

Abstract
Here, we describe an approach towards analogs of the potent antibiotic Bactobolin A. Sulfamate-tethered aza-Wacker cyclization reactions furnish key synthons, which we envision can be elaborated into analogs of Bactobolin A. Docking studies show that the C4 epimer of Bactobolin A and the C4/C6 diastereomer interact with different residues of the 23S rRNA (bacterial ribosome 50S subunit) than the natural product, suggesting that these molecules could be valuable tool compounds for fundamental studies of the bacterial translational machinery.
Keywords: aza-Wacker, Sulfamate, Bactobolin, Antibiotic synthesis
Graphical Abstract
Introduction
Over the last twenty years, analogs-oriented synthetic strategies have gained favor over “make- the-molecule” approaches, which predominated organic chemistry in the twentieth century [1-3]. Our laboratory is deeply invested in developing tethered olefin functionalization methodology, which we envision as key steps in analogs-oriented syntheses of antibiotics [4-7]. Bactobolin A is an unusual dichlorinated polyketide antibiotic that was first isolated from the culture broth of Pseudomonas yoshidomiensis and has been demonstrated to have broad antibacterial activity against a variety of pathogenic gram-positive and gram-negative organisms (Fig. 1) [8-11]. Unfortunately, marked cytotoxicity to eukaryotic cells has precluded its clinical use [12, 13]. Recent work has pinpointed the antibacterial activity of Bactobolin A to inhibition of bacterial protein translation via binding to an unprecedented site on the bacterial ribosome 50S subunit (L2 protein) [9, 14]. A crystal structure shows this interaction [15]. It is hypothesized that binding to the L2 homolog of the eukaryotic ribosome (L8e) is responsible for eukaryotic cytotoxicity. Despite such rich biological activity, there exists only one racemic synthesis (Weinreb) [16-18] and one enantiospecific synthesis (Švenda) [19] of Bactobolin A [20-23]. Furthermore, few studies of Bactobolin derivatives have been reported; thus far, most analyses have focused on modifying side chains attached to the amino and hydroxy functionalities with few attempts at modifying other aspects of the core structures [24-26]. In addition, most exploration of Bactobolin analogs has centered on directly modifying the natural product, obtained from fermentation.
Fig. 1.

A Bactobolin A is a potent antibiotic. B Analogs of Bactobolin A may have useful biological properties and may serve as tool compounds for studying the L2 protein of the bacterial ribosome (50S subunit)
We were attracted to Bactobolin A because of its complex, functional-group laden architecture and its useful antibacterial activity. We were most interested in designing a route to access the C4 epimer and the C4/C6 diastereomer of Bactobolin A (Fig. 1). Docking studies (see Results and Discussion section) establish that these compounds interact with bacterial 23S rRNA differently from Bactobolin A, suggesting that these compounds may have divergent biological properties relative to the natural isomer. Furthermore, these molecules and their derivatives could serve as biological tool compounds for fundamental studies of the bacterial L2 protein, a rare and unique target for antibacterial action. It is unlikely that these unnatural isomers could be accessed from modifications of fermented Bactobolin A, making de novo synthesis necessary. We envisioned our laboratory’s sulfamate-tethered aza-Wacker cyclization would serve as a key step in the construction of the 1,3-amino alcohol motif found in these analogs [4, 27, 28].
Results and discussion
Our synthesis commenced from D-(-)-quinic acid, a commercially available cyclitol chiron (Scheme 1). Following literature precedent, quinic acid was transformed into tricyclic lactone 2, which was reduced into triol 3 using NaBH4 [29]. Cleavage with NaIO4-impregnated silica gel, mesylation, and elimination afforded enone 5 [29]. Removal of the double bond proceeded smoothly with 10% Pd/C under 1 atm of H2. Acetonide elimination coupled with in situ TBS protection of the resulting alkoxide afforded enone 7 [30].
Scheme 1.

Opening sequence of reactions patterned on literature precedent
Diastereoselective conjugate addition of cis-1-propenyl magnesium bromide into enone 7 proved to be a challenge, as, curiously, there is sparse precedent for 1,4-addition with this reagent. We tested a variety of conditions, a portion of which are shown (Scheme 2). We were pleased to find that a combination of super-stoichiometric CuBr•SMe2, LiCl, and cis-1-propenyl-magnesium bromide afforded desired product 8 in 40% yield and as the single trans-diastereomer (Scheme 2, Entry 1). Attempts to further optimize this transformation using variations in temperature were invariably unsuccessful (Scheme 2, Entries 2-3). We observed a modest improvement in yield upon switching to CuCN, eliminating LiCl, and reducing the temperature to −78 °C (Scheme 2, Entry 4). The most dramatic improvement in this reaction was afforded by switching to catalytic CuCl (Scheme 2, Entry 5). With only 0.2 equivalents, the reaction was complete within 1 h at −78 °C. Importantly, these optimized conditions could be used reliably on scales as large as one gram, allowing for more convenient material throughput. We note that the use of cis-1-propenyl-magnesium bromide was critical for achieving high diastereoselectivity in the conjugate addition. Using commercial preparations containing undisclosed ratios of cis- and trans-1-propenyl magnesium bromide afforded intractable mixtures of diastereomers and geometric isomers.
Scheme 2.

