

Abstract
Earlier research has presumed that the male and female biology is similar in most organs except the reproductive system, leading to major misconceptions in research interpretations and clinical implications, with serious disorders being overlooked or misdiagnosed. Careful research has now identified sex differences in the cardiovascular, renal, endocrine, gastrointestinal, immune, nervous, and musculoskeletal systems. Also, several cardiovascular, immunological, and neurological disorders have shown differences in prevalence and severity between males and females. Genetic variations in the sex chromosomes have been implicated in several disorders at young age and before puberty. The levels of the gonadal hormones estrogen, progesterone and testosterone and their receptors play a role in the sex differences between adult males and premenopausal women. Hormonal deficiencies and cell senescence have been implicated in differences between postmenopausal and premenopausal women. Specifically, cardiovascular disorders are more common in adult men vs premenopausal women, but the trend is reversed with age with the incidence being greater in postmenopausal women than age-matched men. Gender-specific disorders in females such as polycystic ovary syndrome, hypertension-in-pregnancy and gestational diabetes have attained further research recognition. Other gender-related research areas include menopausal hormone therapy, the “Estrogen Paradox” in pulmonary arterial hypertension being more predominant but less severe in young females, and how testosterone may cause deleterious effects in the kidney while having vasodilator effects in the coronary circulation. This has prompted the National Institutes of Health (NIH) initiative to consider sex as a biological variable in research. The NIH and other funding agencies have provided resources to establish state-of-the-art centers for women health and sex differences in biology and disease in several academic institutions. Scientific societies and journals have taken similar steps to organize specialized conferences and publish special issues on gender-based research. These combined efforts should promote research to enhance our understanding of the sex differences in biological systems beyond just the reproductive system, and provide better guidance and pharmacological tools for the management of various clinical disorders in a gender-specific manner.
Keywords: endothelium, estrogen, hypertension, testosterone, vascular smooth muscle
Graphical Abstract:
1. Introduction
In earlier research before the 1980s, there was a general assumption that the male and female physiology is largely similar in most organs except the reproductive system. This assumption has led to major research misinterpretations and undesirable clinical implications. In research, most experiments were performed in male or female animals or both sexes as it was presumed that sex would not affect the results. In effect, the Methods section in many of earlier research reports would indicate that animals of either sex or both sexes were used. Also, clinically, some serious disorders were overlooked or misdiagnosed because of gender-based presumptions. For example, early symptoms of angina and myocardial infarction (MI) in women were often misinterpreted as fibromyalgia or psychological stress as they did not present with the typical chest pain and referred pain in the left shoulder and small finger observed in men. Also, many female-specific disorders did not receive adequate research recognition or funding. For instance, preeclampsia, a major hypertensive disorder of pregnancy, has been known in the maternal wards for centuries, but only recently received the appropriate research consideration and resources.
Careful research has now identified sex differences in the cardiovascular, renal, pulmonary, endocrine, gastrointestinal, immune, nervous, and musculoskeletal systems. This has prompted the National Institutes of Health (NIH) and other health and scientific organizations to emphasize the importance of studying sex differences in biology and disease. The purpose of this review is to highlight the sex differences in biological processes and clinical disorders, and to recognize some of the efforts of researchers, funding agencies, and scientific societies to enhance gender-based research. The review will first discuss the cardiovascular system as it has shown marked sex differences and major effects of sex hormones. We will follow with some gender-related controversies in cardiovascular research including menopausal hormone therapy (MHT) and the effects of progesterone and testosterone. We will briefly describe sex differences in other biological systems and draw attention to specific disorders in females that warrant further research. The review will then highlight the NIH initiative to consider sex as a biological variable in research, and provide some recommendations for scientific societies and journals to enhance gender-based research. In this context, it is important to define the terms sex and gender. The term “Sex” refers to a genetically-determined state throughout the animal kingdom, mediated principally by the X and Y chromosomes and reinforced by the gonadal steroid hormones. In other words, “Sex” refers to the reproductive phenotype as male, female or intersex assigned at birth based on the appearance of the external genitalia and, if necessary, by assessment of the chromosomes and gonads. Therefore, sex differences involve biologic factors including genetic and epigenetic factors and sex hormones [1, 2]. The term “Gender”, on the other hand, refers to a psycho-social state unique to humans. In other words, “Gender” refers to one’s internal sense of gender and how it fits into societal categories as man, woman, or nonbinary person. Therefore, gender differences involve psycho-social factors and may change over time [3, 4]. Throughout the review we will describe data in men, premenopausal women (Pre-MW) and postmenopausal women (Post-MW), followed by data in male and female animal models, and different cells and molecules.
2. Gender-based research and sex differences in cardiovascular disease
Cardiovascular diseases (CVD) such as hypertension (HTN) and coronary artery disease (CAD) are some of the most common and costly diseases. In adults (30–50 years old), the incidence of CVD is greater in men vs women, suggesting vascular benefits of estrogen (E2) [5–7]. Among women, the risk of CVD increases with age in Post-MW vs Pre-MW, likely due to decreased plasma E2 levels. E2 is the predominant sex hormone in women, affecting the development/function of the female reproductive system. E2 is also used as a contraceptive, and as a component of MHT for hot flushes, night sweats and vaginal dryness [8]. E2 receptors (ERs) have been identified in the cytosol and nucleus of endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) (Fig. 1). The interaction of E2 with cytosolic/nuclear ERs stimulates genomic effects that affect vascular cell growth and proliferation. E2 also interacts with plasmalemmal ERs to induce non-genomic vascular effects [5, 9]. E2-induced vascular beneficial effects include modification of circulating lipoproteins, inhibition of lipoprotein oxidation, attenuation of atherosclerotic lesions, modulation of homocysteine, changes in blood coagulation, inhibition of vascular accumulation of collagen, and modulation of vascular tone [7, 10].
Fig. 1.
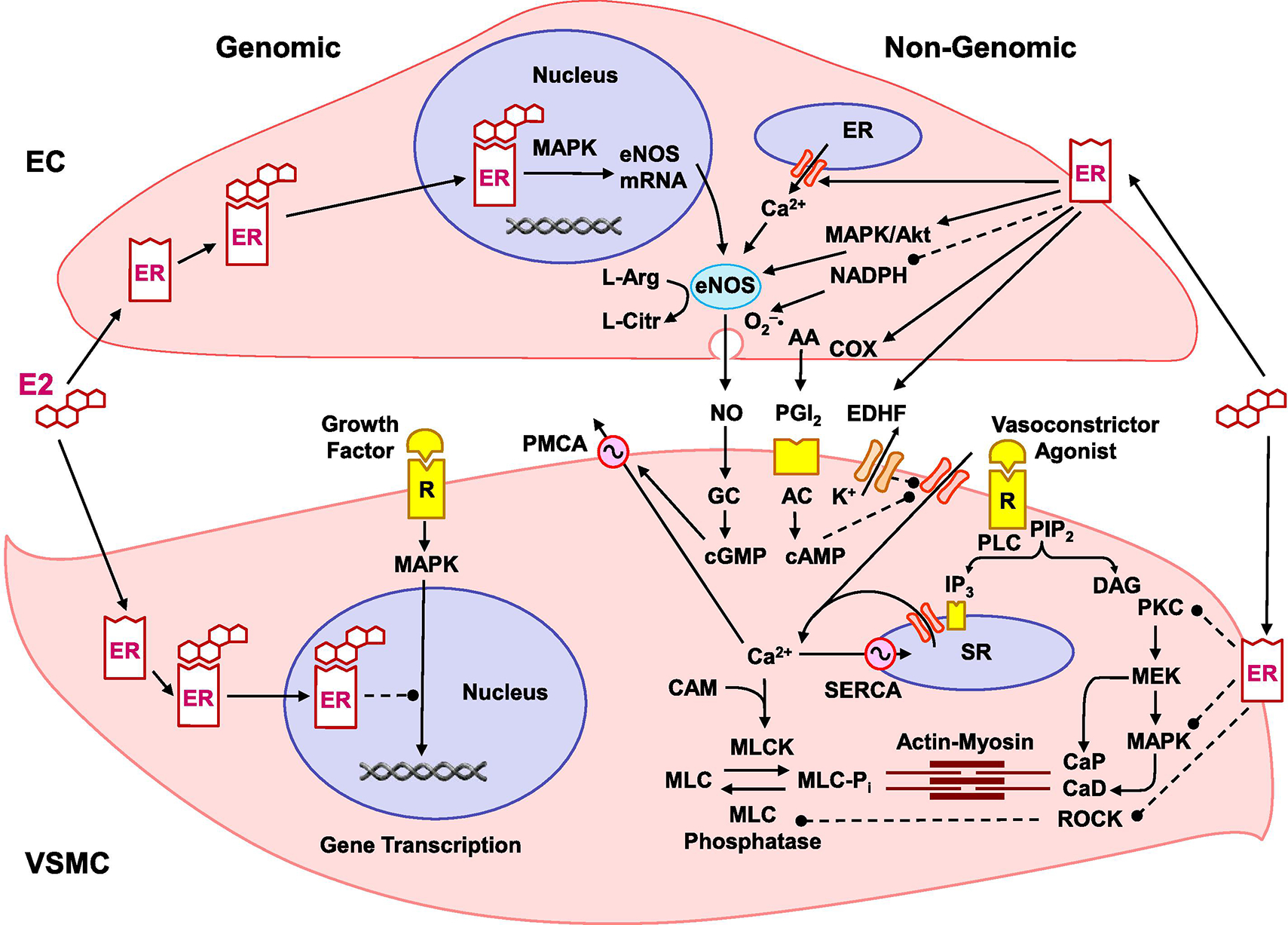
Genomic and nongenomic estrogen (E2)/estrogen receptor (ER) mediated pathways in endothelial cells (ECs) and vascular smooth muscle cells (VSMC). In ECs, E2 binds to cytosolic/nuclear ERα and ERβ and stimulates genomic pathways involving MAPK activation, gene transcription, EC growth and eNOS expression. E2 also binds plasmalemmal ERα, ERβ and GPER in ECs and activates nongenomic pathways and phospholipase C (PLC) to produce inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 releases Ca2+ from the endoplasmic reticulum (ER) to form a complex with calmodulin (CAM) and initiate eNOS activation. E2 also activates phosphatidylinositol 3-kinase (PI3K) to transform phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3) and activate Akt. ER-mediated activation of MAPK/Akt causes phosphorylation and full activation of eNOS, which transforms L-arginine to L-citrulline and produces NO. NO diffuses to VSMC, activates guanylate cyclase (GC) to form cyclic guanosine monophosphate (cGMP), which decreases [Ca2+]c by stimulating plasmalemmal Ca2+ extrusion pump (PMCA) and sarcoplasmic reticulum (SR) Ca2+ uptake pump (SERCA), and decreases the actin-myosin myofilaments force sensitivity to [Ca2+]c leading to VSM relaxation. E2 also inhibits NADPH and peroxynitrite (ONOO−) formation thus promoting antioxidant effects that prevent NO inactivation. E2 also activates cyclooxygenases (COX) to produce prostacyclin (PGI2) which binds PGI2 receptor in VSMC, activates adenylate cyclase (AC) to form cyclic adenosine monophosphate (cAMP), which similar to cGMP decreases [Ca2+]c and myofilament force sensitivity to Ca2+ leading to VSM relaxation. E2 also increases endothelium-derived hyperpolarizing factor (EDHF), causing activation of VSM K+ channels, hyperpolarization, inhibition of Ca2+ influx through Ca2+ channels and VSM relaxation. In VSM, E2 binds cytosolic/nuclear ERα and ERβ to activate genomic pathway, and inhibit MAPK, gene transcription and VSMC proliferation. Also, in VSM, a vasoconstrictor agonist such as ET-1, TXA2 or Ang II activates its specific receptor (R) to stimulate PLC and generate IP3 and DAG. IP3 stimulates Ca2+ release from SR. Agonists also stimulate Ca2+ entry through Ca2+ channels. Ca2+ binds CAM to activate myosin light chain kinase (MLCK), causing MLC phosphorylation, actin-myosin interaction and VSM contraction. DAG activates PKC to phosphorylate calponin (CaP) or activate MAPK kinase (MEK) and MAPK cascade, leading to phosphorylation of caldesmon (CaD) and increased myofilament sensitivity to [Ca2+]c. E2 binds plasmalemmal ERα, ERβ and GPER to activate non-genomic pathways and inhibit agonist-activated mechanisms of VSM contraction including PKC, MAPK and ROCK [Ca2+]c sensitization pathways. Dashed lines indicate inhibition.
2.1. Sex differences in vascular tone
Vascular tone is the degree of constriction of a blood vessel relative to its maximal diameter in the dilated state. Vascular tone is influenced by the combined activities of ECs and VSMCs as well as a balance between vasoconstrictors such as norepinephrine, angiotensin II (Ang II), vasopressin and 5-hydroxytryptamine, and vasodilators such as nitric oxide (NO), prostacyclin (PGI2) and bradykinin. Factors that impact vascular tone can be either extrinsic or intrinsic. Extrinsic factors originating from outside the vessel regulate vascular resistance and arterial pressure while intrinsic factors originating from the vessel itself allow small microvessels to respond to changes in intraluminal pressure and regulate regional blood flow to an organ [11]. Active myogenic tone originating from VSMCs is Ca2+-dependent and is regulated by mechanosensitive cation channels [12, 13]. Passive intrinsic response is Ca2+-independent and involves structural proteins and remodeling enzymes.
Sex differences in vascular tone have been described in several vascular beds in human and animal models [7, 14]. For example, the α-adrenergic agonist norepinephrine causes less forearm vasoconstriction in women than in men. Oxidized low-density lipoprotein enhances 5-hydroxytryptamine-induced contraction to a greater extent in coronary arteries from male than female pigs. Also, contraction in response to norepinephrine or phenylephrine is greater in the aorta of male than female rats [14]. Of note, the pressor response to vasopressin infusion in vivo is greater in male than female rat, although vasopressin-induced contraction in isolated rat aorta is almost twice in females vs males, likely due to tachyphylactic effects ex vivo [14]. Vascular contraction in response to phenylephrine is not different in the aorta of castrated vs intact male rats, but enhanced in ovariectomized (OVX) vs intact females, suggesting that the sex differences in vascular tone are related to E2 levels and ERs in the vasculature [7].
