugin. If a particular antibody signal had high background or non-specific nuclear labeling, regions of interest were defined prior to analysis, typically using VE-cadherin signal to make a junctional mask. For threshold analysis, MIPs were made, and an equal threshold applied to all images for individual experiments. Using the “Analyze Particles” tool, a map was generated of regions that passed the threshold and collected area measurements of individual objects. Image enhancement for publication was applied linearly and equally to all images within a given figure but did not alter actual values used for analysis.
2.14 |. Actin fiber analysis
FiberFit Software™, created by the Northwest Tissue Mechanics Laboratory at Boise State University, was used to measure actin fiber organization.44 Images were cropped into square ROIs as required by the software, which also limited analysis to junctional regions. Once converted to 8-bit color depth, images were uploaded into FiberFit Software™ and analyzed for fiber distribution.
2.15 |. Super resolution radial fluctuation analysis
To optimize images of actin fibers prior to analysis by FiberFit Software™, we utilized super resolution radial fluctuation (SRRF). This method, established by the Henriques lab, allowed us to overcome limitations of standard confocal imaging by accounting for fluorophore movement, blinking, and the refraction limit which blur the image.45 By capturing 100 confocal images of the same field and Z plane, SRRF generates a sharpened representative image of fiber signal with increased resolution and contrast.
2.16 |. Sample preparation for lipidomics
One T75 of HUVEC was transduced with shβ2M or shANXA2 lentivirus seeded onto a T175 and cultured for 4 days until confluent. After serum starving for 1 h, cells were activated with 1 μM S1P for an additional hour. Media was removed and cells were rinsed with HEPES buffer before being collected with trypsin, which was neutralized in equal parts FBS. Cells were diluted with M199, spun down at 300g for 5 min at 4°C, suspended in 1 ml of new M199, transferred to a 1.7 ml tube, and spun down again. The media was removed and cell pellets of approximately 12 million cells were frozen using a methanol/dry ice slurry and stored at −80°C. Lipidomic analyses were performed by Creative Proteomics (Shirley, NY, USA). Cell pellets were thawed and suspended in 1.5 ml Chloroform/MeOH at 2:1 (v/v), vortexed for 1 min, and sonicated at 4°C for 30 min. Samples were centrifuged at 3000 rpm at 4°C for 10 min. The lower phase was transferred to a new tube and dried under nitrogen. Dried extract was suspended in 200 μl isopropyl alcohol/MeOH at 1:1 (v/v). 10 μl LPC (12:0) was added as an internal standard. After a final centrifuge at 12 000 rpm at 4°C for 10 min, the super-natant was transferred for liquid chromatography–mass spectrometry (LC–MS) analysis.
2.17 |. LC–MS analysis
Lipid separation was accomplished using an Ultimate 3000 LC combined with Q Exactive MS (Thermo) and screened via ESI-MS. The LC system utilized ACQUITY LPC BEH C18 (100 × 2.1 mm × 1.7 μm) with Ultimate 3000 LC. Mobile phase comprised of solvent A (60% ACN + 40% H2O + 10 mM HCOONH4) and solvent B (10% ACN + 90% isopropyl alcohol +10 mM HCOONH4) with a gradient elution (0–10.5 min, 30%–100% B; 10.5–12.5 min, 100% B; 12.5–12.51 min, 100–30% B; 12.51–16.0 min, 30% B). Flow rate was 0.3 ml × min−1. Column temperature was maintained at 40°C with sample manager set to 4°C. Mass spectrometry parameters for ESI+ and ESI− mode were as follows:
ESI+: Heater Temp 300°C; Sheath Gas Flow rate, 45 arb; Aux Gas Flow Rate, 15 arb; Sweep Gas Flow Rate, 1 arb; spray voltage, 3.0 KV; Capillary Temp, 350°C; S-Lens RF Level, 30%.
ESI−: Heater Temp 300°C, Sheath Gas Flow rate, 45 arb; Aux Gas Flow Rate, 15 arb; Sweep Gas Flow Rate, 1 arb; spray voltage, 3.2 KV; Capillary Temp, 350°C; S-Lens RF Level, 60%.
2.18 |. Multivariate statistical analysis
Peak data were merged and imported into SIMCA-P software for analysis. Principal Component Analysis was used followed by Partial Least Squares-Discriminant Analysis and Orthogonal Projections to Latent Structures Discriminant Analysis to analyze observations acquired in both ion modes. Significantly different values between groups were then filtered out using VIP values >1.5.
2.19 |. Single variable statistical analysis and clustering
Lipids with a VIP >1.5, fold change >2.0, and p-value < .05 were selected as significant values for hierarchical clustering analysis (HCA). Mean values of lipid contents from biological replicates were used to calculate lipid ratios between shβ2M and shANXA2 samples. Data were adjusted to log scale and median centered ratios were normalized. HCA was performed using Cluster 3.0 (Stanford University) and visualized using Treeview (Stanford University). Lipid ratios from two independent experiments were used for HCA, where color intensity correlates with increase and decrease relative to mean ratios.
2.20 |. Pathway analysis
A correlation network diagram using the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases was constructed (Creative Proteomics), enabling the categorization of lipids to identify potential pathways impacted by loss of ANXA2.
2.21 |. Figure creation
All figures were made via a combination of ImageJ, Microsoft PowerPoint, and GraphPad Prism.
2.22 |. Lipid supplementation
DPPC and dioleoylphosphatidylcholine (DOPC) were purchased from Avanti Polar Lipids (Alabaster, AL, USA) (cat. #850355 and #850375). Lipids were dried with a nitrogen stream and suspended as a 100 mM stock in 0.7 mM BSA and water. Stocks were sonicated in a water bath at 37°C for 90 min and stored at −20°C. To avoid possible effects from lipid oxidation, 100 mM stocks were used within 1 week.
2.23 |. Lipid isolation
Lipids were extracted from cell pellets by the Folch (2:1, v/v, chloroform/methanol) method.46,47 Briefly, cell pellets were resuspended in 300 μl of ice-cold H2O (Fisher Scientific, W7–4), transferred into 1.5 ml Bioruptor microtubes (Diagenode, C30010016), and kept on ice. Microtubes were then loaded into a Bioruptor Pico sonication device (Diagenode, B01060010) and sonicated for 10 cycles of 30 s on and 30 s off at 4°C. After sonication, 920 μl of ice-cold H2O was added to each of the samples and samples were gently vortexed. A 20 μl aliquot of sample was separated and utilized for protein quantification. The remaining 1.2 ml of sample was transferred into a 11 ml glass tube (Pyrex, 9825–16) which had been rinsed with Folch and dried prior to use. Subsequently, 2 ml of ice-cold methanol (Sigma, MX0488) was added to the glass tubes followed by 4 ml of ice-cold chloroform (Sigma, CX1059). Samples were then vortexed for 1 min and allowed to sit on ice for 30 min before being centrifuged at 800g for 5 min at 4°C. After centrifugation, the organic-rich (lower) phase was collected in a 3.7 ml glass vial and set aside. An additional 2 ml of Folch was added to the remaining aqueous-rich (upper) phase, and the extraction was repeated. The organic-rich phase was once again collected and combined with the first collection. The combined lower phases were then dried under nitrogen gas and the dried lipid was resuspended in 1 ml Folch and stored at −20°C.
2.24 |. Amplex red-based cholesterol quantification
Free and total cholesterol concentrations were measured by Amplex Red cholesterol assay kit (Invitrogen, A12216) according to the manufacturer’s instructions as previously described.47 In brief, 100 μl (out of 1 ml) of each sample was dried down under a stream of nitrogen gas and resuspended in 210 μl of the 1X reaction buffer included with the kit. Samples were run in duplicate, and fluorescence was measured on a CLARIOstarPlus microplate reader. Background signal was subtracted from each standard and sample prior to plotting the standard curve. Cholesteryl esters were determined by subtracting free cholesterol from total cholesterol.
2.25 |. Protein concentration
Protein concentration was measured by Pierce BCA protein assay kit (Thermo Scientific, 23225) according to the manufacturer’s instructions. In brief, 5 μl of each sample was mixed with 200 μl of the BCA working reagent and incubated at 60°C for 30 min. Samples were run in duplicate, and absorbance was measured on a CLARIOstarPlus microplate reader (BMG LABTECH). Background signal was subtracted from each standard and sample prior to plotting the standard curve.
2.26 |. Statistics
Within a given experiment, at least three fields per treatment were obtained and measured according to experiment design. When comparing one treatment against a control, statistical significance was determined using Student’s t-test (two-tailed) where p < .05 was considered significant under the assumption of equal variance. When comparing multiple treatments against a control, ANOVA with Dunnett’s multiple comparisons test was performed where p < .05 was considered significant. All experiments were independently repeated at least three times.
2.27 |. Supplemental materials
Additional images provide clarity regarding immunofluorescence experiments, the utility of SRRF improving the accuracy of FiberFit Software™, and information regarding lipidomic analyses.
3 |. RESULTS
3.1 |. ANXA2 knock-down results in excess sprout initiation, but failed sprout maturation
As ANXA2 stabilizes adherens junctions and interacts with F-actin, we wanted to better visualize why ANXA2 is fundamental for endothelial sprouting in collagen matrices. We therefore performed a time series of invasion and observed localization of VE-cadherin and F-actin at 1, 5, 10, and 20 h. Experimental treatment groups included expression of short hairpin RNAs directed to ANXA2 (shANXA2) and beta 2 microglobulin (shβ2M). The latter was shown previously to be a suitable negative control, because invasion responses in shβ2M expressing cells were identical to non-transduced controls.34,41,48–52 After 1 h of activation, shANXA2 cells extended twice as many initial F-actin processes into the collagen matrix compared to shβ2M controls (Figure 1A,B). Despite excess sprout initiation with loss of ANXA2 (1 h), there is also failed sprout maturation over time (20 h) as determined by formation of fewer multicellular structures that possess distinct VE-cadherin-positive junctions in shANXA2 cells (Figure 1C–G), indicating that ANXA2 functions to temper the initiation of sprouts. In addition, because the Arp2/3 complex has been established as a critical component of sprout maturation,53 we also visualized localization of Arp2. Loss of ANXA2 caused reduced Arp2 in sprouts between 10 and 20 h of invasion, so we reasoned ANXA2 may regulate localization of Arp2 and organization of F-actin to stabilize junctions. Further examination of junctional changes in the absence of ANXA2 reveal wider, destabilized junctions, which is consistent with a role for ANXA2 in junctional stabilization (Figure S1).
FIGURE 1.
