

Abstract
Existing therapeutics for autoimmune diseases remain problematic due to low efficacy, severe side effects, and difficulties to reach target tissues. Herein, we design multifunctional fusion nanovesicles that can target lesions for the treatment of autoimmune skin diseases. The grapefruit‐derived exosome‐like nanovesicles (GEVs) with anti‐inflammatory and antioxidant effects are first encapsulated with CX5461, an immunosuppressant with anti‐proliferative properties to form GEV@CX5461. In order to enhance therapeutic efficiency and safety, GEV@CX5461 are then fused with CCR6+ nanovesicles derived from membranes of engineered gingiva‐derived mesenchymal stem cells (GMSCs). The resulting FV@CX5461 not only maintain the bioactivity of GEVs, CX5461, and GMSC membranes but also home to inflamed tissues rich in chemokine CCL20 through the chemotaxis function of CCR6 on FVs. Moreover, FV@CX5461 reduce the secretion of inflammatory factors, calm down Th17 cell activation, and induce Treg cell infiltration. Finally, impressive therapeutic efficiency in both psoriasis and atopic dermatitis disease models is demonstrated using FV@CX5461 to reshape the unbalanced immune microenvironment. A nanotherapeutic drug delivery strategy is developed using fusion nanovesicles derived from plant and animal cells with high clinical potential.
Keywords: autoimmune diseases, CCR6, CX5461, exosome‐like nanovesicles, fusion vesicles
1. INTRODUCTION
Autoimmune diseases are a result of the immune system failing to distinguish self‐antigen from non/altered‐self antigens and thus causing immune cells to destroy tissues and organs. With a steady rise in the global average annual rate of incidence and prevalence, autoimmune diseases have become the third‐largest chronic disease ranked after cardiovascular disease and cancer. (Fugger et al., 2020). Currently, dozens of types have been discovered, including atopic dermatitis (AD, incidence rate: 3.5%) (Weidinger & Novak, 2016), psoriasis(incidence rate: 2%−3%) (Ghoreschi et al., 2021), rheumatoid arthritis (incidence rate: 0.5%−1.0%) (Smolen et al., 2016), ulcerative colitis (incidence rate: 0.4%) (Kobayashi et al., 2020), and so on (Wang et al., 2015). Among autoimmune diseases, psoriasis and AD are two major skin disorders becoming the major issues threatening public health with increasing prevalence. Commonly used therapeutic drugs for autoimmune disease treatment are glucocorticoids, immunosuppressants, and monoclonal antibodies. However, they have limitations of insufficient efficacy and large side effects (Cifuentes‐Rius et al., 2021). The shortcomings of mainstay treatments and surging cases of autoimmune diseases across the world meant safer and more effective therapeutic strategies were now urgently needed.
During the development and progression of psoriasis and AD, the abnormal activation of immune responses is commonly characterized as a result of the aberrant proliferation of keratinocytes, the activation and massive infiltration of inflammatory immune cells, and the abundant accumulation of proinflammatory cytokines in both dermis and epidermis involving the innate and adaptive immune systems (Ghoreschi et al., 2021; Weidinger & Novak, 2016). Mesenchymal stem cells (MSCs) have been demonstrated to exert their therapeutic effects on various immune‐related diseases by secreting small extracellular vesicles (sEVs) due to their excellent immunomodulatory functions (Gao et al., 2016; Lai et al., 2019). MSC‐derived sEVs have been utilized to load target proteins by genetic modification, and encapsulate drugs for alleviating autoimmune diseases without side effects (Tsai et al., 2022; Xu et al., 2022). Although this approach is promising, the high cost of large‐scale production of MSC nanovesicles impairs their clinical applications. Excitingly, exosome‐like nanovesicles secreted from plant cells also are emerging as the next‐generation nanotherapeutics with the merits of low toxicity, low immunological risk, biocompatible, eco‐friendly, low cost, and easy mass production (Kim et al., 2022). There are several reports demonstrating the nanovesicles derived from grape (Ju et al., 2013), grapefruit (Wang et al., 2014), ginger (Teng et al., 2018; Zhang et al., 2016), and ginseng (Xu et al., 2021) exert anti‐inflammatory and anti‐oxidation effects through the natural lipids, microRNAs, or other substances they contain. Previous research confirmed that the nanoparticles made of total lipids of grapefruit could deliver chemotherapeutic agents, siRNA, and proteins to different types of cells (Wang et al., 2013). We hypothesize that plant‐derived exosome‐like nanovesicles (PLEVs), could present themselves as a natural biological vector to deliver encapsulated drugs and endogenous miRNAs. However, PLEVs are less effective in immunomodulation and lack targeting ability compared with MSC‐derived exosomes. Given the fact that EVs from animal and plant cells share similar biophysical properties of bi‐layer lipid membranes (Zhang et al., 2017), and cell‐derived nanovesicles prepared by membrane extrusion are very similar in morphology and composition to natural exosomes (Wen et al., 2022), we proposed a new approach to prepare engineered hybrid vesicles by fusing plant‐derived vesicles and engineered MSCs‐derived vesicles to take advantage of both types of vesicles and to generate personalized delivery nanovectors that would the lesional sites for the treatment of autoimmune diseases.
We have identified recently that CX5461, the rDNA‐specific transcription inhibitor approved for clinical trials in patients with solid tumours (Ismael et al., 2019), has stronger immunosuppressive effects with lower toxicity than the widely used immunosuppressive agent FK506 (Tsai et al., 2021). In this study, we fused CX5461‐loaded grapefruit‐derived exosome‐like nanovesicles (GEVs) with CCR6‐NVs derived from genetically engineered gingiva‐derived mesenchymal stem cells (GMSCs) to obtain multifunctional fusion vesicles (FV@CX5461) for the treatment of autoimmune skin diseases (Figure 1). We validated that FV@CX5461 not only maintains the bioactivity of GEVs and the CCR6‐NVs but also has a potent immunomodulatory function. In psoriasis and atopic dermatitis models, FV@CX5461 significantly targeted and restored tissue lesions by inhibiting the inflammatory immune cells. Our results indicated that our strategy might provide a universal nanovector platform for the targeted immunotherapy of autoimmune diseases.
FIGURE 1.
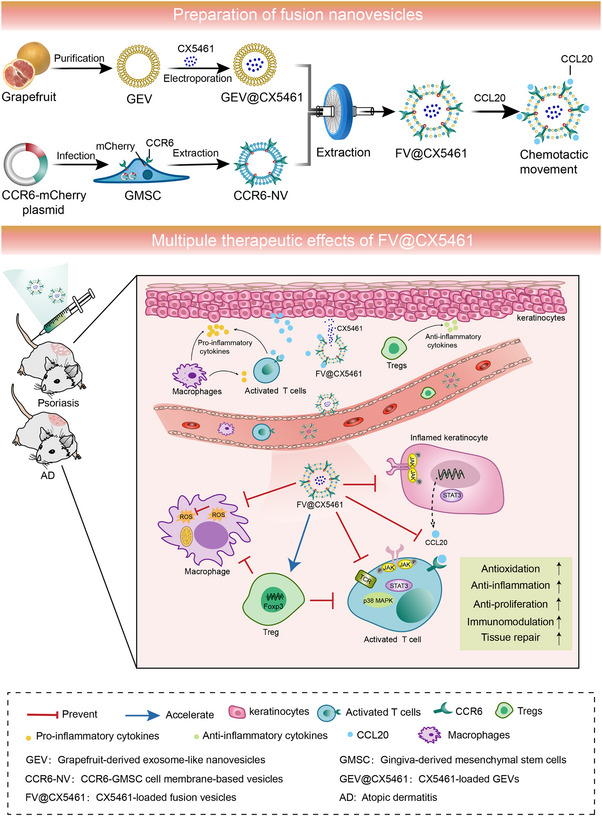
Schematic illustration showing the preparation process of FV@CX5461 and their therapeutic effects for autoimmune skin disorders.
2. RESULTS
2.1. GEVs had anti‐inflammatory and antioxidant effects
In order to identify potential PLEVs for the treatment of autoimmune skin disorders, we first investigated the literature as well as Chinese medicine prescriptions and determined ten promising tissues from edible plants to isolate PLEVs (Figure 2a). We next examined the effect of these ten PLEVs on cell proliferation of inflamed HacaT (human immortalized keratinocytes), which contribute to the development of psoriasis and atopic dermatitis. These natural PLEVs at low doses might be treated as nutrient substances, so presented a mild promoting effect on cell growth. With the increased concentration, only ginger and grapefruit‐derived nanovesicles inhibited cell proliferation, as well as mRNA expression of inflammatory factors such as IL‐1β, IL‐6, and TNF‐α of inflamed HaCaT cells (Figure 2b,c), which was consistent with the result of the bibliometrics (Figure S1A, Supporting Information). We then detected the intracellular ROS level to compare the antioxidant capacity of both PLEVs. The results obtained by the DCFH‐DA probe showed that only grapefruit‐derived nanovesicles (GEVs) significantly reduced the intracellular ROS level (Figure 2d). Given that cell metabolism may influence the antioxidant effects observed, we further assessed the generation of mitochondrial ROS and found both GEVs and ginger‐derived nanovesicles reduced the production of the mitochondrial ROS, and the antioxidant effect of GEVs was slightly better than ginger‐derived nanovesicles (Figure S1B, Supporting Information). The combination of these results suggested GEVs were the best candidate among selected PLEVs to treat autoimmune skin diseases. We assessed the quality of GEVs by transmission electron microscopy (TEM), zeta potential measurement, and nanoparticle tracking analysis (NTA). Similar to mammal‐derived sEVs, GEVs had spherical‐like morphology with membrane structure (Figure 2e), the zeta potential was around −25 mV (Figure S1C; Supporting Information), and the average diameter was around 163.4 nm (Figure 2f). Subsequent lipidomic analysis revealed the lipid species of GEVs displayed almost no difference from that of grapefruit tissues (Figure 2g; Figure S1D, Supporting Information). Interestingly, GEVs contained a higher percentage of sphingolipids, especially hexosylceramides, than grapefruit tissues (Figure 2g; Figure S1E, Supporting Information). Further analysis of the lipid composition of GEVs revealed abundant hexosylceramides and ceramides in GEVs (Figure 2h), which are indispensable in epidermal barrier maintenance and immune modulation. (Li et al., 2020) Ceramides can promote the proliferation of normal HacaT cells, and our further study indicated that ceramides have a mild inhibitory effect on LPS‐induced keratinocytes at high concentrations (Figure S1F, Supporting Information), and slightly reduced ROS levels in LPS‐activated macrophages (Figure S1G, Supporting Information). The metabolites composition of GEVs was analysed by Ultra High Performance Liquid Chromatography and Mass spectrometry (UHPLC–MS) and found that flavonoids accounted for a relatively high proportion (Figure S1H, Supporting Information), which play a role in many traditional Chinese medicines and have been widely reported to have anti‐inflammatory, anti‐bacterial, and antioxidant effects (Farhadi et al., 2019; Hendrich, 2006). We tested naringin, a flavonoid extracted from grapefruit, and found it had significant anti‐inflammatory, antioxidant, and antiproliferative effects at high concentrations (Figure S1I–K, Supporting Information). We further explored whether GEVs can affect the proliferation of T cells, especially CD4+T cells, which play a key role in the pathogenesis of psoriasis and atopic dermatitis (Guttman‐Yassky et al., 2018). However, no significant inhibitory effect on CD3+T cell proliferation (Figure 2i,k) and only a mild inhibitory effect on CD4+T cells (Figure 2j,l) were observed. In summary, GEVs had anti‐inflammatory and antioxidant effects but lacked immunosuppressive function in vitro.
FIGURE 2.
