

Abstract
Nα-2-thiophenoyl-d-phenylalanine-2-morpholinoanilide [MMV688845, Pathogen Box®; Medicines for Malaria Venture; IUPAC: N-(1-((2-morpholinophenyl)amino)-1-oxo-3-phenylpropan-2-yl)thiophene-2-carboxamide)] is a hit compound which shows activity against Mycobacterium abscessus (MIC90 6.25 - 12.5 μM) and other mycobacteria. This work describes a derivatization of MMV688845 with focus on the thiomorpholine moiety. The molecular structure of these oxidation products is confirmed by X-ray crystallography. Conservation of the essential R configuration during the synthesis sequence was proven by chiral HPLC for an exemplary compound. Structure activity relationship information is provided for four different substituents of the molecular scaffold. The S-oxides show lower MIC90 values (M. abscessus: 0.78 μM, 8-fold more active) and higher aqueous kinetic solubility. MIC90 determination was conducted on M. abscessus, M. intracellulare, M. smegmatis and M. tuberculosis. Assessment of bactericidal effects showed a 2-log reduction of viable bacteria at just 2X their MIC90 for new derivatives. No cytotoxic effects against a variety of mammalian cell lines were observed.
Graphical Abstract
1. Introduction
Infections with mycobacteria are difficult to treat, as they often require prolonged antibiotic therapy, which can be accompanied by severe side effects. Whereas drug-susceptible tuberculosis can usually be treated successfully within six months1-4, the therapy of infections with multi-drug resistant (MDR) Mycobacterium tuberculosis (Mtb) strains and non-tuberculous mycobacteria (NTM)5,6 is protracted and cure rates are often low (25 - 58 % for Mycobacterium abscessus7,8 (Mabs) infections9-11). For infections with MDR Mtb, progress has already been made in recent years. Scientific efforts have led to the approval of two new drugs: bedaquiline and pretomanid12-14. Bedaquiline works by inhibiting mycobacterial ATP synthase, while pretomanid has a rather complex mechanism of action. Under normoxic conditions, it is converted into reactive intermediates that interfere with mycolic acid synthesis, while in anaerobic environments it acts as an NO donor that effectively poisons the respiration of mycobacteria15,16. By combining bedaquiline, pretomanid and linezolid, a new therapeutic regimen for treatment of MDR tuberculosis could be established17.
NTM have come into scientific focus in recent years, owing to the fast-growing, multidrug-resistant Mabs and the Mycobacterium avium complex (MAC) which have emerged as problematic opportunistic pathogens18-21. They possess numerous intrinsic resistance mechanisms, which render classical antitubercular drugs and many other common antibiotics ineffective.
Currently, the RNA polymerase (RNAP) inhibitor rifampicin (RIF) is a cornerstone of antimycobacterial therapy. As a result, mycobacteria with acquired or intrinsic resistance to rifamycins, e.g., multi drug resistant Mtb or Mabs, are a particular threat. Mycobacteria use different mechanisms to counteract the effect of RIF. In Mtb, 95 % of the resistant strains possess acquired mutations in the target of RIF, the RpoB subunit of the RNAP. Less frequently, the expression of efflux pumps influences RIF resistance22. In contrast, RIF resistance in Mabs is caused by other mechanisms. Mabs has the ability to covalently modify RIF through the enzyme ADP ribosylase23 and thereby inactivate it. In addition, naphthohydroquinone oxidation increases resistance24. Certain rifamycins, such as rifabutin, are less susceptible to these mechanisms of resistance, which is reflected in improved efficacy in vitro and in vivo25,26. Recent advances in rifamycin research made it possible to completely block ADP-ribosylation while retaining its anti-mycobacterial activity. The redesign of the C25-O-acyl position was crucial for these improvements of the rifamycin structures27,28.
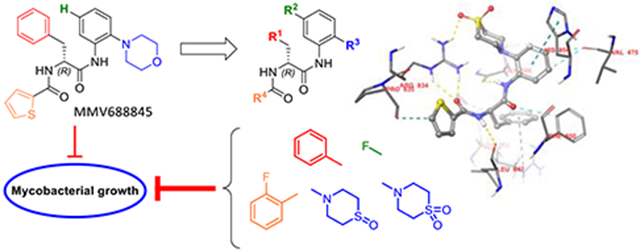
Despite the promising attributes of rifabutin, a synthetic RNAP inhibitor that exhibits no cross-resistance with rifamycins is of interest for anti-mycobacterial drug development. In particular, the Nα-aroyl-N-aryl-phenylalanine amide (AAP) MMV688845 (Nα-2-thiophenoyl-d-phenylalanine-2 morpholinoanilide), discovered as an anti-Mtb hit29, has been shown to be active against Mabs (ATCC19977) by screening of the Pathogen Box® library30 (Medicines for Malaria Ventures, MMV). Analogs of MMV688845 have been shown to be inhibitors of Mtb RNAP that bind to the RpoB subunit but address a different binding site than rifamycins31. In addition, derivatives of MMV688845 are described in a patent32, although these were not investigated against NTM including Mabs. Based on the promising screening experiments, we have developed a synthesis for MMV688845, yielding the active enantiomer with ee values of >99 % and investigated its stereospecific activity against Mabs, as well as its cytotoxicity33. In parallel, the RNAP was validated as target in Mabs, and a detailed in vitro profiling of the hit compound was performed34. Motivated by these encouraging results, a series of MMV688845 derivatives was synthesized with the aim of increasing in vitro activity against NTM, improving physicochemical properties such as solubility and getting a first insight into in vitro metabolism properties of this compound class.
2. Results and Discussion
2.1. Docking studies
To explore which structural alternations of the hit molecule MMV688845 can be favourable for target binding, a molecular docking experiment was conducted based on the published crystallographic structure of Mtb RNAP (PDB code: 5UHE). We used the X-ray structure of the crystallized protein with the model compound D-AAP1 as a reference for our modelling approaches31. The structures of both substances are given in Scheme 1.
Scheme 1.

Structures of the hit compound MMV688845 and D-AAP1
According to Lin et al., D-AAP1 offers space for six additional non-hydrogen atoms at the position of the methyl group while it is coordinated to its target enzyme. Therefore, this position offers potential for structure-based hit to lead optimization31. In MMV688845, the methyl group is replaced by a morpholine ring, introducing five additional non-hydrogen atoms while retaining activity30. The docking study of MMV688845 and RNAPMtb (Figure 1A) indicates a hydrogen bond between the morpholine oxygen atom and the arginine residue R834 of the β’ subunit. Motivated by this finding, we were eager to find other hydrogen bond acceptor groups, which exploit the potential additional hydrogen bond for drug-target interactions and possibly lead to different physiochemical properties.
Figure 1.

Docking poses of MMV688845 (A), compound 20 (B), compound 14 (C) and the β’ subunit of RNAPMtb, and compound 39 after exchange of phenylalanine for tyrosine (D). Hydrogen bonds are displayed as yellow dashed lines. 2D visualizations of the shown interactions can be found in the Supporting Information (Figure S2). Visualization generated with Maestro graphical interface (Schrödinger Release 2022-3: Maestro, Schrödinger, LLC, New York, NY, 2021).
As the results of Lin et al. suggest only limited space for derivatization (five of the six proposed non-hydrogen atoms are already part of MMV688845), we chose to investigate structures that offer the desired properties while keeping the size of the relevant part of the molecule almost unchanged with only one or two additional atom(s). Our docking studies indicate that substitution of the morpholine part with a thiomorpholine and its respective oxidation products (sulfoxide and sulfone) will lead to a similar binding mode as observed for MMV688845. Figure 1B and Figure 1C depict the modelled complexes with MMV688845 derivatives containing sulfone and sulfoxide groups. The modelling data suggest that the oxygen atom(s) of the sulfone or sulfoxide can act as hydrogen bond acceptors for R834, whereas clashes with the target protein could not be observed. A superimposed visualization of D-AAP1 and substance 20 can be found in the Supporting Information (Figure S1).
In addition, we found in our docking experiments that a p-hydroxy group in a tyrosine analog of MMV688845 could form an additional hydrogen bond to the backbone of the target protein at P477 (Figure 1D) without any calculated clashes.
To check whether the proposed binding modes also have relevance in the RNAP of Mabs a protein-protein BLAST was used to align and compare the Mtb β and the β’ RNAP subunits with their respective relatives in Mabs. The sequence homology was found to be 90.4 % for the β subunits and 89.9 % for the β’ subunits, while the amino acids that are relevant for drug-target interaction in Mtb RNAP are highly conserved (only exception: A563 in Mtb to G512 in Mabs, a list of the relevant amino acids can be found in the Supporting Information, Table S1), making a similar binding mode in Mabs RNAP probable.
2.2. Chemistry
The objective of the synthetic work described in this study is the derivatization of MMV688845. For this purpose, a synthetic route based on a published synthesis of the hit compound33 was designed. Starting from Boc-protected amino acids, the preparation of analogs is possible in three steps, as shown in Scheme 2. This modified synthetic route allows targeted derivatization of MMV688845 without racemization of the amino acid stereocenter.
Scheme 2.

Synthetic pathway for derivatization of MMV688845 analogs (T3P: propanephosphonic acid anhydride, TFA: trifluoroacetic acid, DEPBT: 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one, DIPEA: N,N-diisopropylethylamine)
2.2.1. Synthesis
In the first step (general procedure A), an amide bond is synthesized using the phosphonic acid anhydride T3P in a mixture of EtOAc and pyridine35, followed by removal of the Boc protecting group using TFA. The second amide bond in the molecule is formed using the coupling reagent DEPBT36, for which the amine is reacted with an aromatic carboxylic acid.
The synthesis and modification of aniline building blocks were crucial for the desired derivatization, for which the 2-morpholinoaniline in MMV688845 was the starting point. Using the syntheses shown in Scheme 3, thiomorpholine and its oxidation products, i.e., sulfone and sulfoxide, were introduced as substituents for derivatization. The introduction of thiomorpholine was achieved by a nucleophilic substitution on 2-bromonitrobenzene (general procedure C) and subsequent reduction of the nitro group by hydrazine (general procedure D). In case of IS11 we tried to couple commercially available thiomorpholine dioxide to 1-bromo-2-nitrobenzene to avoid the use of oxidizing agents completely, because oxidization could also occur at the aniline nitrogen atom to form its N-oxide. General procedure C did not yield the desired product in this case. A reason for this could be the electron withdrawing effect of the two oxygen atoms which makes the nitrogen of thiomorpholine dioxide less nucleophilic preventing the substitution. A BINAP and transition metal aided coupling was investigated which produced the desired aniline in yield of 32 %. Thus, we eventually decided against this latter approach as it is more expensive and less efficient than the nucleophilic substitution approach37.
Scheme 3.

Preparation of the aniline building block (BINAP: 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; TFA: trifluoroacetic acid)
For the synthesis of 2-(tetrahydro-2H-thiopyran-4-yl) anilines, a different synthetic strategy was necessary (general procedure H in Scheme 3). The desired derivatives were prepared by C-C coupling of 4-oxothiane and Boc-2-aminophenylboronic acid pinacol ester38. For this synthetic method, 4-oxothiane was transformed to a sulfonylhydrazone using tosylhydrazide. In the subsequent step the C-C coupling was carried out by reaction of the sulfonylhydrazone with the boronic acid pinacol ester in the presence of cesium carbonate. The final Boc cleavage was performed in TFA/DCM. Mechanistically, Barluenga et al. postulated that the tosylhydrazones so formed decompose thermally with formation of a diazo intermediate (Bamford-Stevens reaction) which reacts with the boronic acid or its ester.39
The oxidation of the thioether moiety was carried out according to general procedure E and F as shown in Scheme 4. By using different oxidizing agents, sulfone and sulfoxides were selectively obtained. The oxidation was conducted on the products of general procedure A or on the substituted nitro building blocks. Starting from the thiomorpholine derivatives, sulfoxides were prepared using NaIO4, while the analogous sulfones were obtained by oxidation with m-CPBA. The molecular structure of the oxidation products was unambiguously determined by X-ray crystallography, as described below. Oxidizing the sulfur in the 4-(4-R2-2-nitro-phenyl)thiomorpholines intermediates is an alternative way to achieve oxidation (for example see synthesis of 47 (sulfoxide) or 25 (sulfone)). S-Oxidation of the nitro building block has the disadvantage that the more polar groups (especially the sulfoxides) are present in subsequent synthetic steps, which complicates purification of the products by normal phase column chromatography. S-Oxidation after the final coupling step has not been investigated since the thiophene groups are also prone to oxidation40.
Scheme 4.

