

Abstract
The mammalian target of rapamycin (mTOR) is a protein kinase of the PI3K/Akt signaling pathway that regulates cell growth and division and is an attractive target for cancer therapy. Many reports on finding alternative mTOR inhibitors available in a database contain a mixture of active compound data with different mechanisms, which results in an increased complexity for training the machine learning models based on the chemical features of active compounds. In this study, a deep learning model supported by principal component analysis (PCA) and structural methods was used to search for an alternative mTOR inhibitor from mushrooms. The mTORC1 active compound data set from the PubChem database was first filtered for only the compounds resided near the first-generation inhibitors (rapalogs) within the first two PCA coordinates of chemical features. A deep learning model trained by the filtered data set captured the main characteristics of rapalogs and displayed the importance of steroid cores. After that, another layer of virtual screening by molecular docking calculations was performed on ternary complexes of FKBP12–FRB domains and six compound candidates with high “active” probability scores predicted by the deep learning models. Finally, all-atom molecular dynamics simulations and MMPBSA binding energy analysis were performed on two selected candidates in comparison to rapamycin, which confirmed the importance of ring groups and steroid cores for interaction networks. Trihydroxysterol from Lentinus polychrous Lev. was predicted as an interesting candidate due to the small but effective interaction network that facilitated FKBP12–FRB interactions and further stabilized the ternary complex.
1. Introduction
The mammalian target of rapamycin (mTOR) is an atypical serine/threonine (S/T) protein kinase that belongs to the phosphoinositide 3-kinase (PI3K)-related kinase family. mTOR is frequently referred to as the master regulator of cell growth, proliferation, metabolism, and survival.1−4 It interacts with several proteins to form two distinct protein complexes called mTOR complexes 1 (mTORC1) and 2 (mTORC2). Dysregulation of mTOR is associated with many diseases, such as fibrosis, obesity, type 2 diabetes, neurodegeneration, and cancer, attracting great interest in developing drugs targeting mTOR.5,6 The potent and specific mTOR inhibitor is rapamycin, a 31-membered macrocyclic lactone produced by Streptomyces hygroscopicus.7,8 It plays a critical role in inhibiting cell growth, cell cycle progression, and proliferation.9−11 Upon entering the cells, rapamycin specifically forms a gain-of-function complex with the 12 kDa FK506-binding protein (FKBP12),12,13 interacting and inhibiting the part of mTORC1. This inhibitory complex binds to the FRB (FKB12–rapamycin binding) domain from the C terminus of TOR proteins, leading to cell growth inhibitory and cytotoxic effects. At the molecular level, an experimental study exhibited the key role of FKBP12–rapamycin complex binding to the FRB domain as a ternary complex as rapamycin itself possessed only a modest affinity for the FRB domain in the absence of FKBP12.14 Despite being poorly soluble in water and unstable, rapamycin analogues, also known as rapalogs, have been created with enhanced biopharmaceutical properties.2,10 These rapalogs have been approved by the FDA as the first generation of mTOR inhibitors for combatting cancer malignancies and other diseases.
In addition to rapamycin and rapalogs, several small molecules have also demonstrated the potential to inhibit the mTOR pathway in some cancer cells.10,11,15 As the second-generation mTOR inhibitors, the small molecules bind to the ATP-binding site in the mTOR kinase domain, resulting in the downregulation of both mTORC1 and mTORC2.16−18 Examples of the second generation of mTOR inhibitors include AZD8055,19 AZD2014,19−21 CC-115,22 CC214-2,23 CC-223,24 WAY-600, WYE-354, WYE-687,25 WYE-125132,26 OSI-027,27 PP242,28 Torin 1,29 and Torin 2.30 In addition, the dual mTOR/PI3K inhibitors that effectively inhibit mTORC1 and mTORC2 are GDC-0980,31 GSK2126458, SF1126,32 NVP-BEZ235,33,34 PKI-402,35 and XL765.36 However, these second-generation inhibitors exhibit lower specificity and are likely to cause a correspondingly higher toxicity when compared to rapamycin and rapalogs.37 Increasing evidence suggested that natural compounds may exert their antiproliferative effects by inhibiting mTOR signaling, thus representing new mTOR inhibitor alternatives. For example, numerous studies have shown that curcumin inhibited the growth of various cancer cells and showed effectiveness as a chemopreventive agent in animal carcinogenesis models.38−40 Furthermore, many natural compounds, including epigallocatechin gallate (EGCG),41 caffeine,42 and resveratrol,43 have also been found to downregulate mTOR signaling in vitro. Additionally, some natural products or nutraceuticals isolated from plants also inhibit the PI3K/Akt/mTOR pathway, such as apigenin,44 fisetin,45 indoles (indole-3-carbinol and 3,3′-diindolylmethane),46 quercetin,47 and tocotrienol.48
Despite the progress reported by these studies, there remains a pressing need to find novel active compounds that have improved pharmacokinetic properties and specificity toward the mTOR complex or more potent dual PI3–mTOR inhibitors. According to the large quantity of available data, it is possible to utilize computational techniques, including deep learning as the recently most successful machine learning technique in drug discovery research, to predict and screen for active compounds and mTOR-targeting drugs. Several computational approaches and recent machine learning techniques have been utilized to search for chemical scaffolds to engender druglike properties as well as the selectivity of allosteric mTOR inhibitors.49−52 For example, one recently published work used the support vector machine (SVM) as a virtual screening strategy to identify novel mTOR inhibitors, which can supply some candidates for mTOR anticancer drugs.50 Kumari et al. also suggested that an imbalance between the numbers of active and inactive compounds in a public database could impair the result of machine learning prediction. Therefore, they used the technique called SMOTE to balance the mTOR inhibitor data from ChEMBL and demonstrated the improved prediction by various machine learning models.53 Anderson et al. applied a Bayesian machine learning model to search for inhibitors of chordoma, a rare type of cancer, and found that the mTOR inhibitor AZD2014 is the most effective candidate.54 Another work employed pharmacophore modeling to create a structure-based pharmacophoric model, which is a useful tool for discovering small molecules. Then, combined with molecular dynamics (MD) simulations, the most important interactions occurring in the ternary complex FKPB12–rapamycin–FRB were revealed as an essential step for the mTOR inhibition.51
It should be noted that there is considerable inconsistency in the data associated with the chemical structures, as they have been contributed by more than a hundred organizations. Moreover, the positive data contained substances from several subgroups, including the first, second, and other generations of mTOR inhibitors with different inhibition mechanisms that should cause uncertainty for the training set. Therefore, the difficulty could be circumvented by preprocessing the data with principal component analysis (PCA). Based on the filtered data through PCA, a deep learning model was constructed and trained to identify novel bioactive compounds from BACMUSHBASE, a database of compounds from mushrooms found in Thailand (http://bacmushbase.sci.ku.ac.th/), as potent mTOR inhibitors, analogous to rapamycin and the first-generation mTOR inhibitors (rapalogs). Then, molecular docking and atomistic molecular dynamics simulations were performed on the predicted compound candidates with high active probability to obtain the structural detail of ternary complex formation with FKPB12 and FRB domains and to verify the deep learning prediction. Therefore, this study aims to identify the natural compounds with excellent mTOR kinase inhibitory potency and to provide a basic understanding of the molecular mechanism for one of the downstream regulations of the mTOR pathway to support the discovery of new kinase-targeting drugs.
2. Computational Methods
2.1. mTOR Inhibitor Data Set
A list of compounds tested against the activity of mTOR from PubChem55,56 (NCBI gene ID: 2475) contains 4866 compounds as “active” (has an inhibitory effect against mTOR) and 48328 compounds as “inactive” (has no inhibitory effect against mTOR) as of 18 November 2020. The “active” compound list contains all generations of mTOR inhibitors. To select only compounds whose structures are similar to the first-generation mTOR inhibitors (rapalogs), 75 per-atom features were extracted from each mTOR active compound using the ConvMolFeaturizer() command from the DeepChem package. Chemical features presented in the binary vectors, e.g., atom type, degree, implicit valence, formal charges, hybridization type, aromaticity, and total number of hydrogens, enabled us to interpret the outcomes from screened data. The per-atom feature matrix was then incorporated into the structural information by calculating
| 1 |
where A is the adjacency matrix N × N of the molecule with N atoms, I is the identity matrix, F is the per-atom feature matrix N × 75, and H is the matrix N × 75, whose representation of each atom is a sum of its neighboring atoms’ features. The atom-level features in the matrix H were then pooled by averaging feature values over all atoms in the molecule, resulting in a vector 1 × 75 that contains a molecule-level fingerprint of each mTOR active compound. Finally, principal component (PC) analysis was performed on the fingerprint vectors to project the 75-dimensional features onto the first and second principal components (PC1 and PC2). The initial data set of active compounds from PubChem contains all generations of mTOR inhibitors. Here, only compounds whose structures are similar to the first-generation mTOR inhibitors based on the Euclidean distance on the PC coordinates were selected as the “positive” data set for the deep learning model (DL). Specifically, 900 compounds with the nearest distances to the centroid of 10 known rapalogs on the PC coordinates were chosen. Meanwhile, the mTOR-inactive compound data from the PubChem database (as of 18 November 2020) was used as the “negative” data set for the DL.
