

Abstract
Iodide-bound ruthenium-JOSIPHOS complexes catalyze the redox-neutral C-C coupling of primary alcohols 2a-2r with the gaseous allene (propadiene) 1a to form enantiomerically enriched homoallylic alcohols 3a-3r with complete atom-efficiency. Using formic acid as reductant, aldehydes dehydro-2a and dehydro-2c participate in reductive C-C coupling with allene to deliver adducts 3a and 3c with comparable levels of asymmetric induction. Deuterium labeling studies corroborate a mechanism in which alcohol dehydrogenation triggers allene hydroruthenation to form transient allylruthenium-aldehyde pairs that participate in carbonyl addition. Notably, due to a kinetic preference for primary alcohol dehydrogenation, chemoselective C-C coupling of 1°,2°-1,3-diols occurs in the absence of protecting groups. As illustrated by the synthesis of C7-C15 of spirastrellolide B and F (7 vs 17 steps), C3-C10 of cryptocarya diacetate (3 vs 7 or 9 steps), and a fragment common to C8′-C14′ of mycolactone F (1 vs 4 steps) and C22-C28 marinomycin A (1 vs 9 steps), this capability streamlines type I polyketide construction.
Keywords: Ruthenium, allylation, enantioselective, alcohol dehydrogenation, polyketide, green chemistry, allene
Graphical Abstract
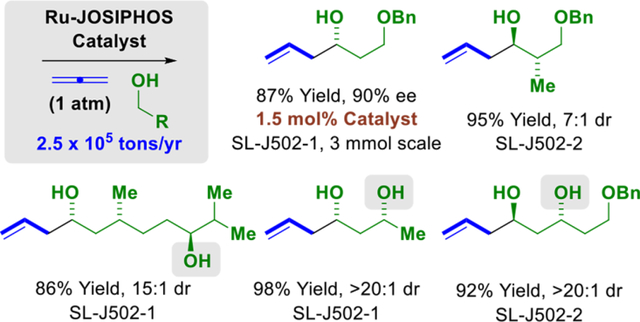
Introduction
Polyketide natural products play a prominent role in human and veterinary medicine, as well as crop protection.1 Manufacturing routes to commercial polyketides and their semi-synthetic congeners nearly all rely on fermentation.2 De novo chemical synthesis potentially offers entry to otherwise inaccessible polyketide derivatives, yet despite advances in acyclic stereocontrol, especially in the area of aldol addition3 and carbonyl allylation,4 the classical lexicon of synthetic methods often does not deliver concise/scalable routes to these structurally complex compounds.2 Aiming to address this deficiency, our laboratory has developed a family of catalytic C-C couplings for the enantioselective conversion of lower alcohols to higher alcohols via hydrogen auto-transfer and related metal-catalyzed carbonyl reductive couplings mediated by 2-propanol.5,6 Of relevance to polyketide construction, these processes encompass iridium-catalyzed allylations, crotylations and propargylations.5a
Motivated by the prospect of exploiting a more abundant and cost-effective metal catalyst, our laboratory developed a new class of iodide-bound JOSIPHOS complexes.7 These catalysts affect highly enantioselective couplings of primary alcohols with 1-aryl-1-propynes (to furnish products of carbonyl α-aryl-allylation7a or 2-butyne (to furnish tiglyl alcohols)7b and, most recently, reactions of 1,2- and 1,3-butadiene (to furnish products of carbonyl crotylation).7c The latter transformation suggests the feasibility of exploiting gaseous allene (propadiene) as a pronucleophile for the allylation of alcohol proelectrophiles. A racemic iridium-catalyzed reaction of this type was first reported by our laboratory in 2007.8a Enantioselective allene-mediated reductive allylations of aldehydes8b and ketones8c catalyzed by chiral iridium and copper complexes were disclosed in 2019 (Figure 1).
Figure 1.

The importance of carbonyl allylation vis-à-vis polyketide construction and allene as an allylmetal pronucleophile.