Optimization of a conjugate addition reaction of cis-1-propenyl magnesium bromide
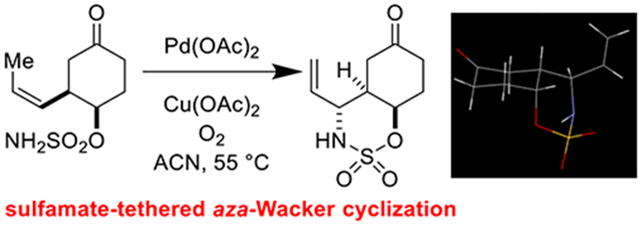
Removal of the TBS group proceeded smoothly with aqueous HF affording alcohol 9 (Scheme 3). Sulfamoyl chloride was generated upon treatment of chlorosulfonyl isocyanate with formic acid [31] after which a solution of 9 in DMA was added, forming sulfamate 10. To our delight (and perhaps some relief), the sulfamate-tethered aza-Wacker cyclization of 10 afforded oxathiazinane 11 in synthetically useful yields and as a single diastereomer. We anticipate that 11 will serve as a key synthon for future elaboration into C4-epi-Bactobolin.
Scheme 3.

Tethered aza-Wacker technology furnishes an anticipated synthon for the C4 epimer of Bactobolin A
In order to invert the C6 stereocenter, a Mitsunobu protocol involving pre-treatment of 9 with PPh3 and 3,5-dinitrobenzoic acid followed by addition of DEAD was developed (Scheme 4) [32]. Rapid hydrolysis of the 3,5-dinitrobenzoate ester occurred upon addition of K2CO3 in anhydrous MeOH.
Scheme 4.

Mitsunobu inversion of the C6 alcohol
Condensation of 13 with sulfamoyl chloride in a mixture of DMA/acetonitrile yielded primary sulfamate 14 (Scheme 5). Treatment with 40 mol% of Pd(OAc)2 and 1 equivalent of Cu(OAc)2 under 1 atmosphere of O2 afforded bicyclic oxathiazinane 15 in a modest yield of 30%. X-ray crystallography (CCDC Number: 2045973) established its absolute configuration. Our previous work has shown that primary sulfamates perform poorly in the aza-Wacker cyclization relative to secondary sulfamates [4]. Accordingly, we prepared N-pOMe-phenyl sulfamate 16 via condensation of 13 with pOMe-phenylsulfamoyl chloride and NEt3. In line with our previous results, we found a dramatic improvement in reaction performance when sulfamate 16 was subject to the aza-Wacker protocol, with oxathiazinane 17 forming in 65% yield. There is ample precedent for oxidative removal of pOMe-phenyl groups from amines [33, 34] and amides [35, 36]; we anticipate analogous deprotection at some later stage of the synthesis of the C4/C6 diastereomer.
Scheme 5.

C4/C6 diastereomer synthons. A aza-Wacker cyclization reaction of primary sulfamate 14. B Using pOMe-phenyl sulfamate 16 dramatically improves the yield of the tethered aza-Wacker cyclization reaction
Docking studies (Fig. 2) of Bactobolin, the C4 epimer, and the C4/C6 diastereomer show that the three molecules interact with the 23S rRNA (bacterial ribosome 50S subunit) in different regions. All three coordinate to a Mg2+ ion and form van der Waals interactions with the bacterial 23S rRNA. With Bactobolin A, the alanine side chain is oriented towards a small sub-pocket (Fig. 2A). With both the C4 epimer (Fig. 2B, C) and the C4/C6 diastereomer (Fig. 2D), the alanine residue is pointed towards a larger open space. Thus, appending longer side chains onto the nitrogen would allow for interrogation of different regions of the 23S rRNA. This suggests that both analogs could have divergent biological properties relative to Bactobolin A and that they could serve as valuable tool compounds for fundamental studies of the bacterial ribosome L2 protein, an unusual and rare antibiotic target. Such data gives us the impetus to continue pursuing analogs-oriented syntheses of the Bactobolin family.
Fig. 2.

A Bactobolin A binds in a cleft between the 23S rRNA (gray sticks) and the tRNA (orange sticks), coordinating with a magnesium ion (blue sphere) and forming several internal and external hydrogen bonds (yellow dashes) and salt bridge interactions (magenta dashes). B The Bactobolin epimer is oriented away from the sub-pocket, interacting with different regions of the rRNA. C The space fill representation with the Bactobolin epimer suggests the potential to interrogate other sub-pockets. D The C4/C6 diastereomer regains interactions that were lost in the C4 epimer
Conclusion
In summary, we present our progress towards analogs of Bactobolin A. Stereospecific sulfamate-tethered aza-Wacker cyclization reactions furnish key synthons, which we envision can be elaborated into the C4 epimer and the C4/C6 diastereomer. Docking studies show that these isomers interact with the bacterial 23S rRNA in disparate ways relative to Bactobolin A, suggesting that access to these molecules could provide valuable tool compounds for fundamental biological studies of the bacterial ribosome.