2.2. Deep Learning Model and Prediction on the mTOR Bioactivity
The positive and negative data sets were combined, shuffled, and used to train a deep learning model which was created by the commands in the Deepchem package.57 The model consisted of a graph convolutional layer (GraphConv) of 64 nodes with batch normalization (layers.BatchNormalization) and graph pooling (GraphPool), followed by a hidden layer of 128 nodes with batch normalization (layers.BatchNormalization). A data dropout (layers_dropout) was performed for each training with a dropout rate of 0.2. The model was trained with an optimized learning rate of 0.001. A 5-fold cross-validation was performed to evaluate the performance of the deep learning model. Finally, due to the stochastic nature of the learning algorithm, the whole data set (900 positive and 900 negative data points) was used to train three replicas of deep learning models. Then, the three independently trained deep learning models were employed to predict “active” probability scores for compounds from the BACMUSHBASE database. Compounds that achieved top-ranking consensus across all three models were selected for further validation by structural methods.
2.3. Molecular Docking
From the previous step, our deep learning model screened a number of compounds based on the similarity of chemical properties to those of the mTORC1 active compounds from the BACMUSHBASE database. However, the deep learning model could not confirm whether the screened bioactive compounds could bind to the active site of the target protein. Therefore, molecular docking calculations were performed to verify that the screened compounds could bind to the target protein at the native binding site. In this research, the AutoDock 458,59 was used to perform all molecular docking calculations, including the calculations of binding energy between the target protein and the compounds screened by the deep learning model with a predicted probability greater than 95%. Atomistic coordinates of an FKBP12–FRB complex with rapamycin were obtained from the protein databank (PDB 1FAP) before the coordinates of rapamycin were removed. Meanwhile, the atomistic coordinates of the compounds screened by the deep learning models were obtained from the PubChem database. During each molecular docking calculation, steric repulsion and hydrophobic or hydrogen bonding interactions were classified from the PDBQT coordinate files of protein target mTOR, and a screened bioactive compound with a combined knowledge-based and empirical binding energy score was provided. Then, the Iterated Local Search global optimizer implemented within AutoDock 4 was used to search for the most favorable binding mode of the bioactive compound on a protein target mTOR with the lowest binding energy score. For each docking calculation, a 30 × 30 × 30 Å3 dimension box was defined at the midpoint of the FKBP12–FRB interfacial region to limit the configuration space for only productive binding modes. After that, each docked configuration was visualized for the details of hydrogen bonding and hydrophobic interaction networks by the LigPlot software.60,61
2.4. Molecular Dynamics Simulations
A series of all-atom molecular dynamics (MD) simulations were performed for ternary complexes, each involving FKBP12–FRB domains and rapamycin or a screened compound docked within a binding cleft formed at the interface between FKBP12 and FRB to fully investigate the intermolecular interaction network, where protein dynamics and solvation effects were taken into account. Atomistic coordinates of each ternary complex were obtained from the best binding mode obtained from the molecular docking calculation. All molecular structures were parametrized by the GROMOS54A7 force field.62 Partial charge distribution on each atom within a screened compound was determined by the Automated Topology Builder (ATB) web server tool,63 where a semiempirical QM calculation by the MOPAC program was performed. After that, each system of ternary complexes was explicitly solvated by simple point-charge (SPC) solvent molecules64 and a number of Na+ or Cl– counterions so that the total charge of the system was zero. Then, energy minimization was performed, followed by 1 ns simulated annealing that linearly increased the temperature from 100 to 300 K. After that, a 50 ns productive MD run was performed within an NPT ensemble under a constant temperature of 300 K regulated by the velocity-rescale algorithm and constant pressure of 1 atm regulated by the Parrinello–Rahman barostat.65 Particle mesh Ewald (PME)72 treatment with a periodic boundary condition (PBC) and the 10 Å cutoff distance was employed. Holonomic constraints by the P-LINC algorithm66 were applied for covalent bonding with hydrogen atoms, which allowed the use of 2 fs time step. All MD simulations were performed by the GROMACS 5.1.2 package.67 After all simulations were finished, root-mean-square deviation (RMSD) for global conformational change of proteins and for the motion of docked compounds relative to the proteins were calculated along the trajectory, and per residue root-mean-square fluctuations (RMSF) of C-α atomic positions were calculated from the last 50 ns of each simulation. Binding free energy between the protein complex and the docked compound contributed by electrostatics, van der Waals, and solvation was further investigated through the MM/PBSA calculation by the g_mmpbsa package68 along each MD trajectory of ternary complexes. For each calculation, dielectric constants of proteins and water were set to 4 and 80, respectively, and the surface tension of the solvent was set to 0.0226778 kJ/(mol Å2). Also, the contribution of each amino acid residue was considered to identify the important binding sites within the FKBP12–FRB protein interface.
3. Results
3.1. Data Filtering via Principal Component Analysis for Deep Learning Model Training
The overall virtual screening workflow in this study is illustrated in Figure 1. First, known inhibitors and noninhibitors of mTOR were retrieved from PubChem (NCBI gene ID: 2475). The primary data set revealed 4866 compounds as active (has an inhibitory effect against mTOR) and 48328 compounds as inactive (has no inhibitory effect against mTOR) as of 18 November 2020. Principal component analysis (PCA) was performed on the data set consisting of 75 molecular descriptor features extracted by the graph convolution technique for all 4866 active compounds. The projection of the 75-feature molecule fingerprint of the active compounds on the first two principal components (PC1 and PC2) in Figure 2 clearly displayed the clustering of 10 known rapalogs (red dots in Figure 2). Then, the projected data of 900 compounds (blue dots in Figure 2) with shortest distances from the centroid of 10 rapalogs (red cross in Figure 2) in the PC1–PC2 space were classified as the positive data set with relatively high structural similarity to the rapalogs. Meanwhile, the negative data set contains the randomly selected 900 compounds out of 48 328 compounds labeled as inactive mTOR inhibitors.
Figure 1.

Combined deep learning and molecular modeling workflow to identify candidate mTOR inhibitors from natural compounds found in Thai mushrooms.
Figure 2.
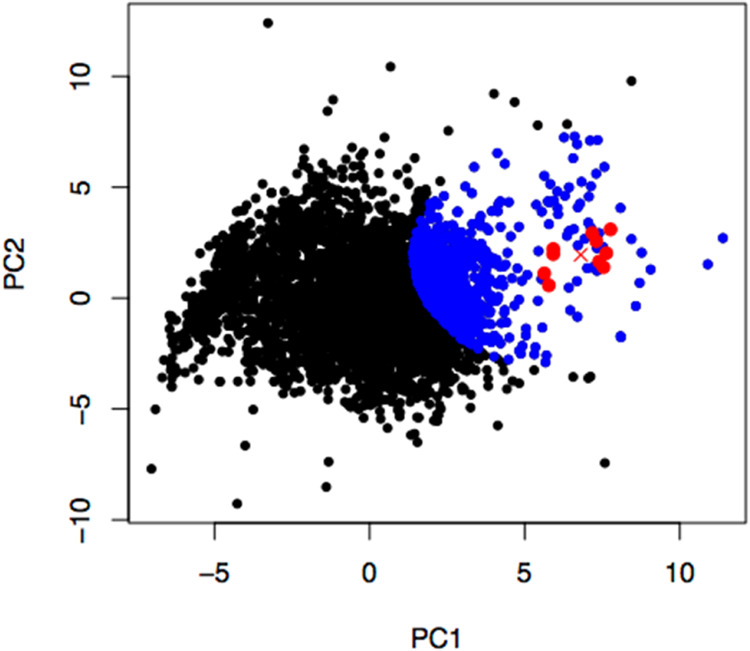
Projection of the 75-feature molecule fingerprint of the active mTOR inhibitors from PubChem on the first two principal axes. Red dots, a red cross, and blue dots are 10 rapalog data points, the centroid of the red dots, and 890 nearest data points from the centroid, respectively.