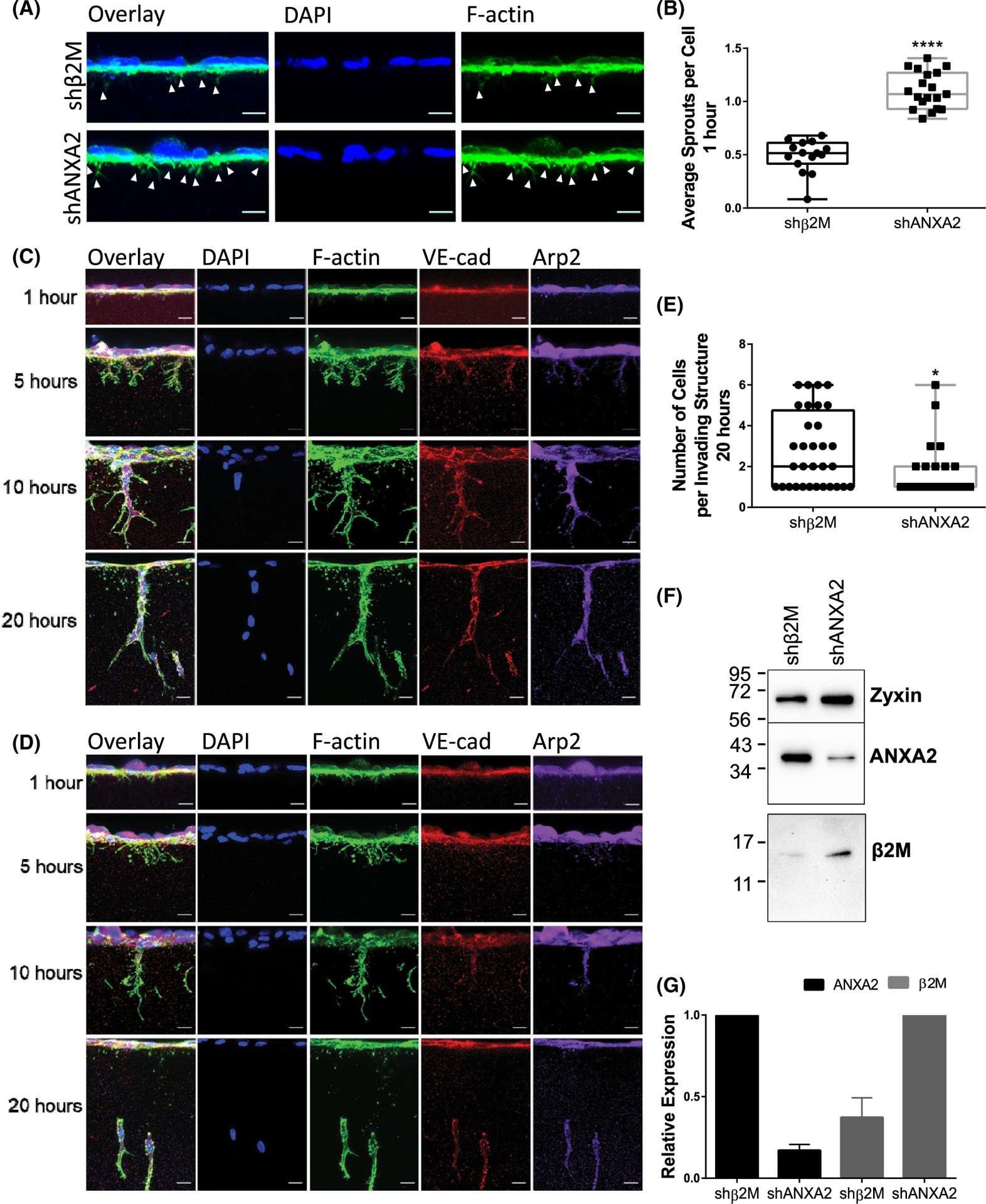
ANXA2 knock-down results in excess sprout initiation, but failed maturation. (A) Representative side views of shβ2M and shANXA2 human umbilical vein endothelial cell (HUVEC) on collagen matrices after 1 h with sphingosine 1-phosphate (S1P) + growth factors (GFs). Arrowheads denote F-Actin positive (green) processes entering the matrix. Cell nuclei are labeled with DAPI (blue). Scale bar: 10 μm. (B) Average number of initiating sprouts per cell at 1 h for fields of equal cell density. Student’s t-test, ****p < .0001. n = 15, 40× fields per treatment. Representative side views of (C) shβ2M and (D) shANXA2 HUVEC on collagen matrices after 1, 5, 10, or 20 h of invasion with S1P + GFs. Samples were stained for nuclei with DAPI (blue), F-Actin (green), VE-cadherin (red), and Arp2 (purple). Scale bar: 10 μm. (E) Quantified average cell count per invading structure after 20 h. Student’s t-test, *p = .0104. n = 25 sprout structures. shANXA2 cells fail to incorporate into multicellular structures that possess stabilized, linear junctions as labeled by VE-cadherin. (F) Western blot of ANXA2, β2M, and Zyxin (loading control). (G) Normalized signal of ANXA2 and β2M from western blots of three independent experiments as confirmation of knock-down. Error bar denotes standard deviation. All experiments repeated three times with representative data shown in panels (A–F).
3.2 |. Endothelial sprout initiation favors junctions
To understand why junctional destabilization associated with loss of ANXA2 would result in excess sprouting initiation, we imaged VE-cadherin and F-actin in wild-type cells during the early stages of sprout initiation (Figure 2A,B). Examination of initiating sprouts relative to junctions revealed an average of 89% of sprouts initiating from adherens junctions (Figure 2C), indicating junctions are strongly favored to support sprout development. Interestingly, the junctions where sprouts initiated (arrowheads, Figure 2A, Figure S2) often appeared destabilized, reminiscent of junctions in shANXA2 cells after 1 h of S1P treatment (Figure S1). This supports the concept that adherens junctions must locally destabilize prior to sprout formation, taking on a reticular morphology as junctional complexes remodel to initiate invasion.
FIGURE 2.
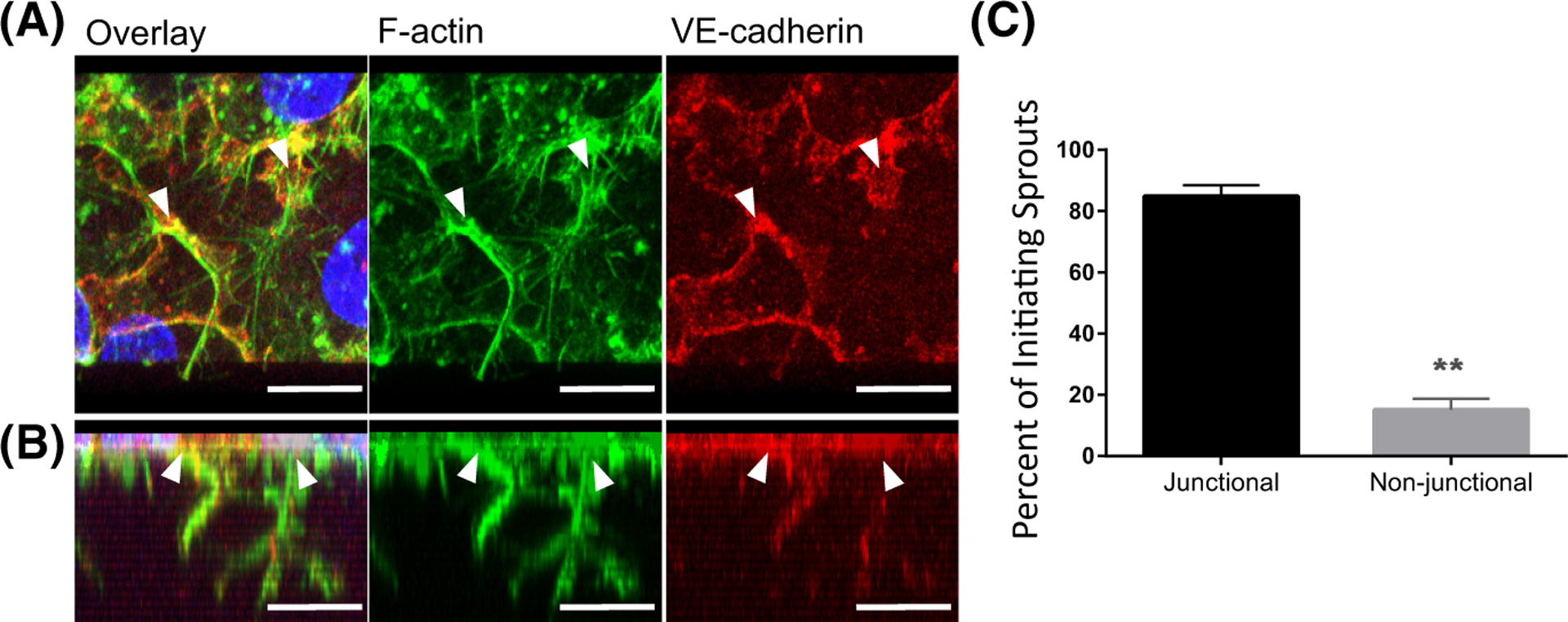
Endothelial sprout initiation favors junctions. (A) Top view of human umbilical vein endothelial cell (HUVEC) seeded on collagen matrices, activated with S1P for 1 h, then vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) for an additional hour. Cells were labeled for F-Actin (green), VE-cadherin (red). Nuclei stained with DAPI (blue). (B) Side view of 3D reconstruction from the Z-stack shown in A. White arrowheads denote origin points of sprouts. Scale bars: 10 μm. (C) Bar graph of average sprout origins (percent) of three independent experiments, +/− standard deviation. Student’s t-test: **p = .0036, n = 375 sprouts total.
3.3 |. ANXA2 knock-down causes disorder of actin fibers at junctions following S1P activation
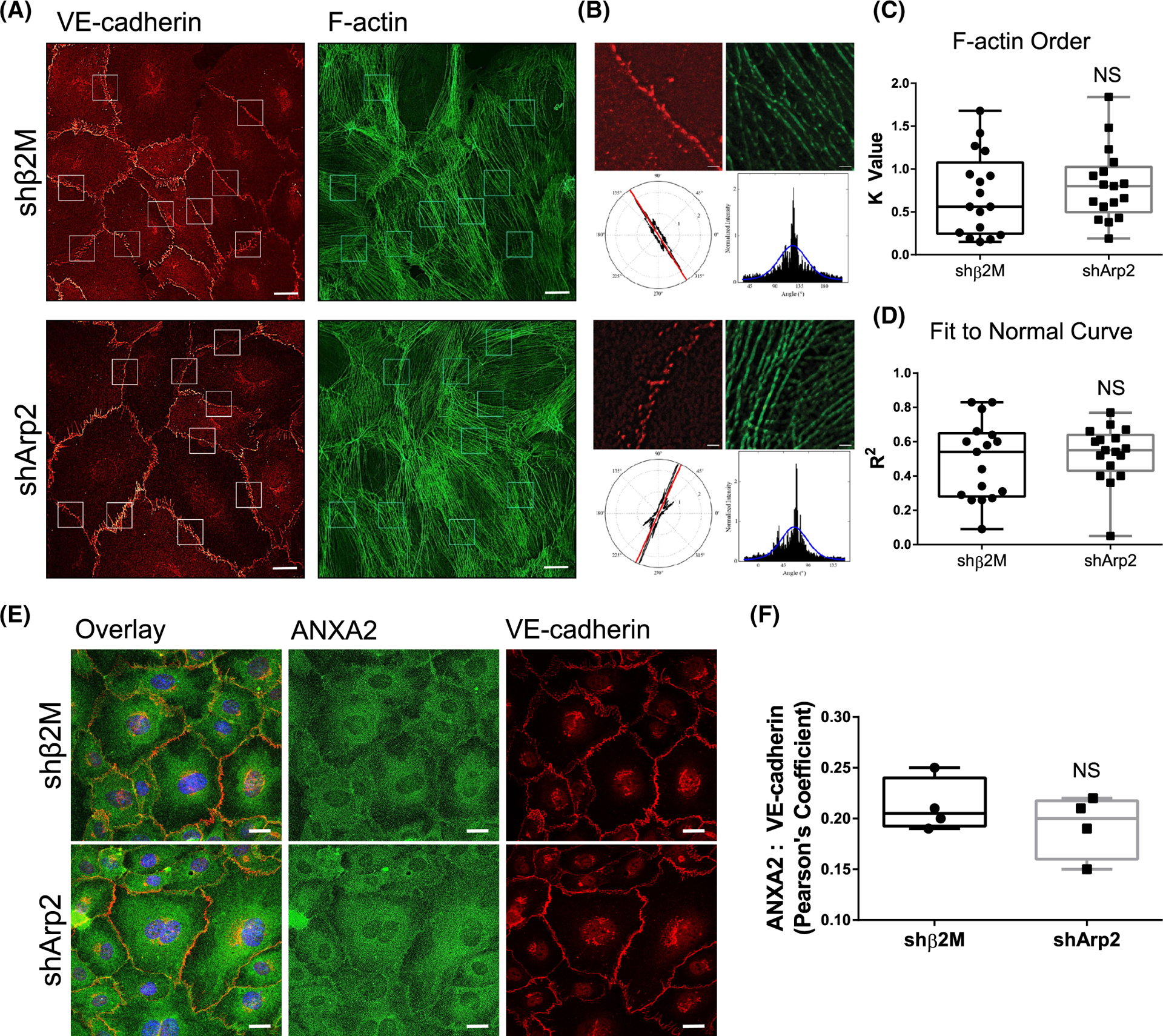
Because ANXA2 is known to assist with actin bundling,54 and bundled actin is associated with stable junctions,55 we considered that loss of ANXA2 may result in alterations of F-actin organization which would further contribute to junctional destabilization. We compared F-actin organization relative to VE-cadherin-positive junctions in shβ2M and shANXA2 cells after 1 h of S1P activation (Figure 3). To quantify actin organization, we utilized FiberFit Software™ to analyze the relative degree of order within established junctional regions between exactly two cells (Figure 3B).44 We also used SRRF analysis to optimize the clarity of phalloidin signal for the purposes of increasing image contrast.45 This allowed the threshold application by FiberFit Software™ to detect individual fibers more accurately (Figure S3). The analysis revealed that shANXA2 cells show decreased order of F-actin fibers (K Value) and decreased fit to normal curve (R2), indicating increased disorganization of F-actin fibers compared to the parallel cortical fibers that are characteristic of shβ2M controls (Figure 3C,D). We also observed a decrease in the size of focal adhesions at junctions (Figure S4), which suggests there is reduced tensional force at the cell periphery to support junctions.56,57 Altogether, this validates that shANXA2 disorganization of F-actin and destabilization of junctions favors a state of remodeling for sprout initiation.