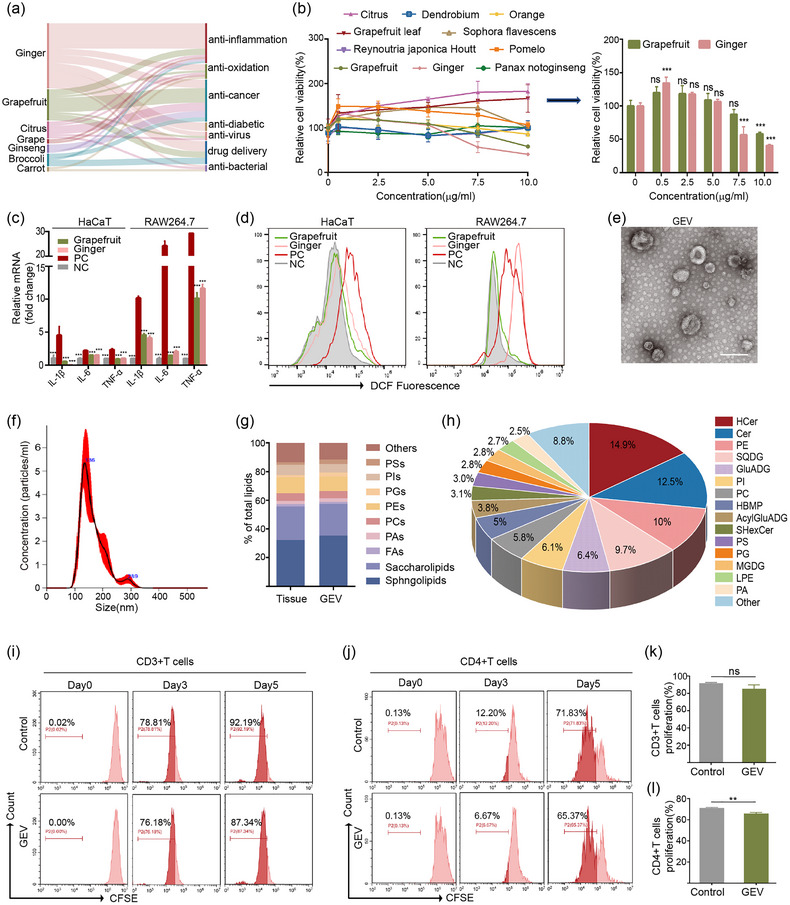
Grapefruit EVs had anti‐inflammatory and antioxidant effects. (a) Sankey plot showing PLEVs (left) with various therapeutic functions (right) via bibliometrics. (b) Cell viability curve of LPS‐stimulated HaCaT cells treated with the indicated different PLEVs (n = 3). (c) mRNA expression of IL‐1β, IL‐6 and TNF‐α in LPS‐induced HaCaT cells and RAW264.7 cells (n = 3). NC, cells without LPS treatment. (d) Flow cytometric analysis of intracellular ROS levels in LPS‐stimulated HaCaT cells and RAW264.7 cells treated with grapefruit‐derived nanovesicles (Grapefruit), and ginger‐derived nanovesicles (Ginger), respectively. (e,f) Representative TEM image and size distribution of GEVs. Scale bar: 200 nm. (g) Distribution of major types of lipids in grapefruit tissue and GEV. PAs: phosphatidic acids; PCs: phosphatidylcholines; PEs: phosphatidylethanolamines; PSs: phosphatidylserines; PIs: phosphatidylinositols; FAs: fatty acids. (h) Lipid composition of GEV. HCer: hexosylceramide; Cer: ceramide. (i–l) Flow cytometry analysis and corresponding quantitative analysis of CFSE‐labelled CD3+ or CD4+ T cells treated with GEV for 5 days (n = 3). Error bar, mean ± SD. n.s represents no significant, **P < 0.01; ***P < 0.001.
2.2. CCL20 expression rose in the lesions of autoimmune diseases, and CCR6‐NVs had an immunosuppressive function in vitro
In order to improve the therapeutic efficacy of GEVs, more efficient and targeted strategies are required to calm down the overactivated inflammatory immune responses in the affected tissue in autoimmune diseases. However, bioengineering plant‐derived nanovesicles remains a considerable challenge compared to mammalian cells‐derived EVs. We and others have previously reported that MSCs‐derived sEVs can be used as cell‐free vectors in alternative therapies for a variety of inflammatory diseases, due to their potency to regulate immune responses (Tsai et al., 2022; Wang et al., 2022). Thus we hypothesize that fusing GEVs and nanovesicles (NVs) derived from genetically modified mesenchymal stem cells (MSCs) would be an efficient strategy to take advantage of the unlimited availability of GEVs and the potent immunosuppressive function of MSC‐NVs to generate delivery vectors to target inflammatory sites. To test this hypothesis, we first compared the immunomodulatory ability of NVs derived from a variety of MSCs and found that NVs derived from gingiva‐derived mesenchymal stem cells (GMSCs) induce significant expression of immunosuppressive cytokines IDO1 and IL‐10 when compared with umbilical cord mesenchymal stem cells (UC‐MSCs), bone marrow‐derived mesenchymal stem cells (BMSCs), and adipose‐derived stem cells (ADSCs) (Figure 3a). As shown in Figure S2, the GMSCs were displayed as spindle‐shaped under the microscope, and the purity of MSCs was assessed by marker expression with CD105, CD90 positively, and CD19, CD34, CD45 negatively (Figure S2A‐B, Supporting Information). Meanwhile, GMSC‐NVs reduced the ROS levels in RAW 264.7 cells under inflammatory conditions (Figure 3b), further indicating that GMSCs were the appropriate MSCs for bioengineering targeted delivery NVs. CCL20, also known as macrophage inflammatory protein 3α (MIP‐3α), is a well‐known high‐affinity chemokine binding to CCR6 on leukocytes, especially Th17 cells (Martin‐Orozco et al., 2009), playing an indispensable role in the attraction of T cells homing to inflammatory sites (Meitei et al., 2021). We confirmed the correlation of CCL20 overexpression in the inflamed tissue in the context of many autoimmune diseases. GEO (Gene Expression Omnibus) data analysis of patients’ lesional tissue including psoriasis, atopic dermatitis, colitis, rheumatoid arthritis, and Sjogren's Syndrome correlated with a higher expression of the CCL20 gene compared to healthy tissue (Figure 3c). Therefore, we aim to generate CCR6‐overexpressed GMSCs‐derived NVs (CCR6‐NVs) to fuse with GEVs as an innovative treatment for autoimmune diseases. CCR6‐overexpressed GMSCs were established by lentivirus infection (Figure 3c), and CCR6 was expressed and localized on the cell membrane indicated by the colocalization of mCherry‐CCR6 (red), and the cell membrane dye WGA488 (green) (Figure 3c).
FIGURE 3.
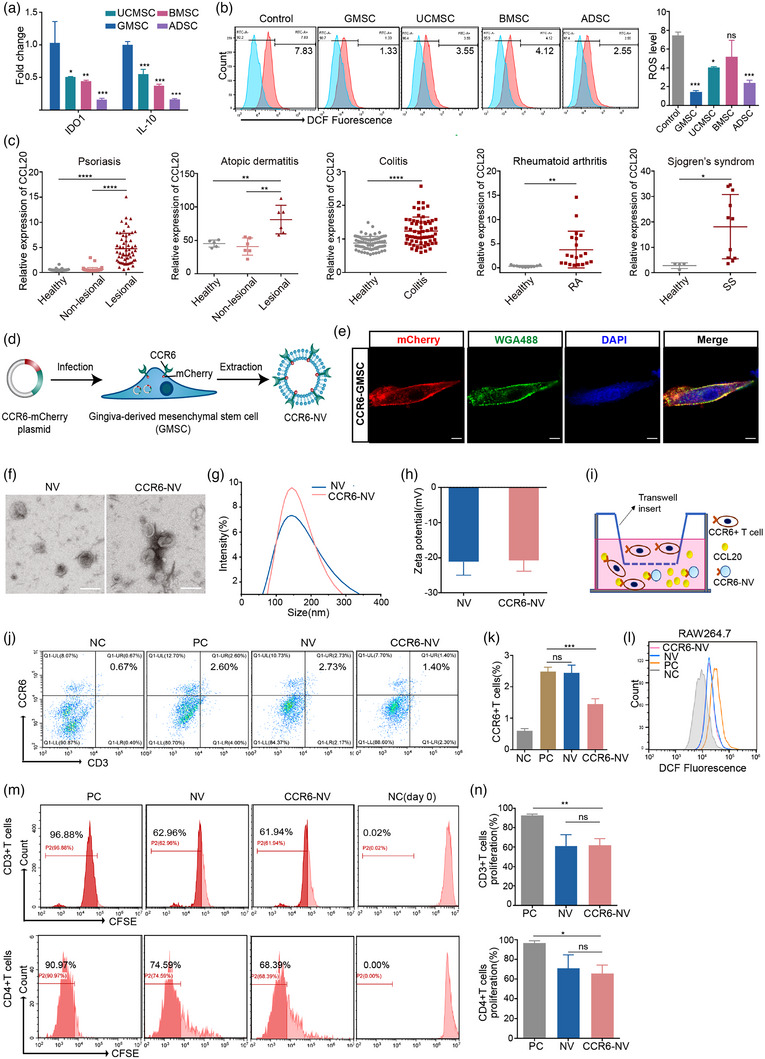
Characterizations of CCR6‐NVs derived from GMSCs. (a) mRNA expression of IDO1 and IL‐10 in NVs from different stem cells (n = 3). (b) Flow cytometric analysis of intracellular ROS levels in RAW264.7 cells pretreated with LPS for 12 h, then treated with indicated PBS or different NVs. (c) GEO data analysis of CCL20 gene expression in skin punch biopsies of psoriasis patients (GDS4602), skin biopsies of atopic dermatitis (AD) patients (GDS2381), colon epithelial biopsies of ulcerative colitis patients (GDS3268), synovial fluid mononuclear cells of rheumatoid arthritis patients (GDS711), and minor salivary glands of Sjogren's syndrome (SS) patients (GDS3940). (d) Schematic diagram of the preparation of CCR6‐NV. (e) Representative confocal images of CCR6 expression on GMSC cell membranes. Scale bar: 10 μm. (f–h) Representative TEM image, zeta potential, size distribution of control NVs and CCR6‐NVs. Scale bar: 200 nm. (i) Schematic illustration of transwell transmigration assay. Mouse lymphocytes were seeded in the upper chamber of the transwell apparatus and serum‐free culture medium containing PBS (NC) or CCL20 recombinant protein in a concentration gradient (PC) was added to the basolateral chamber. After 3 h of co‐culture of NVs or CCR6‐NVs at 37°C, the migrated cells were harvested from the basolateral chamber and counted by flow cytometry. (j,k) Flow cytometric analysis of transmigrated cells and corresponding quantitative analysis of CCR6+ T cells (n = 3). (l) Flow cytometric analysis of intracellular ROS levels in RAW264.7 cells pretreated with LPS for 12 h then treated with PBS (PC), and with no treatment (NC), CCR6‐NVs, and control NVs, respectively. (m,n) Flow cytometry analysis and corresponding quantitative analysis of CFSE‐labelled CD3+ or CD4+T cells treated with PBS, NVs, and CCR6‐NVs for 5 days (n = 3). Error bar, mean ± SD. n.s represents no significant, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We next prepared bioengineering CCR6‐NVs to compete with immune cells for binding CCL20 in vitro. TEM imaging, DLS analysis, and NTA analysis showed spherical morphology of CCR6‐NVs (Figure 3f) with an average diameter of 186.2 nm (Figure 3g; Figure S2C, Supporting Information), and the average zeta potential was −20 mV, which had no significant difference compared to control NVs (Figure 3h). Furthermore, we identified the existence of extracellular vesicles‐associated proteins Alix, TSG101, CD63, CD81, and high level of CCR6 on CCR6‐NVs (Figure S4A, Supporting Information), confirming that we successfully prepared CCR6 overexpressed GMSC‐derived NVs. To study the targeting and neutralizing potential of CCR6‐NVs to CCL20, we first performed a transwell transmigration assay. Mouse lymphocytes were seeded in the upper chamber of the transwell apparatus and serum‐free culture medium containing CCL20 recombinant protein in a concentration gradient was added to the basolateral chamber. After 3 h of co‐culture at 37°C, the migrated cells were harvested from the basolateral chamber and counted by flow cytometry (Figure 3i–k). As expected, transmigration of CCR6‐expressing T cells was dramatically decreased when that CCR6‐NVs competed with CCR6 on the T cell membrane for binding CCL20 in the culture medium (Figure 3j,k), supporting a potent targeting and chemokine neutralizing capability of CCR6‐NVs. To assess the immunomodulatory effects of CCR6‐NVs, we used DCFH probes to detect the ROS level and found that similar to control GMSCs‐derived NVs, CCR6‐NVs eliminated ROS in LPS‐induced RAW264.7 cells (Figure 3l), and blocked the proliferation of activated CD3+ and CD4+T cells in vitro (Figure 3m,n). Taken together, these results showed that CCR6‐NVs exhibit an impressive ability to target chemokine CCL20 but have a very mild effect in altering the phenotype of activated immune cells to the immunosuppressed state.