3.2. Active Compound Selection by a Deep Neural Network Classification and Molecular Docking Calculations
3.2.1. Deep Learning Model Exhibited the Importance of Steroid Core Structures
The combined and shuffled positive and negative data sets of 1800 molecules with 75-feature molecule fingerprints were used for training a deep learning model that predicts the probability of finding an individual compound within the “active” class for mTOR inhibition. A 5-fold cross-validation was performed to estimate the model performance, for which the ROC-AUC score was 0.9970 ± 0.0009 for the training data sets and 0.9917 ± 0.0019 for the testing data sets. Then, due to the stochastic nature of the learning algorithm, three independent deep learning models were trained by the same data set (1800 data points) to predict the probability score for finding each of 589 compounds from the BACMUSHBASE database (http://bacmushbase.sci.ku.ac.th/) in the “active” class. Fifty compounds with the highest “active” probability were retrieved from each model. Among the top 50 compounds from each model, six compounds appeared across all three models (Figure 3 and Table 1). Structural representations of rapamycin and the six potential alternatives in Figure 3 showed that all predicted candidates consisted of the steroid core structures. Moreover, 5 of 6 compounds were found in Ganoderma lucidum.
Figure 3.

Structural representation of six compounds with predicted “active” class whose probability scores were ranked within the top 50 in consensus among three deep learning models.
Table 1. List of Six Compounds with Consensus Predictions as Top-Ranked “Active” Class by Three Deep Learning Modelsa.
| binding
energy (kcal/mol) |
|||||
|---|---|---|---|---|---|
| name | source | log (prob) | FKBP12–FRB | FKBP12 | FRB (mTOR) |
| rapamycin | known inhibitor | N/A | –21.17 | –12.19 | –10.12 |
| trihydroxysterolb | Lentinus polychrous Lev. | –1.50 × 10–5 | –10.08 | –7.39 | –9.37 |
| butyl ganoderic acid A | G. lucidum | –1.57 × 10–5 | –10.02 | –7.75 | –7.18 |
| butyl ganoderic acid B | G. lucidum | –1.71 × 10–5 | –11.29 | –6.66 | –7.11 |
| ganoderic acid C2 | G. lucidum | –2.78 × 10–5 | –9.81 | –6.33 | –7.16 |
| cerevisterol | G. lucidum | –3.64 × 10–5 | –10.85 | –7.54 | –9.57 |
| methyl ganoderate A | G. lucidum | –3.73 × 10–5 | –11.31 | –7.64 | –7.15 |
The table displays the log probability of being an active compound (log Prob) predicted by deep learning and the binding free energy predicted by molecular docking calculations of the six compounds in comparison with rapamycin.
IUPAC name: (3β,5α,6β,22E,24R)-Ergost-22-ene-3,5,6-triol.
3.2.2. Molecular Docking Underestimated the Binding Energy of Compounds Within Ternary Complexes: Relaxation Needed
In order to verify the prediction results from deep learning, molecular docking calculations were performed to search for the favorable binding posture between the protein complex and the screened compounds (FKBP12–FRB compound). Moreover, docking calculations were also performed to investigate the interactions of FKBP12 compound and FRB compound for each of the six candidate molecules. From the docking calculations results in Table 1, six candidate compounds with the steroid core structure were with the predicted binding affinity between the compound and the FKBP12–FRB complex from −10.02 to −11.31 kcal/mol. Meanwhile, the binding affinity with only the FKBP12 part ranged from −6.66 to −7.77 kcal/mol, and the binding affinity with only the FRB part ranged from −7.11 to −9.57 kcal/mol.
An additional molecular docking calculation was performed for rapamycin. It was found that the binding affinity to the FKBP12–FRB complex (−21.17 kcal/mol) and both isolated proteins (−12.19 and −10.12 kcal/mol) was significantly higher than all of the predicted compounds. It could be explained that the crystallographic structure of a rapamycin-bound FKBP12–FRB complex was with the most suitable binding pocket conformation for rapamycin. Therefore, atomistic molecular dynamics simulations should be performed to better accommodate the binding of other predicted compounds by allowing for conformational relaxation at the interfacial region of the FKBP12–FRB complex.
3.3. MD Simulations to Elucidate the Binding Mechanisms of Selected Compounds
To investigate the binding mechanisms of the predicted compounds at the interfacial region of the FKBP12–FRB complex by atomistic MD simulations, a representative compound with the highest probability score/binding energy score was chosen from each of the two different organisms: butyl ganoderic acid B from G. lucidum and trihydroxysterol from L. polychrous Lev. (IUPAC name: (3β,5α,6β,22E,24R)-Ergost-22-ene-3,5,6-triol). Butyl ganoderic acid B was preferred over butyl ganoderic acid A due to its significantly better binding energy score from molecular docking. A reference simulation was performed for binding of rapamycin with the protein complex. For each compound, the best-docked configuration with the highest binding affinity was chosen as the starting structure for the atomistic MD simulations. After a 50 ns production MD run, conformational analysis was performed for each trajectory file. Figure 4a,b displays the root-mean-square deviation (RMSD) calculated for global conformational changes of the FKBP12 and FRB structures, respectively, from all three simulations of ternary complexes. The RMSD of FKBP12 structures from all three simulations was found in the range between 0.2 and 0.3 nm, while the RMSD of FRB domains was found between 0.18 and 0.25 nm, signifying global structural changes from X-ray crystallography. Fluctuation of the protein RMSD values observed near the beginning and the end of the simulation with trihydroxysterol was contributed by flexible loops far from the ligand-binding site, as shown in the root-mean-square fluctuation (RMSF) profiles from Figure 4c. From Figure 4c,4d, RMSF profiles were calculated for each amino acid of FKBP12 (Figure 4c) and FRB (Figure 4d) from all three simulations. Regions highlighted in both figures represented important binding regions for rapamycin and other compounds (FKBP12: residues 36, 37, 46, 53–59, 90, and 91; FRB: residues 2039 and 2098–2108). Butyl ganoderic acid B displayed relatively higher RMSF values around the binding residues compared to those of trihydroxysterol and rapamycin. Interestingly, despite a relatively high RMSF profile of trihydroxysterol at a number of flexible regions (especially around residues 2060–2080 in the FRB domain), trihydroxysterol had a relatively low RMSF profile at all binding regions, signifying high binding stability. This was confirmed through the RMSD calculation for ligand positions shown in Figure 4e. The RMSD for each compound in Figure 4e was calculated relative to the starting structure from the best docking mode, while the translational and rotational motions of the proteins were removed. Changes observed in the RMSD profile of butyl ganoderic acid B signified the continuous movement of the ligand from the docked regions. Meanwhile, RMSD profiles of rapamycin and trihydroxysterol displayed significant changes only at the beginning of production runs and shortly became equilibrated, signifying that the ligands were displaced from the binding sites predicted by molecular docking to another stable binding site when exposed to explicit solvent. Furthermore, the smaller radius of gyration when the two predicted compounds were introduced into the FKBP12–FRB complex instead of rapamycin (Figure 4f) suggested that the ternary complex became more compact, which could promote protein–protein interactions.
Figure 4.

Conformational analysis from three 50 ns MD trajectories of rapamycin and the two selected compounds binding with the FRB–FKBP12 complex. (a) RMSD of the FKBP12 protein compared to the starting structure, (b) RMSD of the FRB protein compared to the starting structure, (c) RMSF per residue of the FKBP12 protein, (d) RMSF per residue of the FRB protein, (e) RMSD of each compound compared to the starting binding configuration, and (f) radius of gyration representing the compactness of each system.
