

Abstract
Highly functionalized skeletons of macrolide natural products gain access to rare spatial arrangements of atoms, where changes in stereochemistry can have profound impact on structure and function. Spliceosome modulators present a unique consensus motif, with the majority targeting a key interface within the SF3B spliceosome complex. Our recent preparative-scale synthetic campaign of 17S-FD-895 provided unique access to stereochemical analogues of this complex macrolide. Here, we report on the preparation and systematic activity evaluation of multiple FD-895 analogues. These studies examine the effects of modifications at specific stereocenters within the molecule and highlight future directions for medicinal chemical optimization of spliceosome modulators.
Graphical Abstract
INTRODUCTION
In 1994, a team at Taisho Pharmaceutical Co. Ltd reported the discovery of a 12-membered macrolide, FD-895 (1) (Fig. 1) from strain A-9561 isolated from a soil sample collected at Iromote Island, Japan.1 Nearly a decade later, efforts at the Tsukuba Research Laboratories of Eisai Co. Ltd reported the discovery of a set of 17-deoxy-31-demethyl analogues of FD-895 from a strain of Streptomyces platensis Mer-11107 and renamed the materials pladienolides, as exemplified by pladienolide B (2a) in Fig. 1.2–4
Figure 1.
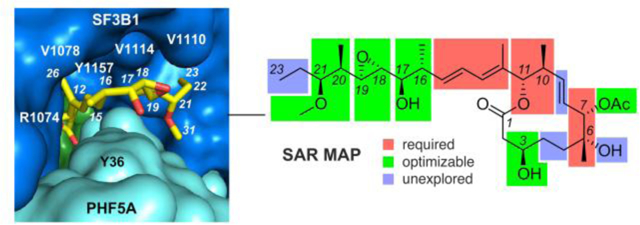
Splice modulatory natural products families, including FD-895 (1), pladienolide B (2a), FR901464 (2b), meayamycin (2c), and thailanstatin A (2d), and herboxadiene (2e).
While not recognized at the time, efforts by Fujisawa Pharmaceutical Co. Ltd. and Monsanto Co. led to the parallel discoveries of FR901464 (2b)5–7 (Fig. 1) from Pseudomonas sp. and herboxadiene (2e) (Fig. 1)8 from Streptomyces chromofuscus, respectively. All three families of natural products (1–2a, 2b-2d and 2e, Fig. 1) would later be united by a similar antitumor mode of action (MOA)9–11 targeting the SF3B complex of the human spliceosome and are referred to as spliceosome modulators (SPLMs).12
The X-ray crystal structure of the core SF3B complex was solved by Pena in 2016,13 followed soon after with its elucidation by cryo-EM.14 The SF3B complex bound to pladienolide B (2a) was subsequently reported15,16 and has since been furthered to provide detailed structures with additional analogues, including FD-895 (1).17 These SPLMs occupy a two-sphere shaped pocket, where the diene (C12-C15) occupies a tunnel between pockets bound to the core (C1-C11) and side chain (C12-C23) of 1. Here, FD-895 (1) adopts a conformation that enables the hydrophilic side chain to position itself between V1100 and F1153 in SF3B and Y35 in PHF5A.17 The 12-membered lactone core of 1 adopts a conformation that is held within the pocket by hydrogen bonding interactions between R1047 (with the C1 carbonyl) and K1071 (to the C3 OH) of SF3B and R38 of PHF4A (to the C29 acetate carbonyl).17
The importance of these classes of SPLMs is now well recognized through significant synthetic effort, including that on the FD-895 (1), pladienolides (2a, Fig. 1), and spliceostatins including FR901464 (2b), meayamycin (2c) and thailanstatin A (2d), and herboxidiene (2e), as recently reviewed.18,19 These studies have led to a remarkable level of structure activity relationship (SAR) data that defines how each of these molecules adopts a consensus motif that uniquely positions itself into the core SF3B complex of the spliceosome.20,21
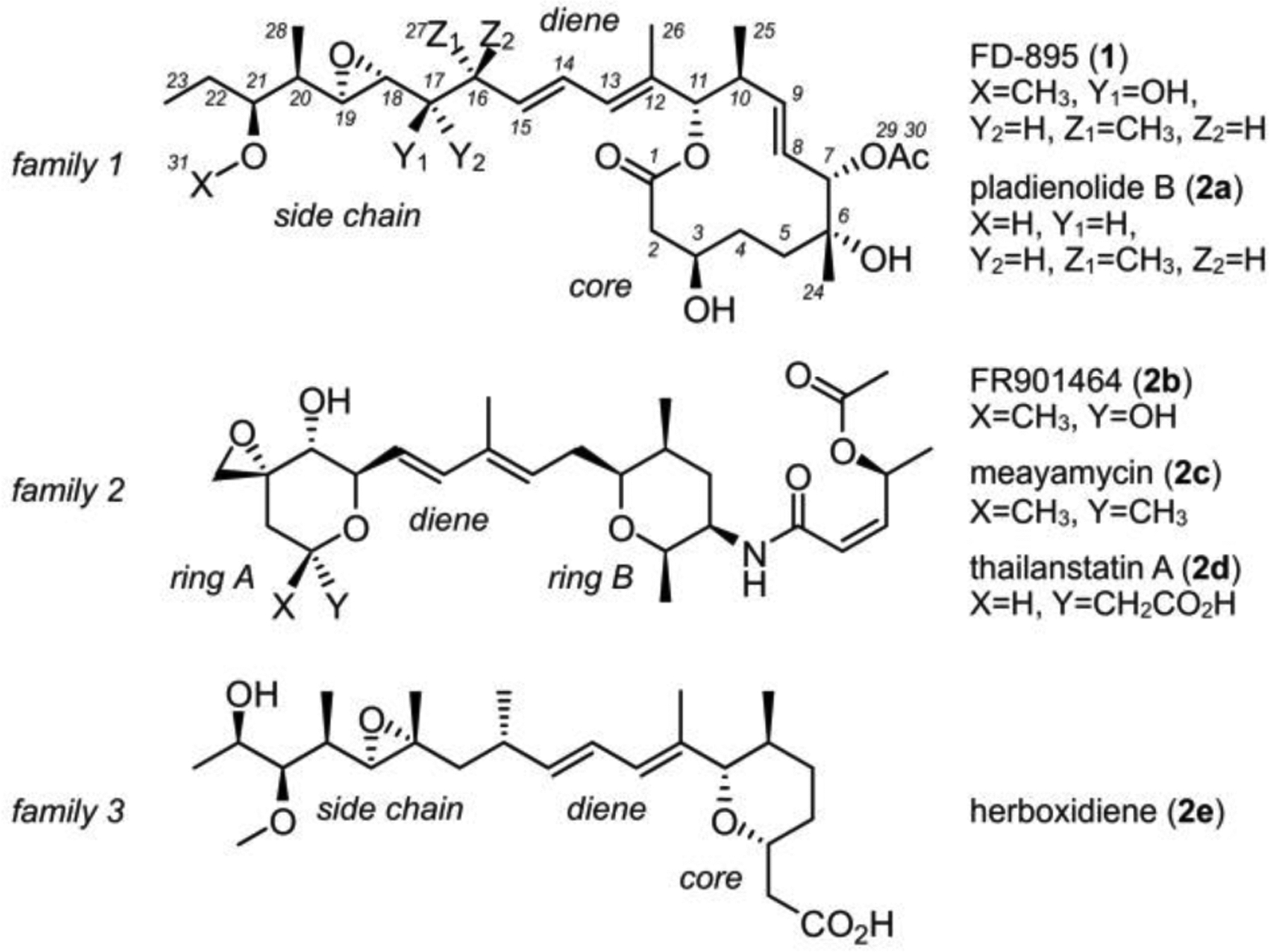
Recognized since their discovery, the high potency and tumor cell selectivity of these analogues has ultimately lead to the entry of two Phase I clinical trials on semi-synthetic analogues E7107 (3a, Fig. 2) and H3B-8800 (3b).22,23 While both trials were unsuccessful, the promise of this class remains, and medicinal chemical efforts have continued to tune metabolic stability and splice modulatory activity of this class, with the ultimate goal of realizing active analogues that can serve either as primary therapeutics or as the active agents in antibody drug conjugates.24 We have continued to explore the SAR within FD-895 (1, Fig. 3), as there remains a need to retain potent activity while providing the necessary pharmacological properties for human use. Our recent efforts on the preparative synthesis of one potential therapeutic, 17S-FD-895 (1d, Fig. 3),25 resulted in isolation of multiple stereoisomeric precursors of the pharmacophore, providing an excellent opportunity for SAR evaluation. Here, we report the systematic evaluation of 10 of the 11 stereochemical positions within FD-895 (1).
Figure 2.
Two semi-synthetic analogues E7107 (3a) and H3B-8800 (3b) have been advanced into Phase I clinical trials and were unsuccessful due to a combination of a lack in efficacy and off-target effects.
Figure 3.
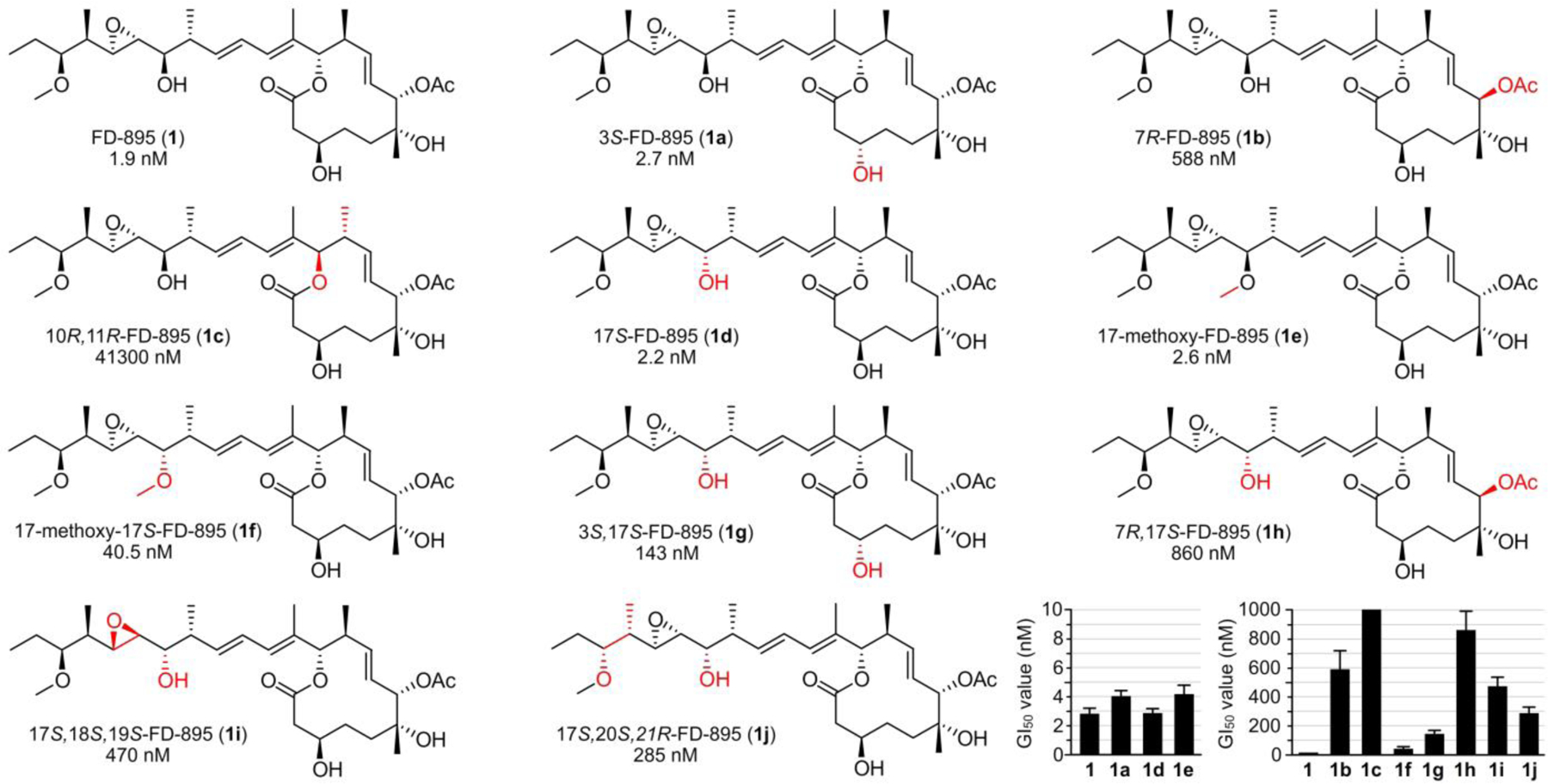
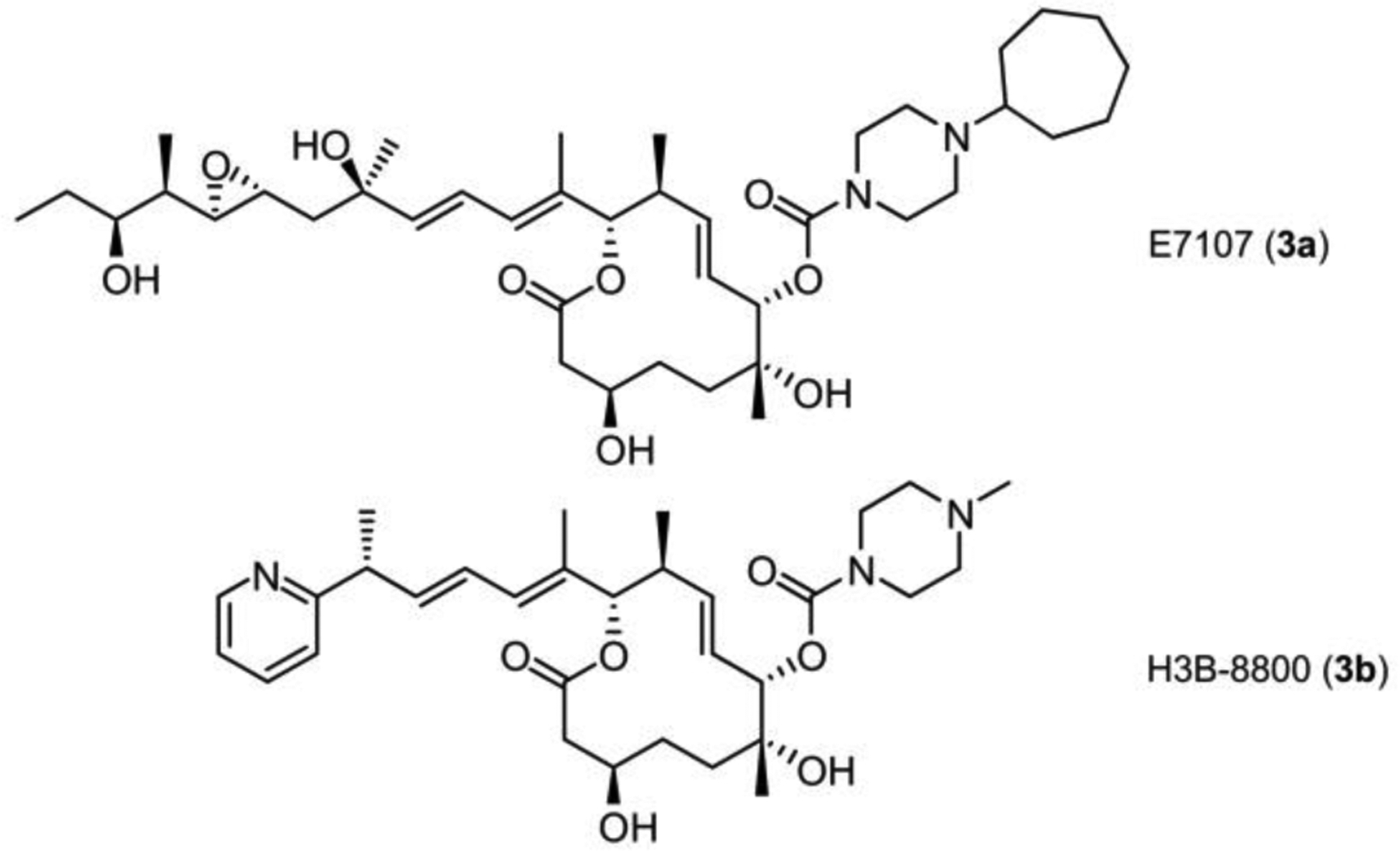
A panel of FD-895 analogues 1a-1j, including stereochemical and functional modifications in the core at C3, C7, or C10, C11 and side chain at C17-C21. Red indicates the position of modification. Cellular activity (GI50 values in HCT-116 cells at 72 h) are provided under each compound number and graphically at the bottom right. GI50 values (Supporting Table S12) were calculated from dose curves.