Experimental section
All reagents were obtained commercially unless otherwise noted. Solvents were purified by passage under 10 psi N2 through activated alumina columns. Infrared (IR) spectra were recorded on a Thermo Scientific™ Nicolet™ iS™5 FT-IR Spectrometer; data are reported in frequency of absorption (cm−1). Optical rotations were determined by a Rudolph Polarimeter AUTOPOL IV. NMR spectra were recorded on a Bruker Avance 400 operating at 400 and 100 MHz. 1H NMR spectra were recorded at 400 MHz. Data are recorded as: chemical shift in ppm referenced internally using residue solvent peaks, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or overlap of nonequivalent resonances), integration, coupling constant (Hz). 13C NMR spectra were recorded at 100 MHz. Exact mass spectra were recorded using an electrospray ion source (ESI) either in positive mode or negative mode and with a time-of-flight (TOF) analyzer on a Waters LCT PremierTM mass spectrometer and are given in m/z. TLC was performed on pre-coated glass plates (Merck) and visualized either with a UV lamp (254 nm) or by dipping into a solution of KMnO4–K2CO3 in water followed by heating. Flash chromatography was performed on silica gel (230–400 mesh). (−)-Quinic acid was purchased from ACROS and cis-1-bromo propene was purchased from Sigma-Aldrich. Compounds 2-7 were prepared using reported procedures [29, 30].
(3R,4S)-4-((tert-butyldimethylsilyl)oxy)-3-((Z)-prop-1-en-1-yl)cyclohexan-1-one (8)
To a solution of CuCl (87.4 mg, 0.884 mmol, 0.2 equiv.) in dry THF (5 mL) was added freshly prepared (Z)-prop-1-en-1-ylmagnesium bromide (0.44M in THF, 20 mL, 8.8 mmol, 2.4 equiv.) at −78 °C. A solution of 7 (1 g, 4.42 mmol) in THF (3 mL) was added to the reaction mixture at −78 °C and the resulting mixture was stirred for 1 h at same temperature. After 1 h, the reaction was quenched with saturated aqueous ammonium chloride (40 mL) at 0 °C. The mixture was transferred to a separatory funnel. The organic layer was removed, and the aqueous layer was extracted with ethyl acetate (2 × 100 mL). The combined organic layers were washed with brine solution (50 mL), dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. The residue was purified through silica gel column chromatography (hexane/ethyl acetate = 95:5) to afford 700 mg (60%) of 8 as a colorless oil. [α]D = +39.82 (c 0.625, CHCl3); FT-IR (KBr): 3012, 2958, 2886, 1717, 1471 cm−1; 1H NMR (400 MHz, CDCl3): δ 5.52 (ddd, J = 10.8, 6.9, 1.2 Hz, 1H), 5.14 (ddd, J = 11.0, 9.5, 1.8 Hz, 1H), 3.78 (td, J = 5.8, 2.9 Hz, 1H), 2.99 (dddd, J = 11.8, 6.5, 5.4, 4.2 Hz, 1H), 2.70 (ddd, J = 14.1, 5.4, 1.6 Hz, 1H), 2.61 (dddd, J = 14.3, 9.9, 5.9, 1.7 Hz, 1H), 2.25 (dddd, J = 14.2, 6.9, 5.3, 1.9 Hz, 1H), 2.18 – 1.98 (m, 2H), 1.85 (dqd, J = 13.7, 5.9, 1.3 Hz, 1H), 1.62 (dd, J = 6.9, 1.8 Hz, 3H), 0.90 (s, 9H), 0.09 (d, J = 2.3 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 211.4, 130.6, 126.3, 70.4, 43.1, 42.9, 37.4, 31.7, 25.9, 18.1, 13.3 −4.5. HRMS calculated for C15H28O2SiNa+ 291.1756 Found 291.1745 (MNa+).
(3R,4S)-4-hydroxy-3-((Z)-prop-1-en-1-yl)cyclohexan-1-one (9)
48% aq. HF (4 mL) was added dropwise to a solution of compound 8 (500 mg, 1.86 mmol) in acetonitrile (40 mL) (Note: teflon tube) at 0 °C. The resulting solution was stirred at room temperature for 16 h. After 16 h, the reaction mixture was cooled to 0 °C and quenched with saturated NaHCO3 (20 mL). The reaction mixture was transferred to a separatory funnel. The organic layer was removed, and the aqueous layer was extracted with DCM (3 × 20 mL). The combined organic layers were washed with brine solution (2 × 20 mL), dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. The residue was purified through silica gel column chromatography (hexane/ethyl acetate = 60:40) to afford 250 mg (1.62 mmol, 88%) of 9 as a white solid. [α]D = +124 (c 0.5, CHCl3); FT-IR (KBr): 3416, 3017, 2825, 1705, 1420 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.74 (dqd, J = 10.8, 6.9, 1.0 Hz, 1H), 5.22 (ddt, J = 12.6, 9.7, 1.8 Hz, 1H), 3.75 (ddd, J = 10.1, 8.9, 3.9 Hz, 1H), 2.88 – 2.77 (m, 1H), 2.52 – 2.40 (m, 2H), 2.40 – 2.35 (m, 1H), 2.32 – 2.26 (m, 1H), 2.22 (ddd, J = 14.7, 11.8, 1.1 Hz, 1H), 1.86 – 1.76 (m, 2H), 1.68 (dd, J = 6.9, 1.8 Hz, 3H).; 13C NMR (101 MHz, CDCl3) δ 209.4, 130.1, 128.9, 71.6, 44.1, 43.6, 38.7, 32.0, 13.5. HRMS calculated for C9H14O2Na+ 177.0892 Found 177.0896 (MNa+).