3.4. Interaction Analysis: Roles of Steroid Cores and Other Functional Groups
3.4.1. Contributions of Extra Hydrophobic Contacts and Polar Solvation
Table 2 displays the binding free energy calculated by the MM/PBSA method between pairs of molecules within all three MD trajectories of ternary complexes with rapamycin and two selected compounds: butyl ganoderic acid B from G. lucidum and trihydroxysterol from L. polychrous Lev. The “protein–compound” binding energy for each ligand molecule was contributed by van der Waals, electrostatics, polar solvation, and nonpolar solvation terms. The strongest total binding free energy was found for rapamycin and trihydroxysterol from L. polychrous Lev., and the slightly weaker binding was found for butyl ganoderic acid B. According to Table 2, the major interactions for all three simulations of FKBP12–FRB compounds were the van der Waals interactions contributed by hydrophobic contacts. From the decomposition of MM/PBSA binding free energy at some important residues (with binding energy <−3 kJ/mol) shown in Table 3, three common binding sites were found between the compounds and the FRB domain of the mTORC1 complex at the hydrophobic residues Phe2039, Trp2101, and Tyr2105 for all simulations, while the common binding sites were found between the compound and three hydrophobic residues Phe46, Ile56, and Trp59 of the FKBP12 protein. Despite missing Tyr26 and Ile90 for additional hydrophobic contacts, extra binding sites were found at residues Tyr82, Lys52, and Arg2036 for rapamycin, which contributed to its strongest hydrophobic interactions along with the strongest FKBP12 interaction at Ile56 and the FRB interaction at Phe2039. Meanwhile, trihydroxysterol from L. polychrous Lev. displayed the strongest FKBP12 interaction at Phe46 and Ile90.
Table 2. Contribution of van der Waals, Electrostatics, Polar Solvation, and Nonpolar Solvation to the Binding Free Energy of the Compounds to the FRB–FKBP12 Complex Calculated by the MM/PBSA Method for All Three Atomistic MD Simulationsa.
| MM/PBSA energy (kJ/mol): protein–compound |
||||||
|---|---|---|---|---|---|---|
| name | van der Waals | electrostatics | polar solvation | nonpolar solvation | total | MM/PBSA energy (kJ/mol): FKBP12–FRB domain |
| rapamycin | –321 ± 15 | –36 ± 23 | 176 ± 25 | –41 ± 2 | –222 ± 21 | –192 ± 70 |
| butyl ganoderic acid B | –238 ± 16 | –27 ± 14 | 101 ± 15 | –27 ± 2 | –191 ± 18 | –289 ± 140 |
| trihydroxysterol | –263 ± 12 | –22 ± 10 | 82 ± 10 | –25 ± 1 | –227 ± 15 | –393 ± 106 |
MM/PBSA binding free energy for the FRB–FKBP12 interaction was also calculated.
Table 3. Per Residue Binding Free Energy from the Decomposition of MM/PBSA Results from All Three MD Simulationsa.
| FKBP12–FRB residue | butyl ganoderic acid B (kJ/mol) | trihydroxysterol (kJ/mol) | rapamycin (kJ/mol) | |
|---|---|---|---|---|
| TYR | 26 | –5.99 | –3.88 | –2.93 |
| PHE | 36 | –3.57 | –2.51 | –4.07 |
| PHE | 46 | –5.37 | –8.01 | –4.31 |
| LYS | 52 | 1.52 | –1.01 | 3.84 |
| GLU | 54 | –1.87 | 3.99 | 7.83 |
| VAL | 55 | –6.04 | –4.00 | –6.53 |
| ILE | 56 | –5.91 | –4.46 | –10.75 |
| ARG | 57 | 2.60 | 1.59 | 5.06 |
| TRP | 59 | –4.81 | –4.25 | –3.43 |
| TYR | 82 | –1.77 | –1.08 | –3.43 |
| ILE | 90 | –6.51 | –9.60 | –1.88 |
| PHE | 99 | –2.88 | –2.73 | –2.47 |
| GLU | 107 | –1.34 | –0.01 | 0.92 |
| LEU | 2031 | –0.04 | –2.71 | -3.66 |
| ARG | 2036 | –0.46 | –0.18 | -6.42 |
| PHE | 2039 | –6.45 | –6.47 | –12.31 |
| GLY | 2040 | –0.38 | –0.05 | -3.37 |
| THR | 2098 | –1.63 | –3.43 | –0.60 |
| TRP | 2101 | –4.59 | –6.90 | –5.66 |
| TYR | 2105 | –9.22 | –10.51 | –11.24 |
| PHE | 2108 | –1.04 | –3.56 | –5.13 |
Only residues with binding free energy lower than <−3 kJ/mol are highlighted through the bold and italic text.
Despite the strongest van der Waals interaction between the FKBP12–FRB complex and rapamycin due to the largest number of hydrophobic contacts, the largest surface contact between rapamycin and the proteins resulted in the loss of polar solvation energy. The binding free energy loss of 135 ± 27 kJ/mol for rapamycin (combining both polar and nonpolar solvation terms) was significantly higher than the 57 ± 11 kJ/mol binding free energy loss for trihydroxysterol from L. polychrous Lev. so that the total binding free energies of rapamycin and trihydroxysterol became similar. Moreover, the binding of the FKBP12–FRB complex with trihydroxysterol with the lowest molecular mass and binding surface area corresponded to the strongest protein–protein interaction between FKBP12 and FRB, which could positively affect the mTORC1 kinase inhibition.
3.4.2. Chemical Features of Rapamycin and Selected Compounds Affect Interaction Networks
Figure 5 illustrates the interactions between a FKBP12–FRB complex and a rapamycin molecule. A schematic representation of rapamycin in Figure 5a shows that rapamycin is a macrocyclic lactone containing an ester group (–(C=O)O–) at the carbon position 1, along with three aliphatic rings about the ester group capable of hydrophobic contacts, and three ketone groups (–(C=O)–) capable of hydrogen bonding. The opposite side of the macrocyclic ring from the ester group contains a long strand of a terpene-like hydrophobic chain (carbon positions 17–25) of three methyl groups and three alkene groups between two methoxy groups (–O–CH3) at carbon positions 16 and 27. The X-ray crystallographic data of the FKBP12–FRB–rapamycin tertiary complex (PDB ID: 1FAP(69)) in Figure 5a showed that FKBP12 was bound with the three aliphatic rings of rapamycin and that FRB was bound with the long hydrophobic strand (see Figure 5b). Figure 5c displays the important binding residues containing 13 hydrophobic contacts and four hydrogen bonds formed between the FKBP12–FRB complex and rapamycin in the crystal structure.
Figure 5.

(a) 3D-structure of the FRB–FKBP12–rapamycin tertiary complex from X-ray crystallographic data, (b) schematic representation of a rapamycin molecule, and (c) interaction network between the FRB–FKBP12 complex and rapamycin analyzed from X-ray crystallographic data.
After the starting structure from X-ray crystallographic data was subjected to explicit solvation under a 50 ns MD simulation (see Figure 6a), all interactions from the crystallographic data were conserved, and additional hydrophobic contacts at Ile90 and Leu2031 were observed. An important binding characteristic of rapamycin observed both directly from the crystallographic data and after an MD simulation is the binding of an aliphatic ring at carbon positions 37–42 containing a hydroxy and a methoxy group with both FRB and FKBP12. For the two selected compounds in Figure 6b,c, it was found that one of the ring structures from each compound served as common binding sites to both FRB and FKBP12, while the steroid core of both butyl ganoderic acid B and trihydroxysterol was also found interacting with the FKBP12, analogous with the three aliphatic rings within the rapamycin. The terpene-like hydrophobic chain of rapamycin interacting with the FRB side was, in turn, mimicked by the tail structure of each selected compound in Figure 6b,6c. Considering the interaction networks along with the MM/PBSA energy decomposition in Table 3, it was found that there was a greater number of binding residues between the FRB side (residues 2031–2108) and trihydroxysterol than between butyl ganoderic acid B. The tail structure of trihydroxysterol was the shortest among other compounds and contained no hydrogen bonding sites; however, it corresponded to the strongest binding free energy as the steroid core was involved in the interactions with both FRB and FKBP12 sides.
Figure 6.

Interaction networks between the FRB–FKBP12 complex with (a) rapamycin, (b) butyl ganoderic acid B, and (c) trihydroxysterol after 50 ns MD simulations.
