

Abstract
Protein disulfide isomerase (PDI) and its superfamilies are mainly endoplasmic reticulum (ER) resident proteins with essential roles in maintaining cellular homeostasis, via thiol oxidation/reduction cycles, chaperoning, and isomerization of client proteins. Since PDIs play an important role in ER homeostasis, their upregulation supports cell survival and they are found in a variety of cancer types. Despite the fact that the importance of PDI to tumorigenesis remains to be understood, it is emerging as a new therapeutic target in cancer. During the past decade, several PDI inhibitors has been developed and commercialized, but none has been approved for clinical use. In this review, we discuss the properties and redox regulation of PDIs within the ER and provide an overview of the last 5 years of advances regarding PDI inhibitors.
1. Introduction
Despite the 27% drop the cancer death rate in the last two decades, it remains the second leading cause of death in the United States with toll of 602,350 lives in 2020 (Richardson, 2022). Initially, cancer cells are more vulnerable to chemo- or radiotherapy than normal cells, but they often develop resistance to stresses including treatments as disease progresses. While the exact causes of cancer are not fully understood, oxidative stress is thought to play a role in the development and progression of cancer (Hayes, Dinkova-Kostova, & Tew, 2020). Oxidative stress, resulted from imbalance of the reactive oxygen species (ROS) production and cell’s own antioxidant defenses, promotes tumor formation by initiating an aberrant induction of signaling networks that cause tumorigenesis. Approximately one-third of the cellular proteome passes through the secretory pathway before they arrive at their destined subcellular localizations. The endoplasmic reticulum (ER) constitutes the first compartment of the secretory pathway (Rahman et al., 2022), and specializes in folding and maturation of co-translationally inserted into it client proteins of the secretory pathway. This is achieved by coordinated function of ER-resident chaperones, foldases and isomerases. Correctly folded proteins are exported to the Golgi complex for extensive glycosylation and lipidation, and further intracellular sorting (Barlowe & Miller, 2013). Misfolded proteins are recognized by ER quality control machinery translocated to the cytoplasm and targeted for degradation by ER-associated degradation ubiquitin–proteasome system (Ellgaard & Helenius, 2003). A major component of ER-resident proteins and chaperones includes protein disulfide isomerases (PDI). PDI and its superfamilies responsible for transfer of oxidative disulfide bond equivalents to their respective client protein, as well as protein isomerisation. PDI’s themselves are redox sensitive proteins that have essential roles in maintaining cellular homeostasis (Grek & Townsend, 2014). Here we provide overview of selected examples that illustrate how redox state and modifications of PDI family members are linked to cancer progression.
2. Classification and Structure of PDI
21 members of PDI family have been identified since the first protein folding catalyst was discovered in 1960s (Venetianer & Straub, 1963) (Goldberger, Epstein, & Anfinsen, 1963). Several protein names were given, including PDI, endoplasmic reticulum protein (ERP), thioredoxin related transmembrane protein (TMC), thioredoxin domain containing (TXNDC), anterior gradient (AGR), DnaJ heat shock protein family member C (DNAJC) and calsequestrin (CASQ). According to the HUGO gene nomenclature committee (HGNG) database, the human PDI gene family is classified into PDIA1 to PDIA19 and PDIB1 to PDIB2 (https://www.genenames.org/data/genegroup/#!/group/692), each of which share a common structure feature, the thioredoxin (TRX)-like domain, but vary in length, substrate specificity and domain arrangement (Table 1). The discovery in 2006 of full-length crystal structure of PDI revealed detailed architecture and allowed mechanistic insights into their function. The yeast PDI is folded into four domains that form an overall flexible ‘U’ shape, with two movable redox active thiols in the each of distal catalytic domains. This flexibility suggested the mechanism for positioning of its substrate proteins relative to the two catalytic domains (Tian, Xiang, Noiva, Lennarz, & Schindelin, 2006). Most PDI family members contain both catalytic (with redox active thiols) and non-catalytic TRX-like domains that are identified as either a or b based on the presence or absence of a catalytic motif, with the use of the prime symbol to indicate their position in the protein. PDI has four such domains, a, b, b′ and a′ (Alanen et al., 2003) (Fig. 1). The a and a′ domains share 33.6% sequence identity functionally resemble thioredoxin, and each contains catalytic Cys-x-x-Cys motifs that react with thiols of client proteins to transfer redox equivalents to them. The b and b′ domains, although structurally like TRX, do not contain catalytically active cysteines and only share 16.5% sequence identity. The b and b′ domains act as spacers and are often responsible for substrate recruitment (Kozlov, Maattanen, Thomas, & Gehring, 2010; Xu, Sankar, & Neamati, 2014). The most conserved active site motif is CGHC (Cys-Gly-His-Cys), can be found in PDIA1-PDIA6, PDIA13/TMX3, PDIA15/TXNDC5 and PDIA16/TXNDC12. In several PDI proteins, such as PDIA10/ERP44, PDIA17/AGR2 and PDIA18/AGR2, one cysteine of “a” domains is replaced with serine. In PDIA7/PDILT, both cysteines of the N-terminal “a” domain are replaced by serine. PDIA11/TMX1, PDIA12/TMX2, PDIA14/TMX4, PDIA15/TXNDC5, PDIA16/TXNDC12, PDIA17/AGR2, and PDIA18/AGR3 have only “a” domains, while PDIA8/ERP27, PDIA9/ERP29, PDIB1/CASQ1 and PDIB2/CASQ2 have only “b” domains. It was postulated that those PDI members function as molecular chaperones rather than as oxidoreductases (Lee & Lee, 2017). Other notable structural motifs found in PDIs include x-linker (connecting a′ and b′-domains) and ER-retention motifs. The most common being highly acidic tetrapeptide KDEL (Lys-Asp-Glu-Leu). The x-linker is a substrate binding 19-amino acid residue located between a′ and b′ domains (Wang et al., 2010). KDEL is a recognition sequence for KDEL receptors which mediate retrieval of ER-resident proteins that escaped into a post-ER compartment. Additional ER-retention motifs (e.g., KTEL, EDEL, QSEL, KEEL, KTEL, KVEL and QEDL) were found within the PDI family members. Those have different affinities to KDEL receptors. This is a suggestive of the mechanism to regulate escape of different PDI from ER (Jordan & Gibbins, 2006). PDIA11/TMX1, PDIB1/CASQ1 and PDIB2/CASQ2 are lacking C-terminal ER retention sequences, the mechanism of ER retention for those is not clear, however, the association with ER resident protein calnexin has been suggested for retaining TMX on the ER membrane (Matsuo, Masutani, Son, Kizaka-Kondoh, & Yodoi, 2009) (Table 1).
Table 1.
The human PDI family members and their domain representation, including a and b type domain arrangements and ER retention motifs of all 21 PDI family members.

|
Fig. 1.

Domain organization of PDI and Ribbon diagram of PDI [2B5E].