2.2. Estrogen receptors in the vasculature
E2 binds ERs with high affinity and specificity. ERα, ERβ and several ER variants have been described [15, 16]. E2 diffuses through the plasma membrane and complexes with cytosolic/nuclear ER, which binds to chromatin and promotes transcription of genes with specific ER-responsive regulatory element [17] (Fig. 1). E2 also binds to plasmalemmal ER to induce rapid non-genomic effects [5, 18, 19]. ERα knock-out (KO), ERβ KO and ERα/ERβ double KO mice survive, but show reproductive malfunction [20]. ERα and ERβ have been identified not only in the female reproductive organs, but also in the heart, blood vessels, lungs, brain, hypothalamus, and bones, causing multiple effects in the cardiovascular, immune, nervous, and musculoskeletal system [21, 22]. ERα and ERβ have been detected in ECs and VSMCs [23, 24]. There is a strong association between ER number and normal EC function [25]. Upon binding to E2, plasmalemmal ERα or ERβ largely form homodimers, but could also form ERα/ERβ heterodimers [26]. ERα and ERβ are expressed in VSMCs of human coronary artery, iliac artery, aorta, and saphenous vein, with greater density in females vs males [27]. In rats, ERα is found in the uterine artery, and ERβ is abundant in the aorta, tail and uterine arteries [28]. ERα could mediate the protective effects of E2 against vascular injury [29], but ERβ is more predominantly expressed than ERα in all wall layers of human coronary arteries and most avidly in the media and VSMCs [30]. ERβ mRNA expression is upregulated following aortic balloon injury in rats. In transfected HeLa cells stimulated with E2, ERα is a stronger transactivator than ERβ at low ER concentrations, but at higher concentrations, ERα activity self-suppresses, and ERβ becomes the stronger transactivator [27]. GPER or GPR30 is a 7-transmembrane G protein-coupled ER that binds E2 and mediates some of its rapid non-genomic effects [31]. GPER is widely distributed in the brain, peripheral tissues and human mammary artery and saphenous vein [32], and has been localized in the plasma membrane and endoplasmic reticulum [31, 33]. Adult female mice display lower blood pressure (BP) and pulse wave velocity (indicator of arterial stiffness) than adult males, and the female carotids are more distensible and show greater acetylcholine (ACh)-induced relaxation than male carotids. Interestingly, GPER deletion eliminates the sex differences in pulse wave velocity, BP and arterial stiffness in adult mice, and is associated with endothelial dysfunction in females., suggesting that nongenomic E2 signaling through GPER could impact the vascular phenotype differently in females vs males [34].
2.3. Effects of E2 on ECs
The interaction of E2 with cytosolic/nuclear receptors trigger a host of genomic effects involving phosphorylation of mitogen-activated protein kinase (MAPK) and promoting EC proliferation [35]. E2 also alters several signaling pathways in ECs causing non-genomic effects and vasodilation. Acute administration of E2 in healthy Post-MW promoted endothelium-dependent flow-mediated vasodilation, and potentiated the forearm vasodilation induced by ACh [36]. Also, acute administration of E2 in dogs and in isolated rat and rabbit hearts lowers coronary vascular resistance and enhances blood flow [37]. In the aorta of spontaneously hypertensive rat (SHR), endothelium-dependent relaxation is greater in females vs males [38, 39]. E2 promotes vasodilation by increasing the synthesis, release and bioactivity of relaxing factors such as NO, PGI2 and endothelium-derived hyperpolarizing factor (EDHF), as well as decreasing contracting factors such as endothelin-1 (ET-1) and thromboxane A2 (TXA2) [40].
NO is a major vasodilator produced from the transformation of L-arginine to L-citrulline by neuronal NO synthase (nNOS, NOS I), inducible iNOS (NOS II), and endothelial eNOS (NOS III) [41]. While eNOS is Ca2+-dependent and rapidly regulates vascular tone, iNOS is Ca2+-independent and involved in long term regulation of vascular function. Total NO production is greater in Pre-MW vs men [42]. Small arteries isolated from healthy Post-MW not receiving MHT show impaired EC function, which is improved when the vessels are treated with E2 [43]. Pretreatment of human coronary ECs with E2 increases both basal and stimulated NO release [7]. In OVX female guinea pigs, E2 replacement increases endothelial NO production and enhances the sensitivity of coronary arteries to vasodilators [44]. Also, endothelial NO release is greater in female vs male rat arteries, largely due to differences in E2 levels [38, 45].
The effects of E2 on endothelial NO production involves ER-mediated activation of both genomic and non-genomic effects. E2 activation of nuclear ERs causes upregulation of eNOS [25, 45] (Fig. 1). E2 increases eNOS mRNA expression in cultured human coronary ECs [46], and prevent destabilization of eNOS mRNA by tumor necrosis factor-α (TNFα) [47]. E2 also regulates NOS activity through interaction with EC plasmalemmal ERs and activation of non-genomic pathways [45, 48]. Under basal conditions eNOS is firmly attached to the scaffolding protein caveolin in the plasma membrane caveolae. Like other vasodilators such as ACh and bradykinin, E2/ER activation in ECs causes increases in cytosolic free Ca2+ concentration ([Ca2+]c), and initial intracellular translocation of eNOS away from caveolin. E2 also activates MAPK and protein kinase B/Akt which promote phosphorylation and translocation of cytosolic eNOS back to the plasma membrane where it undergoes myristoylation/palmitoylation and full activation [49]. In support of E2 non-genomic effects, membrane-impermeant E2 bind ERs at the cell surface and stimulate NO release from human ECs [50]. E2 administration in OVX female mice causes rapid dilation of elastic and muscular arteries through ER-mediated NO production [51]. In human ECs, E2 stimulates eNOS independent of cytosolic Ca2+ mobilization [52], likely through eNOS phosphorylation by MAPK or Akt [50, 53] In ovine ECs, E2 causes activation of MAPK and eNOS, which are blocked by MAPK inhibition [54]. Also, E2 induces eNOS phosphorylation through the phosphatidylinositol-3 (PI3)-kinase-Akt pathway and thereby reduces its [Ca2+]c requirement for activation [55]. Human umbilical vein ECs (HUVECs) express both ERα and ERβ [23]. ERα gene transfer into bovine aortic ECs induces eNOS gene expression [56]. The acute response of eNOS to E2 was reconstituted in COS-7 cells co-transfected with wild-type ERα and eNOS [54]. Selective ERα agonists ameliorate EC dysfunction in blood vessels of OVX female SHR [57]. Also, basal NO production was increased in thoracic aorta of ERβ KO mice and abolished in ERα KO mice, suggesting that ERα, but not ERβ, mediates the beneficial effect of E2 on basal NO production [58]. However, overexpression of ERβ in COS-7 cells enhances E2-induced eNOS activity and ERβ association to plasma membrane caveolae independent of ERα, supporting ERβ-mediated eNOS activation [59]. Other studies suggest that E2-induced vasodilation and activation of MAPK or PI3-kinase are absent in ERα and ERβ double-KO mice [51].
E2 could promote NO activity through antioxidant effects. Superoxide anion (O2−) levels are greater in the aorta of male vs female rats [60]. In OVX female rats, increases in BP are associated with increased plasma lipoperoxides and free radicals and lower levels of antioxidants and, and E2 replacement decreases free radicals and increases the levels of nitrites/nitrates [61]. Also, E2 inhibits NADPH oxidase expression and the generation of reactive oxygen species (ROS) and peroxynitrite (ONOO−) in HUVECs [62] and enhances NO bioactivity in bovine aortic ECs [63]. E2 also reduces the expression of Ang II type 1 receptor in ECs, and thereby prevents Ang II-induced increases in NADPH oxidase expression, oxidative stress, and ONOO− formation [64]. E2 through activation of ERs in the mitochondria could also regulate mitochondria-encoded genes and decrease mitochondrial O2− production and oxidative stress [10].
PGI2 is an endothelium-derived relaxing factor produced from the metabolism of arachidonic acid by cyclooxygenase-1 (COX-1) and COX-2. Studies suggest a role of COX-2 in the rapid E2-induced enhancement of cholinergic vasodilation in the cutaneous vasculature of Post-MW [65]. Arteries from OVX female monkeys with induced atherosclerosis show reduced PGI2 levels that are inversely related to plaque size, and treatment of the arteries with E2 increases PGI2 production [66]. E2 stimulates urinary excretion of PGI2 metabolites in ERβ but not ERα KO mice, supporting a role for ERα in PGI2 production [67]. E2 upregulates COX-1 expression and promotes PGI2 synthesis in HUVECs and fetal ovine pulmonary artery ECs [45, 68], partly through rapid activation of ERβ-mediated Ca2+-dependent pathway [69]. However, the COX inhibitor indomethacin does not affect E2-induced relaxation in rabbit coronary artery, suggesting a little role of prostanoids in E2-induced coronary vasodilation [70]. E2 may initiate a cross-talk between NOS and COX such that an increase in NO-mediated vasodilation would decrease COX-mediated vascular relaxation [71].
The greater endothelium-dependent relaxation in females vs males could also involve EDHF and activation of K+ channels [39]. ACh-induced hyperpolarization and relaxation of mesenteric arteries are reduced in male and OVX female vs intact female rats, and the differences are eliminated by the K+ channel blockers apamin and charybdotoxin. Also, E2 replacement improves ACh-induced vascular hyperpolarization and relaxation in OVX female rats [72, 73].
The sex differences in vascular tone may also involve endothelium-derived contracting factors such as ET-1 and TXA2. ET-1 interacts with ETAR and ETB2R in VSM to induce contraction, and with ETB1R in ECs to induce vascular relaxation. Acute intracoronary administration of E2 decreases ET-1 levels in coronary sinus plasma of Post-MW with CAD [74]. ET-1 release from ECs along with vascular tone and BP are reduced in female vs male SHR rats [39, 75]. ET-1 causes greater contraction in mesenteric arteries of male vs female DOCA-salt hypertensive rats, likely due to differences in ETBR [76]. ET-1 and ETB2R mRNA expression and the vasoconstriction induced by the ETBR agonist IRL-1620 are greater in mesenteric arteries of OVX vs intact females, and these increases are reversed in E2-replaced OVX females, supporting that E2 attenuates ET-1 and ETB2R expression [77]. Also, aortic strips from male rats exposed to trauma-hemorrhage show increases in ET-1 induced contraction, and treatment with E2 and the ERβ agonist diarylpropionitrile (DPN) counteracts the vascular contraction [78]. E2 also inhibits basal and serum, TNFα, transforming growth factor β1, Ang II, and thrombin-induced ET-1 release in ECs [79, 80]. The release of COX-derived vasoconstricting prostanoids such as TXA2 is also greater in male vs female SHR [39].
2.4. Effects of E2 on VSMCs
Although E2 enhances EC growth through nuclear ERs and genomic pathways, it inhibits VSMC proliferation through inhibition of MAPK [81] or activation of protein phosphatases [82] (Fig. 1). NO inhibits VSMC proliferation, and E2-induced NO production contributes to VSMC growth inhibition. E2 also stimulates cyclic adenosine monophosphate (cAMP) and adenosine production which can regulate VSMC growth [83]. The different effects of E2 on growth of ECs vs VSMCs could be related to the ER subtype or the E2 response element in the promoters of various genes.
E2 also causes rapid relaxation of endothelium-denuded rabbit and porcine coronary arteries, suggesting endothelium-independent effects on plasmalemmal ERs in VSMCs [84, 85]. The vasodilator effects of E2 vary with the vascular bed, estrous cycle, and exposure to E2. For example, in rats, E2-induced vasodilation is the largest in the tail and mesenteric arteries from females at the pro-estrous stage. Also, E2 causes less microvascular relaxation in E2-replaced vs nonreplaced OVX rats, suggesting that chronic E2 exposure downregulates ERs [86]. Importantly, the rapid vascular effects of E2 in ex vivo studies are observed at micromolar concentrations, which are several-fold higher than the physiological picomolar plasma levels in vivo. This is likely because E2 is lipophilic and the E2 tissue levels may exceed the plasma levels. Also, the responses of large conduit vessels such as the aorta or left anterior descending coronary artery generally require higher concentrations of sex hormones to produce vasodilation, while similar vasodilatory effects are seen in microvascular preparations at much lower concentrations. It is likely the greater structural complexity of the large vessels with their several layers of VSMCs and collagens that require much higher concentrations of sex hormones to produce an effect.
VSMC contraction is triggered by increases in [Ca2+]c due to Ca2+ release from the sarcoplasmic reticulum and Ca2+ entry from the extracellular space [87]. Activation of myosin light chain (MLC) kinase and Rho-kinase (ROCK) as well as inhibition of MLC phosphatase also contribute to VSMC contraction. Also, the interaction of vasoconstrictors with their receptors increases the production of diacylglycerol which activates protein kinase C (PKC) and increases the myofilament force sensitivity to [Ca2+]c [88]. In isolated VSMCs, the resting cell length is longer and basal [Ca2+]c is less in female vs male rats, and in E2-replaced vs non-replaced OVX females, suggesting sex differences and marked effects of E2 on the Ca2+ handling mechanisms [87].
In VSMCs incubated in Ca2+-free solution, phenylephrine or caffeine causes cell contraction and transient increase in [Ca2+]c that are not different between intact and gonadectomized male and female rats, suggesting that the Ca2+ release mechanism is not involved [87]. However, in VSMCs incubated in Ca2+-containing solution the maintained [Ca2+]c induced by phenylephrine or high KCl is greater in intact male and OVX female vs intact female or E2-replaced OVX female rats, suggesting sex differences in the Ca2+ entry mechanism of VSMC contraction [87]. In support, the expression of the L-type Ca2+ channel is increased in cardiac muscle of ER-deficient mice and in coronary VSMCs of male vs female swine [89, 90]. Also, E2 inhibits the maintained PGF2α and TXA2 induced VSM contraction, Ca2+ influx and [Ca2+]c, supporting inhibition of Ca2+ entry mechanisms [84, 91]. E2 could activate BKCa channel in coronary VSMCs, causing hyperpolarization and inhibition of Ca2+ entry through voltage-gated channels [92]. Also, E2 directly blocks Ca2+ channel activity in cultured A7r5 and aortic VSMCs [93]. E2 also inhibits vascular contraction, Ca2+ influx and [Ca2+]c in isolated vessels and VSMCs stimulated by high KCl (which prevents K+ efflux through K+ channels), supporting direct inhibition of Ca2+ channels [84, 91]. E2 may also decrease [Ca2+]c by stimulating Ca2+ extrusion via plasmalemmal Ca2+ pump [94]. However, the rate of decay of caffeine- and carbachol-induced contraction and [Ca2+]c transient in VSMCs incubated in Ca2+-free solution, an indicative of Ca2+ extrusion, is not affected by E2 [84, 87].
Studies have also examined whether the sex differences in vascular contraction involve PKC and ROCK. PKC activation by phorbol esters causes greater contraction in the aorta of male vs female rats [95]. Phenylephrine and phorbol ester-induced activation/translocation of α- and δ-PKC from the cytosolic to the particulate fraction are greater in male vs female rats and in OVX vs intact or E2-replaced OVX females, suggesting E2 related sex differences in PKC activity [95]. ROCK inhibits MLC phosphatase and enhances VSM sensitivity to [Ca2+]c. ROCK is upregulated in CVD and coronary vasospasm. The vasodilator response to ROCK inhibitor Y-27632 is reduced in male and OVX female rats and restored in E2-replaced OVX females. E2 also decreases ROCK mRNA expression in human coronary VSMCs [96]. MLC kinase, tyrosine kinases and MLC phosphatase also regulate VSMC contraction, and whether the expression/activity of these protein kinases/phosphatases is affected by gender or E2 levels should be examined.