Synthesis of thiomorpholine sulfoxides and sulfones (m-CPBA: meta-chloroperoxybenzoic acid)
2.2.2. Stereochemical evaluation of the synthetic pathway
The synthetic pathway was designed to meet a series of requirements as previous synthetic approaches harbored certain disadvantages as highlighted below. It is known that only the R enantiomer of MMV688845 shows activity against Mabs, whereas the S enantiomer is inactive31,33. The synthetic procedure described by Ebright et. al.32 required preparative chiral HPLC separation of the enantiomers in the racemic mixture of the final compounds. To avoid the necessity of enantiomer separation, an important goal in the design of the synthesis was to start with reactants containing the required R configuration and preserve that configuration throughout the synthesis. Boc-protected (R)-phenylalanine is a readily available and inexpensive starting material for most compounds. In our previous study, we made important steps towards the realization of this goal33. To determine the stereochemistry of the reactions, (R)-MMV88845, (S)-MMV88845 and (rac)-MMV88845 were synthesized starting from the (R)-, (S)- or (rac)-phenylalanine using the new synthetic procedure.
Although phenylalanine is not known for its susceptibility to racemization, like e.g., phenylglycine containing peptides41, literature shows that phenylalanine racemization can occur even in usual and mild amidation procedures33,42. This is why we chose to investigate on the issue of stereochemistry to avoid this uncertainty and find a straightforward synthetic procedure that allows an efficient and exclusive production of the active R enantiomer. The synthetic step bearing the highest risk of phenylalanine racemization is the first amide coupling with the aniline building block yielding an anilide. The necessary activation of the carboxylic acid of the phenylalanine induces a strong electron-withdrawing effect on the α-H atom of the phenylalanine. As a result, the α-H atom can be subtracted more easily than in any other step of the synthesis, especially under the basic conditions present during most amide coupling reactions43-45. To achieve coupling conditions as mild as possible, we adapted the T3P (n-propanephosphonic acid anhydride46) coupling method published by Dunetz et al35. The authors showed that a mixture of EtOAc and pyridine at 0 °C yielded the lowest degree of racemization. Another advantage of T3P is that the side products formed in the coupling reactions are water-soluble and hence can be readily separated by extraction during work up. The intermediate and final compounds were analyzed by chiral HPLC (see Supporting Information). The ee-values are listed in Table 1.
Table 1.
ee-values of the starting materials (Boc protected amino acids), the intermediates 1-(R), 1-(S) and 1-(rac) being the intermediates after anilide formation; 2A-(R), 2A-(S) and 2A-(rac) being the Boc deprotected intermediate and final compounds 2B-(R), 2B-(S) and 2B-(rac) during synthetic route.
| Boc-Phe- | 1- | 2A- | 2B- | |
|---|---|---|---|---|
| ee-value [%] | ||||
| (R) | 99.82 | 99.80 | 99.92 | 99.70 |
| (S) | 100.00 | 99.34 | 99.84 | 99.79 |
| (rac) | 0.24 | 0.64 | 0.47 | 0.06 |
All products that were synthesized from enantiopure Boc-phenylalanine derivatives showed ee-values higher than 99 % indicating that virtually no racemization occurs under the reaction conditions used. Specific optical rotations have been determined for the compounds in Table 1 and can be found in the Supporting Information.
2.2.3. Structural elucidation
To prove that oxidation indeed occurred at the S atom of the thiomorpholine moiety only, an X-ray crystal structure analysis was conducted on a selection of compounds, viz. 6, 14 and 20 , because the formation of thiomorpholine N-oxide is also conceivable under the conditions used47. We recently described the crystal and molecular structure of racemic MMV688845 (CSD refcode: BALNUB)33, while the investigated compound set for this study represents the direct thiomorpholine analog of MMV688845 6, its sulfoxide derivative 14 and its sulfone derivative 20.
We obtained colourless crystals of enantiopure 6 and 20 suitable for X-ray diffraction from a chloroform/heptane solvent system. Interestingly, two crystallographically independent homochiral molecules of 6 (Figure 2A) form a hydrogen-bonded dimer about a pseudo center of symmetry in the crystal structure (triclinic system, space group P1) analogous to the crystallographic center of symmetry observed for racemic MMV688845 (triclinic system, space group P-1). The crystal structure of 20 is isomorphous with that of 6.
Figure 2.


Molecular structures of 6 (A), 20 (B) and 14 (C), as determined by X-ray crystallography. Displacement ellipsoids are drawn at the 50 % probability level. Hydrogen atoms on nitrogen and the chirality center, are represented by small spheres of arbitrary radius otherwise omitted for clarity. Dashed lines present N─H⋯O hydrogen bonds. Minor positional disorder of thiophene rings (ca. 6 %) in 6 (A) and 20 (B) is also not shown for the sake of clarity. Solvent water in the crystal structure of 14 (C)· 1.5 H2O is not shown. The structure of 14 (C) was refined with aspherical atomic scattering factors using NoSpherA248,49.
The corresponding sulfoxide 14 crystallized as a sesquihydrate from aqueous methanol. The homochiral molecules likewise constitute a hydrogen-bonded dimer through N─H⋯O hydrogen bonds formed between the phenylalanine amide moieties. The dimer is, however, not pseudo centrosymmetric as in 6 and 20 but exhibits a crystallographic twofold rotation axis (monoclinic system, space group I2). N─H⋯O hydrogen bond parameters within dimers in are essentially comparable in 6, 20 and 14 (see Supporting Information).
Bearing in mind that single-crystal X-ray diffraction does not prove the homogeneity of the bulk material, the molecular structures and absolute configurations of the sulfone 20 (Figure 2B) and the sulfoxide 14 (Figure 2C) were also confirmed by X-ray crystallography, indicating oxidation of the sulphur atom in the isolated products without formation of thiomorpholine N-oxides.
2.3. Microbiology
2.3.1. In vitro activity determination against NTM and Mtb
The in vitro activity of MMV688845 analogs described in this study was determined as MIC90 against a panel of NTM (Mabs, Mycobacterium intracellulare and Mycobacterium smegmatis) and Mtb – the most prevalent mycobacterial pathogen worldwide. The mycobacteria were selected to ensure comparability of our data (where applicable) to other publications31,33,50,51, for their clinical relevance52 in both pulmonary53 and extrapulmonary54 clinical presentations, and in view of the severity and treatability55 of the infections.
Since Mabs has become a serious problem in the clinics19, we particularly focused on this species. The activity was therefore analyzed against two different Mabs strains, namely the reference strain ATCC 19977 and the clinical isolate Mabs Bamboo. To assess possible effects of the growth medium on the in vitro activity, the assays were performed in both Middlebrook 7H9 and cation-adjusted Mueller-Hinton II broth medium.
As Mabs is capable of infecting human macrophages and actively proliferates intracellularly by evading certain immune defense mechanisms55, activity was also determined in a macrophage infection model assay. The macrophages used were derived from THP1 cells, infected with an RFP labelled Mabs strain prior to treatment with the test substances. After incubating for three days the macrophages were stained with DAPI and internal growth inhibition was determined by measuring RFP fluorescence (for detailed method description see Supporting Information).
M. intracellulare ATCC 35761 (Mintra) was chosen to represent the clinically relevant M. avium complex (MAC)56. Studies in the United States have shown that MAC is the most frequently isolated NTM in both pulmonal lung disease53 and extrapulmonary infections57.
M. smegmatis (Msmeg) is a fast-growing mycobacterium generally considered non-pathogenic and often used as a surrogate organism for M. tuberculosis50,51. To demonstrate a broad spectrum of activity against fast-growing mycobacteria and to analyze species-dependent differences in efficacy, M. smegmatis mc2 155 was included in the selection of mycobacteria. We report the results of broth microdilution MIC90 assays for each substance in Table 2.
Table 2.
Antimycobacterial activity of MMV688845 derivativesA.

|
MIC90 | MIC50 | MIC90 | MIC50 | MIC90 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Msmeg mc2 155B |
Mtb H37RvC |
Mabs
ATCC19977 B |
Mabs ATCC19977B |
Mintra ATCC 35761B |
Mabs BambooD |
Mabs ATCC 19977 + THP1E |
|||||||
| 7H9 | 7H9 | 7H9 | MHII | 7H9 | 7H9 | RPMI | |||||||
| R1 | R2 | X, Y | R3 | RFP | OD | RFP | RFP | RFP | OD | OD | RFP | RFP | |
| MMV845 | Phenyl | H | O, N | 2-Thiophenyl | 0.78 | 0.78 | 6.25 | 12.5 | 0.78 | 1.7 | 4.4 | 1.56 | 12.5 |
| 6 | Phenyl | H | S, N | 2-Thiophenyl | 3.13 | 6.25 | 12.5 | 50 | 0.78 | 2.5 | 14 | 12.5 | > 100 |
| 7 | Phenyl | H | S, N | 2-F-Phenyl | 3.13 | 6.25 | 12.5 | 50 | 0.78 | 3 | 18 | 12.5 | > 100 |
| 11 | Phenyl | F | S, N | 2-Thiophenyl | 3.13 | 1.56 | 25 | 50 | 0.78 | 1.4 | 14 | 3.13 | > 100 |
| 12 | Phenyl | F | S, N | 2-F-Phenyl | 6.25 | 1.56 | 25 | 25 | 0.78 | 1.15 | 12 | 3.13 | 100 |
| 14 | Phenyl | H | S=O, N | 2-Thiophenyl | 1.56 | 0.4 | 3.13 | 6.25 | 0.1 | 0.33 | 2 | 3.13 | 25 |
| 15 | Phenyl | H | S=O, N | 2-F-Phenyl | 0.78 | 0.78 | 6.25 | 3.13 | 0.1 | 0.4 | 2.1 | 0.78 | 12.5 |
| 17 | Phenyl | F | S=O, N | 2-Thiophenyl | 1.56 | 0.4 | 1.56 | 3.13 | 0.05 | 0.28 | 1.9 | 0.78 | 25 |
| 18 | Phenyl | F | S=O, N | 2-F-Phenyl | 1.56 | 0.78 | 3.31 | 3.13 | 0.2 | 0.52 | 2 | 0.39 | 12.5 |
| 20 | Phenyl | H | O=S=O, N | 2-Thiophenyl | 1.56 | 0.2 | 3.13 | 3.13 | 0.1 | 0.24 | 1.7 | 0.78 | 6.25 |
| 24 | Phenyl | H | O=S=O, N | 2-F-Phenyl | 0.78 | 0.2 | 0.78 | 1.56 | 0.05 | 0.23 | 4.3 | 0.39 | 6.25 |
| 28 | Phenyl | F | O=S=O, N | 2-Thiophenyl | 1.56 | 0.78 | 6.25 | 3.13 | 0.2 | 0.43 | 6.5 | 0.39 | 3.13 |
| 29 | Phenyl | F | O=S=O, N | 2-F-Phenyl | 1.56 | 0.78 | 3.13 | 3.13 | 0.1 | 0.4 | 4.5 | 0.39 | 3.13 |
| 32 | Phenyl | H | S, CH | 2-Thiophenyl | 6.25 | > 25 | 50 | 100 | 6.25 | 21 | 44 | > 100 | > 100 |
| 34 | Phenyl | H | S=O, CH | 2-Thiophenyl | 6.25 | > 25 | 25 | 50 | 3.13 | 6 | 20 | > 100 | > 100 |
| 36 | p-Hydroxyphenyl | F | S, N | 2-Thiophenyl | 12.5 | 0.78 | 12.5 | 50 | 0.1 | 1.6 | 15 | 12.5 | 50 |
| 37 | p-Hydroxyphenyl | F | S, N | 2-F-Phenyl | 6.25 | 1.56 | 25 | 50 | 0.2 | 1.7 | 15 | 6.25 | 25 |
| 39 | p-Hydroxyphenyl | F | O=S=O, N | 2-Thiophenyl | 6.25 | 0.78 | 6.25 | 3.13 | 0.05 | 0.4 | 9 | 6.25 | 25 |
| 40 | p-Hydroxyphenyl | F | O=S=O, N | 2-F-Phenyl | 6.25 | 0.78 | 6.25 | 6.25 | 0.2 | 1.1 | 13 | 3.13 | 25 |
| 42 | 3-Thiophenyl | H | O, N | 2-Thiophenyl | 1.56 | 3.13 | 12.5 | 12.5 | 1.56 | 2.3 | 7 | 12.5 | 50 |
| 43 | 3-Thiophenyl | H | O, N | 2-F-Phenyl | 1.56 | 3.13 | 12.5 | 12.5 | 0.78 | 1.9 | 7 | 6.25 | 25 |
| 45 | 2-Thiophenyl | H | O, N | 2-Thiophenyl | 1.56 | 6.25 | 12.5 | 12.5 | 1.56 | 2.4 | 9 | 12.5 | > 100 |
| 46 | 2-Thiophenyl | H | O, N | 2-F-Phenyl | 1.56 | 3.13 | 6.25 | 12.5 | 0.78 | 2 | 7 | 6.25 | 25 |
| 50 | 3-Thiophenyl | H | S=O, N | 2-Thiophenyl | 3.13 | 6.25 | 6.25 | 12.5 | 0.2 | 3.4 | 15 | 12.5 | 100 |
| 51 | 3-Thiophenyl | H | S=O, N | 2-F-Phenyl | 1.56 | 3.13 | 6.25 | 12.5 | 0.2 | 1.5 | 4.4 | 6.25 | 50 |
| 53 | 2-Thiophenyl | H | S=O, N | 2-Thiophenyl | 3.13 | 3.13 | 12.5 | 12.5 | 0.2 | 1.3 | 5.8 | 12.5 | 100 |
| 54 | 2-Thiophenyl | H | S=O, N | 2-F-Phenyl | 1.56 | 1.56 | 6.25 | 12.5 | 0.2 | 1.8 | 6.5 | 6.25 | 50 |
Color coding: For each tested mycobacterial strain the colors show the difference to the average activity value, dark green: lowest MIC value; light green: below average; white: closest to average; light red: above average, dark red: highest MIC value. AFor detailed information on the methodology of the assays see the Supporting Information. BIncubated for three days at 37 °C (Msmeg and Mabs) or five days (Mintra). Performed in duplicate, results were averaged. Data was obtained via RFP measurement (shown here) or OD measurement (see SI). CIncubated for seven days at 37 °C. Data was obtained via OD measurement. Performed in duplicate, results were averaged. DIncubated for three days at 37 °C. Data was obtained via OD measurement. Performed in duplicate, results were averaged. EInfected cells were incubated for 4 days (37 °C, 5 % CO2). Performed in duplicate, results were averaged.
Optical density derived and RFP-fluorescence derived MIC90 values were similar. Although OD measurement is a well-established method to analyze bacterial growth, RFP assay data often offer higher sensitivity and specificity58,59. Nevertheless, in our case the OD and RFP values correlate well and the MIC90 values of the reference compound do not differ substantially.
The MIC90 values for MMV688845 determined in this study are consistent with values found in the literature. Low et al. determined a MIC90 value against Mabs of 7 μM, and we found 6.25 μM in Middlebrook 7H9 medium. To investigate culture media independency the growth inhibitory effect was analysed in MHII and a MIC90 value of 12.5 μM was determined indicating culture medium independency. The MIC90 value we determined against Mtb is slightly lower (0.78 μM) than that of Low et. al. (1.2 μM).
Derivatization of the parent compound MMV845 was performed at the residues R1 to R4 as shown in Scheme 5.
Scheme 5.