Prior to this study, the SAR data on the pladienolides and FD-895 reported primarily on the GI50 values (GI50 values correspond to a global growth decrease by 50% induced by a compound) values of analogues at C3, C7 and C16-C18.20 Within this dataset, we reported the total synthesis of FD-895 (1) along with its three C16-C17 isomers, including 17S-FD-895, in 2012 (1d).26,27 In this study, we identified differential activity within each of the four C16-C17 analogues25 and discovered both an increased activity and stability for 1d. Ultimately, this led to the completion of a 17 g production of 1d25 and associated in vitro and in vivo pharmacological evaluation for IND-enabling studies.28,29 These methods were used to prepare isomeric materials at C3 (1a) and C7 (1b). For the current study, we began by adapting the synthesis to prepare analogues of FD-895 (1) with inversion of the stereochemistry at 10 of the 11 stereocenters (C3, C7, C10, C11, C16, C17, C18, C19, C20 and C21).
RESULTS AND DISCUSSION
Analogue Synthesis.
Our synthetic approach (Supporting Figs. S1–S11) developed through the Stille coupling of macrolide core (C1-C11) and side chain (from C12-C28) components. Isomers with inversion at C3 (Supporting Figs. S2 and S8), C7 (Supporting Figs. S3 and S9), or C10-C11 (Supporting Figs. S4) were prepared by synthesis of the corresponding isomeric macrolide core and coupling it to a desired side chain in a final step. Side chain analogues at C17, C18, C19, C20, C21 were prepared in an analogous manner (Supporting Fig. S5–S11). This along with methylation at C17 enabled the preparation the set of 11 analogues 1 and 1a-1i (Fig. 3).
Cell growth inhibition activity.
With milligram quantities of 1 and 1a-1i in hand, we began to evaluate the relative efficacy of each analogue using cell growth inhibition studies with HCT116 colorectal carcinoma cells. As reported in Fig. 3, GI50 values were collected for each analogue after a 72 h treatment using the MTS cell viability assay (lower right, Fig. 3, Supporting Table S12). Under these conditions, the natural FD-895 (1), 3S-FD-895 (1a) 17S-FD-895 (1d), 17-methoxy-FD-895 (1e) provided activities of < 10 nM, indicating that functionalization or stereochemical inversion at C17 and stereochemical inversion at C3 were not critical to growth inhibitory activity. While 1d and 1e suggested that functionalization and inversion was tolerated at C16, a 21-fold loss in activity of 17-methoxy-17S-FD-895 (1f) when compared to 1 suggested that this was a small window for functional manipulation.
This data (Fig. 3) also highlighted the importance of the C7 position, as 7R-FD-895 (1b) was 300-fold less active than 1. Even more pronounced was the loss from stereochemical inversion at C10 and C11, as 10R,11R-FD-895 (1c) displayed a GI50 value of 41.3 μM (21,700-fold less than 1). Methylation at C17 was tolerated, as illustrated by 17-methoxy-FD-895 (1e) and 17-methoxy-17S-FD-895 (1f), with GI50 values of 2.6 nM and 40.5 nM, respectively.
As noted, the combination of inversion and methylation at C17 in 1f was apparently less tolerated than direct methylation in 1e. Double modifications in general were less tolerated, as further illustrated by 3S,17S-FD-895 (1g) and 7R-17S-FD-895 (1h), with GI50 values of 143 nM and 860 nM, respectively. Doubly modified 1g and 1h were 52-fold and 318-fold less effective, respectively, when compared to 17S-FD-895 (1a), bearing a single inversion. Comparable losses of activity were also observed in modifications within the side chain, as 17S,18S,19S-FD-895 (1i) and 17S,20S,21R-FD-895 (1j) displayed GI50 values of 470 nM and 285 nM, respectively. Here, we learned that inversion of the epoxide in 1i or inversion of the two stereocenters at C20 and C21 in 1j also contributed to a loss of activity when compared to 1d. The fact that multiple modifications were present in 1i and 1j contributed to their activity loss. Moreover, this illustrated the importance of the stereochemical configuration within the terminus of the side chain.
Splice Modulatory Activity.
Next, we turned our efforts to explore the selectivity of these analogues in modulating splicing. Our prior work identified that even small changes in SPLM structure can influence spliceosome activity in terms of spliced mRNA identity.30 This phenomenon offers the potential to discover SPLMs that could target select gene products over others and could be leveraged for targeted and synergistic chemotherapeutic approaches.31
For these studies, we refined our studies to analogues with GI50 values ≤ 500 nM. Here, we treated HCT116 cells with 1 at 34 nM, 1a at 44 nM, 1b at 8.6 μM 1b, 1d at 42 nM, 1e at 44 nM, 1f at 750 nM, 1g at 2.6 μM, 1i at 9 μM and 1j at 5.6 μM allowing us to normalize each analogue relative to each other and thereby evaluate changes in gene response due to splicing selectivity rather than compounding effects due to differences in potency. These concentrations were selected at ~20 times their GI50 values (Fig. 3) by using 1 to identify the optimal correlation of activity at 72 h to that observed by qRT-PCR analyses at 4 h (see additional discussion within the Experimental Section). Using this design, cells were treated for 4 h to evaluate early changes in splicing activity, and then cellular RNA was isolated, purified and analyzed by qRT-PCR (Fig. 4). Importantly, none of the concentrations tested led to significant changes in cell viability, ensuring that modifications in gene expression were not attributable to cell death (Supporting Fig. S12a).
Figure 4.
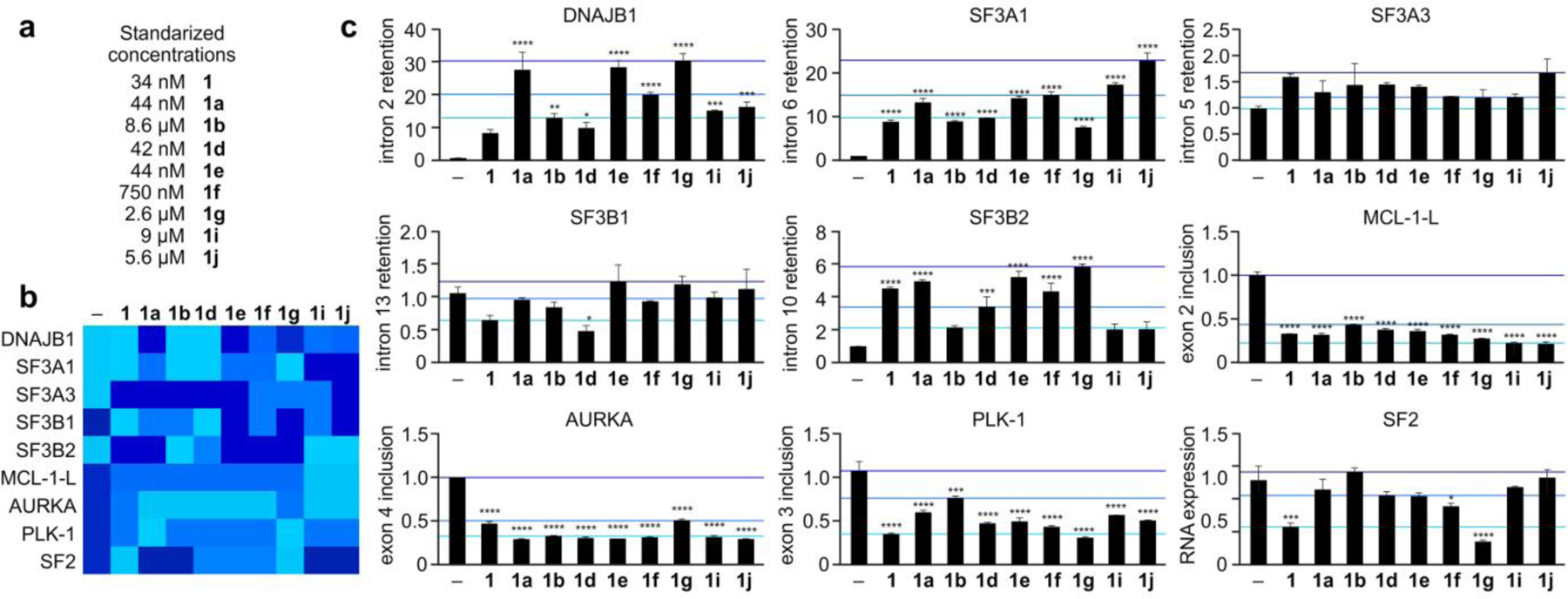
Structure-splicing profiles for mRNA splice modulation after 4 h treatment. a) Experiments were conducted with standardized concentrations at ~20 times their 72 h GI50 values (see discussion in the Experimental Section). Data is presented in b) heat map format and c) bar graph format, where intensity in the heat map in c) represent activities above the dark blue (high), light blue (medium) and cyan (low) levels as depicted by colored lines in b). HCT116 tumor cells were treated with each analogue at a standardized concentration (upper left) based on their GI50 values (see Fig. 3 and further discussion within the Experimental Section) for 4 h and then cellular RNA was isolated, purified and analyzed by qRT-PCR. Primers were designed to evaluate intron retention or exon skipping, direct responses to splice modulator treatment. Genes evaluated included those involved in splicing regulation (SF3A1, SF3A3, SF3B1, SF3B2), apoptosis (MCL-1L), protein folding (DNAJB1), and cell cycle regulation (AURKA, PLK-1) relative to the unspliced control GAPDH. For alternative splicing regulator (SF2), primers were designed to evaluate gene expression, which changes in response to splice modulator treatment for SF2 are provided relative to the unspliced control GAPDH. (–) denotes untreated cells. Numerical values represent the relative levels of intron retention with respect to negative control (–) at 1.
For these studies, we evaluated the expression of select genes involved in splicing regulation (SF3A1, SF3A3, SF3B1, SF3B2), apoptosis (MCL-1L), protein folding (DNAJB1), and cell cycle regulation (AURKA, PLK-1) relative to the unspliced control GAPDH. These mRNAs were selected based upon prior RNA sequencing studies correlating their splicing to oncogenesis.27–31 Primers were designed to evaluate intron retention or exon skipping, direct responses to splice modulator treatment. The master splicing regulator alternative splicing factor (SF2) was also evaluated, with the primers for this gene designed to evaluate overall RNA expression, which changes in response to splice modulator treatment.
As shown in Fig. 4, each analogue displayed an individualized efficacy against each pre-mRNA further validating prior observations that structural modifications alter the gene selectivity of each analogue.30 While some genes, such as SF3A3 or SF3B1, showed modest intron retention (levels of intron retention < 3, Fig. 4), as well as differentiation over the analogue panel, others, such as DNAJB1, SF3A1 and SF3B2, displayed significant efficacy (level of intron retention > 3, Fig. 4). For DNAJB1, analogues 1a, 1e and 1g (provided the highest level of intron retention, indicating that inversion at C3 in 1a and 1g and methylation at C17 in 1e played a beneficial role in enhancing DNAJB1 intron 2 retention as compared to 1.
For the splicing factor SF3A1, 1a, 1e, 1f, 1i and 1j were more effective than 1, further confirming the ability of C3 and C17 methylation as a tool to increase efficacy against this gene. While higher concentrations were required, this study also showed that side chain modifications can also lead to comparable intron retention, as illustrated by the effects of 1i and 1j on intron 6 retention in SF3A1. Analogue selectivity was also observed in the other splicing factors explored, including SF3B1, and SF3B2. While only modest selectivity was observed for SF3A3 or SF3B1, statistically relevant selectivity was observed for SF3B2, indicating improved efficacy for 1a, 1e, and 1g, as compared to 1. While the data obtained in Fig. 4 was collected from the same cells (each compound was applied to the same cell culture individually, and intron retention, exon skipping and relative RNA expression data was collected from the same sample), the efficacy of each analogue was different for each gene as best illustrated by comparing the effects on DNAJB1, SF3A1 and SF3B2 (Fig. 4). These observations provide further evidence for the medicinal chemical optimization of analogues with targeted gene selectivity.
In addition to intron retention, we also confirmed the ability of these analogues to induce exon skipping, as illustrated by exon 2 in MCL-1L, exon 3–4 in AURKA and exon 3 in PLK-1. While the levels of exon skipping was comparable for each analogue in MCL-1-L (Fig. 4), analogues 1a and 1b displayed the most potent effect on exon 3 inclusion in PLK-1, while 1a-1f and 1i-1j displayed effects greater than 1 on exon 4 skipping in AURKA, again furthering the fact that inversion at C3 or C17 suggests utility at tuning gene selectivity. This was further supported by the fact that 1g and 1 (also displayed the highest efficacy in down regulating the expression of SF2.
From this 4 h study (Fig. 4), analogues 1, 1b, 1g, 1d, 1e, 1i and 1j were selected to evaluate for effects on splicing when presented for a longer period (24 h). While our prior data suggested that 4 h treatment was sufficient for response in many tumor cell lines, we wanted to fully evaluate if time could play a role in gene selectivity. Using identical procedures as that used for 4 h (Fig. 4), HCT116 cells were treated with three concentrations (100 nM, 250 nM, and 500 nM) of each analogue for 24 h. Multiple concentrations were used to further validate the use of the GI50 concentration in our prior study (Fig. 4) as compared to cell growth inhibition at the highest dose at 24 h (Supporting Fig. S12b).
We found that splicing activity (Fig. 5) typically corresponded to growth inhibitor activity for each analogue (Fig. 3), with the caveat that each analogue displayed a unique gene-selective signature (Fig. 5). The natural product FD-895 (1), a potent cell growth inhibitor, altered the splicing of nearly all genes tested (weak efficacy noted for PLK-1 and SF2, Fig. 5). Epimerization of the C17 center given by 1d (a comparable inhibitor of cell growth as 1) resulted in a decreased level of intron retention in DNAJB1, dose dependent-increase intron retention of SF3B2 and SF3A1 and decreased effect at reducing PLK-1 and SF2 expression when compared to 1.
Figure 5.