2.3. Construction and characterization of fusion vesicles loaded with a novel immunosuppressant CX5461
In order to compensate insignificant immunosuppression effects of GEVs and CCR6‐NVs, we proposed to encapsulate CX5461, a specific rRNA synthesis inhibitor we previously reported to have a better immunosuppressive effect than FK506, with our therapeutic nanovector system (Drygin et al., 2014; Tsai et al., 2021). Both activated immune cells and hyperproliferating keratinocytes contribute to the skin lesions of psoriasis and atopic dermatitis. We validated that CX5461 inhibited the proliferation of CD4+T cells in vitro (Figure 4a,b), and decreased the IL‐2 level of cell culture supernatant (Figure 4c). We also found that CX5461 was able to inhibit proliferation and induce G2/M phase arrest on inflamed HaCaT cells and primary murine keratinocytes (Figure 4d–g; Figure S3A,B, Supporting Information). JAK‐STAT3 signalling pathway plays a crucial role in autoimmune skin diseases (Palanivel et al., 2014; Villarino et al., 2015). Strikingly, we found that CX5461 downregulated the protein level of p‐JAK1, p‐JAK2, and p‐STAT3 (Figure 4h–i; Figure S3C,D, Supporting Information) accompanied by decreased nuclear translocation of p‐STAT3 (Figure 4j). Taken together, these data demonstrated that CX5461 can be employed for autoimmune skin disease treatment. Inspired by the research that exosomes could be fused with membranes of leukocytes by extrusion (Wang et al., 2015), we prostitute the membrane fusion and encapsulating approaches (Figure 5a) to enhance the targeting ability and treatment efficiency of the plant and MSC‐derived NVs. As plant‐derived vesicles have been reported to be excellent nanovectors for drug delivery (Wang et al., 2013), we first loaded poorly soluble CX5461 into GEVs by electroporation to form GEV@CX5461 and then fused GEV@CX5461 and CCR6‐NVs to generate FV@CX5461. The confocal images verified the co‐existence of the GEV@CX5461 and CCR6‐NVs in FV@CX5461(Figure 5b), which was consistent with the result of the western blot assay (Figure S4A, Supporting Information). We next confirmed that GEV@CX5461 and FV@CX5461 had similar morphology (Figure S4B, Supporting Information), diameter (Figure 5c), and zeta potential (Figure S4C, Supporting Information) to GEVs, suggesting that drug loading and fusion processes had no impact on the basic properties of GEVs. Furthermore, we evaluated the encapsulation rate (Figure S4D, Supporting Information), in vitro drug release curve (Figure S4E, Supporting Information), and validated that FV@CX5461 could be uptaken by PBMC and HaCaT cells (Figure S4F,G, Supporting Information). These results indicated that we successfully prepared FV@CX5461 using the strategy of membrane fusion to deliver CX5461 to effector cells.
FIGURE 4.
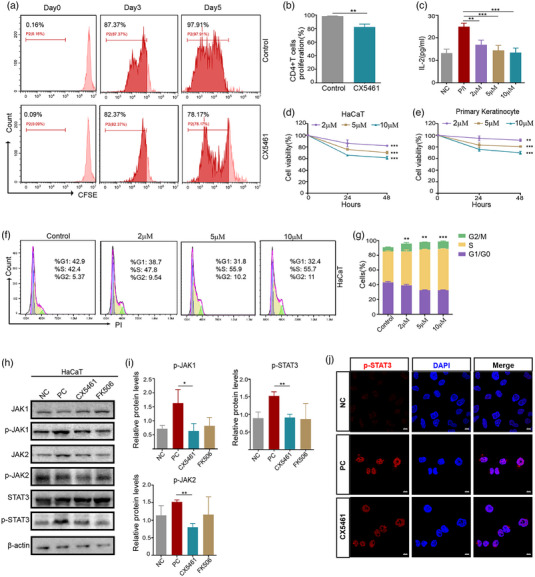
CX5461 inhibited the activation of CD4+T cells and keratinocytes in vitro. (a,b) Flow cytometry analysis of CFSE‐labelled CD4+ T cells treated with control DMSO or CX5461 for 5 days and corresponding quantitative analysis (n = 3). (c) ELISA assay to detect IL‐2 in cell culture supernatants of CD4+T cells stimulated by PMA/Ionomycin (P/I) for 12 h or with no treatment (NC), then treated with DMSO or CX5461 (n = 3). (d,e) Cell viability curve of LPS‐stimulated HaCaT cells and primary murine keratinocytes treated with the indicated different concentrations of CX5461 (n = 3). (f,g) Flow cytometry analysis of HaCaT cell cycle distribution and corresponding quantitative analysis (n = 3). (h) Western blot analysis of the protein levels of JAK1, p‐JAK1, JAK2, p‐JAK2, STAT3, and p‐STAT3 in LPS‐induced HaCaT cells treated with DMSO (PC), CX5461, and FK506. (i) Quantitative analysis of Figure 4h (n = 3). (j) Confocal images of the subcellular localization of p‐STAT3 in LPS‐induced HaCaT cells treated with DMSO (PC) or CX5461, and in HaCaT cells with no treatment (NC). Scale bar: 10 μm. Error bar, mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
FIGURE 5.
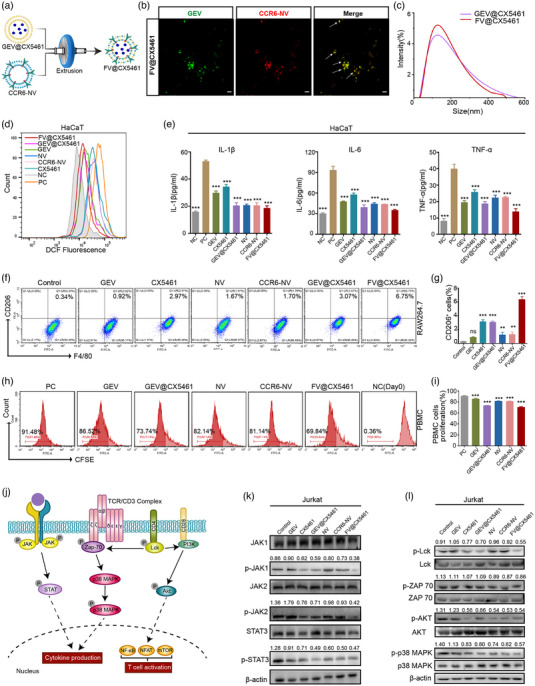
FV@CX5461 had enhanced immunosuppressive and anti‐inflammatory effects in vitro. (a) Schematic diagram of the preparation process of the FV@CX5461. (b) Confocal images of the co‐existence of CCR6‐NV and GEV in FV@CX5461. Scale bar: 2 μm. (c) Size distribution of GEV@CX5461 and FV@CX5461. (d) Flow cytometric analysis of intracellular ROS levels in HaCaT cells treated with indicated different vesicles (protein concentration: 10 μg/mL), pretreated with LPS for 12 h, then treated with PBS (PC), and with no treatment (NC), respectively. (e) ELISA assay to detect IL‐1β, IL‐6, and TNF‐α in the supernatant of LPS‐induced HaCaT cells (n = 3). (f,g) Flow cytometry analysis of the percentage of M2 macrophages (CD206 and F4/80 double positive) and corresponding quantitative analysis (n = 3). (h,i) Flow cytometry analysis of CFSE‐labelled PBMCs treated with indicated different vesicles for 7 days and corresponding quantitative analysis (n = 3). (j) Schematic diagram of the JAK‐STAT signalling pathway and TCR signalling pathway. (k,l) Western blot analysis of the JAK‐STAT pathway‐related protein levels and TCR pathway‐related protein levels in Jurkat cells, pre‐stimulated by PMA/Ionomycin (P/I) for 12 h, then treated with PBS, GEVs, CX5461, GEV@CX5461, NVs, CCR6‐NVs, and FV@CX5461. Error bar, mean ± SD. n.s represents no significant, **P < 0.01; ***P < 0.001.
2.4. FV@CX5461 had an immunosuppressive function in vitro
We further confirmed that FV@CX5461 significantly inhibited ROS levels in HaCaT (Figure 5d) and decreased the expression including inflammatory cytokines IL‐6, IL‐1β and TNF‐α compared to other groups (Figure 5e; Figure S4H, Supporting Information). Next, we demonstrated the immunomodulation efficacy of FV@CX5461 in vitro. Macrophages can polarize to pro‐inflammatory M1 type by LPS stimulation or anti‐inflammatory M2 type under certain conditions (Orecchioni et al., 2019). We found that the expression of the M2 macrophage surface marker CD206 was increased by the treatment of FV@CX5461, suggesting that FV@CX5461 could promote M2 polarization (Figure 5f,g). To investigate the influence of FV@CX5461 on human peripheral blood mononuclear cells (PBMC) proliferation, CFSE‐labelled PBMCs were cultured with GEVs, GEV@CX5461, NVs, CCR6‐NVs and FV@CX5461 respectively for 7 days. The flow cytometry data showed that GEV@CX5461, CCR6‐NVs, and FV@CX5461 inhibited the proliferation of PBMCs by 17.74%, 10.34% and 21.64% respectively, suggesting that the immunosuppressive effect of FV@CX5461 was more pronounced than GEV@CX5461 or CCR6‐NVs alone (Figure 5h,i). Moreover, FV@CX5461 decreased the transmigration of CCR6‐expressing T cells similar to CCR6‐NVs (Figure S4I–K, Supporting Information), and had an enhanced inhibition function on CD4+T cell proliferation (Figure S4L,M, Supporting Information) and ROS production in macrophages (Figure S4N, Supporting Information). All the above results indicated the enhanced treatment efficiency of fusing GEV@CX5461 with CCR6‐NVs to form FV@CX5461. To understand the molecular mechanisms of how FV@CX5461 affected immune cells, we performed the western blot assay and found that protein levels of p‐JAK1, p‐JAK2, p‐STAT3, p‐LCK, p‐AKT, p‐ZAP70 and p‐p38 MAPK were significantly down‐regulated in CX5461, GEV@CX5461, NVs, CCR6‐NVs and FV@CX5461 groups, suggesting that they could influence the T cell receptor pathway and JAK‐STAT pathway, and thus inhibit T cell proliferation, activation, and inflammatory cytokines production (Figure 5j–l).
MicroRNAs (miRNAs) play important gene‐regulatory roles in animal and plant cells majorly by blocking the translation of target mRNA, (Bartel, 2009) which may be involved in immunomodulation of FV@CX5461. miRNAs derived from plants can not only perform biological functions in plants but also have been reported to regulate gene expression across species borders (Chin et al., 2016; Zhang et al., 2012; Zhou et al., 2015). Therefore, we want to explore whether miRNAs from GEVs could influence mammalian cells and the retention rate of miRNAs after membrane fusion. The result of miRNA‐sequencing showed the FV@CX5461 retained abundant miRNAs from GEVs and CCR6‐NVs, although some miRNAs were lost (Figure 6a–c). As the target genes of plant‐derived miRNAs in cross‐species regulation are still unknown, we performed gene enrichment analysis on the target genes of human miRNAs in FV@CX5461. GO enrichment analyses showed that target genes were enriched in vesicular transport, histone modification, and neural‐related biological process (Figure 6d). KEGG enrichment analysis and Reactome enrichment analyses indicated the great potential of FV@CX5461 to regulate the PI3K‐AKT signalling pathway and the MAPK signalling pathway (Figure 6e; Figure S5A, Supporting Information), which was consistent with the result of the western blot assay (Figure 5l). Interestingly, cis‐miR159a‐3p not only showed the highest expression level in GEVs (Figure S5B, Supporting Information) but also in FV@CX5461 (Figure S5C, Supporting Information). Some miRNAs were lost during fusion but cis‐miR159a‐3p was partially retained in FV @CX5461 (Figure 6f). To identify whether cis‐miR159a‐3p contributes to the anti‐inflammatory effect of GEVs and FV@CX5461, we applied miR159a mimics to LPS‐induced HaCaT cells, the cell proliferation assay demonstrated that miR159a mimics reduced proliferation of the inflamed HaCaT cells (Figure 6g). Moreover, the qPCR results showed the reduction of inflammatory cytokines including IL‐6, IL‐1β, TNF‐α, IFN‐γ, TSLP and CCL20 (Figure 6h). These results illustrated that plant miR159a may exert cross‐border regulatory effects on human cells and may serve as a potential therapeutic agent to improve current treatment for autoimmune skin diseases.
FIGURE 6.