(1S,2R)-4-oxo-2-((Z)-prop-1-en-1-yl)cyclohexyl sulfamate (10)
Formic acid (61 μL, 1.62 mmol, 2.5 equiv.) was added to neat Chlorosulfonyl isocyanate (140 μL, 1.62 mmol, 2.5 equiv.) at 0 °C and the mixture was stirred for 5 minutes. Vigorous gas evolution was observed during the addition, and the mixture turned into a white solid after 5 to 10 minutes. Anhydrous acetonitrile (2 mL) was added to the solid. The resulting solution was stirred at room temperature for 12 h. After 12 h, the reaction mixture was cooled to 0°C, and alcohol 9 (100 mg, 0.649 mmol) dissolved in 0.5 mL of DMA was added dropwise using a syringe. The resulting solution was stirred at room temperature for 5 h. The reaction was quenched with ice water (10 mL). The reaction mixture was transferred to a separatory funnel, the organic layer was separated, and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were washed with brine solution (2 × 10 mL), dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. The residue was purified through silica gel column chromatography (hexane/ethyl acetate = 50:50) to afford 82 mg (54%) of 10 as a colorless oil. [α]D = +18.5 (c 0.4, CHCl3); FT-IR (KBr): 3260, 2910, 1710, 1210 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.65 (dqd, J = 10.7, 6.9, 1.2 Hz, 1H), 5.20 (ddq, J = 11.0, 9.3, 1.8 Hz, 1H), 4.97 (s, 2H), 4.70 (td, J = 6.4, 3.1 Hz, 1H), 3.42 – 3.30 (m, 1H), 2.74 – 2.53 (m, 2H), 2.45 – 2.28 (m, 2H), 2.22 (tddd, J = 13.8, 7.3, 4.7, 1.4 Hz, 2H), 1.68 (dd, J = 6.9, 1.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 208.6, 128.7, 128.1, 81.1, 42.9, 40.1, 37.1, 28.8, 13.3. HRMS calculated for C9H15NO4SNa+ 256.0620 Found 256.0617 (MNa+).
(1R,2R)-4-oxo-2-((Z)-prop-1-en-1-yl)cyclohexyl 3,5-dinitrobenzoate (12)
A mixture of 9 (0.4 g, 2.59 mmol), PPh3 (3.4 g, 12.98 mmol, 5 equiv.) and 3,5-dinitro-benzoic acid (2.75 g, 12.98 mmol, 5 equiv.) in dry toluene (40 mL) was stirred at room temperature for 30 min under N2 atmosphere. After 30 min, diethyl azo-dicarboxylate (2 mL, 12.74 mmol, 5 equiv.) was added drop-wise to the reaction mixture at 0 °C. The resulting mixture was stirred at room temperature for 24 h under N2 atmosphere. After 24 h, the reaction mixture was concentrated under reduced pressure. The residue was taken in 20% EtOAc/hexanes (200 mL) and filtered through a celite pad. The filtrate was concentrated under reduced pressure. The resulting residue was purified through silica gel column chromatography (hexane/ethyl acetate = 85:15) to afford 588 mg (1.69 mmol, 65%) of 12 as a white solid. [α]D = −91.0 (c 0.5, CHCl3); FT-IR (KBr): 3110, 3023, 1712, 1719, 1460 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.26 (t, J = 2.1 Hz, 1H), 9.14 (d, J = 2.2 Hz, 2H), 5.68 – 5.59 (m, 1H), 5.52 (dt, J = 5.8, 3.1 Hz, 1H), 5.37 (ddt, J = 12.7, 9.3, 1.8 Hz, 1H), 3.30 (tt, J = 9.4, 4.5 Hz, 1H), 2.69 – 2.52 (m, 3H), 2.47 (dddd, J = 14.8, 13.3, 8.1, 3.5 Hz, 2H), 2.17 (ddt, J = 16.3, 9.6, 3.9 Hz, 1H), 1.65 (dd, J = 6.9, 1.8 Hz, 3H); 13C NMR (126 MHz, CDCl3): δ 208.2, 162.1, 148.9, 133.9, 129.4, 127.9, 127.3, 122.8, 74.2, 43.3, 38.6, 36.5, 28.9, 13.4; HRMS calculated for C16H16N2O7Na+ 371.0855 Found 371.0855 (MNa+).