FIGURE 3.
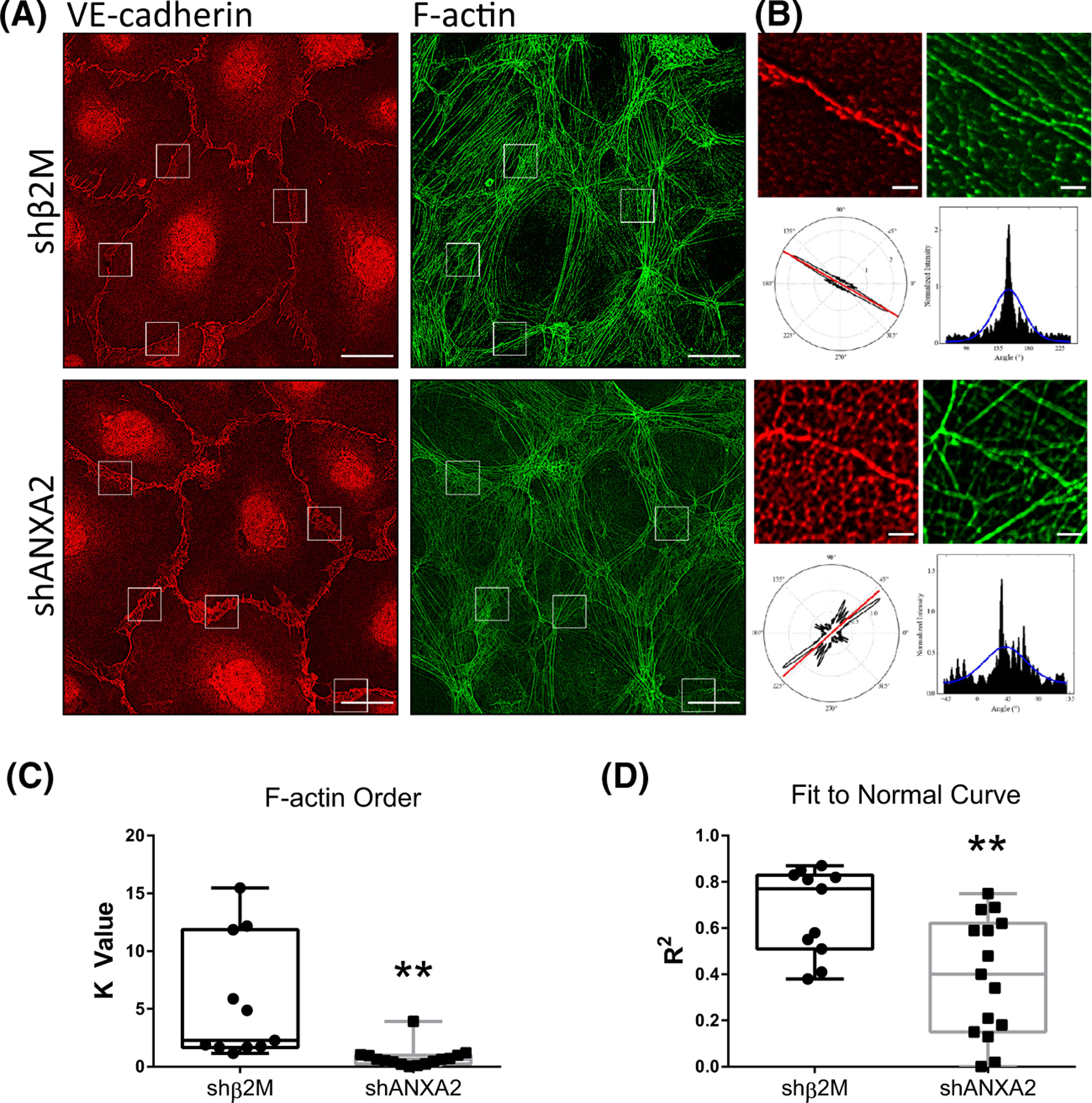
ANXA2 knock-down results in disorder of Actin fibers at junctions following sphingosine 1-phosphate (S1P) treatment. (A) Whole field of shβ2M and shANXA2 cells on coverslips following 1 h S1P treatment, labeled for VE-cadherin (red) and F-Actin (green). White boxes denote ROIs chosen for analysis in FiberFit Software™. Scale bar: 10 μm. (B) Example ROIs that were loaded into FiberFit Software™ with corresponding VE-cadherin channel as reference (only F-Actin channel was analyzed). Lower left panel denotes radial graph of F-Actin signal. Lower right graph is the radial graph plotted as a histogram. Blue line denotes Gaussian curve. Scale bar: 1 μm. (C) Averaged results from B. Graph shows the relative degree of order, quantified as K value. n = 3 fields, at least 12 junctions per treatment. **p = .002, Student’s t-test. (D) Graph shows fit to normal curve, reflecting average fit of the blue line in B. **p = .0053, Student’s t-test. All experiments independently repeated at least three times with representative data shown.
3.4 |. Localization of Arp2, but not α-actinin 1 or Vinculin, is altered with ANXA2 knock-down
Since the organization of F-actin is disrupted with ANXA2 knock-down, we considered whether other actin-regulating proteins fail to localize to junctions in response to S1P. To evaluate actin nucleation/branching, bundling, and organization, we analyzed Arp2,58,59 α-actinin 1,60,61 and Vinculin,62 respectively. In shANXA2 cells, we observed significantly less localization of Arp2 at junctions, as quantified by Pearson’s coefficient (Figure 4A,B). α-actinin 1 localized at junctions similarly to control cells; however, its organization and distribution reflected the reticular pattern of VE-cadherin and disorganization of actin fibers from loss of ANXA2 (Figure 4C,D). We also found that Vinculin localization did not change relative to VE-cadherin, maintaining similarly disorganized junctions with loss of ANXA2 (Figure 4E,F). It is interesting to note that loss of the Arp2/3 complex at junctions implies that the generation of new cortical fibers is reduced, as there would be reduced assembly of localized lamellipodia, resulting in reduced structural reinforcement of junctions.63 Altogether, these findings indicate that the organization of actin fibers relative to junctions fails, which agrees with Figure 3.
FIGURE 4.
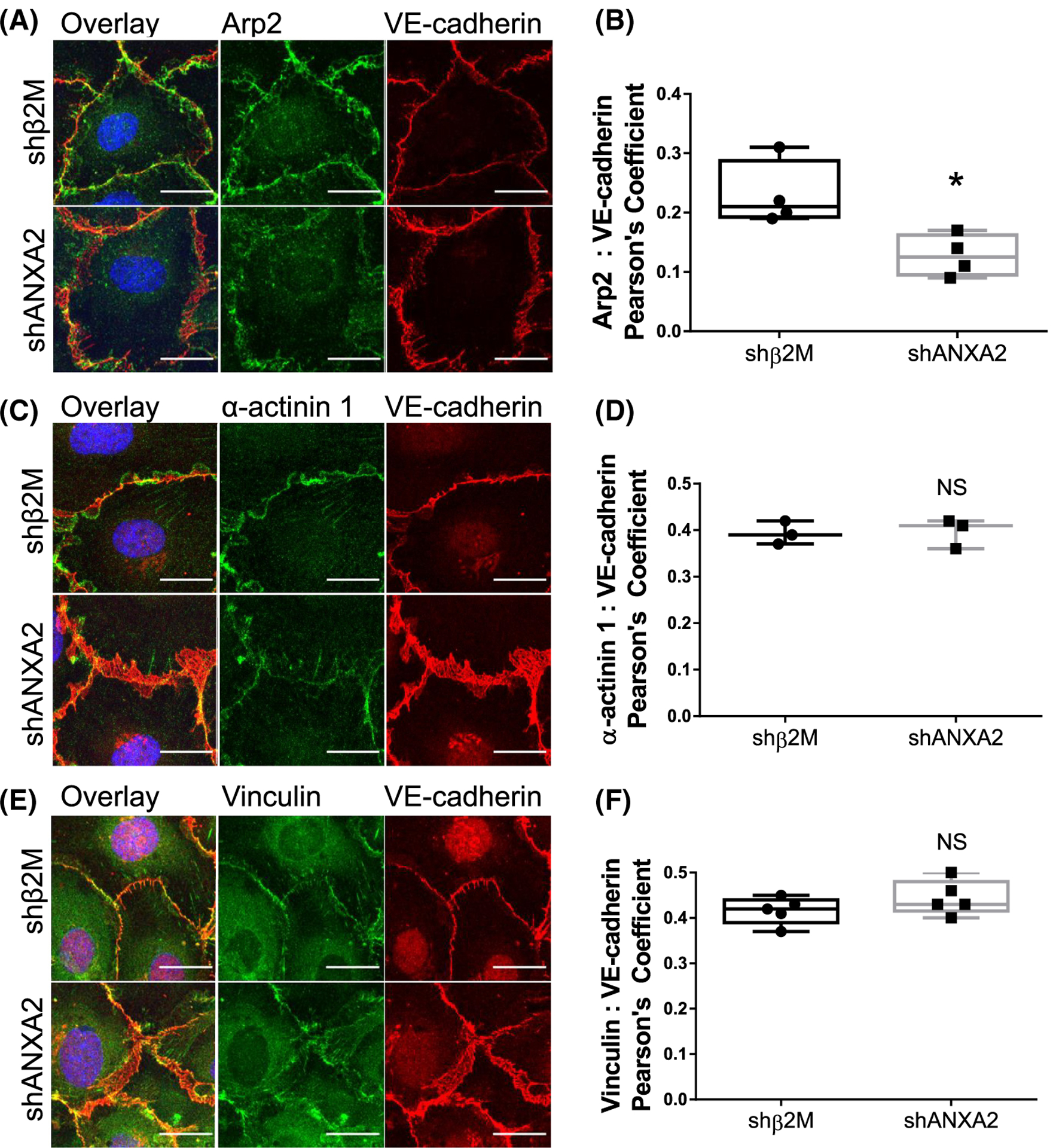
Junctional localization of Arp2, α-actinin 1, and Vinculin with ANXA2 knock-down. In all experiments, shβ2M and shANXA2 cells seeded on coverslips received 1 h sphingosine 1-phosphate (S1P) treatment prior to analysis. (A) Cells were labeled for Arp2 (green), VE-cadherin (red), and DAPI (blue). (B) Quantified colocalization between Arp2 and VE-cadherin as measured by Pearson’s Coefficient. N = 4 fields per treatment; Student’s t-test: *p = .0197. (C) Cells were then labeled for α-actinin 1 (green), VE-cadherin (red), and DAPI (blue). (D) Pearson’s Coefficient as quantification of colocalization between α-actinin 1 and VE-cadherin. N = 3 fields per treatment; Student’s t-test: NS = not significant. (E) Cells were labeled for Arp2 (green), VE-cadherin (red), and DAPI (blue). (F) Colocalization of Vinculin and VE-cadherin, measured by Pearson’s Coefficient. N = 5 fields per treatment; Student’s t-test: NS = not significant. Scale bars: 10 μm. All experiments repeated independently three times with representative data shown.
3.5 |. Loss of Arp2 impairs sprouting maturation, but does not affect initiation, cortical F-Actin order, or ANXA2 localization
To better understand the interplay between ANXA2, Arp2, and actin dynamics, we reduced Arp2 expression using a short-hairpin RNA (shArp2). After confirmation of knock-down, we performed invasion assays and found that loss of Arp2 results in impaired sprouting density and reduced invasion distance (Figure 5A–F). Unlike shANXA2 cells, we saw no change in sprout initiation at 1 h (Figure 5G,H); however, we observed a change in formation of multicellular structures after overnight invasion. The shArp2 group contained less continuous VE-cadherin-positive junctions (Figure 5I) and fewer cells compared to the shβ2M group (Figure 5J). In 2D immunofluorescence experiments, after 1 h of S1P activation, we surprisingly saw no significant change in junctional morphology with loss of Arp2, nor increased disorder of cortical F-actin (Figure 6A–D). Finally, localization of ANXA2 was not altered with loss of Arp2 (Figure 6E,F); altogether, this suggests that ANXA2 functions in a manner upstream of Arp2 organization, and Arp2 is vital for the maturation of multicellular invading structures but not sprout initiation or cortical actin organization in endothelial cell monolayers.
FIGURE 5.