3. Unexplained, paradoxical, and under-investigated vascular effects of sex hormones
While gender-based research has highlighted important sex differences in the cardiovascular system and CVD, some of the observations remain controversial, and several questions regarding the effects of E2 vs other sex hormones on the vascular system remain unanswered. Some questions center on the vascular benefits of MHT, the E2 paradox in pulmonary arterial hypertension (PAH) and the vascular effects of progesterone (P4) and testosterone (T3).
3.1. Does menopausal hormone therapy (MHT) confer vascular benefits?
E2 is commonly used as a contraceptive and as a component of MHT for hot flushes, night sweats and vaginal dryness [8]. Since E2 promotes vasodilation and modulates VSM Ca2+ channels, MHT was also proposed as a pharmacological approach to decrease the severity of certain forms of HTN and CVD. Earlier observational studies such as the Nurses’ Health Study (NHS) in the 1970s showed reduced risk of cardiovascular events in Post-MW using oral E2 [6, 97]. Also, a meta-analysis of observational studies showed that MHT was associated with 33% reduction in fatal CVD [8]. A study in 337 women undergoing elective coronary angioplasty showed that MHT users had fewer cardiovascular events (12% vs 35%) and better survival (93% vs 75%) than nonusers [98]. However, in these studies participants were not well-randomized, women who took MHT might have been healthier than those who did not (“healthy user” bias), and different MHT preparations were used for different periods of time. Of note, randomized clinical trials (RCTs) such as the Women’s Health Initiative (WHI) and Heart and Estrogen/progestin Replacement Study (HERS) showed no cardiovascular benefits and even increased risk for cerebrovascular events with MHT [99–101]. Clinical studies also raised concerns about possible relationship between MHT and the risk of venous thromboembolism (VTE) [102, 103]. Also, ~35% increase in the risk of cerebral ischemia and stroke has been associated with MHT in the NHS [104], WHI [105] and the Women’s Estrogen for Stroke Trial (WEST) [106]. Although E2 may reduce some vascular risk factors, it may promote proinflammatory effects on neurons and increase sensitivity to ischemia by modulating the excitatory effects of glutamate or the inhibitory effects of γ-aminobutyric acid [106].
MHT failure to elicit vascular benefits in RCTs could be related to the biosynthesis and levels of natural estrogens and their metabolites, and age-related changes in ER subtypes, variants, polymorphisms, tissue distribution, and post-ER signaling in ECs, VSMCs and extracellular matrix (ECM). MHT failure could also be due to the design of RCTs, the subjects’ age, pre-existing CVD, timing of MHT, the hormone environment and interaction with other hormones such as P4 and T3.
The outcome of MHT in Post-MW could be affected by the type of estrogen used. Natural estrogens include estrone (E1), estradiol (E2) and estriol (E3). Conjugated equine estrogen (CEE) is a common form of MHT derived from urine of pregnant mares that was approved by the FDA for treatment of menopausal symptoms. CEE contains saturated estrogens e.g. E1, 17β-E2 and 17α-E2, and unsaturated estrogens e.g. equilin. Because of low bioavailability of oral estrogens and their first-pass metabolism, synthetic estrogens such as diethylstilbestrol, esterified E2, and ethinyl-E2 may provide cardiovascular benefits. In Post-MW, treatment with esterified E2 showed lower risk of ischemic stroke and MI, and no increase in VTE risk compared to CEE [102]. Phytoestrogens such as flavonoids, isoflavonoids, stillbenes and lignans bind ERs and produce estrogenic effects. Food containing phytoestrogens such as soybeans, red clover, wheat grains, and peas is consumed in large quantities in cultures with lower rate of menopausal symptoms, osteoporosis, cancer, and CVD particularly in Japan, China, and Southeast Asia. However, phytoestrogens have not been tested for their estrogenic effects in well-designed RCTs. Selective estrogen receptor modulators (SERMs) such as raloxifene, tamoxifen, toremifene, and idoxifene are non-steroidal molecules that bind ERs with a wide range of activity from purely estrogenic, purely anti-estrogenic, to partial estrogenic. SERMs reduce the levels of total cholesterol, LDL-c, triglycerides, fibrinogen and the cytokines interleukin-6 (IL-6) and TMFα. The Raloxifene Use for The Heart (RUTH) trial showed that raloxifene treatment for a median of 5.6 years reduced the risk of invasive breast cancer in Post-MW with CVD but did not affect the risk of primary coronary events, while increasing the risk of fatal stroke and VTE [107]. As in WHI, the advanced age of the participants could have affected the estrogenic response. Also, the Multiple Outcomes of Raloxifene Evaluation (MORE) study showed that raloxifene therapy for 4 years did not affect the risk of cardiovascular events in the overall cohort, but increased the risk of VTE and did not alter the risk of stroke [108]. Selective ER agonists have also been developed. Propylpyrazole trisphenol (PPT) is 400-fold more potent on ERα than ERβ and has been shown to increases flow-mediated relaxation in mesenteric microvessels of female mice [109]. Diarylpropionitrile (DPN) is a potent ERβ agonist with a 30- to 70-fold selectivity over ERα that causes rapid NO-dependent vasodilation [41]. G1 is a selective GPER agonist. Further research is needed to develop specific estrogenic compounds with high ER and cardiovascular selectivity in Post-MW.
The adverse effects of MHT observed in RCTs may be related to the relatively high dose of estrogens used. Estrogens are highly lipophilic, and their circulating levels may not reflect their levels in the vascular wall. Also, E2 regulates fat mass, adipose deposition and metabolism, and E2 deficiency in Post-MW could increase visceral fat. Adipose tissue has P450 aromatase activity, which converts androstenedione into E1, and increases E1 levels in aging Post-MW with obesity. Also, adipose tissue secretes inflammatory cytokines such as IL-6 and TNFα, that could affect vascular ERs and their signaling pathways. Estrogens are also extensively bound to plasma proteins particularly globulin, and the hormone pharmacokinetics and volume of distribution may change in Post-MW with liver and kidney disease. Thus, a dose of E2 that seems to be normal in Pre-MW could produce supraphysiological levels in Post-MW, and lead to side-effects [40]. In effect, the Women’s Health, Osteoporosis, Progestin, Estrogen (Women’s HOPE) trial showed improved benefit/risk ratio of lower-dose CEE for prevention of bone loss, relief of vaginal atrophy and vasomotor symptoms, and reduction in endometrial bleeding [110].
The route of administration could be a factor in successful MHT. Estrogen can be administered orally, or as percutaneous gel, transdermal patch or subcutaneous implant. Transdermal E2 avoids exaggerated peaks in plasma E2 levels that occur during oral administration, undergoes a rate of conversion to E1 that is similar to that during the menstrual cycle, and may have less adverse effects than oral preparations [111]. Also, oral E2 may increase C-reactive protein (CRP) levels in the vascular wall [112], while transdermal E2 increases brachial artery flow-mediated vasodilation without affecting CRP levels in healthy Post-MW [113]. The route of administration of MHT should be an active area of research to identify treatment modalities with the least adverse effects.
The failure of vascular benefits of MHT to materialize in RCTs is likely linked to age-related changes in ER distribution, integrity, and post-ER signaling pathways as well as structural changes in the vasculature. ER genes are located on separate chromosomes; ESR1 is on chromosome 6q(25.1) and encodes ERα, while ESR2 is on chromosome 14q(23–24.1) and encodes ERβ [114]. Polymorphisms in ERα at positions c.454–397 T>C (PvuII) and c.454–351 A>G (XbaI) have been associated with the severity of CAD in Post-MW [115]. On the other hand, HDL and apolipoprotein A-1 levels, forearm blood flow, brachial artery diameter, and endothelium-dependent dilation are greater in Post-MW with the recessive ERα IVS1–397 polymorphism C/C genotype vs the dominant phenotype [116]. Also, a 2 times greater increase in HDL levels was found in Post-MW with the ERα IVSI-401 polymorphism C/C genotype vs control women with CAD who were being treated with E2 alone or E2+progestin [117]. ER isoforms or splice variants have been described, and their biological functions remain an important area for research [22]. Also, ERβ expression is regulated by methylation of CpG islands in the ERβ gene promoter, and may represent an epigenetic mechanism of vascular aging and atherosclerosis [118].
Although E2 levels decrease with age in Post-MW, little is known about age-related changes in ER subtypes, distribution, and function in blood vessels. In other tissues. ERα is detected in the retina of young women, but not in men or Post-MW. Experimental data suggest that ER expression is not different in the aorta of aging vs adult OVX SHR [119]. ERβ mRNA is decreased in specific regions of the brain of old vs young and middle-aged female rats and is not altered by E2 treatment. Aging may also affect the subcellular distribution of ERα in the nucleus and cytoplasm of mouse cholinergic neurons [120]. Further elucidation of the changes in E2-ER interactions with age should help in the design of selective estrogenic drugs for aging women.
In general, women live longer than men, with life expectancy at 76.6 years for men and 82.2 years for women [121], although women are frailer and have worse health at the end of life. While men still perform better on physical function examinations, women outlive men. The survival benefit in females is also seen in nonhuman mammals [122]. However, the cardiovascular system deteriorates with age and the responsiveness of the vasculature to E2 are altered by age-related EC senescence and dysfunction, VSMC proliferation, and vascular remodeling, inflammation, and atherosclerosis. With age, NO production decreases and ET-1 production increases, which promotes VSMC proliferation and favors a procoagulant state. Aging also affects the blood vessel architecture and mechanical properties. Elastin fibers determine the mechanical strength of the vessels at lower pressures and collagen fibers bear most of the strength at higher pressures. During vascular aging, there is progressive arterial stiffening and arteriosclerosis due to decreased elastin/collagen ratio. E2 could affect vascular ECM proteins and remodeling through modulation of matrix metalloproteinases (MMPs). At low doses, E2 inhibits MMPs and decreases collagen deposition, whereas high E2 doses activate MMPs and promote vascular lesion formation and plaque destabilization. In cultured human coronary artery and umbilical artery VSMCs increasing E2 from physiological to supraphysiological levels increases MMP-2 levels, supporting different effects of low and high E2 doses on vascular integrity [123]. MMP-induced ECM degradation within the atherosclerotic plaque also affects plaque instability, and changes in the levels of MMP-2, -9 and -10 in Post-MW receiving MHT may contribute to the risk of cardiovascular events [124].
Because of the age-related vascular changes, the time of initiation of MHT could be critical in determining the vascular outcome [125]. The mean age of menopause in the United States (U.S.) is 51 years, which corresponds to the age of women in the NHS receiving MHT for menopausal symptoms [126]. Women who participated in NHS were 30 to 55 years of age, and ~80% of participants initiated MHT within 2 years of menopause [104]. On the other hand, women in HERS were on average 67 years of age and had been postmenopausal for many years at the time of enrollment [125]. Likewise, the participants in WHI were older (50–79 years) with only 10% of the participants between 50 and 54 years and 20% between 54 and 59 years. Even younger healthy participants (aged 50 to 59) in WHI had been menopausal ~6 years before enrollment [6]. Because atherosclerosis progresses with age, many of the women in WHI likely had subclinical atherosclerosis. The timing of MHT was considered in the Kronos Early Estrogen Prevention Study (KEEPS) and Early vs Late Intervention Trial with Estradiol (ELITE) to evaluate the effects of MHT on progression of atherosclerosis in early menopausal women [126]. In KEEPS, women were randomized to daily low dose oral CEE or weekly transdermal E2, both in combination with cyclic oral micronized P4 or placebo for 12 days/month. Participants were 42–58 years of age and 6 to 36 months postmenopausal with E2 levels <40 pg/mL [126]. He ELITE study randomized 643 women, < 6 years or > 10 years postmenopausal, to receive oral E2 for 2 to 5 years to test for differences between early and late initiation of MHT. The results from these have helped to address the benefits of MHT timing, and low dose, and the differences between oral and transdermal E2 [40].
The pre-existing cardiovascular condition could also affect the outcome of MHT in Post-MW. One criticism of HERS was that the participants had CVD at baseline [126]. Also, while WHI was designed as a primary prevention RCT in “healthy” women, 36% of the women assigned MHT had HTN, 49% were smokers, 4.4% had diabetes, 34% were obese, and 13% being treated for hypercholesterolemia, suggesting an active atherosclerotic process. Arteries of Post-MW have some degree of atherosclerosis that could impede the vasodilator effects of E2. The Cardiovascular Health Study in women older than 65 years of age showed positive association between MHT and flow-mediated vasodilation only in healthy Post-MW, but had no effect on brachial artery blood flow in women older than 80 years or with CVD or multiple cardiovascular risk factors [117]. Also, In OVX cynomolgus monkeys fed an atherosclerotic diet, early CEE administration simultaneously with the atherosclerotic diet caused a 70% reduction in coronary artery plaque size. A delay in hormone therapy until after the development of moderate atherosclerosis resulted in only 50% protection. In primates who received CEE after being on atherosclerotic diet for 2 years, no protection was observed [127]. Similarly, in OVX female rats with carotid artery balloon injury, administration of E2 resulted in inhibition of intimal hyperplasia, and VSMC proliferation when given on the day of injury, 15 minutes before, and continuing 3 days after injury, but did not show beneficial effects when administered 7 days after balloon injury [128]. Thus, proper timing of initiation of MHT may prevent or delay the progression of atherosclerosis and CVD. Around the time of menopause, women still have relatively healthy arteries, allowing a “window” for MHT to produce vascular benefits. However, as arterial disease increases with time after menopause, the diseased arteries become less responsive to the beneficial effects of E2.
3.2. The “Estrogen Paradox” in pulmonary arterial hypertension (PAH)
PAH is a progressive disease involving impaired pulmonary vascular structure and function, and causing right ventricular failure that is ultimately fatal [129, 130]. PAH can be idiopathic, familial, or associated with other cardiovascular disorders, and its underlying pathology involves pulmonary vasoconstriction, progressive fibro-proliferative remodeling of the pulmonary arterioles, as well as inflammation, thrombosis, and right ventricular hypertrophy and failure. Although only a small percentage of patients have demonstrable pulmonary vasoconstriction when they undergo cardiac catheterization, the mainstay of current pharmacologic therapies includes vasodilators such as the phosphodiesterase-5 (PDE-5) inhibitor sildenafil and the ET-1 receptor antagonist bosentan [131]. While many vasodilators also have anti-proliferative effects on VSMCs, current vasodilator therapies are not universally successful in altering PAH progression and improving survival. Therefore, novel approaches that directly target pulmonary vessel wall pathology are needed to reverse the established pathology in PAH.