AAP base scaffold and derivatized residues
First, we compared analogs that only differ at the R1 substitution site. Figure 3 gives an overview. Four different moieties for R1 (phenyl, p-hydroxyphenyl, 2-thiophenyl, 3-thiophenyl) were included in our study. As a patent by Ebright et al. reveals32, MIC90 values are particularly sensitive to changes at the R1 position. The most active compounds against Mtb and M. avium that were synthetized have a phenyl group in position R1. p-Hydroxyphenylgroups and their respective acetyl or benzoic acid esters substitutions at R1 were also investigated in this patent and resulted in a loss in activity against both Mtb and M. avium. The other variations in R1 that are shown (e.g., m-hydroxyphenyl) resulted in severe activity loss32. Bearing this in mind, we decided to include both phenyl and p-hydroxyphenyl substituents for derivatization and screening. Despite knowing that even minor changes result in activity loss, 2- and 3- thiophenyl substituents were included because these structures are isosteric to phenyl60 and therefore offer a hopefully tolerable change to the molecule without activity loss.
Figure 3.

Comparison of MIC90 values of compounds that only differ in R1. Compounds that only differ in R1 are shown in the same color. Displayed values were generated by RFP measurement. Only Mtb MIC90 values were generated by OD measurement. Compounds that only differ in R1are displayed in the same color; the symbols represent the respective R1 substituent.
For Mabs no differences in activities can be seen, but there is a trend that phenyl groups perform better than p-OH-phenyl, e.g., 29 has a twofold increased activity compared to 40. The same trend can also be seen in Mabs Bamboo. It is remarkable that the difference becomes more pronounced when looking at the MIC90 values determined in the macrophage infection model assay. In this case the compounds suffer from a four- to eightfold decrease in activity when a p-OH-phenyl group is present. Possibly these results are based on the higher polarity of the p-OH-phenyl group leading to lower permeability through the membranes of both the macrophages and bacteria.
In Msmeg, MIC90 values show a fourfold increase when a p-OH-phenyl group is introduced into the molecule.
Interestingly, the MIC90 values against Mintra show an opposite trend. A comparison of 28 and 39 shows that the p-OH-phenyl group has a positive effect on the MIC90 values with a two- to fourfold increase in activity. Currently, we have no well-founded explanation for this result, though structural differences between the RNAPs of Mabs and Mintra and the interaction of R1 with the target may play a role.
The MIC90 values determined against Mtb show that p-OH-phenyl as a substituent is never inferior to phenyl. In the case of compounds 11 and 36 p-OH-phenyl substitution shows a better activity against Mtb.
Although the change from phenyl to 2- or 3-thiophenyl is very small regarding the chemical and electronic properties of the molecule, it leads to activity loss in every single species investigated, showing that R1 is indeed sensitive to even minor changes in the substituent R1.
The primary intent of derivatizing R2 was to sterically shield the anilide to preventhydrolysis and to slow down or prevent metabolism of the morpholino-anilide moiety of the molecule. To this end, we introduced a fluorine atom in para position of the morpholine moiety and determined whether this resulted in lower MIC90 values. An overview of compounds only different at R2 is given in Figure 4.
Figure 4.

Comparison of MIC90 values of compounds that only differ in R2. Compounds that only differ in R2 are shown in the same color. Displayed values were generated by RFP measurement. Only Mtb MIC90 values were generated by OD measurement. Compounds that only differ in R2are displayed in the same color; the symbols represent the respective R2 substituent.
The introduction of the fluorine atom results in a tendency for higher MIC90 values. It is worth noting that for Mabs a two- to fourfold increase in MIC90 values was observed (e.g., 6/11 or 24/29). Interestingly, this behaviour is reversed when R3 contains a thiomorpholine sulfoxide group. Both thiomorpholine sulfoxide structures (14/17, 15/18) show twofold increased MIC90 values against Mabs. This effect is not observed for the respective sulfones. For example, the Mabs and Mtb MIC90 values of the most active compound 24 are increased fourfold on addition of the fluorine substituent. A similar effect was observed in the other sulfones, 20/28.
The MIC90 values determined for Mabs in the macrophage infection model decreased upon introduction of the fluorine substituent. The effect is more pronounced in the MIC50 values. Here a two- to fourfold decrease for all substances with the fluorine substituent was found.
The main R3 variation of the compound set presented here is the exchange of the morpholine group of MMV688845 by a thiomorpholine group and the respective oxides 1λ4-thiomorpholin-1-one (thiomorpholine-sulfoxide) and 1λ6-thiomorpholine-1,1-dione (thiomorpholine-sulfone). Tetrahydrothiopyran and its sulfone were included to check on the necessity of an aromatic amine structure within R3. An overview of the MIC values is given in Figure 5.
Figure 5.

Comparison of MIC90 values of compounds that only differ in R3. Compounds that only differ in R3 are shown in the same color. Displayed values were generated by RFP measurement. Only Mtb values were generated by OD measurement. Compounds that only differ in R3are displayed in the same color; the symbols represent the respective R3 substituent. Compounds 32 and 34 were excluded in this figure.
MIC90 determination against Msmeg revealed a decrease (fourfold) in activity when morpholine was exchanged for thiomorpholine in MMV688845. An increase of activity was seen for the respective oxides (twofold increase). For all other comparison groups, S-oxidation had no strong effect on the MIC90 values.
In Mtb, however, oxidation to the sulfone in 20 resulted in a fourfold decrease in the MIC90 values (20, MIC90 down to 200 nM) in comparison to MMV688845. The respective sulfoxide also showed better activity than the hit compound (twofold decrease in the MIC90 values).
For Mabs a similar behaviour was observed: In comparison to MMV688845 a two- to fourfold increase in activity was observed, the sulfone leading to lower MIC values. The same is true for all other compared groups that contain either phenyl or p-hydroxyphenyl as R1. In these groups, improvements of MIC90 values of down to 780 nM (24) were achieved. Groups that contain 2- or 3-thiophenyl as R1 did not benefit from the oxidation. The same effects are seen with the clinical isolate Mabs Bamboo, even though the most active substances do not reach as low MIC90 values as those that we observed with the Mabs ATCC 19977 strain. Nevertheless, the respective sulfones and sulfoxides offer sub-micromolar MIC50 values and low micromolar MIC90 values. Within the macrophage infection model, we noted that the oxides are still more potent than MMV688845 but with less of a difference. A difference between the sulfoxides used and their respective sulfones is remarkable: Sulfoxides show a strong decrease in activity when tested in the macrophage infection model. This could be due to their high polarity which could negatively affect their ability to cross biological bilayer membranes. The need to pass two membranes (macrophage and bacterium) rather than one may have magnified this effect.
Against Mintra, thiomorpholine oxidation also increases activity. MIC90 values are decreased by the factor of 4 in comparison to MMV688845. In other groups, oxidation resulted in compounds that have MIC90 values down to 50 nM (24 and 39), which is equivalent to an eightfold increase in activity.
For varying R4, we considered the thiophene-2-carboxamide (same as in the hit compound MMV688845) and the 2-fluorobenzoic acid amide for initial testing, since Ebright et al. had demonstrated that both these residues are present in the most active compounds32.
Figure 6 shows the comparison of MIC90 values against the different mycobacterial strains of compounds that differ only in R4. MIC90 values of the displayed compounds do not differ substantially depending on the R4 substituent. In most of the cases, 2-fluorophenyl and 2-thiophenyl demonstrate similar activity levels within a maximum twofold difference with the tendency that the 2-fluorobenzoic acid compounds achieve slightly lower MIC90 values. Only in 17 of 84 direct comparisons (throughout all bacterial species tested) 2-thiophenyl compounds exhibit lower values than their respective 2-fluorophenyl compounds. In the macrophage infection assay the 2-thiophenyl compounds were always inferior to the 2-fluorophenylcompounds.
Figure 6.

Comparison of MIC90 values of compounds that only differ in R4. Compounds that only differ in R4 are shown in the same color. Displayed values were generated by RFP measurement. Only Mtb values were generated by OD measurement. Compounds that only differ in R4 are displayed in the same color; the symbols represent the respective R4 substituent.
2.3.2. Bactericidality
The ability to kill mycobacteria instead of forcing them into a dormant or quiescent state is a crucial property of effective anti-mycobacterial treatments, in particular for the therapy of Mabs infections61. To find out whether the AAP derivatives kill Mabs, a minimum bactericidal concentration (MBC) evaluation was conducted. Typically, an MBC is defined as the concentration at which the level of colony forming units in an inoculum is reduced by three logarithmic units, or in other words a killing rate of 99.9 % of the bacteria that were present at the start of the experiment. In mycobacteriology though, this threshold was redefined for reasons of reproducibility and accuracy and is currently set at 99.0 % as mycobacteria need inconveniently longer incubation times for high killing rates of 99.9 %62.
MBC testing against Mabs ATCC19977 was conducted for a selection of the most active compounds as well as MMV688845 Figure 7. MMV688845 did not reduce viable bacteria at a concentration below 12.5 μM. At its MIC90 value of 6.25 μM, no reduction of viable bacteria was observed. It achieves its MBC99 at a concentration of 50 μM (MBC99 at 8X MIC90). In contrast, the new derivatives reported here show a higher reduction of viable bacteria at lower concentration levels. 14 and 15 (both including a sulfoxide moiety) show a reduction at 3.13 μM but stay below 90 % reduction (58 % and 81 % respectively) while the sulfones 20 and 24 both reach a reduction of 95 % at the same concentration levels. For all tested derivatives, concentration levels of 6.25 μM and higher result in a reduction of 99 % or only slightly above that threshold making them about 4 times more bactericidal than the hit compound. Compounds 20 and 15 achieved their respective MBC99 at 2X MIC90. This is a significant improvement in cidal activity over MMV845.
Figure 7.

MBC determination against Mabs ATCC19977 of a selection of AAP derivatives. Bacteria reduction levels are displayed as dashed, horizontal lines. Substances were analysed in triplicates as indicated by the white dots.
2.3.3. Testing against MMV688845-resistent mutants
To verify on-target the activity of the analogs described in this study, compounds 14, 15 and 24 were tested against the MMV688845 resistant strain Mabs Bamboo 845R-2.1. The strain and its resistance to MMV688845 were previously described34. The data presented in Table 3 show evidence that the target is the RpoB subunit of the bacterial RNAP, because Mabs Bamboo 845R-2.1. harbors a P473L mutation of RpoB subunit, which is located directly at the binding site of the phenylalanine amides that was proposed by Lin et al.31 and used for the herein described modelling study (2.1).
Table 3.
Activity data against the MMV688845 resistant strain Mabs Bamboo 845R-2.1. (generated by selecting at 50 μM MMV688845 using Mabs Bamboo). This strain is resistant to MMV688845 and has a RpoB P473L mutation34. The observed frequency of resistance (FoR) was 5.83 x 10−8 CFU−1. CLR: clarithromycin; RFB: rifabutin)
| MIC90 (μM) | |
|---|---|
| Mabs Bamboo 845R-2.1 | |
| 7H9 | |
| CLR | 0.27 |
| RFB | 1.1 |
| MMV688845 | >100 |
| 24 | 42 |
| 14 | >100 |
| 15 | 100 |
Similar to MMV688845, the three tested analogs suffered from a severe activity loss by at least factor 10 indicating the bacterial RNAP is involved in the molecular activity of the newly synthesized phenylalanine amides. Rifabutin, a RNAP inhibitor with a different binding site, achieved a MIC90 value of 3 μM in a former activity determination against the MMV688845 sensitive Mabs Bamboo25. The results of the present study show no decline in activity against the resistant strain which indicates that the probability of cross resistance between the two RNAP inhibitors is low.
2.4. In vitro plasma and microsomal stability
Former investigations on MMV688845 revealed low drug plasma levels after oral administration in Sprague Dawley Male rats30 and CD 1 mice34, which could be due to low metabolic stability. To narrow down the reasons why MMV688845 seems to exhibit low plasma levels, we conducted plasma stability and microsomal stability assays on the hit compound and the most active derivative, compound 24. The substances were tested in both human and murine plasma and microsomes. The plasma stability is given as a %-remaining versus time plot in Figure 8. Microsomal stability is displayed in Table 4.
Figure 8.