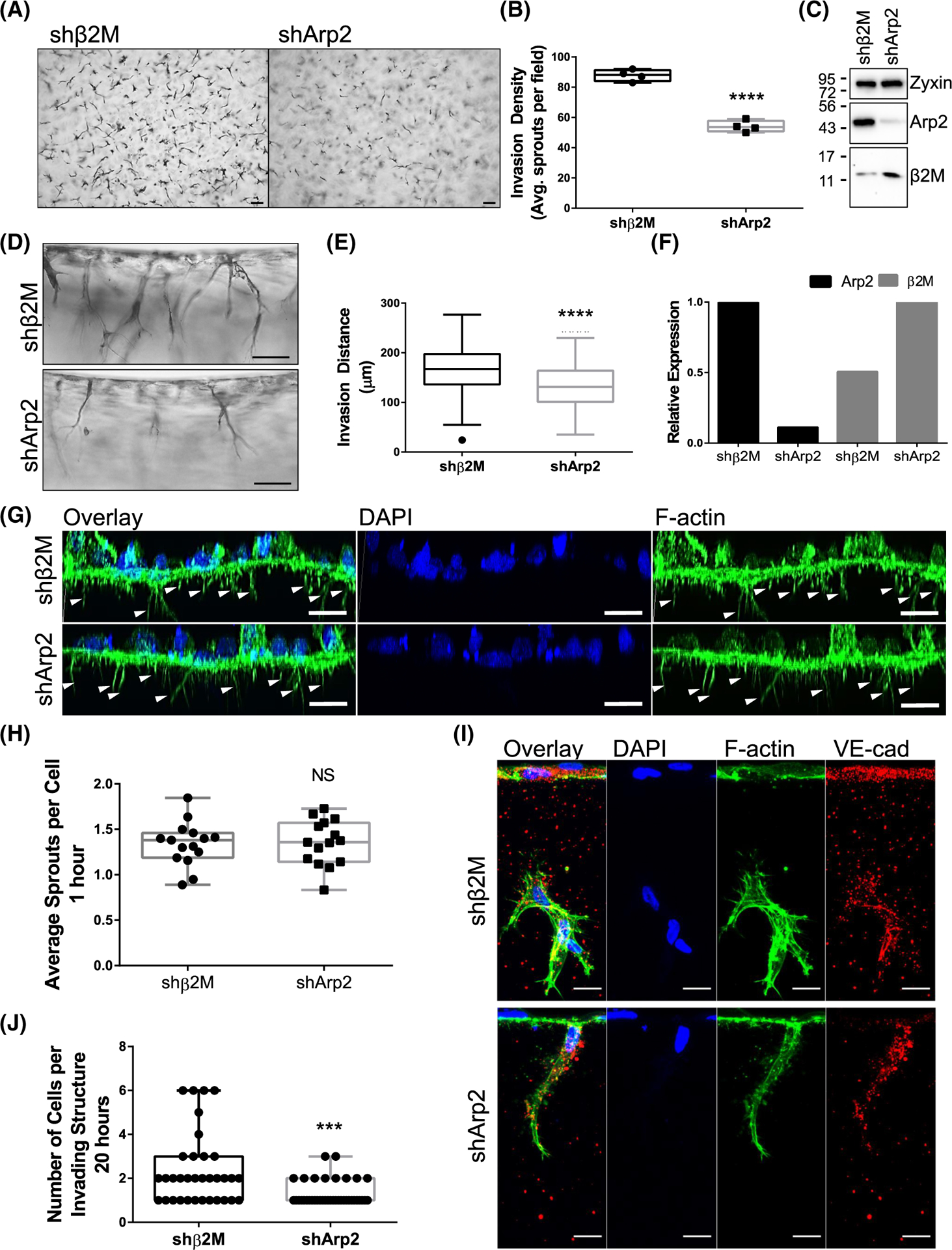
Arp2 is required for sprout maturation in 3D collagen matrices but does not affect sprout initiation. (A) Top view of sprouts in collagen matrices after 21 h. Scale bar: 100 μm. (B) Quantified sprouts per field; n = 4 fields per group. Student’s t-test: ****p < .0001. (C) Western confirmation of Arp2 and β2M knock-down, along with Zyxin loading control. (D) Side view of sprouts in collagen matrices after 21 h. Scale bar: 100 μm. (E) Quantified invasion distance. n = 100 sprouts per group; Student’s t-test: ****p < .0001. (F) Relative protein expression of Arp2 and β2M from western blots of three independent experiments. (G) 3D reconstructed Z-stack of F-Actin (green) in shβ2M or shArp2 cells after 1 h invasion. Nuclei were counterstained with DAPI (blue). Arrowheads denote initiating processes. (H) Average number of sprouts per cell after 1 h. n = 15 fields per treatment. Student’s t-test: p > .05, NS, not significant. (I) Immunofluorescence labeling of F-Actin (green), VE-cadherin (VE-cad; red), and DAPI (blue) in shβ2M and shArp2 cells after overnight invasion. Scale bar = 10 μm. (J) Average number of cells per invading structure at 20 h. n ≥ 35 sprouts per treatment; Student’s t-test: ***p = .0003.
FIGURE 6.
Loss of Arp2 does not impact organization of cortical F-Actin or localization of ANXA2. (A) Immunofluorescence of shβ2M and shArp2 cells on coverslips, labeled for F-Actin (green) and VE-cadherin (red). Scale bar: 10 μm. (B) Example output from FiberFit analysis. Scale bar: 1 μm. (C) Quantified order of cortical F-Actin from FiberFit. n = 17 junctional regions of interest from at least three fields per treatment. Student’s t-test: p > .05, NS, not significant. (D) Quantified Gaussian distribution of cortical F-Actin from FiberFit. n = 17 junctional regions of interest from at least three fields per treatment. Student’s t-test: p > .05, NS, not significant. (E) Immunofluorescence of ANXA2 (green), VE-cadherin (red), and DAPI (blue) after 1 h of S1P activation. Scale bar: 10 μm. (F) Quantified colocalization between ANXA2 and VE-cadherin in shβ2M and shArp2 cells. Student’s t-test: p > .05, NS, not significant. All experiments were performed three times, and representative results are shown.
3.6 |. The complex between filipin III and cholesterol is greatly reduced with loss of ANXA2 in intact cells
Membrane cholesterol is known to be a critical regulator of lipid rafts involving PI (4,5)P2, adherens junction stability, and actin dynamics.64,65 Though it has been shown in vitro that ANXA2 does not directly bind cholesterol66 and the presence of cholesterol enhances ANXA2 binding to membranes,67,68 to our knowledge, an ANXA2-dependent mechanism to regulate cholesterol function has not been identified. Initial experiments showed that the localization of filipin III, which specifically labels cholesterol, was enhanced at junctions in response to S1P (Figure S5), so we next considered whether localization of cholesterol is dependent on ANXA2, as loss of ANXA2 results in junctional destabilization. We observed loss of junctional filipin III signal in shANXA2 cells, suggesting either the total levels of cholesterol were reduced, or cholesterol was no longer accessible to complex with filipin III (Figure 7A,B). Disruption of the plasma membrane with TX-100 resulted in similar filipin III staining between shANXA2 and control shβ2M cells (Figure 7A,C), suggesting that cholesterol is present; however, its presentation within the membrane is altered and inaccessible by filipin III. This finding was corroborated via the Amplex-Red assay. Cholesterol concentration was normalized to ug protein, and we found no significant change in total cholesterol, free cholesterol, or cholesteryl esters between shANXA2 and shβ2M cells (Figure 7D–F). This agrees with filipin III signal in permeabilized cells, suggesting ANXA2 maintains orientation of cholesterol within intact membranes.
FIGURE 7.
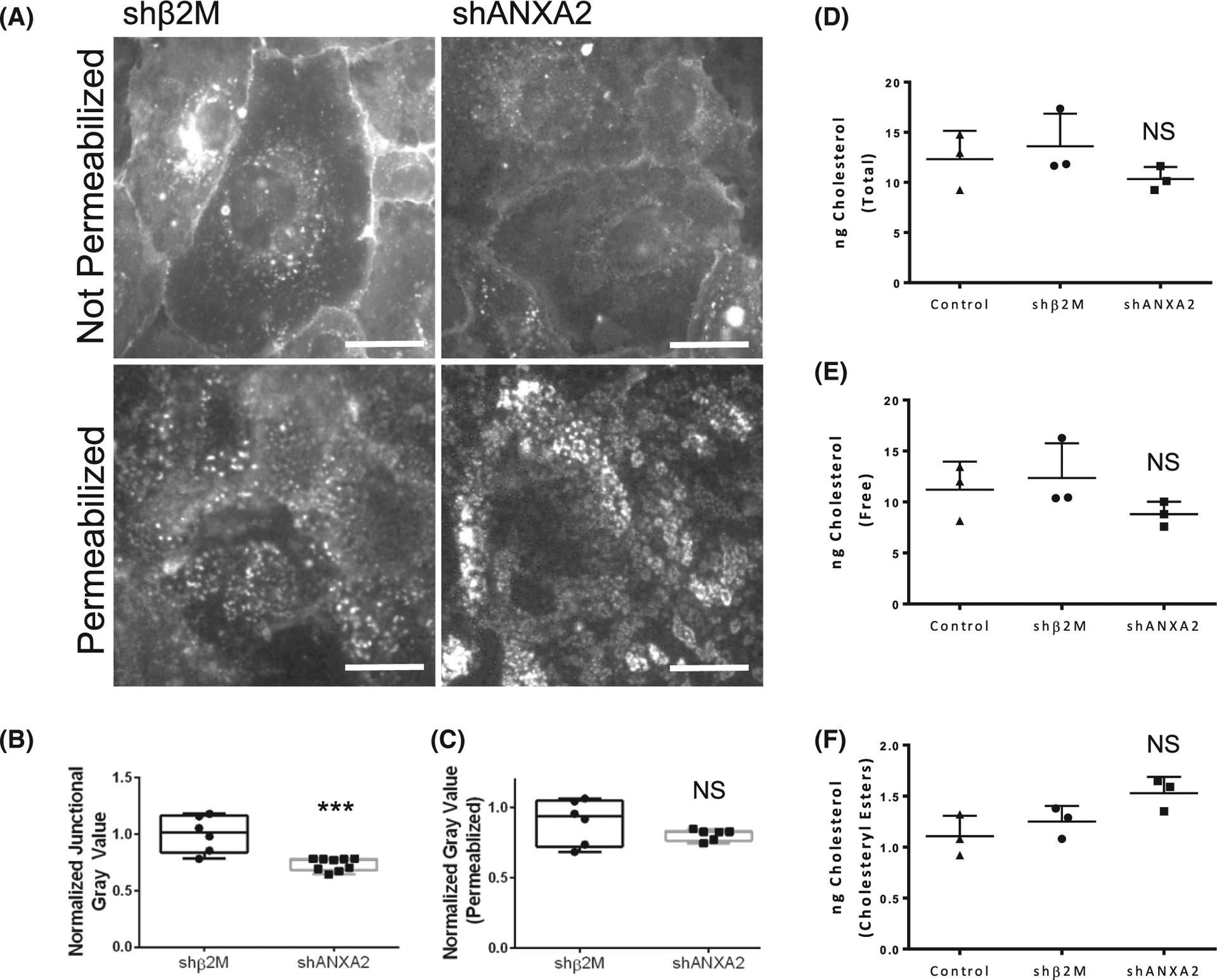
Loss of ANXA2 impairs filipin III labeling of cholesterol within intact membranes, without reducing total cholesterol. (A) shβ2M and shANXA2 cells on coverslips after 1 h of sphingosine 1-phosphate (S1P) treatment, stained with filipin III with or without permeabilization. (B) Quantified junctional filipin III signal in shβ2M and shANXA2 without permeabilization. Student’s t-test, ***p = .0004. n ≥ 6 fields per treatment. (C) Quantified whole field filipin III signal in cells with permeabilization. Student’s t-test, p > .05, NS = not significant. n = 6 fields per treatment. The Amplex-Red assay was used to quantify ng of (D) Total Cholesterol (E) Free Cholesterol, and (F) Cholesterol Esters. Cholesterol levels were normalized relative to μg protein for non-treated control, shB2M, and shANXA2 human umbilical vein endothelial cell (HUVEC). Statistical significance was determined using the Mann–Whitney test. NS, not significant.