PAH affects all age groups and both genders. There is a striking preponderance of young females affected by heritable PAH, but among patients with PAH, women show better prognosis and right ventricular function and higher survival rates compared with men (the E2 Paradox) [132]. The preponderance of PAH in females is intriguing because it is opposite of the preponderance of systemic CVD in males. Attempts have been made to define the role of sex steroids in pulmonary vascular disease, but significant knowledge gaps exist. One study showed that sex hormone depletion in castrated male and OVX female rats did not alter pulmonary artery reactivity to acute hypoxia [133]. However, E2 is a pulmonary vasodilator that increases endothelial NO production [134] and promotes PGI2 synthesis in ovine pulmonary artery ECs [68, 69]. Also, the E2 active metabolite 2-methoxy-E2 shows anti-proliferative effects in VSMCs [135]. Interestingly, 2-methoxy-E2 is as efficacious as sildenafil and bosentan in ameliorating monocrotaline (MCT)-induced PAH in male and female rats [136, 137]. Also, 2-methoxy-E2 when combined with either sildenafil or bosentan confers additional protection and improves survival in MCT rat model. Importantly, these pharmacological interventions were initiated 12 days after administering MCT and establishing pulmonary vascular remodeling, supporting effective reversal of established PAH. 2-methoxy-E2 likely exerts anti-proliferative and anti-inflammatory actions in the pulmonary vasculature and the lung, making it important to further examine its effects and other E2 metabolites in PAH.
3.3. Vascular effects of progesterone (P4)
P4 is a steroid hormone produced by the female ovary and adrenal cortex, and by the placenta during pregnancy. P4 receptors (PRα and PRβ) have been identified in ECs and VSMCs of human, primates, rabbit, rat, and mouse [7, 138, 139]. While the effects of P4 often require previous E2 priming in the reproductive system, P4 could have separate effects or modify the effects of E2 in the cardiovascular system. Similar to E2, P4 has anti-atherosclerotic effects, decreases LDL-c, and increases HDL-c [140]. Intravenous infusion of P4 in pigs induces endothelium-dependent coronary vasodilation through release of NO [140]. Also, chronic administration of P4 upregulates eNOS in ovine uterine artery ECs [141]. P4 also stimulates endothelium-dependent NO-mediated relaxation in ovine uterine artery and rat aorta [142]. Additionally, P4 rapidly activates COX and increases PGI2 production. P4 and combined E2/P4 inhibit serum- and Ang II-stimulated ET-1 release from bovine aortic ECs [79]. P4 via PRβ inhibits VSMC proliferation and facilitates the inhibitory effects of E2, likely through inhibition of MAPK [79, 143]. Also, P4 causes endothelium-independent relaxation of primate, porcine, rabbit and rat coronary arteries [84]. P4 also rapidly decreases Ca2+ influx and [Ca2+]c in porcine and rabbit coronary artery VSM [84, 91]. Furthermore, P4 inhibits phorbol ester-induced VSMC contraction and PKC activation and translocation in VSM likely through increasing cAMP levels [144].
However, P4 produces less vasorelaxation than E2, and may even antagonize the vasoprotective effects of E2. P4 counteracts the stimulatory effects of E2 on NO production and vascular relaxation in canine and porcine coronary artery [145]. Also, P4 antagonizes the antioxidant effect of E2, and enhances NADPH oxidase activity and production of ROS in OVX mice [146]. P4 could also promote vasoconstriction by upregulating vascular AT1R [147]. P4 may also diminish E2-induced anti-inflammatory effects and its attenuation of ischemic brain injury [40].
P4 could modify the vascular effects of E2 in MHT. Medroxy-P4 acetate (MPA) antagonizes the inhibitory effects of CEE on coronary atherosclerosis, and may attenuate E2-induced NO production in Post-MW [148]. A study comparing the effect of sequential E2/progestin treatment cycle, two weeks with E2 alone followed by two weeks with E2/norethisterone acetate (NETA), in Post-MW showed increased excretion of cGMP in both phases, but found an increase in PGI2 metabolite during the E2 phase, and an increase of TXA2 metabolite during the E2/NETA phase [149]. Another study showed an increase in TXA2 metabolites with oral but not transdermal E2/MPA after 12 months [150]. Also, treatment of human female coronary artery ECs with MPA or NETA enhances E2-induced reduction of MMP-1, an action that could affect plaque stability. However, CEE alone or CEE+MPA increased MMP-9 levels after 4 weeks in Post-MW with established CAD [148]. These complex interactions of E2 and P4 on the vasculature highlight the need to determine the benefits vs risk of combined E2/P4 in postmenopausal CVD.
Drospirenone (DRSP) is an 17α-spironolactone derivative with similar affinity to PR and pharmacodynamics as natural P4. DRSP is used in combination with ethinyl-E2 as oral contraceptive and in combination with E2 as MHT. Of note, DRSP has anti-mineralocorticoid effects that counteract the salt-retaining actions of E2 and greater anti-androgenic effects than P4. E2/DRSP lowers BP in hypertensive Post-MW, particularly when administered in combination with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers [151]. Angeliq, an MHT composed of DRSP/E2, reduced carotid intima–media thickness and vertigo/dizziness in Post-MW possibly through its anti-androgenic and anti-mineralocorticoid effects [152].
In women with intact uterus, P4 is given with E2 to reduce the risk of endometrial cancer. The negative findings of HERS and one arm of WHI may have been caused by concomitant use of MPA, as evidenced by increased stroke risk in women taking CEE+MPA vs women never using MHT. Also, in Postmenopausal Estrogen/Progestin Interventions (PEPI) trial, CEE caused beneficial effects on LDL and HDL levels that were attenuated by MPA [6]. However, other studies did not find any attenuation of CEE-induced vasodilation by MPA or micronized P4 [153]. Also, NHS showed a similar risk reduction for CAD among women taking CEE alone or CEE+MPA [104].
In cynomolgus monkeys, E2 or E2+P4 had similar anti-atherosclerotic effects. However, these vascular protective effects were lost in monkeys administered CEE+MPA vs CEE alone [154]. Also, in aging postmenopausal cynomolgus monkeys MPA abrogates the vascular benefits of E2 [155]. Furthermore, ACh caused vasoconstriction in E2-deprived monkeys not receiving MHT, and the vasodilatory response was restored in monkeys treated with E2 alone, but by only 50% during co-administration of MPA [155]. Also, MPA abrogated the ability of E2 to attenuate balloon injury-induced intimal thickening in rat model [156]. However, in rabbits, the anti-atherosclerotic actions of CEE or E2 were not prevented by MPA or other progestins [6]. Thus, whether MPA is responsible for the lack of vascular protective actions of MHT in RCTs needs to be further examined.
3.4. Vascular effects of testosterone (T3)
In men, androgens such as T3 maintain masculine secondary sexual characteristics. In women, androgens are important for maintaining bone mass, secondary sexual characteristics and libido [40]. Androgens are produced in the testis, adrenal glands, and ovaries. Normally, 10% of circulating T3 is irreversibly converted by 5-α-reductase in peripheral tissues such as the liver, prostate, genital skin and hair follicles into dihydrotestosterone (DHT3), with higher binding affinity to androgen receptor. T3 is also aromatized to E1 or E2 mainly in adipose tissue.
During the female reproductive years, ~25% of circulating T3 originates from the ovaries, 25% from the adrenal glands and 50% from conversion of peripheral androstenedione. Serum T3 levels are markedly lower in women than in men. Like E2, serum T3 levels decrease with age from 1.0 to 1.5 nmol/L in Pre-MW to 0.3–0.5 nmol/L in Post-MW [157, 158]. However, serial measurements of sex hormones in Post-MW for 10 years showed age-related increase in serum T3 and androstenedione and decrease in E2 and DHT3 [159]. Other studies showed that serum T3 decreased immediately after menopause, but then increased with age, reaching Pre-MW levels at 70–79 years of age. In women with surgical menopause, serum T3 levels did not increase with age, but were 40–50% lower than in women with natural menopause, suggesting that natural postmenopause is a relatively hyperandrogenic state [157].
Androgen receptors have been identified in ECs and VSMCs. The androgen receptor levels are lower in aortic VSMCs from female vs male rats [160]. In primates VSMCs, androgen receptor expression is upregulated by combined E2+T3 while E2 alone had minimal effect, suggesting a collaborative action of E2 and T3 on androgen receptor expression [161].
Some studies suggest that T3 causes deleterious effects in the kidney and could promote cardiovascular-renal disease in males. The increased cardiovascular-renal disease in Post-MW could be related to increased T3/E2 ratio secondary to the decreased ovarian function and E2 production. Because men have higher BP and develop CVD at an earlier age than women, it is thought that androgens promote CVD in men. Although serum T3 levels are lower in men with chronic CVD than in healthy men, the decreased androgen synthesis may be a compensatory mechanism in response to CVD. Of note, BP is higher in male vs female SHR, reduced in castrated males to levels observed in females, and increased in OVX females treated with T3, suggesting a role of increased T3/E2 ratio in promoting HTN post-menopause [157]. Some studies suggest that T3 enhances vascular contraction either by inhibiting endothelium-dependent relaxation or by directly stimulating VSM contraction [161]. For example, pretreatment of porcine coronary artery with T3 impairs bradykinin-induced endothelium-dependent vascular relaxation and NO production [162]. T3 also enhances TXA2-induced coronary vasoconstriction in guinea pigs. Also, the androgen receptor antagonist flutamide causes relaxation of rat vessels [163].
However, some studies suggest beneficial cardiovascular effects of T3 through modulating lipid metabolism, stimulating hepatic lipoprotein lipase activity and reducing serum triglycerides and atherogenic lipids [180]. In men, low circulating T3 is positively correlated with risk factors for CAD [164]. In a study of middle-aged women (50–59 years) T3 was positively associated with low LDL and high HDL levels in all women, and women with CVD had lower T3 levels [165]. T3 may also promote coronary vasodilation in young men, and the increased risk for angina and MI in older men may be related to decreased T3 levels. Interestingly, in men with established CVD, intracoronary administration of T3 increases coronary blood flow within minutes and rapidly improves myocardial ischemia [166]. In healthy Post-MW, plasma T3 levels positively correlate with brachial artery flow-mediated vasodilation, suggesting protective effects on ECs [167]. Epidemiology studies have shown that low T3 levels are associated with more atherosclerosis, CAD, and cardiovascular events. Also, emerging studies suggest that T3 may alter the course of heart failure, angina, and myocardial ischemia. However, treating hypogonadism in aging men has produced discrepant effects and cardiovascular events [168]. The T3 Trials (TTrials) were a coordinated set of seven placebo-controlled, double-blind trials in 788 men with a mean age of 72 years to determine the efficacy of increasing T3 levels in older men with low T3. In the Cardiovascular Trial, T3 increased the coronary artery noncalcified plaque volume as assessed by computed tomographic angiography. Although T3 was not associated with more cardiovascular or prostate adverse events than placebo, a much larger, prospective, long-term studies of T3 replacement, with primary endpoints of adverse cardiovascular events including MI, stroke, and cardiovascular death as well as prostate cancer, are needed to determine whether T3 increases cardiovascular or prostate risk [169]. In effect, the Food and Drug Administration recently announced additional restrictions on T3 replacement therapy labeling and called for further studies to determine its cardiac safety [168].
Nevertheless, experimental studies have supported T3 effects on vascular function. T3 induces relaxation of rabbit and rat aorta, and canine, porcine and rabbit coronary artery [84, 170]. T3 may also promote endothelium-dependent vascular relaxation [171, 172]. Intracoronary administration of T3 in canine induces vasodilation and NO release [171]. T3-induced vascular relaxation is reduced by K+ channel blockers, suggesting a role of voltage-dependent (delayed-rectifier) K+ channel [170]. In SHR aorta, T3 may release EDHF and activate voltage-dependent and BKCa channels, but a significant component of tT3-induced vasorelaxation in both WKY and SHR is endothelium-independent and may involve ATP-sensitive K+ channels in VSMCs [173]. In VSMCs, some studies suggest that androgens stimulate VSMC proliferation, while other studies show androgen-induced inhibition [161, 174]. In human umbilical VSMCs, DHT3 modulate cell proliferation in a dose-dependent manner, with low concentrations stimulating while high concentrations inhibiting [3H]thymidine incorporation [174]. Also, a significant portion of T3-induced vascular relaxation appears to be endothelium-independent. T3 and DHT3, which cannot be aromatized to E2, cause relaxation of porcine coronary artery and decrease VSMC [Ca2+]c by inhibiting Ca2+ entry from the extracellular space [84, 91, 170]. In porcine coronary artery, T3 causes greater inhibition of PGF2α-induced contraction than KCl-induced contraction [84], suggesting additional effects on contraction mechanisms activated by PGF2α such as PKC.
In addition to E2 and P4, androgens may play a role in determining the cardiovascular risk in Post-MW. Studies suggest that the relationship between circulating free E2, free T3, and sex hormone-binding globulin (SHBG) may be more predictive of changes in carotid intimal thickening than the levels of any of these hormones alone. Also, conversion of T3 to E2 may contribute to the regulation of circulation in men, and administration of an aromatase inhibitor to young men results in a decrease in EC vasodilator function. Additionally, circulating levels of E2 and T3 may not reflect those at the tissue level, as both aromatase and 5-α-reductase are found in various tissues including blood vessels [175]. Steroid hormone metabolism is a complex process that allows inter-conversion of sex hormones precursors and metabolites. With the growing use of steroid metabolism inhibitors, including aromatase and 5-α-reductase inhibitors, e.g. in women with history of breast cancer, cardiovascular side effects are predicted [40].
Conditions associated with hormone imbalance could provide insights into the effects of the hormone environment on cardiovascular function. Polycystic Ovary Syndrome (PCOS) is characterized by polycystic ovaries, amenorrhea, hirsutism, anovulatory infertility, elevated androgens and estrogens, as well as obesity, insulin resistance, and abnormal lipid metabolism. In PCOS, both insulin and luteinizing hormone stimulate androgen production from ovarian theca cells, leading to elevated levels of T3 and androstenedione. Because women with PCOS are overweight or obese, elevated androgen levels and conversion of androgens to E1 in adipose tissue by aromatase may contribute to increased E1 levels. Although women with PCOS have risk factors for CVD including low HDL-c, elevated BMI, high BP, hypertriglyceridemia, and high fasting glucose levels, a retrospective study from the United Kingdom failed to show increased cardiovascular morbidity or mortality in PCOS women [176]. Importantly, Post-MW and women with PCOS have similar sex hormone profiles, including lack of P4, E1 being the major estrogen, and relative increase in androgens. Further studies in PCOS women and animal models could provide insights into the role of other sex hormones in modulating the vascular effects of E2.