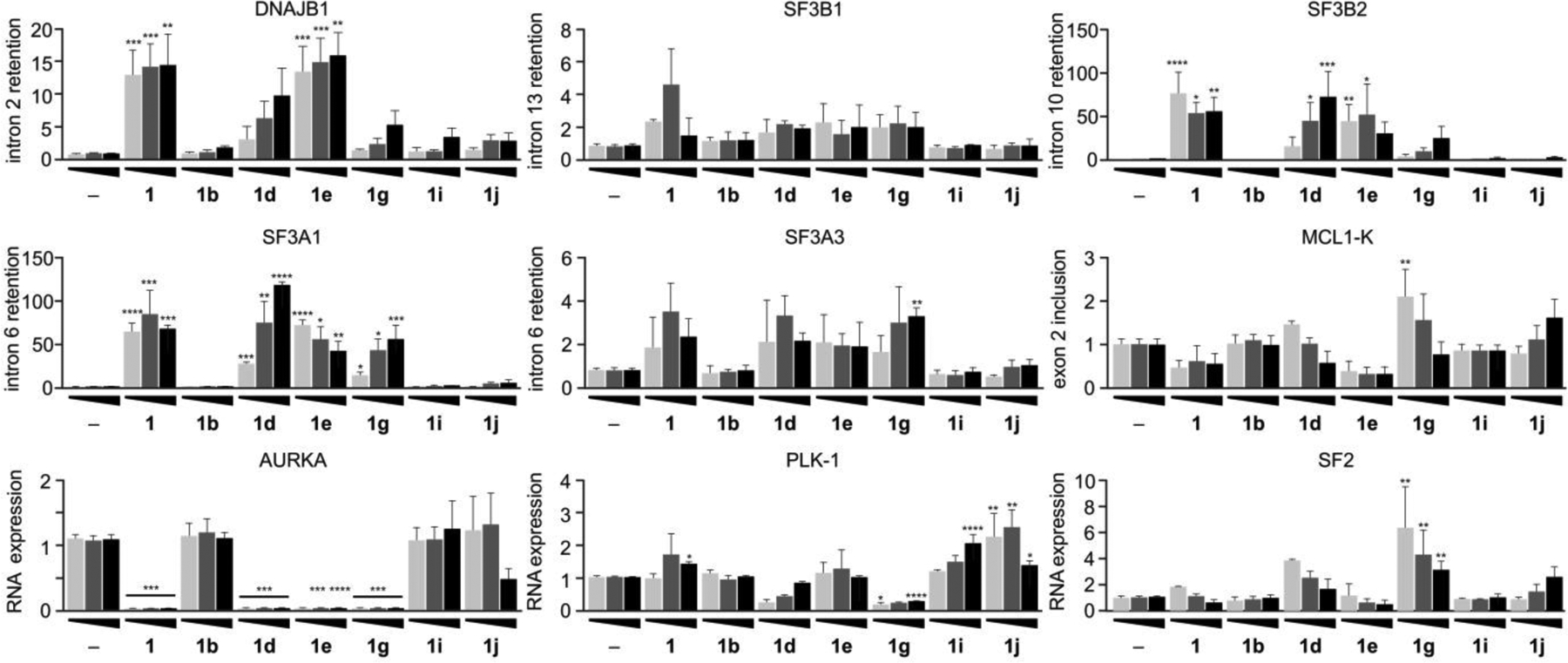
Structure-splicing profiles for RNA splice modulation after 24 h treatment. HCT116 tumor cells were treated with analogues at 100 nM (light grey), 250 nM (grey), or 500 nM (black) for 24 h, and then cellular RNA was isolated, purified and analyzed by qRT-PCR. Primers were designed to evaluate intron retention or exon skipping, direct responses to splice modulator treatment. The same genes are evaluated in this study as that at 4 h in Fig. 4. (–) denotes untreated cells. Numerical values represent the relative levels of intron retention with respect to negative control (–) at 1.
Epimerization of C3 in 17S-FD-895 (1d) led to 1g with 75-fold loss in cell growth inhibitory activity when compared to 1 (Fig. 3). While 1g displayed the expected loss in intron retention of DNAB1, SF3B2, SF3A1 and reduced ability to inhibit SF2 expression (Fig. 5) when compared to 1, it displayed comparable intron retention of SF3A3 and reduced PLK-1 expression when compared to 1. In addition, 1g induced intron retention of SF3A1 and altered the expression of AURKA comparably to 1.
Not all analogues provided productive splice modulatory activity. As illustrated in Fig. 5, epimerization at C7 in 1b (GI50 value 250-fold less than 1) displayed a loss of splicing activity when compared to 1. In contrast, side chain modified 17S,18S,19S-FD-895 (1i) showed a drastic reduction in splicing activity, correlating with its decrease in cell growth inhibition activity, for all genes tested except for PLK-1, for which 1i increased the RNA expression relative to negative controls. Comparably, 17S,20S,21R-FD-895 (1j) provided a splicing profile comparable to 1i with the exception of its activity at reducing the expression of AURKA at 500 nM. Finally, methylation as illustrated by 1e, which had comparable cell growth inhibition activity as 1, was found have comparable or slightly reduced splice modulatory activity as 1. In summary, these observations clearly show the complex interplay between gene selectivity and the overall effect on cell viability.
CONCLUSION
Overall, RNA splice modulation through inhibition of the SF3B complex provides a complicated response, where SARs are evaluated not only by cell growth inhibition activity, but also by discrete changes in splice isoforms of specific genes. While not comprehensive, this study illustrated the unique ability of stereochemical changes within FD-895 (1) to modulate intron retention, exon skipping and overall RNA expression within specific genes. These studies not only provide an important window into understanding the unique medicinal chemical aspects of splice modulation, but also, and perhaps most importantly, they illustrate how inversion of single or sets of stereocenters can lead to changes in gene selectivity.
Outside modifications at C7 in 1b, C10,C11 in 1c and the side chain terminus in 1i and 1j, stereochemical inversion within the core in 1a, side chain in 1d or both in 1e, were not only tolerated but served to modulate their splice activity in a gene-selective manner. Here, we found that inversion of the stereochemistry at C7 in 1b and 1h and C10 and C11 in 1c resulted in loss of activity, indicating a key structural requirement for the core unit. This, along with the fact that modifications at the terminus of the side chain in 1i or 1j resulted in 100-fold loss in activity, would be consistent with activity losses in clinic for 2b (Fig. 2).
Studies are now underway to understand the role of these stereochemical inversions on the three dimensional structure of 1. Here, our effort will be focused on understanding how these stereochemical modifications regulate the conformational states within each analogue. Already observed by Floreancig,32 the vinyl methyl moiety at C12 plays a critical role in regulating the solution state of this family of splicing modulators.33 The question remains whether other stereochemical factors are similarly critical for guiding the ground state of this natural product family.
Few natural products with >10 stereocenters have been interrogated at each stereocenter. As described herein, these studies offer an insight into how nature can uniquely adapt stereochemistry for such a broad variety of downstream targets. Most critically, this updated SAR data (SAR map17 provided in the Table of Contents graphic) provides a resource to guide the identification of analogues with single digit nanomolar activity34 as well as guide the design of linkages for ADC applications.35
EXPERIMENTAL SECTION
General experimental methods:
Chemical reagents were obtained from Acros Organics, Alfa Aesar, Chem-Impex Int., CreoSalus, Fischer Scientific, Fluka, Oakwood Chemical, Sigma-Aldrich, Spectrum Chemical Mfg. Corp., or TCI Chemicals. Deuterated NMR solvents were obtained from Cambridge Isotope Laboratories. All reactions were conducted with rigorously dried anhydrous solvents that were obtained by passing through a column composed of activated A1 alumina or purchased as anhydrous. Anhydrous N,N-dimethylformamide was obtained by passage over activated 3Å molecular sieves and a subsequent NaOCN column to remove traces of dimethylamine. Triethylamine (Et3N) was dried over Na and freshly distilled. Ethyl-N,N-diisopropylamine (EtNi-Pr2) was distilled from ninhydrin, then from KOH. Anhydrous CH3CN was obtained by distillation from CaH2. All reactions were performed under positive pressure of argon in oven-dried glassware sealed with septa, with stirring from a Teflon coated stir bars using an IKAMAG RCT-basic stirrer (IKA GmbH). Solutions were heated on adapters for IKAMAG RCT-basic stirrers. Analytical Thin Layer Chromatography (TLC) was performed on Silica Gel 60 F254 precoated glass plates (EM Sciences). Preparative TLC (pTLC) was conducted on Silica Gel 60 plates (EM Sciences). Visualization was achieved with UV light and/or an appropriate stain (I2 on SiO2, KMnO4, bromocresol green, dinitrophenylhydrazine, ninhydrin, and ceric ammonium molybdate). Flash chromatography was carried out on Fischer Scientific Silica Gel, 230–400 mesh, grade 60 or SilaFlash Irregular Silica Gel P60, 40–63 μm mesh, grade 60. Yields correspond to isolated, chromatographically, and spectroscopically homogeneous materials. 1H NMR and 13C NMR spectra were recorded on a Varian VX500 spectrometer equipped with an Xsens Cold probe. This probe was subject to radio frequency interference (RFI) at 154±3 and is reported as noise on each 13C spectrum. Chemical shift δ values for 1H and 13C spectra are reported in parts per million (ppm) and multiplicities are abbreviated as s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. All 13C NMR spectra were recorded with complete proton decoupling. FID files were processed using MestraNova 12.0.3. (MestreLab Research). Electrospray (ESI) mass spectrometric analyses were performed using a ThermoFinnigan LCQ Deca spectrometer, and high-resolution analyses were conducted using a ThermoFinnigan MAT900XL mass spectrometer with electron impact (EI) ionization. A ThermoScientific LTQ Orbitrap XL mass spectrometer was used for high-resolution electrospray ionization mass spectrometry analysis (HR-ESI-MS). FTIR spectra were obtained on a Nicolet magna 550 series II spectrometer as thin films on either KBr or NaCl discs, and peaks are reported in wavenumbers (cm−1). Optical rotations [α]D were measured using a Perkin-Elmer Model 241 polarimeter with the specified solvent and concentration and are quoted in units of deg cm2 g−1.
Spectral data tabulation:
Spectral data and procedures are provided for all new compounds and copies of select spectra have been provided. NMR tables were computed by hand using assigned spectra in MestraNova 12.0.3. Each peak was checked one peak at a time through Zoom meetings (W.C. and J. J. L). Copies of these assignment files can be provided upon email request.
Compound purity:
Samples of 1 and 1a-1i used for testing was over >95% pure as evident by the NMR analyses as given by 1D and 2D datasets herein provided for each compound. These datasets were conducted on the exact materials used for screening. HPLC analyses were conducted in January 2023 (studies in Figures 3 and 4 were complete in 12/2020) and showed trace <5% decomposition over this period of storage. Supporting Figures S1–S11 provide full routes for the preparation of 1 and 1a-1i. NMR data for 1 and 1a-1i has been tabulated in Supporting Tables S1–S11.
General Stille Coupling Procedure:
following protocol was used to couple 2-2j and 3-3j for the synthesis of 1-1j. Vinylstannanes 4 and 4a-4j (1.1 to 1.5 eq) and cores 5 and 5a-5f (1 eq) were combined in a 50 mL flask and dried via rotary evaporation of benzene. To the mixture was then sequentially added CuCl (1.5 to 2.0 eq), KF (1.5 to 2.0 eq) and XPhos Pd G2 (0.01 to 0.05 eq) and anhydrous t-BuOH (1 to 10 mL). The reaction vessel was purged under Ar, heated to 50 °C and stirred overnight, at which point solution turns into a gray cloudy mixture. The mixture was then filtered through a plug of Celite and eluted with acetone (20 mL when scaled under 1 g and then 20 mL/g when at larger scale). The elutants were concentrated on a rotary evaporator to yield a crude brown semi-solid. Pure 1-1j was obtained as a white semi-solid by flash chromatography over neutral silica gel, eluting with a gradient of hexanes to 1:1 acetone/hexanes.
(1R,2R)-1-((2R,3R)-3-((2R,3S)-3-Methoxypentan-2-yl)oxiran-2-yl)-2-methylbut-3-yn-1-ol (17).
Aldehyde 15 (0.701 g, 4.07 mmol, 1.0 eq) and allenylstannane 16 (2.10 g, 6.12 mmol, 1.5 eq) in a 100 mL flask were dissolved in anhydrous CH2Cl2 (40 mL) and purged with an Ar atmosphere. The mixture was cooled to −78 °C and BF3•Et2O (0.75 mL, 4.74 mmol, 1.2 eq) was added dropwise over 5 min. The reaction was stirred for 1 h at −78 °C. A mixture of MeOH (5 mL) and satd. NaHCO3 (1 mL) was added, and the solution was warmed to rt. The phases were separated, and the aqueous phases were extracted with Et2O (3 × 100 mL). The organic phases were combined, dried with Na2SO4, and concentrated on a rotary evaporator. Alkyne 17 (0.692 g, 75%) was obtained in a 10:1 mixture of diastereomers (by NMR) as a colorless oil by flash chromatography, eluting with a gradient of hexanes to 1:3 Et2O/hexanes. TLC (2:1 hexanes/EtOAc): Rf = 0.50; 1H NMR (CDCl3, 400 MHz) δ 3.55 (s, 3H), 3.41 (m, 1H), 3.21 (ddd, J = 10.4, 6.4, 3.9 Hz, 1H), 3.07 (dd, J = 4.6, 2.3 Hz, 1H), 3.0 (dd, J = 8.1, 2.2 Hz, 1H), 2.67 (m, 1H), 2.16 (d, J = 2.5 Hz, 1H), 2.07 (bs, 1H), 1.57 (m, 3H), 1.31 (d, J = 6.9 Hz, 3H), 0.97 (d, J = 7.0 Hz, 3H), 0.90 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 83.8, 77.4, 73.7, 71.2, 59.5, 59.4, 58.3, 39.0, 31.1, 23.9, 17.2, 10.4, 10.2; ESI–MS m/z 249.14 [M+Na]+; FTIR (film) νmax 3430, 3310, 2967, 2935, 2878, 1457, 1379, 1260, 1093 cm−1; HR– ESI–MS m/z calcd. for C13H22O3Na [M+Na]+: 249.1467, found 249.1462. [α]25D = +10.2 ° (c = 1.0, CH2Cl2).
(1R,2R,E)-1-((2R,3R)-3-((2R,3S)-3-Methoxypentan-2-yl)oxiran-2-yl)-2-methyl-4-(tributylstannyl)but-3-en-1-ol (4).
PdCl2(PPh3)2 (0.155 g, 0.221 mmol, 0.1 eq) was added to a solution of alkyne 17 (0.501 g, 2.21 mmol, 1.0 eq) in a 50 mL flask in anhydrous THF (20 mL). The mixture was cooled to 0 °C and n-Bu3SnH (1.58 mL, 5.08 mmol, 2.3 eq) was added dropwise. The mixture was stirred for 45 min at 0 °C, at which point the resulting mixture was concentrated to yield a black crude oil. The material was extracted into hexanes, filtered through a pad of Celite and was eluted with hexanes. The elutant was concentrated on a rotary evaporator, and this process was repeated twice until a clear black solution was achieved. Pure vinylstannane 4 (0.571 g, 50%) was obtained as a 5:1 mixture of diastereomers by flash chromatography, eluting with a gradient of hexanes to CH2Cl2 to 1:20 Et2O/CH2Cl2. The desired regioisomer and diastereomer can be obtained in 95+% purity by additional flash chromatography, eluting with a gradient of hexanes to CH2Cl2 to 1:20 Et2O/CH2Cl2. TLC (10:1 hexanes/Et2O): Rf = 0.28 (CAM stain); 1H NMR (C6D6, 500 MHz) δ 6.20 (m, 2H), 6.19 (d, J = 6.8 Hz, 1H), 3.34 (td, J = 4.9, 1.8 Hz, 1H), 3.23 (s, 3H), 3.16 (td, J = 6.3, 4.2 Hz, 1H), 2.98 (dd, J = 8.0, 2.3 Hz, 1H), 2.84 (dd, J = 4.3, 2.3 Hz, 1H), 2.51 (td, J = 6.9, 5.2 Hz, 1H), 1.61 (m, 6H), 1.40 (m, 7H), 1.19 (d, J = 6.9 Hz, 3H), 0.97 (m, 19H), 0.86 (t, J = 7.4 Hz, 3H); 13C NMR (C6D6, 500 MHz) δ 154.5, 150.5, 150.4, 150.4, 150.3, 83.8, 83.3, 72.8, 59.0, 57.5, 57.3, 57.2, 39.0, 39.0, 29.3, 27.4, 23.5, 15.9, 15.8, 13.4, 10.5, 9.6, 9.4; HR–ESI–MS m/z calcd. for C25H51O3Sn [M+H]+ 519.2861, found 519.2839; [α]25D = −2.3° (c = 1.0, CH2Cl2).