3. Redox regulation: Thiol–disulfide exchange and PDI
The ER hosts a network of highly regulated mechanisms governed by folding enzymes and chaperones that facilitate the translation and folding of proteins. Many cancer-related pathologies are associated with imbalance of ER homeostasis and unfolded protein response under ER stress. The increased ER stress in cancer may result from high protein folding demands with increased rates of cancer cell proliferation, or the tumor micro-environment (Grek & Townsend, 2014). The main function of PDI members is to facilitate transfer of disulfide bonds to client proteins. Formation of disulfide bonds in client proteins ensures that they are folded properly and become functional before they allowed to exit ER. As an oxidoreductase, PDI transfers existing but does not create de novo disulfide bonds. Transfer is achieved by repeated cycles of thiol oxidation and reduction until the mature configuration of client proteins is acquired (Schwaller, Wilkinson, & Gilbert, 2003). Mature configuration is sensed by chaperone machinery as an absence of exposed hydrophobic regions. Flavoprotein oxidase ERO1, forms disulfide bonds de novo. It uses a FAD-dependent reaction to transfer electrons from reduced PDI to molecular oxygen (O2), which results in the production of reactive oxygen species in the form of H2O2 and oxidized PDI (Appenzeller-Herzog et al., 2010). The oxidative environment of the ER is optimized for the formation and transfer of disulfide bonds.
PDI is a redox-regulated chaperone, that acts as oxidoreductase and isomerase. The 3D conformation changes depending on its oxidative state (Wang et al., 2013). The 3D structure of reduced PDI shows extensive interactions between domain b′ and the regions around the active site in domain a′ with a conformational plasticity of the b′-x-a′ region mediated by the x linker. It has been shown that the linker can adopt alternative conformations (Wang, et al., 2010). In a reduced form, a linker allows narrow open angle of the PDI cleft (~15), and the inner volume of the cleft is ~6816 Å. This form will preferentially interact with Ero1 to accept newly formed disulfide bonds or act as an isomerase for mis-oxidized folding intermediates, and acceptance of disulfide bonds occurs via direct hydrophobic interactions with its b′ domain and electron transfer from the a′ domain (Wang et al., 2009). Isomerase function involves a reduced PDI active site attacking a substrate disulfide. This takes place with cycles of reduction (breaking of mis-oxidized folding intermediate disulfide bonds) and oxidation (to introduce correct pairing of cysteines) to eventually form the native proteins (Schwaller, et al., 2003). In its oxidized form, the cleft opens wider (~30 Å), and the inner volume becomes larger (~14,453 Å). The hydrophobic inner surface of the U-shaped oxidized PDI is extensively exposed, allowing large substrates to enter and accept oxidized disulfide bonds from PDI. During this process, PDI 3D conformation changes to narrow opening, and client protein is expelled. PDI now is again in a favorable conformation to capture ERO1 and accept newly formed disulfide bonds from it. Therefore, ERO1 regulates the balance of oxidized and reduced PDI. The general mechanistic model for PDI catalyzed reactions is schematically presented in (Fig. 2).
Fig. 2.
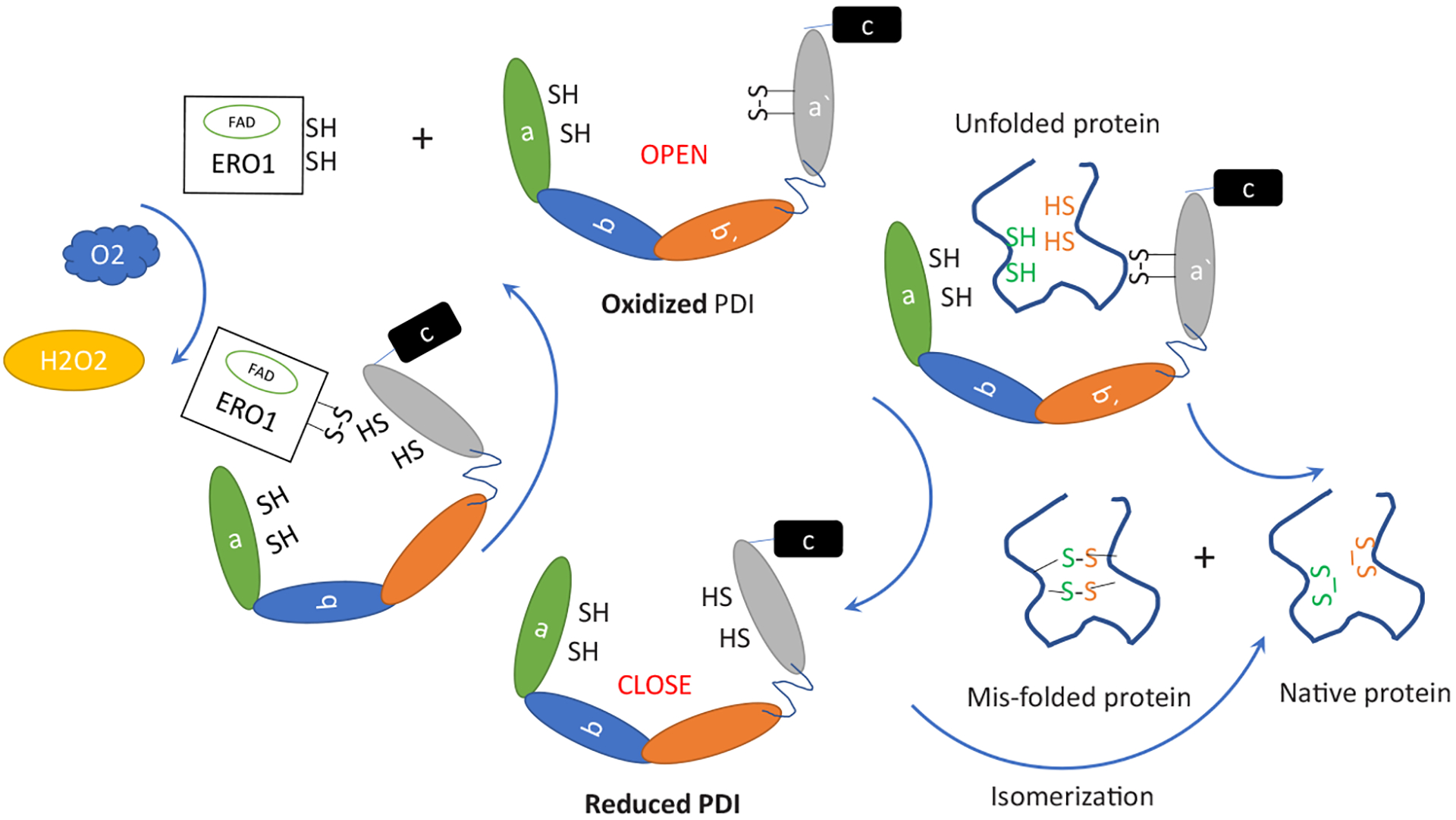
Mechanism of PDI-catalyzed disulfide exchange. Oxidation of PDI increases the distance between the two active sites, exposing a larger surface area of the hydrophobic region, enables the binding of unfolded proteins. After oxidation of unfolded proteins, the reduced PDI with closed conformation release the oxidized native proteins or mis-oxidized folding intermediate in some cases. The latter may be captured by reduced PDI to start a direct isomerization, the step of isomerization involves a reduced PDI active site attacking a substrate disulfide. Reduced PDI is then re-oxidized by ERO1 via direct hydrophobic interactions with its b′ domain and electron transfer from the a′ domain, resulting the production of H2O2.