3.7 |. ANXA2 does not broadly impact lipid classes or acyl chain metabolism
Composition of the lipid bilayer dramatically impacts the behavior of membrane cholesterol,69 so we hypothesized that loss of ANXA2 will change membrane lipid composition. To determine this, whole cell, untargeted lipidomics was used to measure changes in lipid composition with loss of ANXA2. After 1 h of S1P treatment, shβ2M and shANXA2 endothelial cells were analyzed by LC–MS. This process identified and quantified 1894 lipid species, which were organized according to general lipid class, acyl chain length, and saturation. Overall, shβ2M and shANXA2 cells were clustered separately, however lipid classes between the two groups were not significantly changed (Figure S6). We observed both shβ2M and shANXA2 cells contained equal parts phosphatidylethanolamine (PE) and PC at approximately 40% each, while phosphatidic acid (PA), PI, phosphatidylserine (PS), and phosphatidylglycerol ranged between 1% and 2% (Figure 8A). When considering acyl chain composition, there was very little change between shβ2M and shANXA2 cells (Figure 8B). The majority of stereospecific numbered-1 (sn-1) acyl chains were 16 and 18 carbons long and either saturated or monounsaturated. Sn-2 fatty acids were mainly 16, 18, 20, or 22 carbons long and were mostly monounsaturated, followed by saturated and polyunsaturated. There was a small, yet significant decrease detected in sn-2 acyl chains of 6 and 15 carbons, while an increase in 16 carbon sn-2 chains was detected with loss of ANXA2 (Figure 8B). These results indicate that most phospholipid synthesis/modification events are still occurring in the absence of ANXA2, leading us to consider that specific lipid species might be impacted.
FIGURE 8.
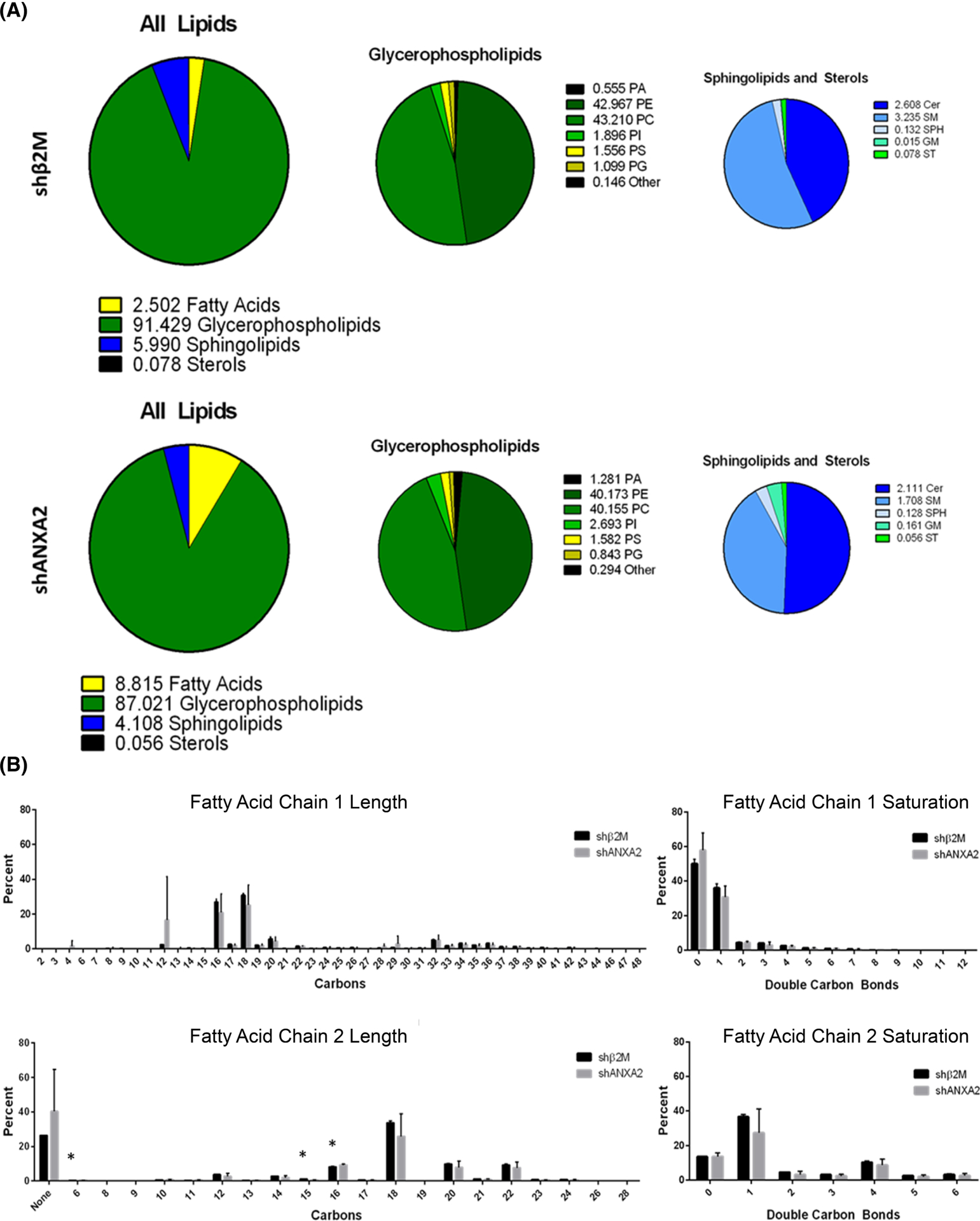
Overview of lipid profile in shβ2M versus shANXA2 cells. (A) Breakdown of total lipid profile by lipid class, averaged between three independent lots of primary human umbilical vein endothelial cell (HUVEC; 28, 38, and 72). No significance detected according to Student’s t-test across the three cell lots. (B) Breakdown of acyl chain length and saturation with loss of ANXA2. Data shown are averaged between three cell lots. Error bars represent standard deviation. Student’s t-test: *p = .021, .027, and .033 for 6, 15, and 16 carbons on sn-2, respectively. Very long chains (>28 carbons) likely include both sn-1 and sn-2 chains, which liquid chromatography–mass spectrometry (LC–MS) could not identify at higher resolution under the given parameters.
3.8 |. Loss of ANXA2 alters specific lipid species
HCA identified significant changes in 42 lipid species with loss of ANXA2 (Figure 9). This included reduction in several species of PC, PE, and PS, as well as minor species of sphingomyelin, ceramide, and diacylglycerol (Figure 9A, Table S1). Of the identified lipids, only two increased with loss of ANXA2, identified as PS (42:9) and PC (16:0_16:0), also known as DPPC. Despite the analysis in Figure 8 suggesting there were no significant shifts in lipid class, a correlation network was established using KEGG databases. Input of the significantly altered lipid species into categorical annotations allowed for pathway enrichment analysis to highlight potentially relevant biomarker networks (Figure S7).
FIGURE 9.
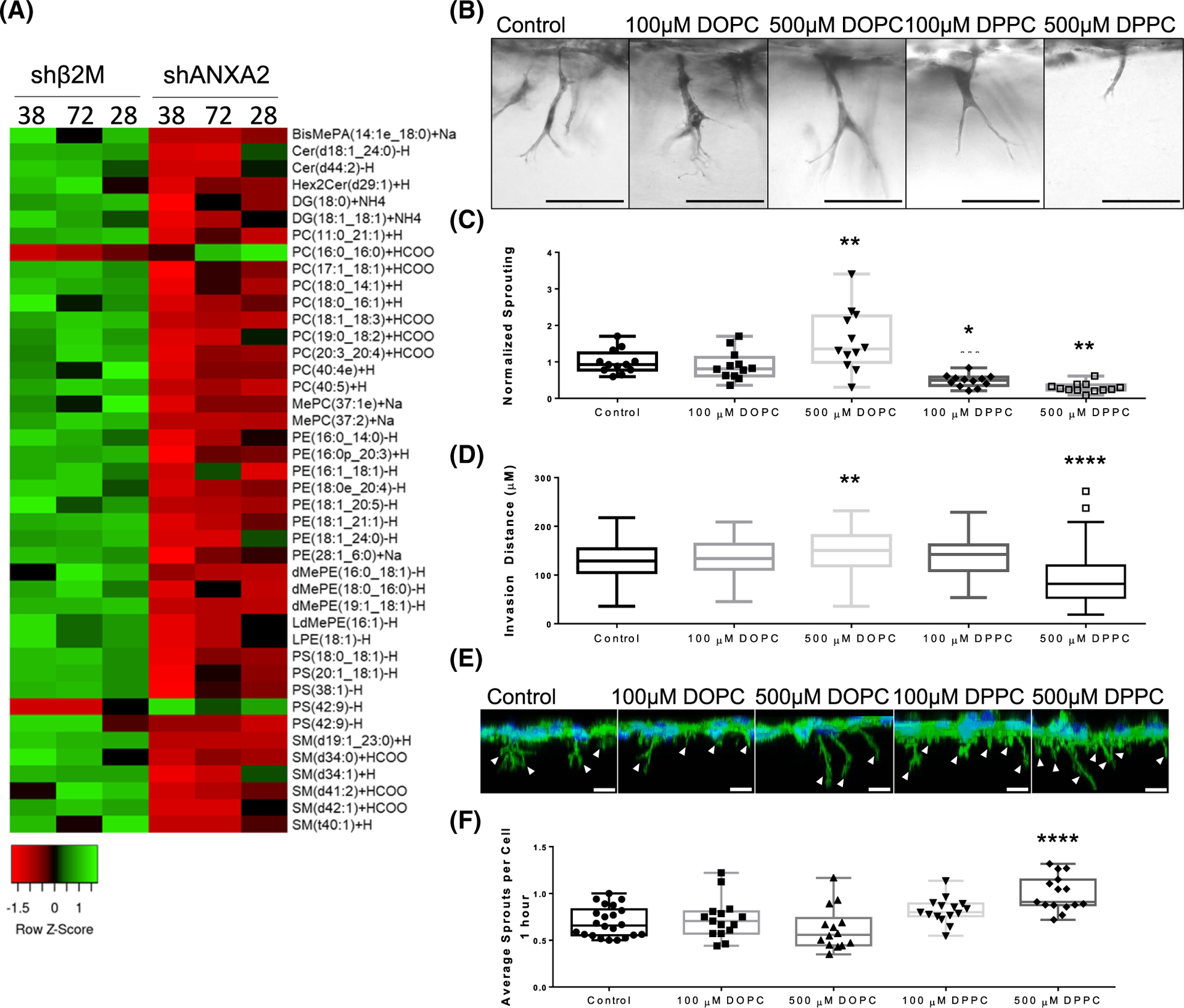
Supplementation with PC (16:0_16:0) (DPPC) impairs sprouting. (A) Heat map of lipid species altered by loss of ANXA2 in three independent lots of primary human umbilical vein endothelial cell (HUVEC; 38, 72, and 28). (B) Side view of invasion after 21 h while supplemented with DPPC or dioleoylphosphatidylcholine (DOPC). (C) Sprouting density normalized to the average control density. ANOVA, Dunnett’s multiple comparisons: **p = .0088, *p = .0241, **p = .0013 versus Control. n = 12 fields per treatment across three independent experiments. (D) Quantified average invasion distance with lipid supplementation. ANOVA, Dunnett’s multiple comparisons: **p = .0067, ****p < .0001 versus Control. n = 100 sprouts per treatment. (E) 3D reconstruction from Z-stack of HUVEC after 1 h of invasion. F-Actin (green) and DAPI (blue) was used to reveal initiating sprouts, denoted by white arrowheads. Scale bar: 10 μm. (F) Quantified initiating sprouts per cell after 1 h. ANOVA, Dunnett’s multiple comparisons: ****p < .0001 versus Control. Experiments were repeated independently at least three times with representative data shown.
3.9 |. Supplementation of PC (16:0_16:0) mimics the shANXA2 phenotype
To determine if excess PC (16:0_16:0), referred from here on as DPPC, is causal or symptomatic of endothelial dysfunction, we evaluated HUVEC on coverslips and in 3D invasion assays after supplementation with DPPC. As a positive control, we used PC (18:1_18:1), referred from here on as DOPC, which was one of the most abundant PCs detected in our system that was unaffected by loss of ANXA2. Cells were seeded onto collagen-coated coverslips or collagen matrices in the presence of these lipids to determine if there were lipid-dependent effects. Similar to ANXA2 knock-down, supplementation with DPPC decreased overnight sprouting responses, yet promoted excess sprout initiation at 1 h (Figure 9B–F). Furthermore, DPPC supplementation increased junction width (Figure 10A,B) and impaired cholesterol labeling with filipin III (Figure 10C,D). Interestingly, elevated DOPC also slightly reduced filipin III signal, despite not having an effect on junctional morphology. DOPC did not impact the organization of F-actin, while DPPC treatment resulted in decreased order and reduced normal distribution (Figure 10E–G). Lastly, DPPC treatment impaired the recruitment of Arp2 to junctions, as measured by colocalization with VE-cadherin (Figure 10H,I). In contrast, we observe no ability of DPPC to alter ANXA2 localization to junctions (Figure S8). Together, these findings indicate that DPPC treatment mimics loss of ANXA2, inducing excessive sprout initiation, but failed sprout maturation. This is concurrent with wider adherens junctions, disrupted cortical actin organization, failed recruitment of Arp2, and disruption of cholesterol within membranes.