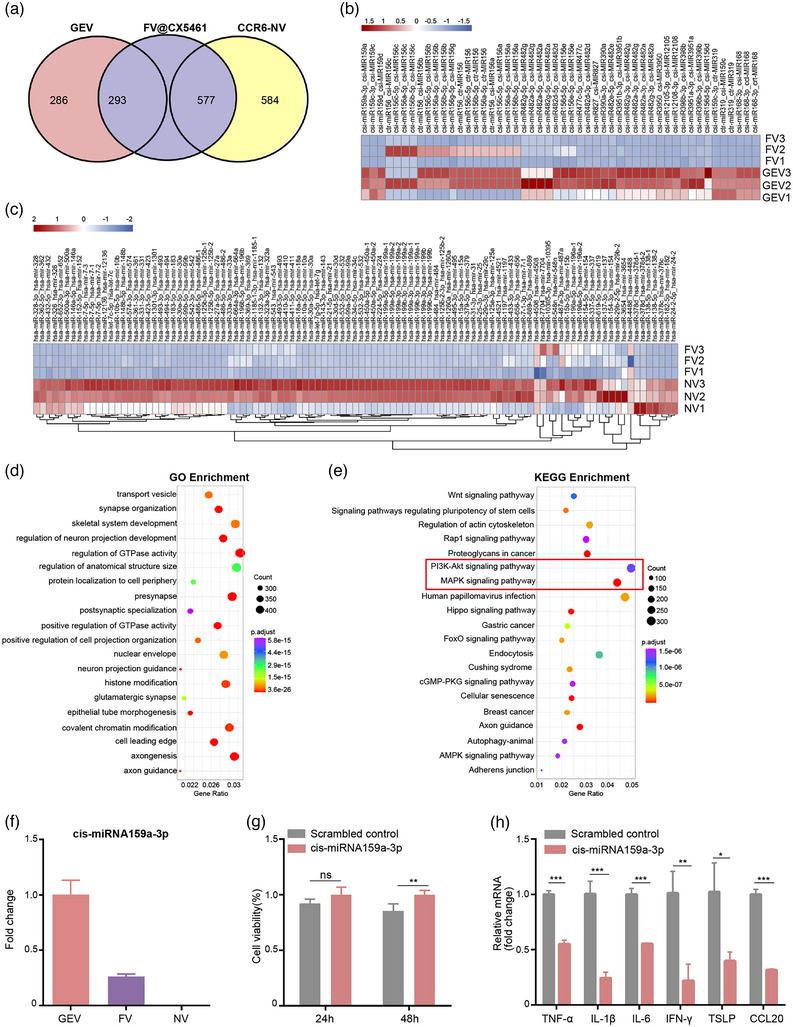
FV@CX5461 contained miRNAs from GEVs and CCR6‐NVs. (a) Venn diagram of miRNAs contained in the GEV, CCR6‐NV, and FV@CX5461. (b) Heatmap of differentially expressed miRNAs in GEVs and FV@CX5461 matched to the miRbase of Citrus species. (c) Heatmap of differentially expressed miRNAs in CCR6‐NVs and FV@CX5461 matched to the miRbase of Homo species. (d,e) GO enrichment analyses and KEGG enrichment analyses of the target genes of miRNAs expressed in FV@CX5461. (f) Relative mRNA Expression of csi‐miR159a‐3p in GEVs, FVs, and NVs. (g) Cell viability of LPS‐induced HaCaT cells transfected with csi‐miR159a‐3p mimics or scrambled control (n = 5). (h) Relative mRNA expression of TNF‐α, IL‐1β, IL‐6, IFN‐γ, TSLP and CCL20 in LPS‐induced HaCaT cells transfected with csi‐miR159a‐3p mimics or scrambled control (n = 3). Error bar, mean ± SD. n.s represents no significant, *P < 0.05; **P < 0.01; ***P < 0.001.
2.5. FV@CX5461 alleviated IMQ‐induced psoriasis and DNCB‐induced atopic dermatitis
To explore the therapeutic effect of the FV@CX5461 against autoimmune skin disease, we first performed the verification experiment in the imiquimod (IMQ)‐induced psoriasis models (Figure 7a). We analysed the distribution of drugs using the IVIS Spectrum imaging system and found that CCR6‐NVs and FV@CX5461 mainly accumulated in lesional skin, and NVs partly distributed in lesional skin while GEVs seldom appeared on the skin (Figure 7b). And these vesicles were metabolized after 48 h (Figure S6A, Supporting Information). The result demonstrated that FV@CX5461 and CCR6‐NVs could target inflamed sites. Compared with the NC group, all other groups showed an increased PASI (psoriasis area and severity index) score with psoriasis‐like pathological changes, including erythema, scaling, and epidermal thickening (Figure S6B, Supporting Information). After treatments, the PASI of the FV@CX5461 group rapidly descended, with ameliorated psoriasis symptoms, and other groups also showed varying degrees of decline (Figure 7c). All the mice were sacrificed on day 7, the peak of psoriasis‐like changes in the IMQ group, including erythema, scales and thickened epidermis. We observed that FV@CX5461 significantly alleviated these symptoms (Figure 7d), along with relief of splenomegaly (Figure S6C, Supporting Information). In addition, CX5461, GEV@CX5461, NVs, CCR6‐NVs and Dexamethasone (Dex) also ameliorated psoriasis symptoms. The result of Hematoxylin‐Eosin (H&E) staining and Immunohistochemical analysis of skin tissues showed FV@CX5461 decreased epidermal thickness and inflammatory cell infiltration and inhibited the excessive proliferation of keratinocytes (Figure 7e; Figure S6D, Supporting Information). Flow analysis further confirmed the effectiveness of FV@CX5461. We observed that FV@CX5461 significantly decreased the counts of CD3+T cells and the percentage of CD4+T cells compared with other groups. In addition, the proportion of splenic Th17 cells was reduced while the percentage of CD4+CD25+Foxp3+Treg cells was increased by the treatment of FV@CX5461 (Figure 7f,g). Correspondingly, the ELISA assay revealed the serum level of inflammatory cytokines IL‐1β, TNF‐α, IL‐17, IL‐22, and IL‐12/23 p40 was significantly decreased in the FV@CX5461 group, as well as the decrease of CCL20 in skin tissues (Figure 7h; Figure S6F, Supporting Information), and the RT‐qPCR analysis showed reduced mRNA expression of IFN‐γ, IL‐6, IL‐17F, IL‐23 and CCL20 in skin tissues (Figure S6E, Supporting Information). Dex as a clinically proven effective drug generated an unsatisfactory effect compared with FV@CX5461, as reflected by these above results. Furthermore, we validated in vivo that GEVs and FV@CX5461 can reduce ROS production caused by psoriasis inflammation (Figure S6H,I, Supporting Information). Collectively, these results unveiled that the FV@CX5461 in vivo can effectively mitigate psoriasis disease severity, and suppress the inflammatory response by reducing immune cell infiltration.
FIGURE 7.
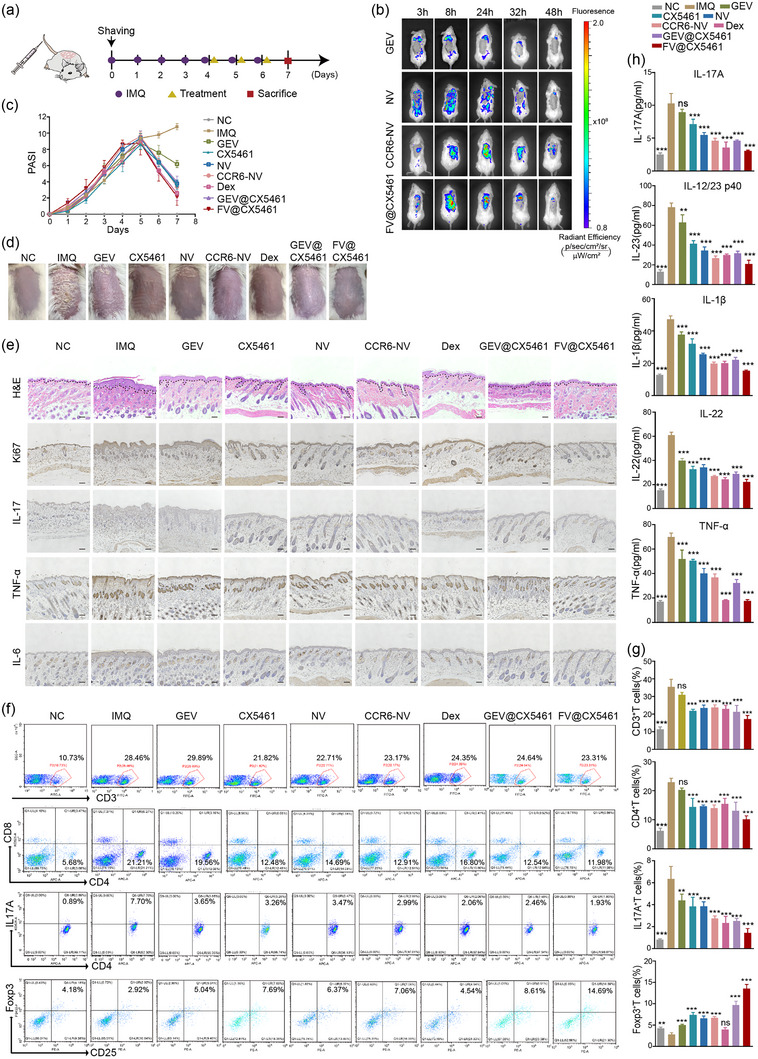
FV@CX5461 alleviated IMQ‐induced psoriasis. (a) Schematic diagram for i.v. administration of drugs on day 4, day 5, and day 6 during the application of IMQ on a daily basis. (b) In vivo distribution of GEVs, NVs, CCR6‐NVs, and FV@CX5461 in IMQ‐induced psoriasis mice. (c) PASI scores of mice in each group over 7 days of the experiment (n = 5). (d) Representative images of mouse dorsal skin in each group on day 7. (e) H&E staining and immunohistochemistry of lesional skins in each group of mice on day 7. Scale bar: 100 μm. (f,g) Representative flow cytometric analysis of CD3+ T cells, CD4+ T cells, CD4+ IL17+ T cells, and CD4+ CD25+ Foxp3+ Treg cells in spleens (n = 3). (h) ELISA assay to detect IL‐1β, TNF‐α, IL‐17, IL‐22, and IL‐12/23 p40 in the serum of each group of mice on day 7 (n = 3). Error bar, mean ± SD. n.s represents no significant, **P < 0.01; ***P < 0.001.
We next asked whether our strategy could be applied to other autoimmune diseases. We established atopic dermatitis models by 2,4‐dinitrochlorobenzene (DNCB) administration (Figure 8a) which leads to atopic dermatitis‐like lesions and pathological changes. In vivo fluorescence analysis revealed CCR6‐NVs and FV@CX5461 remarkably gathered in skin tissues compared with GEVs and NVs, indicating the targeting ability in atopic dermatitis models (Figure 8b). We noticed that the DNCB group showed severe atopic dermatitis‐like symptoms including erythema, scale, edema, and erosion on day 21 while the skin lesions of the FV@CX5461 group were alleviated significantly (Figure 8c,d; Figure S7C, Supporting Information), along with the reduced spleen size and weight of the mice (Figure S7B, Supporting Information). The result of H&E staining and toluidine blue (TB) staining indicated the decline of skinfold thickness and mast cell infiltration in FV@CX5461‐treated mice (Figure 8e; Figure S7D, Supporting Information). Besides, we found the inflammatory cytokines in the epidermis and dermis of treatment groups were diminished, especially the FV@CX5461 group, by immunohistochemical analysis (Figure 8e). Moreover, quantification of various T cell subsets by flow cytometry demonstrated that FV@CX5461 significantly reduced the proportion of CD4+T cells in T cells, along with the proportion of Th17 cells (IL17+T cells) and Th2 cells (IL4+T cells) in CD4+T cells (Figure 8f,g). Interestingly, we observed the increase of CD4+CD25+Foxp3+ Tregs in atopic dermatitis mice after treatment of FV@CX5461 likewise (Figure 8f). We further detected the level of IgE, IL‐4, IL‐1β, TNF‐α, and IL‐17 from serum, which are indicators reflecting disease severity (Figure 8h). The mRNA expression levels of TSLP (Thymic stromal lymphopoietin), IL‐1β, TNF‐α, and CCL20 were downregulated (Figure S7E, Supporting Information) in lesional skins, providing additional evidence that FV@CX5461 was effective in the treatment of atopic dermatitis. We also found a rise of ROS levels at the lesions of atopic dermatitis, which was mitigated by GEVs and FV@CX5461(Figure S7I,J, Supporting Information), demonstrating that the fusion strategy preserved the antioxidant function of GEVs.
FIGURE 8.