4. Redox regulation: S-Glutathionylation of redox active cysteines as a protective and regulatory mechanism
Apart from their function in disulfide bond transfer and isomerization, redox active cysteines of PDI are targets of oxidation by ROS during different oxidizing conditions. Most common oxidative post-translational modification of active cysteines is sulfenylation, which is reversible, which can be further oxidized into irreversible sulfonylation. Irreversibly oxidized proteins are substrates for degradation. To protect proteins from further oxidation, sulfenylated thiols can be S-glutathionylated making them redox inactive; therefore glutathionylated PDI will not participate efficiently in disulfide bonds oxidoreduction or isomerization (Townsend, Manevich, He, Xiong, et al., 2009). At the same time, glutathionylation can serve as an epitope for so-called redox-mediated signaling, channeling proteins to different cellular destinations and changing their properties (Xiong, Manevich, Tew, & Townsend, 2012).
Increased expression of BiP and PDI are often found in multiple different human cancers (Lee, 2007; Rahman, et al., 2022), and participate in resistance to anticancer therapies. Our previous studies found that as redox sensitive proteins (redox accessible cysteines containing proteins), BiP and PDI are subjected to redox dependent regulation by S-glutathionylation (Tew et al., 2011). S-glutathionylation has been demonstrated to alters structure/function of multiple proteins in various pathways, e.g. cytoskeleton, kinase signaling, energy metabolism, calcium and redox homeostasis pathways within the cell (Grek, Zhang, Manevich, Townsend, & Tew, 2013) To date, proteomic studies of S-glutathionylated proteins have been restricted to tissues and whole cells. However, there is evidence that the sub-cellular organelle compartmentalization of components of the S-glutathionylation cycle may initiate differential regulation of ER functions in normal, tumor and drug resistant cells (Ye et al., 2017).
S-glutathionylation of BiP confers pro-survival properties and determines drug response in multiple myeloma (MM) cells by optimizing the balance between foldase and ATPase activities of BiP in a content-dependent manner (Zhang, Ye, et al., 2020). Cys41-S-glutathionylation increases ATPase activity, and Cys420-S-glutathionylation increases foldase activity. Preventing S-glutathionylation by GSTP specific inhibitor telintra can reverse tumor resistance to VELCADE® (bortezomib). While for PDI, S-glutathionylation induced by nitrosative/oxidative stress caused cell death through activation of the UPR and abrogation of ERα stability and signaling. S-glutathionylation resistant PDI variants can lessen the toxic effects of PABA/NO (Xiong, et al., 2012).
PDI family members are recognized as the most abundant cellular proteins. Nevertheless, analyses using microarray data from Gene expression Atlas datasets (https://www.ebi.ac.uk/gxa/home) or ONCOMINE (www.oncomine.org) demonstrated that they are frequently further upregulated in a variety of cancer types, suggesting that PDI could play an important role in cancer development and survival and could be a potential target for cancer therapy (Powell & Foster, 2021; Rahman, et al., 2022; Xu, et al., 2014). Post translational modifications of PDIs, not detectable by RNAseq, could add another layer of regulation to PDI function in cancer cells. The current proteomic technologies allow us to decipher and analyze the expression and posttranslational modifications of different members of PDI family (Rahman, et al., 2022). Proteomic studies of S-glutathionylated proteins currently restricted to the levels of tissues and whole cells. PDI localization changes during stress and tumorigenesis, and non-ER localized PDI contributes to evasion of immune responses by tumors (Willems et al., 2010). Additional studies are necessary to decipher mechanisms of how PDI localization is regulated, but the sub-cellular organelle compartmentalization of components of the S-glutathionylation machinery may initiate differential regulation of ER functions in normal, tumor and drug resistant cells (Ye, et al., 2017). We showed previously that Glutathione-S-Transferase Pi (GSTP) enzymatically catalyzes S-glutathionylation reactions (Townsend, Manevich, He, Hutchens, et al., 2009). GSTP has historically been classified as a cytosolic protein playing a role in drug metabolism (Townsend & Tew, 2003). However, we discovered that PDI is glutathionylated by GSTP through protein:protein interactions (Ye, et al., 2017) and ~50% of GSTP is localized within the ER, with a putative role in the redox regulation of protein homeostasis.
5. PDI in ER stress and cancer
In mammalian cells, the ER stress response is initiated by activation of the unfolded protein response (UPR). The UPR is mediated through activation of three ER resident trans-membrane proteins, pancreatic ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol requiring enzyme 1(IRE1) (Townsend, 2007). Their association with both ER chaperones, BiP and PDI controls their activity. When BiP and PDI are titrated away to assist in the folding of excess of accumulated unfolded proteins, a massive activation of ER stress effectors can occur (UPR). PERK, which connects ER stress with integrated stress response, phosphorylates the eukaryotic translation initiation factor 2α (eIF2α) and transiently halts translation of newly synthesized proteins, with the exception of ATF4 and possible subsets of other proteins, which have an alternative open reading frame translation and do not require eIF2α (Harding, Zhang, & Ron, 1999). This is the fastest responding branch of ER stress because it is mediated by phosphorylation, and can occur within a minute of the insult. ATF6 is a hidden transcription factor that traffics to the Golgi complex. There the cytoplasmic part is cleaved and relocates to the nucleus, becoming an active transcription factor which boosts the folding capacity of the ER by inducing production of ER foldases and chaperones (Haze, Yoshida, Yanagi, Yura, & Mori, 1999). Action of ATF6 requires more time since it involves intracellular trafficking and transcription/translation of its targets. IRE1 modulates ER stress response via Ire1 dependent decay (RIDD) pathway, which reduces ER bound RNAs (fast action), and via splicing that generates active transcription factor X-box binding protein1 (XBP1) (delayed action, requires splicing, translation of XBP1, followed by it function as transcription factor). C/EBP homologous protein (CHOP) is the late effector in the pathway and mediates recovery of ER function and disulfide bond formation via induction of ERO1 transcription (Hetz, 2012). ER associated degradation (ERAD) and autophagy are also increased during ER stress, and help to clear out misfolded proteins (Rashid, Yadav, Kim, & Chae, 2015). The UPR therefore plays an important role in combatting ER stress and maintaining cellular homeostasis. However, the regulation by UPR is usually short-term, and long-term activation without resolution of protein folding problem can trigger cell death (Perri, Thomas, Parakh, Spencer, & Atkin, 2015). An important link between ER and tumor development has been established recently (Chen & Cubillos-Ruiz, 2021). This suggested that induction of unresolved or lethal ER stress could be exploited to restrain tumor cell growth. Approaches combining standard therapies with UPR modulators have shown remarkable efficacy in preclinical cancer models and highlights future consideration in patients with cancer, especially patients who are resistant to standard therapies. Tumors experience repeatable episodes of UPR and oxidative stress. Upregulation of BiP and PDI are often found in multiple human cancers (Lee, 2007; Rahman, et al., 2022). Those have been demonstrated to promote resistance to anticancer therapies. Small molecules targeting the UPR have been extensively reviewed by Hetz et al. (Hetz, Axten, & Patterson, 2019). These directly target the three main UPR signaling branches and up/down-stream pathways, including IRE1 (Harnoss et al., 2019; Papandreou et al., 2011; Ranatunga et al., 2014; Tang et al., 2014), PERK (Atkins et al., 2013; Axten et al., 2012; Axten et al., 2013), eIF2α (Rabouw et al., 2019), and ATF6 (Gallagher & Walter, 2016).