FIGURE 10.
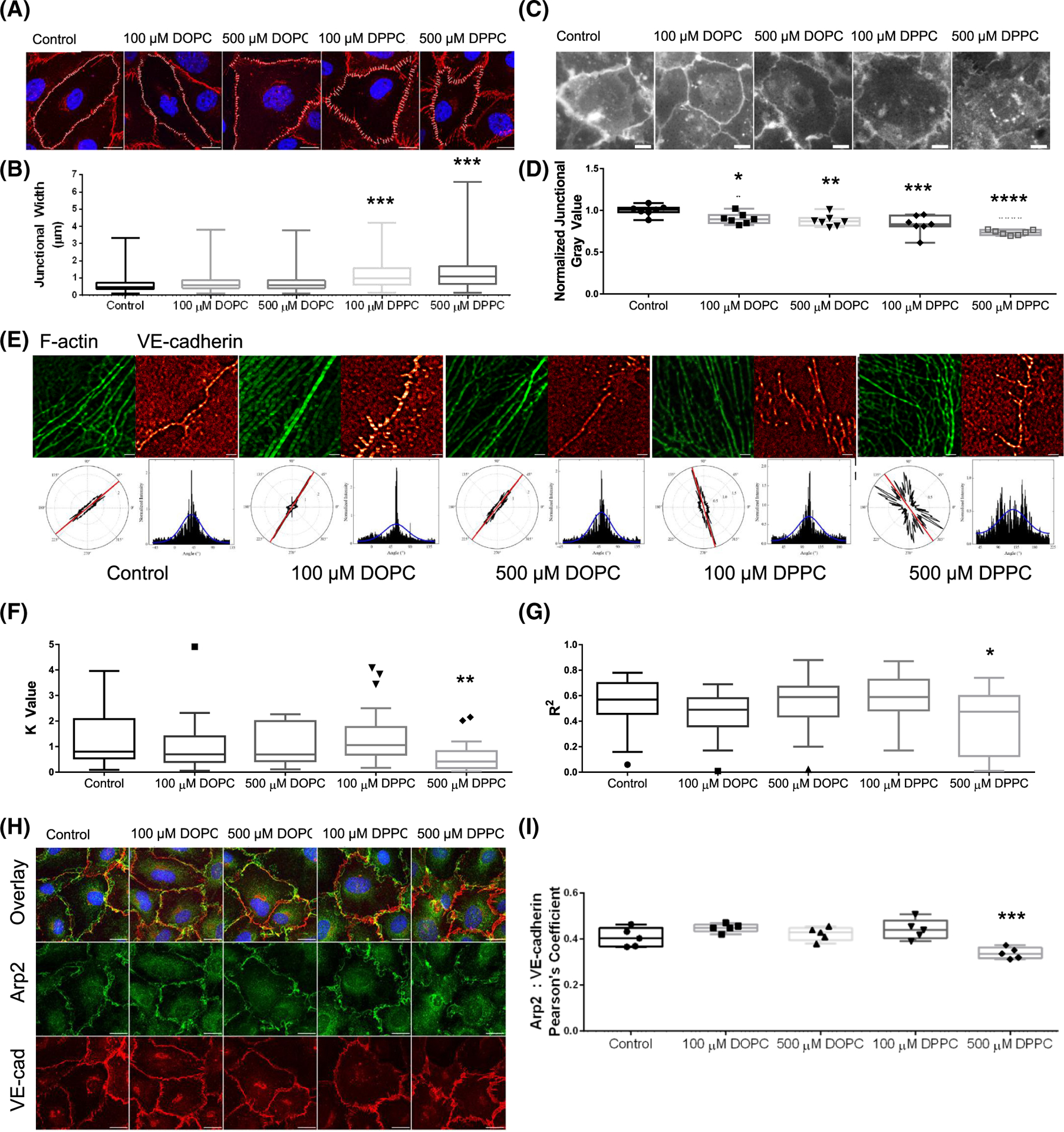
Supplementation with dipalmitoylphosphatidylcholine (DPPC) negatively alters junctional organization. (A) VE-cadherin signal of wild-type cells on coverslips after overnight treatment with DPPC and dioleoylphosphatidylcholine (DOPC), then treated with sphingosine 1-phosphate (S1P) for 1 h. Cells were stained with VE-cadherin (red) and DAPI (blue). Scale bar: 10 μm. (B) Average junctional width from A. ANOVA, Dunnett’s multiple comparisons: ***p = .0009, .0001 for 100 and 500 μM DPPC, respectively versus Control. n = 4 fields per treatment. (C) Filipin III signal of HUVEC after lipid supplementation overnight and 1 h S1P treatment. Scale bar: 10 μm. (D) Average junctional gray value per field, normalized to the control. ANOVA, Dunnett’s multiple comparisons: *p = .0483, **p = .0093, ***p = .0002, ****p < .0001 versus Control. n = 7 fields per treatment. (E) FiberFit™ analysis of human umbilical vein endothelial cell (HUVEC) seeded on coverslips overnight with DPPC or DOPC treatment, followed by 1 h S1P treatment. (F) K value of F-Actin organization from FiberFit™. ANOVA, Dunnett’s multiple comparisons: **p = .0048. n = 20 ROIs from 4 fields per treatment. (G) R2 value of F-Actin distribution from FiberFit™. ANOVA, Dunnett’s multiple comparisons: *p = .0272. n = 20 ROIs from 4 fields per treatment. (H) Immunofluorescence staining of Arp2 (green), VE-cadherin (VE-cad; red), and DAPI (blue) localization after overnight lipid supplementation and 1 h S1P treatment. Scale bar: 10 μm. (I) Quantified colocalization between Arp2 and VE-cadherin using Pearson’s Coefficient. ANOVA, Dunnett’s multiple comparisons: ***p = .0003 versus Control. n = 5 fields per treatment. All experiments were repeated independently at least three times with representative data shown.
4 |. DISCUSSION
For a cell to carry out complex morphological changes, such as sprout formation within an extracellular matrix, coordination between the membrane and protein complexes is required. Dramatic shifts in cell structure inevitably result in regions of enhanced stress, which require targeted reinforcement to overcome energy requirements and remain stable.70 The results shown here highlight the importance of the adaptor protein, ANXA2, in organizing junctional complexes. We found that ANXA2 facilitates organization of actin fibers at junctions, likely through regulation of membrane composition. When cells lacking ANXA2 are activated with S1P, there is accumulation of DPPC, which causes the actin cytoskeleton to become disorganized and adherens junctions to become destabilized. While destabilized junctions are a favored site of sprout initiation, the hyper-initiation seen with ANXA2 knock-down ultimately results in failed sprout maturation, as cells cannot assemble into multicellular structures. These results illustrate that angiogenesis is a balance between opposing dynamic forces on the junctional barrier that requires careful coordination at the membrane.
We report for the first time that ANXA2 is upstream of Arp2 recruitment to junctions, and this has particularly interesting implications for cell invasion in 3D. In cancer cells, the Arp2/3 complex is a critical component of invadopodia, the structures that assemble to promote cell invasion and degradation of the extracellular matrix.71,72 In addition to the Arp2/3 complex, α-actinin and Vinculin have been shown to play critical roles in invadopodia function.73–76 While invadopodia is a term generally reserved for cancer cells, the overlap between proteins localized to invadopodia and these responsible for endothelial cell invasion is undeniable. It is also striking that in our model, S1P selectively recruits these proteins to endothelial junctions. Junctional organization of ANXA2, Arp2, and F-actin allows these assembled protein complexes to simultaneously stabilize junctions and recruit the cellular machinery needed to invade and extend sprouts into the matrix.
Recently, invasive structures in endothelial cells were characterized and termed dactylopodia, based on similar appearance to structures in amoeboid cells.53 A defining attribute of the dactylopodia structure is recruitment of the Arp2/3 complex, which enables expansion of thin filopodia into a trunk that advances angiogenic development. As seen in Figure 1, control cells show enhanced Arp2 signal in sprouts by 10 h of invasion, while shANXA2 cells do not. If the Arp2/3 complex is not recruited appropriately (either to junctions or sprouts extending from junctions), then it follows that actin cytoskeletal dynamics and polymerization would be affected, resulting in a less stable structure. To our knowledge, this is the first report showing ANXA2 is necessary for proper localization of Arp2 to endothelial adherens junctions. It has been shown that ANXA2 works in cooperation with the Arp2/3 complex during F-actin assembly, endosome biogenesis, and autophagosme formation;77–79 however, dependence of the Arp2/3 complex on ANXA2 had not been fully established. While studies on ANXA2 have consistently implicated dynamics of F-actin and membranes, understanding the exact mechanisms remains a challenge due to limits in assaying specific lipid species. While it is possible that ANXA2 limits phospholipid synthesis, since such a process requires guided membrane transport between organelles,80 one would expect to see more dramatic changes in the lipid profile of shANXA2 cells. Because only a small fraction of specific phospholipids was altered by loss of ANXA2, it seems there may be a more subtle mechanism of action. As ANXA2 directly binds membranes in a Ca2+-dependent manner,27 the organization of lipid domains by ANXA2 may directly affect the rate of metabolism for phospholipids, such as processing of DPPC. Conversely, ANXA2 could slow the rate of DPPC production. Although we do not provide mechanistic insights in this study, our findings provide the rationale for investigating the relationship between ANXA2 and DPPC in future studies.
As advances in lipid biology make the field much more accessible for systematic study across disciplines, future studies may identify cell-type specific shifts in lipid profiles under a variety of conditions, which would reveal new targets for identification and treatment of disease. In this project, we provide evidence that ANXA2 regulates specific lipids, particularly DPPC, which impacts positioning of cholesterol and VE-cadherin at endothelial junctions. While it has been shown in vitro that ANXA2 does not directly bind cholesterol,66 we find loss of ANXA2 reduced the ability of filipin III to complex with cholesterol. Membrane-specific reduction of filipin labeling of cholesterol has been reported in vesicle formation81 and is argued to be dependent on proper presentation of cholesterol within the membrane. The misalignment of cholesterol within junctional membranes could be explained by changes in the 42 individual lipids identified by lipidomics in shANXA2 cells, notably DPPC, which likely induces a shift in lipid segregation. If lipid domains at the plasma membrane are not organized appropriately, transmembrane complexes may lose stability, resulting in destabilized adherens junctions. This is mimicked by the exogenous addition of DPPC, where supplementation in HUVEC resulted in a reduced ability of filipin to complex with cholesterol, wider adherens junctions, and impaired sprouting as in shANXA2 cells.
Interestingly, DPPC is the major component of lung surfactant, which significantly alters the properties of the alveolar surface for proper lung function.82 This may be relevant to our observation of the effect of DPPC on endothelial adherens junctions. In the event of damage to the lung epithelium, the endothelium would be exposed to leaking lung surfactant, opening junctions to enable recruitment of immune cells. Similarly, as DPPC has been shown to be increased in parts of the brain in an Alzheimer’s disease mouse model,36 its accumulation may be causal of destabilization of the blood–brain barrier common with neurodegenerative pathologies. Accumulation of palmitic acid, the fatty acid chains found in DPPC, has also been reported to induce inflammation and impair trophoblast migration in obese pregnant women.83 We were able to identify correlations between pathological conditions, expression of DPPC, and expression of ANXA2 in a variety of reports (Table 1). Both increased DPPC and decreased ANXA2 have been associated with esophageal cancer,84,85 osteosarcoma,86,87 and non-malignance of salivary tumors.88,89 Also interesting is a possible association of DPPC and ANXA2 in both lupus and preeclampsia, where increased DPPC can accumulate to form “active hydrophobic spots,” possibly in conjunction with antiphospholipid syndrome and development of autoantibodies against ANXA2.90–94 The connection between ANXA2 and DPPC identified here presents exciting new possibilities for management of disease conditions. As excess DPPC can induce junctional defects in endothelial cells, it will be interesting to determine if that association persists in the conditions listed in Table 1.
TABLE 1.