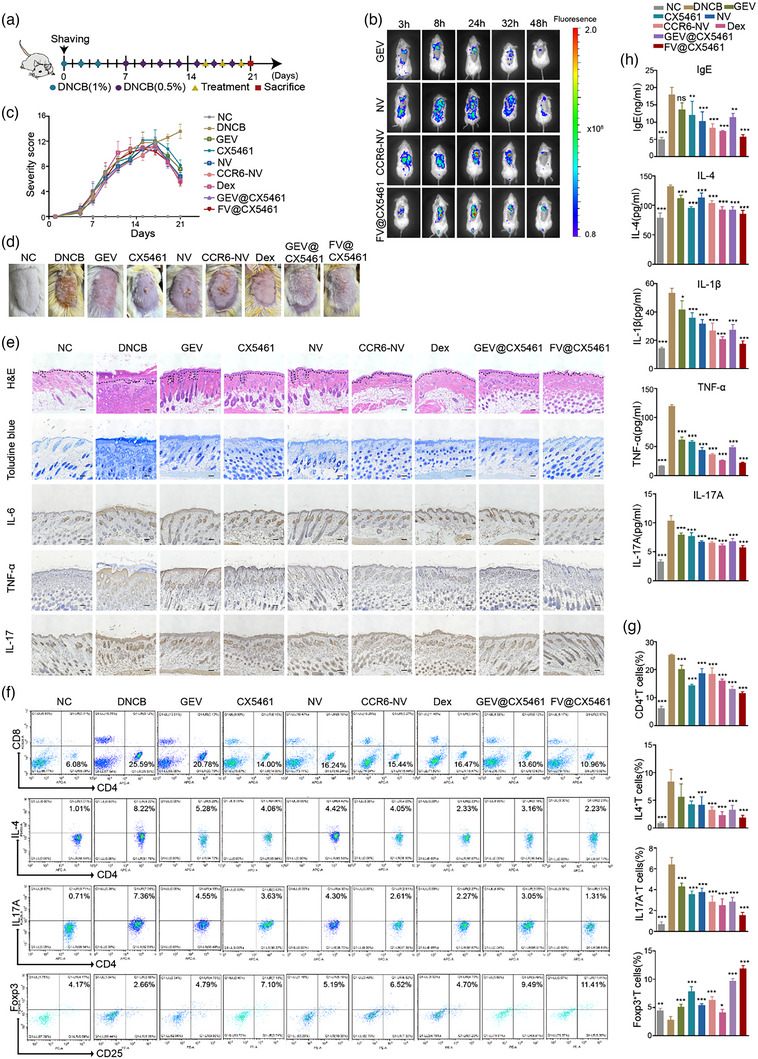
FV@CX5461 mitigated DNCB‐induced atopic dermatitis. (a) Schematic illustration of atopic dermatitis modelling and treatment. 1%DNCB was applied on day 0, day 2, and day 4 in the first week, and 0.5%DNCB was applied every other day for the next two weeks with i.v. administration of drugs on day16, day18, and day20 during the atopic dermatitis modelling. (b) In vivo fluorescence images of DNCB‐induced atopic dermatitis mice treated with GEVs, NVs, CCR6‐NVs, and FV@CX5461. (c) Severity scores of mice in each group (n = 5). (d) Representative images of mouse dorsal skin in each group on the last day. (e) H&E staining, Toluidine blue staining, and immunohistochemistry of lesional skins in each group of mice. Scale bar: 100 μm. (f,g) Representative flow cytometric analysis of CD4+ T cells, CD4+ IL4+ T cells, CD4+ IL17+ T cells, and CD4+ CD25+ Foxp3+ Treg cells in spleens (n = 3). (h) ELISA assay to detect IgE, IL‐4, IL‐1β, TNF‐α, and IL‐17 in the serum (n = 3). Error bar, mean ± SD. n.s represents no significant, *P < 0.05; **P < 0.01; ***P < 0.001.
Lastly, we evaluated the safety of GEVs, CX5461, GEV@CX5461, NVs, CCR6‐NVs and FV@CX5461 in vivo. No obvious changes in body weight were monitored in psoriasis mice and AD mice after the treatment of GEVs, CX5461, GEV@CX5461, NVs, CCR6‐NVs and FV@CX5461 (Figures S6G and S7G, Supporting Information). Histological images of the heart, liver, spleen, lung, and kidney tissues indicated all groups had no noticeable change in tissue structure and cellular morphology (Figure S8A, Supporting Information). The complete blood count test (CBC test) indicated the levels of red blood cell (RBC), white blood cell (WBC), haemoglobin (HGB), platelet (PLT), lymphocytes, monocyte, and neutrophils of blood were within the normal value range and had no marked difference compared to the control group (Figure S8B, Supporting Information). Taken together, although cell‐derived biological NVs carried high complexity, they exhibited great biocompatibility to mice.
3. DISCUSSION AND CONCLUSION
Autoimmune diseases are a general term for many kinds of diseases that are caused by immune cells attacking self‐tissues (Wang et al., 2015). In this work, we focus on psoriasis and atopic dermatitis, whose main lesions are the skin tissues. Compared with conventional glucocorticoid therapy, which has drug resistance and reduces apparent symptoms without fundamentally regulating the disturbed immune system, FV@CX5461 exhibited excellent immunomodulatory capabilities, including inhibition of Th17 cell proliferation and increase of Tregs’ counts. Tregs inhibit immune response and improve the tolerance to self‐antigens by expressing inhibitory signals and secreting inhibitory cytokines, which play a vital function in preventing autoimmune disease (Hu et al., 2021; Raffin et al., 2020). Interestingly, GEVs, NVs, CCR6‐NVs and CX5461 all increased the proportion of Tregs in disease model mice, and FV@CX5461 had a more significant effect while dexamethasone had no obvious impact on Tregs. These results indicating the therapeutic effect of FV@CX5461 was dependent on regulating the immune system instead of suppressing all immune cells, thus circumventing the side effects of excessive immunosuppression.
Currently, most researches focus on mammalian‐derived exosomes. It has been reported that grapefruit‐derived nanovesicles attenuated inflammatory responses in DSS‐induced mouse colitis (Wang et al., 2014), and our work first confirmed the anti‐inflammatory effect of GEVs in mice psoriasis and atopic dermatitis, illustrating that GEVs may be a suitable tool for inflammatory diseases. Plants do not have any reported zoonotic or human pathogens, and we have confirmed that GEVs have low immunological risk and toxicity in vivo, with no induction of pro‐inflammatory cytokines. However, the influence of biomacromolecules in PLEVs in autoimmune diseases has not been systematically reported. MicroRNAs (miRNAs) share a high degree of similarity between animals and plants, along with evolutionary divergence in the composition and biogenesis machinery. Extracellular miRNAs as a new group of messengers and effectors in intercellular communication can be transferred via EVs to neighbouring or distant cells to modulate cell function (Bartel, 2009). In this study, by miRNA sequencing, we found that GEVs and NVs were rich in miRNAs, and abundant miRNAs were still retained in the nanovesicles after membrane fusion, demonstrating that our technique could achieve co‐delivery of active nucleic acids from edible plants to target cells, which may serve as an affordable and powerful therapeutic approach with minimal inconvenience to patients. Among identified miRNAs involved in GEVs and FVs, plant miRNA159 has been recently reported to exist in human serum and inhibit breast cancer growth (Chin et al., 2016). We found that plant miRNA159, by regulating mammalian gene expression, inhibited the proliferation of keratinocytes and reduced the production of inflammatory cytokines, providing new evidence for cross‐species regulation of plant miRNAs. This also provides new evidence that PLEVs are promising vectors for the transfer of plant‐active nucleic acids to mammalian cells. Other biomolecules such as proteins and lipids incorporated in PLEVs might also have effects. We also detected natural metabolites in GEVs, including flavonoids, terpenes, and polysaccharides. These are the active ingredients of many Chinese herbal medicines, inspiring us that exosomes from Chinese herbal medicines may have similar functions to Chinese herbal medicines extracts, and could be further investigated.
In this study, GEVs as biosafety and biodegradable vectors encapsulated CX5461 to protect the immunosuppressant from elimination due to poor solubility. CX5461 is an effective and selective anti‐cancer drug, which has been shown to inhibit rRNA transcription by selectively targeting the RNA polymerase I transcription mechanism (Xu et al., 2017). Excitingly, CX5461 is already in clinical trials to treat malignancies with no obvious genotoxicity (Clinical Study Identifier: ACTRN12613001061729, NCT02719977). In an earlier study, we found that CX5461 showed dramatic inhibitory effects on T cell activation and proliferation in vitro and in vivo, potentially preventing organ transplant rejection (Tsai et al., 2021). This suggests that CX5461 may be also a promising immunosuppressant for autoimmune diseases, but the influence of CX5461 on psoriasis and atopic dermatitis has not been reported. Since specific inhibition of ribosome biogenesis may lead to selective inhibition of proliferating cells (Derenzini et al., 2017), and excessive proliferation of keratinocytes present in both atopic dermatitis and psoriasis, we explored the influence of CX5461 on keratinocytes and found that CX5461 inhibited their proliferation by arresting the cells in G2 phase. CX5461 inhibited the proliferation of CD4+T cells and keratinocytes, and the results of animal experiments suggested that CX5461 has the potential to treat atopic dermatitis and psoriasis clinically.
By GEO data analysis, we confirmed the high expression of CCL20 in lesions of patients who suffer from psoriasis, atopic dermatitis, as well as ulcerative colitis, suggesting the CCL20‐CCR6 axis may be a promising signalling pathway in controlling these autoimmune diseases. In order to successfully target specific tissue, the therapeutic vectors need to meet the goals of tissue penetration, tissue specificity, and release of the therapeutic agent. MSCs can be isolated from human tissues, such as bone marrow, umbilical cord, adipose tissue, gingival tissues, and so on. After comparison, we found that GMSC‐derived NVs had strong immunosuppressive effects on the one hand, and on the other hand, it was more convenient to obtain gingiva tissues, which provides a possibility of autologous treatment. Therefore, we chose GMSCs as the carrier of the CCR6 receptor. It has been reported that CCL20‐CCR6 signalling in the gut‐associated lymphoid tissues inhibited the expression of Foxp3 in Tregs and promoted the differentiation of Th17 cells (Kulkarni et al., 2018). In our study, CCR6‐expressing nanovesicles exhibit an impressive ability to target chemokine CCL20 in vitro. In mouse disease models, enhanced therapeutic efficiency was observed following the CCR6‐NVs/FVs administration. After GEV@CX5461 was coated with the CCR6‐GMSC membrane, the fusion nanovesicle has the targeting ability to inflammatory sites with high expression of CCL20 rather than the non‐specific releasing drug. This indicates that in response to CCL20 chemokines produced in the inflammatory site, CCR6 expressing on NVs/FVs drove the nanovesicles to reach the affected tissue, facilitating CCR6‐FVs and CCL20 interaction in the inflammatory conditions. Together we indicated that CCR6 expression could enhance the efficacy of nanovesicles in autoimmune disease treatment.
In conclusion, we found that CX5461 alleviated psoriasis and atopic dermatitis, and we creatively designed the CX5461‐encapsulated fusion vesicles of grapefruit‐derived exosomes with bioengineered GMSC‐membranes to treat psoriasis and atopic dermatitis. FV@CX5461 has the potential for the targeted immunotherapy of multiple autoimmune diseases. Notably, GEVs, NVs, and FVs were not only used as drug delivery vehicles but also as therapeutic agents to exert synergistic therapeutic effects through the contained lipids, natural metabolites, proteins, and nucleic acids. More significantly, it provides a novel strategy for biomaterials‐based drug delivery: plant‐derived exosomes could be modified by fusing with cell membranes carrying various kinds of disease‐mediating receptors; the source of cell membranes can also be diversified, such as macrophages, lymphocytes, and so on. However, the cross‐border regulatory mechanism of grapefruit‐derived miRNAs was not clearly understood, and whether FV@CX5461 can be used in other autoimmune diseases deserves further study.
3.1. Materials and methods
3.1.1. Materials
CX5461 was purchased from MedChemExpress (MCE); DNCB was purchased from Sigma‐Aldrich; Cell Cycle Detection Kit was purchased from Keygen Biotech; Cell counting kit‐8 was purchased from Dojindo Laboratories, and Imiquimod (IMQ) was purchased from Med‐ shine. Primary antibodies are listed in Table S2, Supporting Information.
3.1.2. Cell culture
HEK293T (human embryonic kidney cell line), RAW267.4 cells (mouse mononuclear macrophage cell line), HaCaT cells (human keratinocyte cell line), were cultured in DMEM supplemented with 10% foetal bovine serum (Gibco) and 1% penicillin‐streptomycin; Jurkat cells (human acute T cell leukaemia cell lines) were cultured in RPMI 1640 supplemented with 10% foetal bovine serum (Gibco) and 1% (v/v) penicillin‐streptomycin; GMSCs were cultured in DMEM‐F12 and supplemented with 10% foetal bovine serum (BI). All cells were grown at 37°C in a 5% CO2 atmosphere. To isolate primary murine keratinocytes, the skin from the neonatal mice was cut into small pieces and then suspended on 0.25% Dispase II (Sigma‐Aldrich) overnight at 4°C. Then the epidermis was peeled and placed into 0.05% trypsin‐EDTA (Gibco) for 10 min at 37°C with gentle shaking. The cell suspension was added to a trypsin neutral medium and then centrifuged at 400 g for 10 min. Primary murine keratinocytes were cultured in EpiLife™ medium (Gibco) supplemented with keratinocyte growth supplement (Gibco).
3.1.3. Bibliometric and visualization analysis
PubMed database is a professional search of data sources, and the citations were published from 1 January 2000 to 31 December 2021. In Figure 2a, the search query was (plant[Title/Abstract]) AND (extracellular vesicles[Title/Abstract]), and a total of 128 citations were retrieved. The inclusion criteria were relevant literature on the study of the therapeutic functions of plant‐derived extracellular vesicles. Save all the citations, then analyse the therapeutic functions of plant‐derived extracellular vesicles studied in the literature and visually analyse the hot plants by Origin.