6. PDI and cancers
Brain Tumors:
PDI is overexpressed in several cancers but most significantly in glioblastoma (Hu, Huang, Tao, & Zhu, 2020; Xu, et al., 2014). Gliomas are aggressive and the most prevalent primary tumors of the central nervous system tumors that develops by infiltrating normal brain tissues, with glioblastoma multiform being the most malignant subtype. Thioredoxin-domain-containing protein (TXNDC) family members were identified as differentially expressed in diffuse gliomas using multiple in silico analyses. High TXNDC5 levels were associated with unfavorable clinicopathological features and poor prognosis (Kocaturk, 2023). TXNDC5, a member of ER localized PDI family also known as ERp46, is potentially regulated by hypoxia and ER stress and shown to protect cells from oxidative stress, promote cell proliferation, and inhibit apoptosis (Wang, Li, & Chang, 2022). Increased levels of PDIA3–5 are highly correlated with glioblastoma multiform and low-grade glioma according to TCGA and CGGA datasets (Wang, Zhang, et al., 2022; Zhang et al., 2021; Zhang, Manevich, et al., 2020). PDIA3 has been postulated to be involved in inflammation and may interact with other immune checkpoint inhibitors such as nivolumab, ipilimumab (Long et al., 2018; Wolchok et al., 2017) and suppression of anti-tumor immunity in the glioma micro-environment (Zhang, Manevich, et al., 2020). PDIA4 stimulated PI3K/AKT/m-TOR pathway dependent proliferation and its downregulation inhibited proliferation and promoted apoptosis. Both correlate with a decline in the Warburg effect, which was suggested to provide redox equivalents for tumor cells proliferation (Wang, Zhang, et al., 2022). Glioma cells with high levels of PDIA5 demonstrated increased tumor cell proliferation and exhausted immune cells (HMC3) (Zhang, et al., 2021). TMX2 (PDI12) has been identified as a sensor in the MAM-regulated redox signaling pathway and a key adaptive regulator of neuronal proliferation, migration, and organization in the developing brain (Vandervore et al., 2019). Functional analysis of PDIP5 (PDIA6) in glioblastoma cells revealed that vimentin was bound to PDIA6, predominantly in U251 glioblastoma cells by immunoprecipitation using an anti-PDIA6 antibody in cancer and normal cells, PDIA6 knockdown in glioblastoma cells by small interfering RNA affected Bip promoter activation during cancer cell growth, and significantly inhibited cancer cell growth and migration, and the expression of epithelial–mesenchymal transition markers, Snail and Slug (Horibe, Torisawa, Masuda, & Kawakami, 2019). These results suggested that PDIA6 may serve an important role in cancer cell growth and may be considered an attractive and potent target for the treatment of glioblastoma.
Colorectal cancer (CRC):
CRC is the third most prevalent cancer in the world, its global incidence rate and mortality are rising (Dekker, Tanis, Vleugels, Kasi, & Wallace, 2019). PDIA1 protein expression was upregulated in CRC. PDIA1 plays a role of inhibition of autophagy via interaction with PHB2 and regulation of AKT-mTOR pathway is pro-survival in CRC cells, reducing their sensitivity to radio/chemo-therapy. Downregulation or use of small molecule inhibitor of PDIA1 resulted in induction of apoptosis in CRC cells (Wang, Shang, et al., 2022). In agreement, overexpression of PDIA1 lessened ER stress response and augmented the viability of CRC cells. The C-terminal catalytic site of PDIA1 was found to be indispensable for those effects. Collectively, PDI protects from endoplasmic reticulum stress and apoptosis of CRC cells through its oxidoreductase activity, thereby promoting the malignancy of CRC (Ma et al., 2021). Additionally, redox dependent regulation of Rac1/Nox1 by PDIA1 has been reported in colorectal cancer. PDIA1 also regulates levels of Ras activity in a context specific manner and maintains Nox1-dependent superoxide production in HEK3 and Caco2 leading to low activity of Ras in those CRC cells. Restricting superoxide production in HCT116 cells leads to high activity of Ras (De Bessa et al., 2019), implying that it is important to understand the context of PDIA1 activity modulation.
PDIA3 can be detected in serum in colon cancer patients. Levels were compared between colorectal cancer patients (n = 88), colorectal polyp patients (n = 77), and normal subjects (n = 36). Serum contents of PDIA3 were higher in colorectal cancer patients and increased alongside disease progression (Niu et al., 2022). Therefore, PDIA3 was suggested as a reliable biological indicator in CRC screening and its reference value as indicator for a progressive stage.
Renal cell carcinoma:
The prognosis of renal cell carcinoma remains poor due to high rate of metastases at the time of diagnosis, and resistance to chemotherapy (Padala et al., 2020). Induction of ferroptosis in resistance to chemotherapy cancers emerges as a potential strategy for treatment (Zhang, Liu, Jin, Chen, & Guo, 2022). Ferroptosis is an iron-catalyzed form of regulated necrosis, which is characterized by iron-dependent over accumulation of lethal levels of lipid hydroperoxides. In renal cell carcinomas, PDIA4 was shown to induce ATF4 and its downstream protein SLC7A11, solute carrier family 7 member 11. SLC7A11 is a cystine transporter upregulated in multiple cancers (Koppula, Zhuang, & Gan, 2021). Its metabolic action results in suppression of ferroptosis, and in promotion of cell growth. Downregulation of PDIA4 inhibits ATF4 and SLC7A11 thereby promoting ferroptosis cell death pathways in cancer cells (Kang et al., 2023).
Lung cancer:
Lung cancer is the leading cause of cancer-related mortality in the world (Sung et al., 2021). Lung cancer can be divided into subtypes based on cellular origins and histology. The main types include non-small cell lung cancers (NSCLC), comprised of adenocarcinomas and squamous cell carcinomas (Chen & Dhahbi, 2021), and less common, small cell lung cancer, which originates from neuroendocrine cell of the lung (Rudin, Brambilla, Faivre-Finn, & Sage, 2021). PDIA4 is implicated in the growth and death of lung cancer cells. Levels of PDIA4 have been found upregulated in a variety of lung cancer cell lines and tumors, negatively correlating with patient survival. Downregulation and overexpression of PDIA4 in tumor cells demonstrated that PDIA4 facilitated cell growth by inhibiting activation of apoptotic caspases 3 and 7 (Kuo et al., 2017). PDIA4 has 2 CGHC motifs in its N-terminus and one in its C-terminus. (Table 1). The CGHC motif of PDIA4 seemed to be crucial for the ability of PDIA4 to inhibit the activation and degradation of procaspases 3 and 7. Both pharmacological inhibition and genetic modification of CGHC interfere withPDIA4 binding to procaspases 3 and 7.