3.5. Druglikeness Evaluation for the Candidate Compounds
Finally, the druglike properties of trihydroxysterol and butyl ganoderic acid B were retrieved from the BACMUSHBASE database and are shown in Table 4. The analysis showed that each candidate violates only one rule (log P of trihydroxysterol = 5.32 and the molecular weight of butyl ganoderic acid B = 572.77 g/mol). The profiles indicate that both compounds have the potential for further drug development.
Table 4. Druglikeness Properties of the Candidate Compounds and Rapamycina.
| property (rule) | trihydroxysterolb | butyl ganoderic acid Bc | rapamycind |
|---|---|---|---|
| molecular weight (<500 g/mol) | 432.68 | 572.77 | 914.19 |
| number of H-bond acceptors (≤10) | 3 | 7 | 14 |
| number of H-bonds donors (≤5) | 3 | 2 | 3 |
| number of rotatable bonds (≤10) | 4 | 10 | 6 |
| log P (≤5) | 5.23 | 4.72 | 4.92 |
| polar surface area (≤140 Å2) | 60.69 | 117.97 | 195.45 |
The properties that violate the rule are marked in bold.
Properties were retrieved from BACMUSHBASE (http://bacmushbase.sci.ku.ac.th/properties.php?CID=34002) (accessed February 6, 2023).
Properties were retrieved from BACMUSHBASE (http://bacmushbase.sci.ku.ac.th/properties.php?CID=1140) (accessed February 6, 2023).
Properties were predicted by the Molinspiration Property Calculation Service (www.molinspiration.com) (accessed February 6, 2023).
4. Discussion
In many Asian countries, mushrooms and their extracts have been long used as traditional medicine with antioxidant, antipathogenic, antitumoral, and immuno-modulating effects.70−72 The present work identified several compounds containing ring groups with steroid cores from G. lucidum and L. polychrous Lev., with possible inhibitory effects on mTOR. A previous study has shown that ergostanoids extracted from L. polychrous Lev. possessed a moderate antiproliferation effect on oestradiol-enhanced T47D breast cancer cells73 and those extracted from G. lucidum have been widely shown to have cytotoxic activities against many cancer types.74
In this study, a combination of machine learning and structural modeling tools was used in a search for potential active compounds for the mTOR complex from natural products. Deep learning models were built based on previous experimental data of the bioactivity. The performance of the deep learning model was enhanced through data filtering after a principal component analysis (PCA) that projected the data with extracted chemical features onto the principal components. The closeness observed between rapamycin and the group of rapalogs in the chemistry space could guide filtering for more compounds with similar chemical properties, reducing the uncertainty of mixing bioactive compounds with different inhibition mechanisms.
Table 5 displays the three most significant loadings or components of eigenvectors of the first three PC modes. Projection of feature vectors extracted from molecules within the data set on the PC1 axis represented the proportion of atoms with the sp3 hybridization, which opposed the proportion of atoms with the sp2 hybridization and hydrophobicity. High proportion of sp3 found for the first-generation mTOR inhibitors was in concurrence with the large proportion of aliphatic structures and the absence of an aromatic ring from the molecular structure of rapamycin. The high score on the PC2 axis represented the high proportion of heavy atoms with only one connection with other heavy atoms (degree = 1), corresponding to the branching groups of rapamycin. As the carbonyl (C=O) group had both degree = 1 for heavy atom connections and no covalently bonded hydrogen atom (total number of hydrogens = 0 and implicit valence = 0), the lower score of zero hydrogen connection corresponded to the lower amount of C=O groups in rapamycin and other first-generation mTOR inhibitors. Finally, the PC3 axis displayed whether the molecules contained a high number of oxygen or nitrogen atoms, for which the first-generation mTOR inhibitors tended to contain a higher oxygen proportion and the second-generation mTOR inhibitors tended to contain a higher nitrogen proportion. Therefore, the first-generation mTOR inhibitors were separated from the others through their aliphaticity (PC1) and hydrocarbon branch structure (PC2) and the training data set was created based on this primary screening scheme.
Table 5. Important Molecular Descriptor Features from the First Three Principal Componentsa.
| feature index (loading) | description | average score from 10 Gen-1 inhibitors | average score from 9 Gen-2 inhibitors | trihydroxysterol (our candidate) | butyl ganoderic acid B (our candidate) | |
|---|---|---|---|---|---|---|
| PC1 | feature 67 (0.378) | hybridization = sp3 | 2.11 ± 0.09 | 0.63 ± 0.39 | 2.97 | 2.29 |
| PC1 | feature 66 (−0.390) | hybridization = sp2 | 0.98 ± 0.11 | 2.59 ± 0.38 | 0.23 | 0.85 |
| PC1 | feature 70 (−0.375) | is aromatic | 0.06 ± 0.13 | 2.21 ± 0.44 | 0.0 | 0.0 |
| PC2 | feature 46 (0.396) | degree = 1 | 0.90 ± 0.07 | 0.44 ± 0.07 | 0.84 | 1.02 |
| PC2 | feature 71 (0.298) | total number of hydrogens = 0 | 0.93 ± 0.06 | 1.72 ± 0.14 | 0.32 | 1.17 |
| PC2 | feature 56 (0.293) | implicit valence = 0 | 0.93 ± 0.06 | 1.73 ± 0.14 | 0.32 | 1.17 |
| PC3 | feature 2 (0.426) | is a N atom | 0.05 ± 0.06 | 0.56 ± 0.20 | 0.0 | 0.0 |
| PC3 | feature 3 (−0.189) | is an O atom | 0.65 ± 0.05 | 0.25 ± 0.13 | 0.26 | 0.63 |
Feature vector components of interest were compared between two generations of inhibitors and the screened candidate in this study.
A remarkable pattern was observed from the virtual screening by the trained deep learning model as most of the compounds with high “active” probability scores were found to involve a “steroid core” as an essential molecular feature. Moreover, the majority of the top candidates were the compounds extracted from G. lucidum, for which each ganoderic acid and its derivatives contained a steroid core. Apart from G. lucidum, trihydroxysterol from L. polychrous Lev. also displayed strong potential as a bioactive compound that facilitates mTOR kinase inhibition. It could also be seen from Table 5 that trihydroxysterol and butyl ganoderic acid B displayed most features in common with the first-generation mTOR inhibitors, except by slightly lower amounts of C=O groups and oxygen proportion.
5. Conclusions
To provide further validation of the prediction results by deep learning in a structural aspect, molecular docking calculations and all-atom molecular dynamics simulations were performed for some top candidate compounds screened by the “active” probability score. It has been known that rapamycin forms a ternary complex with the FK506-binding protein (FKBP12) and the FKBP12–rapamycin binding (FRB) domain, thus inhibiting the kinase activity of an mTORC1 complex. Therefore, intermolecular interactions were investigated for both the compound FKBP12 and compound FRB sides to address how each compound candidate could stabilize the ternary complex during kinase inhibition. Analogously, rapamycin and all of the screened compound candidates contained ring groups and steroid cores that mostly interacted with FKBP12, along with terpene-like chains that interacted with FRB. However, trihydroxysterol from L. polychrous Lev. displayed a rather small but effective interaction network that also facilitated FKBP12–FRB interactions and further stabilized the ternary complex. Therefore, combining information from machine learning, structural modeling, and druglikeness analyses, trihydroxysterol from L. polychrous Lev. and butyl ganoderic acid B from G. lucidum could be interesting alternatives to rapamycin and rapalogs for mTOR kinase inhibition and the downstream regulations of the mTOR pathway.
Acknowledgments
This research project is supported by the Thailand Science Research and Innovation (TSRI) Basic Research Fund: Fiscal year 2022 under Project Number FRB650048/0164. Also, this research was partly supported by the Program Management Unit for Human Resources & Institutional Development, Research, and Innovation [Grant Number B01F630003].
Data Availability Statement
The models for MTOR inhibitor prediction are available at the following link: https://github.com/pribnowbox/virtual-screening-of-new-MTOR-inhibitors.
The authors declare no competing financial interest.