FD-895 (1):
Compound 1 was prepared applying the general Stille coupling method to core 5 (0.125 g, 0.268 mmol, 1.0 eq) and stannane 4 (0.208 g, 0.402 mmol, 1.5 eq) using CuCl (39.8 mg, 0.402 mmol, 1.5 eq), KF (23.4 mg, 0.403 mmol, 1.5 eq) and XPhos Pd G2 (11.0 mg, 0.011 mmol, 0.05 eq) and anhydrous t-BuOH (10 mL) to yield 1 (121.0 mg, 80%). TLC (1:3 acetone/CH2Cl2): Rf = 0.28 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C31H50O9Na [M+Na]+: 589.3353, found 589.3348; [α]25D = +6.8 ° (c = 1.0, CH2Cl2). The complete route used to prepare 1 is provided in Supporting Figure S1. NMR data has been tabulated in Supporting Table S1. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
3S-FD-895 (1a):
Compound 1a was prepared applying the general Stille coupling method to core 5a24 (0.063 g, 0.135 mmol, 1.0 eq) and stannane 4 (0.105 g, 0.203 mmol, 1.5 eq) using CuCl (20.1 mg, 0.203 mmol, 1.5 eq), KF (11.8 mg, 0.203 mmol, 1.5 eq) and XPhos Pd G2 (5.1 mg, 0.006 mmol, 0.05 eq) and anhydrous t-BuOH (10 mL) to yield 1a (65.1 mg, 85%). TLC (1:3 acetone/CH2Cl2): Rf = 0.18 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C31H50O9Na [M+Na]+: 589.3353, found 589.3347; [α]25D = +9.4 ° (c = 1.0, CH2Cl2). The complete route used to prepare 1a is provided in Supporting Figure S2. NMR data has been tabulated in Supporting Table S2. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
(3R,6R,7S)-(3S,4S,E)-1-iodo-2,4-dimethylhexa-1,5-dien-3-yl-3-((tert-butyldimethyl- silyl)oxy)-6,7-dihydroxy-6-methylnon-8-enoate (36b).
Zinc triflate (1.60 g, 4.40 mmol, 5.8 eq) and EtSH (0.95 mL, 17.7 mmol, 23.3 eq) was added to a solution of 35 (0.500 g, 0.761 mmol, 1.0 eq) in CH2Cl2 (50 mL) at 0 °C. The reaction was warmed to rt. After 4 h satd. NaHCO3 (10 mL) was added. The phases were separated, and the organic phases were dried with Na2SO4 and concentrated by a rotary evaporator. Pure diol 37b (356.0 mg, 87%) was obtained as colorless oil by flash chromatography, eluting with a gradient from hexanes to 1:4 EtOAc/hexanes. TLC (1:4 EtOAc/hexanes): Rf = 0.30 (CAM stain); 1H NMR (500 MHz, CDCl3) δ 6.35 (d, J = 1.3 Hz, 1H), 5.62 (dd, J = 15.1, 9.7 Hz, 1H), 5.33 (dd, J = 15.2, 9.9 Hz, 1H), 5.01 (d, J = 10.7 Hz, 1H), 3.72 (m, 1H), 3.69 (d, J = 9.8 Hz, 1H), 2.40 (m, 1H), 2.38 (dd, J = 13.8, 3.3 Hz, 1H), 2.30 (dd, J = 13.8, 4.8 Hz, 1H), 1.81 (s, 3H), 1.68 (d, J = 1.2 Hz, 3H), 1.50 (m, 2H), 1.29 (m, 2H), 1.20 (s, 3H), 1.15 (bs, 1H), 0.81 (d, J = 6.9 Hz, 3H), 0.80 (s, 9H), −0.02 (s, 3H), −0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 168.8, 143.9, 137.1, 130.2, 128.5, 83.9, 80.6, 73.7, 70.6, 40.6, 36.1 30.4, 29.8, 24.8, 25.9, 18.3, 16.6, −4.6, −4.7; [α]25D= −28.1 ° (c = 1.00, CH2Cl2).
(4R,7R,11S,12S,E)-4-((tert-Butyldimethylsilyl)oxy)-7-hydroxy-12-((E)-1-iodoprop-1-en-2-yl)-7,11-dimethyloxacyclododec-9-ene-2,8-dione (38b).
Diol 37b (300.1 mg, 0.557 mmol, 1.0 eq, 1.0 eq) was dissolved in DMSO (3 mL) in a scintillation vial and IBX (389.0 mg, 1.39 mmol, 2.5 eq) was added in one portion. The mixture was stirred at rt for 3 h. EtOAc (50 mL) and H2O (50 mL) were added, and the phases were separated. The organic phase was washed with H2O (3 × 25 mL), dried over Na2SO4, and concentrated by a rotary evaporator. Pure ketone 38b (290 mg, 97%) was obtained as a colorless oil by flash chromatography, eluting with a gradient of hexanes to 1:4 EtOAc/hexanes. TLC (1:4 EtOAc/hexanes): Rf = 0.40 (CAM stain); 1H NMR (500 MHz, C6D6) δ 6.87 (d, J = 15.6 Hz, 1H), 6.37 (dd, J = 15.6, 9.7 Hz, 1H), 6.19 (d, J = 1.2 Hz, 1H), 5.02 (d, J = 10.4 Hz, 1H), 4.25 (tt, J = 8.3, 4.1 Hz, 1H), 2.34 (dd, J = 12.8, 3.6 Hz, 1H), 2.20 (m, 1H), 2.15 (dd, J =12.8, 9.1 Hz, 1H), 1.88 (bs, 1H), 1.79 (ddd, J = 14.0, 9.2, 6.5 Hz, 1H), 1.65 (m, 1H), 1.63 (d, J = 1.7 Hz, 3H), 1.52 (m, 1H), 1.44 (m, 1H), 1.23 (s, 3H), 0.96 (s, 9H), 0.46 (d, J = 6.7 Hz, 3H), 0.10 (s, 3H), 0.05 (s, 3H); 13C NMR (125 MHz, C6D6) δ 202.3, 168.4, 146.7, 143.7, 129.3, 84.3, 79.5, 79.0, 69.0, 44.3, 40.3, 36.9, 32.6, 26.1, 19.1, 18.3, 15.5, −4.3, −4.4; [α]25D= −46.8 ° (c = 1.00, CH2Cl2).
(4R,7R,8R,11S,12S,E)-4-((tert-Butyldimethylsilyl)oxy)-7,8-dihydroxy-12-((E)-1-iodoprop-1-en-2-yl)-7,11-dimethyloxacyclododec-9-en-2-one (39b).
CeCl3•7 H2O (0.298 g, 0.800 mmol, 2.0 eq) was added to a solution of 38b (0.215 g, 0.401 mmol, 1.0 eq) in EtOH (20 mL) and cooled to −20 °C. KBH4 (26.1 mg, 0.484 mmol, 1.2 eq) was added in one portion, and the mixture was stirred for 10 min. The reaction was quenched with water (1 mL) and then satd. NH4Cl (10 mL). The resulting solution was extracted with EtOAc (3 × 100 mL). The combined organic phases were dried over NaSO4 and concentrated by a rotary evaporator. Pure diol 39b (187.5 mg, 87%) was obtained in a 5:1 dr by flash chromatography, eluting with a gradient of hexanes to 1:3 EtOAc/hexanes. TLC (1:4 EtOAc/hexanes): Rf = 0.28 (CAM stain); 1H NMR (500 MHz, C6D6) δ 6.36 (s, 1H), 5.93 (dd, J = 15.6, 2.9 Hz, 1H), 5.30 (dd, J = 15.6, 9.3 Hz, 1H), 5.01 (d, J = 10.1 Hz, 1H), 3.79 (m, 1H), 3.75 (m, 1H), 2.31 (m, 1H), 2.26 (m, 2H), 1.83 (m, 1H), 1.72 (s, 3H), 1.60 (m, 2H), 1.43 (d, J = 5.2 Hz, 1H), 1.29 (m, 1H), 1.18 (s, 3H), 1.01 (s, 9H), 0.65 (d, J = 6.7 Hz, 3H), 0.08 (s, 3H), 0.07 (s, 3H); 13C NMR (125 MHz, C6D6) δ 168.4, 144.8, 132.2, 130.0, 128.6, 83.4, 80.7, 78.3, 74.7, 71.0, 41.7, 40.3, 36.1, 31.6, 26.1, 19.5, 18.4, 16.6, −4.5; [α]25D= +2.5 ° (c = 1.00, CH2Cl2).
(2S,3S,6R,7R,10R,E)-7,10-Dihydroxy-2-((E)-1-iodoprop-1-en-2-yl)-3,7-dimethyl-12-oxooxacyclododec-4-en-6-yl acetate (3b).
Diol 39b (41.1 mg, 0.076 mmol, 1.0 eq) was dissolved in 1:3 MeOH/CH2Cl2 (10 mL) in a 20 mL scintillation vial and (1S)-(+)-10-camphorsulfonic acid (57.5 mg, 0.248 mmol, 3.2 eq) was added as a solid in one portion. The mixture was stirred for 5 h, at which point TLC analyses indicated complete conversion of starting material (TBS deprotection occurred). The solvent was removed under rotary evaporation, and the resulting crude was taken up in anhydrous CH2Cl2 (10 mL) in a 20 mL scintillation vial and cooled to 0 °C. Trimethyl orthoformate (10.4 μL, 0.101 mmol, 1.3 eq) was, and the mixture was stirred at 0 °C for 1 h, at which point satd. NaHCO3 (1 mL) was added. The mixture was extracted into CH2Cl2 (15 mL), and the organics were concentrated on a rotary evaporator. Pure core 5b (31.2 mg, 88%) was obtained as a colorless wax by flash chromatography, eluting with a gradient of CH2Cl2 to 1:3 acetone/CH2Cl2. TLC (1:8 acetone/CH2Cl2): Rf = 0.27 (CAM stain); 1H NMR (500 MHz, C6D6) δ 6.19 (d, J = 1.3 Hz, 1H), 5.87 (dd, J = 15.4, 2.4 Hz, 1H), 5.39 (q, J = 1.9 Hz, 1H), 5.24 (d, J = 10.5 Hz, 1H), 5.24 (m, 1H), 3.54 (bs, 1H), 2.25 (m, 2H), 2.19 (d, J = 14.0 Hz, 1H), 1.71 (m, 1H), 1.66 (s, 3H), 1.65 (d, J = 1.7 Hz, 3H), 1.61 (m, 1H), 1.50 (m, 1H), 1.17 (bs, 1H), 1.01 (s, 3H), 0.96 (m, 1H), 0.56 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, C6D6) δ 171.9, 169.2, 144.1, 129.8, 84.2, 80.0, 77.8, 73.7, 69.5, 41.0, 38.8, 36.4, 30.5, 24.7, 20.3, 19.1, 16.5; HR-ESI-MS m/z calcd. for C18H27IO6Na [M+Na]+: 489.0750, found 489.0744; [α]25D = −14.8 ° (c = 1.00, CH2Cl2).
7R-FD-895 (1b):
Multiple batches of 5b were collected to run this step. Compound 1b was prepared by applying general Stille coupling method to core 5b (0.109 g, 0.234 mmol, 1.0 eq) and stannane 4 (0.127 g, 0.245 mmol, 1.05 eq) using CuCl (46.3 mg, 0.468 mmol, 2.0 eq), KF (27.2 mg, 0.468 mmol, 2.0 eq) and XPhos Pd G2 (18.4 mg, 0.023 mmol, 0.1 eq) and anhydrous t-BuOH (10 mL) to yield 1b (99.1 mg, 75%). TLC (1:3 acetone/CH2Cl2): Rf = 0.28 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C31H50O9Na [M+Na]+: 589.3353, found 589.3347; [α]25D = +10.9 ° (c = 1.0, CH2Cl2). The complete route used to prepare 1b is provided in Supporting Figure S3. NMR data has been tabulated in Supporting Table S3. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
(3R,4R,)-1-Iodo-2,4-dimethylhexa-1,5-dien-3-ol (33c).
(E)-But-2-ene (20.0 mL, 569.4 mmol, 9.2 eq) was condensed and added to a 1 L reaction flask containing anhydrous THF (300 mL) at −78 °C. KOt-Bu (11.4 g, 101.6 mmol, 1.6 eq) was added, and the mixture was stirred at −78 °C for 30 min. n-BuLi (40.0 mL, 100.0 mmol, 1.6 eq) was added dropwise over 15 min, and the resulting yellow mixture was stirred at −78 °C for an additional 30 min. A solution of (+)-B-methoxydiisopinocampheylborane (25.3 g, 80.0 mmol, 1.3 eq) in anhydrous THF (100 mL) was added dropwise over 15 min, and the mixture turned clear. After stirring the mixture for 30 min, BF3•Et2O (13.2 mL, 83.0 mmol, 1.3 eq) was added dropwise over 10 min, and the mixture was stirred for an additional 10 min. After cooling the mixture to −94 °C, a solution of 32 (12.1 g, 61.7 mmol, 1.0 eq) in anhydrous THF (75 mL) was added dropwise over 45 min. The mixture was allowed to warm to rt and stirred for 16 h. H2O (200 mL) was added, and the mixture was concentrated on a rotary evaporator. Vinyl iodide 33c (7.80 g, 50%) was obtained at a 7:1 dr by flash chromatography, eluting with CH2Cl2. TLC (CH2Cl2): Rf = 0.40 (KMnO4); 1H NMR (500 MHz, CDCl3) δ 6.26 (s, 1H), 5.72 (m, 1H), 5.18 (d, J = 16.0 Hz, 1H), 5.18 (d, J = 11.3 Hz, 1H), 3.87 (dd, J = 8.1, 2.9 Hz, 1H), 2.36 (h, J = 7.4 Hz, 1H), 1.88 (d, J = 2.9 Hz, 1H), 1.82 (bs, 3H), 0.92 (d, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 148.1, 140.0, 117.4, 80.2, 80.0, 42.4, 19.4, 16.6; HR-ES-MS m/z calcd. for C8H13IONa [M+Na]+: 274.9909, found 274.9997; [α]25D = +21.4 ° (c = 1.0, CH2Cl2).
(3S,4S,E)-1-Iodo-2,4-dimethylhexa-1,5-dien-3-yl-(3R)-3-((tert-butyldimethylsilyl)oxy)-5-((4R,5S)-2-(4-methoxyphenyl)-4-methyl-5-vinyl-1,3-dioxolan-4-yl)pentanoate (34c).