4. Sex differences in other biological systems and clinical disorders
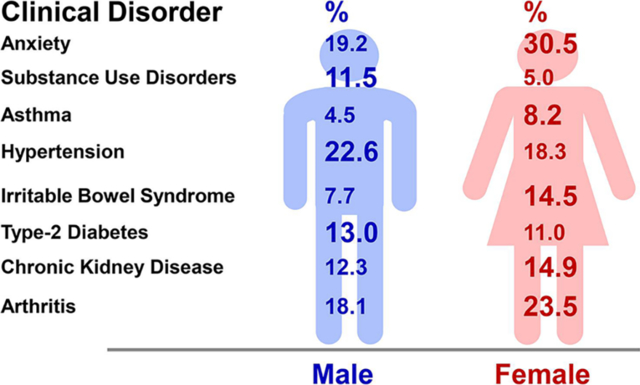
Sex differences have also been identified in other biological systems including the renal, gastrointestinal, endocrine, nervous, musculoskeletal, and immune systems (Fig. 2). Also, sex differences in the prevalence of clinical disorders of these systems have been recognized (Fig. 3).
Fig. 2.
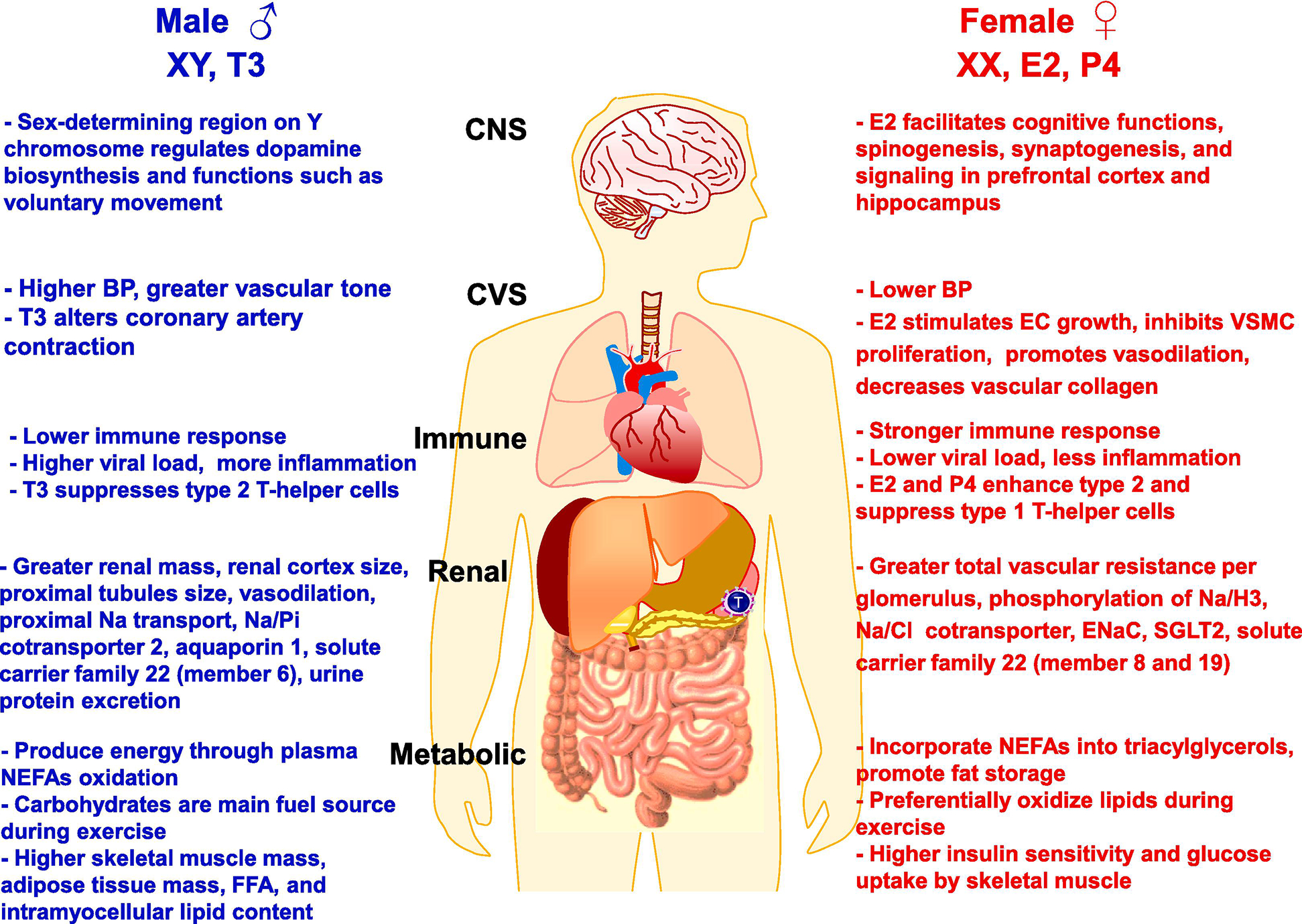
Sex differences in representative biological processes in adult males and females. Sex differences have been observed in the central nervous system (CNS), cardiovascular system (CVS), the immune response in the spleen, lymph nodes and T-helper cells, the kidneys and various ion transport mechanisms, and the metabolic system including the gastrointestinal tract, liver and pancreas. The sex differences have been attributed to genetic differences in the XX and XY chromosomes, and the levels of the sex hormones E2, P4 and T3. FFA, free fatty acids; Na/Cl, sodium chloride cotransporter, Na/H3 sodium/hydrogen exchanger 3; Na/Pi, sodium/phosphate cotransporter, SGL2, sodium-glucose cotransporter 2; NEFAs, non-esterified fatty acids
Fig. 3.
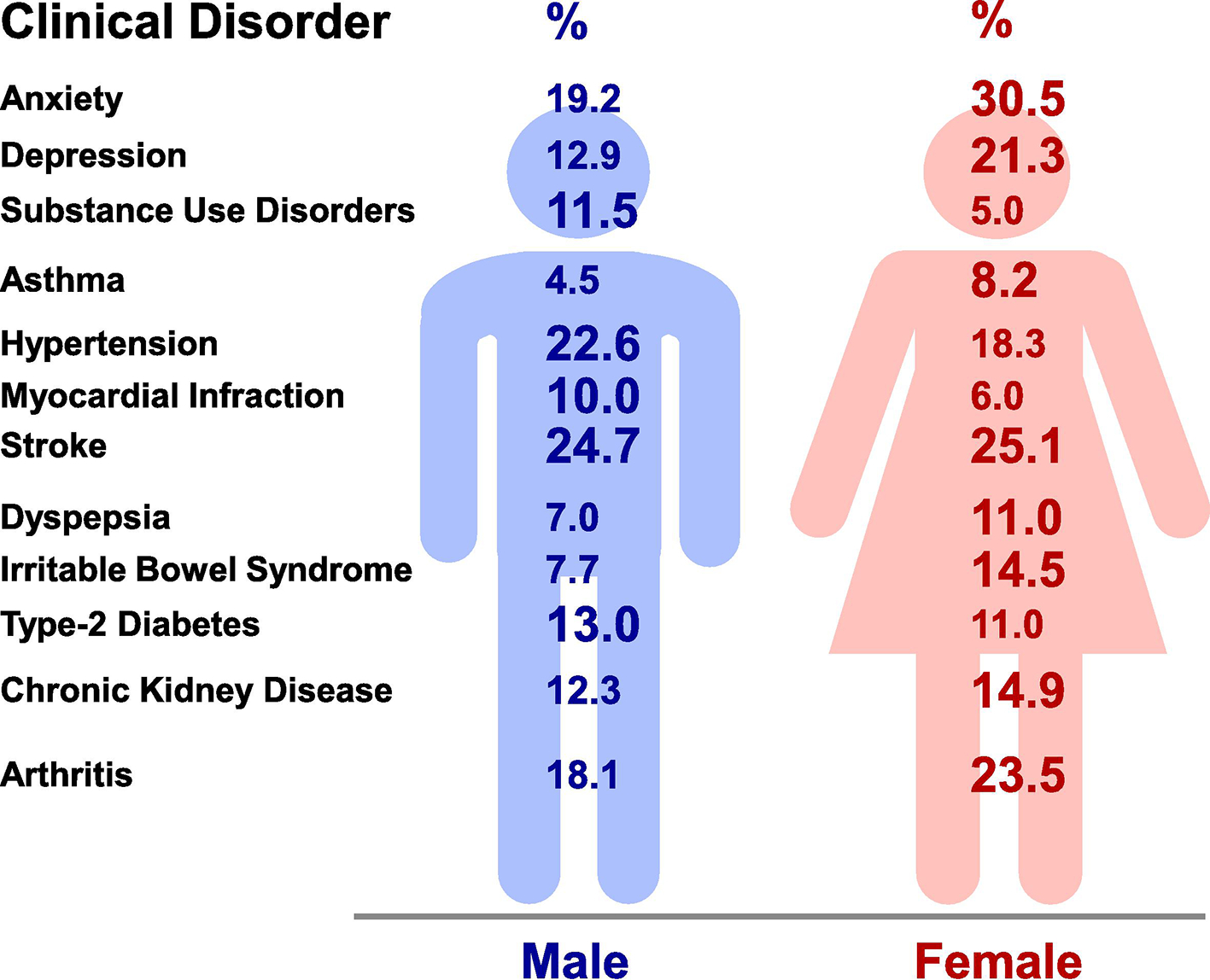
Sex differences in representative clinical disorders. Sex differences are observed in the prevalence of anxiety [237], depression (lifetime, U.S., 1990–1992) [236], substance use disorders (adults, Sweden, 2003–2004) [282], asthma (adults ~40, worldwide, 2018) [227], hypertension (adults ~40, U.S., 2007–2012) [283, 284], myocardial infarction (U.S., 1993), stroke (lifetime, worldwide, 2015) [285, 286], dyspepsia (adults, U.S., Canada, United Kingdom 2018) [197], irritable bowel syndrome (householders, U.S., 1990) [200], diabetes (adults 20–79, U.S., 2013–2016) [213], chronic kidney disease (U.S., 2015–2016) [287], and arthritis (U.S., 2013–2015) [288].
4.1. Sex differences in renal system and kidney disease
Clinical and experimental data demonstrate sex differences in renal anatomy, physiology, and susceptibility to injury and disease. For instance, the renal mass and the size of renal cortex and proximal tubules are greater in males than females. Also, the male kidney shows greater vasodilation, proximal sodium (Na) transport, Na+–Pi cotransporter 2, aquaporin 1, solute carrier family 22 member 6, and urine protein excretion. In contrast, the female kidney shows greater total vascular resistance per glomerulus, phosphorylation of sodium-hydrogen exchanger 3 (NaH3), Na+/Cl− cotransporter (NCC), ENaC (α- and γ-subunits), sodium–glucose cotransporter 2 (SGLT2), and solute carrier family 22 member 8 and 19 [177].
The sex differences in renal anatomy and physiology may affect the incidence of renal injury and progression of kidney disease. Females have an increased prevalence of kidney stone disease, especially in adolescence. Quality of life is also more adversely affected by kidney stone events among females who are also more likely to develop sepsis after endourological surgery [178]. Sexual dimorphism is also evident in the development of ischemic and nephrotoxic acute kidney injury (AKI). Although the female sex is often cited as an independent risk factor for AKI, especially when associated with cardiac surgery, rhabdomyolysis, and nephrotoxic substances [179], some studies suggest that female sex hormones play a protective role [180, 181]. Also, experimental studies consistently show that the female sex protects against renal injury in animal models of ischemic AKI [179]. Other studies suggests that male hormones may be detrimental in renal ischemia-reperfusion injury [180, 181], and that men, especially in old age and with low estimated glomerular filtration rate (eGFR), are at a higher risk for AKI [182]. The disparities between studies highlight the need for research on the impact of sex hormones on AKI.
Population-based studies indicate that chronic kidney disease (CKD) epidemiology differs by sex, affecting more women than men (Fig. 3). The effects of longer life expectancy on the natural decline of GFR with age, as well as potential overdiagnosis of CKD through the inappropriate use of GFR equations, may in part be responsible for the greater prevalence of CKD in women. While women have an increased incidence of CKD and are ~30% more likely to have pre-dialysis CKD, they are less likely than men to progress to End-Stage Kidney Disease [183, 184]. Women are also less likely to have a decline in eGFR of 50% and have a lower risk of death. Possible explanations for this discrepancy include that women may have slower progression of CKD, are more likely to die prior to starting dialysis, and are more likely to opt for conservative care rather than proceed with invasive surgery, kidney transplant or dialysis. The protective effects of E2 in women and/or the damaging effects of T3, together with unhealthier lifestyles, might cause kidney function to decline faster in men than in women. E2 may have nephro-protective effects as it reduces glomerulosclerosis and ischemia-reperfusion injury in animal models. Dissimilarities between the sexes are also apparent in the outcomes of CKD. In patients with predialysis CKD, mortality is higher in men than women; however, this difference disappears for patients on renal replacement therapy [185]. Importantly, women and men should not have the same thresholds for CKD diagnosis and staging as women have smaller kidneys and fewer nephrons, and consequently different impact of a declining GFR on morbidity and mortality. In effect, men have a higher rate of all cause and cardiovascular-renal mortality, increased risk of CKD progression, and a steeper decline in eGFR [186]. The gender disparity is also observed in the progression of membranous nephropathy, immunoglobin A nephropathy, and polycystic kidney disease. The sex differences in renal disease progression may also involve differences in glomerular structure and hemodynamics, diet, levels/activity of cytokines and hormones, and/or direct effects of sex hormones on kidney cells [187]. In animal models of renal disease, the levels of T3 may explain the worse course in males vs females [157].
E2 can be protective during acute infectious processes. Administration of E2 attenuates glomerulosclerosis and tubulointerstitial fibrosis and limits the progression of chronic kidney damage in aging Dahl salt sensitive rat [188] In contrast, T3 has pro-fibrotic and pro-apoptotic properties by promoting the activity and sensitivity of renal cells to TNF-α, and may worsen the symptoms of an acute inflammatory process through its immunosuppressive effects in males, as opposed to females who show a more robust immune response to severe infection [189].
4.2. Sex differences in microbiome, and gastrointestinal and metabolic disorders
Sex differences have been reported in the gastrointestinal and metabolic systems. The gut microbiome stabilizes after 3 years of age. However, sexual dimorphisms and correlations between E2, P4 and T3 levels and gut microbiota composition have been observed in both human and animal studies [190, 191]. In mice, the female gut microbiota more closely resembles that of prepubescent males, or castrated males, rather than age-matched males [192, 193]. Also, the diversity of gut microbiota differs between sexes, with male mice having a lower species richness and evenness compared to females [194]. The differences in the male and female gut microbiota affects various metabolic processes leading to sex differences in dysbiosis and the protection or susceptibility to various gastrointestinal and metabolic disorders [193, 195].