References
- Huang S. Targeting MTOR Signaling for Cancer Therapy. Curr. Opin. Pharmacol. 2003, 3 (4), 371–377. 10.1016/S1471-4892(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Luo Y.; Huang S. Updates of MTOR Inhibitors. Anti-Cancer Agents Med. Chem. 2010, 10 (7), 571–581. 10.2174/187152010793498663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamji A. F.; Nghiem P.; Schreiber S. L. Integration of Growth Factor and Nutrient Signaling. Mol. Cell 2003, 12 (2), 271–280. 10.1016/j.molcel.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Mao B.; Zhang Q.; Ma L.; Zhao D.-S.; Zhao P.; Yan P. Overview of Research into MTOR Inhibitors. Molecules 2022, 27 (16), 5295. 10.3390/molecules27165295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M.; Sabatini D. M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149 (2), 274–293. 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton R. A.; Sabatini D. M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168 (6), 960–976. 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vézina C.; Kudelski A.; Sehgal S. N. Rapamycin (AY-22,989), a New Antifungal Antibiotic. I. Taxonomy of the Producing Streptomycete and Isolation of the Active Principle. J. Antibiot. 1975, 28 (10), 721–726. 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- Sehgal S. N.; Baker H.; Vézina C. Rapamycin (AY-22,989), a New Antifungal Antibiotic. II. Fermentation, Isolation and Characterization. J. Antibiot. 1975, 28 (10), 727–732. 10.7164/antibiotics.28.727. [DOI] [PubMed] [Google Scholar]
- Loewith R.; Jacinto E.; Wullschleger S.; Lorberg A.; Crespo J. L.; Bonenfant D.; Oppliger W.; Jenoe P.; Hall M. N. Two TOR Complexes, Only One of Which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control. Mol. Cell 2002, 10 (3), 457–468. 10.1016/S1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Benjamin D.; Colombi M.; Moroni C.; Hall M. N. Rapamycin Passes the Torch: A New Generation of MTOR Inhibitors. Nat. Rev. Drug Discovery 2011, 10 (11), 868–880. 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- Sun S.-Y. MTOR Kinase Inhibitors as Potential Cancer Therapeutic Drugs. Cancer Lett. 2013, 340 (1), 1–8. 10.1016/j.canlet.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E. J.; Albers M. W.; Bum Shin T.; ichikawa K.; Keith C. T.; Lane W. S.; Schreiber S. L. A Mammalian Protein Targeted by G1-Arresting Rapamycin–Receptor Complex. Nature 1994, 369 (6483), 756–758. 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Sabatini D. M.; Erdjument-Bromage H.; Lui M.; Tempst P.; Snyder S. H. RAFT1: A Mammalian Protein That Binds to FKBP12 in a Rapamycin-Dependent Fashion and Is Homologous to Yeast TORs. Cell 1994, 78 (1), 35–43. 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- Banaszynski L. A.; Liu C. W.; Wandless T. J. Characterization of the FKBP·Rapamycin·FRB Ternary Complex. J. Am. Chem. Soc. 2005, 127 (13), 4715–4721. 10.1021/ja043277y. [DOI] [PubMed] [Google Scholar]
- Bhagwat S. V.; Crew A. P. Novel Inhibitors of MTORC1 and MTORC2. Curr. Opin. Investig. Drugs 2010, 11 (6), 638–645. [PubMed] [Google Scholar]
- Richard D. J.; Verheijen J. C.; Zask A. Recent Advances in the Development of Selective, ATP-Competitive Inhibitors of MTOR. Curr. Opin. Drug Discovery Dev. 2010, 13 (4), 428–440. [PubMed] [Google Scholar]
- García-Echeverría C. Allosteric and ATP-Competitive Kinase Inhibitors of MTOR for Cancer Treatment. Bioorg. Med. Chem. Lett. 2010, 20 (15), 4308–4312. 10.1016/j.bmcl.2010.05.099. [DOI] [PubMed] [Google Scholar]
- Zask A.; Verheijen J. C.; Richard D. J. Recent Advances in the Discovery of Small-Molecule ATP Competitive MTOR Inhibitors: A Patent Review. Expert Opin. Ther. Pat. 2011, 21 (7), 1109–1127. 10.1517/13543776.2011.584871. [DOI] [PubMed] [Google Scholar]
- Pike K. G.; Malagu K.; Hummersone M. G.; Menear K. A.; Duggan H. M. E.; Gomez S.; Martin N. M. B.; Ruston L.; Pass S. L.; Pass M. Optimization of Potent and Selective Dual MTORC1 and MTORC2 Inhibitors: The Discovery of AZD8055 and AZD2014. Bioorg. Med. Chem. Lett. 2013, 23 (5), 1212–1216. 10.1016/j.bmcl.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Fantus D.; Dai H.; Ono Y.; Watson A.; Yokota S.; Mohib K.; Yoshida O.; Ross M. A.; Watkins S. C.; Ramaswami B.; Valusjkikh A.; Rothstein D. M.; Thomson A. W. Influence of the Novel ATP-Competitive Dual MTORC1/2 Inhibitor AZD2014 on Immune Cell Populations and Heart Allograft Rejection. Transplantation 2017, 101 (12), 2830–2840. 10.1097/TP.0000000000001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H.; Huang Y.; Guo B.; Liang B.; Liu X.; Ou H.; Jiang C.; Li X.; Yang D. Dramatic Antitumor Effects of the Dual MTORC1 and MTORC2 Inhibitor AZD2014 in Hepatocellular Carcinoma. Am. J. Cancer Res. 2015, 5 (1), 125–139. [PMC free article] [PubMed] [Google Scholar]
- Mortensen D. S.; Perrin-Ninkovic S. M.; Shevlin G.; Elsner J.; Zhao J.; Whitefield B.; Tehrani L.; Sapienza J.; Riggs J. R.; Parnes J. S.; Papa P.; Packard G.; Lee B. G. S.; Harris R.; Correa M.; Bahmanyar S.; Richardson S. J.; Peng S. X.; Leisten J.; Khambatta G.; Hickman M.; Gamez J. C.; Bisonette R. R.; Apuy J.; Cathers B. E.; Canan S. S.; Moghaddam M. F.; Raymon H. K.; Worland P.; Narla R. K.; Fultz K. E.; Sankar S. Optimization of a Series of Triazole Containing Mammalian Target of Rapamycin (MTOR) Kinase Inhibitors and the Discovery of CC-115. J. Med. Chem. 2015, 58 (14), 5599–5608. 10.1021/acs.jmedchem.5b00627. [DOI] [PubMed] [Google Scholar]
- Mortensen D. S.; Sapienza J.; Lee B. G. S.; Perrin-Ninkovic S. M.; Harris R.; Shevlin G.; Parnes J. S.; Whitefield B.; Hickman M.; Khambatta G.; Bisonette R. R.; Peng S.; Gamez J. C.; Leisten J.; Narla R. K.; Fultz K. E.; Sankar S. Use of Core Modification in the Discovery of CC214–2, an Orally Available, Selective Inhibitor of MTOR Kinase. Bioorg. Med. Chem. Lett. 2013, 23 (6), 1588–1591. 10.1016/j.bmcl.2013.01.110. [DOI] [PubMed] [Google Scholar]
- Mortensen D. S.; Perrin-Ninkovic S. M.; Shevlin G.; Zhao J.; Packard G.; Bahmanyar S.; Correa M.; Elsner J.; Harris R.; Lee B. G. S.; Papa P.; Parnes J. S.; Riggs J. R.; Sapienza J.; Tehrani L.; Whitefield B.; Apuy J.; Bisonette R. R.; Gamez J. C.; Hickman M.; Khambatta G.; Leisten J.; Peng S. X.; Richardson S. J.; Cathers B. E.; Canan S. S.; Moghaddam M. F.; Raymon H. K.; Worland P.; Narla R. K.; Fultz K. E.; Sankar S. Discovery of Mammalian Target of Rapamycin (MTOR) Kinase Inhibitor CC-223. J. Med. Chem. 2015, 58 (13), 5323–5333. 10.1021/acs.jmedchem.5b00626. [DOI] [PubMed] [Google Scholar]
- Yu K.; Toral-Barza L.; Shi C.; Zhang W.-G.; Lucas J.; Shor B.; Kim J.; Verheijen J.; Curran K.; Malwitz D. J.; Cole D. C.; Ellingboe J.; Ayral-Kaloustian S.; Mansour T. S.; Gibbons J. J.; Abraham R. T.; Nowak P.; Zask A. Biochemical, Cellular, and In Vivo Activity of Novel ATP-Competitive and Selective Inhibitors of the Mammalian Target of Rapamycin. Cancer Res. 2009, 69 (15), 6232–6240. 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