Plasma stability of MMV688845 and compound 24 in human and murine plasma over five different time points. Substances were analysed in duplicates. Error bars display standard deviations.
Table 4.
Half-times and intrinsic clearance values after incubation of MMV688845 and compound 24 with human and murine liver microsomes over 40 min. Experiment has been performed in duplicates.
| Substance | t1/2 [min] human microsomes |
Clint [μL/min*mg] human microsomes |
t1/2 [min] murine microsomes |
Clint [μL/min*mg] murine microsomes |
|---|---|---|---|---|
| MMV845 | 1.7 | 1366 | 0.9 | 1837 |
| 24 | 1.5 | 1113 | 1.8 | 1562 |
The results show that both tested substances are stable in human plasma throughout the tested time frame. In murine plasma though, MMV688845 shows a stronger decline in concentration than compound 24. A possible explanation for this behaviour is that the fluorine atom in the carboxylic acid moiety of 24 sterically shields the amide bond of compound 24 from being a target for murine carboxyesterases (CES), leaving it uncleaved. As CES do not seem to be abundant in human plasma63, the stability of both substances in human plasma could be higher.
The determined half-times show that both substances suffer from severe instability in a liver microsome assay, which is probably the reason for the low bioavailability in vivo described above. A rapid degradation of the phenylalanine amides by oxidation (e.g., hydroxylation) or hydrolytic activity of the microsomes seems to us a probable explanation for this observation64-66.
2.5. Cytotoxicity
MMV688845 was tested for cytotoxicity using the HEP 93 liver cancer cell line and found to be non-cytotoxic29,33. In our study we extended cytotoxicity testing of MMV688845 and all synthesized compounds to seven mammalian cell lines using a colorimetric micro-culture assay that utilized sulforhodamine-B as a staining agent. The methodology of this assay is described in various references67-69.
The EC50 values are listed in Table 5. We confirmed the non-cytotoxic properties of MMV688845, which was not cytotoxic for all tested cell lines at concentrations of up to 30 μM. The same was true for most variants of MMV688845. Only a few of the analogs showed very weak cytotoxicity against the tested cell lines. Most potent compounds can be considered as unproblematic since the MIC values are far below the EC50 cytotoxicity values.
Table 5.
EC50 values [μM] of AAP analogs against 7 different mammalian cell lines. A Melanoma; B Colon cancer; C breast cancer; D Ovarian cancer; E Mouse fibroblasts; F Cervical cancer; G Human embryonic kidney cancer. The assay was performed in triplicates. Standard deviations are displayed if applicable-
| A375A | HT29B | MCF7C | A2780D | NIH 2T3E | HeLaF | HEK293G | |
|---|---|---|---|---|---|---|---|
| MMV845 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 6 | > 30 | > 30 | 16.4 ± 1.7 | 19.8 ± 2.0 | > 30 | > 30 | 19.1 ± 2.4 |
| 7 | 18.6 ± 4.2 | > 30 | 13.6 ± 1.4 | 14.9 ± 1.6 | > 30 | > 30 | 6.3 ± 1.2 |
| 11 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 12 | 29.4 ± 1.4 | > 30 | > 30 | 28.3 ± 1.4 | 28.5 ± 2.0 | 28.3 ± 1.5 | n.d. |
| 14 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 |
| 15 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 |
| 17 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 18 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 20 | > 30 | > 30 | > 30 | 23.6 ± 2.2 | > 30 | > 30 | > 30 |
| 24 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 28 | > 30 | > 30 | 23.2 ± 2.8 | 24.0 ± 5.2 | 26.9 ± 5.0 | > 30 | > 30 |
| 29 | > 30 | > 30 | 25.9 ± 2.7 | 23.2 ± 4.4 | 24.2 ± 4.2 | > 30 | > 30 |
| 32 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 34 | > 30 | > 30 | > 30 | > 30 | > 30 | 29.8 ± 1.3 | n.d. |
| 36 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 37 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 39 | > 30 | 29.8 ± 1.7 | > 30 | 29.7 ± 1.2 | > 30 | > 30 | n.d. |
| 40 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 |
| 42 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 43 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 45 | > 30 | 29.8 ± 1.9 | > 30 | 29.5 ± 2.0 | 29.0 ± 1.3 | > 30 | n.d. |
| 46 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 50 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 51 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 53 | > 30 | > 30 | > 30 | > 30 | > 30 | > 30 | n.d. |
| 54 | 29.3 ± 1.5 | > 30 | 28.9 ± 1.8 | 29.0 ± 1.7 | > 30 | > 30 | n.d. |
2.6. Determination of kinetic solubilities and cLogP values
Solubility is a crucial parameter for the development of new drug candidates and their in vivo efficacy, especially for oral administration70. Low bioavailability is often caused by poor aqueous solubility as drug substances must be transferred into a dissolved state so that they can pass through gastro-intestinal barrier and reach the blood circulation. The calculated LogP value can be a good parameter to estimate properties of new drug substances like their solubilities, because it reflects the lipophilicity/hydrophilicity of chemical substances, with high values indicating high lipophilicity. Usually, LogP values between 2 and 3 are desirable for pharmaceutical compounds because within that range a compromise of permeability and first pass clearance is given71.
The kinetic solubilities of the new MMV688845 derivatives in PBS were experimentally determined by a nephelometric method described by Bevan et al.72 (see Supporting Information).
Figure 9A shows the relation between the calculated LogP values and measured solubilities of the synthesized compounds. Compounds with cLogP values higher than 3.5 usually have solubilities lower than 50 μM, making them hard to handle in biological assays and giving them undesirable biopharmaceutical properties. At cLogP values below 3, the solubilities show a much higher variability. In this region six compounds show high aqueous solubilities of up to 431 μM.
Figure 9.

A: Relationship of cLogP values and their corresponding kinetic solubilities of all the synthesized AAP analogs. B: Relationship of cLogP values and their corresponding MIC90 values against Mabs of all the synthesized AAP analogs. C: Relationship of cLogP values and their corresponding MIC90 values against Mabs within the macrophage infection model of all the synthesized AAP analogs. The cLogP values were calculated with ChemDraw (Perkin Elmer Informatics Inc.).
Figure 9B and Figure 9C show the MIC90 values determined against Mabs in the standard micro-dilution assay as well as in the macrophage infection model. Compounds with a cLogP between 2 and 3 achieve the lowest MIC90 values. Within the macrophage infection model, a general increase in MIC90 values is observed as described above. Nevertheless, the most active compounds possess a cLogP value between 2 and 3, verifying a desirable LogP range of 2-3 for the development of the AAP compound class.
Table 6 summarizes the solubilities and cLogP values for the compounds synthesized. Introduction of thiomorpholine lowers the solubility of the compounds in PBS buffer by factor 2 to 3. This may be attributed to the lower electronegativity of the sulfur atom in comparison to oxygen which reduces the polarity within the molecule. A strong improvement in solubility is observed when thiomorpholine sulfoxides are present. For instance, the solubility of 15 is approximately 5 times higher than the solubility of the hit compound MMV688845. The improvement is not as prominent when thiomorpholine is oxidized to the respective sulfone (e.g., 20, 24, 28). In this case, a twofold increase in solubility was observed in comparison to their respective thioethers. The difference in solubility between sulfoxides and sulfones can be explained by the fact that sulfoxide itself is asymmetric since the free electron pair of the sulfur atom is still present. This provides a strong dipole moment to the molecule and increases its polarity. It should be noted that cLogP values are not a good predictor for the high difference in solubility of sulfones and sulfoxides since the cLogP values only differ by a value of 0.05. The results show that the highest increase in solubility is achieved by introduction of sulfoxide group.
Table 6.
Kinetic solubilities and cLogP values of all synthetized AAP derivativesA
| MMV845 | 6 | 7 | 11 | 12 | 14 | 15 | |
|---|---|---|---|---|---|---|---|
| Solubility [μM] | 82 | 34 | 28 | 37 | 31 | 271 | 431 |
| cLogP | 3.72 | 4.45 | 4.62 | 4.6 | 4.78 | 2.49 | 2.67 |
| 17 | 18 | 20 | 24 | 28 | 29 | 32 | |
| Solubility [μM] | 131 | 184 | 62 | 64 | 70 | 32 | 39 |
| cLogP | 2.65 | 2.83 | 2.54 | 2.71 | 2.7 | 2.87 | 4.87 |
| 34 | 36 | 37 | 39 | 40 | 42 | 43 | |
| Solubility [μM] | 85 | 78 | 58 | 170 | 158 | 30 | 31 |
| cLogP | 2.96 | 4.21 | 4.39 | 2.31 | 2.48 | 3.65 | 3.83 |
| 45 | 46 | 50 | 51 | 53 | 54 | ||
| Solubility [μM] | 41 | 39 | 74 | 96 | 60 | 96 | |
| cLogP | 3.53 | 3.71 | 2.42 | 2.59 | 2.3 | 2.48 |
Color coding: The colors show the difference to the average kinetic solubility of all tested compounds, dark blue: highest solubility; light blue: above average; white: closest to average; light red: below average, dark red: lowest kinetic solubilities of the tested compound set.
The aqueous solubility can otherwise be increased when R1 is exchanged for a polar p-hydroxyphenyl group. This resulted in a doubling of the solubility of thiomorpholine containing compounds (11/37 μM to 36/78 μM; 12/31 μM to 37/58 μM). This is also the case for their respective sulfone derivatives (36/78 μM to 39/170 μM; 37/58 μM to 40/158 μM).
In summary, based on a straightforward synthetic route, systematic derivatization made it possible to obtain an overview of the influence of the different substituents on the antimycobacterial activity of AAPs and hence important structure-activity relationship information as summarized in Scheme 6.
Scheme 6.