Dyspepsia is characterized as difficult digestion with symptoms ranging from heartburn to nausea and vomiting, with prevalence between 20% to 25% in Western countries [196]. Among 5,931 adults in the U.S., United Kingdom and Canada, an overall questionnaire-based prevalence of dyspepsia showed a women to men ratio of 11:7. For individuals older than 65 years, the prevalence of dyspepsia was similar in men and women [197]. Females are also more likely to have dysmotility-like symptoms than men, and among individuals with functional dyspepsia women complained more of upper abdominal pain than men. This may be related to E2 direct or indirect actions on immune, endocrine, and neuronal pathways and the motor and sensory functions of the gastrointestinal tract as well as the gut microbiota. E2 also affects visceral pain and gastric motility, suggesting that the female sex could be a risk factor of functional dyspepsia [198].
Irritable bowel syndrome (IBS) is one of the most common functional gastrointestinal disorders, characterized by abdominal pain and abnormal bowel habits with no detectable organic disease. In Western countries including the U.S., IBS is twice as prevalent in women vs men [199]. In the U.S. Householder Survey of functional gastrointestinal disorders, 14.5% of women and 7.7% of men had IBS symptoms [200]. Also, among IBS patients, women are more likely to have severe symptoms, fatigue, coexistent stress sensitivity, anxiety disorders, depression, and lower quality of life than men, [197]. Compared with control subjects, IBS patients are 3-fold more likely to have anxiety, which is more common in women than men. The sex differences in the incidence of IBS could be related to dietary factors, and the effects of E2 on stress, anxiety, depression, and the brain-gut axis. E2 can affect GI motility and sensitivity directly via activation of ERs located throughout the brain-gut axis, and indirectly via modulation of other receptor systems [201]. A more gender-oriented approach in medical care would improve understanding of the heterogeneity of patients with IBS and provide different treatment strategies in men vs women.
Obesity is defined as the excessive accumulation of body fat and is associated with an increased risk of major health problems including diabetes, CVD, and stroke. There are clear sexual dimorphisms in the epidemiology, pathophysiology and sequelae of obesity and its associated metabolic disorders, with females often better protected compared to males. This protection has been attributed to E2 levels, differences in fat distribution, composition of gut microbiota, and the intestinal immune system [202]. Women have more subcutaneous fat (‘gynoid’ pattern), primarily in the gluteofemoral region, while men have more visceral fat (‘android’ pattern) predominantly in the abdominal area [203, 204]. Sexual disparity has also been reported in models of obesity. Male mice on a high fat diet are at a higher risk of developing a pro-inflammatory profile (visceral inflammation, glucose intolerance, insulin resistance and hyperinsulinemia) than their female counterparts [205, 206].
4.3. Sex differences in endocrine disorders and transgender medicine
Sex differences have been observed in the regulation of glucose levels energy homeostasis, and the utilization of carbohydrates and lipids as fuel sources. At rest and during the post-absorptive state, women are more likely to incorporate non-esterified fatty acids (NEFAs) into triacylglycerols, thus promoting fat storage, whereas men are more prone to produce energy through plasma NEFA oxidation. Metabolic adaptation during exercise also differs between sexes as women preferentially oxidize lipids while men use carbohydrates as the main fuel source [207].
Compared to men, women have lower skeletal muscle mass, higher adipose tissue mass, more circulating free fatty acids, and higher intramyocellular lipid content, all factors that could promote insulin resistance. However, in individuals with normal blood glucose levels, insulin sensitivity as assessed by oral glucose insulin sensitivity index is higher in women vs men, even after adjustment for age and BMI. Hyperinsulinemic-euglycemic clamp studies confirm that healthy women are more sensitive to insulin and less prone to free fatty acids-induced insulin resistance than men when matched for physical fitness. This is due to enhanced glucose uptake by skeletal muscle in women [208]. Sex differences in muscle characteristics have also been observed, with a higher proportion of type 1 fibers and capillary density in women, which both favor enhanced insulin action. Women are also protected from NEFA-induced insulin resistance and, thus, more resistant to lipotoxicity, especially in skeletal muscle [209]. In mice, E2 confers protection against insulin resistance through activation of ERα in insulin-sensitive tissues [210].
Among Caucasians, type-1 diabetes (T1D) is more predominant in males vs females. In Sweden, the prevalence of T1D is 16.4/100000 in males and 8.8/100000 in females [211]. Among females, the pubertal period is associated with a decreased incidence of T1D in girls who retain stronger residual β-cell function than boys, but more diabetic women are diagnosed after the age of menopause, suggesting protective effects of female gonadal hormones against T1D [212]. A similar trend is seen in the prevalence of type-2 diabetes (T2D). Males are more likely to develop obesity, insulin resistance and hyperglycemia than females in response to nutritional challenges. The US National Health and Nutrition Examination Survey reported a higher prevalence of diabetes among men vs women (13% vs 11% for the 2013–2016 period, in adults aged 20–79 years) [213]. Global estimates from the International Diabetes Federation also indicate sex differences in worldwide diabetes prevalence in adult populations (9.1% in men vs 8.4% in women) [214]. The peak in diabetes prevalence occurs earlier in men (65–69 years of age) than in women (70–79 years of age). Sex steroids may contribute to sex differences in diabetes susceptibility. The protective role of E2 in women is evidenced by the deleterious impact of menopause on body composition and glucose homeostasis, leading to metabolic disorders. In support, early menopause and premature ovarian insufficiency are associated with an increased risk of T2D, while E2-based MHT is associated with 21–35% reduction in diabetes incidence in Post-MW [214].
The adrenal cortex plays pivotal roles in the maintenance of blood volume, responsiveness to stress and the development of gender characteristics. Gender differences of human adrenal cortex occur from the developing stage, in which female subjects had more activated stem cells with higher renewal capacity resulting in gender divergence in the structure and function of cortical zonation in the human adrenal. Pre-MW have lower BP with lower renin levels and angiotensin-converting enzyme (ACE) activities than age-matched men. Also, the hypothalamic-pituitary-adrenal (HPA) axis is more activated in females than males, which could contribute to gender differences in coping with stressful events. E2 suppresses the renin-angiotensin system but activate HPA axis, whereas T3 had opposite effects. Adrenocortical disorders also occur more frequently in females with more pronounced adrenocortical hormonal abnormalities possibly due to their overactivated Wnt and PRK signaling pathways and abundant activated adrenocortical stem cells in the female adrenal glands [215]. Cushing’s disease is caused by a corticotroph pituitary adenoma that secretes adrenocorticotropic hormone (ACTH), resulting in chronic overproduction of cortisol by the adrenal glands. This chronic hypercortisolism is associated with many metabolic disorders such as central obesity, diabetes, dyslipidemia and HTN [216]. Cushing’s disease occurs up to five times more often in females, but male patients with Cushing’s syndrome have a relatively higher risk of an ectopic ACTH-secreting tumor [217]. Men, compared with women, present higher urinary free cortisol values and ACTH values. In regard to diagnostic tests, men present a lower ACTH response to desmopressin (DDAVP) stimulation. The pituitary tumor itself is less easily visualized by pituitary MRI in males vs females. Furthermore, some complications of disease are more frequent or severe in men, in particular hypokalemia, hypercoagulable state, HTN, dyslipidemia, and osteoporosis at lumbar spine with higher risk for vertebral fractures [218]. Thus, while there is an increased prevalence of Cushing’s disease in females, the prognosis for males and females is markedly different with male patients experiencing a more severe clinical presentation, higher ACTH, and higher cortisol levels [219].. Although males and females with ACTH-dependent Cushing’s syndrome present different clinical patterns, they may not require different sex-based diagnostic strategies or treatment given the similar surgical outcome [217].
Thyroid hormones play a critical role in the functioning of the adult brain, and thyroid diseases impair both mood and cognition. The prevalence of thyroid disease increases with age and is much higher in females than males. Subclinical hypothyroidism is observed in up to 20% of Post-MW. Females also have higher rates of thyroid autoimmunity. Individuals at risk for thyroid disease, such as adult females, may have had less ability to compensate for additional challenges to thyroid metabolism, including lithium treatment [220]. This is likely related to the autoimmune nature of many thyroid disorders, and as the female sex steroids affect the immune system, women have a higher prevalence of autoimmune thyroid disease [221].
Changes in the endocrine system, with consequent effects on kidney health, metabolic disorders and manifestation of disease are not well understood in transgender men, women, and nonbinary individuals. Transgender individuals commonly receive gender-affirming hormone therapy to align their outward appearance with their gender. Recent attention to the differences in fundamental renal and metabolic parameters has shown that transgender individuals may manifest different levels of these biomarkers when compared with their cisgender counterparts [222]. There also appears to be striking differences in the effects of T3 in genetic males vs transgender (female to male) men in terms of cardiovascular function, kidney health and metabolic disorders, making it important to further examine the sex hormone ambience/levels and their differential effects on various biological processes in these populations.
4.4. Sex differences in the immune system, inflammatory cells, cytokines and allergy
Sex differences have been observed in the immune system and the likelihood of atopic disorders. In general, females have stronger innate and adaptive (humoral and cellular) immune responses compared with males due to multiple biologic factors including genetic and epigenetic factors and sex hormones [4, 223]. Sex hormones have different effects on T-helper (Th) cells. E2 and P4 enhance type 2 and suppress type 1 T-helper cells, whereas T3 suppresses type 2 responses and shows an inconsistent pattern for type 1 responses. Also, the X-chromosome may play a role as it encodes some of the genes associated with the immune responses resulting in a lower level of viral load and less inflammation in women when compared to men. Additionally, after infection with viral agents, women produce lower levels of IL-6 compared to men, which is associated with better longevity [224, 225].
In childhood, the risk of being allergic is greater for boys; however, from adolescence onwards women have a clear disadvantage regarding atopic disorders. There is a 60:40 ratio for female to male patients of severe food allergy. Also, among patients with anaphylaxis, females predominate with more than 60% of the documented cases [226].
Asthma is another atopic disorder with differing prevalence and severity in males vs females through various ages (Fig. 4). During childhood, boys have an increased prevalence of asthma compared to girls (11.9% vs. 7.5%), and boys are twice as likely as girls to be hospitalized for an asthma exacerbation. However, during adolescence there is a decline in asthma prevalence and morbidity in males concurrent with an increase in females. By adulthood, women have increased asthma prevalence compared to men (9.6% vs 6.3%), and women are three times more likely than men to be hospitalized for an asthma-related event. By the time of menopause, there is a decrease in asthma prevalence, and the age-adjusted risk of asthma decreases in Post-MW vs Pre-MW. The differing risks can be attributed to fluctuations in sex hormone levels during puberty, the menstrual cycle and pregnancy and their role in asthma pathogenesis. In support, experimental studies have shown that E2 increases Th2-mediated airways inflammation. Yet in the ECRHS II trial, women going through menopause had decreased lung function and had increased asthma symptoms compared with Pre-MW patients [227, 228], making it important to further examine the connection between E2 and asthma pathogenesis
Fig. 4.
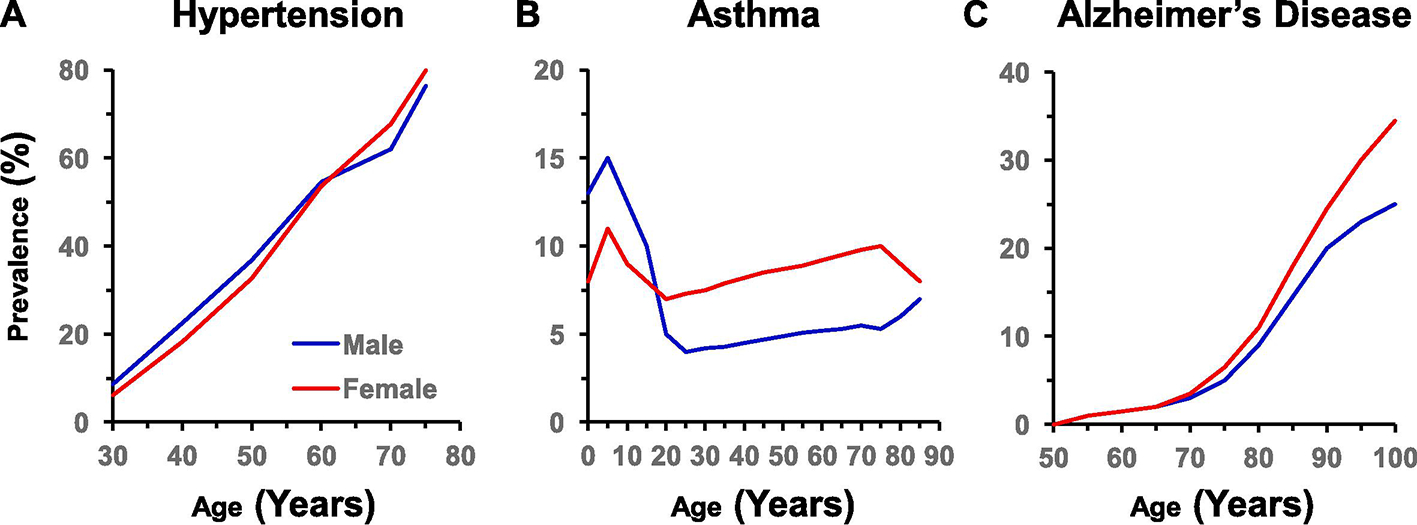
Changes in the sex differences in the prevalence of representative clinical disorders with age. Sex differences are reversed with age in the prevalence of hypertension (U.S., 2007–2012) [283, 284] (A) and asthma (worldwide, 2018) [227] (B), and further exaggerated with age in the prevalence of Alzheimer’s disease and other dementias (worldwide, 1990–2016) [289] (C).
The sex differences in the immune system have also been observed within the COVID-19 data, showing that males have a higher rate of respiratory intubation, longer hospital stay, and higher death rate than females. The Center for Disease Control (CDC) reported that 54% of COVID deaths were men. In a meta-analysis of 3,111,714 global cases, the odds of death were higher in male vs female patients (Odds ratio = 1.4; 95% confidence interval = 1.31, 1.47) [229]. The sex disparity in COVID-19 could be related to the profession type, behavior, and underlying health conditions. Also, female patients have a higher macrophage and neutrophil activity as well as antibody production and response. Immune cells are also more active in women than in men, with greater stimulation of toll-like receptor-7 and interferon production. The association of X-chromosome in women with the biallelic higher expression of immunomodulatory genes may explain their stronger immune response. On the other hand, some Y-chromosome linked genes that are overexpressed in men could lead to the higher viral load and hyperinflammatory response. Also, E2 attenuates the production of pro-inflammatory cytokines IL-6 and IL-12 and thereby prevents the cytokine storm syndrome associated with COVID-19 [230]. On the other hand, T3 could be involved in the progression of COVID-19 through the activation of cytokine storm [231].