(3R,4R)-4-hydroxy-3-((Z)-prop-1-en-1-yl)cyclohexan-1-one (13)
A mixture of 12 (0.350 g, 1 mmol) and K2CO3 (207 mg, 1.5 mmol, 1.5 equiv.) in dry methanol (10 mL) was stirred at room temperature for 1 h. After 1 h, the solvent was evaporated under reduced pressure. The resulting residue was dissolved in DCM and extracted with 1 N HCl (30 mL). The organic layer was separated, and the aqueous layer was extracted with DCM (2 × 40 mL). The combined organic layers were washed with brine solution (2 × 20 mL), dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. The residue was purified through silica gel column chromatography (hexane/ethyl acetate = 1:1) to afford 134 mg (0.87 mmol, 87%) of 13 as a colorless oil. [α]D = +26.4 (c 0.5, CHCl3); FT-IR (KBr): 3450, 3012, 2958, 2886, 1717, 1471 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.60 (ddddd, J = 10.8, 8.1, 6.8, 5.6, 1.3 Hz, 1H), 5.42 (ddq, J = 11.0, 9.2, 1.8 Hz, 1H), 4.02 (dt, J = 4.7, 2.7 Hz, 1H), 2.99 – 2.89 (m, 1H), 2.71 – 2.54 (m, 2H), 2.29 – 2.17 (m, 2H), 2.12 (ddt, J = 14.7, 5.9, 4.4 Hz, 1H), 2.03 (d, J = 9.6 Hz, 1H), 1.91 (tdd, J = 13.8, 5.2, 2.6 Hz, 1H), 1.62 (dt, J = 6.9, 1.5 Hz, 3H);13C NMR (101 MHz, CDCl3) δ 211.3, 129.3, 126.9, 68.3, 42.4, 40.8, 36.0, 32.4, 13.4; HRMS calculated for C9H14O2Na+ 177.0892 Found 177.0894 (MNa+).
(1R,2R)-4-oxo-2-((Z)-prop-1-en-1-yl)cyclohexyl sulfamate (14)
Formic acid (61 μL, 1.62 mmol, 2.5 equiv.) was added to neat chlorosulfonyl isocyanate (140 μL, 1.62 mmol, 2.5 equiv.) at 0 °C and the mixture was stirred for 5 minutes. Vigorous gas evolution was observed during the addition, and the mixture turned into a white solid after 5–10 min. Anhydrous acetonitrile (2 mL) was added to the solid. The resulting solution was stirred at room temperature for 12 h. After 12 h, the reaction mixture was cooled to 0 °C, and alcohol 13 (100 mg, 0.65 mmol) dissolved in 0.5 mL of DMA was added dropwise using a syringe. The resulting solution was stirred at room temperature for 5 h. The reaction was quenched with ice water (10 mL) and transferred to a separatory funnel. The organic layer was separated, and then the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were washed with brine solution (2 × 10 mL), dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. The residue was purified through silica gel column chromatography (hexane/ethyl acetate = 1:1) to afford 82 mg (0.35 mmol, 54%) of 14 as a colorless oil. [α]D = −38.2 (c 0.5, CHCl3); FT-IR (KBr): 3264, 2926, 1708, 1180cm−1; 1H NMR (400MHz, CDCl3) δ 5.63 (dq, J = 10.7, 6.9 Hz, 1H), 5.45 (ddq, J = 10.8, 9.0, 1.8 Hz, 1H), 4.94 (d, J = 6.8 Hz, 2H), 4.83 (d, J = 2.4 Hz, 1H), 3.13 – 3.01 (m, 1H), 2.78 – 2.49 (m, 3H), 2.33 (dddd, J = 29.9, 14.8, 4.9, 2.3 Hz, 2H), 2.00 (dddd, J = 14.8, 12.9, 6.2, 2.3 Hz, 1H), 1.65 (dd, J = 6.9, 1.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 209.0, 128.1, 127.2, 80.3, 42.4, 39.5, 35.6, 30.0,13.3; HRMS calculated for C9H15NO4SNa+ 256.0620 Found 256.0618 (MNa+).
(1R,2R)-4-oxo-2-((Z)-prop-1-en-1-yl)cyclohexyl(4-methoxyphenyl)sulfamate (16)
A mixture of 13 (0.1 g, 0.649 mmol) and (4-methoxyphenyl)sulfamoyl chloride (214 mg, 0.973 mmol, 1.5 equiv.) in dry DCM (3 mL) was added NEt3 (0.135 mL, 0.973 mmol, 1.5 equiv.) at 0 °C. The reaction mixture was stirred at room temperature for 12 h. Then, the reaction was quenched with cold water (20 mL). The mixture was transferred to a separatory funnel, the organic layer was separated, and then the aqueous layer was extracted with DCM (2 × 15 mL). The combined organic layers were washed with brine solution (15 mL), dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. The residue was purified through silica gel column chromatography (hexane/ethyl acetate = 70:30) to afford 82 mg (37%) of 16 as a semi solid. [α]D = −41.2 (c 0.5, CHCl3); FT-IR (KBr): 3288, 2965, 1708, 1511 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.22 – 7.16 (m, 2H), 6.91 – 6.85 (m, 2H), 6.63 (s, 1H), 5.47 (dqd, J = 10.8, 6.9, 1.1 Hz, 1H), 5.25 (ddq, J = 10.8, 9.0, 1.8 Hz, 1H), 4.80 (q, J = 2.7 Hz, 1H), 3.80 (s, 3H), 2.98 (td, J = 8.9, 7.3, 3.9 Hz, 1H), 2.54 – 2.34 (m, 3H), 2.24 (ddd, J = 15.0, 5.3, 3.2 Hz, 2H), 1.95 – 1.80 (m, 1H), 1.57 (dd, J = 6.9, 1.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 208.7, 158.0, 128.6, 127.7, 126.8, 124.1, 114.7, 80.6, 55.5, 42.3, 39.5, 35.4, 29.8, 13.1; HRMS calculated for C16H20NO5S− 338.1068 Found 338.1010 (M-H).