Overview of the influence of the different substituents on the antimycobacterial activity of AAPs. Red arrows indicate a loss in activity, green arrows indicate an increased activity caused by the respective residues.
3. Conclusions
We have shown that synthetic RNAP inhibitors derived from MMV688845 have the potential to reach in vitro MIC90 values of below 1 μM against pathogenic mycobacteria, e.g., Mtb and Mabs. In addition to the improved activity against mycobacteria and the absence of cytotoxicity, the new analogs display bactericidal activity (99 % CFU reduction) against Mabs at concentrations below 10 μM. Compounds 20 and 15 achieved MBC99 at 2X MIC90, giving them higher cidality than MMV845 (MBC99 at 8X MIC90). Systematic derivatization based on a straightforward synthetic route made it possible to obtain important structure-activity relationship information. It is worth noting that the oxidized thiomorpholine derivatives, sulfoxide and sulfone, result in an increase in activity in comparison to MMV688845 reaching sub-micromolar activities against Msmeg, Mintra and even Mabs. Sulfones show higher activities than sulfoxides across the board. Sulfoxides are interesting for further research because of their higher aqueous solubility. The data show that anti-Mabs targeting of MMV688845 is possible and that attractive MIC values can be achieved with this compound class, warranting further preclinical investigation. Since MMV688845 analogs do not exhibit cross-resistance with rifamycin-resistant mycobacteria, further investigation is of scientific interest to develop an RNAP inhibitor that is effective against these resistant pathogens. Particular attention should be paid to NTM, such as Mabs, as there are species-dependent differences in the structure-activity relationships, as shown in this study. In addition to further derivatization with the aim of increasing activity, the investigation of hepatic metabolism, pharmacokinetics, and activity of MMV688845 derivatives in vitro and in vivo should be addressed. In this study it was possible to show that compound 24 has an improved stability against mouse plasma in vitro than the hit compound MMV688845, but the very rapid degradation by liver microsomes is a drawback of the compound class. Elucidation of the hepatic metabolism of phenylalanine amides is desirable in future research, as this should help developing analogs with improved hepatic stability.
4. Materials and Methods
General
Starting materials were purchased and used as received. Solvents used for either synthetical or purification purposes were distilled and stored over 4 Å-molecular sieves. Glassware was oven-dried at 110 °C prior to use. For the determination of Rfvalues and other analytical purposes such as qualitative chromatography Merck TLC silica gel 60 on aluminium sheets with fluorescent indicator F254 were used. Flash chromatography was performed with a puriFlash® 430 instrument (Interchim, Montluçon, France). Columns were packed in either 8 g (v=10 mL/min), 45 g (v=30 mL/min) or 90 g (v=40 mL/min) cartridges with 40 - 63 μm normal phase silica gel produced by Carl Roth. Column loading was performed with the dry load method. NMR spectra were recorded on an Agilent Technologies VNMRS 400 MHz spectrometer. Chemical shifts are reported relative to the residual solvent signal (CDCl3: δH = 7.26 ppm; δC = 77.36 ppm; CD3OD δH = 3.31 ppm). 13C NMR spectral data were generally determined as attached-proton-test spectra (APT). Spectra have been cut, baseline and phase corrected and analyzed utilizing MestreNova 11.0 software (Mestrelab Research, S.L., Spain). APCI-MS (atmospheric pressure chemical ionization) was performed using an expression CMS mass spectrometer (Advion Inc., Ithaca, NY, USA), with both ASAP (atmospheric solids analysis probe) sampling and with the help of the Plate Express TLC-plate extractor. ESI measurements have been conducted on the same expression CMS mass spectrometer with an ESI ionization module and direct injection sampling. HRMS was carried out using a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA).
HPLC
All described final compounds described were confirmed to be of >95 % purity. Purity was measured by UV absorbance at 254 nm. The HPLC apparatus consists of an XTerra RP18 column (3.5 μm, 3.9 mm × 100 mm) from the manufacturer Waters (Milford, MA, USA) and two LC-10AD pumps, a SPD-M10A VP PDA detector, and a SIL-HT autosampler, all from the manufacturer Shimadzu (Kyoto, Japan).
Compounds used for stereochemical analyses (Boc-Phe-(R); 1-(R), 2A-(R) and 2B-(R); Boc-Phe-(S); 1-(S), 2A-(S) and 2B-(S); Boc-Phe-(rac); 1-(rac), 2A-(rac) and 2B-(rac)) were checked for purity using an Agilent 1260 HPLC instrument equipped with UV diode array detection (50 mm Eclipse Plus C18 1.8 μm, i.d. 4.6 mm, v = 1.0 mL min−1, λused = 220 nm). Elution systems: [Boc-Phe-(R); Boc-Phe-(S); Boc-Phe-(rac): acetonitrile/water 35:65; 0.1 % v/v TFA]; [1-(R); 1-(S); 1-(rac): acetonitrile/water 55:45]; [2A-(R); 2A-(S); 2A-(rac): methanol/water 20 mM NH4HCO3 pH9 55:45]; [2B-(R); 2B-(S); 2B-(rac): acetonitrile/water 45:55].
For preparative tasks, a XTerra RP18 column (7 μm, 19 mm × 150 mm) manufactured by Waters (Milford, MA, USA) and two LC-20AD pumps (Shimadzu, Kyoto, Japan) were used. The mobile phase was in all cases a gradient of methanol/water (starting at 95 % v/v water to 5 % v/v water) with 0.05 % v/v TFA added.
The determination of ee values was conducted using a Shimadzu Prominence LC-20A HPLC instrument with diode array detection (v = 1.0 mL min−1, λused = 220 nm). The utilized elution systems and chiral columns differ according to the structure that had to be analyzed: [Boc-Phe-(R); Boc-Phe-(S); Boc-Phe-(rac): 150 mm Chiralpak IA-3, 4.6 mm i.d.; acetonitrile/water 30:70; 0.1 % v/v TFA]; [1-(R); 1-(S); 1-(rac): 150 mm Chiralpak IB N-3, 4.6 mm i.d.; acetonitrile/water 55:45]; [2A-(R); 2A-(S); 2A-(rac): 150 mm Chiralpak IG-3, 4.6 mm i.d.; n-heptane/iso-propanol 90:10]; [2B-(R); 2B-(S); 2B-(rac): 150 mm Chiralcel OJ-3R, 4.6 mm i.d.; acetonitrile/water 40:60].
X-ray crystallography
Details of the X-ray intensity data collections, structure solutions and refinements and hydrogen bond parameters can be found in the Supporting Information. X-ray crystallography structures have been determined for compounds 6; 14 and 20. CCDC 2231845-2231847 contain the supplementary crystallographic data for this paper. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
General Procedure A – Amide coupling T3P
A solution of the Boc-protected α-amino acid (1.1 eq.) and the aniline (1 eq.) in a 1:2 mixture of pyridine (distilled) and EtOAc (distilled) was cooled to −20 °C with an isopropanol/dry ice bath. If the compounds did not dissolve completely at −20 °C additional pyridine/EtOAc mixture was added until complete dissolution. A 50 % m/v solution of T3P in EtOAc (2 eq.) was added to the mixture subsequently. After 10 min the dry ice bath was removed and replaced by an ice/water bath to keep the temperature at 0 °C overnight46. Afterwards a reasonable amount of EtOAc was added to the reaction mixture to wash it with 0.25 M KH2PO4 solution three times in a separation funnel. The organic phase was collected, dried with Na2SO4, and the solvent was removed under reduced pressure. As a general purification procedure, flash chromatography was performed with varying gradient eluting systems on 40-63 μm normal phase silica gel (see respective synthesis documentation in the Supporting Information).
General procedure B – Amide coupling DEPBT
The formation of the anilide bond (General procedure A) was followed by the cleavage of the Boc protecting group using TFA. For this purpose, the purified Boc-α-amino anilides were dissolved in DCM and an equal volume of TFA was added under stirring. The solution was stirred for 30 min at room temperature and checked for complete Boc-cleavage using thin layer chromatography (TLC). The DCM/TFA mixture was removed under reduced pressure after complete conversion. The crude product was reconstituted in EtOAc and washed with saturated NaHCO3 solution three times. The organic phase was collected, and the solvents were evaporated under reduced pressure.
The formation of the second amide bond was achieved with the coupling reagent DEPBT. For this synthetic step the Boc deprotected amino acid was dissolved THF with the respective aromatic carboxylic acid and DEPBT. After complete dissolution, DIPEA was added, and the solution was stirred overnight at room temperature. Afterwards the reaction mixture was washed successively once with 0.25 M KH2PO4, water, saturated NaHCO3, water and brine. The organic phase was dried with Na2SO4 and the solvents were removed under reduced pressure. For purification a flash chromatography was performed with varying gradient eluting systems on 40-63 μm normal phase silica gel (see respective synthesis documentation in the Supporting Information).
General procedure C – Nucleophilic substitution of thiomorpholine
1-Bromo-2-nitrobenzene or the respective derivatives were dissolved in thiomorpholine without addition of solvents in a small glass flask. The mixture was heated to 120 °C and stirred for 2 h. After cooling down to room temperature the precipitated solid was filtered out, the filter was washed thoroughly with DCM and the solid was discarded. DCM was evaporated under reduced pressure to obtain the bright-orange oily crude products. For purification flash chromatography was performed with varying gradient eluting systems on 40-63 μm normal phase silica gel (see respective synthesis documentation in the Supporting Information).
General procedure D – Reduction of 4-(2-nitrophenyl)thiomorpholines
4-(4-R2-2-nitro-phenyl)thiomorpholines were dissolved in ethanol. One third of the total amount of palladium 10 % m/m on active charcoal was added and the mixture was then heated to 50 °C and stirred vigorously. A 50 % v/v solution of hydrazine in water was added very slowly. The remaining amount of catalyst was added to the mixture in portions. Afterwards the temperature was increased to 90 °C and the mixture was refluxed for 30 min. The bright orange-yellow solution turned to a clear and colourless solution. The mixture was cooled down to room temperature and filtered to remove the catalyst. The filter was washed thoroughly with ethanol to extract residual product. Completion of the reaction and purity were checked with TLC. Solvents were then removed under reduced pressure. The crude product was used for the next synthesis without further purification when applicable (for exceptions see respective synthesis documentation in the Supporting Information).
General procedure E – Oxidation of thiomorpholines to sulfoxides
Sodium periodate was dissolved in water and cooled to 0 °C. 4-(4-R2-2-nitro-phenyl)thiomorpholines or their respective Boc-amino acid coupled derivatives were dissolved in methanol. This solution was then added to sodium periodate solution under stirring. If a precipitate formed acetonitrile was added until full dissolution occured. The mixture was then stirred at 0 °C for 4 h, before it was stored in the fridge for 3 days. Subsequently, the mixture was extracted with DCM three times. The organic phase was collected, and the solvents are evaporated under reduced pressure. For purification flash chromatography was performed with varying gradient eluting systems on 40-63 μm normal phase silica gel (see respective synthesis documentation).
General procedure F – Oxidation of thiomorpholines to sulfones
4-(4-R2-2-nitro-phenyl)thiomorpholines or their respective Boc-amino acid coupled derivatives were dissolved in DCM. The mixture was cooled to 0 °C before a solution of m-chloroperbenzoic acid in DCM was added over 30 min with a syringe. The reaction mixture was stirred overnight at room temperature. After completion of the reaction the mixture was washed three times with saturated NaHCO3 solution. The organic phase was collected, the solvent was evaporated under reduced pressure and a dryload was prepared for purification with flash chromatography (see respective synthesis documentation).
General procedure G - Nucleophilic substitution of thiomorpholinedioxides
1-Bromo-2-nitrobenzene and thiomorpholinedioxide were dissolved in dioxane. Then palladium(II) acetate, BINAP and caesium carbonate were added. The mixture was then ultrasonically degassed, the flask was flushed with argon and then refluxed at 100 °C overnight. After the reaction mixture had cooled down to room temperature, it was filtered through celite and the filter was washed thoroughly with EtOAc. The solvents were evaporated under reduced pressure. For purification flash chromatography utilizing an EtOAc/heptane gradient was used (see respective synthesis documentation).
General procedure H – C-C coupling of 4-oxothiane and Boc-2-aminophenylboronic acid pinacol ester
4-Oxothiane was added to a solution of p-toluenesulfonylhydrazide in methanol. The reaction mixture was stirred at room temperature for 1 h and was then monitored until full conversion using TLC. The solvent was removed under reduced pressure and the crude product was used in the next step without further purification.
The sulfonylhydrazone formed in the first step, Boc-2-aminophenylboronic acid pinacol ester and caesium carbonate were added to a 3-neck Schlenk flask attached to a condenser. The apparatus was put under vacuum and backfilled with argon three times while the mixture of solids was stirred with a magnetic stirrer. Dry and ultrasonic-degassed dioxane was added through a septum with a syringe before the reaction was heated to 110 °C for 18 h. After cooling to room temperature, the reaction was quenched with saturated NaHCO3 solution. The mixture was then extracted with DCM three times. Solvents were removed under reduced pressure and the crude product was prepared for purification with flash chromatography.