ACE2 is a key component in the pathogenesis of COVID-19, and may offer an additional explanation of the sex disparities in the susceptibility and progression of related pulmonary disease. Studies showed higher ACE2 expression in the kidneys of male vs female patients [232]. due, in part, to the presence of E2 in the ovarian hormone milieu, and not to the T3 milieu or to differences in sex chromosome dosage (2X versus 1X; 0Y versus 1Y). Experimental studies confirmed higher activity of ACE2 in the mouse male vs female kidney and adipose tissue. Also, E2 treatment decreases ACE2 mRNA expression in differentiated airway epithelial NHBE cells [233]. The regulation of renal ACE2 by E2 could have major implications on women health with the changes in sex hormones during puberty, pregnancy and menopause [234] However, it remains unclear whether ACE2 expression differs in the lungs of male vs female patients [232].
4.5. Sex differences in brain function, pain, and addiction
Sex differences in the nervous system have gained substantial research interest. The rates of stress sensitivity, anxiety disorders, and depression are higher in women vs men [235–237]. However, the net effects of E2 on anxiety could depend on the ER subtype. ERα has a generally anxiogenic effect, ERβ has a generally anxiolytic effect, and GPER both increases and decreases anxiety-like behavior [238]. In addition to gender disparities in mood and anxiety disorders, evidence also suggests sex difference in brain function, pain perception, and addiction [239].
Alzheimer’s disease (AD) is an age-related disease characterized by memory loss, dementia, and steady deterioration in cognitive function, and is one of the leading causes of morbidity in the elderly. Around 5 million people in the U.S. have AD. The AD brain is characterized neuropathologically by diffuse and neuritic extracellular amyloid plaques often circled by dystrophic neurites and intracellular neurofibrillary tangles [240]. Genetically modified animal models have enhanced our knowledge about the influence of E2/ER signaling in the brain on neurological and behavioral regulations. E2 facilitates higher cognitive functions by inducing spinogenesis and synaptogenesis and promotes ER-mediated signaling pathways in the prefrontal cortex and hippocampus [241]. In the brain, E2/GPER1 signaling facilitates neuroprotection, social behaviors and cognition [242]. Additionally, E2 is an important regulator of the blood-brain barrier (BBB) permeability, leading to BBB protection before menopause, and increased BBB permeability with the decline in E2 levels in aging women [243], Studies of sex-related pathophysiological mechanisms of AD risk have implicated the menopause transition and E2 deficiency in Post-MW (Fig. 4). In addition to reproductive senescence, many symptoms of menopausal transition are neurological, including disruption of E2-regulated systems such as thermoregulation, sleep, and circadian rhythms, as well as depression and impairment in multiple cognitive domains. During menopausal transition, the E2 network may uncouple from the brain bioenergetic system, resulting in hypometabolic state and neurological dysfunction. In the brain, the reduction of molecular pathways mediated by ERs favors the progression of AD in post-MW [244]. In support, brain images demonstrate that 40–60 year old perimenopausal and Post-MW exhibit an AD-endophenotype characterized by decreased metabolic activity and increased brain amyloid-β deposition as compared to Pre-MW and age-matched men [245]. In the age group 65–69 years 0.7% of women and 0.6% of men suffer from AD with increasing frequencies to 14.2% and 8.8% in individuals aged 85–89 years [246]. The progressive increases in prevalence of AD in aging women compared with men could be related to their longer lifespan and stronger immune response. It is possible that the amyloid plaques are part of the brain’s immune system to fight infections, and because women have a stronger immune response, they develop more plaques [243, 247]
Sex differences have also been observed in the perception and prevalence of pain [248]. Chronic pain is a major burden on the society and is more prevalent and severe in females. Chronic pain is linked to persistent alterations in the peripheral and the central nervous system. One of the main peripheral pain transducers are the transient receptor potential channels (TRP), also known as thermoTRP channels, which participate in the perception of hot and cold external stimuli. The TRP vanilloid 1 (TRPV1), TRPA1 and TRPM8 channels function not only as thermosensors but also as regulators of acute and chronic pain, and they are differently modulated by male and female sex hormones, thus contributing to the sex differences in chronic pain [249].
Sex hormones influence pain sensitivity, and the pain threshold/tolerance in women vary with the stage of the menstrual cycle [250]. E2 affects brain function through modulation of several targets, including TRPV1 and P2X purinoceptor 3 (P2X3), two important nociceptive receptors. E2 may upregulate TRPV1 expression resulting in pro-nociceptive effect. However, E2 could alleviate pain by downregulating nerve growth factor-activated pathways or inhibiting P2X3 receptors and ATP-signaling. Understanding the E2-dependent regulation of nociceptive receptors would provide insight into the mechanisms of sex difference in pain perception [251].
Somatic symptom disorder is characterized as excessive anxiety towards persistent symptoms that do not have an identifiable physical origin. Fibromyalgia syndrome is a stress-related illness characterized by widespread musculoskeletal pain, fatigue, poor sleep, and tenderness on palpation at multiple sites (tender points). The majority of fibromyalgia patients seeking medical care are women, and only about 10% of patients are men [252]. Women have more common fatigue, morning fatigue, “hurt all over” pain, a greater total number of symptoms, and a greater number of tender points [253]. Gender differences have also been reported in related disorders such as tension headache, migraine, temporomandibular disorder, chronic fatigue syndrome, and IBS [254]. Imaging studies of the brain have shown differences between men and women in the spatial pattern and intensity of response to acute pain. Among rodents, females are more sensitive than males to noxious stimuli and have lower levels of stress-induced analgesia [255].
There is also evidence of sex-based differences in the likelihood and prognosis of addiction and substance use disorders [256]. Addiction is a major socioeconomical and health problem, with 15–20% of the U.S. population most at-risk. A widely held dogma in the preclinical addiction field is that females are more vulnerable than males to drug craving and relapse. Preclinical studies suggest sex differences in reinstatement and incubation of cocaine seeking but not for reinstatement or incubation of methamphetamine or opioid seeking [257]. In rodents, repeated psychostimulant exposure enhances incentive sensitization to a greater extent in females than males. E2 increases females’ motivation to attain psychostimulants and enhances the value of drug related cues, which ultimately increases their susceptibility towards spontaneous relapse [258]. However, clinical studies do not support the notion that women are more vulnerable to psychostimulant and opioid craving and relapse [257]. In general, males still consume more alcohol and experience and cause more alcohol-related injuries and deaths than females do, but the gaps are narrowing [259]. Alcohol-associated liver disease is becoming increasingly prevalent throughout the U.S. Although alcohol-associated liver disease was believed to affect men more often than women, the gap between the sexes is narrowing, and women could develop liver disease with lesser alcohol exposure and suffer worse disease as compared with men [260].
There are sex differences in all stages of addiction including acquisition, escalation, maintenance, withdrawal and relapse. During acquisition women experience more pleasurable responses to drugs than men do and are more likely to self-medicate. On the other hand, men take drugs and engage in risky behaviors to be part of the group more than women do. Among those at risk of addiction, escalation of use of drugs and alcohol is more rapid in women than in men. For maintenance, females stabilize at higher doses of drug than do males, leading to greater side effects in women. In females there is the tendency to experience the shift in loss of voluntary control of drug intake to compulsive drug intake to the point of addiction more rapidly. During withdrawal, female smokers show increased negative effects and greater stress response, while men exhibit greater symptoms of withdrawal from alcohol. Also, women are more likely to relapse than men and do so more sporadically. The sex differences in addiction could be related to the effects of E2 on dopaminergic function and sensory processing in the dorsolateral striatum, as the acute subjective effects of drugs of abuse vary with the level of E2 over the female menstrual cycle. Differences in socioeconomic factors and the reward system can also be involved [261].
5. Gender-specific disorders in females that have received recent research consideration
The increased awareness of sex differences in biological systems and diseases has led to increased interest in women’s health. Researchers have shown more interest in studying gender-specific disorders in females such as PCOS and their influence on other systems and the general health of afflicted females. Studies have sought further understanding of the relationships between infertility and in vitro fertilization and the link to parity, ectopic pregnancy, placental disruption, and preterm delivery. Important studies have also examined the mechanisms underlying pregnancy-related disorders such as gestational diabetes and hypertension-in-pregnancy (HTN-Preg).
In contrast with the slight decrease in BP during normal pregnancy, some women may show HTN-Preg in one of four forms: chronic HTN that predates pregnancy, preeclampsia (PE)-eclampsia, chronic HTN with superimposed PE, and non-proteinuric gestational HTN [262]. PE is a major complication affecting 5–8% of pregnancies in the U.S. and ~8 million pregnancies worldwide [263]. PE is characterized by new-onset HTN-Preg at or after 20 wk of gestation and frequently near term, with or without proteinuria [264, 265]. PE could progress to eclampsia, with severe HTN, cerebral edema, seizures, and maternal death, accounting for 15–20% of maternal mortality [266]. PE is also often associated with fetal intrauterine growth restriction, accounting for 10–15% of preterm pregnancy and small-for-gestational-age birth weight [267]. PE may also be a part of hemolysis, elevated liver enzymes, and low platelet count (HELLP) syndrome [268]. However, the pathophysiological mechanisms of PE are unclear. Predisposing genetic and environmental factors could cause placental maladaptations leading to defective placentation, apoptosis of invasive cytotrophoblasts, inadequate expansive remodeling of the spiral arteries, reduced uteroplacental perfusion pressure, and placental ischemia. Placental ischemia promotes the release of bioactive factors into the maternal circulation, causing an imbalance between antiangiogenic soluble fms-like tyrosine kinase-1 and soluble endoglin and proangiogenic vascular endothelial growth factor, placental growth factor, and transforming growth factor-β. Placental ischemia also stimulates the release of proinflammatory cytokines, hypoxia-inducible factor, ROS, and AT1R agonistic autoantibodies. These circulating factors target vascular ECs, causing generalized endotheliosis in the systemic, renal, cerebral, and hepatic vessels, leading to decreases in endothelium-derived vasodilators such as NO, PGI2 and EDHF, and increases in vasoconstrictors such as ET-1 and TXA2. The bioactive factors also target VSMCs and enhance the mechanisms of vascular contraction, including [Ca2+]c, PKC, and ROCK. The bioactive factors could also target MMPs and ECM causing inadequate vascular remodeling, increased arterial stiffening, and further increases in vascular resistance and HTN. As therapeutic options are limited, further understanding of the underlying vascular mechanisms and molecular targets should help design new tools for the detection and management of HTN-Preg and PE [269].
6. In-uetro developmental programming and origins of adult diseases
Early-life exposures to suboptimal environmental factors, including intrauterine growth restriction due to maternal malnutrition or pregnancy disorders such as PE could lead to in utero developmental programming and increased incidence of adulthood disease such as HTN, kidney disease, metabolic disorders and diabetes [270]. Mounting evidence suggests differences in the growth and metabolism of male and female fetuses. Sexual dimorphism in the response to pre-conception or early-life exposures could contribute to known sex differences in susceptibility to poor metabolic health in adulthood [271]. Exposure to states of both under- and over-nutrition during early life predisposes both sexes to develop metabolic disease. Females are particularly susceptible to develop increased adiposity and disrupted glucose homeostasis as a result of exposure to in utero undernutrition or high sugar environments, respectively. The male placenta is particularly vulnerable to damage by adverse nutritional state, and this may underlie some of the metabolic phenotypes observed in adulthood [272].
There are also sex differences in vulnerability to prenatal stress [273]. Compared to females, male fetuses are larger by the second trimester of pregnancy (based on ultrasound data) [274, 275], show a more pro-inflammatory immune response across gestation, and are at a higher risk of infection leading to preterm birth and other pregnancy complications [276, 277]. This in turn may contribute to sex differences in early susceptibility to childhood conditions including neurodevelopmental disorders [278, 279]. More males are diagnosed with autism spectrum disorder than females. Genes on the sex chromosomes and differential regulation by sex steroid hormones and their receptors especially ERβ may promote maternal immune activation and induce transcription and subsequent inflammation in myeloid-lineage cells that contribute to the etiology of autism [280]. On the other hand, throughout life, females remain at lower risk of infection, but are more likely than males to develop adult-onset autoimmune diseases [281].
7. Recommendations to further promote gender-based research
Gender-based research can be enriched by joint efforts from funding agencies, scientific societies, researchers, and journals. The NIH has taken several steps to advance research into sex differences. The NIH initiative to consider sex as a biological variable in research requires that all grant applications should include studies in both male and female animals or human subjects, and identify any sex differences in the research outcome. The NIH also issues periodic request for applications (RFAs) for grants that focus on sex differences. Additionally, the NIH provides supplemental funds to already awarded grants if they add a research component that specifically addresses sex differences. Importantly, the NIH has provided resources to help establish state-of-the-art Centers for Women Health and Centers for Studying Sex Differences in major US teaching hospitals and academic institutions The NIH and other funding agencies should continue to encourage gender-based research, periodically initiate RFAs on sex differences, and help in establishing and maintaining centers for women health and research on sex differences.
The scientific societies should enhance the career of both male and female scientists and provide them with equal opportunities to achieve prominent positions in the societies’ councils and presidents, who have considerable influence in shaping the direction of the societies. Scientific conferences should include sessions on sex differences. Also, specialized conferences on sex differences could provide a venue for new trends in gender-related pathobiology and medicine.
The excellent research work from investigators in various fields have identified sex differences in multiple biological systems and disorders. Researchers are encouraged to conduct studies on both sexes to further identify the genetic, cellular, and molecular mechanisms underlying sex differences in biology and disease.
Scientific journals can support and highlight gender-based research by periodically organizing a special issue on gender differences in biology and medicine. The journal may initiate a call for papers on gender differences, and when receiving a positive response, perhaps consider including a new category in the journal submission categories under gender-based research. Also, the journal classification terms should include specific terms such as gender medicine, sex differences, and sex hormones. Following the NIH guidelines for grant proposals, the journal instructions to the authors may include a question in the checklist asking if the authors considered sex as a biological variable and justify their reasoning if they did not do so. The journal may also request that the authors include a statement regarding studies of gender differences in the article text. For the appointment of new editors and editorial board members, the publication record and evidence of previous service and review for the journals are paramount, but adequate representation of both females and males on the editorial board should be considered. The editors and reviewers could also highlight articles addressing gender-based research particularly from female authors through editorials, commentaries, or editor’s pick/choice.