Here, we report the first ruthenium-catalyzed reactions of primary alcohols with gaseous allene to form homoallylic alcohols. Unlike enantioselective allene-mediated carbonyl allylations catalyzed by iridium8b or copper,8c the present ruthenium-catalyzed processes are applicable to primary alcohol proelectrophiles. Furthermore, due to a kinetic preference for primary alcohol dehydrogenation, chemoselective C-C coupling of 1°,2°-diols occurs in the absence of protecting groups.9 The impact of this capability on the efficiency of polyketide construction was explored, as illustrated in preparations of several previously described polyketide fragments. Specifically, C7-C15 of spirastrellolide B and F (7 vs 17 steps),10a C3-C10 of cryptocarya diacetate (3 vs 7 or 9 steps),10b,c C8′-C14′ of mycolactone F (1 vs 4 steps),10d and C22-C28 of marinomycin A (1 vs 9 steps)10e were each made in significantly fewer steps than previously possible. These data, along with prior work from our laboratory,5 highlight how abundant π-unsaturated petrochemical feedstocks can function as surrogates to stoichiometric organometallic reagents, unlocking pathways for chemical synthesis of greater step- and atom-efficiency.5b,11
Results and Discussion
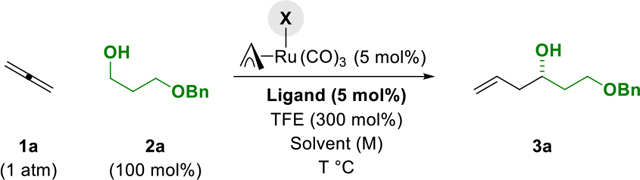
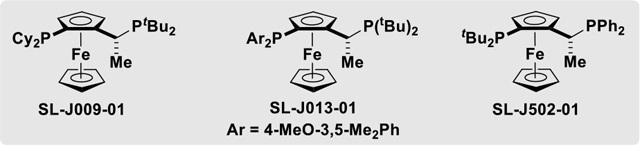
Our optimization experiments began with conditions identified for the reaction of primary alcohols with 1,2- and 1,3-butadiene (to furnish products of carbonyl crotylation).7c Thus, a pressure tube charged with alcohol 2a, the iodide-containing complex RuI(CO)3(η3-C3H5) (5 mol%), the JOSIPHOS ligand SL-J009-01 (5 mol%), trifluoroethanol (TFE) (300 mol%) (which is inert to ruthenium-catalyzed dehydrogenation)12 and and THF (0.5 M) was back-filled with gaseous allene 1a, sealed and placed in a 100 °C oil bath. To our surprise, only trace quantities of the targeted homoallylic alcohol 3a were observed (Table 1, entry 1). By increasing the reaction temperature to 110 °C, alcohol 3a could be formed in 20% yield and 74% ee (Table 1, entry 2). In an effort to improve the yield of 3a and its level of enantiomeric enrichment, the JOSIPHOS ligands SL-J013-01 and SL-J502-01 were evaluated (Table 1, entries 3, 4). The JOSIPHOS SL-J502-01 proved to be superior, providing the alcohol 3a in 75% yield and 85% ee (Table 1, entry 4). As revealed by a concise screening of solvent (Table 1, entries 4–7), reactions run in 1,2-diethoxyethane (DEE) at 0.1 M concentration enabled access to alcohol 3a in 86% yield and 90% ee (Table 1, entry 7). In the absence of TFE, the yield of alcohol 3a was dramatically diminished (Table 1, entry 8). Finally, as revealed in reactions of the corresponding chloride- and bromide-bound ruthenium catalysts (Table 1, entries 9, 10), iodide counterions are essential for maintaining optimal yields and enantioselectivities.7a,13
Table 1.
Selected optimization experiments in the ruthenium-JOSIPHOS-catalyzed C-C coupling of gaseous allene 1a with alcohol 2a to form homoallylic alcohol 3a.a
| |||||
|---|---|---|---|---|---|
| Entry | T °C | X | Ligand | Solvent | Yield, ee |
|
| |||||
| 1 | 100 | I | SL-J009-01 | THF (0.5 M) | Trace |
| 2 | 110 | I | SL-J009-01 | THF (0.5 M) | 20%, 74% |
| 3 | 110 | I | SL-J013-01 | THF (0.5 M) | 65%, 69% |
| 4 | 110 | I | SL-J502-01 | THF (0.5 M) | 75%, 85% |
| 5 | 110 | I | SL-J502-01 | MTBE (0.5 M) | 78%, 87% |
| 6 | 110 | I | SL-J502-01 | DEE (0.5 M) | 88%, 88% |
|
|
110 | I | SL-J502-01 | DEE (0.1 M) | 86%, 90% |
| 8b | 110 | I | SL-J502-01 | DEE (0.1 M) | 42%, 90% |
| 9 | 110 | CI | SL-J502-01 | DEE (0.1 M) | Trace |
| 10 | 110 | Br | SL-J502-01 | DEE (0.1 M) | 29%, 75% |
| |||||
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by HPLC analysis.