The purified product was then dissolved in DCM and an equal volume of TFA was added under stirring. The solution was stirred for 30 min at room temperature and checked for complete Boc-cleavage by TLC. The DCM/TFA mixture was removed under reduced pressure after complete conversion. The crude product was reconstituted in EtOAc and washed with saturated NaHCO3 solution three times. The organic phase was collected, and solvents were evaporated under reduced pressure. The crude product was used for the subsequent reactions without further purification.
Microbiological assays
Bacteria and culture media
M. smegmatis mc2 155 pTEC27, M. intracellulare ATCC 35761 pTEC27 and M. abscessus ATCC 19977 pTEC27 expressing tomato RFP were used for the activity assays. Stocks of the bacteria grown in Middlebrook 7H9 medium + 10 % v/v ADS + 0,05 % v/v polysorbate 80 + 400 μg/mL hygromycin were stored in approximately 15 % v/v glycerol at −80°C. Using an inoculation loop, bacteria were streaked on 7H10 plates (containing hygromycin 400 μg/mL) and grown for 5 days in an incubator at 37°C.
Bacteria were grown in complete 7H9 broth supplemented with 10 % v/v ADS and 0.05 % v/v Tween 80, respectively, in MHII broth supplemented with 0.05 % v/v Tween 80. The culture volume was 10 mL in a 50 mL Falcon tube. The tubes were covered to protect the photosensitive hygromycin and shaken in an incubator at 37°C. Solid cultures were grown on 7H10 medium supplemented with 0.5 % v/v glycerol and 10 % v/v ADS containing 400 μg/mL hygromycin.
M. abscessus Bamboo was isolated from the sputum of a patient with amyotrophic lateral sclerosis and bronchiectasis and was provided by Wei Chang Huang, Taichung Veterans General Hospital, Taichung, Taiwan. M. abscessus Bamboo whole-genome sequencing showed that the strain belongs to M. abscessus subsp. abscessus and harbours an inactive clarithromycin-sensitive erm41 C28 sequevar. M. tuberculosis H37Rv (ATCC 27294) was obtained from the American Type Culture Collection.
For general bacteria culturing and certain MIC experiments, Middlebrook 7H9 broth (BD Difco) was supplemented with 0.5 % m/v albumin, 0.2 % m/v glucose, 0.085 % m/v sodium chloride, 0.0003 % m/v catalase, 0.2 % v/v glycerol, and 0.05 % v/v Tween 80. Unless otherwise stated, solid cultures were grown on Middlebrook 7H10 agar (BD Difco) supplemented with 0.5 % m/v albumin, 0.2 % m/v glucose, 0.085 % m/v sodium chloride, 0.5 % v/v glycerol, 0.0003 % m/v catalase, and 0.006 % v/v oleic acid. All drugs were prepared as 10 mM stocks in dimethyl sulfoxide (DMSO).
THP-1 cells and culture media
THP-1 cells were put in 90 % v/v FBS + 10 % v/v DMSO and stored in liquid nitrogen. THP-1 cells were grown in complete RPMI medium. The cells were grown in a tissue culture flask with a minimum volume of 30 mL and a maximum volume of 40 mL and were incubated in an atmosphere of 95 % air and 5 % carbon dioxide (CO2) at a temperature of 37 °C. The cell density was kept between 0.25 million and 1 million cells/mL. Every two or three days the cells were counted and diluted to 0.25 million cells/mL. The cells doubled every 48 h. A culture from nitrogen stock could be subcultured for up to 3 months; after this time, a change in morphology and growth behaviour was observed. For culturing of THP-1 cells, RPMI 1640 medium supplemented with 5 % v/v fetal bovine serum (FBS), 2 % m/v glutamine, and 1 % v/v nonessential amino acids.
MIC determination against M. smegmatis mc2 155 pTEC27, M. intracellulare ATCC 35761 pTEC27 and M. abscessus ATCC 19977 pTEC27
MIC values were determined by the broth microdilution method. 96-well flat bottom tissue culture plates (Sarstedt, 83.3924.500) were used73. In the third well of each row two times the desired highest concentration of each compound was added in 7H9 medium supplemented with 10 % v/v ADS and 0.05 % v/v Tween 80. Each compound was diluted twofold in a nine-point serial dilution. The concentration of the starting inoculum was 5 x 105 cells/mL. The starting inoculum was diluted from a preculture at the mid-log phase (OD600 0.3 to 0.7) and an OD600 of 0.1 was correlated to 1 x 108 CFU/mL. The plates were sealed with parafilm, placed in a container with moist tissue and incubated for three days at 37 °C (M. smegmatis and M. abscessus) or five days (M. intracellulare). Each plate had eight negative controls (1 % v/v dimethyl sulfoxide) and eight positive controls (100 μM amikacin). After incubation the plates were monitored by OD measurement at 550 nm (BMG labtech Fluostar Optima) and by measurement of fluorescence (λex = 544 nm λem = 590 nm). The assay was performed in duplicate and the results were averaged.
Data analysis:
Every assay plate contained eight wells with dimethyl sulfoxide (1 % v/v) as negative control, which corresponds to 100 % bacterial growth and eight wells with amikacin (100 μM) as positive control in which 100 % inhibition of bacterial growth was reached. Controls were used to monitor the assay quality through determination of the Z’ score. The Z’ factor was calculated as follows:
The percentage of growth inhibition was calculated by the equation:
MIC determination against M. abscessus Bamboo
MIC value determination by optical density at 600 nm [OD600] was carried out in 96-well plate format. 96-well plates were initially set up with 100 μl of 7H9 per well. For each compound, a 10-point twofold dilution series starting at twice the desired highest concentration was dispensed onto the 96-well plates using a Tecan D300e Digital Dispenser, with the DMSO concentration normalized to 2 % v/v. A bacteria culture grown to mid-log-phase (OD600,0.4 to 0.6) was diluted to OD600 = 0.1 (1*107 CFU/mL). 100 μl of the resulting bacteria suspension was dispensed onto the 96-well plates containing the sample compounds to give a final volume of 200 μl per well with an initial OD600 = 0.05 (5*106 CFU/mL) and a final DMSO concentration of 1 % v/v. Final compound concentration ranges were typically 50 to 0.098 μM or 6.25 to 0.012 μM. Untreated control wells, which contained bacteria suspension and 1 % v/v DMSO, were included on each plate. Plates were sealed with parafilm, stored in boxes with wet paper towels, and incubated at 37°C with shaking (110 rpm) and were incubated for 3 days.
To determine growth, OD600 was measured using a Tecan Infinite M200 plate reader on day 0 and day 3. Two biological replicates were performed. Clarithromycin was included in each experiment as a positive control. For each well on the 96-well plate, bacterial growth was calculated by subtracting the day 0 OD600 value from the day 3 OD600 value. For each compound series, the bacterial growth values for the untreated control wells were averaged to give the average drug-free bacterial growth. For compound-containing wells, percentage growth was calculated by dividing their growth values by the average drug-free bacterial growth for the compound series and multiplying by 100. For each compound series, we plotted percentage growth versus compound concentration. By visual inspection of the dose-response curve, we determined the MIC of a compound as the compound concentrations that would result in 90 % growth inhibition. The MIC determination was performed two times with different starter cultures. The MIC values shown in the script are the averaged results of biological duplicates.
MIC determination against M. tuberculosis H37Rv
MICs were determined as described previously with slight modifications74. Briefly, compounds were serially diluted in flat-bottom 96-well plates, and a mid-log-phase culture was mixed with the compound-containing broth (final OD600 = 0.05). Plates were sealed with Breathe-Easy sealing membrane (Sigma), placed in humidified plastic boxes and incubated at 37oC for 7 days, shaking at 80 rpm. Growth was monitored by measuring turbidity at 600 nm using a Tecan Infinite 200 Pro microplate reader (Tecan). MIC90 values were deduced from the generated dose–response curves. The MIC values shown in the script are the averaged results of the two biological replicates.
MIC determination in the macrophage infection model
For the infection assay, an M. abscessus pTEC27 culture (OD, 0.2 to 0.8, mid-log phase) was centrifuged (4,000 rpm, RT, 10 min), washed with 7H9 medium with 0.05 % v/v Tween 80 (about 10 mL) and vortexed. After a second centrifugation (4,000 rpm, RT, 10 min), 7H9 medium was replaced by RPMI medium (same volume or a little less to concentrate the bacteria), vortexed and incubated at RT for 5 min. After incubation, the bacterial suspension was filtered through a 5 μM-pore-size filter to remove the clumps. The OD600 was determined after filtration (OD = 0.1 corresponds to 1 x 108 CFU/mL). The appropriate number of bacteria were incubated in the presence of 10 % v/v human serum at 37 °C for 30 min for opsonization. A cell suspension of THP-1 cells (1 x 106 cells/mL) in RPMI incomplete medium was incubated with the opsonized M. abscessus single-cell suspension (MOI, 5:1) and PMA (40 ng/mL) for 4 h at 37 °C under constant agitation. After infection, the THP-1 cell suspension was centrifuged (750 rpm, RT, 10 min) and washed with RPMI medium. A ten-point twofold serial dilution of each compound was then prepared in 96-well flat bottom plates that had been tissue culture treated (Sarstedt, 3924). Column one of the 96-well plate included eight negative controls (1 % v/v dimethyl sulfoxide [DMSO]) and column two eight positive controls (1.5 μM bedaquiline). The plates were then inoculated with the infected cells (1x105 THP-1 cells/well), sealed with parafilm, and incubated for 4 days (37 °C, 5 % CO2)). After incubation, the cells were fixed with para-formaldehyde (PFA; 4 % m/v in PBS) for 30 min. After removal of the PFA, the cells were stained with DAPI readymade solution (Sigma, MBD0015). The plates were washed twice with RPMI medium. Image acquisition (DAPI 386-23 nm, RFP 560-25 nm, brightfield) and analysis were done with a CellInsight™ CX5 (Thermofisher). The mean CircSpotAvgArea of the RFP channel was used for calculation of growth inhibition.
MIC values were determined against M. abscessus ATCC 19977 pTEC27 by the microdilution method in RPMI incomplete media (RPMI 1640 medium supplemented with 5 % v/v FBS, 1 % m/v glutamine, and 1 % v/v nonessential amino acids) with 70 μM amikacin in the assay medium. A ten-point twofold serial dilution of each compound was prepared in 96-well flat bottom plates that had been tissue culture treated (Sarstedt, 3924). Row one of the 96-well plate included eight negative controls (1 % v/v DMSO) and column two eight positive controls (1.5 μM bedaquiline).
Data analysis:
Every assay plate contained eight wells with DMSO (1 % v/v) as a negative control, which correspond to 100 % bacterial growth, and eight wells with bedaquiline (1.5 μM) as a positive control, in which 100 % inhibition of bacterial growth was reached. The controls were used to monitor assay quality through determination of the Z-score and for normalizing the data on a plate basis. The Z-factor was determined using the following equation:
Percent inhibition was calculated as follows:
MBC determination against M. abscessus ATCC 19977 pTEC27
For MBC determination M. abscessus ATCC19977 was incubated in a microplate macrophage infection dilution assay for 4 days as described below. Subsequently, the MBC was determined by CFU counting: For this purpose, 6-well plates were used, each filled with 4 mL 7H10 agar supplemented with 0.5 % v/v glycerol, 10 % v/v ADS and 400 μg/mL hygromycin. From the drug concentrations where growth inhibition was detected in the microplate dilution assay, 10 μl (undiluted or diluted 1:100) were plated into one well of the 6-well plates. The colonies were counted after 4 days of incubation at 37 °C and the experiment was carried out in triplicate. Based on the result, the concentration of CFUs per mL was calculated. The number of CFUs was also determined in the inoculum prior to the 4-day incubation.
Plasma stability
Plasma stability in human and murine plasma was determined at five different time points over 120 min using HPLC-MS/MS analytic. Pooled human plasma, anti-coagulated with trisodium citrate, and non-sterile murine plasma, anti-coagulated with Li-heparin, were used. The plasma stability is given as the percentage of substance remaining in plasma over time. All measurements were performed using the Shimadzu Prominence HPLC system including vacuum degasser, gradient pumps, reverse phase column (ZORBAX Extend-C18 column, 2.1x50 mm, 5 μm), column oven and autosampler. The HPLC system was coupled with tandem mass API 3000 (AB Sciex). Both positive and negative ion modes of the TurboIonSpray ion source were used. Acquisition and analysis of the data were performed using Analyst 1.6.3 software (PE Sciex).
Incubations of every compound + references (Verapamil and Propantheline) were carried out in 5 aliquots of 60 μL each (one for each time point), in duplicates. Plasma was spiked with test compounds from a 10 mM DMSO stock solution to yield a test concentration of 1 μM, final DMSO concentration 1 % v/v. The aliquots were incubated at 37 °C with shaking at 100 rpm. Five time points over 120 min were analyzed. The reactions were stopped by adding 300 μL of methanol containing internal standard with subsequent plasma proteins sedimentation by centrifuging at 6000 rpm for 4 minutes. Supernatants were analyzed by the HPLC system that was coupled with a tandem mass spectrometer. The percentage of the test compounds remaining after incubation in plasma and their half-lives (t1/2) were calculated.