3.1.4. Isolation and purification of GEVs
Fresh grapefruits were peeled and then homogenized in a high‐speed blender at 4°C for 1 min. The juice was collected and filtered through paper filters. The supernatant was collected after centrifugation at 500 × g for 15 min, 2000 × g for 20 min, 10,000 × g for 1 h, and 40,000 × g for 1 h. The centrifugal precipitation containing GEVs was spun down at 150,000 × g for 2 h at 4°C. Then, the isolated exosomes were further purified in a sucrose gradient (8%, 30%, 45%, 60% sucrose in 20 mM Tri‐CI, pH 7.2) (Wang et al., 2013).
3.1.5. Isolation of human gingival mesenchymal stem cells (GMSCs)
Human gingiva‐derived mesenchymal stem cells (GMSCs) were isolated from discarded gingival tissues from healthy human subjects who underwent a tooth extraction procedure following informed consent. Briefly, the gingival tissues were cut into 1 mm3 pieces, dispersedly transferred into a petri dish with DMEM‐F12 containing 10% foetal bovine serum, 1% P/S and incubated at 37°C with 5% CO2. Adherent cells were further cultured for another 7 days under the same conditions mentioned above and GMSCs at the 2nd−5th passages were used for subsequent experiments.
3.1.6. Preparation of CCR6‐NVs and NVs
To access GMSCs stably expressing chemokine receptor CCR6, the target plasmids (plv‐cmv‐mcs‐mcherry‐6xhis‐ires‐bla, Sino Biological Inc) together with lentiviral packaging and envelope plasmids were transfected into HEK‐293T cells using Lipofectamine 3000 (ThermoFisher, USA). The supernatant containing lentivirus was harvested after 48–72 h, filtered through 0.45 μm pore‐sized polycarbonate membrane filters, and stored at −80°C. GMSCs were infected with lentivirus and selected with blasticidin (6 μg/mL) to obtain target cells. Then CCR6‐GMSCs or GMSCs were incubated with homogenization medium buffer (containing 0.25 M sucrose, 1 mM EDTA, 20 mM Hepes‐NaOH, pH 7.4, and protease inhibitor cocktail) on ice. After grinding, and centrifugation at 5000 rpm for 10 min and then 12000 rpm for 10 min at 4°C, the sediments containing cell membranes were filtered using 0.22 μm pore‐sized polycarbonate membrane filters and stored at −80°C.
3.1.7. Loading of CX5461 into GEVs
To encapsulate CX5461 into GEVs, 1 mg CX5461 was mixed with 2 mg GEVs at the total protein level and diluted with electroporation buffer (1.15 mM potassium phosphate, 25 mM potassium chloride, 21% OptiPrep working solution) to 1 mL at 4°C. The mixtures were added to 0.4 cm electroporation cuvettes and subjected to electroporation at 300 V and 150 μF for 1 s using a Bio‐Rad Gene Pulser Xcell Electroporation System. The mixtures were incubated on ice for 1 h and then repeat electroporation. After electroporation, the mixtures were incubated on ice for 30 min for membrane recovery. Following centrifugated at 100,000 × g for 70 min to remove unloaded CX5461, the resulting pellets (GEV@CX5461) were resuspended in cold PBS for further application. The supernatants were detected by UV–VIS at 300 nm for calculating loading efficiency.
3.1.8. Preparation of FV@CX5461
To prepare the FV@CX5461, CX5461‐encapsulated GEVs were mixed with CCR6‐NVs at a concentration ratio of 1:1 (the concentration was measured with Nanosight NS300, unit: particles/ml) and extruded 10 times through 0.22 μm pore‐sized polycarbonate membrane filters. After centrifugation, deposits were resuspended in cold PBS and stored at −80°C.
3.1.9. Cellular uptake of FV@CX5461
GEVs were pre‐stained with WGA488 for 15 min and washed with PBS, then fused with CCR6‐GMSCs membranes to obtain FV@CX5461. Next, FV@CX5461 were respectively added to PBMCs and HaCaT cells which were seeded in the confocal plate. After 30 min of incubation, cells were fixed with 4% paraformaldehyde and stained with DAPI. Results were observed using a confocal microscope (Zeiss, LSM880). To detect the cellular uptake rate of CX5461, HaCaT cells were cultured with CX5461, GEV@CX5461, and FV@CX5461, respectively, then HaCaT cells were lysed and detected by UV–VIS at 300 nm.
3.1.10. Characterization experiments
Purified vesicles were dripped onto 230 mesh copper and incubated for 10 min. Then, the residual liquid was removed and washed with PBS. vesicles on the copper were then stained with 2% uranyl acetate and incubated for 1 min, washed with distilled water, and air‐dried in the dark. TEM images were acquired using transmission electron microscopy (JEM‐1400, 120 kV). The size distributions and zeta potentials of GEVs, NVs, CCR6‐NVs, GEV@CX5461, and FV@CX5461 were evaluated by a NanoBrook 90Plus PALS (Brookhaven) instrument.
3.1.11. Isolation of PBMCs from human peripheral blood and CFSE staining
PBMCs were isolated from the peripheral blood of healthy volunteers, using Ficoll lymphocyte isolation medium (Sigma‐Aldrich), and stained with carboxyfluorescein succinimidyl amino ester (CFSE, 5 μM, BioLegend) for 20 min at 37°C. Culture medium was used to quench staining and washing by centrifuging at 1000 × g for 5 min five times. Finally, CFSE‐labelled PBMCs were cultured in RPMI 1640 with 10% FBS, and the proliferation of PBMCs was analysed by flow cytometry (Cytoflex, Beckman, USA) on day 0, day 3, day 5, and day 7.
3.1.12. Isolation of CD3+ and CD4+ T cells from mice spleens
CD3+ and CD4+ T cells were purified (>98%) from single‐cell suspensions of mice spleen by negative selection with MojoSortTM Mouse CD3+ T cell Isolation Kit and MojoSortTM Mouse CD4+ T cell Isolation Kit (Biolegend, USA), respectively. CFSE‐labelled CD3+T cells or CD4+T cells were cultured in CD3‐coated plates (Biolegend, USA) and medium (RPMI1640 with 10% FBS, 1% penicillin/streptomycin, 2 μg/mL anti‐CD28, and 2 ng/mL IL‐2). The proliferation of cells was analysed by flow cytometry (Cytoflex, Beckman, USA).
3.1.13. Cell viability assay and detection of the cell cycle
Cell viability was evaluated using the Cell Counting Kit‐8. HaCaT cells or HDF cells were seeded in 96‐well plates, then added CX5461 with different concentrations for 24 and 48 h. Then the culture medium was replaced with serum‐free DMEM containing 10% CCK‐8 and incubated for an hour. The results were presented as absorbance at 450 nm using a microplate reader. The cell cycle of HaCaT cells was detected by Cell Cycle Detection Kit (KeyGen Biotech, China). In brief, cells were collected and fixed in 70% cold ethanol overnight at 4°C. After washing with PBS twice, cells were incubated with PI/RNase A staining buffer for 30 min and subsequently analysed by Beckman flow cytometry and FlowJo.
3.1.14. Immunofluorescence assay
HaCaT cells attached to the slide glass coverslips were fixed with 4% paraformaldehyde for 15 min and then blocked with 3% BSA containing 0.4% Triton X‐100 for 1 h. After incubation with p‐stat3 antibody (CST, USA) for 2 h at room temperature, cells were incubated with fluorescein‐conjugated secondary antibodies (CST, USA) for 1 h at room temperature, then the cell nucleus was counterstained with DAPI. The localization of p‐stat3 was observed using a confocal microscope (Zeiss, LSM880).
3.1.15. Cell migration assay
Transwell assay was used to verify the blocking effect of CCR6‐NVs on T cell migration induced by chemokine CCL20. Briefly, mouse spleen lymphocytes (1 × 106/well) were suspended in serum‐free RPMI media and loaded in the upper chamber of 5.0 μm pore size transwell apparatus (Corning, USA). Serum‐free RPMI containing CCL20 recombinant protein (MCE, USA) in a concentration gradient was added to the basolateral chamber. After 3 h of co‐culture at 37°C, the migrated cells were harvested from the basolateral chamber and counted by flow cytometry using anti‐CD3 and anti‐CCR6 monoclonal antibodies (BioLegend).
3.1.16. ROS level detection
HKSOX‐1 m(MCE), a mitochondria‐targeting fluorescent probe, was used to analyse the mitochondrial ROS level. More specifically, cells and HKSOX‐1 m (10 μM) were co‐incubated for 30 min at room temperature, then washed twice with PBS, 5 min each time, and the cells were collected and analysed by flow cytometry (Cytoflex, Beckman, USA). 2′,7‐Dichlorofluorescin diacetate probe (DCFH‐DA, Sigma) was used to analyse the intracellular ROS level. Cells and DCFH‐DA(5μM)were co‐incubated for 30 min at 37°C, then washed three times with PBS to remove extracellular DCFH‐DA, and the cells were collected and analysed by flow cytometry. To analyse the in vivo ROS levels, mice were intradermally injected with DCFH‐DA probe (150 μL, 100 μM) and sacrificed after different treatments. The probe was incubated in vivo for 1 h, and then all the mice were dissected and an equal amount of skin tissues were clipped and ground for the detection of the fluorescence intensity of DCF by the fluorescence microplate reader (Ex/Em = 488/525 nm). The rest of the skin tissues were embedded for the preparation of frozen sections and photographed with the confocal microscope (LSM 880 NLO, ZEISS).
3.1.17. Lipidomic analysis
The sample (100 uL) was incubated with 480 μL of extract solution (MTBE:methanol1 = 5:1) and sonicated and lysed in ice water for 10 min. After incubating at −40°C for 1 h, and centrifuging at 3000 rpm for 15 min at 4°C, the supernatant was dried in a rotary vacuum concentratorand reconstituted in 100 μL solution (DCM: MeOH = 1:1). After sonication in ice water for 10 min, the sample was centrifuged at 13,000 rpm for 15 min at 4°C, and the supernatant was introduced to a vial for UPLC/MS analysis. LC–MS/MS analyses were performed by UHPLC (Vanquish, Thermo) with a UPLC HSS T3 column (2.1 mm × 100 mm, 1.8 μm) coupled to Exactive HFX mass spectrometer (Orbitrap MS, Thermo). The column temperature = 55°C, the auto‐sampler temperature = 4°C, and the injection volume = 2 μL. Mobile phase A consisted of 40% water, 60% acetonitrile, and 10 mmol/L ammonium formate, and mobile phase B consisted of 10% acetonitrile, 90% isopropanol, and 10 mmol/L ammonium formate (50 mL for every 1000 mL mixed solvent). The analysis was carried out with elution gradient as follows: 0∼1.0 min,40% B;1.0∼12.0 min,40%∼100% B; 12.0∼13.5 min, 100% B; 13.5∼13.7 min, 100%∼40% B; 13.7∼18.0 min, 40% B.
3.1.18. Chemical composition detection
To detect the chemical composition of GEVs, the sample was dried under nitrogen and incubated with an extraction solution (methanol:water = 3:1, pre‐cooled at −40°C, with internal standard 2‐Chloro‐DL‐Phenylalanine). After sonication and centrifugation, the supernatant was analysed by Exion LC System (Sciex) with Waters Acquity UPLC HSS T3 column (1.8 μm 2.1 × 100 mm) coupled to Sciex QTrap 6500+ (Sciex Technologies) system. Mobile phase A was water (with 0.1% formic acid), and mobile phase B was acetonitrile. The column temperature was 40°C, and the injection volume was 2 μL.
3.1.19. miRNA sequencing
RNA was extracted using an RNA miRNeasy Micro Kit (Qiagen, Germany), and the quantity and purity were quantified by Quantus Fluorometer (Promega, USA) and Qsep100 (BIOptic, China). Library construction was performed using Multiplex Small RNA Library Prep Set for Illumina (NEB, USA), and subsequent sequencing was performed on the Illumina Novaseq System (Illumina, USA). After aligning the Rfam library to remove ncRNAs (such as rRNA, tRNA, etc.), the reading of each miRNA‐seq sample was mapped to specific species precursors in miRbase 22.0 to identify known miRNAs. Quantitative statistics of miRNAs were performed using miRDeep2, and differential expression analysis was performed with DESeq2. Sequencing data supporting these studies can be found in the Gene Expression Omnibus database under accession numbers GSE223353 and GSE223577.