TFE was omitted. See Supporting Information for further details.
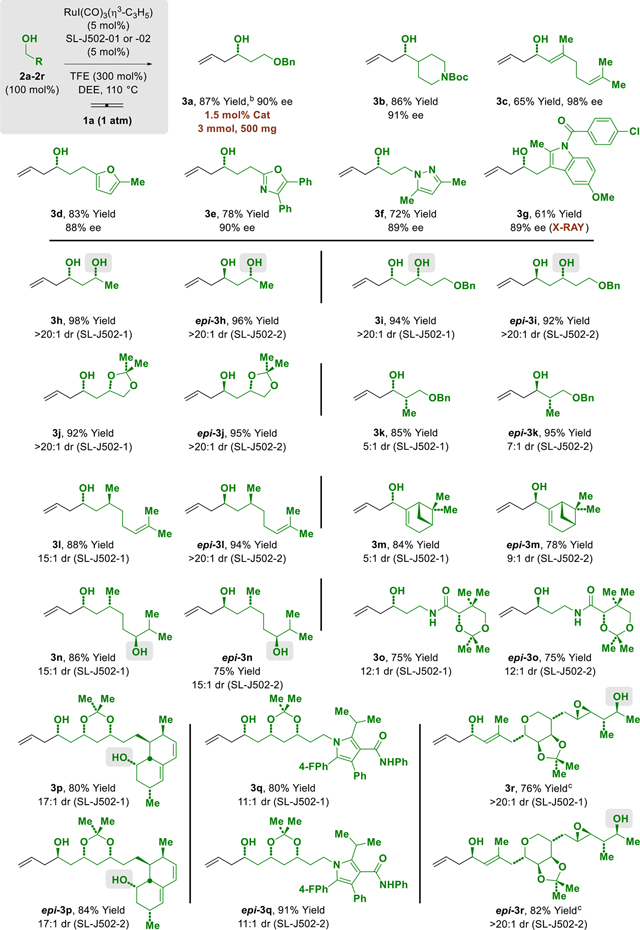
Optimal conditions identified for the ruthenium-JOSIPHOS-catalyzed C-C coupling of gaseous allene 1a were applied to alcohols 2a-2g (Table 2). The resulting homoallylic alcohols 3a-3g were formed in good to excellent yields and enantioselectivities. As demonstrated by the formation of 3b, primary alcohols with adjacent branched alkyl groups are tolerated. Geraniol 2c is converted to adduct 3c bearing 3 olefinic functional groups without competing alkene reduction or isomerization.14 Heteroaromatic moieties are compatible with the conditions for catalytic C-C coupling, as demonstrated by the formation of adducts 3d-3g. Finally, it is most notable that the reaction of 2a at 3 mmol scale could be conducted at a catalyst loading of 1.5 mol% without any erosion in yield or enantioselectivity. The absolute stereochemical assignment of these adducts is made in analogy to that determined for compound 3g, which was established by single crystal X-ray diffraction analysis.
Table 2:
Ruthenium-JOSIPHOS-catalyzed C-allylation of primary alcohols 2a-2r mediated by gaseous allene.a
|
Yields of material isolated by silica gel chromatography. Enantioselectivities determined by HPLC analysis. Diastereoselectivities determined by 1H NMR analysis of crude reaction mixtures.
Ru-ligand (1.5 mol%).
Ru-ligand (10 mol%), 80 °C. See Supporting Information for further details.
To further explore the scope of this process, the reaction of alcohols 2h-2r, which contain preexisting stereogenic centers and, in many cases, unprotected secondary hydroxyl groups, were explored (Table 2). Remarkably, exposure of (R)-1,3-butane diol 2h to the optimized conditions for allene-mediated allylation delivered the homoallylic alcohol 3h as single diastereomer in 98% yield. Use of the enantiomeric catalyst provided epi-3h in 96% yield as single diastereomer, thus revealing high levels of catalyst directed diastereoselectivity. Of direct relevance to polyketide total synthesis, the “Roche alcohol”15 2k could be converted to adducts 3k and epi-3k upon exposure to the enantiomeric ruthenium-JOSIPHOS catalysts without any epimerization of the methyl-bearing stereocenter, as determined by HPLC.16 Compared to epi-3k, diastereoselectivity associated with the formation of adduct 3k is lower, as the diastereofacial bias of the catalyst opposes the intrinsic Felkin-Anh selectivity of the aldehyde.17 The reactions of alcohol 2r (derived from pseudomonic acid A) to form 3r and epi-3r, again underscore the applicability of this method to polyketide construction.