In wild-type and PDIA4−/−deficient mice, host PDIA4 promoted lung cancer development via stromal cells. Tumor development was enhanced due to the presence of immunologically suppressive T and B cells. Mechanistic studies showed that host PDIA4 positively regulates the Stat3/Vegf pathway in T and B lymphocytes by stabilizing activated Stat3. Furthermore, the first two CGHC motifs (a.a. 1–279) of PDIA4 are important for STAT33 stabilization. The N-terminally truncated PDIA4280–646 loses its ability to stabilize STAT3, and lowered development of immunologically suppressive T and T lymphocytes. Additional studies demonstrated that active thiols of cysteines in CGHC motifs are important for STAT3 binding, because mutations in any of the cysteines in the CGHC caused PDIA4(SGHS)x3 to lose its ability to bind STAT3 (Chen et al., 2022). These data suggest that PDIA4-STAT3 interaction are redox dependent, and those redox regulated interactions serve as negative regulators of cancer cell apoptosis, making PDIA4 a potential therapeutic target in certain lung cancers.
PDIA6 expression was upregulated in non-small cell lung cancers (NSCLC) compared with adjacent normal tissues. Expression levels correlated with prognosis; specifically, the higher PDIA6 expression, the poorer prognosis. Downregulation of PDIA6 slowed lung cancer cell proliferation and enhanced cisplatin-induced intrinsic apoptosis. PDIA6 was found to interact with MAP4K and thereby inhibited JNK/c-Jun signaling pathways (Bai et al., 2019). Thus, PDIA6 may serve as a potential biomarker for NSCLC diagnosis and prognosis. Collectively, these data suggest that targeting PDIA6 may be a promising therapeutic strategy for NSCLC patients. The expression of PDIA1 and ERO1A were evaluated individually and in context of co-expression in 198 NSCOC patients and controls based on their smoking status and prognosis. PDI and ERO1 expression levels were individually statistically associated with shorter overall survival. Increased levels of PDIA1 expression and in co-expression of PDIA1 and ERO1 were found as independent poor prognostic factors for overall survival in patients with NSCLC. The same correlations between increase in individual or co-expression of PDIA1 and ERO1A and poor prognosis were found for non-small cell lung cancers (Kim et al., 2018).
Breast cancer:
Upwards of 81% of breast cancer reach the invasive stage at the time of diagnosis. They can be classified into four subtypes: Luminal A, Luminal B, HER2-enriched, and Basal-like (commonly known as triple-negative breast cancer, TNBC). TNBC lead to more aggressive disease with fewer identified at early stages where treatment options are more promising. PDIs are frequently found upregulated in breast cancer, with PDIA1, PDIA3, PDIA4, and PDIA6 being most commonly increased at the mRNA level (Yang et al., 2022). High expression of those PDIs closely associate with cell proliferation, adhesion, invasion, and migration, acquisition of chemoresistance and poor clinical outcomes. Stojak et al., reported that PDIA1 is a major isoform of PDI, identified by proteomics in human breast cancer cells (MDA-MB-231 and MCF-7) (Stojak et al., 2020). The hypoxic tumor microenvironment plays critical a role in induction of cancer stem cell-like phenotype in multiple cancers, including breast cancer. Tumor stem cells are thought to be responsible for tumor progression, survival, metastasis, and chemoresistance (Emami Nejad et al., 2021). Chronic activation of UPR is a feature of aggressive breasts cancers and has been demonstrated to contribute to stemness of cancer cells (Liang et al., 2022). Yan et. al. demonstrated that hypoxia induced HIF1alpha, increases SOD2 levels and activity as a part of the mechanism to resolve hypoxia. Activities of SOD2 create less mtROS than those transported to the ER, leading to decreases in PDI levels and activity, accumulation of misfolded proteins, which in turn titrate BiP from UPR sensors, and activate the UPR (Yan et al., 2022). Therefore, increases in SOD2 → decrease in mtROS → decrease in PDIA1, increase in unfolded proteins → BiP titration from UPR sensors → UPRER → stemness and chemoresistance pathways—eventually conferring resistance and stem-like properties to breast cancers. Roles mediated by PDIs employ different molecular mechanisms in ERα (+) and ERα (−) breast cancers with dual tumorigenic/anti-tumorigenic functions of some PDIs founds in ERα (+) tumors. For example, in ERα (−) MDA-MB-231 human breast cancer cells, downregulation or inhibition of PDIA1 suppresses ferroptosis via inhibition of iNOS dimerization and catalytic activation, required for lipid oxidation (Lei, Zhuang, & Gan, 2022; Wang, Wang, & Zhu, 2022). In the ERα (+) breast cancers, PDIA1 suppresses carcinogenesis via modulating NF-κB and p53 activity (Bakker, Fujii, Krstic-Demonacos, Demonacos, & Alhammad, 2022). PDI is a molecular chaperone for the estrogen receptor alpha (ERα) and plays a key role in estrogenic signaling. PDI shares homology with the estrogen-binding domain of ERα. S-glutathionylation of PDI disrupts protein interactions with ERα (Xiong, et al., 2012). Drug induced S-glutathionylation of PDI reversed estradiol-induced upregulation of gene products involved in cell proliferation, suggesting that mediating cell death through abrogation of ERα stability and signaling may be a promising therapeutic opportunity (Xiong, Uys, Tew, & Townsend, 2011).
7. PDI inhibitors
Different PDI family members are implicated in tumor progression, metastases, immune invasion and chemoresistance. The therapeutic potential for PDI inhibitors in cancer treatment does seem promising. Currently, various PDI inhibitors have been developed (Powell & Foster, 2021; Xu, et al., 2014) (Table 2). Analoguing and new development of PDI inhibitors obtained by chemical screening or natural products are listed in Table 3. However, presently there is no late stage, or FDA-approved, PDI inhibitor for cancer treatment. The development of PDI as a therapeutic target in cancers have been complicated by several factors that require additional investigation. (i) Screening studies of PDI inhibitors were conducted using limited PDI isoforms with overlapping substrate specificities and were primarily focused on PDIA1. The possibility exists that the other isoforms can at least partially compensate for the loss of PDIA1 activity as they mediate the same general processes of reduction, oxidation or isomerization (Rutkevich, Cohen-Doyle, Brockmeier, & Williams, 2010). Although, it has been demonstrated that different PDI isoforms have separate substrate specificities, compensation due to inhibition or unconventional ER locations was not investigated. (ii) Development of better PDI activity screening methods is required. The current screening methods are based on measuring PDI activities such as reductase or oxidase or isomerase assays based on use of purified proteins. Multiple functions of PDI make it difficult to mimic the in vivo environment using in vitro approaches. In particular, the reduction assays require the inclusion of reducing equivalents such as DTT or β-ME, which can interfere with the activities of potential screening hits, especially redox sensitive molecules (Watanabe, Laurindo, & Fernandes, 2014). (iii) Low sensitivity existing PDI assays, which underestimate in vitro assays compared to in vivo studies, e.g. E61 induces biomarkers of PDI inhibition in cells at low micromolar concentrations but shows a high micromolar IC50 in vitro (Robinson et al., 2019). (iv) PDI and its superfamilies have multiple roles in maintaining cellular homeostasis. Approaches that target only PDI isoforms expressed in cancer cells need to be considered. Therefore, although PDIs are crucial for cancer progression, metastases, evasion of immune response and contribute to resistance, numerous technical and conceptual challenges need to be addressed before patients can benefit.