3.1.20. In vitro experiments of miRNA159 mimics
miRNA159 mimic oligos for cell experiments consisting of the sense strand and the antisense strand to create a double‐stranded miRNA duplex: miR159 sense 5′‐UUUGGAUUGAAGGGAGCUCUmA‐3 (“m” denotes 2′‐O‐methylation), and miR159a anti‐sense 5′‐UAGAGCUCCCUUCAAUCCAAmA‐3′ (“m” denotes 2′‐O‐methylation). miR159 mimics and scrambled control were synthesized by Sino Biological Inc. To explore the effect of miRNA159 on LPS‐induced HaCaT cells, cells were transfected with either miRNA159 mimics or scrambled control using lipo3000 (Thermo Fisher Scientific). For cell proliferation assays, at 24 and 48 h post‐transfection, the cells were seeded at a 96‐well dish for CCK‐8 assay. For qPCR assays, at 48 h post‐transfection, cells were harvested and extracted RNA for subsequent experiments. miR159a primer 5′‐TTTGGATTGAAGGGAGCTC‐3′.
3.1.21. Mouse psoriasis model induction and treatment
The use of laboratory animals and all animal experiments were reviewed and approved by the Animal Ethics Committee of Sun Yat‐sen University, China. The approval number is SYSU‐IACUC‐2021‐000007. The 6–8 weeks healthy male BALB/c mice were purchased from the laboratory animal centre of Sun Yat‐Sen University and kept in a pathogen‐free facility. Following the anaesthesia with 1% pentobarbital, the back hair of mice was shaved with a razor and mild depilation cream, then mice were randomly divided into nine groups: NC, IMQ, GEVs (20 mg/kg), CX5461(2 mg/kg), NVs (20 mg/kg), CCR6‐NVs (20 mg/kg), Dex, GEV@CX5461 (20 mg/kg@2 mg/kg), and FVs@CX5461(20 mg/kg@2 mg/kg). Except for the NC group, all other groups established the psoriasis model by applying 62.5 mg imiquimod (IMQ) ointment on the depilated skin on the back of mice for 7 consecutive days. On day4, the psoriasis mice were respectively treated with PBS, GEVs, CX5461, CCR6‐NVs, NVs, GEV@CX5461, and FVs@CX5461 by intravenous administration for 3 consecutive days. The dexamethasone (Dex) group was used as a positive control. The severity of skin inflammation in the psoriasis‐like mouse model was scored with three clinical signs (erythema, scale, and thickening) based on the human clinical Psoriasis Area and Severity Index (PASI) on a scale from 0 to 4: 0, none symptoms; 1, slight; 2, moderate; 3, marked; 4, very marked. Mice were scored objectively daily and the total score was obtained by accumulating the three index scores (score 0–12). After 7 days, mice were sacrificed to obtain blood, skin tissues, and spleen for the following experiments.
3.1.22. Mouse atopic dermatitis model induction and treatment
The 6–8 weeks healthy male BALB/c mice were anesthetised with 1% pentobarbital and their back hair was shaved, then mice were randomly divided into nine groups: NC, DNCB, GEVs (20 mg/kg), CX5461 (2 mg/kg), CCR6‐NVs (20 mg/kg), NVs (20 mg/kg), Dex, GEV@CX5461 (20 mg/kg@2 mg/kg), and FVs@CX5461 (20 mg/kg@2 mg/kg). To induce the atopic dermatitis mouse model, 1%DNCB was applied three times in the first week (200 μL of a 3:1 mixture of acetone/olive), and 0.5%DNCB (200 μL of a 3:1 mixture of acetone/olive) was applied every other day for the next two weeks. The NC group was treated with the same dose of solvent. The treatment started in the third week when symptoms of atopic dermatitis in mice were evident. PBS, GEVs, CX5461, CCR6‐NVs, NVs, GEV@CX5461, and FVs@CX5461 were administered separately via tail vein injection on day 16, day 18, and day 20. The Dex group was used as a positive control. The severity of skin inflammation in the atopic dermatitis mouse model was scored with four clinical signs (erythema, scale/dryness, oedema, and erosion) based on the scoring system as previously described on a scale from 0 to 4: 0, no symptoms; 1, mild; 2, moderate; 3, severe; 4, very serve. The sum of the individual scores was used as the Severity score (score 0–16). After 21 days, mice were sacrificed to obtain blood, skin tissues, and spleen for the following experiments.
3.1.23. In vivo imaging and biodistribution analysis
Cy5.5‐labelled GEVs, NVs, CCR6‐NVs, and FV@CX5461(20 mg/kg) were injected into psoriasis and atopic dermatitis mice, respectively, through the tail vein. At 3 h, 8 h, 24 h, 32 h, and 48 h after the injection, mice were detected by the Xenogen IVIS Spectrum imaging system. And after 48 h, major organs and skin tissues were harvested and the fluorescence was detected by the Xenogen IVIS Spectrum imaging system. Fluorescence emission was measured at an excitation of 640 nm and an emission of 680 nm.
3.1.24. Flow cytometry analysis
The spleen of mice was ground on 100 μm cell sieves to obtain a single‐cell suspension and treated with red blood cell lysis buffer (Solarbio). Then, cells were resuspended in cell staining buffer (BioLegend) and incubated with FITC anti‐mouse CD3 antibody (Biolegend), APC anti‐mouse CD4 antibody, and Brilliant violet 510™ anti‐mouse CD8 (BioLegend) on ice for 15 min in dark. For intracellular staining, cells were fixed in fixation buffer (Biolegend) in dark for 20 min at room temperature, then permeabilized and stained with Intracellular staining perm wash buffer (Biolegend) in dark for 15 min at room temperature with the following antibodies: Brilliant Violet 510™ anti‐mouse IL‐17A (Biolegend) and Brilliant Violet 421™ anti‐mouse IL‐4 (Biolegend). For the analysis of Tregs, cells were first stained with APC anti‐mouse CD4 antibody and PE anti‐mouse CD25 antibody, then fixed and permeabilized with a FOXP3 Fix/Perm Buffer Set (Biolegend). Next, cells were stained with Pacific Blue anti‐mouse Foxp3 antibody (Biolegend) for 30 min at room temperature. Subsequently, cells were washed twice with cell staining buffer and subjected to flow cytometry analysis (Cytoflex, Beckman, USA).
3.1.25. RNA isolation and quantitative real‐time PCR
Total RNA was extracted from skin tissues or cells using Trizol reagent (Takara Biotech, Japan), and the concentration was measured by Nanodrop One (Thermo Fisher Scientific). Then total RNA samples were reversely transcribed into cDNA with EasyScript cDNA Synthesis SuperMix (TransGen Biotech, China) and followed by qPCR using PerfectStart Green qPCR Mix (TransGen Biotech, China) with LightCycler 96 (Roche). miR159 was specifically reverse transcribed with the RransScript miRNA First‐Strand cDNA Synthesis SuperMix (TransGen Biotech, China) according to the manufacturer's instructions. Fold changes of relative gene expression were calculated with the 2−ΔΔCt method, which normalized to the internal controls U6 (for miRNA) or GAPDH (for mRNA). Primers used in this study are listed in Table S1, Supporting Information.
3.1.26. Western blot
Cells were lysed with RIPA lysis buffer (Thermo Scientific) containing protease inhibitor and phosphatase inhibitor (Roche). Protein concentrations were measured by Bradford assay. The protein samples (10 μg of protein per well) were separated by 10%SDS‐PAGE and were transferred to PVDF membranes (Bio‐Rad Laboratories). Then the PVDF membranes were blocked with 5% non‐fat milk for 1.5 h at room temperature and incubated with primary antibodies overnight at 4°C. After that, the membranes were incubated with HRP‐conjugated anti‐mouse or anti‐rabbit secondary antibodies for 1.5 h at room temperature, Protein bands were visualized using the Enhanced Chemiluminescence Reagent Kit (ECL, Protein Tech), and Chemiluminescence imaging system (BG‐gdsAUTO730, Bay Gene). Quantification of the target protein was normalized to β‐actin by grey value.
3.1.27. Enzyme‐linked immunosorbent assay (ELISA)
To detect the concentrations of cytokines in serum and CCL20 in skin tissues, blood samples collected from mice were separated by centrifugation (12,000 rpm, 10 min, 4°C) and skin tissues were homogenized. Levels of cytokines IgE, IL‐4, IL‐1β, TNF‐α, IL‐17A, IL‐22 and IL‐12/23 p40 in serum were measured by ELISA kits (Dakewe Biotech, China) following the manufacturer's instructions. CCL20 in skin tissues was detected by Elisa kits purchased from MultiSciences, China.
3.1.28. Histology and immunohistochemistry analysis
Skin tissues were collected and submerged with 4% paraformaldehyde for fixation. Then thinly sliced skin tissues were embedded with paraffin and stained with haematoxylin and eosin (H&E) . The condition of inflammatory cell infiltration was observed by microscopy with 10X magnification. For immunohistochemistry analysis, paraffin‐embedded tissues were deparaffinized and incubated with primary antibodies including Ki67, IL‐6, IL‐17A and TNF‐α, followed by incubation with secondary antibodies and visualization by DAB detection IHC Kit. Quantification of IHC was performed by optical density value determination using Image J software.
3.1.29. Statistical analysis
All data were presented as the mean ± standard deviation (SD) of at least three independent experiments and analysed with GraphPad Prism Ver 8.0. Statistical significance was measured using the unpaired two‐tailed Student's t‐test for comparisons between two groups and one‐way analysis of variance (ANOVA) followed by relevant post hoc tests for comparisons in multiple groups.
AUTHOR CONTRIBUTIONS
Rufan Huang: Conceptualization; Data curation; Formal analysis; Methodology; Validation; Visualization; Writing—original draft. Bo Jia: Conceptualization; Data curation; Formal analysis; Funding acquisition; Resources; Validation. Dandan Su: Formal analysis; Validation; Visualization. Manchun Li: Formal analysis; Visualization. Zhanxue Xu: Data curation; Formal analysis. Chao He: Data curation; Formal analysis; Validation. Yisheng Huang: Resources; Validation. Hang Fan: Resources. Hongbo Chen: Funding acquisition; Project administration; Resources; Supervision; Writing—review and editing. Fang Cheng: Funding acquisition; Project administration; Resources; Supervision; Writing—review and editing
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
This research was supported by the National Natural Science Foundation of China (grant number 81970145); Natural Science Foundation of Guangdong Province (grant numbers 2020A1515011465, 2022A1515012214 and 2020A151501467, China); International Collaboration of Science and Technology of Guangdong Province (No. 2020A0505100031, China); Guangdong Provincial Key Laboratory of Digestive Cancer Research (No. 2021B1212040006, China); Science, Technology & Innovation Commission of Shenzhen Municipality (grant numbers JCYJ20200109142605909 and JCYJ20210324120007020, China); Sun Yat‐sen University (No. 20ykzd17, China).
Huang, R. , Jia, B. , Su, D. , Li, M. , Xu, Z. , He, C. , Huang, Y. , Fan, H. , Chen, H. , & Cheng, F. (2023). Plant exosomes fused with engineered mesenchymal stem cell‐derived nanovesicles for synergistic therapy of autoimmune skin disorders. Journal of Extracellular Vesicles, 12, e12361. 10.1002/jev2.12361
Rufan Huang and Bo Jia contributed equally to this work.
Contributor Information
Hongbo Chen, Email: chenhb7@mail.sysu.edu.cn.
Fang Cheng, Email: chengf9@mail.sysu.edu.cn.