The ability to affect enantioselective allylation from the alcohol or carbonyl oxidation levels would lend further flexibility to this method. Whereas primary alcohols serve dually as reductant and carbonyl proelectrophile, direct use of aldehydes requires an exogenous reductant. It was found that reactions conducted in the presence of formic acid (200 mol%) at lower temperatures (80° C), but under otherwise identical conditions, enabled efficient allene-mediated allylation of aldehydes dehydro-2a and dehydro-2c to form adducts 3a and 3c, respectively, with good levels of enantiocontrol (eq. 1 and 2).
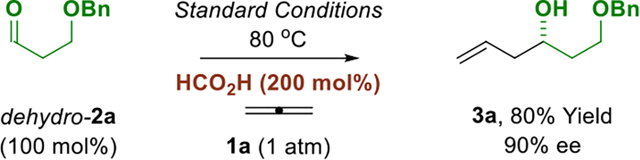 |
(eq. 1) |
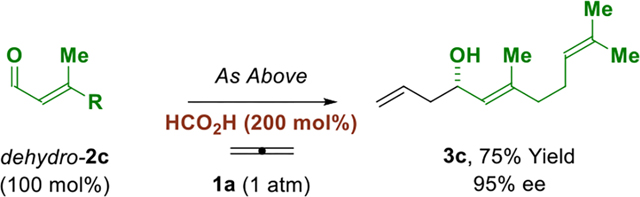 |
(eq. 2) |
To demonstrate how the protecting group-free asymmetric allylation of primary alcohols in the presence of secondary alcohols streamlines de novo polyketide synthesis, the present method was used to prepare a known C7-C15 substructure of spirastrellolide B and F (Scheme 1).10a Thus, alcohol 2a was subjected to standard conditions for enantioselective allene-mediated carbonyl allylation on 500 mg scale at low loadings of catalyst (1.5 mol%) to form the homoallylic alcohol 3a in 87% yield and 90% ee. Ozonolysis of 3a accompanied by treatment of the ozonide with NaBH4 delivered the 1°,2°-1,3-diol 2i. Allene-mediated carbonyl allylation of diol 2i delivered the 2°,2°-1,3-diol 3i in 92% yield. Conversion of 3i to the corresponding acetonide followed by treatment with ozone and NaBH4 provided primary alcohol 4, which upon asymmetric crotylation mediated by butadiene 1b furnished compound 5.7c Finally, successive treatment of 5 with ozone and NaBH4 provides diol 6, which encompasses C7-C15 of spirastrellolide B and F. Compound 6, which was previously prepared by Sabitha and coworkers in 17 steps (longest linear sequence, LLS), is now accessible in 7 steps (LLS) via iterative alcohol-mediated C-C coupling. Further underscoring the increase in step-efficiency enabled by the present method, compound 3i is itself a known C3-C10 substructure of cryptocarya diacetate that has been prepared on two prior occasions in 7 or 9 steps (LLS).10b,c Compound 3i is now accessible in only 3 steps (LLS). Finally, compound 3h is known fragment common to C8′-C14′ of mycolactone F10d and C22-C28 marinomycin A,10e which were prepared in 4 steps (LLS) and 9 steps (LLS), respectively. Compound 3h is now accessible in only 1 step (LLS) via direct allylation of (R)-butanediol (Table 2).
Scheme 1.