General procedure for sulfamate-tethered aza-Wacker cyclization
A 10 mL microwave vial containing a magnetic stir-bar was charged with sulfamate [10, 14 or 16] (0.1 mmol), Pd(OAc)2 (0.04 mmol, 0.4 equiv.), and Cu(OAc)2 (0.1 mmol, 1 equiv.) in 2 mL of acetonitrile (final concentration 0.05 M). The reaction vial was sealed and then evacuated and backfilled with oxygen three times. The reaction vessel was then submerged in an oil bath preheated to 55 °C and kept at this temperature under a balloon of O2 (~1 atm) for 16 h. After 16 h, the reaction mixture was cooled to room temperature and filtered through a small plug of silica. The silica plug was further rinsed with DCM (10 mL). The solvent was removed under reduced pressure. The resulting residue was purified with silica gel chromatography (gradient of 20 to 30% ethyl acetate in hexanes) to afford the cyclized product [11, 15 and 17] in pure form.
(4R, 4aR,8aS)-4-vinylhexahydrobenzo[e][1,2,3]oxathiazin-6 (5H)-one 2,2-dioxide (11)
Following the general procedure, compound 10 (23 mg, 0.1 mmol) furnished cyclized product 11 (11.5 mg, 0.05 mmol, 50%) as an off white solid. [α]D = + 12.11 (c 1, CHCl3); FT-IR (KBr): 3260, 2910, 1710, 1210 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.69 – 5.56 (m, 1H), 5.44 – 5.35 (m, 2H), 4.88 (ddd, J = 11.6, 10.4, 4.1 Hz, 1H), 4.50 (d, J = 9.3 Hz, 1H), 4.08 – 3.96 (m, 1H), 2.63 – 2.43 (m, 3H), 2.40 (dddd, J = 14.9, 6.6, 4.0, 2.3 Hz, 1H), 2.14 – 2.02 (m, 1H), 2.05 – 1.83 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 205.4, 131.9, 121.7, 82.9, 62.1, 42.2, 40.4, 38.6, 30.2; HRMS calculated for C9H12NO4S− 230.0487 Found 230.0461 (M-H).
(4S,4aR,8aR)-4-vinylhexahydrobenzo[e][1,2,3]oxathiazin-6 (5H)-one 2,2-dioxide (15)
Following the general procedure, compound 10 (46 mg, 0.2 mmol) furnished cyclized product 11 (13.6 mg, 0.06 mmol, 30%) as an off white solid [α]D = +10.0 (c 0.35, CHCl3); FT-IR (KBr): 3264, 2926, 1708, 1180 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.31 (ddd, J = 17.2, 10.6, 6.3 Hz, 1H), 5.38 – 5.28 (m, 2H), 5.22 (q, J = 2.9 Hz, 1H), 5.02 (d, J = 6.0 Hz, 1H), 3.82 – 3.70 (m, 1H), 2.92 – 2.79 (m, 1H), 2.73 – 2.60 (m, 1H), 2.49 (dddd, J = 15.0, 6.2, 3.7, 2.3 Hz, 1H), 2.44 – 2.31 (m, 2H), 2.25 – 2.12 (m, 1H), 2.03 (dddd, J = 15.0, 13.8, 5.1, 2.7 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 207.7, 134.9, 118.8, 77.5, 61.4, 40.4, 38.8, 35.4, 30.0; HRMS calculated for C9H12NO4S− 230.0487 Found 230.0476 (M-H).
(4S,4aR,8aR)-3-(4-methoxyphenyl)-4-vinylhexahydrobenzo[e][1,2,3]oxathiazin-6(5H)-one-2,2-dioxide (17)
Following the general procedure, compound 10 (34 mg, 0.1 mmol) furnished cyclized product 17 (22 mg, 0.065 mmol, 65%) as an off-white solid. [α]D = +18.6 (c 0.5, CHCl3); FT-IR (KBr): 2954, 2922, 1708, 1509, 1169 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.26 – 7.23 (m, 2H), 6.91 – 6.86 (m, 2H), 6.69 (ddd, J = 17.1, 10.3, 9.0 Hz, 1H), 5.45 (d, J = 3.1 Hz, 1H), 5.32 – 5.27 (m, 1H), 5.05 – 4.99 (m, 1H), 3.80 (s, 3H), 3.72 – 3.64 (m, 1H), 3.29 (t, J = 13.8 Hz, 1H), 2.76 (td, J = 14.4, 6.5 Hz, 1H), 2.62 – 2.36 (m, 3H), 2.31 – 2.23 (m, 1H), 2.05 (tdd, J = 14.3, 5.1, 2.5 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 207.8, 159.7, 133.8, 131.9, 129.7, 120.7, 114.9, 77.9, 72.2, 55.6, 41.9, 41.0, 35.4, 30.0. HRMS calculated for C16H18NO5S− 3636.0911 Found 336.0872 (M-H).
Docking studies
The structure of Bactobolin A bound to the T. thermophilus 70S ribosome-tRNA complex [15] was downloaded from the PDB (PDB ID 4WT8). Because of the size of the complex relative to the size of the interaction site, no residues/bases within 10 Å of Bactobolin were removed, leaving the 23S rRNA, the tRNA, the L2 protein subunit, and the L27 protein subunit. The resulting reduced complex was prepared for modeling using Schrodinger’s Protein Preparation Wizard, which adds hydrogens, removes H2O molecules, optimizes hydrogen bonds, and performs a constrained minimization [37]. A dockable “receptor” was created and centered on Bactobolin A, with a metal coordination constraint with the magnesium ion. Ligands were prepared using LigPrep, which calculates potential ionization and tautomerization states [37], and these were docked using Glide [38-40] with XP precision. In these docking studies, a core constraint of the fused ring substructure and a metal binding constraint were applied.