DMAP (0.15 g, 1.23 mmol, 0.1 eq) and pivalic anhydride (3.71 mL, 21.7 mmol, 1.8 eq) were added sequentially to a 250 mL flask containing 27 (5.51 g, 12.2 mmol, 1.0 eq) and alcohol 33c (3.23 g, 12.8 mmol, 1.05 eq). The mixture was purged with Ar and stirred neat at 50 °C for 8 h. Pivalic anhydride was removed from the mixture under airflow. Crude material was then loaded directly onto silica gel in hexanes and eluted with a gradient of hexanes to 1:10 Et2O/hexanes. Pure esters 34c (6.44 g, 73%) were obtained as a clear oil. TLC (1:4 Et2O/hexanes): Rf = 0.40 and 0.38 (CAM stain); 1H NMR (500 MHz, C6D6) δ 7.57 (d, J = 8.7 Hz, 2H), 7.55 (d, J = 8.7 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.6 Hz, 2H), 6.22 (s, 1H), 6.20 (s, 1H), 5.93 (s, 1H), 5.83 (m, 1H), 5.79 (m, 2H), 5.63 (m, 1H), 5.60 (m, 1H), 5.33 (dt, J = 17.2, 1.6 Hz, 1H), 5.22 (d, J = 8.1 Hz, 1H), 5.19 (d, J = 8.1 Hz, 1H), 5.09 (dq, J = 10.4, 1.4 Hz, 1H), 4.96 (m, 2H), 4.27 (m, 1H), 4.22 (m, 1H), 4.12 (dt, J = 6.6, 1.3 Hz, 1H), 3.29 (s, 3H), 3.26 (s, 3H), 2.46 (dd, J = 15.0, 6.3 Hz, 1H), 2.42 (dd, J = 15.0, 6.6 Hz, 1H), 2.30 (dd, J = 15.0, 5.6 Hz, 1H), 2.25 (m, 1H), 2.22 (dd, J = 15.0, 5.7 Hz, 1H), 1.98 (dt, J = 13.0, 4.0 Hz, 1H), 1.87 (m, 1H), 1.79 (m, 1H), 1.71 (d, J = 1.1 Hz, 3H), 1.69 (d, J = 1.1 Hz, 3H), 1.67 (m, 1H), 1.25 (s, 3H), 1.24 (m, 2H), 1.21 (s, 3H), 1.01 (s, 9H), 0.98 (s, 9H), 0.67 (d, J = 5.3 Hz, 3H), 0.65 (d, J = 5.3 Hz, 3H), 0.14 (s, 3H), 0.14 (s, 3H), 0.12 (s, 3H), 0.09 (s, 3H); 13C NMR (125 MHz, C6D6) δ 170.3, 170.3, 160.8, 160.6, 144.8, 144.7, 139.8, 133.6, 132.9, 131.2, 128.6, 128.4, 128.2, 128.1, 127.6, 118.0, 117.9, 115.8, 115.8, 114.0, 113.9, 102.7, 102.3, 87.9, 86.0, 83.3, 82.4, 82.1, 80.4, 80.3, 69.9, 69.8, 54.8, 42.9, 42.6, 40.5, 40.5, 32.9, 31.9, 31.4, 28.9, 26.2, 26.2, 22.7, 22.1, 20.0, 18.3, 18.3, 16.5, 16.4, −4.4, −4.4, −4.5; FTIR (film) νmax 2956, 2929, 2856, 1739, 1616, 1517, 1378, 1249, 1170, 1070 cm−1; HR-ES-MS m/z calcd. for C32H49IO6SiNa [M+Na]+: 707.2241, found 707.2199; [α]25D = −13.1 ° (c = 1.0, CH2Cl2).
(2R,3R,6S,7R,10R,E)-7,10-dihydroxy-2-((E)-1-iodoprop-1-en-2-yl)-3,7-dimethyl-12-oxooxacyclododec-4-en-6-yl acetate (5c).
Three steps were conducted without isolation of the intermediates. Esters 34c (5.15 g, 7.52 mmol) in a two-necked 3 L flask equipped with a 1 L addition funnel were dissolved into anhydrous, degassed toluene (700 mL). The mixture was purged with Ar and heated to reflux. 2nd Generation Hoyveda-Grubbs catalyst (706.0 mg, 1.13 mmol) in anhydrous, degassed toluene (700 mL) purged under Ar was dropwise added to the solution of 34c in boiling toluene. After stirring for 20 min the mixture turned from a clear green color into a black solution and was further stirred at reflux for 5 h. The mixture 34c was then cooled to rt and concentrated by a rotary evaporator. The crude black semi-solid was then suspended in hexanes and filtered through a pad of Celite and eluted with hexanes. The elutants were concentrated on a rotary evaporator to yield a crude green oil 35c.
Crude lactones 35c were then dissolved in 1:3 MeOH/CH2Cl2 (300 mL) in a 1 L flask. (1S)-(+)-10-Camphorsulfonic acid (3.45 g, 14.9 mmol) was added as a solid in one portion. The mixture was stirred for 5 h, at which point TLC analyses indicated complete conversion of starting material. Satd. NaHCO3 (50 mL) was added, and the mixture was extracted into CH2Cl2 (3 × 200 mL). The organics were collected and concentrated on a rotary evaporator to a crude oil. Crude triol 36c was subjected to dry column vacuum chromatography over silica and the column was washed with CH2Cl2 (500 mL).
Crude triol 36c was eluted with acetone, concentrated, and carried forward without further purification. Trimethyl orthoformate (400 μL, 3.9 mmol) was added dropwise as a solution of CH2Cl2 (20 mL) to a mixture of crude triol 36c and (1S)-(+)-10-camphorsulfonic acid (120 mg, 0.52 mmol). The mixture was stirred at 0 °C for 1 h, at which point satd. NH4Cl (5 mL) was added. The mixture was stirred for 20 min and extracted into CH2Cl2 (150 mL). The organics were concentrated on a rotary evaporator. Pure core 5c (820.0 mg, 23%) was obtained as a mixture of two isomers by flash chromatography, eluting with a gradient of CH2Cl2 to 1:3 acetone/CH2Cl2. TLC (1:8 acetone/CH2Cl2): Rf = 0.328 (CAM stain); 1H NMR (500 MHz, C6D6) δ 6.19 (s, 1H), 5.82 (dd, J = 15.3, 9.8 Hz, 1H), 5.45 (dd, J = 15.3, 10.1 Hz, 1H), 5.18 (d, J = 9.8 Hz, 1H), 5.09 (d, J = 10.6 Hz, 1H), 4.11 (bs, 1H), 2.30 (m, 2H), 2.21 (m, 1H), 2.08 (d, J = 14.9 Hz, 1H), 1.76 (bs, 1H), 1.64 (m, 1H), 1.62 (s, 3H), 1.61 (d, J = 1.1 Hz, 3H), 1.55 (m, 1H), 1.44 (m, 1H), 1.22 (m, 2H), 1.04 (s, 3H), 0.51 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, C6D6) δ 171.7, 169.0, 143.8, 139.8, 126.9, 84.4, 80.0, 79.0, 73.2, 69.3, 41.1, 38.4, 35.8, 30.2, 24.7, 20.8, 19.1, 16.1; FTIR (film) νmax 3502, 3058, 2959, 2873, 1733, 1616, 1368, 1243, 1168, 1021 cm−1; HR-ESI-MS m/z calcd. for C18H27IO6Na [M+Na]+: 489.0750, found 489.0742; [α]25D = −38.5 ° (c = 1.0, CH2Cl2).
10R,11R-FD-895 (1c):
Compound 1c was prepared applying the general Stille coupling method to core 5c (10.9 mg, 0.023 mmol, 1.0 eq) and stannane 4 (18.0 mg, 0.035 mmol, 1.5 eq) using CuCl (3.5 mg, 0.035 mmol, 1.5 eq), KF (2.1 mg, 0.036 mmol, 1.5 eq) and XPhos Pd G2 (1.9 mg, 0.002 mmol, 0.1 eq) and anhydrous t-BuOH (2 mL) to yield of 1 (6.6 mg, 50%). TLC (1:3 acetone/CH2Cl2): Rf = 0.28 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C31H50O9Na [M+Na]+: 589.3353, found 589.3348; [α]25D = +10.9 ° (c = 1.0, CH2Cl2). The complete route used to prepare 1c is provided in Supporting Figure S4. NMR data has been tabulated in Supporting Table S4. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
17S-FD-895 (1d):
Compound 1d was prepared applying the general Stille coupling method to core 5 (108.0 mg, 0.232 mmol, 1.0 eq) and stannane 4d (185.0 mg, 0.358 mmol, 1.5 eq) using CuCl (35.1 mg, 0.355 mmol, 1.5 eq), KF (20.7 mg, 0.0356 mmol, 1.5 eq) and XPhos Pd G2 (19.0 mg, 0.024 mmol, 0.1 eq) and anhydrous t-BuOH (10 mL) to yield of 1d (66.4 mg, 51%). TLC (1:3 acetone/CH2Cl2): Rf = 0.28 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C31H50O9Na [M+Na]+: 589.3353, found 589.3347; [α]25D = +8.8 ° (c = 1.0, CH2Cl2). The complete route used to prepare 1d is provided in Supporting Figure S5. NMR data has been tabulated in Supporting Table S5. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
Tributyl((3R,4R,E)-4-methoxy-4-((2S,3R)-3-((2R,3S)-3-methoxypentan-2-yl)oxiran-2-yl)-3-methylbut-1-en-1-yl)stannane (4e).
MeI (0.0586 mL, 0.1810 mmol, 1.9 eq) was added at rt to a solution of stannane 4 (50.0 mg, 0.0966 mmol, 1.0 eq) in a mixture of anhydrous THF (10 mL) and anhydrous DMF (3 mL) in a 50 mL flask. The mixture was cooled to 0 °C and NaH (60% in mineral oil, 8.9 mg, 0.221 mmol, 2.3 eq) was added in portions ensuring the mixture remained at 0 °C. The mixture was slowly warmed to rt and stirred for 16 h. After cooling the mixture to 0 °C, a solution of phosphate buffered saline pH 7 (5 mL) was added dropwise. The volatiles were concentrated on a rotary evaporator. The mixture was extracted with hexane (3 × 50 mL). The combined organic phases were washed with brine, dried over Na2SO4, filtered, and concentrated on a rotary evaporator. Pure stannane 4e (55.6 mg, 65%) was obtained as a colorless oil by flash chromatography, eluting with a gradient of hexanes to 10% Et2O/hexanes. TLC (1:10 Et2O/hexanes): Rf = 0.50 (CAM stain); 1H NMR (500 MHz, C6D6) δ 6.35 (dd, J = 19.1, 6.8 Hz, 1H), 6.16 (d, J = 19.1 Hz, 1H), 3.50 (s, 3H), 3.45 (m, 1H), 3.23 (s, 3H), 3.19 (m, 1H), 2.83 (dd, J = 4.4, 2.3 Hz, 1H), 2.75 (dd, J = 4.4, 2.3 Hz, 1H), 2.60 (td, J = 6.9, 5.2 Hz, 1H), 1.61 (m, 8H), 1.39 (m, 8H), 1.19 (d, J = 6.9 Hz, 3H), 1.01 (d, J = 7.1 Hz, 3H), 1.00 (d, J = 8.1 Hz, 3H), 0.95 (t, J = 7.4 Hz, 12H), 0.84 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, C6D6) δ 151.7, 127.3, 86.2, 83.5, 60.2, 58.4, 58.4, 57.6, 56.9, 44.9, 39.7, 32.0, 29.6, 27.8, 23.1, 23.1, 15.6, 14.4, 14.0, 10.9, 10.0, 9.8; FTIR (film) νmax 3454, 3310, 2973, 2937, 2890, 1459, 1101, 840 cm−1; HR-ESI-MS m/z calcd. for C26H53O3Sn [M+H]+ 533.3017, found 533.2994; [α]25D = +10.4 ° (c = 1.0, CH2Cl2).
17-O-Methyl-FD-895 (1e):
Compound 1e was prepared applying the general Stille coupling method to core 5 (9.0 mg, 0.0193 mmol, 1.0 eq) and stannane 4e (15.2 mg, 0.0286 mmol, 1.5 eq) using CuCl (2.8 mg, 0.0286 mmol, 1.5 eq), KF (1.7 mg, 0.0293 mmol, 1.5 eq) and XPhos Pd G2 (1.7 mg, 0.0019 mmol, 0.1 eq) and anhydrous t-BuOH (2 mL) to yield of 1d (8.7 mg, 78%). TLC (1:3 acetone/hexanes): Rf = 0.40 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C32H52O9Na [M+Na]+: 603.3509, found 603.3506; [α]25D = +36.9 ° (c = 1.0, CH2Cl2). The complete route used to prepare 1d is provided in Supporting Figure S6. NMR data has been tabulated in Supporting Table S6. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
Tributyl((3R,4R,E)-4-methoxy-4-((2S,3R)-3-((2R,3S)-3-methoxypentan-2-yl)oxiran-2-yl)-3-methylbut-1-en-1-yl)stannane (2f).
MeI (0.0586 mL, 0.1810 mmol, 1.9 eq) was added at rt to a solution of stannane 4d (50.0 mg, 0.0966 mmol, 1.0 eq) in a mixture of anhydrous THF (10 mL) and anhydrous DMF (3 mL) in a 50 mL flask. The mixture was cooled to 0 °C and NaH (60% in mineral oil, 8.9 mg, 0.221 mmol, 2.3 eq) was added in portions ensuring the mixture remained at 0 °C. The mixture was slowly warmed to rt and stirred for 16 h. After cooling the mixture to 0 °C, a solution of phosphate buffered saline pH 7 (5 mL) was added dropwise. The volatiles were concentrated on a rotary evaporator. The mixture was extracted with hexane (3 × 50 mL). The combined organic phases were washed with brine, dried over Na2SO4, filtered, and concentrated on a rotary evaporator. Pure stannane 4f (31.1 mg, 61%) was obtained as a colorless oil by flash chromatography, eluting with a gradient of hexanes to 10% Et2O/hexanes. TLC (1:10 Et2O/hexanes): Rf = 0.50 (CAM stain); 1H NMR (500 MHz, C6D6) δ 6.39 (dd, J = 19.1, 6.8 Hz, 1H), 6.26 (d, J = 19.1 Hz, 1H), 3.25 (s, 3H), 3.23 (s, 3H), 3.21 (m, 1H), 3.04 (dd, J = 4.4, 2.3 Hz, 1H), 2.83 (m, 2H), 2.75 (dd, J = 4.4, 2.3 Hz, 1H), 1.61 (m, 8H), 1.39 (m, 8H), 1.19 (d, J = 6.9 Hz, 3H), 1.01 (d, J = 7.1 Hz, 3H), 1.00 (d, J = 8.1 Hz, 3H), 0.95 (t, J = 7.4 Hz, 12H), 0.84 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, C6D6) δ 150.7, 127.5, 84.7, 83.3, 59.7, 58.3, 57.6, 57.4, 45.3, 39.4, 29.4, 27.5, 23.5, 16.7, 13.8, 10.3, 9.8, 9.5; FTIR (film) νmax 3454, 3310, 2973, 2937, 2890, 1459, 1101, 840 cm−1; HR-ESI-MS m/z calcd. for C26H53O3Sn [M+H]+ 533.3017, found 533.2998; [α]25D = −8.6° (c = 1.0, CH2Cl2).