Streamlined polyketide construction via iterative allene-mediated allylation and crotylation.a
A catalytic cycle has been posited and corroborated by a deuterium labelling experiment (Scheme 2). β-Hydride elimination from the pentacoordinate ruthenium(II) alkoxide I forms an aldehyde and a ruthenium(II) hydride II. Ruthenium(II) complexes such as alkoxide I are octahedral d6 metal ions with vacant dx2−y2 orbitals, which makes alkoxide β-hydride elimination especially facile. Allene 1a suffers hydroruthenation by the ruthenium(II) hydride II to form the π-allylruthenium(II) complex III. Related stoichiometric reactions of HXRu(CO)(PR3)3 (X = Cl, Br) with allenes or dienes to form isolable π-allylruthenium species have been described.18 Aldehyde addition from the σ-allylruthenium haptomer (not shown) through a six-centered transition structure provides the homoallylic ruthenium(II) alkoxide IV. Substitution of the homoallylic ruthenium(II) alkoxide IV with the reactant alcohol catalyzed by TFE closes the catalytic cycle. Entry into the catalytic cycle is envisioned to occur via protonation of the π-allylruthenium(II) precatalyst at carbon by TFE to furnish the transient ruthenium trifluoroethoxide, which is inert with respect to dehydrogenation due to inductive stabilization of the transition state for β-hydride elimination.12 Incomplete transfer of deuterium from the reactant alcohol deuterio-2a to the interior vinylic position of the reaction product deuterio-3a is due (in part) to 2H-1H exchange of ruthenium(II) deuteride II with the hydroxyl protons of the reactant alcohol, TFE and adventitious water.19 The persistence of deuterium at the carbinol position of deuterio-3a suggests that dehydrogenation of the secondary hydroxyl group is suppressed by chelation of the homoallylic olefin (as in complex IV), which blocks the otherwise open coordinate site required for β-hydride elimination.
Scheme 2.

Proposed catalytic cycle as corroborated by a deuterium labeling experiment.
Conclusion
In summary, polyketides have played a pervasive, longstanding role in medicine and agrochemistry, which, in turn, has inspired decades of research on the development of methods for their synthesis. Asymmetric protocols for carbonyl allylation figure prominently among these methods, yet until work from our laboratory,5 such methods have uniformly relied upon premetalated reagents or metallic reductants.4 Here, using an iodide-bound ruthenium-JOSIPHOS complex recently developed in our laboratory, we report a catalytic method for enantioselective carbonyl allylation via hydrogen auto-transfer that is not only byproduct-free, but can be conducted from primary alcohol proelectrophiles in the presence of secondary alcohols. As illustrated in concise syntheses of multiple known polyketide substructures, these capabilities significantly enhance the efficiency of polyketide construction. More broadly, these data demonstrate how the native reducing ability of alcohols can be harnessed for the generation of transient organometallic nucleophiles to provide a path for chemical synthetic beyond stoichiometric organometallic reagents.
Supplementary Material
ACKNOWLEDGMENT
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (R01 GM093905) are acknowledged for partial support of this research.
Footnotes
The authors declare no competing financial interest.
Supporting Information. Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra and HPLC traces for racemic and enantiomerically enriched compounds. Single-crystal X-ray diffraction data for the 3,5-dinitrobenzoate derived from compound 3g (CCDC Deposition Number: 2172182). This information is available free of charge on the ACS Publications website.
REFERENCES
- (1).For selected reviews on polyketide natural products, see:O’Hagan D The Polyketide Metabolites; Ellis Horwood: Chichester, 1991.Rohr J A New Role for Polyketides. Angew. Chem. Int. Ed. 2000, 39, 2847–2849.Weissman KJ; Leadlay F Combinatorial Biosynthesis of Reduced Polyketides. Nat. Rev. Microbiol. 2005, 3, 925–936.Rimando AM; Baerson SR Eds., Polyketides: Biosynthesis, Biological Activity, and Genetic Engineering (ACS Symp. Ser.,Vol. 955), American Chemical Society, Washington DC, 2007.Dayan FE; Cantrell CL; Duke SO Natural Products in Crop Protection. Bioorg. Med. Chem. 2009, 17, 4022–4034.Dechert-Schmitt A-MR; Schmitt DC; Gao X; Itoh T; Krische MJ Polyketide Construction via Hydrohydroxyalkylation and Related Alcohol CH Functionalizations: Reinventing the Chemistry of Carbonyl Addition. Nat. Prod. Rep. 2014, 31, 504–513.Feng J; Kasun ZA; Krische MJ Enantioselective Alcohol C-H Functionalization for Polyketide Construction: Unlocking Redox-Economy and Site-Selectivity for Ideal Chemical Synthesis. J. Am. Chem. Soc. 2016, 138, 5467–5478.