Supplementary Material
Acknowledgements
This work was supported by start-up funding provided jointly by the University of Kansas Office of the Provost and the Department of Medicinal Chemistry as well as an NIH COBRE Chemical Biology of Infectious Diseases Research Project Grant to S. S. (P20GM113117). We thank Dr. Victor Day (University of Kansas) for X-ray crystallography analysis. Funding for the X-ray dif-fractometer was provided by an NSF-MRI grant (CHE-0923449).
Footnotes
Conflict of interest The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1007/s00044-021-02724-7.
References
- 1.Baran PS. Natural product total synthesis: as exciting as ever and here to stay. J Am Chem Soc. 2018;140:4751–5. 10.1021/jacs.8b02266. [DOI] [PubMed] [Google Scholar]
- 2.Denmark SE. Organic synthesis: wherefrom and whither? (Some very personal reflections). Isr J Chem. 2018;58:61–72. 10.1002/ijch.201700085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wender PA. Toward the ideal synthesis and molecular function through synthesis-informed design. Nat Prod Rep. 2014;31:433–40. 10.1039/C4NP00013G. [DOI] [PubMed] [Google Scholar]
- 4.Shinde AH, Sathyamoorthi S. Oxidative cyclization of sulfamates onto pendant alkenes. Org Lett. 2020;22:896–901. 10.1021/acs.orglett.9b04448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shinde AH, Nagamalla S, Sathyamoorthi S. N-arylated oxathiazinane heterocycles are convenient synthons for 1,3-amino ethers and 1,3-amino thioethers. Medicinal Chem Res. 2020;29:1223–9. 10.1007/s00044-020-02556-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas AA, Nagamalla S, Sathyamoorthi S. Salient features of the aza-Wacker cyclization reaction. Chem Sci. 2020;11:8073–88. 10.1039/D0SC02554B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shinde AH, Sathyamoorthi S. Tethered silanoxymercuration of allylic alcohols. Org Lett. 2020;22:8665–9. 10.1021/acs.orglett.0c03257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondo S, Horiuchi Y, Hamada M, Takeuchi T, Umezawa H. A new antitumor antibiotic, bactobolin produced by pseudomonas. J Antibiot. 1979;32:1069–71. [DOI] [PubMed] [Google Scholar]
- 9.Chandler JR, Truong TT, Silva PM, Seyedsayamdost MR, Carr G, Radey M, et al. Bactobolin resistance is conferred by mutations in the L2 ribosomal protein. mBio. 2012;3:e00499–12. 10.1128/mBio.00499-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seyedsayamdost MR, Chandler JR, Blodgett JAV, Lima PS, Duerkop BA, Oinuma K-I, et al. Quorum-sensing-regulated bactobolin production by Burkholderia thailandensis E264. Org Lett. 2010;12:716–9. 10.1021/ol902751x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klaus JR, Coulon PML, Koirala P, Seyedsayamdost MR, Déziel E, Chandler JR. Secondary metabolites from the Burkholderia pseudomallei complex: structure, ecology, and evolution. J Ind Microbiol Biotechnol. 2020;47:877–87. 10.1007/s10295-020-02317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munakata T, Okumoto T. Some structure-activity relationships for bactobolin analogs in the treatment of mouse leukemia P388. Chem Pharm Bull. 1981;29:891–4. 10.1248/cpb.29.891. [DOI] [PubMed] [Google Scholar]
- 13.Okumoto T, Kontani M, Hoshino H, Nakanishi M. Antitumor activity of newly isolated antibiotics, 3-dichloromethylactinobolins. J Pharmacobio-Dyn. 1980;3:177–82. 10.1248/bpb1978.3.177. [DOI] [PubMed] [Google Scholar]
- 14.Greenberg EP, Chandler JR, Seyedsayamdost MR. The chemistry and biology of bactobolin: a 10-year collaboration with natural product chemist extraordinaire jon clardy. J Nat Prod. 2020;83:738–43. 10.1021/acs.jnatprod.9b01237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amunts A, Fiedorczuk K, Truong TT, Chandler J, Greenberg EP, Ramakrishnan V. Bactobolin A binds to a site on the 70S ribosome distinct from previously seen antibiotics. J Mol Biol. 2015;427:753–5. 10.1016/j.jmb.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garigipati RS, Tschaen DM, Weinreb SM. Stereoselective total syntheses of the antitumor antibiotics (+)-actinobolin and (−)-bactobolin from a common bridged lactone intermediate. J Am Chem Soc. 1990;112:3475–82. 10.1021/ja00165a035. [DOI] [Google Scholar]
- 17.Weinreb SM, Garigipati RS. Design of an efficient strategy for total synthesis of the microbial metabolite (−)-bactobolin. Pure Appl Chem. 1989;61:435 [Google Scholar]