Table 2.
Commercially available PDI inhibitors.
| Inhibitor | Chemical name | IC50 (μM) | PDI members | PDI inhibition activity | Membrane permeable reversibility | Refs. |
|---|---|---|---|---|---|---|
| 35G8 | 1,3,6-Trimethylpyrimido [5,4-e] [1,2,4] triazine-5,7-dione | 0.17 | PDIA1 | Induces Nrf2 antioxidant response, endoplasmic reticulum stress response, autophagy, and may induce cell death via ferroptosis | Cell permeable | Kyani et al. (2018) |
| E64FC26 | (lE)-1-nonylidene-3-(trifluoromethyl) indene-5,6-diol | 1.9 20.9 25.9 16.3 25.4 |
PDIA1 PDIA3 PDIA4, TXNDC5 PDIA6 |
anti-myeloma activity | Cell permeable | Robinson et al. (2019) |
| PACMA 31 | N-(2,4-Dimethoxyphenyl)-N-(1-oxo-2-propyn-1-yl)-2-(2-thienyl) glycyl-glycine ethyl ester | 10 | PDIA1 | Cytotoxic in ovarian cancer cells | Cell permeable irreversible | Xu et al. (2012) |
| Bacitracin | Commercial bacitracin is a mixture of at least 22 structurally related peptides. | 150 | PDIA1 | Inhibits cell death through enhanced apoptosis in melanoma cells | Cell permeable reversible | Karala and Ruddock (2010) |
| 16F16 | 2-(2-Cbloroacetyl)-2,3,4,9-tetrahydro-1-methyl-1H-pyrido [3,4-b] indole-1-carboxylic acid methyl ester, Methyl 2-(2-chloroacetyl)-1-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b] indole-1-carboxylate | 63 | PDIA1 PDIA3 |
Inhibits apoptosis in PC12 cell model of HD | Cell permeable irreversible | Kaplan et al. (2015) |
| P1 | N-[(1,1-Dimethylethoxy)carbonyl]-l-phenylalanyl-O-(ethenylsulfonyl) -N-4-pentyn-1-yl-l-tyrosinamide | 1.7 | PDIA1 | Inhibits proliferation of MCF7, MDA-MB-231, HepG2 and UACC257 and T47D | Cell permeable irreversible | Ge et al. (2013) |
| CCF 642 | Spyietqlovdjcf-xyokqwhbsa-n | 2.9 | PDIA1 | Induce apoptosis and cytotoxic effects in multiple myeloma cells | Cell permeable irreversible | Vatolin et al. (2016) |
| LOC14 | 2-((4-(Cyclopropanecarbonyl)piperazin-1-yl) methyl) benzo[d]isothiazol-3(2H-f)-one, 2-((4-(Cyclopropylcarb onyl)-1-piperazinyl) methyl)-1,2-benzothiazol-3 (2H)-one | 0.5 | PDIA3 | Antiapoptotic neuroprotective | Cell permeable reversible |
Kaplan, et al. (2015); O’Sullivanet al. (2022) |
| BAP2 | (E)-3-(3-(4-Hydroxyphenyl)-3-oxoprop-1-en-1-yl)benzonitrile | 0.9 | PDIA1 PDIA2 |
Inhibits tumor growth in glioblastoma | Cell permeable | Xu et al. (2019) |
| KSC-34 | 2-Chloro-N-[2-[[4-[methyl(4-phenylbutyl)amino]-6-(prop-2-ynylamino)-l,3,5-triazin-2-yl]amino]ethyl]acetamide | 3.5 | PDIA1 | Reduce the extracellular pathogenic load of the amyloidogenic antibody light chain | Cell Permeable | Cole et al. (2018) |
Table 3.
Recently identified PDI inhibitors.
| Inhibitor | Activity assay | IC50 (μM) | PDI members | In vitro | In vivo | Ref. |
|---|---|---|---|---|---|---|
| Aziridine-2-carboxylic acid derivatives | Insulin turbidity assay | 0.6 1 |
PDIA1 PDIA3 |
HT-29 CaCo-2 cytotoxicity MCF-7 and MDA-MB-231 cell lines |
In vivo in rat model of arterial thrombosis |
Kurpinska et al. (2022); Zelencova-Gopejenko et al. (2023) |
| Piericones A Piericones B | Insulin turbidity assay | 0.15 0.23 |
PDIA1 PDIA3 PDIA4 PDIA5 |
In vitro platelet aggregation and fibrin formation | In vivo thrombus formation | Zheng et al. (2023) |
| Galangin | Tryptophan fluorescence-based assay | 5.9 | PDIAl | N/A | Delayed the time of blood vessel occlusion in an electricity-induced mouse thrombosis model | Liang, et al. (2022) |
| Quercetin-3-rutinoside | Tryptophan displacement assay | 0.43 | PDIAl | Quercetin-3-rutinoside at 10μM effectively reduced chemotherapy-induced TF activity to its baseline | N/A |
Beckmann et al. (2021); Liao et al. (2022) |
| tcyDTDO derivatives | Binding assay | N/A | PDIA1 PDIA10 PDIA17 |
DDAs disrupt disulfide bonding between PDIA1 and ERp44 and their client proteins | N/A | Law et al. (2022) |
| Origamicin | Protein disulfide isomerase activity assay | 32.9 | PDIA1 | Induces oxidative stress response, downregulates MYC, and activates the p53 signaling pathway in neuroblastoma cells | N/A | Ozcelik and Pezacki (2019) |
| BAP2 derivatives | PDI reductase assay | 0.12–11 | PDIA1 PDIA2 |
Inhibit glioblastoma cell growth, induce ER stress, increase expression of G2M checkpoint proteins, and reduce expression of DNA repair proteins | N/A | Yang et al. (2019) |
8. Conclusions
The PDI family consists of 21 proteins with “all in one” functions: dithiol–disulfide oxidation, reduction, isomerization, and chaperone activities. As redox folding catalysts from the ER, the functions in ER related redox homeostasis and signaling have been well studied. In the past decade, PDI structure and 3D architecture, thiol based redox reactions, physiological roles in multiple diseases, particularly in cancer have been demonstrated. PDI is highly expressed in several cancer types, including gliomas, colon, breast, lung and renal cancers. Cancer cells require high levels of PDI proteins to overcome significant ER stress and high demand of protein folding to sustain rapid rate of cell proliferation. These may render cancer cells more vulnerable to the inhibition of PDI than normal cells. Investigation that will decipher the mechanisms of PDI mediated adaptor functions in redox signaling, will provide further advances in our understanding of cellular redox signaling and provide novel potential drug targets of cancer therapy.