- (2).To our knowledge, eribulin remains the sole example of an FDA-approved polyketide drug made via de novo chemical synthesis. Over 60 total steps are required for its preparation: Yu MJ; Zheng W; Seletsky BM From Micrograms to Grams: Scale-Up Synthesis of Eribulin Mesylate. Nat. Prod. Rep. 2013, 30, 1158–1164.
- 3.For selected reviews on catalytic enantioselective aldol additions, see: Trost BM; Brindle CS The Direct Catalytic Asymmetric Aldol Reaction. Chem. Soc. Rev. 2010, 39, 1600–1632.Dias LC; de Lucca EC Jr.; Ferreira MAB; Polo EC Metal-Catalyzed Asymmetric Aldol Reactions. J. Braz. Chem. Soc. 2012, 23, 2137–2158.Yamashita Y; Yasukawa T; Yoo W-J; Kitanosono T; Kobayashi S Catalytic Enantioselective Aldol Reactions. Chem. Soc. Rev. 2018, 47, 4388–4480.Meyer CC; Ortiz E; Krische MJ Catalytic Reductive Aldol and Mannich Reactions of Enone, Acrylate and Vinyl Heteroaromatic Pronucleophiles. Chem. Rev. 2020, 120, 3721–3748.
- (4).For selected reviews on catalytic enantioselective carbonyl allylation, see: Denmark SE; Fu J Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev. 2003, 103, 2763–2794.Hall DG Lewis and Brønsted Acid Catalyzed Allylboration of Carbonyl Compounds: From Discovery to Mechanism and Applications. Synlett 2007, 1644–1655.Yus M; González-Gómez JC; Foubelo F Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev. 2011, 111, 7774–7854.Huo H-X; Duvall JR; Huang M-Y; Hong R Catalytic Asymmetric Allylation of Carbonyl Compounds and Imines with Allylic Boronates. Org. Chem. Front. 2014, 1, 303–320.Spielmann K; Niel G; de Figueiredo RM; Campagne J-M Catalytic Nucleophilic ‘Umpoled’ π-Allyl Reagents. Chem. Soc. Rev. 2018, 47, 1159–1173.
- 5.For selected reviews, see: Kim SW; Zhang W; Krische MJ Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res. 2017, 50, 2371–2380.Doerksen RS; Meyer CC; Krische MJ Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target-Oriented Synthesis. Angew. Chem. Int. Ed. 2019, 58, 14055–14064.Santana CG; Krische MJ From Hydrogenation to Transfer Hydrogenation to Hydrogen Auto-Transfer in Enantioselective Metal-Catalyzed Carbonyl Reductive Coupling: Past, Present and Future. ACS Catal. 2021, 11, 5572–5585.
- (6).The hydrogen auto-transfer processes we report result in carbonyl addition and, hence, are distinct from so-called borrowing hydrogen reactions, which result in hydroxyl substitution. For selected reviews, see: Hamid MHSA; Slatford PA; Williams JMJ Borrowing Hydrogen in The Activation of Alcohols. Adv. Synth. Catal. 2007, 349, 1555–1575.Guillena G; Ramón DJ; Yus M Alcohols as Electrophiles in C-C Bond-Forming Reactions: The Hydrogen Autotransfer Process. Angew. Chem. Int. Ed. 2007, 46, 2358–2364.Dobereiner GE; Crabtree RH Dehydrogenation as a Substrate-Activating Strategy in Homogeneous Transition Metal Catalysis. Chem. Rev. 2010, 110, 681–703.Bähn S; Imm S; Neubert L; Zhang M; Neumann H; Beller M The Catalytic Amination of Alcohols. ChemCatChem 2011, 3, 1853–1864.Yang Q; Wang Q; Yu Z Substitution of Alcohols by N-Nucleophiles via Transition Metal-Catalyzed Dehydrogenation. Chem. Soc. Rev. 2015, 44, 2305–2329.Aitchison H; Wingad RL; Wass DF Homogeneous Ethanol to Butanol Catalysis - Guerbet Renewed. ACS Catal. 2016, 6, 7125–7132.Quintard A; Rodriguez J Catalytic Enantioselective OFF ↔ ON Activation Processes Initiated by Hydrogen Transfer: Concepts and Challenges. Chem. Commun. 2016, 52, 10456–10473.Reed-Berendt BG; Polidano K; Morrill LC Recent Advances in Homogeneous Borrowing Hydrogen Catalysis Using Earth-Abundant First Row Transition Metals. Org. Biomol. Chem. 2019, 17, 1595–1607.Kwok T; Hoff O; Armstrong RJ; Donohoe TJ Control of Absolute Stereochemistry in Transition-Metal-Catalysed Hydrogen-Borrowing Reactions. Chem. Eur. J. 2020, 26, 12912–12926.