17-O-Methyl-FD-895 (1f):
Compound 1f was prepared applying the general Stille coupling method to core 5 (4.4 mg, 0.0094 mmol, 1.0 eq) and stannane 4f (7.6 mg, 0.0143 mmol, 1.5 eq) using CuCl (1.4 mg, 0.0141 mmol, 1.5 eq), KF (0.9 mg, 0.0155 mmol, 1.6 eq) and XPhos Pd G2 (0.8 mg, 0.0010 mmol, 0.1 eq) and anhydrous t-BuOH (1 mL) to yield of 1f (4.2 mg, 77%). TLC (1:3 acetone/hexanes): Rf = 0.40 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C32H52O9Na [M+Na]+: 603.3509, found 603.3502; [α]25D = +36.9 ° (c = 1.0, CH2Cl2). The complete route used to prepare 1f is provided in Supporting Figure S7. NMR data has been tabulated in Supporting Table S7. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
3S,17S-FD-895 (1g):
Compound 1g was prepared applying the general Stille coupling method to core 5a (14.9 mg, 0.0320 mmol, 1.0 eq) and stannane 4e (25.2 mg, 0.0487 mmol, 1.5 eq) using CuCl (4.7 mg, 0.0475 mmol, 1.5 eq), KF (2.8 mg, 0.0482 mmol, 1.5 eq) and XPhos Pd G2 (2.5 mg, 0.0032 mmol, 0.1 eq) and anhydrous t-BuOH (1 mL) to yield of 1e (14.2 mg, 77%). TLC (1:3 acetone/CH2Cl2): Rf = 0.17 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C31H50O9Na [M+Na]+: 589.3353, found 589.3350; [α]25D= +12.4° (c = 1.0, CH2Cl2). The complete route used to prepare 1g is provided in Supporting Figure S8. NMR data has been tabulated in Supporting Table S8. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
7R,17S-FD-895 (1h):
Compound 1h was prepared applying the general Stille coupling method to core 5b (5.2 mg, 0.0112 mmol, 1.0 eq) and stannane 4d (8.6 mg, 0.0166 mmol, 1.5 eq) using CuCl (1.7 mg, 0.0172 mmol, 1.5 eq), KF (1.0 mg, 0.0172 mmol, 1.5 eq) and XPhos Pd G2 (0.9 mg, 0.0011 mmol, 0.1 eq) and anhydrous t-BuOH (1 mL) to yield of 1e (5.0 mg, 77%). Yield: 75%, 5.01 mg; TLC (1:3 acetone/CH2Cl2): Rf = 0.28 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C31H50O9Na [M+Na]+: 589.3353, found 589.3347; [α]25D= +20.1° (c = 1.0, CH2Cl2). The complete route used to prepare 1g is provided in Supporting Figure S9. NMR data has been tabulated in Supporting Table S9. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
((2S,3S)-3-((2R,3S)-3-Methoxypentan-2-yl)oxiran-2-yl)methanol (14i).
t-Butylhydroperoxide (3.3 M, 19.2 mL, 63.4 mmol, 2.0 eq) was added to a 500 mL flask containing a stirring solution of Ti(Oi-Pr)4 (0.650 mL, 2.44 mmol, 0.08 eq), (−)-diethyl tartrate (0.550 mL, 2.21 mmol, 0.07 eq) and powdered 4Å molecular sieves (1 g) in anhydrous CH2Cl2 (100 mL). The mixture was cooled to −20 °C and stirred for 30 min. A solution of alcohol 13 (5.0 g, 31.6 mmol, 1.0 eq) in CH2Cl2 (25 mL) was added dropwise. The reaction was stirred at −20 °C for 4 h. The reaction was quenched via addition of 10% NaOH (10 mL). The mixture was then extracted into CH2Cl2 and concentrated on a rotary evaporator. Pure epoxyalcohol 14i (5.5 g, 87%) was obtained as a 6:1 mixture of diastereomers by flash chromatography, eluting with a gradient of hexanes to 1:1 EtOAc/hexanes. TLC (1:2 EtOAc/hexanes): Rf = 0.10 (CAM stain); 1H NMR (500 MHz, C6D6) δ 3.59 (tq, J = 15.9, 12.6, 2.7 Hz, 1H), 3.37 (m, 12.0, 7.1, 4.4 Hz), 3.02 (s, 3H), 2.77 (dd, J = 7.1, 2.3 Hz, 1H), 2.74 (dd, J = 4.7, 2.5 Hz, 1H), 2.70 (dt, J = 7.3, 4.8 Hz, 1H), 2.36 (t, J = 6.1Hz, 1H), 1.44 (m, 1H), 1.37 (m, 1H), 1.30 (dtd, J = 14.0, 7.5, 5.2 Hz, 1H), 0.96 (d, J = 7.0 Hz, 3H) 0.77 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, C6D6) δ 83.8, 62.2, 58.0, 57.9, 57.7, 38.8, 24.0, 10.4, 10.1; FTIR (film) νmax 3422, 2972, 2930, 2879, 1468, 1103 cm−1; HR-ESI-MS m/z calcd. for C9H18O3 [M]+: 174.1256, found 174.1250.
(2R,3S)-3-((2R,3S)-3-Methoxypentan-2-yl)oxirane-2-carbaldehyde (15i).
A solution of KBr (0.242 g, 2.03 mmol, 0.08 eq) in H2O (10 mL), satd. NaHCO3 (20 mL) and TEMPO (0.266 g, 1.70 mmol, 0.07 eq) were added sequentially to a 500 mL flask containing a solution of epoxyalcohol 14i (4.42 g, 25.4 mmol, 1.0 eq) in CH2Cl2 (150 mL). The mixture was cooled to 0 °C and a solution of NaOCl (2 M, 17.0 mL, 34.0 mmol, 1.3 eq) and satd. NaHCO3 (20 mL) were added dropwise via an addition funnel. The mixture was allowed to warm to rt and stirred for 2 h. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 100 mL). The combined organic phases were washed with brine (100 mL), dried over Na2SO4, filtered, and concentrated on a rotary evaporator. Aldehyde 15i (4.32 g, 99%) was obtained without further purification and was carried on directly to the next step. TLC (1:2 EtOAc/hexanes): Rf = 0.55 (CAM stain); 1H NMR (500 MHz, C6D6) δ 8.67 (d, J = 6.3 Hz, 1H), 2.93 (s, 3H), 2.89 (dd, J = 6.4, 2.0 Hz, 1H), 2.82 (dd, J = 6.4, 2.0 Hz, 1H), 2.60 (dt, J = 7.2, 4.7 Hz, 1H), 1.30 (m, 2H), 1.15 (dqd, J = 14.6, 7.4, 5.3 Hz, 1H), 0.75 (d, J = 7.0 Hz, 3H), 0.67 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, C6D6) δ 198.0, 84.2, 58.4, 58.4, 57.3, 37.6, 23.6, 11.4, 10.2; FTIR (film) νmax 2972, 2930, 2879, 2828, 1732, 1468, 1103 cm−1; HR-ESI-MS m/z calcd. for C9H16O3 [M+H]+: 173.1178, found 173.1174; [α]25D = +36.1 ° (c = 1.0, CH2Cl2).
(1R,2R)-1-((2S,3S)-3-((2R,3S)-3-methoxypentan-2-yl)oxiran-2-yl)-2-methylbut-3-yn-1-ol (17i).
Aldehyde 15i (70.1 mg, 0.407 mmol, 1.0 eq) and allenylstannane 16d (210.0 mg, 0.612 mmol, 1.5 eq) in a 50 mL flask were dissolved in anhydrous CH2Cl2 (10.0 mL) and purged with an Ar atmosphere. The mixture was cooled to −78 °C and BF3•Et2O (75.3 μL, 0.474 mmol, 1.2 eq) was added dropwise over 5 min. The reaction was stirred for 1 h at −78 °C. A mixture of MeOH (5 mL) and satd. NaHCO3 (1 mL) was added, and the solution was warmed to rt. The phases were separated, and the aqueous phases were extracted with Et2O (3 × 50 mL). The organic phases were combined, dried with Na2SO4, and concentrated on a rotary evaporator. Alkyne 17i (69.2 mg, 75%) was obtained in a 5:1 mixture of diastereomers (by NMR) as a colorless oil by flash chromatography, eluting with a gradient of hexanes to 1:3 Et2O/hexanes. TLC (2:1 hexanes/EtOAc): Rf = 0.40; 1H NMR (500 MHz, C6D6,) δ 3.55 (td, J = 8.3, 7.6, 4.4 Hz, 1H), 3.18 (q, J = 2.7 Hz, 1H), 3.14 (s, 3H), 3.03 (ddd, J = 6.8, 4.6, 2.3 Hz, 1H), 2.92 (dt, J = 7.5, 4.8 Hz, 1H), 2.55 (dqd, J = 10.0, 7.1, 3.4 Hz, 1H), 1.86 (m, 1H), 1.54 (m, 2H), 1.39 (m, 1H), 1.30 (d, J = 7.1 Hz, 3H), 1.06 (d, J = 6.8 Hz, 3H), 0.83 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, C6D6,) δ 85.4, 84.6, 71.9, 71.2, 59.0, 58.9, 57.4, 57.3 38.4, 31.0, 23.7, 17.4, 12.2, 10.3; ESI–MS m/z 249.14 [M+Na]+; FTIR (film) νmax 3430, 3310, 2967, 2935, 2878, 1457, 1379, 1260, 1093 cm−1; HR–ESI–MS m/z calcd. for C13H22O3Na [M+Na]+: 249.1467, found 249.1462. [α]25D = +24.2 ° (c = 1.0, CH2Cl2).
(1R,2R,E)-1-((2R,3R)-3-((2R,3S)-3-Methoxypentan-2-yl)oxiran-2-yl)-2-methyl-4-(tributylstannyl)but-3-en-1-ol (4i).
PdCl2(PPh3)2 (15.5 mg, 0.0221 mmol, 0.1 eq) was added to a solution of alkyne 17i (50.1 mg, 0.221 mmol, 1.0 eq) in a 10 mL flask in anhydrous THF (5 mL). The mixture was cooled to 0 °C and n-Bu3SnH (0.179 mL, 0.568 mmol, 2.6 eq) was added dropwise. The mixture was stirred for 45 min at 0 °C, at which point the resulting mixture was concentrated to yield a black crude oil. The material was extracted into hexanes, filtered through a pad of Celite and was eluted with hexanes. The elutant was concentrated on a rotary evaporator, and this process was repeated twice until a clear black solution was achieved. Pure vinylstannane 4i (22.0 mg, 19%) was obtained as a mixture of 1:5 α:β regioisomers by flash chromatography, eluting with a gradient of hexanes to CH2Cl2 to 1:20 Et2O/CH2Cl2. The desired regioisomer and diastereomer can be obtained in 95+% purity by additional flash chromatography, eluting with a gradient of hexanes to CH2Cl2 to 1:20 Et2O/CH2Cl2. This reaction was run once and unoptimized for yield and reaction conditions. TLC (10:1 hexanes/Et2O): Rf = 0.25 (CAM stain); 1H NMR (C6D6, 500 MHz) δ 6.16 (m, 2H), 3.54 (m, 1H), 3.16 (s, 3H), 3.07 (d, J = 7.2, Hz, 1H), 2.93 (m, 2H), 2.49 (m, 2H), 1.98 (s, 1H), 1.60 (m, 9H), 1.39 (dt, J = 15.5, 8.5 Hz, 6H), 1.28 (d, J = 7.0 Hz, 3H), 1.07 (d, J = 6.9 Hz, 3H), 0.96 (m, 12H), 0.87 (t, J = 7.4 Hz, 3H); 13C NMR (C6D6, 500 MHz) δ 151.5, 84.8, 72.7, 59.4, 57.4, 46.5, 38.4, 29.6, 27.7, 23.8, 16.2, 14.0, 12.2, 10.5, 9.8; HR–ESI–MS m/z calcd. for C25H51O3Sn [M+H]+ 519.2861, found 519.2839; [α]25D = +10.1° (c = 1.0, CH2Cl2).
17S,18S,19S-FD-895 (1i):
Compound 1i was prepared applying the general Stille coupling method to core 5 (7.2 mg, 0.0154 mmol, 1.0 eq) and stannane 4d (12.1 mg, 0.0234 mmol, 1.5 eq) using CuCl (2.3 mg, 0.0232 mmol, 1.5 eq), KF (1.3 mg, 0.0224 mmol, 1.5 eq) and XPhos Pd G2 (0.6 mg, 0.0008 mmol, 0.05 eq) and anhydrous t-BuOH (1 mL) to yield of 1i (6.3 mg, 80%). TLC (1:3 acetone/CH2Cl2): Rf = 0.28 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C31H50O9Na [M+Na]+: 589.3353, found 589.3348; [α]25D = +4.2 ° (c = 1.0, CH2Cl2). The complete route used to prepare 1g is provided in Supporting Figure S10. NMR data has been tabulated in Supporting Table S10. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
(2S,3R)-1-((S)-4-Benzyl-2-thioxothiazolidin-3-yl)-3-hydroxy-2-methylpentan-1-one (8j).
(S)-1-(4-Benzyl-2-thioxothiazolidin-3-yl)propan-1-one (7) (23.5 g, 88.5 mmol, 1.0 eq) was added to a 2 L reaction flask and dissolved in CH2Cl2 (700 mL) with mechanical stirring. The mixture was cooled below 0 °C. TiCl4 (10.1 mL, 30.8 mmol, 0.3 eq) was added dropwise over 1 h, while maintaining the temperature below 0 °C, at which point the mixture turned orange. EtN(i-Pr)2 (13.9 mL, 144.9 mmol, 1.6 eq) was added dropwise over 30 min, at which point the resulting black mixture was stirred at 0 °C for 15 min. After cooling the reaction to −94 °C, a solution of propionaldehyde (7.10 mL, 150.9 mmol, 1.7 eq) in anhydrous CH2Cl2 (50 mL) was added dropwise over 1 h. The mixture was stirred at −94 °C for 30 min before being slowly warmed to rt overnight. The mixture was cooled to 0 °C and satd. NaHCO3 (200 mL) was slowly added. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 300 mL). The combined organic phases were washed with brine (300 mL), dried over Na2SO4, filtered, and concentrated on a rotary evaporator. Pure adduct 8j (25.0 g, 87%) was obtained in a 9.5:1 dr mixture by flash chromatography, eluting with a gradient of hexanes to 1:3 EtOAc/hexanes. TLC (1:3 EtOAc/heptane): Rf = 0.63 (CAM stain); 1H-NMR (400 MHz, CDCl3) δ 7.35 (m, 2H), 7.28 (m, 3H), 5.37 (ddd, J = 4.1, 7.0, 10.7 Hz, 1H), 4.52 (dq, J = 2.3, 7.1 Hz, 1H), 3.97 (ddd, J = 2.2, 5.3, 8.0 Hz, 1H), 3.38 (ddd, J = 1.1, 7.2, 11.6 Hz, 1H), 3.25 (dd, J = 4.0, 13.2 Hz, 1H), 3.05 (dd, J = 10.5, 13.2 Hz, 1H), 2.89 (d, J = 11.6 Hz, 1H), 2.05 (m, 1H), 1.59 (m, 1H), 1.45 (dqd, J = 14.7, 7.4, 5.2 Hz, 1H), 1.18 (d, J = 7.1 Hz, 3H), 0.98 (t, J = 7.4 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ 201.7, 178.7, 136.5, 129.6, 129.1, 127.4, 72.6, 69.0, 42.2, 37.0, 31.9, 26.7, 10.6, 10.5. FTIR (film) νmax 3444, 3027, 2964, 2937, 2876, 1689, 1455, 1352, 1258, 1191, 1164, 1041, 1029, 960 cm−1; HR-ESI-MS m/z calcd. for C16H21NO2S2Na [M+Na]+: 346.0912, found 346.0902; [α]25D = 36.2 ° (c = 1.0 CH2Cl2).
(2S,3R)-3-Hydroxy-N-methoxy-N,2-dimethylpentanamide (9j).