- Yu K.; Shi C.; Toral-Barza L.; Lucas J.; Shor B.; Kim J. E.; Zhang W.-G.; Mahoney R.; Gaydos C.; Tardio L.; Kim S. K.; Conant R.; Curran K.; Kaplan J.; Verheijen J.; Ayral-Kaloustian S.; Mansour T. S.; Abraham R. T.; Zask A.; Gibbons J. J. Beyond Rapalog Therapy: Preclinical Pharmacology and Antitumor Activity of WYE-125132, an ATP-Competitive and Specific Inhibitor of MTORC1 and MTORC2. Cancer Res. 2010, 70 (2), 621–631. 10.1158/0008-5472.CAN-09-2340. [DOI] [PubMed] [Google Scholar]
- Tan D. S.; Dumez H.; Olmos D.; Sandhu S. K.; Hoeben A.; Stephens A. W.; Poondru S.; Gedrich R.; Kaye S. B.; Schoffski P. First-in-Human Phase I Study Exploring Three Schedules of OSI-027, a Novel Small Molecule TORC1/TORC2 Inhibitor, in Patients with Advanced Solid Tumors and Lymphoma. J. Clin. Oncol. 2010, 28 (15_suppl), 3006. 10.1200/jco.2010.28.15_suppl.3006. [DOI] [Google Scholar]
- Zeng Z.; Shi Y. X.; Tsao T.; Qiu Y.; Kornblau S. M.; Baggerly K. A.; Liu W.; Jessen K.; Liu Y.; Kantarjian H.; Rommel C.; Fruman D. A.; Andreeff M.; Konopleva M. Targeting of MTORC1/2 by the MTOR Kinase Inhibitor PP242 Induces Apoptosis in AML Cells under Conditions Mimicking the Bone Marrow Microenvironment. Blood 2012, 120 (13), 2679–2689. 10.1182/blood-2011-11-393934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen C. C.; Kang S. A.; Chang J. W.; Liu Q.; Zhang J.; Gao Y.; Reichling L. J.; Sim T.; Sabatini D. M.; Gray N. S. An ATP-Competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-Resistant Functions of MTORC1. J. Biol. Chem. 2009, 284 (12), 8023–8032. 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Wang J.; Kang S. A.; Thoreen C. C.; Hur W.; Ahmed T.; Sabatini D. M.; Gray N. S. Discovery of 9-(6-Aminopyridin-3-Yl)-1-(3-(Trifluoromethyl)Phenyl)Benzo[ h ][1,6]Naphthyridin-2(1 H)-One (Torin2) as a Potent, Selective, and Orally Available Mammalian Target of Rapamycin (MTOR) Inhibitor for Treatment of Cancer. J. Med. Chem. 2011, 54 (5), 1473–1480. 10.1021/jm101520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherlin D. P.; Bao L.; Berry M.; Castanedo G.; Chuckowree I.; Dotson J.; Folks A.; Friedman L.; Goldsmith R.; Gunzner J.; Heffron T.; Lesnick J.; Lewis C.; Mathieu S.; Murray J.; Nonomiya J.; Pang J.; Pegg N.; Prior W. W.; Rouge L.; Salphati L.; Sampath D.; Tian Q.; Tsui V.; Wan N. C.; Wang S.; Wei B.; Wiesmann C.; Wu P.; Zhu B.-Y.; Olivero A. Discovery of a Potent, Selective, and Orally Available Class I Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (MTOR) Kinase Inhibitor (GDC-0980) for the Treatment of Cancer. J. Med. Chem. 2011, 54 (21), 7579–7587. 10.1021/jm2009327. [DOI] [PubMed] [Google Scholar]
- Garlich J. R.; De P.; Dey N.; Su J. D.; Peng X.; Miller A.; Murali R.; Lu Y.; Mills G. B.; Kundra V.; Shu H.-K.; Peng Q.; Durden D. L. A Vascular Targeted Pan Phosphoinositide 3-Kinase Inhibitor Prodrug, SF1126, with Antitumor and Antiangiogenic Activity. Cancer Res. 2008, 68 (1), 206–215. 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- Manara M. C.; Nicoletti G.; Zambelli D.; Ventura S.; Guerzoni C.; Landuzzi L.; Lollini P.-L.; Maira S.-M.; García-Echeverría C.; Mercuri M.; Picci P.; Scotlandi K. NVP-BEZ235 as a New Therapeutic Option for Sarcomas. Clin. Cancer Res. 2010, 16 (2), 530–540. 10.1158/1078-0432.CCR-09-0816. [DOI] [PubMed] [Google Scholar]
- Marone R.; Erhart D.; Mertz A. C.; Bohnacker T.; Schnell C.; Cmiljanovic V.; Stauffer F.; Garcia-Echeverria C.; Giese B.; Maira S.-M.; Wymann M. P. Targeting Melanoma with Dual Phosphoinositide 3-Kinase/Mammalian Target of Rapamycin Inhibitors. Mol. Cancer Res. 2009, 7 (4), 601–613. 10.1158/1541-7786.MCR-08-0366. [DOI] [PubMed] [Google Scholar]
- Mallon R.; Hollander I.; Feldberg L.; Lucas J.; Soloveva V.; Venkatesan A.; Dehnhardt C.; Delos Santos E.; Chen Z.; dos Santos O.; Ayral-Kaloustian S.; Gibbons J. Antitumor Efficacy Profile of PKI-402, a Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor. Mol. Cancer Ther. 2010, 9 (4), 976–984. 10.1158/1535-7163.MCT-09-0954. [DOI] [PubMed] [Google Scholar]
- Yu P.; Laird A. D.; Du X.; Wu J.; Won K.-A.; Yamaguchi K.; Hsu P. P.; Qian F.; Jaeger C. T.; Zhang W.; Buhr C. A.; Shen P.; Abulafia W.; Chen J.; Young J.; Plonowski A.; Yakes F. M.; Chu F.; Lee M.; Bentzien F.; Lam S. T.; Dale S.; Matthews D. J.; Lamb P.; Foster P. Characterization of the Activity of the PI3K/MTOR Inhibitor XL765 (SAR245409) in Tumor Models with Diverse Genetic Alterations Affecting the PI3K Pathway. Mol. Cancer Ther. 2014, 13 (5), 1078–1091. 10.1158/1535-7163.MCT-13-0709. [DOI] [PubMed] [Google Scholar]
- Guduru S. K. R.; Arya P. Synthesis and Biological Evaluation of Rapamycin-Derived, next Generation Small Molecules. Med. Chem. Commun. 2018, 9 (1), 27–43. 10.1039/C7MD00474E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. G.; Kunnumakkara A. B.; Nair A.; Merritt W. M.; Han L. Y.; Armaiz-Pena G. N.; Kamat A. A.; Spannuth W. A.; Gershenson D. M.; Lutgendorf S. K.; Aggarwal B. B.; Sood A. K. Curcumin Inhibits Tumor Growth and Angiogenesis in Ovarian Carcinoma by Targeting the Nuclear Factor-KB Pathway. Clin. Cancer Res. 2007, 13 (11), 3423–3430. 10.1158/1078-0432.CCR-06-3072. [DOI] [PubMed] [Google Scholar]
- Johnson S. M.; Gulhati P.; Arrieta I.; Wang X.; Uchida T.; Gao T.; Evers B. M. Curcumin Inhibits Proliferation of Colorectal Carcinoma by Modulating Akt/MTOR Signaling. Anticancer Res. 2009, 29 (8), 3185–3190. [PMC free article] [PubMed] [Google Scholar]
- Beevers C. S.; Li F.; Liu L.; Huang S. Curcumin Inhibits the Mammalian Target of Rapamycin-Mediated Signaling Pathways in Cancer Cells. Int. J. Cancer 2006, 119 (4), 757–764. 10.1002/ijc.21932. [DOI] [PubMed] [Google Scholar]
- Van Aller G. S.; Carson J. D.; Tang W.; Peng H.; Zhao L.; Copeland R. A.; Tummino P. J.; Luo L. Epigallocatechin Gallate (EGCG), a Major Component of Green Tea, Is a Dual Phosphoinositide-3-Kinase/MTOR Inhibitor. Biochem. Biophys. Res. Commun. 2011, 406 (2), 194–199. 10.1016/j.bbrc.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Reinke A.; Chen J. C.-Y.; Aronova S.; Powers T. Caffeine Targets TOR Complex I and Provides Evidence for a Regulatory Link between the FRB and Kinase Domains of Tor1p. J. Biol. Chem. 2006, 281 (42), 31616–31626. 10.1016/S0021-9258(19)84075-9. [DOI] [PubMed] [Google Scholar]