References
- Alanen HI, Salo KE, Pekkala M, Siekkinen HM, Pirneskoski A, & Ruddock LW (2003). Antioxidants & Redox Signaling, 5, 367–374. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Riemer J, Zito E, Chin KT, Ron D, Spiess M, & Ellgaard L (2010). The EMBO Journal, 29, 3318–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, & Kumar R (2013). Cancer Research, 73, 1993–2002. [DOI] [PubMed] [Google Scholar]
- Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, & Gampe RT (2012). Journal of Medicinal Chemistry, 55, 7193–7207. [DOI] [PubMed] [Google Scholar]
- Axten JM, Romeril SP, Shu A, Ralph J, Medina JR, Feng Y, & Kumar R (2013). ACS Medicinal Chemistry Letters, 4, 964–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Liu X, Qi X, Liu X, Peng F, Li H, & Shao S (2019). EBioMedicine, 42, 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker EY, Fujii M, Krstic-Demonacos M, Demonacos C, & Alhammad R (2022). International Journal of Oncology, 60(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe CK, & Miller EA (2013). Genetics, 193, 383–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann L, Rolling CC, Voigtlander M, Mader J, Klingler F, Schulenkorf A, & Langer F (2021). Cancers (Basel), 13, 3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, & Dhahbi J (2021). Scientific Reports, 11, 13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TY, Yang CY, Yang MT, Kuo TF, Chang CL, Chen CL, & Yu AY (2022). Clinical and Translational Medicine, 12, e606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, & Cubillos-Ruiz JR (2021). Nature Reviews. Cancer, 21, 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole KS, Grandjean JMD, Chen K, Witt CH, O’Day J, Shoulders MD, & Weerapana E (2018). Biochemistry, 57, 2035–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bessa TC, Pagano A, Moretti AIS, Oliveira PVS, Mendonca SA, Kovacic H, & Laurindo FRM (2019). Cell Death and Disease, 10, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, & Wallace MB (2019). Lancet, 394, 1467–1480. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, & Helenius A (2003). Nature Reviews. Molecular Cell Biology, 4, 181–191. [DOI] [PubMed] [Google Scholar]
- Emami Nejad A, Najafgholian S, Rostami A, Sistani A, Shojaeifar S, Esparvarinha M, & Manian M (2021). Cancer Cell International, 21, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher CM, & Walter P (2016). Elife, 5, e11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Zhang CJ, Li L, Chong LM, Wu X, Hao P, & Yao SQ (2013). ACS Chemical Biology, 8, 2577–2585. [DOI] [PubMed] [Google Scholar]
- Goldberger RF, Epstein CJ, & Anfinsen CB (1963). The Journal of Biological Chemistry, 238, 628–635. [PubMed] [Google Scholar]
- Grek C, & Townsend DM (2014). Endoplasmic Reticulum Stress in Diseases, 1, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grek CL, Zhang J, Manevich Y, Townsend DM, & Tew KD (2013). The Journal of Biological Chemistry, 288, 26497–26504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, & Ron D (1999). Nature, 397, 271–274. [DOI] [PubMed] [Google Scholar]
- Harnoss JM, Le Thomas A, Shemorry A, Marsters SA, Lawrence DA, Lu M, & Ashkenazi A (2019). Proceedings of the National Academy of Sciences of the United States of America, 116, 16420–16429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Dinkova-Kostova AT, & Tew KD (2020). Cancer Cell, 38, 167–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, & Mori K (1999). Molecular Biology of the Cell, 10, 3787–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C (2012). Nature Reviews. Molecular Cell Biology, 13, 89–102. [DOI] [PubMed] [Google Scholar]
- Hetz C, Axten JM, & Patterson JB (2019). Nature Chemical Biology, 15, 764–775. [DOI] [PubMed] [Google Scholar]
- Horibe T, Torisawa A, Masuda Y, & Kawakami K (2019). Oncology Reports, 41, 961–972. [DOI] [PubMed] [Google Scholar]
- Hu Q, Huang K, Tao C, & Zhu X (2020). Journal of Cellular and Molecular Medicine, 24, 5888–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan PA, & Gibbins JM (2006). Antioxidants & Redox Signaling, 8, 312–324. [DOI] [PubMed] [Google Scholar]
- Kang L, Wang D, Shen T, Liu X, Dai B, Zhou D, & Tan X (2023). Cell Death and Disease, 14, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A, Gaschler MM, Dunn DE, Colligan R, Brown LM, Palmer AG 3rd, & Stockwell BR (2015). Proceedings of the National Academy of Sciences of the United States of America, 112, E2245–E2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karala AR, & Ruddock LW (2010). The FEBS Journal, 277, 2454–2462. [DOI] [PubMed] [Google Scholar]
- Kim KM, An AR, Park HS, Jang KY, Moon WS, Kang MJ, & Chung MJ (2018). Oncology Letters, 16, 5753–5760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocaturk B (2023). Cancer Medicine, 12, 3830–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppula P, Zhuang L, & Gan B (2021). Protein & Cell, 12, 599–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov G, Maattanen P, Thomas DY, & Gehring K (2010). The FEBS Journal, 277, 3924–3936. [DOI] [PubMed] [Google Scholar]
- Kuo TF, Chen TY, Jiang ST, Chen KW, Chiang YM, Hsu YJ, & Yang WC (2017). Oncogene, 36, 5484–5496. [DOI] [PubMed] [Google Scholar]
- Kurpinska A, Suraj-Prazmowska J, Stojak M, Jarosz J, Mateuszuk L, Niedzielska-Andres E, … Chlopicki S (2022). Cancer Cell International, 22, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyani A, Tamura S, Yang S, Shergalis A, Samanta S, Kuang Y, & Neamati N (2018). ChemMedChem, 13, 164–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law ME, Yaaghubi E, Ghilardi AF, Davis BJ, Ferreira RB, Koh J, & Law BK (2022). Cancer Letters, 534, 215604. [DOI] [PubMed] [Google Scholar]
- Lee AS (2007). Cancer Research, 67, 3496–3499. [DOI] [PubMed] [Google Scholar]
- Lee E, & Lee DH (2017). BMB Reports, 50, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei G, Zhuang L, & Gan B (2022). Nature Reviews. Cancer, 22, 381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Cai M, Xu Y, Fu W, Wu J, Liu Y, & Yuan C (2022). Journal of Natural Products, 85, 1332–1339. [DOI] [PubMed] [Google Scholar]
- Liao X, Zhuang X, Liang C, Li J, Flaumenhaft R, Yuan C, & Huang M (2022). Journal of Agricultural and Food Chemistry, 70, 4475–4483. [DOI] [PubMed] [Google Scholar]
- Long GV, Atkinson V, Lo S, Sandhu S, Guminski AD, Brown MP, & McArthur GA (2018). The Lancet Oncology, 19, 672–681. [DOI] [PubMed] [Google Scholar]
- Ma YS, Feng S, Lin L, Zhang H, Wei GH, Liu YS, & Fu D (2021). Cellular Signalling, 86, 110076. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Masutani H, Son A, Kizaka-Kondoh S, & Yodoi J (2009). Molecular Biology of the Cell, 20, 4552–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y, Xue J, Wu X, Qu M, Wang L, Liang W, & Li T (2022). Frontiers in Pharmacology, 13, 935500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan ED, Mylonas KJ, Bell R, Carvalho C, Baird DP, Cairns C, & Ferenbach DA (2022). JCI Insight, 7, e154124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcelik D, & Pezacki JP (2019). ACS Chemical Neuroscience, 10, 4068–4075. [DOI] [PubMed] [Google Scholar]