Microsomal stability
Microsomal stability was determined at five different time points over 40 min using HPLC-MS/MS analytic. Pooled, mixed gender human liver microsomes (XenoTech, H0630/lot N#2010065) and murine liver microsomes (pooled, male BALB/c mice, XenoTech, M3000/lot #1810163) were used.
Microsomal incubations were carried out in 96-well plates in 5 aliquots of 30 μL each (one for each time point). Liver microsomal incubation medium comprised of phosphate buffer (100 mM, pH 7.4), MgCl2 (3.3 mM), NADPH (3 mM), glucose-6-phosphate (5.3 mM), glucose-6-phosphate dehydrogenase (0.67 units/ml) with 0.42 mg of liver microsomal protein per ml. In the control reactions, the NADPH-cofactor system was substituted with phosphate buffer. Test compounds (2 μM, final solvent concentration 1.6 % v/v) were incubated with microsomes at 37°C, shaking at 100 rpm. Five time points over 40 minutes were analyzed. The reactions were stopped by adding 5 volumes of 90 % v/v acetonitrile with internal standard to incubation aliquots, followed by protein sedimentation by centrifuging at 5500 rpm for 3 minutes. Each reaction was performed in duplicates.
Analysis of supernatants was performed using a Shimadzu HPLC system including vacuum degasser, gradient pumps, reverse phase HPLC column, column oven, and autosampler. Mass spectrometric analysis was performed using an API 3000 mass spectrometer from Applied Biosystems/ MDS Sciex (AB Sciex) with Turbo V ion source and TurboIonspray interface. The TurboIonSpray ion source was used in both positive and negative ion modes. The data acquisition and system control were performed using Analyst 1.6.3 software from AB Sciex.
The microsomal stability is presented as the substances half-life times and intrinsic clearance calculated from their respective elimination constants with the following formula:
Eliminations constants were determined in a ln(AUC) versus time plot using linear regression analysis.
Determination of cytotoxicity
Cytotoxicity was determined using a colorimetric micro-culture test with Sulforhodamine-B (Kiton-Red S, ABCR) as the staining agent. Cell lines were obtained from the institute of oncology (MLU Halle-Wittenberg). Cells were transferred to a 96-well plate in order to treat them with increasing compound concentrations (1; 3; 7; 12; 20; and 30 μM) after 24 h. DMSO/DMF concentrations never exceeded 0.5 % v/v which is non-toxic for the cells. The supernatant was discarded after 72 h, the cells were fixed with 10 % v/v TFA and left to rest for 24 h at 4 °C. After washing the cells with a strip, washed cells were stained with a 10 μM Sulforhodamine-B solution (200 μL) for 20 min before they were washed again with 1 % v/v acetic acid solution to remove excess dye. Cells were air-dried overnight. Absorption was determined using a 96-well plate reader (Tecan spectra). Three independent experiments in triplicate for each were conducted for the given compounds. The averaged data resulted in semi-logarithmic dose-effect curves which were fitted with the non-linear Hill-slope equation (GraphPad Prism 5).
Nephelometry solubility screen
The solubilities of the final compounds were determined nephelometrically using a NEPHELOstar Plus (BMG Labtech GmbH, GER) device. 25 mM stock solutions in DMSO of the compounds were prediluted in pure DMSO. Then 5 μL of the predilutions were furtherly diluted in flat-bottom 96-well plates in 245 μL of phosphate buffered saline (PBS) and mixed and measured immediately (2 % v/v DMSO in measured sample). In this way, concentrations of 0.78; 1.56; 3.13; 6.25; 12.5; 25; 50; 100; 150; 200; 250; 300; 350; 400; 450 and 500 μM were measured. The blank corrected raw data was interpreted with a segmental regression fit utilizing the Omega software (BMG Labtech GmbH, GER). Each compound was analyzed as a quadruplet prepared from the same stock solution.
Supplementary Material
Acknowledgments
We would like to thank Dr. Nadine Taudte and Dr. Jens-Ulrich Rahfeld for providing and maintaining the biosafety level 2 facility, Professor Christian W. Lehmann for providing access to the X-ray diffraction facility at the Max-Planck-Institut für Kohlenforschung (Mülheim an der Ruhr, Germany), Elke Dreher and Heike Schucht for technical assistance, Dr. Christian Ihling and Antje Herbrich-Peters for measuring the HRMS spectra and Gudrun Hahn for specific rotation measurements. We are grateful to Wei Chang Huang (Taichung Veterans General Hospital, Taichung,Taiwan) for providing M. abscessus Bamboo. Thanks are due to Dr. Dieter Ströhl for the NMR spectroscopy.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—432291016, the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI132374 and Mukoviszidose Institut gGmbH (Bonn, Germany) project number 2202, the research and development arm of the German Cystic Fibrosis Association Mukoviszidose e. V.
Abbreviations
- AAP
Nα-aroyl-N-aryl-phenylalanine amide
- ADP
adenosine diphosphate
- APT
attached proton test
- ATCC
American type culture collection
- BINAP
(2,2′-bis(diphenylphosphino)-1,1′-binaphthyl)
- Boc
tert-Butoxycarbonyl
- CES
carboxylesterase
- CFU
Colony forming units
- CLR
clarithromycin
- cLogP
calculated logarithmic octanol-water partition coefficient
- DCM
dichloromethane
- DIPEA
N,N-Diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- ee values
enantiomeric excess values
- EtOAc
ethyl acetate
- EtOH
ethanol
- FBS
fetal bovine serum
- LogP
logarithmic octanol-water partition coefficient
- Mabs
Mycobacterium abscessus
- MAC
Mycobacterium avium complex
- MBC
minimum bactericidal concentration
- m-CPBA
m-chloroperbenzoic acid
- MDR
multi drug resistent
- MHII
Miller-Hinton broth II
- MIC
minimum inhibitory concentration
- Mintra
Mycobacterium intracellulare
- MMV
medicines for malaria ventures
- MMV845
MMV688845
- MOI
multiplicity of infection
- Msmeg
Mycobacterium smegmatis
- Mtb
Mycobacterium tuberculosis
- NTM
non-tuberculous mycobacteria
- OD
optical density
- PBS
phosphate buffered saline
- PFA
para-formaldehyde
- PMA
phorbol-12-myristate-13-acetate
- RFB
rifabutin
- RFP
red fluorescent protein
- RIF
rifampicin
- RNAP
RNA polymerase
- RT
room temperature
- SAR
structure activity relationship
- T3P
n-propanephosphonic acid anhydride
- TFA
trifluoro acetic acid
- THF
tetrahydrofurane
- TLC
thin layer chromatography
Footnotes
Supporting Information
Additional information on the molecular docking studies; 2D visualizations of 3D structures; experimental details of X-ray crystallography; antimycobacterial activity of all MMV688845 derivatives derived from OD measurements (not RFP measurements); chemical structures; synthetic protocols, molar equivalents of all reactants, purification procedures and percentage yields for each synthetic experiment; TLC Rf values for all synthesized compounds, 1H-NMR spectra for all synthesized compounds and respective written documentation; APT-NMR spectra for all in vitro tested compounds and respective written documentation; HPLC-purity and respective HPLC traces for all in-vitro tested; atmospheric pressure chemical ionization mass spectrometry data for all synthesized compounds; high resolution mass spectrometry data for all in-vitro tested compounds; Molecular formula strings for all described substances; PDB files for the presented crystal structures
References
- (1).World Health Organization. Module 4: Treatment Drug-Susceptible Tuberculosis Treatment; World Health Organization, 2022. https://www.who.int/publications/i/item/9789240048126 (accessed 2023-02-21). [Google Scholar]
- (2).World Health Organization. Global Tuberculosis Report 2022; 2022. https://www.who.int/publications/i/item/9789240061729 (accessed 2023-01-02). [Google Scholar]
- (3).Dartois VA; Rubin EJ Anti-Tuberculosis Treatment Strategies and Drug Development: Challenges and Priorities. Nature Reviews Microbiology 2022 20:11 2022, 20 (11), 685–701. 10.1038/s41579-022-00731-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sharma A; de Rosa M; Singla N; Singh G; Barnwal RP; Pandey A Tuberculosis: An Overview of the Immunogenic Response, Disease Progression, and Medicinal Chemistry Efforts in the Last Decade toward the Development of Potential Drugs for Extensively Drug-Resistant Tuberculosis Strains. J Med Chem 2021, 64 (8), 4359–4395. 10.1021/ACS.JMEDCHEM.0C01833. [DOI] [PubMed] [Google Scholar]
- (5).Ahmed I; Tiberi S; Farooqi J; Jabeen K; Yeboah-Manu D; Migliori GB; Hasan R Non-Tuberculous Mycobacterial Infections—A Neglected and Emerging Problem. International Journal of Infectious Diseases 2020, 92, S46–S50. 10.1016/J.IJID.2020.02.022. [DOI] [PubMed] [Google Scholar]
- (6).Dartois V; Dick T Drug Development Challenges in Nontuberculous Mycobacterial Lung Disease: TB to the Rescue. Journal of Experimental Medicine 2022, 219 (6). 10.1084/JEM.20220445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gupta RS; Lo B; Son J Phylogenomics and Comparative Genomic Studies Robustly Support Division of the Genus Mycobacterium into an Emended Genus Mycobacterium and Four Novel Genera. Front Microbiol 2018, 9 (FEB), 67. 10.3389/FMICB.2018.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tortoli E; Brown-Elliott BA; Chalmers JD; Cirillo DM; Daley CL; Emler S; Andres Floto R; Garcia MJ; Hoefsloot W; Koh WJ; Lange C; Loebinger M; Maurer FP; Morimoto K; Niemann S; Richter E; Turenne CY; Vasireddy R; Vasireddy S; Wagner D; Wallace RJ; Wengenack N; van Ingen J Same Meat, Different Gravy: Ignore the New Names of Mycobacteria. European Respiratory Journal 2019, 54 (1). 10.1183/13993003.00795-2019. [DOI] [PubMed] [Google Scholar]
- (9).Koh WJ; Jeong BH; Kim SY; Jeon K; Park KU; Jhun BW; Lee H; Park HY; Kim DH; Huh HJ; Ki CS; Lee NY; Kim HK; Choi YS; Kim J; Lee SH; Kim CK; Shin SJ; Daley CL; Kim H; Kwon OJ Mycobacterial Characteristics and Treatment Outcomes in Mycobacterium Abscessus Lung Disease. Clinical Infectious Diseases 2017, 64 (3), 309–316. 10.1093/CID/CIW724. [DOI] [PubMed] [Google Scholar]
- (10).Jarand J; Levin A; Zhang L; Huitt G; Mitchell JD; Daley CL Clinical and Microbiologic Outcomes in Patients Receiving Treatment for Mycobacterium Abscessus Pulmonary Disease. Clinical Infectious Diseases 2011, 52 (5), 565–571. 10.1093/CID/CIQ237. [DOI] [PubMed] [Google Scholar]
- (11).Jeon K; Kwon OJ; Nam YL; Kim BJ; Kook YH; Lee SH; Young KP; Chang KK; Koh WJ Antibiotic Treatment of Mycobacterium Abscessus Lung Disease. Am J Respir Crit Care Med 2009, 180 (9), 896–902. https://doi.org/ 10.1164/rccm.200905-0704OC. [DOI] [PubMed] [Google Scholar]
- (12).Stover CK; Warrener P; VanDevanter DR; Sherman DR; Arain TM; Langhorne MH; Anderson SW; Towell JA; Yuan Y; McMurray DN; Kreiswirth BN; Barry CE; Baker WR A Small-Molecule Nitroimidazopyran Drug Candidate for the Treatment of Tuberculosis. Nature 2000 405:6789 2000, 405 (6789), 962–966. 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- (13).Diacon AH; Pym A; Grobusch MP; de los Rios JM; Gotuzzo E; Vasilyeva I; Leimane V; Andries K; Bakare N; de Marez T; Haxaire-Theeuwes M; Lounis N; Meyvisch P; de Paepe E; van Heeswijk RPG; Dannemann B Multidrug-Resistant Tuberculosis and Culture Conversion with Bedaquiline. New England Journal of Medicine 2014, 371 (8), 723–732. 10.1056/NEJMOA1313865. [DOI] [PubMed] [Google Scholar]
- (14).Diacon AH; Pym A; Grobusch M; Patientia R; Rustomjee R; Page-Shipp L; Pistorius C; Krause R; Bogoshi M; Churchyard G; Venter A; Allen J; Palomino JC; de Marez T; van Heeswijk RPG; Lounis N; Meyvisch P; Verbeeck J; Parys W; de Beule K; Andries K; Neeley DFM The Diarylquinoline TMC207 for Multidrug-Resistant Tuberculosis. New England Journal of Medicine 2009, 360 (23), 2397–2405. 10.1056/NEJMOA0808427. [DOI] [PubMed] [Google Scholar]
- (15).Mudde SE; Upton AM; Lenaerts A; Bax HI; de Steenwinkel JEM Delamanid or Pretomanid? A Solomonic Judgement! Journal of Antimicrobial Chemotherapy 2022, 77 (4), 880–902. 10.1093/JAC/DKAB505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Manjunatha U; Boshoff HIM; Barry CE The Mechanism of Action of PA-824. Commun Integr Biol 2009, 2 (3), 215–218. 10.4161/CIB.2.3.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Conradie F; Diacon AH; Ngubane N; Howell P; Everitt D; Crook AM; Mendel CM; Egizi E; Moreira J; Timm J; McHugh TD; Wills GH; Bateson A; Hunt R; van Niekerk C; Li M; Olugbosi M; Spigelman M Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. New England Journal of Medicine 2020, 382 (10), 893–902. 10.1056/NEJMOA1901814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Brode SK; Daley CL; Marras TK The Epidemiologic Relationship between Tuberculosis and Non-Tuberculous Mycobacterial Disease: A Systematic Review. International Journal of Tuberculosis and Lung Disease 2014, 18 (11), 1370–1377. 10.5588/IJTLD.14.0120. [DOI] [PubMed] [Google Scholar]
- (19).Lopeman RC; Harrison J; Desai M; Cox JAG Mycobacterium Abscessus: Environmental Bacterium Turned Clinical Nightmare. Microorganisms 2019, Vol. 7, Page 90 2019, 7 (3), 90. 10.3390/MICROORGANISMS7030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Boudehen YM; Kremer L Mycobacterium Abscessus. Trends Microbiol 2021, 29 (10), 951–952. 10.1016/J.TIM.2021.06.006. [DOI] [PubMed] [Google Scholar]