List of conditions associated with decreased expression of ANXA2 and increased levels of PC (16:0_16:0)
| Disease | ANXA2 | PC (16:0_16:0) | Citation |
|---|---|---|---|
| Esophageal Cancer | Decreased | Increased | [84, 85] |
| Lupus | Increased (Autoantibodies) | Increased (Active Hydrophobic Spots) | [90, 91] |
| Non-malignant Salivary Tumor | Decreased | Increased | [88, 89] |
| Osteosarcoma | Decreased | Increased | [86, 87] |
| Preeclampsia | Decreased | Possibly Increased (Identified as PC (32:0)) | [93, 94] |
The status of the junctional barrier remains a critical determinant of successful angiogenesis. As shown here, junctional destabilization facilitates sprout initiation, while maintenance of stabilized junctions is necessary for sprout maturation. This is likely modulated through proper phospholipid composition, which provides a foundation from which to organize multicellular sprouting structures. The data presented here show that when this foundation is lacking and actin cytoskeletal organization is decreased, the nascent sprout is unable to achieve coordinated cell movement. ANXA2 is crucial for junctional maturation, which, in turn, is necessary for sprout maturation. As ANXA2 knock-down results in excess DPPC, the result is destabilized junctions, followed by hyper-initiation of sprouting. However, the lack of junctional reinforcement ultimately causes failed sprout maturation at later stages of the process, involving Arp2 recruitment.
Supplementary Material
ACKNOWLEDGMENTS
We would like to extend our thanks to Dr. Stanislav Vitha at the Texas A&M Microscopy and Imaging Center for helpful advice on imaging analysis. This project was funded by the Department of Molecular and Cellular Medicine, Texas A&M Health Science Center.
Footnotes
DISCLOSURES
The authors have no competing interests in this work, financial or otherwise.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
Full lipidomics data available from the corresponding author upon request.
REFERENCES
- 1.Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26(5):441–454. doi: 10.1016/j.devcel.2013.08.020 [DOI] [PubMed] [Google Scholar]
- 2.Fonseca CG, Barbacena P, Franco CA. Endothelial cells on the move: dynamics in vascular morphogenesis and disease. Vasc Biol. 2020;2(1):H29–H43. doi: 10.1530/vb-20-0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. doi: 10.1038/nature10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dejana E, Vestweber D. Chapter six—the role of VE-cadherin in vascular morphogenesis and permeability control. In: van Roy F, ed. The Molecular Biology of Cadherins. Vol 116. Academic Press; 2013:119–144. doi: 10.1016/B978-0-12-394311-8.00006-6 [DOI] [PubMed] [Google Scholar]
- 5.Duran CL, Howell DW, Dave JM, et al. Molecular regulation of sprouting angiogenesis. Compr Physiol. 2018;8(1):153–235. doi: 10.1002/cphy.c160048 [DOI] [PubMed] [Google Scholar]
- 6.Szymborska A, Gerhardt H. Hold me, but not too tight—endothelial cell–cell junctions in angiogenesis. Cold Spring Harb Perspect Biol. 2018;10(8):1–16. doi: 10.1101/cshperspect.a029223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan SY, Rigor RR. Regulation of endothelial barrier function. Colloquium Series on Integrated Systems Physiology: From Molecule to Function. Vol. 3. Morgan and Claypool; 2011:1–146. doi: 10.4199/C00025ED1V01Y201101ISP013 [DOI] [PubMed] [Google Scholar]
- 8.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science (1979). 1983;219(4587):983 LP-985. doi: 10.1126/science.6823562 [DOI] [PubMed] [Google Scholar]
- 9.Ferrara N Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney Int. 1999;56(3):794–814. doi: 10.1046/j.1523-1755.1999.00610.x [DOI] [PubMed] [Google Scholar]
- 10.Sato Y, Rifkin DB. Autocrine activities of basic fibroblast growth factor: regulation of endothelial cell movement, plasminogen activator synthesis, and DNA synthesis. J Cell Biol. 1988;107(3):1199–1205. doi: 10.1083/jcb.107.3.1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pepper MS, Ferrara N, Orci L, Montesano R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun. 1992;189(2):824–831. doi: 10.1016/0006-291X(92)92277-5 [DOI] [PubMed] [Google Scholar]
- 12.Murakami M, Nguyen LT, Zhang ZW, et al. The FGF system has a key role in regulating vascular integrity. J Clin Investig. 2008;118(10):3355–3366. doi: 10.1172/JCI35298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatanaka K, Lanahan AA, Murakami M, Simons M. Fibroblast growth factor signaling potentiates VE-cadherin stability at adherens junctions by regulating SHP2. PLoS One. 2012;7(5):1–10. doi: 10.1371/journal.pone.0037600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kriaučiūnaitė K, Pociūtė A, Kaušylė A, Pajarskienė J, Verkhratsky A, Pivoriūnas A. Concentration-dependent duality of bFGF in regulation of barrier properties of human brain endothelial cells. J Cell Physiol. 2021;236:7642–7654. doi: 10.1002/jcp.30410 [DOI] [PubMed] [Google Scholar]
- 15.Yatomi Y, Ruan F, Hakomori S, Igarashi Y. Sphingosine-1-phosphate: a platelet-activating sphingolipid released from agonist-stimulated human platelets. Blood. 1995;86(1):193–202. [PubMed] [Google Scholar]
- 16.Murata N, Sato K, Kon J, et al. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J. 2000;352(3):809–815. doi: 10.1042/0264-6021:3520809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hänel P, Andréani P, Gräler MH. Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J. 2007;21(4):1202–1209. doi: 10.1096/fj.06-7433com [DOI] [PubMed] [Google Scholar]
- 18.Lee OH, Kim YM, Lee YM, et al. Sphingosine 1-phosphate induces angiogenesis: its angiogenic action and signaling mechanism in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 1999;264(3):743–750. doi: 10.1006/bbrc.1999.1586 [DOI] [PubMed] [Google Scholar]
- 19.Lee MJ, Thangada S, Claffey KP, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99(3):301–312. doi: 10.1016/s0092-8674(00)81661-x [DOI] [PubMed] [Google Scholar]
- 20.Garcia JGN, Liu F, Verin AD, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Investig. 2001;108(5):689–701. doi: 10.1172/JCI12450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bayless KJ, Davis GE. Sphingosine-1-phosphate markedly induces matrix metalloproteinase and integrin-dependent human endothelial cell invasion and lumen formation in three-dimensional collagen and fibrin matrices. Biochem Biophys Res Commun. 2003;312(4):903–913. doi: 10.1016/j.bbrc.2003.11.017 [DOI] [PubMed] [Google Scholar]
- 22.Nguyen DHT, Stapleton SC, Yang MT, et al. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc Natl Acad Sci. 2013;110(17):6712–6717. doi: 10.1073/pnas.1221526110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paatero I, Sauteur L, Lee M, et al. Junction-based lamellipodia drive endothelial cell rearrangements in vivo via a VE-cadherin-F-Actin based oscillatory cell-cell interaction. Nat Commun. 2018;9(1):3545. doi: 10.1038/s41467-018-05851-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao J, Ehling M, März S, et al. Polarized Actin and VE-cadherin dynamics regulate junctional remodelling and cell migration during sprouting angiogenesis. Nat Commun. 2017;8(1):1–20. doi: 10.1038/s41467-017-02373-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zobiack N, Rescher U, Laarmann S, Michgehl S, Schmidt MA, Gerke V. Cell-surface attachment of pedestal-forming enteropathogenic E. coli induces a clustering of raft components and a recruitment of Annexin 2. J Cell Sci. 2002;115(Pt 1):91–98. [DOI] [PubMed] [Google Scholar]
- 26.Rescher U, Ruhe D, Ludwig C, Zobiack N, Gerke V. Annexin 2 is a phosphatidylinositol (4,5)-bisphosphate binding protein recruited to Actin assembly sites at cellular membranes. J Cell Sci. 2004;117(16):3473–3480. doi: 10.1242/jcs.01208 [DOI] [PubMed] [Google Scholar]
- 27.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6(6):449–461. doi: 10.1038/nrm1661 [DOI] [PubMed] [Google Scholar]
- 28.Hayes MJ, Shao D, Bailly M, Moss SE. Regulation of Actin dynamics by annexin 2. EMBO J. 2006;25(9):1816–1826. doi: 10.1038/sj.emboj.7601078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin-Belmonte F, Gassama A, Datta A, et al. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128(2):383–397. doi: 10.1016/j.cell.2006.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uhlén M, Fagerberg L, Hallström BM, et al. Tissue-based map of the human proteome. Science (1979). 2015;347(6220):1–10. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 31.Human Protein Atlas. Published 2021. http://www.proteinatlas.org/
- 32.Heyraud S, Jaquinod M, Durmort C, et al. Contribution of annexin 2 to the architecture of mature endothelial adherens junctions. Mol Cell Biol. 2008;28(5):1657–1668. doi: 10.1128/mcb.00695-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo M, Flood EC, Almeida D, et al. Annexin A2 supports pulmonary microvascular integrity by linking vascular endothelial cadherin and protein tyrosine phosphatases. J Exp Med. 2017;214(9):2535–2545. doi: 10.1084/jem.20160652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su SC, Maxwell SA, Bayless KJ. Annexin 2 regulates endothelial morphogenesis by controlling AKT activation and junctional integrity. J Biol Chem. 2010;285(52):40624–40634. doi: 10.1074/jbc.M110.157271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura T, Jennings W, Epand RM. Roles of specific lipid species in the cell and their molecular mechanism. Prog Lipid Res. 2016;62:75–92. doi: 10.1016/j.plipres.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 36.Román EG d S, Llorente-Ovejero A, Martínez-Gardeazabal J, et al. Modulation of neurolipid signaling and specific lipid species in the triple transgenic mouse model of alzheimer’s disease. Int J Mol Sci. 2021;22(22):1–23. doi: 10.3390/ijms222212256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marsh D. Handbook of Lipid Bilayers. Taylor & Francis Group; 2013. [Google Scholar]
- 38.Avraham-Davidi I, Grunspan M, Yaniv K. Lipid signaling in the endothelium. Exp Cell Res. 2013;319(9):1298–1305. doi: 10.1016/j.yexcr.2013.01.009 [DOI] [PubMed] [Google Scholar]
- 39.Stratman AN, Farrelly OM, Mikelis CM, et al. Anti-angiogenic effects of VEGF stimulation on endothelium deficient in phosphoinositide recycling. Nat Commun. 2020;11(1):1204. doi: 10.1038/s41467-020-14956-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bayless KJ, Il KH, Su SC. Investigating endothelial invasion and sprouting behavior in three-dimensional collagen matrices. Nat Protoc. 2009;4(12):1888–1898. doi: 10.1038/nprot.2009.221 [DOI] [PubMed] [Google Scholar]
- 41.Dave JM, Abbey CA, Duran CL, Seo H, Johnson GA, Bayless KJ. Hic-5 mediates the initiation of endothelial sprouting by regulating a key surface metalloproteinase. J Cell Sci. 2016;129(4):743–756. doi: 10.1242/jcs.170571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su SC, Mendoza EA, Il KH, Bayless KJ. Molecular profile of endothelial invasion of three-dimensional collagen matrices: insights into angiogenic sprout induction in wound healing. Am J Physiol Cell Physiol. 2008;295(5):C1215–C1229. doi: 10.1152/ajpcell.00336.2008 [DOI] [PubMed] [Google Scholar]
- 43.Kang H, Bayless KJ, Kaunas R. Fluid shear stress modulates endothelial cell invasion into three-dimensional collagen matrices. Am J Physiol Heart Circ Physiol. 2008;295(5):H2087–H2097. doi: 10.1152/ajpheart.00281.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morrill EE, Tulepbergenov AN, Stender CJ, Lamichhane R, Brown RJ, Lujan TJ. A validated software application to measure fiber organization in soft tissue. Biomech Model Mechanobiol. 2016;15(6):1467–1478. doi: 10.1007/s10237-016-0776-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gustafsson N, Culley S, Ashdown G, Owen DM, Pereira PM, Henriques R. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat Commun. 2016;7:1–9. doi: 10.1038/ncomms12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497–509. [PubMed] [Google Scholar]