- Padala SA, Barsouk A, Thandra KC, Saginala K, Mohammed A, Vakiti A, & Barsouk A (2020). World Journal of Oncology, 11, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, & Koong AC (2011). Blood, 117, 1311–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perri ER, Thomas CJ, Parakh S, Spencer DM, & Atkin JD (2015). Frontiers in Cell and Developmental Biology, 3, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell LE, & Foster PA (2021). Cancer Medicine, 10, 2812–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouw HH, Langereis MA, Anand AA, Visser LJ, de Groot RJ, Walter P, & van Kuppeveld FJM (2019). Proceedings of the National Academy of Sciences of the United States of America, 116, 2097–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman NSA, Zahari S, Syafruddin SE, Firdaus-Raih M, Low TY, & Mohtar MA (2022). Cell Biosci, 12, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranatunga S, Tang CH, Kang CW, Kriss CL, Kloppenburg BJ, Hu CC, & Del Valle JR (2014). Journal of Medicinal Chemistry, 57, 4289–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid HO, Yadav RK, Kim HR, & Chae HJ (2015). Autophagy, 11, 1956–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson LC (2022). Centers for Disease Control and Prevention, Division of Cancer Prevention and Control, Atlanta GA. [Google Scholar]
- Robinson RM, Reyes L, Duncan RM, Bian H, Reitz AB, Manevich Y, & Dolloff NG (2019). Leukemia: Official Journal of the Leukemia Society of America, Leukemia Research Fund, U. K 33, 1011–1022. [Google Scholar]
- Rudin CM, Brambilla E, Faivre-Finn C, & Sage J (2021). Nature Reviews Disease Primers, 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkevich LA, Cohen-Doyle MF, Brockmeier U, & Williams DB (2010). Molecular Biology of the Cell, 21, 3093–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller M, Wilkinson B, & Gilbert HF (2003). The Journal of Biological Chemistry, 278, 7154–7159. [DOI] [PubMed] [Google Scholar]
- Stojak M, Milczarek M, Kurpinska A, Suraj-Prazmowska J, Kaczara P, Wojnar-Lason K, & Chlopicki S (2020). Cancers (Basel), 12, 2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, & Bray F (2021). CA: A Cancer Journal for Clinicians, 71, 209–249. [DOI] [PubMed] [Google Scholar]
- Tang CH, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, & Hu CC (2014). The Journal of Clinical Investigation, 124, 2585–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, & Townsend DM (2011). Free Radical Biology & Medicine, 51, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Xiang S, Noiva R, Lennarz WJ, & Schindelin H (2006). Cell, 124, 61–73. [DOI] [PubMed] [Google Scholar]
- Townsend DM (2007). Molecular Interventions, 7, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, & Tew KD (2009). The Journal of Biological Chemistry, 284, 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, Manevich Y, He L, Xiong Y, Bowers RR Jr., Hutchens S, & Tew KD (2009). Cancer Research, 69, 7626–7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend DM, & Tew KD (2003). Oncogene, 22, 7369–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandervore LV, Schot R, Milanese C, Smits DJ, Kasteleijn E, Fry AE, & Mancini GMS (2019). American Journal of Human Genetics, 105, 1126–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatolin S, Phillips JG, Jha BK, Govindgari S, Hu J, Grabowski D, & Reu FJ (2016). Cancer Research, 76, 3340–3350. [DOI] [PubMed] [Google Scholar]
- Venetianer P, & Straub FB (1963). Biochimica et Biophysica Acta, 67, 166–168. [DOI] [PubMed] [Google Scholar]
- Wang C, Chen S, Wang X, Wang L, Wallis AK, Freedman RB, & Wang CC (2010). The Journal of Biological Chemistry, 285, 26788–26797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Li W, Ren J, Fang J, Ke H, Gong W, & Wang CC (2013). Antioxidants & Redox Signaling, 19, 36–45. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang P, & Zhu BT (2022). Molecular and Cellular Biology, 42, e0052221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li SJ, Sidhu A, Zhu L, Liang Y, Freedman RB, & Wang CC (2009). The Journal of Biological Chemistry, 284, 199–206. [DOI] [PubMed] [Google Scholar]
- Wang M, Zhang W, Liu Y, Ma Z, Xiang W, Wen Y, & Zhou J (2022). Biochemical and Biophysical Research Communications, 597, 83–90. [DOI] [PubMed] [Google Scholar]
- Wang R, Shang Y, Chen B, Xu F, Zhang J, Zhang Z, & Zhao G (2022). Cell Death and Disease, 13, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li H, & Chang X (2022). European Journal of Medical Research, 27, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe MM, Laurindo FR, & Fernandes DC (2014). Frontiers in Chemistry, 2, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems SH, Tape CJ, Stanley PL, Taylor NA, Mills IG, Neal DE, & Murphy G (2010). The Biochemical Journal, 428, 439–450. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, & Larkin J (2017). The New England Journal of Medicine, 377, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Manevich Y, Tew KD, & Townsend DM (2012). International Journal of Cell Biology, 2012, 273549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Uys JD, Tew KD, & Townsend DM (2011). Antioxidants & Redox Signaling, 15, 233–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, & Neamati N (2012). Proceedings of the National Academy of Sciences of the United States of America, 109, 16348–16353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Liu Y, Yang K, Wang H, Shergalis A, Kyani A, & Neamati N (2019). Theranostics, 9, 2282–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Sankar S, & Neamati N (2014). Drug Discovery Today, 19, 222–240. [DOI] [PubMed] [Google Scholar]
- Yan Y, He M, Zhao L, Wu H, Zhao Y, Han L, & Wei M (2022). Cell Death and Differentiation, 29, 1769–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Jackson C, Karapetyan E, Dutta P, Kermah D, Wu Y, & Vadgama JV (2022). Cancers (Basel), 14, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Shergalis A, Lu D, Kyani A, Liu Z, Ljungman M, & Neamati N (2019). Journal of Medicinal Chemistry, 62, 3447–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZW, Zhang J, Ancrum T, Manevich Y, Townsend DM, & Tew KD (2017). Antioxidants & Redox Signaling, 26, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelencova-Gopejenko D, Andrianov V, Domracheva I, Kanepe-Lapsa I, Milczarek M, Stojak M, & Kalvins I (2023). Journal of Enzyme Inhibition and Medicinal Chemistry, 38, 2158187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu X, Jin S, Chen Y, & Guo R (2022). Molecular Cancer, 21, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, He J, Dai Z, Wang Z, Liang X, He F, & Cheng Q (2021). Frontiers in Immunology, 12, 628966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhou Y, Cheng Q, Dai Z, Wang Z, Liu F, … Cao H (2020). Aging (Albany NY), 12, 15392–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ye ZW, Chen W, Culpepper J, Jiang H, Ball LE, & Townsend DM (2020). Free Radical Biology & Medicine, 160, 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Lv K, Wang H, Huang L, Feng Y, Gao B, & Yao G (2023). Journal of the American Chemical Society, 145, 3196–3203. [DOI] [PubMed] [Google Scholar]