- (21).Victoria L; Gupta A; Gómez JL; Robledo J Mycobacterium Abscessus Complex: A Review of Recent Developments in an Emerging Pathogen. Front Cell Infect Microbiol 2021, 11, 338. 10.3389/FCIMB.2021.659997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Malenfant JH; Brewer TF Rifampicin Mono-Resistant Tuberculosis—A Review of an Uncommon But Growing Challenge for Global Tuberculosis Control. Open Forum Infect Dis 2021, 8 (2). 10.1093/OFID/OFAB018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rominski A; Roditscheff A; Selchow P; Böttger EC; Sander P Intrinsic Rifamycin Resistance of Mycobacterium Abscessus Is Mediated by ADP-Ribosyltransferase MAB_0591. Journal of Antimicrobial Chemotherapy 2017, 72 (2), 376–384. 10.1093/JAC/DKW466. [DOI] [PubMed] [Google Scholar]
- (24).Ganapathy US; Lan T; Krastel P; Lindman M; Zimmerman MD; Ho HP; Sarathy JP; Evans JC; Dartois V; Aldrich CC; Dick T Blocking Bacterial Naphthohydroquinone Oxidation and Adpribosylation Improves Activity of Rifamycins against Mycobacterium Abscessus. Antimicrob Agents Chemother 2021, 65 (9). https://doi.org/ 10.1128/AAC.00978-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Aziz DB; Low JL; Wu ML; Gengenbacher M; Teo JWP; Dartois V; Dick T Rifabutin Is Active against Mycobacterium Abscessus Complex. Antimicrob Agents Chemother 2017, 61 (6). 10.1128/AAC.00155-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dick T; Shin SJ; Koh WJ; Dartois V; Gengenbacher M Rifabutin Is Active against Mycobacterium Abscessus in Mice. Antimicrob Agents Chemother 2020, 64 (2). 10.1128/AAC.01943-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lan T; Ganapathy US; Sharma S; Ahn Y-M; Zimmerman M; Molodtsov V; Hegde P; Gengenbacher M; Ebright RH; Dartois V; Freundlich JS; Dick T; Aldrich CC Redesign of Rifamycin Antibiotics to Overcome ADP-Ribosylation-Mediated Resistance. Angewandte Chemie 2022, 134 (45), e202211498. 10.1002/ANGE.202211498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Paulowski L; Beckham KSH; Johansen MD; Berneking L; Van N; Degefu Y; Staack S; Sotomayor FV; Asar L; Rohde H; Aldridge BB; Aepfelbacher M; Parret A; Wilmanns M; Kremer L; Combrink K; Maurer FP C25-Modified Rifamycin Derivatives with Improved Activity against Mycobacterium Abscessus. PNAS Nexus 2022, 1 (4), 1–13. 10.1093/PNASNEXUS/PGAC130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ballell L; Bates RH; Young RJ; Alvarez-Gomez D; Alvarez-Ruiz E; Barroso V; Blanco D; Crespo B; Escribano J; González R; Lozano S; Huss S; Santos-Villarejo A; Martín-Plaza JJ; Mendoza A; Rebollo-Lopez MJ; Remuiñan-Blanco M; Lavandera JL; Pérez-Herran E; Gamo-Benito FJ; García-Bustos JF; Barros D; Castro JP; Cammack N Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem 2013, 8 (2), 313–321. 10.1002/CMDC.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Low JL; Wu ML; Aziz DB; Laleu B; Dick T Screening of TB Actives for Activity against Nontuberculous Mycobacteria Delivers High Hit Rates. Front Microbiol 2017, 8 (AUG), 1539. 10.3389/FMICB.2017.01539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lin W; Mandal S; Degen D; Liu Y; Ebright YW; Li S; Feng Y; Zhang Y; Mandal S; Jiang Y; Liu S; Gigliotti M; Talaue M; Connell N; Das K; Arnold E; Ebright RH Structural Basis of Mycobacterium Tuberculosis Transcription and Transcription Inhibition. Mol Cell 2017, 66 (2), 169–179.e8. 10.1016/J.MOLCEL.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ebright Richard H.; Ebright Yon W.; Mandal Soma; Wilde Richard; Li S Preparation of N-Alpha-Aroyl-N-Aryl-Phenylalaninamides as Inhibitors of Bacterial RNA Polymerase and as Antibacterials. WO2015120320 A1 2015-August-13, 2015. [Google Scholar]
- (33).Mann L; Lang M; Schulze P; Halz JH; Csuk R; Hoenke S; Seidel RW; Richter A Racemization-Free Synthesis of Nα-2-Thiophenoyl-Phenylalanine-2-Morpholinoanilide Enantiomers and Their Antimycobacterial Activity. Amino Acids 2021, 53 (8), 1187–1196. 10.1007/S00726-021-03044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mann L; Ganapathy US; Abdelaziz R; Lang M; Zimmerman MD; Dartois V; Dick T; Richter A In Vitro Profiling of the Synthetic RNA Polymerase Inhibitor MMV688845 against Mycobacterium Abscessus. Microbiol Spectr 2022, 10 (6). 10.1128/SPECTRUM.02760-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Dunetz JR; Xiang Y; Baldwin A; Ringling J General and Scalable Amide Bond Formation with Epimerization-Prone Substrates Using T3P and Pyridine. Org Lett 2011, 13 (19), 5048–5051. 10.1021/OL201875Q/SUPPL_FILE/OL201875Q_SI_001.PDF. [DOI] [PubMed] [Google Scholar]
- (36).Li H; Jiang X; Ye Y-H; Fan C; Romoff T; Goodman M 3-(Diethoxyphosphoryloxy)-1,2,3-Benzotriazin-4(3H)-One (DEPBT): A New Coupling Reagent with Remarkable Resistance to Racemization. 1999. 10.1021/ol990573k. [DOI] [PubMed] [Google Scholar]
- (37).Brewster WK THIENO-PYRIMIDINE COMPOUNDS HAVING FUNGICIDAL ACTIVITY. US 2006/0089370 A1, 2006.
- (38).Allwood DM; Blakemore DC; Brown AD; Ley S v. Metal-Free Coupling of Saturated Heterocyclic Sulfonylhydrazones with Boronic Acids. Journal of Organic Chemistry 2014, 79 (1), 328–338. 10.1021/JO402526Z. [DOI] [PubMed] [Google Scholar]
- (39).Barluenga J; Tomás-Gamasa M; Aznar F; Valdés C Metal-Free Carbon–Carbon Bond-Forming Reductive Coupling between Boronic Acids and Tosylhydrazones. Nature Chemistry 2009 1:6 2009, 1 (6), 494–499. 10.1038/nchem.328. [DOI] [PubMed] [Google Scholar]
- (40).Thiemann T. Thiophene S-Oxides. Chalcogen Chemistry 2018. 10.5772/INTECHOPEN.79080. [DOI] [Google Scholar]
- (41).Elsawy MA; Hewage C; Walker B Racemisation of N-Fmoc Phenylglycine under Mild Microwave-SPPS and Conventional Stepwise SPPS Conditions: Attempts to Develop Strategies for Overcoming This. Journal of Peptide Science 2012, 18 (5), 302–311. 10.1002/PSC.2398. [DOI] [PubMed] [Google Scholar]
- (42).Motta L; Sturabotti E; Vetica F; Toscano G; Calcaterra A; Martinelli A; Migneco LM; Leonelli F N-Acetyl-l-Phenylalanine Racemization during TBTU Amidation: An In-Depth Study for the Synthesis of Anti-Inflammatory 2-(N-Acetyl)-l-Phenylalanylamido-2-Deoxy-d-Glucose (NAPA). Molecules 2023, Vol. 28, Page 581 2023, 28 (2), 581. 10.3390/MOLECULES28020581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Al-Warhi TI; Al-Hazimi HMA; El-Faham A Recent Development in Peptide Coupling Reagents. Journal of Saudi Chemical Society 2012, 16 (2), 97–116. 10.1016/J.JSCS.2010.12.006. [DOI] [Google Scholar]
- (44).Bodanszky M. Principles of Peptide Synthesis. [Google Scholar]
- (45).Antonovics I; Young GT Amino-Acids and Peptides. Part XXV. The Mechanism of the Base-Catalysed Racemisation of the p-Nitrophenyl Esters of Acylpeptides. Journal of the Chemical Society C: Organic 1967, 99 (0), 595–601. 10.1039/J39670000595. [DOI] [PubMed] [Google Scholar]
- (46).García ALL T3P: A Convenient and Useful Reagent in Organic Synthesis. Synlett 2007, 2007 (08), 1328–1329. 10.1055/S-2007-980339. [DOI] [Google Scholar]
- (47).Harada H. AZOLECARBOXAMIDE DERIVATIVE. WO 2007/123269 A1, 2017. [Google Scholar]
- (48).Midgley L; Bourhis LJ; Dolomanov O. v.; Grabowsky S; Kleemiss F; Puschmann H; Peyerimhoff N Vanishing of the Atomic Form Factor Derivatives in Non-Spherical Structural Refinement - a Key Approximation Scrutinized in the Case of Hirshfeld Atom Refinement. Acta Crystallogr A Found Adv 2021, 77 (6), 519–533. 10.1107/S2053273321009086. [DOI] [PubMed] [Google Scholar]
- (49).Kleemiss F; Dolomanov O. v.; Bodensteiner M; Peyerimhoff N; Midgley L; Bourhis LJ; Genoni A; Malaspina LA; Jayatilaka D; Spencer JL; White F; Grundkötter-Stock B; Steinhauer S; Lentz D; Puschmann H; Grabowsky S Accurate Crystal Structures and Chemical Properties from NoSpherA2. Chem Sci 2021, 12 (5), 1675–1692. 10.1039/D0SC05526C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Chaturvedi V; Dwivedi N; Tripathi RP; Sinha S Evaluation of Mycobacterium Smegmatis as a Possible Surrogate Screen for Selecting Molecules Active against Multi-Drug Resistant Mycobacterium Tuberculosis. J Gen Appl Microbiol 2007, 53 (6), 333–337. 10.2323/JGAM.53.333. [DOI] [PubMed] [Google Scholar]
- (51).T JAS; J R; Rajan A; Shankar V Features of the Biochemistry of Mycobacterium Smegmatis, as a Possible Model for Mycobacterium Tuberculosis. J Infect Public Health 2020, 13 (9), 1255–1264. 10.1016/J.JIPH.2020.06.023. [DOI] [PubMed] [Google Scholar]
- (52).Tortoli E. Microbiological Features and Clinical Relevance of New Species of the Genus Mycobacterium. Clin Microbiol Rev 2014, 27 (4), 727–752. 10.1128/CMR.00035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Weiss CH; Glassroth J Pulmonary Disease Caused by Nontuberculous Mycobacteria. Expert Rev Respir Med 2014, 6 (6), 597–613. 10.1586/ERS.12.58. [DOI] [PubMed] [Google Scholar]
- (54).Henkle E; Hedberg K; Schafer SD; Winthrop KL Surveillance of Extrapulmonary Nontuberculous Mycobacteria Infections, Oregon, USA, 2007–2012. Emerg Infect Dis 2017, 23 (10), 1627–1630. 10.3201/EID2310.170845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).To K; Cao R; Yegiazaryan A; Owens J; Venketaraman V General Overview of Nontuberculous Mycobacteria Opportunistic Pathogens: Mycobacterium Avium and Mycobacterium Abscessus. Journal of Clinical Medicine 2020, Vol. 9, Page 2541 2020, 9 (8), 2541. 10.3390/JCM9082541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Koh WJ; Jeong BH; Jeon K; Lee NY; Lee KS; Woo SY; Shin SJ; Kwon OJ Clinical Significance of the Differentiation Between Mycobacterium Avium and Mycobacterium Intracellulare in M Avium Complex Lung Disease. Chest 2012, 142 (6), 1482–1488. 10.1378/CHEST.12-0494. [DOI] [PubMed] [Google Scholar]
- (57).Ricotta EE; Adjemian J; Blakney RA; Lai YL; Kadri SS; Prevots DR Extrapulmonary Nontuberculous Mycobacteria Infections in Hospitalized Patients, United States, 2009–2014 - Volume 27, Number 3—March 2021 - Emerging Infectious Diseases Journal - CDC. Emerg Infect Dis 2021, 27 (3), 845–852. 10.3201/EID2703.201087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ollinger J; Kumar A; Roberts DM; Bailey MA; Casey A; Parish T A High-Throughput Whole Cell Screen to Identify Inhibitors of Mycobacterium Tuberculosis. PLoS One 2019, 14 (1), e0205479. 10.1371/JOURNAL.PONE.0205479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Richter A; Shapira T; Av-Gay Y THP-1 and Dictyostelium Infection Models for Screening and Characterization of Anti-Mycobacterium Abscessus Hit Compounds. Antimicrob Agents Chemother 2020, 64 (1). 10.1128/AAC.01601-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Martin-Smith M; Reid ST Biological Activity in Compounds Possessing Thiophen Rings. J Med Chem 1959, 1 (6), 507–564. 10.1021/JM50007A001. [DOI] [PubMed] [Google Scholar]
- (61).Maurer FP; Bruderer VL; Ritter C; Castelberg C; Bloemberg G. v.; Böttger EC Lack of Antimicrobial Bactericidal Activity in Mycobacterium Abscessus. Antimicrob Agents Chemother 2014, 58 (7), 3828–3836. 10.1128/AAC.02448-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Heifets L Qualitative and Quantitative Drug-Susceptibility Tests in Mycobacteriology. American Review of Respiratory Disease 1987, 137 (5). 10.1164/AJRCCM/137.5.1217. [DOI] [PubMed] [Google Scholar]
- (63).Bahar FG; Ohura K; Ogihara T; Imai T Species Difference of Esterase Expression and Hydrolase Activity in Plasma. J Pharm Sci 2012, 101 (10), 3979–3988. 10.1002/JPS.23258. [DOI] [PubMed] [Google Scholar]
- (64).Bathelt CM; Ridder L; Mulholland AJ; Harvey JN Aromatic Hydroxylation by Cytochrome P450: Model Calculations of Mechanism and Substituent Effects. J Am Chem Soc 2003, 125 (49), 15004–15005. 10.1021/JA035590Q. [DOI] [PubMed] [Google Scholar]
- (65).Cerny MA Prevalence of Non-Cytochrome P450-Mediated Metabolism in Food and Drug Administration-Approved Oral and Intravenous Drugs: 2006-2015. Drug Metabolism and Disposition 2016, 44 (8), 1246–1252. 10.1124/DMD.116.070763/-/DC1. [DOI] [PubMed] [Google Scholar]
- (66).Bradshaw PR; Wilson ID; Gill RU; Butler PJ; Dilworth C; Athersuch TJ Metabolic Hydrolysis of Aromatic Amides in Selected Rat, Minipig, and Human In Vitro Systems. Scientific Reports 2018 8:1 2018, 8 (1), 1–8. 10.1038/s41598-018-20464-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Sommerwerk S; Heller L; Kuhfs J; Csuk R Selective Killing of Cancer Cells with Triterpenoic Acid Amides - The Substantial Role of an Aromatic Moiety Alignment. Eur J Med Chem 2016, 122, 452–464. 10.1016/J.EJMECH.2016.06.053. [DOI] [PubMed] [Google Scholar]
- (68).Heller L; Knorrscheidt A; Flemming F; Wiemann J; Sommerwerk S; Pavel IZ; Al-Harrasi A; Csuk R Synthesis and Proapoptotic Activity of Oleanolic Acid Derived Amides. Bioorg Chem 2016, 68, 137–151. 10.1016/J.BIOORG.2016.08.004. [DOI] [PubMed] [Google Scholar]
- (69).Csuk R; Barthel A; Sczepek R; Siewert B; Schwarz S Synthesis, Encapsulation and Antitumor Activity of New Betulin Derivatives. Arch Pharm (Weinheim) 2011, 344 (1), 37–49. 10.1002/ARDP.201000232. [DOI] [PubMed] [Google Scholar]
- (70).Savjani KT; Gajjar AK; Savjani JK Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm 2012, 2012, 1–10. 10.5402/2012/195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Congreve M; Carr R; Murray C; Jhoti HA “Rule of Three” for Fragment-Based Lead Discovery? Drug Discov Today 2003, 8 (19), 876–877. 10.1016/S1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- (72).Bevan CD; Lloyd RS A High-Throughput Screening Method for the Determination of Aqueous Drug Solubility Using Laser Nephelometry in Microtiter Plates. 2000. 10.1021/ac9912247. [DOI] [PubMed] [Google Scholar]
- (73).Richter A; Strauch A; Chao J; Ko M; Av-Gay Y Screening of Preselected Libraries Targeting Mycobacterium Abscessus for Drug Discovery. Antimicrob Agents Chemother 2018, 62 (9), 1–11. 10.1128/AAC.00828-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Yang X; Wedajo W; Yamada Y; Dahlroth SL; Neo JJL; Dick T; Chui WK 1,3,5-Triazaspiro[5.5]Undeca-2,4-Dienes as Selective Mycobacterium Tuberculosis Dihydrofolate Reductase Inhibitors with Potent Whole Cell Activity. Eur J Med Chem 2018, 144, 262–276. 10.1016/J.EJMECH.2017.12.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.