- Park D.; Jeong H.; Lee M. N.; Koh A.; Kwon O.; Yang Y. R.; Noh J.; Suh P.-G.; Park H.; Ryu S. H. Resveratrol Induces Autophagy by Directly Inhibiting MTOR through ATP Competition. Sci. Rep 2016, 6 (1), 21772 10.1038/srep21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X.; Pelling J. Targeting the PI3K/Akt/MTOR Axis by Apigenin for Cancer Prevention. Anti-Cancer Agents Med. Chem. 2013, 13 (7), 971–978. 10.2174/18715206113139990119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed D.; Adhami V.; Khan M.; Mukhtar H. Inhibition of Akt/MTOR Signaling by the Dietary Flavonoid Fisetin. Anti-Cancer Agents Med. Chem. 2013, 13 (7), 995–1001. 10.2174/18715206113139990129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A.; Biersack B.; Li Y.; Kong D.; Bao B.; Schobert R.; Padhye S.; Sarkar F. Targeted Regulation of PI3K/Akt/MTOR/NF-κB Signaling by Indole Compounds and Their Derivatives: Mechanistic Details and Biological Implications for Cancer Therapy. Anti-Cancer Agents Med. Chem. 2013, 13 (7), 1002–1013. 10.2174/18715206113139990078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning A. Inhibition of MTOR Signaling by Quercetin in Cancer Treatment and Prevention. Anti-Cancer Agents Med. Chem. 2013, 13 (7), 1025–1031. 10.2174/18715206113139990114. [DOI] [PubMed] [Google Scholar]
- Sylvester P.; Ayoub N. Tocotrienols Target PI3K/Akt Signaling in Anti-Breast Cancer Therapy. Anti-Cancer Agents Med. Chem. 2013, 13 (7), 1039–1047. 10.2174/18715206113139990116. [DOI] [PubMed] [Google Scholar]
- Kumari C.; Abulaish M.; Subbarao N. Exploring Molecular Descriptors and Fingerprints to Predict MTOR Kinase Inhibitors Using Machine Learning Techniques. IEEE/ACM Trans. Comput. Biol. Bioinf. 2021, 18 (5), 1902–1913. 10.1109/TCBB.2020.2964203. [DOI] [PubMed] [Google Scholar]
- Wang P.; Xu X.; Li Y.; Li B.; Pei Q.; Yu P.; Jing C.; Lu M. Discovery of Novel Mammalian Target of Rapamycin (MTOR) Inhibitors by Support Vector Machine. IOP Conf. Ser.: Earth Environ. Sci. 2021, 692 (3), 032028 10.1088/1755-1315/692/3/032028. [DOI] [Google Scholar]
- Vittorio S.; Gitto R.; Adornato I.; Russo E.; De Luca L. In Silico Strategy for Targeting the MTOR Kinase at Rapamycin Binding Site by Small Molecules. Molecules 2021, 26 (4), 1103. 10.3390/molecules26041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Wang L. Discovery and Development of ATP-Competitive MTOR Inhibitors Using Computational Approaches. Curr. Pharm. Des. 2017, 23 (29), 4321–4331. 10.2174/1381612823666170710150604. [DOI] [PubMed] [Google Scholar]
- Kumari C.; Abulaish M.; Subbarao N. Using SMOTE to Deal with Class-Imbalance Problem in Bioactivity Data to Predict MTOR Inhibitors. SN Comput. Sci. 2020, 1 (3), 150. 10.1007/s42979-020-00156-5. [DOI] [Google Scholar]
- Anderson E.; Havener T. M.; Zorn K. M.; Foil D. H.; Lane T. R.; Capuzzi S. J.; Morris D.; Hickey A. J.; Drewry D. H.; Ekins S. Synergistic Drug Combinations and Machine Learning for Drug Repurposing in Chordoma. Sci. Rep. 2020, 10 (1), 12982 10.1038/s41598-020-70026-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Thiessen P. A.; Bolton E. E.; Chen J.; Fu G.; Gindulyte A.; Han L.; He J.; He S.; Shoemaker B. A.; Wang J.; Yu B.; Zhang J.; Bryant S. H. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44 (D1), D1202–D1213. 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers E. W.; Bolton E. E.; Brister J. R.; Canese K.; Chan J.; Comeau D. C.; Connor R.; Funk K.; Kelly C.; Kim S.; Madej T.; Marchler-Bauer A.; Lanczycki C.; Lathrop S.; Lu Z.; Thibaud-Nissen F.; Murphy T.; Phan L.; Skripchenko Y.; Tse T.; Wang J.; Williams R.; Trawick B. W.; Pruitt K. D.; Sherry S. T. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50 (D1), D20–D26. 10.1093/nar/gkab1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsundar B.; Eastman P.; Walters P.; Pande V.. Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery and More, 1st ed.; O’Reilly Media: Sebastopol, CA, 2019. [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19 (14), 1639–1662. . [DOI] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; Swindells M. B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51 (10), 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Wallace A. C.; Laskowski R. A.; Thornton J. M. LIGPLOT: A Program to Generate Schematic Diagrams of Protein-Ligand Interactions. Protein Eng. Des. Sel. 1995, 8 (2), 127–134. 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- Schmid N.; Eichenberger A. P.; Choutko A.; Riniker S.; Winger M.; Mark A. E.; van Gunsteren W. F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40 (7), 843–856. 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Malde A. K.; Zuo L.; Breeze M.; Stroet M.; Poger D.; Nair P. C.; Oostenbrink C.; Mark A. E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7 (12), 4026–4037. 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Tepper H. L.; Voth G. A. Flexible Simple Point-Charge Water Model with Improved Liquid-State Properties. J. Chem. Phys. 2006, 124 (2), 024503 10.1063/1.2136877. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45 (14), 1196–1199. 10.1103/PhysRevLett.45.1196. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J.; Fraaije J. G. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18 (12), 1463–1472. 10.1002/(sici)1096-987x(199709)18:123.0.co;2-h. [DOI] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Kumari R.; Kumar R.; Lynn A. G_mmpbsa —A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54 (7), 1951–1962. 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Choi J.; Chen J.; Schreiber S. L.; Clardy J. Structure of the FKBP12-Rapamycin Complex Interacting with Binding Domain of Human FRAP. Science 1996, 273 (5272), 239–242. 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- Panda S. K.; Sahoo G.; Swain S. S.; Luyten W. Anticancer Activities of Mushrooms: A Neglected Source for Drug Discovery. Pharmaceuticals 2022, 15 (2), 176. 10.3390/ph15020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasser S. P. Medicinal Mushroom Science: History, Current Status, Future Trends, and Unsolved Problems. Int. J. Med. Mushrooms 2010, 12 (1), 1–16. 10.1615/IntJMedMushr.v12.i1.10. [DOI] [PubMed] [Google Scholar]
- Zaidman B.-Z.; Yassin M.; Mahajna J.; Wasser S. P. Medicinal Mushroom Modulators of Molecular Targets as Cancer Therapeutics. Appl. Microbiol. Biotechnol. 2005, 67 (4), 453–468. 10.1007/s00253-004-1787-z. [DOI] [PubMed] [Google Scholar]
- Fangkrathok N.; Sripanidkulchai B.; Umehara K.; Noguchi H. Bioactive Ergostanoids and a New Polyhydroxyoctane from Lentinus Polychrous Mycelia and Their Inhibitory Effects on E2-Enhanced Cell Proliferation of T47D Cells. Nat. Prod. Res. 2013, 27 (18), 1611–1619. 10.1080/14786419.2012.742079. [DOI] [PubMed] [Google Scholar]
- Yuen J. W. M.; Gohel M. D. I. Anticancer Effects of Ganoderma Lucidum: A Review of Scientific Evidence. Nutr. Cancer 2005, 53 (1), 11–17. 10.1207/s15327914nc5301_2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The models for MTOR inhibitor prediction are available at the following link: https://github.com/pribnowbox/virtual-screening-of-new-MTOR-inhibitors.