N,O-Dimethylhydroxylamine hydrochloride (8.7 g, 142.4 mmol, 3.2 eq) and imidazole (9.1 g, 133.7 mmol, 3.0 eq) were added in succession to a solution of 8j (14.4 g, 44.5 mmol, 1.0 eq) in CH2Cl2 (500 mL) in a 2 L reaction vessel at rt. The mixture was stirred at rt for an additional 16 h. H2O (300 mL) was added, and the mixture was separated followed by extraction of the aqueous phase with CH2Cl2 (3 × 500 mL). The combined organic phases were washed with brine (500 mL), dried over Na2SO4, filtered, and concentrated on a rotary evaporator to afford a yellow oil. Pure amide 9j (6.60 g, 85%) was obtained by flash chromatography, eluting with a gradient of hexanes to 3:1 EtOAc/hexanes. TLC (3:1 EtOAc/heptane): Rf = 0.17 (KMnO4); 1H NMR (500 MHz, CDCl3) δ 3.77 (m, 1H), 3.70 (s, 3H), 3.19 (s, 3H), 3.16 (m, 1H), 2.90 (m, 1H), 1.77 (bs, 1H), 1.58 (dp, J = 5.1, 7.7 Hz, 1H), 1.39 (dt, J = 14.3, 7.2 Hz, 1H), 1.15 (d, J = 7.1 Hz, 3H), 0.96 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 178.5, 73.1, 61.7, 38.0, 32.0, 26.8, 10.6, 10.1; FTIR (film) νmax 2969, 2917, 2855, 1719, 1449, 1265, 1178, 1108, 1020, 715 cm−1; HR-ESI-MS m/z calcd. for C8H17NO3Na [M+Na]+: 198.1106, found 198.1101; [α]25D = +7.3 ° (c = 1.0, CH2Cl2).
(2S,3R)-N,3-Dimethoxy-N,2-dimethylpentanamide (10j).
MeI (35.8 mL, 110.6 mmol, 3.9 eq) was added at rt to a solution of amide 9j (5.0 g, 28.5 mmol, 1.0 eq) in a mixture of anhydrous THF (200 mL) and anhydrous DMF (50 mL) in a 1 L reaction vessel. The mixture was cooled to 0 °C and NaH (60% in mineral oil, 2.8 mg, 70.8 mmol, 2.5 eq) was added in portions ensuring the mixture remained at 0 °C. The mixture was slowly warmed to rt and stirred for 16 h. After cooling the mixture to 0 °C, a solution of phosphate buffered saline pH 7 (200 mL) was added dropwise. The volatiles were concentrated on a rotary evaporator. H2O (100 mL) was added to the residue, and the obtained mixture was extracted with t-butyl methyl ether (3 × 300 mL). The combined organic phases were washed with brine (300 mL), dried over Na2SO4, filtered, and concentrated on a rotary evaporator. Pure amide 10j (4.1 g, 76%) was obtained as a colorless oil by flash chromatography, eluting with a gradient of hexanes to 1:1 EtOAc/hexanes. TLC (3:1 EtOAc/heptane): Rf = 0.27 (KMnO4); 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 3.40 (s, 3H), 3.29 (ddd, J = 8.1, 7.0, 3.8 Hz, 1H), 3.17 (s, 3H), 3.0 (bs, 1H), 1.57 (dqd, J = 14.8, 7.5, 3.9 Hz, 1H), 1.41 (dp, J = 14.3, 7.2 Hz, 1H), 1.20 (d, J = 6.9 Hz, 3H), 0.91 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 176.5, 83.9, 61.6, 58.7, 39.6, 32.2, 25.3, 14.5, 9.6; FTIR (film) νmax 3581, 3502, 2969, 2934, 2882, 2820, 1658, 1457, 1379 cm−1; HR-ESI-MS m/z calcd. for C9H19NO3Na [M+Na]+: 212.1263, found 212.1257; [α]25D = +16.1 ° (c = 1.0 CHCl3).
Ethyl (4R,5R,E)-5-methoxy-4-methylhept-2-enoate (12j).
Amide 10j (4.00 g, 21.10 mmol, 1.0 eq) was dissolved in anhydrous CH2Cl2 (100 mL) in a 500 mL flask. The mixture was cooled to −78 °C. DIBAL-H (1.0 M, 32.8 mL, 32.8 mol, 1.6 eq) was added dropwise over 45 min at −78 °C and stirred for 1 hr. Acetone (10 mL) was added dropwise over 10 min, and the mixture was warmed to 0 °C. Satd. Rochelle’s salt (100 mL) was added over 30 min, and the mixture was stirred at rt for 1.5 h. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 150 mL). The combined organic phases were dried over Na2SO4, filtered, and concentrated on a rotary evaporator. The residue was then dried via azeotropic removal of toluene to deliver aldehyde 11j, which was used immediately after preparation.
A solution of triethyl phosphonoacetate (21.2 mL, 83.7 mmol, 4.0 eq) in anhydrous 2-methyltetrahydrofuran (40 mL) was added dropwise over 30 min to a 500 mL reaction flask containing a suspension of NaH (60% in mineral oil, 3.6 g, 90.0 mol, 4.3 eq) in anhydrous 2-methyltetrahydrofuran (150 mL) cooled to 0 °C. The mixture was stirred at 0 °C for 15 min and a solution of 11j in 2-methyltetrahydrofuran (100 mL) was added dropwise over 30 min. The mixture was stirred at rt for 16 h, cooled to 0 °C and quenched with satd. NH4Cl (250 mL). The organics were concentrated on a rotary evaporator. The mixture was extracted with EtOAc (2 × 300 mL), and the combined organic phases were dried over Na2SO4, filtered, and concentrated on a rotary evaporator. Pure ester 12j (3.27 g, 77% over two steps) was obtained as a colorless oil by flash chromatography, eluting with a gradient of CH2Cl2 to 1:10 EtOAc/CH2Cl2. TLC (CH2Cl2): Rf = 0.14 (CAM stain); 1H NMR (500 MHz, CDCl3) δ 6.94 (ddd, J = 15.8, 7.7, 1H), 5.81 (dd, J = 15.7, 1.4 Hz, 1H), 4.17 (q, J = 7.2 Hz, 2H), 3.36 (s, 3H), 3.00 (ddd, J = 7.5, 5.6, 4.4 Hz, 1H), 2.56 (dddd, J = 7.9, 6.8, 5.6, 1.3 Hz, 1H), 1.51 (dqd, J = 14.9, 7.5, 3.3 Hz, 1H), 1.40 (dp, J = 14.6, 7.3 Hz, 1H), 1.28 (t, J = 7.1 Hz, 3H), 1.05 (d, J = 6.9 Hz, 3H), 0.90 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.9, 151.3, 121.1, 85.6, 60.4, 57.9, 39.3, 23.9, 14.9, 14.4, 9.9; FTIR (film) νmax 2978, 2934, 2882, 2820, 1719, 1650, 1466 cm−1; HR-ESI-MS m/z calcd. for C11H20O3Na [M+Na]+: 223.1310, found 223.1305; [α]25D = +44.9 ° (c = 1.0, CH2Cl2).
(4R,5R,E)-5-Methoxy-4-methylhept-2-en-1-ol (13j).
DIBAL-H (1.0 M, 37.1 mL, 37.1 mmol, 2.5 eq) was added dropwise over 60 min to a 5 L reaction flask containing a solution of ester 12j (3.00 g, 14.98 mmol, 1.0 eq) in anhydrous CH2Cl2 (150 mL) cooled to −78 °C. The mixture was stirred for 1 h at −78 °C. Acetone (30 mL) was then added dropwise. The mixture was warmed to 0 °C, satd. Rochelle’s salt (50 mL) was added, and the mixture was stirred at rt for 2 h. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 100 mL). The combined organic phases were washed with brine (100 mL), dried over Na2SO4, filtered, and concentrated on a rotary evaporator. Pure alcohol 13j (1.93 g, 81%) was obtained by flash chromatography, eluting with a gradient of heptane to 1:1 EtOAc/heptane. TLC (1:3 EtOAc/heptane): Rf = 0.26 (CAM stain); 1H NMR (500 MHz, CDCl3) δ 5.65 (dd, J = 5.6, 2.3 Hz, 1H), 4.09 (m, 1H), 3.35 (s, 3H), 2.92 (ddd, J = 7.6, 5.7, 4.1 Hz, 1H), 2.44 (m, 1H), 1.86 (bs, 1H), 1.51 (dqd, J = 14.9, 7.5, 4.2 Hz, 1H), 1.39 (dp, J = 14.7, 7.4 Hz, 1H), 1.00 (d, J = 6.9 Hz, 3H), 0.90 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 135.2, 129.0, 86.4, 63.9, 57.7, 38.9, 23.4, 16.0, 10.0; FTIR (film) νmax 3388, 2968, 2932, 2876, 2826, 1460, 1375 cm−1; HR-ESI-MS m/z calcd. for C9H18O2Na [M+Na]+: 181.1205, found 181.1199.
17S,18S,19S-FD-895 (1j):
Analogue 1j was prepared once in a five-step manner from 13j without detailed spectroscopic characterization of the intermediates 14j, 15j, 17j and 4j. Due to its lack of activity, we did not resynthesize this material and characterized each intermediate as we did for 1, and 1a-1j. t-Butylhydroperoxide (3.3 M, 4.80 mL, 15.8 mmol, 2.0 eq) was added to a 100 mL flask containing a stirring solution of Ti(Oi-Pr)4 (0.16 mL, 0. 0.60 mmol, 0.1 eq), (−)-diethyl tartrate (0.55 mL, 3 2.21 mmol, 0.3 eq) and powdered 4Å molecular sieves (1 g) in anhydrous CH2Cl2 (100 mL). The mixture was cooled to −20 °C and stirred for 30 min. A solution of alcohol 13j (1.25 g, 7.90 mmol, 1.0 eq) in CH2Cl2 (25 mL) was added dropwise. The reaction was stirred at −20 °C for 4 h. The reaction was quenched via addition of 10% NaOH (10 mL). The mixture was then extracted into CH2Cl2 and concentrated on a rotary evaporator.
Pure epoxyalcohol 14j (1.21 g, 88%) was obtained as a 6:1 mixture of diastereomers by flash chromatography, eluting with a gradient of hexanes to 1:1 EtOAc/hexanes. Next, a solution of KBr (60.5 mg, 0.51 mmol, 0.08 eq) in H2O (3 mL), satd. NaHCO3 (5 mL) and TEMPO (66.5 mg, 0.43 mmol, 0.07 eq) were added sequentially to a 125 mL flask containing a solution of epoxyalcohol 14j (1.11 g, 6.37 mmol, 1.0 eq) in CH2Cl2 (50 mL). The mixture was cooled to 0 °C and a solution of NaOCl (2 M, 4.25 mL, 8.50 mmol, 1.3 eq) and satd. NaHCO3 (5 mL) were added dropwise via an addition funnel. The mixture was allowed to warm to rt and stirred for 2 h. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 50 mL). The combined organic phases were washed with brine (30 mL), dried over Na2SO4, filtered, and concentrated on a rotary evaporator.
Aldehyde 15j (1.10 g, 99%) was obtained without further purification and was carried on directly to the next step. Aldehyde 15j (70.1 mg, 0.407 mmol, 1.0 eq) and allenylstannane 16 (210.0 g, 0.612 mmol, 1.5 eq) in a 50 mL flask were dissolved in anhydrous CH2Cl2 (10.0 mL) and purged with an Ar atmosphere. The mixture was cooled to −78 °C and BF3•Et2O (75.3 μL, 0.474 mmol, 1.2 eq) was added dropwise over 5 min. The reaction was stirred for 1 h at −78 °C. A mixture of MeOH (5 mL) and satd. NaHCO3 (1 mL) was added, and the solution was warmed to rt. The phases were separated, and the aqueous phases were extracted with Et2O (3 × 50 mL). The organic phases were combined, dried with Na2SO4, and concentrated on a rotary evaporator. Alkyne 17j (69.2 mg, 75%) was obtained in a 5:1 mixture of diastereomers (by NMR) as a colorless oil by flash chromatography, eluting with a gradient of hexanes to 1:3 Et2O/hexanes.
PdCl2(PPh3)2 (130.0 mg, 0.19 mmol, 0.1 eq) was added to a solution of alkyne 17j (420.0 mg, 1.86 mmol, 1.0 eq) in a 50 mL flask in anhydrous THF (25 mL). The mixture was cooled to 0 °C and n-Bu3SnH (1.50 mL, 4.76 mmol, 2.6 eq) was added dropwise. The mixture was stirred for 45 min at 0 °C, at which point the resulting mixture was concentrated to yield a black crude oil. The material was extracted into hexanes, filtered through a pad of Celite and was eluted with hexanes. The elutant was concentrated on a rotary evaporator, and this process was repeated twice until a clear black solution was achieved. Pure vinylstannane 4j (611.0 mg, 64%) was obtained as a mixture of 1:5 α:β regioisomers by flash chromatography, eluting with a gradient of hexanes to CH2Cl2 to 1:20 Et2O/CH2Cl2. The desired regioisomer and diastereomer can be obtained in >95% purity by additional flash chromatography, eluting with a gradient of hexanes to CH2Cl2 to 1:20 Et2O/CH2Cl2.
Finally, compound 1j was prepared applying the general Stille coupling method to core 5 (4.0 mg, 0.0086 mmol, 1.0 eq) and stannane 4d (6.5 mg, 0.0126 mmol, 1.5 eq) using CuCl (1.3 mg, 0.0131 mmol, 1.5 eq), KF (0.7 mg, 0.0138 mmol, 1.6 eq) and XPhos Pd G2 (0.7 mg, 0.0009 mmol, 0.1 eq) and anhydrous t-BuOH (1 mL) to yield of 1i (3.5 mg, 70%). TLC (1:3 acetone/CH2Cl2): Rf = 0.28 (CAM stain); FTIR (film) νmax 3447, 2963, 2930, 2875, 1739, 1457, 1374, 1239, 1176, 1089, 1021 cm−1; HR-ESI-MS m/z calcd. for C31H50O9Na [M+Na]+: 589.3353, found 589.3350; [α]25D = −2.3 ° (c = 1.0, CH2Cl2). The complete route used to prepare 1g is provided in Supporting Figure S11. NMR data has been tabulated in Supporting Table S11. Copies of 1H-NMR, 13C-NMR, 1H-1H-COSY, 1H-1H-NOESY, 1H-13C-HSQC and 1H-13C-HMBC spectra for 1 are provided in the Supporting Information.
Cell culture.
The HCT-116 cell line was cultured in McCoy’s 5a media (Life Technologies) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL of penicillin and streptomycin at 37 °C in an atmosphere of 5% CO2. Cell splitting was conducted every other day and culturing was not conducted past five generations of the parent cell line. HCT-116 cells were obtained from ATCC and were cultured once and stored. Test for mycoplasma were routinely conducted using a Mycoplasma Detection Kit (Lonza).
Cellular treatments.
Compounds 1 or 1a-1j were dissolved in DMSO (Millipore Sigma) and applied to cells in media so that the DMSO concentration was ≤0.5% DMSO. To achieve these solutions of 1 and 1a-1j were prepared at 1 mg/mL in DMSO and stored at −80 °C for up to 6 months. These solutions were diluted accordingly in DMSO prior to use. Microscaled 1.7 mm NMR studies were conducted to test the solubility of these materials. Here, 5 μL of a 1 mg/mL stock of 1 or 1a-1j in d6-DMSO (same bottle used for all samples) was added to D2O (45 μL). NMR data indicated that each material integrated within 5% deviation when compared to the solvent (d6-DMSO). Copies of spectral data from this study can be provided upon email request.
Cell viability assays.