- (7).(a) Ortiz E; Shezaf JZ; Chang Y-H; Gonçalves TP; Huang K-W; Krische MJ Understanding Halide Counterion Effects in Enantioselective Ruthenium-Catalyzed Carbonyl (α-Aryl)allylation: Alkynes as Latent Allenes and Trifluoroethanol-Enhanced Turnover in The Conversion of Ethanol to Higher Alcohols via Hydrogen Auto-Transfer. J. Am. Chem. Soc. 2021, 143, 16709–16717. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ortiz E; Chang Y-H; Shezaf JZ; Shen W; Krische MJ Stereo- and Site-Selective Conversion of Primary Alcohols to Allylic Alcohols via Ruthenium-Catalyzed Hydrogen Auto-Transfer Mediated by 2-Butyne. J. Am. Chem. Soc. 2022, 144, 8861–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ortiz E; Spinello BJ; Cho Y; Wu J; Krische MJ Stereo- and Site-Selective Crotylation of Alcohol Proelectrophiles via Ruthenium-Catalyzed Hydrogen Auto-Transfer Mediated by Methylallene and Butadiene. Angew. Chem. Int. Ed. 2022, 61, e202212814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Bower JF; Skucas E; Patman RL; Krische MJ Catalytic C-C Coupling via Transfer Hydrogenation: Reverse Prenylation, Crotylation and Allylation from the Alcohol or Aldehyde Oxidation Level. J. Am. Chem. Soc. 2007, 129, 15134–15135. [DOI] [PubMed] [Google Scholar]; (b) Kim SW; Meyer CC; Mai BK; Liu P; Krische MJ Inversion of Enantioselectivity in Allene Gas versus Allyl Acetate Reductive Aldehyde Allylation Guided by Metal-Centered Stereogenicity: An Experimental and Computational Study. ACS Catal. 2019, 9, 9158–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu RY; Zhou Y; Yang Y; Buchwald SL Enantioselective Allylation Using Allene, a Petroleum Cracking Byproduct. J. Am. Chem. Soc. 2019, 141, 2251–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For iridium-catalyzed stereo- and site-selective allylation of 1,3-diols using allyl acetate as a pronucleophile, see: Dechert-Schmitt A-MR; Schmitt DC; Krische MJ Protecting-Group-Free Diastereoselective C-C Coupling of 1,3-Glycols and Allyl Acetate through Site-Selective Primary Alcohol Dehydrogenation. Angew. Chem. Int. Ed. 2013, 52, 3195–3198.Shin I; Wang G; Krische MJ Catalyst-Directed Diastereo- and Site-Selectivity in Successive Nucleophilic and Electrophilic Allylations of Chiral 1,3-Diols: Protecting Group-Free Synthesis of Substituted Pyrans. Chem. Eur. J. 2014, 20, 13382–13389.
- (10).(a) Sabitha G; Rao AS; Yadav JS Synthesis of The C1–C25 Southern Domain of Spirastrellolides B and F. Org. Biomol. Chem. 2013, 11, 7218–7231. [DOI] [PubMed] [Google Scholar]; (b) Krishna PR; Reddy VVR Stereoselective Total Synthesis of (+)-Cryptocarya Diacetate by an Iterative Jacobsen’s Hydrolytic Kinetic Resolution Protocol. Tetrahedron Lett. 2005, 46, 3905–3907. [Google Scholar]; (c) Sabitha G; Reddy NM; Prasad MN, Yadav JS Stereoselective Routes for the Total Synthesis of (+)-Cryptocarya Diacetate. Helv. Chim. Acta. 2009, 92, 967–976. [Google Scholar]; (d) Kim H-J; Kishi Y Total Synthesis and Stereochemistry of Mycolactone F. J. Am. Chem. Soc. 2008, 130, 1842–1844. [DOI] [PubMed] [Google Scholar]; (e) Nishimaru T; Kondo M; Takeshita K; Takahashi K; Ishihara J; Hatakeyama S Total Synthesis of Marinomycin A Based on a Direct Dimerization Strategy. Angew. Chem. Int. Ed. 2014, 53, 8459–8462. [DOI] [PubMed] [Google Scholar]
- (11).For selected reviews on the use of allenes and dienes in enantioselective metal-catalyzed reductive C=X (X = O, NR) allylation, see: Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes and Enynes with Carbonyl Compounds and Imines. Chem. Rev. 2018, 118, 6026–6052.Xiang M; Pfaffinger DE; Krische MJ Allenes and Dienes as Chiral Allylmetal Pronucleophiles in Catalytic Enantioselective C=X Addition: Historical Perspective and State-of-The-Art Survey. Chem. Eur. J. 2021, 27, 13107–13116.