- 18.Garigipati RS, Weinreb SM. Stereoselective total synthesis of the antitumor antibiotic (−)-bactobolin. J Org Chem. 1988;53:4143–5. 10.1021/jo00252a060. [DOI] [Google Scholar]
- 19.Vojackova P, Michalska L, Necas M, Shcherbakov D, Bottger EC, Sponer J, et al. Stereocontrolled synthesis of (−)-Bactobolin A. J Am Chem Soc. 2020;142:7306–11. 10.1021/jacs.0c01554. [DOI] [PubMed] [Google Scholar]
- 20.Askin D, Angst C, Danishefsky S. An approach to the synthesis of bactobolin and the total synthesis of N-acetylactinobolamine: some remarkably stable hemiacetals. J Org Chem. 1987;52:622–35. 10.1021/jo00380a025. [DOI] [Google Scholar]
- 21.Ward DE, Gai Y, Kaller BF.Synthetic studies on actinobolin and bactobolin: synthesis of N-Desalanyl-N-[2-(trimethylsilyl)ethanesulfonyl] derivatives from a common intermediate and attempted removal of the SES protecting group.J Organic Chem.1996;61:5498–505. 10.1021/jo960579c. [DOI] [Google Scholar]
- 22.Ward DE, Gai Y, Kaller BF. Synthesis of (−)-bactobolin from D-glucose and from (+)-actinobolin. Tetrahedron Lett. 1994;35:3485–8. 10.1016/S0040-4039(00)73216-3. [DOI] [Google Scholar]
- 23.Underwood R, Fraser-Reid B. Support studies for the conversion of actinobolin into bactobolin. J Chem Soc Perkin Trans 1. 1990:731–8. 10.1039/P19900000731. [DOI] [Google Scholar]
- 24.Adachi H, Nishimura Y, Takeuchi T. Synthesis and activities of bactobolin derivatives having new functionality at C-3. J Antibiot. 2009;55:92–8. [DOI] [PubMed] [Google Scholar]
- 25.Adachi H, Nishimura Y. Synthesis and biological activity of bactobolin glucosides. Nat Prod Res. 2003;17:253–7. 10.1080/1057563021000060112. [DOI] [PubMed] [Google Scholar]
- 26.Adachi H, Usui T, Nishimura Y, Kondo S, Ishizuka M, Takeuchi T. Synthesis and activities of bactobolin derivatives based on the alteration of the functionality at C-3 position. J Antibiot. 1998;51:184–8. [DOI] [PubMed] [Google Scholar]
- 27.Kotov V, Scarborough CC, Stahl SS. Palladium-catalyzed aerobic oxidative amination of alkenes: development of intra- and intermolecular aza-Wacker reactions. Inorg Chem. 2007;46:1910–23. 10.1021/ic061997v. [DOI] [PubMed] [Google Scholar]
- 28.Wang D, Weinstein AB, White PB, Stahl SS. Ligand-promoted palladium-catalyzed aerobic oxidation reactions. Chem Rev. 2018;118:2636–79. 10.1021/acs.chemrev.7b00334. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z-X, Miller SM, Anderson OP, Shi Y. A class of C2 and pseudo C2 symmetric ketone catalysts for asymmetric epoxidation. Conformational effect on catalysis. J Org Chem. 1999;64:6443–58. 10.1021/jo9908849. [DOI] [Google Scholar]
- 30.Kawashima H, Sakai M, Kaneko Y, Kobayashi Y. Further study on synthesis of the cyclobakuchiols. Tetrahedron. 2015;71:2387–92. 10.1016/j.tet.2015.02.092. [DOI] [Google Scholar]
- 31.Espino CG, Wehn PM, Chow J, Du Bois J. Synthesis of 1,3-difunctionalized amine derivatives through selective C─H bond oxidation. J Am Chem Soc. 2001;123:6935–6. 10.1021/ja011033x. [DOI] [Google Scholar]
- 32.Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. Mitsunobu and related reactions: advances and applications. Chem Rev. 2009;109:2551–651. 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- 33.Takaya J, Kagoshima H, Akiyama T. Mannich-type reaction with trifluoromethylated N,O-hemiacetal: facile preparation of β-amino-β-trifluoromethyl carbonyl compounds. Org Lett. 2000;2:1577–9. 10.1021/ol005812e. [DOI] [PubMed] [Google Scholar]
- 34.Fustero S, Soler JG, Bartolomé A, Roselló MS. Novel approach for asymmetric synthesis of fluorinated β-amino sulfones and allylic amines. Org Lett. 2003;5:2707–10. 10.1021/ol034892u. [DOI] [PubMed] [Google Scholar]
- 35.Kronenthal DR, Han CY, Taylor MK. Oxidative N-dearylation of 2-azetidinones. p-Anisidine as a source of azetidinone nitrogen. J Org Chem. 1982;47:2765–8. 10.1021/jo00135a016. [DOI] [Google Scholar]
- 36.Ha D-C, Hart DJ. Syntheses of methyl N-benzoylacosaminide and methyl N-(benzyloxyoxalyl)-daunosaminide from (S)-ethyl 3-hydroxybutyrate. Tetrahedron Lett. 1987;28:4489–92. 10.1016/S0040-4039(00)96544-4. [DOI] [Google Scholar]
- 37.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–34. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 38.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 39.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein—ligand complexes. J Med Chem. 2006;49:6177–96. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 40.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–9. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.