- 47.Salinas ML, Fuentes NR, Choate R, Wright RC, McMurray DN, Chapkin RS. AdipoRon attenuates Wnt signaling by reducing cholesterol-dependent plasma membrane rigidity. Biophys J. 2020;118(4):885–897. doi: 10.1016/J.BPJ.2019.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang H, Kwak HI, Kaunas R, Bayless KJ. Fluid shear stress and sphingosine 1-phosphate activate calpain to promote membrane type 1 matrix metalloproteinase (MT1-MMP) membrane translocation and endothelial invasion into three-dimensional collagen matrices. J Biol Chem. 2011;286(49):42017–42026. doi: 10.1074/jbc.M111.290841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwak H, Kang H, Dave JM, et al. Calpain-mediated vimentin cleavage occurs upstream of MT1-MMP membrane translocation to facilitate endothelial sprout initiation. Angiogenesis. 2012;15(2):287–303. doi: 10.1007/s10456-012-9262-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dave JM, Kang H, Abbey CA, Maxwell SA, Bayless KJ. Proteomic profiling of endothelial invasion revealed receptor for activated C kinase 1 (RACK1) complexed with vimentin to regulate focal adhesion kinase (FAK). J Biol Chem. 2013;288(42):30720–30733. doi: 10.1074/jbc.M113.512467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duran CL, Abbey CA, Bayless KJ. Establishment of a three-dimensional model to study human uterine angiogenesis. Mol Hum Reprod. 2018;24(2):74–93. doi: 10.1093/molehr/gax064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abbey CA, Bayless KJ. Matrix density alters zyxin phosphorylation, which limits peripheral process formation and extension in endothelial cells invading 3D collagen matrices. Matrix Biol. 2014;38:36–47. doi: 10.1016/j.matbio.2014.06.006 [DOI] [PubMed] [Google Scholar]
- 53.Figueiredo AM, Barbacena P, Russo A, et al. Endothelial cell invasion is controlled by dactylopodia. Proc Natl Acad Sci U S A. 2021;118(18):1–10. doi: 10.1073/pnas.2023829118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerke V, Weber K. Identity of p36K phosphorylated upon Rous sarcoma virus transformation with a protein purified from brush borders; calcium-dependent binding to non-erythroid spectrin and F-Actin. EMBO J. 1984;3(1):227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brückner BR, Nöding H, Skamrahl M, Janshoff A. Mechanical and morphological response of confluent epithelial cell layers to reinforcement and dissolution of the F-Actin cytoskeleton. Prog Biophys Mol Biol. 2019;144:77–90. doi: 10.1016/j.pbiomolbio.2018.08.010 [DOI] [PubMed] [Google Scholar]
- 56.Sun X, Shikata Y, Wang L, et al. Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc Res. 2009;77(3):304–313. doi: 10.1016/j.mvr.2008.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oakes PW, Beckham Y, Stricker J, Gardel ML. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J Cell Biol. 2012;196(3):363–374. doi: 10.1083/jcb.201107042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by Arp2/3 protein complex at the surface of listeria monocytogenes. Nature. 1997;385(6613):265–269. doi: 10.1038/385265a0 [DOI] [PubMed] [Google Scholar]
- 59.Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with Actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci U S A. 1998;95(11):6181–6186. doi: 10.1073/pnas.95.11.6181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maruyama K, Ebashi S. α-Actinin, a new structural protein from striated muscle: II. Action on Actin. J Biochem. 1965;58(1):13–19. doi: 10.1093/oxfordjournals.jbchem.a128158 [DOI] [PubMed] [Google Scholar]
- 61.Meyer RK, Aebi U. Bundling of Actin filaments by alpha-actinin depends on its molecular length. J Cell Biol. 1990;110(6):2013–2024. doi: 10.1083/jcb.110.6.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huveneers S, Oldenburg J, Spanjaard E, et al. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol. 2012;196(5):641–652. doi: 10.1083/jcb.201108120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Breslin JW, Zhang XE, Worthylake RA, Souza-Smith FM. Involvement of local lamellipodia in endothelial barrier function. PLoS One. 2015;10(2):1–31. doi: 10.1371/journal.pone.0117970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pike LJ, Casey L. Localization and turnover of phosphatidylinositol 4,5-bisphosphate in caveolin-enriched membrane domains. J Biol Chem. 1996;271(43):26453–26456. doi: 10.1074/jbc.271.43.26453 [DOI] [PubMed] [Google Scholar]
- 65.Baumgartner W, Weth A, Gutberlet J, Harms G, Groschner K. Localization of VE-cadherin in plasmalemmal cholesterol rich microdomains and the effects of cholesterol depletion on VE-cadherin mediated cell-cell adhesion. Biochim Biophys Acta Mol Cell Biol Lipids. 2014;1841(12):1725–1732. doi: 10.1016/j.bbalip.2014.08.016 [DOI] [PubMed] [Google Scholar]
- 66.Gokhale NA, Abraham A, Digman MA, Gratton E, Cho W. Phosphoinositide specificity of and mechanism of lipid domain formation by annexin A2-p11 heterotetramer. J Biol Chem. 2005;280(52):42831–42840. doi: 10.1074/jbc.M508129200 [DOI] [PubMed] [Google Scholar]
- 67.Ayala-Sanmartin J, Henry JP, Pradel LA. Cholesterol regulates membrane binding and aggregation by annexin 2 at submicromolar Ca2+ concentration. Biochim Biophys Acta Biomembr. 2001;1510(1–2):18–28. doi: 10.1016/S0005-2736(00)00262-5 [DOI] [PubMed] [Google Scholar]
- 68.Ayala-Sanmartin J Cholesterol enhances phospholipid binding and aggregation of annexins by their core domain. Biochem Biophys Res Commun. 2001;283(1):72–79. doi: 10.1006/bbrc.2001.4748 [DOI] [PubMed] [Google Scholar]
- 69.Marquardt D, Kučerka N, Wassall SR, Harroun TA, Katsaras J. Cholesterol’s location in lipid bilayers. Chem Phys Lipids. 2016;199:17–25. doi: 10.1016/j.chemphyslip.2016.04.001 [DOI] [PubMed] [Google Scholar]
- 70.Kessels MM, Qualmann B. Interplay between membrane curvature and the Actin cytoskeleton. Curr Opin Cell Biol. 2021;68:10–19. doi: 10.1016/j.ceb.2020.08.008 [DOI] [PubMed] [Google Scholar]
- 71.Lorenz M, Yamaguchi H, Wang Y, Singer RH, Condeelis J. Imaging sites of N-WASP activity in lamellipodia and invadopodia of carcinoma cells. Curr Biol. 2004;14(8):697–703. doi: 10.1016/j.cub.2004.04.008 [DOI] [PubMed] [Google Scholar]
- 72.Murphy DA, Courtneidge SA. The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12(7):413–426. doi: 10.1038/nrm3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.David-Pfeuty T, Singer SJ. Altered distributions of the cytoskeletal proteins vinculin and α-actinin in cultured fibroblasts transformed by Rous sarcoma virus. Proc Natl Acad Sci U S A. 1980;77(11 I):6687–6691. doi: 10.1073/pnas.77.11.6687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marchisio PC, Cirillo D, Teti A, Zambonin-Zallone A, Tarone G. Rous sarcoma virus-transformed fibroblasts and cells of monocytic origin display a peculiar dot-like organization of cytoskeletal proteins involved in microfilament-membrane interactions. Exp Cell Res. 1987;169(1):202–214. doi: 10.1016/0014-4827(87)90238-2 [DOI] [PubMed] [Google Scholar]
- 75.Zallone AZ, Teti A, Gaboli M, Marchisio PC. Beta3 subunit of vitronectin receptor is present in osteoclast adhesion structures and not in other monocyte-macrophage derived cells. Connect Tissue Res. 1989;20(1–4):143–149. doi: 10.3109/03008208909023882 [DOI] [PubMed] [Google Scholar]
- 76.Hirooka S, Akashi T, Ando N, et al. Localization of the invadopodia-related proteins actinin-1 and cortactin to matrix-contact-side cytoplasm of cancer cells in surgically resected lung adenocarcinomas. Pathobiology. 2011;78(1):10–23. doi: 10.1159/000322734 [DOI] [PubMed] [Google Scholar]
- 77.Hayes MJ, Shao DM, Grieve A, Levine T, Bailly M, Moss SE. Annexin A2 at the interface between F-Actin and membranes enriched in phosphatidylinositol 4,5,-bisphosphate. Biochim Biophys Acta Mol Cell Res. 2009;1793(6):1086–1095. doi: 10.1016/j.bbamcr.2008.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morel E, Parton RG, Gruenberg J. Annexin A2-dependent polymerization of Actin mediates endosome biogenesis. Dev Cell. 2009;16(3):445–457. doi: 10.1016/j.devcel.2009.01.007 [DOI] [PubMed] [Google Scholar]
- 79.Moreau K, Ghislat G, Hochfeld W, et al. Transcriptional regulation of annexin A2 promotes starvation-induced autophagy. Nat Commun. 2015;6:1–12. doi: 10.1038/ncomms9045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vance JE. Phospholipid synthesis and transport in mammalian cells. Traffic. 2015;16(1):1–18. doi: 10.1111/tra.12230 [DOI] [PubMed] [Google Scholar]
- 81.Steer CJ, Bisher M, Blumenthal R, Steven AC. Detection of membrane cholesterol by filipin in isolated rat liver coated vesicles is dependent upon removal of the clathrin coat. J Cell Biol. 1984;99(1 I):315–319. doi: 10.1083/jcb.99.1.315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Holm BA, Wang Z, Egan EA, Notter RH. Content of dipalmitoyl phosphatidylcholine in lung surfactant: ramifications for surface activity. Pediatr Res. 1996;39(5):805–811. doi: 10.1203/00006450-199605000-00010 [DOI] [PubMed] [Google Scholar]
- 83.Rampersaud AM, Dunk CE, Lye SJ, Renaud SJ. Palmitic acid induces inflammation in placental trophoblasts and impairs their migration toward smooth muscle cells through plasminogen activator inhibitor-1. Mol Hum Reprod. 2020;26(11):850–865. doi: 10.1093/molehr/gaaa061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yuan Y, Zhao Z, Xue L, et al. Identification of diagnostic markers and lipid dysregulation in oesophageal squamous cell carcinoma through lipidomic analysis and machine learning. Br J Cancer. 2021;125(3):351–357. doi: 10.1038/s41416-021-01395-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qi YJ, Wang LD, Jiao XY, et al. Dysregulation of annexin II expression in esophageal squamous cell cancer and adjacent tissues from a high-incidence area for esophageal cancer in Henan province. Ai Zheng. 2007;26(7):730–736. [PubMed] [Google Scholar]
- 86.Gillette JM, Chan DC, Nielsen-Preiss SM. Annexin 2 expression is reduced in human osteosarcoma metastases. J Cell Biochem. 2004;92(4):820–832. doi: 10.1002/jcb.20117 [DOI] [PubMed] [Google Scholar]
- 87.Roy J, Dibaeinia P, Fan TM, Sinha S, Das A. Global analysis of osteosarcoma lipidomes reveal altered lipid profiles in metastatic versus nonmetastatic cells. J Lipid Res. 2019;60(2):375–387. doi: 10.1194/jlr.M088559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.He Q, Takizawa Y, Hayasaka T, et al. Increased phosphatidylcholine (16:0/16:0) in the folliculus lymphaticus of Warthin tumor. Anal Bioanal Chem. 2014;406(24):5815–5825. doi: 10.1007/s00216-014-7890-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu JS, Chen YT, Chiang WF, et al. Saliva protein biomarkers to detect oral squamous cell carcinoma in a high-risk population in Taiwan. Proc Natl Acad Sci U S A. 2016;113(41):11549–11554. doi: 10.1073/pnas.1612368113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arieli R, Khatib S. Dipalmitoylphosphatidylcholine in the heart of mice with lupus might support the hypothesis of dual causes of autoimmune diseases. Respir Physiol Neurobiol. 2022;300(January):103871. doi: 10.1016/j.resp.2022.103871 [DOI] [PubMed] [Google Scholar]
- 91.Salle V, Mazière JC, Brulé A, et al. Antibodies against the N-terminal domain of annexin A2 in antiphospholipid syndrome. Eur J Intern Med. 2012;23(7):665–668. doi: 10.1016/j.ejim.2012.04.006 [DOI] [PubMed] [Google Scholar]
- 92.Moroni G, Ponticelli C. Pregnancy in women with systemic lupus erythematosus (SLE). Eur J Intern Med. 2016;32:7–12. doi: 10.1016/j.ejim.2016.04.005 [DOI] [PubMed] [Google Scholar]
- 93.Garrido-Gomez T, Quiñonero A, Dominguez F, et al. Preeclampsia: a defect in decidualization is associated with deficiency of annexin A2. Am J Obstet Gynecol. 2020;222(4):376. e1–376.e17. doi: 10.1016/j.ajog.2019.11.1250 [DOI] [PubMed] [Google Scholar]
- 94.Zhang L, Bi S, Liang Y, et al. Integrated metabolomic and lipidomic analysis in the placenta of preeclampsia. Front Physiol. 2022;13(February):1–11. doi: 10.3389/fphys.2022.807583 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Full lipidomics data available from the corresponding author upon request.