HCT-116 cells were plated at 5 × 103 cells/well in supplemented McCoy’s 5a media (see Cell culture section above). Cells were cultured at 37 °C in an atmosphere of 5% CO2 for 24 h and then treated with 1 or 1a-1j for a given time period (4h to 72 h were used within the study). After incubation, the cells were washed twice with 100 μL of PBS, charged with 100 μL of PBS followed by 20 μL of Cell Titer Aqueous One Solution (Promega). After 2 h at 37 °C, absorbance readings were taken at 490 nm (test wavelength) and 690 nm (reference wavelength). GI50 values were calculated using Prism (GraphPad) using ≥3 biological replicates. Values and 95% confidence intervals are provided in Supplementary Table S12.
Compound dosing for qRT-PCR studies at 4 h.
In prior studies, we learned that splice modulation occurs very readily (observable as low as 15 minutes after treatment) while cell growth inhibition (GI50 values) occurs slowly (requiring >24 h, 72 h used herein).28 We also learned from this work that 4 h treatments provided an ideal time point to obtain statistically relevant data on a compounds ability to modulate the spicing of individual genes.28 Based on this data and reconfirming this result with a small set of the panel, we standardize the concentrations to ~20 times their 72 h GI50 values. These concentrations (Fig. 4a) were used for all qRT-PCR analyses conducted at 4 h.
Compound dosing for qRT-PCR studies at 24 h.
To further the rigor of our studies, we conducted concentration gradient at 24 h. This time point was chosen to reflect a point where splice modulation by the compound and cell growth inhibitor processes were both ongoing. Here, three concentrations were evaluated for each probe 100 nM (light grey, Fig. 5), 250 nM (grey, Fig. 5), and 500 nM (black, Fig. 5). This data is shown in Fig. 5. It is important to realize that these compounds target the spliceosome and do so in a structure-based manner. The efficacy of each analogue is therefore different for each intron/exon pair within each gene. Currently, we have selected 9 genes in Figure 5 and this data along with that at 4 h represents the complex selectivity modulated by stereochemical inversion and/or functionalization. The use of both 4 h (Fig. 4) and 24 h (Fig. 5) time points, standardization in Fig. 4 and concentration gradients represents our state of the art in analogue screening.
Quantitative real time PCR (qRT-PCR).
HCT-116 cells were treated with 1 or 1a-1j in supplemented McCoy’s 5a with 0.5% DMSO for 4 h or 24 h. Untreated cells were used as a control. Total RNA was isolated using a mirVana miRNA isolation kit (Life Technologies). A 1 μg sample of RNA was subjected to DNAseI from a TURBO DNA free kit (Life Technologies). The cDNA was prepared by using a SuperScript III reverse transcriptase kit (Life Technologies). The amount of unspliced RNA for different genes was determined using Power SYBR Green PCR master mix (Applied Biosciences) by qPCR with specific primers for each gene (Supplementary Table 13). qPCR data was collected on QuantStudio3 96-welled Real Time PCR System (Applied Biosystems). qPCR using 2.5 μM of each primer was performed on 5 ng of the obtained cDNA. qPCR conditions were as follows: 95 °C for 10 min for one cycle, the 95 °C for 30 s, 55 °C for 60 s, 72 °C for 60 s, for 40 cycles using MXPro QPCR software (Agilent). Quantification cycle (Qc) values were identified for each sample, and then RNA levels were calculated using 2−ΔΔCT method. GAPHDH was used as a control for normalization. At least, three biological replicates were conducted for each treatment. Statistical analyses were conducted using a standard one-way ANOVA; p-values were presented such that * denotes p<0.0001. Copies of the raw data and/or tabulated data can be provided upon request.
Supplementary Material
ACKNOWLEDGMENT
This work was funded by a grant from the California Institute for Regenerative Medicine translational stage research project (CIRM-TRAN1) entitled: A Splicing Modulator Targeting Cancer Stem Cells in Acute Myeloid Leukemia. We thank Dr. Anthony Mrse, Dr. Brendan Duggan for enabling the NMR studies, and Dr. Yongxuan Su for LC/MS and HRMS analyses.
ABBREVIATIONS
- CAM
Cerium Ammonium Molybdate stain
- EI
electron impact ionization
- EM
electron microscopy
- ESI
electrospray ionization
- FTIR
Fourier transform infrared
- HR-ESI-MS
high-resolution electrospray ionization mass spectrometry analysis
- MOA
mode or mechanism of action
- ppm
parts per million
- pTLC
preparative thin layer chromatography
- Qc
quantification cycle
- RFI
radio frequency interference
- SAR
structure activity relationship
- SPLM
spliceosome modulator
- TLC
thin layer chromatography
Footnotes
Supporting Information
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
Synthetic schemes for the preparation of 1 and 1a-1i; Tabulated NMR data on 1 and 1a-1i; and NMR spectra from 1 and 1a-1i (PDF)
Cell viability data at 1 h (CSV)
Cell viability data at 4 h (CSV)
Cell viability data at 24h (CSV)
qRT-PCR data at 4 h (CSV)
qRT-PCR data at 24 h (CSV)
Molecular formula strings (CSV)
The authors declare the following competing financial interest: M.D.B and C.H.M.J. are founders, shareholders, and scientific advisors of Aspera Biomedicines.
REFERENCES
- (1).Seki-Asano M; Okazaki T; Yamagishi M; Sakai N; Takayama Y; Hanada K; Morimoto S; Takatsuki A; Mizoue K Isolation and characterization of a new 12-membered macrolide FD-895. J. Antibiot. (Tokyo) 1994, 47, 1395–1401. [DOI] [PubMed] [Google Scholar]
- (2).Sakai T; Sameshima T; Matsufuji M; Kawamura N; Dobashi K; Mizui Y Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. I. Taxonomy, fermentation, isolation and screening. J. Antibiot. (Tokyo) 2004, 57, 173–179. [DOI] [PubMed] [Google Scholar]
- (3).Sakai T; Asai N; Okuda A; Kawamura N; Mizui Y Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. II. Physico-chemical properties and structure elucidation. J. Antibiot. (Tokyo) 2004, 57, 180–187. [DOI] [PubMed] [Google Scholar]
- (4).Mizui Y; Sakai T; Iwata M; Uenaka T; Okamoto K; Shimizu H; Yamori T; Yoshimatsu K; Asada M Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J. Antibiot. (Tokyo) 2004, 57, 188–196. [DOI] [PubMed] [Google Scholar]
- (5).Nakajima H; Sato B; Fujita T; Takase S; Terano H; Okuhara M New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J. Antibiot. (Tokyo) 1996, 49, 1196–1203. [DOI] [PubMed] [Google Scholar]
- (6).Nakajima H; Hori Y; Terano H; Okuhara M; Manda T; Matsumoto S; Shimomura K New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J. Antibiot. (Tokyo) 1996, 49, 1204–1211. [DOI] [PubMed] [Google Scholar]
- (7).Nakajima H; Takase S; Terano H; Tanaka H New antitumor substances, FR901463, FR901464 and FR901465. III. Structures of FR901463, FR901464 and FR901465. J. Antibiot. (Tokyo) 1996, 49, 96–99. [DOI] [PubMed] [Google Scholar]
- (8).Miller-Wideman M; Makkar N; Tran M; Isaac B; Biest N; Stonard R Herboxidiene, a new herbicidal substance from Streptomyces chromofuscus A7847. Taxonomy, fermentation, isolation, physico-chemical and biological properties. J Antibiot (Tokyo).. J. Antibiot. (Tokyo) 1992, 45, 914–921. [DOI] [PubMed] [Google Scholar]
- (9).Kotake Y; Sagane K; Owa T; Mimori-Kiyosue Y; Shimizu H; Uesugi M; Ishihama Y; Iwata M; Mizui Y Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol 2007, 3, 570–575. [DOI] [PubMed] [Google Scholar]
- (10).Kaida D; Motoyoshi H; Tashiro E; Nojima T; Hagiwara M; Ishigami K; Watanabe H; Kitahara T; Yoshida T; Nakajima H; Tani T; Horinouchi S; Yoshida M Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA Nat. Chem. Biol 2007, 3, 576–583. [DOI] [PubMed] [Google Scholar]
- (11).Hasegawa M; Miura T; Kuzuya K; Inoue A; Won Ki S; Horinouchi S; Yoshida T; Kunoh T; Koseki K; Mino K; Sasaki R; Yoshida M; Mizukami T Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem. Biol 2011, 18, 229–233. [DOI] [PubMed] [Google Scholar]
- (12).Larsen NA The SF3b Complex is an Integral Component of the Spliceosome and Targeted by Natural Product-Based Inhibitors. Subcell. Biochem 2021, 96, 409–432 [DOI] [PubMed] [Google Scholar]
- (13).Cretu C; Schmitzová J; Ponce-Salvatierra A; Dybkov O; De Laurentiis EI; Sharma K; Will CL; Urlaub H; Lührmann R; Pena V; Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 2016, 64, 307–319. [DOI] [PubMed] [Google Scholar]
- (14).Finci LI; Zhang X; Huang X; Zhou Q; Tsai J; Teng T; Agrawal A; Chan B; Irwin S; Karr C; Cook A; Zhu P; Reynolds D; Smith PG; Fekkes P; Buonamici S; Larsen NA; The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018, 32, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cretu C; Agrawal AA; Cook A; Will CL; Fekkes P; Smith PG; Lührmann R; Larsen N; Buonamici S; Pena V Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol. Cell 2018, 70, 265–273. [DOI] [PubMed] [Google Scholar]
- (16).Cretu C; Gee P; Liu X; Agrawal A; Nguyen TV; Ghosh AK; Cook A; Jurica M; Larsen NA; Pena V Structural basis of intron selection by U2 snRNP in the presence of covalent inhibitors. Nat. Commun 2021, 12, 4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chan WC; León B; Krug KA; Patel A; La Clair JJ; Burkart MD Daedal Facets of Splice Modulator Optimization. ACS Med. Chem. Lett 2018, 9, 1070–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sim J; Jang E; Jin Kim HJ; Jeon H Total Syntheses of Pladienolide-Derived Spliceosome Modulators. Molecules. 2021, 26, 5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ghosh AK; Mishevich JL; Jurica MS Spliceostatins and Derivatives: Chemical Syntheses and Biological Properties of Potent Splicing Inhibitors. J. Nat. Prod 2021, 84, 1681–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).León B; Kashyap MK; Chan WC; Krug KA; Castro JE; La Clair JJ; Burkart MD A Challenging Pie to Splice: Drugging the Spliceosome. Angew. Chem. Int. Ed. Engl 2017, 56, 12052–12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang D; Meng F A Comprehensive Overview of Structure-Activity Relationships of Small-Molecule Splicing Modulators Targeting SF3B1 as Anticancer Agents. ChemMedChem. 2020, 15, 2098–2120. [DOI] [PubMed] [Google Scholar]
- (22).Hong DS; Kurzrock R; Naing A; Wheler JJ; Falchook GS; Schiffman JS; Faulkner N; Pilat MJ; O’Brien J; LoRusso P A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest. New Drugs 2014, 32, 436–444. [DOI] [PubMed] [Google Scholar]
- (23).Seiler M; Yoshimi A; Darman R; Chan B; Keaney G; Thomas M; Agrawal AA; Caleb B; Csibi A; Sean E; Fekkes P; Karr C; Klimek V; Lai G; Lee L; Kumar P; Lee SC; Liu X; Mackenzie C; Meeske C; Mizui Y; Padron E; Park E; Pazolli E; Peng S; Prajapati S; Taylor J; Teng T; Wang J; Warmuth M; Yao H; Yu L; Zhu P; Abdel-Wahab O; Smith PG; Buonamici S H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med 2018, 24, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Puthenveetil S; Loganzo F; He H; Dirico K; Green M; Teske J; Musto S; Clark T; Rago B; Koehn F; Veneziale R; Falahaptisheh H; Han X; Barletta F; Lucas J; Subramanyam C; O’Donnell CJ; Tumey LN; Sapra P; Gerber HP; Ma D; Graziani EI Natural Product Splicing Inhibitors: A New Class of Antibody-Drug Conjugate (ADC) Payloads. Bioconjug. Chem 2016, 27, 1880–1888. [DOI] [PubMed] [Google Scholar]
- (25).Chan WC; La Clair JJ; León B; Trieger KA; Slagt MQ; Verhaar MT; Bachera DU; Rispens MT; Hofman RM; de Boer VL; der Hulst R; Bus R; Hiemstra P; Neville ML; Mandla KA; Figueroa JS; Jamieson CHM; Burkart MD Scalable Synthesis of 17S-FD-895 Expands the Structural Understanding of Splice Modulatory Activity. Cell Rep. Phys. Sci, 2020, 100277. [Google Scholar]
- (26).Villa R; Mandel AL; Jones BD; La Clair JJ, Burkart MD Structure of FD-895 revealed through total synthesis. Org. Lett 2012, 14, 5396–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Crews LA; Balaian L; Delos Santos NP; Leu HS; Court AC; Lazzari E; Sadarangani A; Zipeto MA; La Clair JJ; Villa R; Kulidjian A; Storb R; Morris SR; Ball ED; Burkart MD; Jamieson CHM RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML Cell Stem Cell. 2016, 19, 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Crews LA; Ma W; Ladel L; Pham J; Balaian L; Steel SK; Mondala PK; Diep RH; Wu CN; Mason CN; van der Werf I; Oliver I; Reynoso E; Pineda G; Whisenant TC; Wentworth P; La Clair JJ; Jiang Q; Burkart MD; Jamieson CHM Reversal of malignant ADAR1 splice isoform switching with Rebecsinib. Cell Stem Cell. 2023, 30, 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).van der Werf I; Mondala PK; Steel SK; Balaian L; Ladel L; Mason CN; Diep RH; Pham J; Cloos J; Kaspers GJL; Chan WC; Mark A; La Clair JJ; Wentworth P; Fisch KM; Crews LA; Whisenant TC; Burkart MD; Donohoe ME, Jamieson CHM Detection and targeting of splicing deregulation in pediatric acute myeloid leukemia stem cells. Cell Rep. Med 2023, 100962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kumar D; Kashyap MK; La Clair JJ; Villa R; Spaanderman I; Chien S; Rassenti LZ; Kipps TJ; Burkart MD; Castro JE Selectivity in Small Molecule Splicing Modulation. ACS Chem. Biol 2016, 11, 2716–2723. [DOI] [PubMed] [Google Scholar]
- (31).Trieger KA; La Clair JJ; Burkart MD Splice Modulation Synergizes Cell Cycle Inhibition. ACS Chem. Biol 2020, 15, 669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Rohrs TM; Qin Q; Floreancig PE Re2 O7-Mediated Dehydrative Cyclization Reactions: Total Synthesis of Herboxidiene and Its 12-Desmethyl Analogue. Angew. Chem. Int. Ed. Engl 2017, 56, 10900–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gamboa Lopez A; Allu SR; Mendez P; Chandrashekar Reddy G; Maul-Newby HM; Ghosh AK; Jurica MS Herboxidiene Features That Mediate Conformation-Dependent SF3B1 Interactions to Inhibit Splicing. ACS Chem. Biol 2021, 16, 520–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sim J; Jang E; Kim HJ; Jeon H Total Syntheses of Pladienolide-Derived Spliceosome Modulators. Molecules. 2021, 26, 5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Puthenveetil S; He H; Loganzo F; Musto S; Teske J; Green M; Tan X; Hosselet C; Lucas J; Tumey LN; Sapra P; Subramanyam C; O’Donnell CJ; Graziani EI Multivalent peptidic linker enables identification of preferred sites of conjugation for a potent thialanstatin antibody drug conjugate. PLoS One. 2017, 12, e0178452. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.