- (12).Sam B; Luong T; Krische MJ Ruthenium-Catalyzed C-C Coupling of Fluorinated Alcohols with Allenes: Dehydrogenation at the Energetic Limit of β-Hydride Elimination. Angew. Chem. Int. Ed. 2015, 54, 5465–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For reviews of halide effects in transition metal-catalyzed reactions, see: Maitlis PM; Haynes A; James BR;Catellani M; Chiusoli GP Iodide Effects in Transition Metal Catalyzed Reactions. Dalton Trans. 2004, 3409–3419.Fagnou K; Lautens M Halide Effects in Transition Metal Catalysis. Angew. Chem. Int. Ed. 2002, 41, 26–47.
- (14).For selected reviews on metal-catalyzed olefin isomerization, see: Larionov E; Li H; Mazet C Chem. Commun. 2014, 50, 9816–9826.Massad I; Marek I Alkene Isomerization through Allylmetals as a Strategic Tool in Stereoselective Synthesis. ACS Catal. 2020, 10, 5793–5804.
- (15).Cohen N; Eichel WF; Lopresti RJ; Neukom C; Saucy G Synthetic Studies on (2R,4’R,8’R)-α-Tocopherol. An Approach Utilizing Side Chain Synthons of Microbiological Origin. J. Org. Chem. 1976, 41, 3505–3511. [DOI] [PubMed] [Google Scholar]
- (16).Direct allylation of the “Roche alcohol” to form adducts 3k and epi-3k avoids discrete formation of the configurationally unstable “Roche aldehyde,” which suffers 5–7% racemization upon purification by silica gel chromatography: Roush WR; Palkowitz AD; Ando K Acyclic Diastereoselective Synthesis Using Tartrate Ester-Modified Crotylboronates. Double Asymmetric Reactions with α-Methyl Chiral Aldehydes and Synthesis of the C(19)-C(29) Segment of Rifamycin S. J. Am. Chem. Soc. 1990, 112, 6348–6359.
- (17).For a related study, see: Schmitt DC; Dechert-Schmitt A-MR; Krische MJ Iridium-Catalyzed Allylation of Chiral β-Stereogenic Alcohols: Bypassing Discrete Formation of Epimerizable Aldehydes. Org. Lett. 2012, 14, 6302–6305.
- (18).(a) Hiraki K; Ochi N; Sasada Y; Hayashida H; Fuchita Y; Yamanaka S Organoruthenium(II) Complexes Formed by Insertion Reactions of Some Vinyl Compounds and Conjugated Dienes into a Hydrido–Ruthenium Bond. J. Chem. Soc., Dalton Trans. 1985, 873–877. [Google Scholar]; (b) Hill AF; Ho CT; Wilton-Ely JDET The Coupling of Methylene and Vinyl Ligands at a Ruthenium(II) Centre. Chem. Commun. 1997, 2207–2208. [Google Scholar]; (c) Xue P; Bi S; Sung HHY; Williams ID; Lin Z; Jia G Isomerism of [Ru(η3-allyl)Cl(CO)(PPh3)2] Organometallics 2004, 23, 4735–4743. [Google Scholar]
- (19).(a) Tse SKS; Xue P; Lin Z; Jia G Hydrogen/Deuterium Exchange Reactions of Olefins with Deuterium Oxide Mediated by the Carbonylchlorohydrido-tris(triphenylphosphine)ruthenium(II) Complex. Adv. Synth. Catal. 2010, 352, 1512–1522. [Google Scholar]; (b) Isbrandt ES; Vandavasi JK; Zhang W; Jamshidi MP; Newman SG Catalytic Deuteration of Aldehydes with D2O. Synlett 2017, 28, 2851–2854. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.