REFERENCES
- Bartel, D. P. (2009). MicroRNAs: Target recognition and regulatory functions. Cell, 136(2), 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, A. R. , Fong, M. Y. , Somlo, G. , Wu, J. , Swiderski, P. , Wu, X. , & Wang, S. E. (2016). Cross‐kingdom inhibition of breast cancer growth by plant miR159. Cell Research, 26(2), 217–228. 10.1038/cr.2016.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes‐Rius, A. , Desai, A. , Yuen, D. , Johnston, A. P. R. , & Voelcker, N. H. (2021). Inducing immune tolerance with dendritic cell‐targeting nanomedicines. Nature Nanotechnology, 16(1), 37–46. 10.1038/s41565-020-00810-2 [DOI] [PubMed] [Google Scholar]
- Derenzini, M. , Montanaro, L. , & Trere, D. (2017). Ribosome biogenesis and cancer. Acta Histochemica, 119(3), 190–197. [DOI] [PubMed] [Google Scholar]
- Drygin, D. , O'Brien, S. E. , Hannan, R. D. , McArthur, G. A. , & Von Hoff, D. D. (2014). Targeting the nucleolus for cancer‐specific activation of p53. Drug Discovery Today, 19(3), 259–265. 10.1016/j.drudis.2013.08.012 [DOI] [PubMed] [Google Scholar]
- Farhadi, F. , Khameneh, B. , Iranshahi, M. , & Iranshahy, M. (2019). Antibacterial activity of flavonoids and their structure‐activity relationship: An update review. Phytotherapy Research: PTR, 33(1), 13–40. 10.1002/ptr.6208 [DOI] [PubMed] [Google Scholar]
- Fugger, L. , Jensen, L. T. , & Rossjohn, J. (2020). Challenges, progress, and prospects of developing therapies to treat autoimmune diseases. Cell, 181(1), 63–80. [DOI] [PubMed] [Google Scholar]
- Gao, F. , Chiu, S. M. , Motan, D. A. , Zhang, Z. , Chen, L. , Ji, H. L. , Tse, H. F. , Fu, Q. L. , & Lian, Q. (2016). Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death & Disease, 7(1), e2062. 10.1038/cddis.2015.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi, K. , Balato, A. , Enerbäck, C. , & Sabat, R. (2021). Therapeutics targeting the IL‐23 and IL‐17 pathway in psoriasis. The Lancet, 397(10275), 754–766. 10.1016/s0140-6736(21)00184-7 [DOI] [PubMed] [Google Scholar]
- Guttman‐Yassky, E. , Krueger, J. G. , & Lebwohl, M. G. (2018). Systemic immune mechanisms in atopic dermatitis and psoriasis with implications for treatment. Experimental Dermatology, 27(4), 409–417. [DOI] [PubMed] [Google Scholar]
- Hendrich, A. B. (2006). Flavonoid‐membrane interactions: Possible consequences for biological effects of some polyphenolic compounds. Acta Pharmacologica Sinica, 27(1), 27–40. [DOI] [PubMed] [Google Scholar]
- Hu, W. , Wang, Z. M. , Feng, Y. , Schizas, M. , Hoyos, B. E. , van der Veeken, J. , Verter, J. G. , Bou‐Puerto, R. , & Rudensky, A. Y. (2021). Regulatory T cells function in established systemic inflammation and reverse fatal autoimmunity. Nature Immunology, 22(9), 1163–1174. 10.1038/s41590-021-01001-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismael, M. , Webb, R. , Ajaz, M. , Kirkby, K. J. , & Coley, H. M. (2019). The targeting of RNA polymerase I transcription using CX‐5461 in combination with radiation enhances tumour cell killing effects in human solid cancers. Cancers, 11(10), 1429. 10.3390/cancers11101429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju, S. , Mu, J. , Dokland, T. , Zhuang, X. , Wang, Q. , Jiang, H. , Xiang, X. , Deng, Z. B. , Wang, B. , Zhang, L. , Roth, M. , Welti, R. , Mobley, J. , Jun, Y. , Miller, D. , & Zhang, H. G. (2013). Grape exosome‐like nanoparticles induce intestinal stem cells and protect mice from DSS‐induced colitis. Molecular Therapy: The Journal of the American Society of Gene Therapy, 21(7), 1345–1357. 10.1038/mt.2013.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , Li, S. , Zhang, S. , & Wang, J. (2022). Plant‐derived exosome‐like nanoparticles and their therapeutic activities. Asian Journal of Pharmaceutical Sciences, 17(1), 53–69. 10.1016/j.ajps.2021.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T. , Siegmund, B. , & Le Berre, C. (2020). Ulcerative colitis. Nature Reviews Disease Primers, 6(1), 74. [DOI] [PubMed] [Google Scholar]
- Kulkarni, N. , Meitei, H. T. , Sonar, S. A. , Sharma, P. K. , Mujeeb, V. R. , Srivastava, S. , Boppana, R. , & Lal, G. (2018). CCR6 signaling inhibits suppressor function of induced‐Treg during gut inflammation. Journal of Autoimmunity, 88, 121–130. 10.1016/j.jaut.2017.10.013 [DOI] [PubMed] [Google Scholar]
- Lai, P. , Weng, J. , Guo, L. , Chen, X. , & Du, X. (2019). Novel insights into MSC‐EVs therapy for immune diseases. Biomarker Research, 7, 6. 10.1186/s40364-019-0156-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Fang, H. , Dang, E. , & Wang, G. (2020). The role of ceramides in skin homeostasis and inflammatory skin diseases. Journal of Dermatological Science, 97(1), 2–8. 10.1016/j.jdermsci.2019.12.002 [DOI] [PubMed] [Google Scholar]
- Martin‐Orozco, N. , Muranski, P. , Chung, Y. , Yang, X. O. , Yamazaki, T. , Lu, S. , Hwu, P. , Restifo, N. P. , Overwijk, W. W. , & Dong, C. (2009). T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity, 31(5), 787–798. 10.1016/j.immuni.2009.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitei, H. T. , Jadhav, N. , & Lal, G. (2021). CCR6‐CCL20 axis as a therapeutic target for autoimmune diseases. Autoimmunity Reviews, 20(7), 102846. [DOI] [PubMed] [Google Scholar]
- Orecchioni, M. , Ghosheh, Y. , Pramod, A. B. , & Ley, K. (2019). Macrophage polarization: Different gene signatures in M1(LPS+) vs. classically and M2(LPS‐) vs. alternatively activated macrophages. Frontiers in Immunology, 10, 1084. 10.3389/fimmu.2019.01084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanivel, J. A. , Macbeth, A. E. , Chetty, N. C. , & Levell, N. J. (2014). An insight into JAK‐STAT signalling in dermatology. Clinical and Experimental Dermatology, 39(4), 513–518. 10.1111/ced.12273 [DOI] [PubMed] [Google Scholar]
- Raffin, C. , Vo, L. T. , & Bluestone, J. A. (2020). T‐reg cell‐based therapies: Challenges and perspectives. Nature Reviews Immunology, 20(3), 158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolen, J. S. , Aletaha, D. , & McInnes, I. B. (2016). Rheumatoid arthritis. Lancet, 388(10055), 2023–2038. [DOI] [PubMed] [Google Scholar]
- Teng, Y. , Ren, Y. , Sayed, M. , Hu, X. , Lei, C. , Kumar, A. , Hutchins, E. , Mu, J. , Deng, Z. , Luo, C. , Sundaram, K. , Sriwastva, M. K. , Zhang, L. , Hsieh, M. , Reiman, R. , Haribabu, B. , Yan, J. , Jala, V. R. , Miller, D. M. , … Zhang, H. G. (2018). Plant‐derived exosomal microRNAs shape the gut microbiota. Cell Host & Microbe, 24(5), 637–652. e8. 10.1016/j.chom.2018.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, H. I. , Wu, Y. , Liu, X. , Xu, Z. , Liu, L. , Wang, C. , Zhang, H. , Huang, Y. , Wang, L. , Zhang, W. , Su, D. , Khan, F. U. , Zhu, X. , Yang, R. , Pang, Y. , Eriksson, J. E. , Zhu, H. , Wang, D. , Jia, B. , … Chen, H. (2022). Engineered small extracellular vesicles as a FGL1/PD‐L1 dual‐targeting delivery system for alleviating immune rejection. Advanced Science (Weinheim, Baden‐Wurttemberg, Germany), 9(3), e2102634. 10.1002/advs.202102634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, H. I. , Zeng, X. , Liu, L. , Xin, S. , Wu, Y. , Xu, Z. , Zhang, H. , Liu, G. , Bi, Z. , Su, D. , Yang, M. , Tao, Y. , Wang, C. , Zhao, J. , Eriksson, J. E. , Deng, W. , Cheng, F. , & Chen, H. (2021). NF45/NF90‐mediated rDNA transcription provides a novel target for immunosuppressant development. EMBO Molecular Medicine, 13(3), e12834. 10.15252/emmm.202012834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino, A. V. , Kanno, Y. , Ferdinand, J. R. , & O'Shea, J. J. (2015). Mechanisms of Jak/STAT signaling in immunity and disease. Journal of Immunology (Baltimore, Md.: 1950), 194(1), 21–27. 10.4049/jimmunol.1401867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Zhuang, X. , Deng, Z. B. , Jiang, H. , Mu, J. , Wang, Q. , Xiang, X. , Guo, H. , Zhang, L. , Dryden, G. , Yan, J. , Miller, D. , & Zhang, H. G. (2014). Targeted drug delivery to intestinal macrophages by bioactive nanovesicles released from grapefruit. Molecular Therapy: The Journal of the American Society of Gene Therapy, 22(3), 522–534. 10.1038/mt.2013.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Wang, F. S. , & Gershwin, M. E. (2015). Human autoimmune diseases: A comprehensive update. Journal of Internal Medicine, 278(4), 369–395. [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Li, T. , Yang, J. , Zhao, Z. , Tan, K. , Tang, S. , Wan, M. , & Mao, C. (2022). Engineered exosomes with independent module/cascading function for therapy of Parkinson's disease by multistep targeting and multistage intervention method. Advanced Materials (Deerfield Beach, Fla.), 34(27), e2201406. 10.1002/adma.202201406 [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Ren, Y. , Mu, J. , Egilmez, N. K. , Zhuang, X. , Deng, Z. , Zhang, L. , Yan, J. , Miller, D. , & Zhang, H. G. (2015). Grapefruit‐derived nanovectors use an activated leukocyte trafficking pathway to deliver therapeutic agents to inflammatory tumor sites. Cancer Research, 75(12), 2520–2529. 10.1158/0008-5472.CAN-14-3095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Zhuang, X. , Mu, J. , Deng, Z. B. , Jiang, H. , Zhang, L. , Xiang, X. , Wang, B. , Yan, J. , Miller, D. , & Zhang, H. G. (2013). Delivery of therapeutic agents by nanoparticles made of grapefruit‐derived lipids. Nature Communications, 4, 1867. 10.1038/ncomms2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidinger, S. , & Novak, N. (2016). Atopic dermatitis. Lancet, 387(10023), 1109–1122. [DOI] [PubMed] [Google Scholar]
- Wen, Y. , Fu, Q. , Soliwoda, A. , Zhang, S. , Zheng, M. , Mao, W. , & Wan, Y. (2022). Cell‐derived nanovesicles prepared by membrane extrusion are good substitutes for natural extracellular vesicles. Extracellular Vesicle, 1, 100004. 10.1016/j.vesic.2022.100004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, F. , Fei, Z. , Dai, H. , Xu, J. , Fan, Q. , Shen, S. , Zhang, Y. , Ma, Q. , Chu, J. , Peng, F. , Zhou, F. , Liu, Z. , & Wang, C. (2022). Mesenchymal stem cell‐derived extracellular vesicles with high PD‐L1 expression for autoimmune diseases treatment. Advanced Materials (Deerfield Beach, Fla.), 34(1), e2106265. 10.1002/adma.202106265 [DOI] [PubMed] [Google Scholar]
- Xu, H. , Di Antonio, M. , McKinney, S. , Mathew, V. , Ho, B. , O'Neil, N. J. , Santos, N. D. , Silvester, J. , Wei, V. , Garcia, J. , Kabeer, F. , Lai, D. , Soriano, P. , Banáth, J. , Chiu, D. S. , Yap, D. , Le, D. D. , Ye, F. B. , Zhang, A. , … Aparicio, S. (2017). CX‐5461 is a DNA G‐quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nature Communications, 8, 14432. 10.1038/ncomms14432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. H. , Yuan, T. J. , Dad, H. A. , Shi, M. Y. , Huang, Y. Y. , Jiang, Z. H. , & Peng, L. H. (2021). Plant exosomes as novel nanoplatforms for microrna transfer stimulate neural differentiation of stem cells in vitro and in vivo. Nano Letters, 21(19), 8151–8159. 10.1021/acs.nanolett.1c02530 [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Hou, D. , Chen, X. , Li, D. , Zhu, L. , Zhang, Y. , Li, J. , Bian, Z. , Liang, X. , Cai, X. , Yin, Y. , Wang, C. , Zhang, T. , Zhu, D. , Zhang, D. , Xu, J. , Chen, Q. , Ba, Y. , Liu, J. , … Zhang, C. Y. (2012). Exogenous plant MIR168a specifically targets mammalian LDLRAP1: Evidence of cross‐kingdom regulation by microRNA. Cell Research, 22(1), 107–126. 10.1038/cr.2011.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Xiao, B. , Wang, H. , Han, M. K. , Zhang, Z. , Viennois, E. , Xu, C. , & Merlin, D. (2016). Edible ginger‐derived nano‐lipids loaded with doxorubicin as a novel drug‐delivery approach for colon cancer therapy. Molecular Therapy: The Journal of the American Society of Gene Therapy, 24(10), 1783–1796. 10.1038/mt.2016.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, P. , Liu, G. , & Chen, X. (2017). Nanobiotechnology: Cell membrane‐based delivery systems. Nano Today, 13, 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z. , Li, J. X. , Liu, J. X. , Dong, L. , Chen, Q. , Liu, J. , Kong, H. , Zhang, Q. , Qi, X. , Hou, D. , Zhang, L. , Zhang, G. , Liu, Y. , Zhang, Y. , Li, J. , Wang, J. , Chen, X. , Wang, H. , Zhang, J. , … Zhang, C. Y. (2015). Honeysuckle‐encoded atypical microRNA2911 directly targets influenza A viruses. Cell Research, 25(1), 39–49. 10.1038/cr.2014.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information