8. Conclusions and perspectives
Gender-based research has identified sex differences not only in the reproductive system but also in the cardiovascular, renal, gastrointestinal, endocrine, immune, nervous and musculoskeletal systems. Epidemiological and clinical studies have also shown sex differences in the incidence and severity of cardiovascular, renal, immunological and neurological disorders in males vs females and in Post-MW vs Pre-MW. The sex differences could be related to the influence of the sex X- and Y-chromosome on the expression of various genes. Changes in the sex hormones E2, P4 and T3 during puberty, in adult males and females, and with age could also play a role. Gender-based research has shown that CVD is more common in adult males and post-MW vs Pre-MW, and earlier observational clinical studies have suggested potential benefits of MHT. Additionally, gender-based research has demonstrated sex differences in cardiovascular function, and supported beneficial genomic and nongenomic effects of E2 on specific ERs leading to vasodilation, decreased VSMC proliferation and reduced vascular remodeling. On the contrary, RCTs have shown little benefits of MHT, and some studies suggested that it may increase the risk of VTE and stroke. However, the final chapter on MHT has not yet been written. Careful evaluation of estrogens, route of administration, dose and timing, as well as consideration of age-related changes in the ER subtype, distribution, sensitivity and signaling, the preexisting health conditions and the potential interactions with other sex hormones and metabolites could improve the cardiovascular benefits of MHT. The use of natural vs synthetic E2, phytoestrogens, specific ER agonists, selective estrogen-receptor modulators (SERMs) and other estrogenic compounds should be further evaluated in Post-MW. As newer forms of MHT become available, and if used at the proper dose, route of administration and timing depending on the subjects’ age and preexisting cardiovascular condition, the vascular effects of E2 could be translated into beneficial outcomes of MHT in post-menopausal CVD. The sex differences and the effects of E2 and MHT in the renal, gastrointestinal, endocrine, immune, nervous and musculoskeletal systems and related clinical disorders should also be further assessed in more detailed experimental studies as well as randomized clinical trials. Gender-based research should also further examine the effects of other sex hormones including P4 in females and T3 in males as well as gender-specific disorders in females such as polycystic ovary syndrome, the E2 paradox in PAH, HTN-Preg and gestational diabetes. Gender-based research can be further enhanced by combined efforts from the funding agencies that could provide resources to establish and maintain state-of-the-art centers for women health and sex differences in biology and disease in all academic institutions and teaching hospitals. Investigators, scientific societies and journals can also contribute by conducting and highlighting gender-based research, and participating in specialized conferences and journals’ special issues on sex differences. These collective efforts should enhance our understanding of the genetic and hormonal under-pinning of sex differences in the biological systems and provide better pharmacological tools for management of gender-related disorders.
Acknowledgements
This work was supported by grants from National Heart, Lung, and Blood Institute (R56HL147889, R01HL147889-A1).
List of Abbreviations:
- ACE
angiotensin converting enzyme
- ACh
acetycholine
- ACTH
adrenocorticotropic hormone
- AD
Alzheimer’s disease
- AKI
acute kidney injury
- Ang II
angiotensin I
- BBB
blood brain barrier
- BP
blood pressure
- [Ca2+]c
cytosolic free calcium concentration
- CAD
coronary artery disease
- CEE
conjugated equine estrogen
- CKD
chronic kidney disease
- CRP
C-reactive protein
- CVD
cardiovascular disease
- DHT3
dihydrotestosterone
- DRSP
drospirenone
- E2
estrogen (17β-estradiol)
- EC
endothelial cell
- ECM
extracellular matrix
- EDHF
endothelium-derived hyperpolarizing factor
- eGFR
estimated glomerular filtration rate
- ELITE
Early versus Late Intervention Trial with Estradiol
- eNOS
endothelial nitric oxide synthase
- ER
estrogen receptor
- ET-1
endothelin-1
- GPR30
GPER, G protein-coupled receptor 30
- HERS
Heart and Estrogen/progestin Replacement Study
- HTN
hypertension
- HTN-Preg
hypertension-in-pregnancy
- HUVECs
human umbilical vein ECs
- IBS
irritable bowel syndrome
- IL-6
interleukin-6
- KEEPS
Kronos Early Estrogen Prevention Study
- KO
knock-out
- MAPK
mitogen-activated protein kinase
- MCT
monocrotaline
- MHT
menopausal hormone therapy
- MI
myocardial infarction
- MMP
matrix metalloproteinase
- MLC
myosin light chain
- MPA
medroxyprogesterone acetate
- NEFAs
non-esterified fatty acids
- NETA
norethisterone acetate
- NIH
National Institutes of Health
- Na
sodium
- NO
nitric oxide
- NHS
Nurses’ Health Study
- ONOO−
peroxynitrite
- OVX
ovariectomized
- P4
progesterone
- PAH
pulmonary arterial hypertension
- PE
preeclampsia
- PCOS
polycystic ovary syndrome
- PGI2
prostacyclin
- PKC
protein kinase C
- Pre-MW
premenopausal women
- Post-MW
postmenopausal women
- RCT
randomized clinical trial
- ROCK
Rho-kinase
- SERMs
selective estrogen receptor modulators
- SHR
spontaneously hypertensive rat
- T2D
type-2 diabetes
- T3
testosterone
- TNFα
tumor necrosis factor-α
- TRP
transient receptor potential
- TXA2
thromboxane A2
- U.S.
United States
- VSMC
vascular smooth muscle cell
- VTE
venous thrombo-embolism
- WHI
Women’s Health Initiative
Footnotes
Conflict of Interest
None
Credit Statement
Conceptualization: RAK
Supervision: RAK
Resources: RAK
Methodology: SRB, CK, RAK
Data analysis: SRB, CK, RAK
Writing: SRB, CK, RAK
Validation: SRB, CK, RAK
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Vaidakis N Conceptual controversies regarding the terms Gender and Sex. Psychiatriki 2020;31:271–4. [DOI] [PubMed] [Google Scholar]
- [2].Ratnu VS, Emami MR, Bredy TW. Genetic and epigenetic factors underlying sex differences in the regulation of gene expression in the brain. J Neurosci Res 2017;95:301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liszewski W, Peebles JK, Yeung H, Arron S. Persons of Nonbinary Gender - Awareness, Visibility, and Health Disparities. N Engl J Med 2018;379:2391–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ortona E, Pierdominici M, Rider V. Editorial: Sex Hormones and Gender Differences in Immune Responses. Front Immunol 2019;10:1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Farhat MY, Lavigne MC, Ramwell PW. The vascular protective effects of estrogen. FASEB J 1996;10:615–24. [PubMed] [Google Scholar]
- [6].Dubey RK, Imthurn B, Zacharia LC, Jackson EK. Hormone replacement therapy and cardiovascular disease: what went wrong and where do we go from here? Hypertension 2004;44:789–95. [DOI] [PubMed] [Google Scholar]
- [7].Orshal JM, Khalil RA. Gender, sex hormones, and vascular tone. Am J Physiol Regul Integr Comp Physiol 2004;286:R233–49. [DOI] [PubMed] [Google Scholar]
- [8].Stefanick ML. Estrogens and progestins: background and history, trends in use, and guidelines and regimens approved by the US Food and Drug Administration. Am J Med 2005;118 Suppl 12B:64–73. [DOI] [PubMed] [Google Scholar]
- [9].Gerhard M, Ganz P. How do we explain the clinical benefits of estrogen? From bedside to bench. Circulation 1995;92:5–8. [DOI] [PubMed] [Google Scholar]
- [10].Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev 2008;60:210–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 1999;79:387–423. [DOI] [PubMed] [Google Scholar]
- [12].Bender SB, Klabunde RE. Altered role of smooth muscle endothelin receptors in coronary endothelin-1 and alpha1-adrenoceptor-mediated vasoconstriction in Type 2 diabetes. Am J Physiol Heart Circ Physiol 2007;293:H2281–8. [DOI] [PubMed] [Google Scholar]
- [13].Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res 2002;90:248–50. [DOI] [PubMed] [Google Scholar]
- [14].Stallone JN, Crofton JT, Share L. Sexual dimorphism in vasopressin-induced contraction of rat aorta. Am J Physiol 1991;260:H453–8. [DOI] [PubMed] [Google Scholar]
- [15].Kos M, Denger S, Reid G, Gannon F. Upstream open reading frames regulate the translation of the multiple mRNA variants of the estrogen receptor alpha. J Biol Chem 2002;277:37131–8. [DOI] [PubMed] [Google Scholar]
- [16].Scobie GA, Macpherson S, Millar MR, Groome NP, Romana PG, Saunders PT. Human oestrogen receptors: differential expression of ER alpha and beta and the identification of ER beta variants. Steroids 2002;67:985–92. [DOI] [PubMed] [Google Scholar]
- [17].Guiochon-Mantel A [Regulation of the differentiation and proliferation of smooth muscle cells by the sex hormones]. Rev Mal Respir 2000;17:604–8. [PubMed] [Google Scholar]
- [18].Dan P, Cheung JC, Scriven DR, Moore ED. Epitope-dependent localization of estrogen receptor-alpha, but not -beta, in en face arterial endothelium. Am J Physiol Heart Circ Physiol 2003;284:H1295–306. [DOI] [PubMed] [Google Scholar]
- [19].Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER. ERs associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol Endocrinol 2002;16:100–15. [DOI] [PubMed] [Google Scholar]
- [20].Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci U S A 1998;95:15677–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Harris HA. Estrogen receptor-beta: recent lessons from in vivo studies. Mol Endocrinol 2007;21:1–13. [DOI] [PubMed] [Google Scholar]
- [22].Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev 2007;87:905–31. [DOI] [PubMed] [Google Scholar]
- [23].Mendelsohn ME. Genomic and nongenomic effects of estrogen in the vasculature. Am J Cardiol 2002;90:3F–6F. [DOI] [PubMed] [Google Scholar]
- [24].Kim-Schulze S, McGowan KA, Hubchak SC, Cid MC, Martin MB, Kleinman HK, et al. Expression of an estrogen receptor by human coronary artery and umbilical vein endothelial cells. Circulation 1996;94:1402–7. [DOI] [PubMed] [Google Scholar]
- [25].Rubanyi GM, Freay AD, Kauser K, Sukovich D, Burton G, Lubahn DB, et al. Vascular estrogen receptors and endothelium-derived nitric oxide production in the mouse aorta. Gender difference and effect of estrogen receptor gene disruption. J Clin Invest 1997;99:2429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics 2006;7:497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hodges YK, Tung L, Yan XD, Graham JD, Horwitz KB, Horwitz LD. Estrogen receptors alpha and beta: prevalence of estrogen receptor beta mRNA in human vascular smooth muscle and transcriptional effects. Circulation 2000;101:1792–8. [DOI] [PubMed] [Google Scholar]
- [28].Andersson C, Lydrup ML, Ferno M, Idvall I, Gustafsson J, Nilsson BO. Immunocytochemical demonstration of oestrogen receptor beta in blood vessels of the female rat. J Endocrinol 2001;169:241–7. [DOI] [PubMed] [Google Scholar]
- [29].Pare G, Krust A, Karas RH, Dupont S, Aronovitz M, Chambon P, et al. Estrogen receptor-alpha mediates the protective effects of estrogen against vascular injury. Circ Res 2002;90:1087–92. [DOI] [PubMed] [Google Scholar]
- [30].Christian RC, Liu PY, Harrington S, Ruan M, Miller VM, Fitzpatrick LA. Intimal estrogen receptor (ER)beta, but not ERalpha expression, is correlated with coronary calcification and atherosclerosis in pre- and postmenopausal women. J Clin Endocrinol Metab 2006;91:2713–20. [DOI] [PubMed] [Google Scholar]
- [31].Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005;307:1625–30. [DOI] [PubMed] [Google Scholar]
- [32].Haas E, Meyer MR, Schurr U, Bhattacharya I, Minotti R, Nguyen HH, et al. Differential effects of 17beta-estradiol on function and expression of estrogen receptor alpha, estrogen receptor beta, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension 2007;49:1358–63. [DOI] [PubMed] [Google Scholar]
- [33].Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, et al. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 2007;148:3236–45. [DOI] [PubMed] [Google Scholar]
- [34].Ogola BO, Abshire CM, Visniauskas B, Kiley JX, Horton AC, Clark-Patterson GL, et al. Sex differences in vascular aging and impact of GPER deletion. Am J Physiol Heart Circ Physiol 2022;323:H336–H49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Geraldes P, Sirois MG, Bernatchez PN, Tanguay JF. Estrogen regulation of endothelial and smooth muscle cell migration and proliferation: role of p38 and p42/44 mitogen-activated protein kinase. Arterioscler Thromb Vasc Biol 2002;22:1585–90. [DOI] [PubMed] [Google Scholar]
- [36].Gilligan DM, Badar DM, Panza JA, Quyyumi AA, Cannon RO 3rd. Acute vascular effects of estrogen in postmenopausal women. Circulation 1994;90:786–91. [DOI] [PubMed] [Google Scholar]
- [37].Eckstein N, Nadler E, Barnea O, Shavit G, Ayalon D. Acute effects of 17 beta-estradiol on the rat heart. Am J Obstet Gynecol 1994;171:844–8. [DOI] [PubMed] [Google Scholar]
- [38].Kauser K, Rubanyi GM. Gender difference in endothelial dysfunction in the aorta of spontaneously hypertensive rats. Hypertension 1995;25:517–23. [DOI] [PubMed] [Google Scholar]
- [39].Kahonen M, Tolvanen JP, Sallinen K, Wu X, Porsti I. Influence of gender on control of arterial tone in experimental hypertension. Am J Physiol 1998;275:H15–22. [DOI] [PubMed] [Google Scholar]
- [40].Smiley DA, Khalil RA. Estrogenic compounds, estrogen receptors and vascular cell signaling in the aging blood vessels. Curr Med Chem 2009;16:1863–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wilcox JN, Subramanian RR, Sundell CL, Tracey WR, Pollock JS, Harrison DG, et al. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol 1997;17:2479–88. [DOI] [PubMed] [Google Scholar]
- [42].Forte P, Kneale BJ, Milne E, Chowienczyk PJ, Johnston A, Benjamin N, et al. Evidence for a difference in nitric oxide biosynthesis between healthy women and men. Hypertension 1998;32:730–4. [DOI] [PubMed] [Google Scholar]
- [43].Kublickiene K, Svedas E, Landgren BM, Crisby M, Nahar N, Nisell H, et al. Small artery endothelial dysfunction in postmenopausal women: in vitro function, morphology, and modification by estrogen and selective estrogen receptor modulators. J Clin Endocrinol Metab 2005;90:6113–22. [DOI] [PubMed] [Google Scholar]
- [44].Thompson LP, Pinkas G, Weiner CP. Chronic 17beta-estradiol replacement increases nitric oxide-mediated vasodilation of guinea pig coronary microcirculation. Circulation 2000;102:445–51. [DOI] [PubMed] [Google Scholar]
- [45].Geary GG, Krause DN, Duckles SP. Estrogen reduces mouse cerebral artery tone through endothelial NOS- and cyclooxygenase-dependent mechanisms. Am J Physiol Heart Circ Physiol 2000;279:H511–9. [DOI] [PubMed] [Google Scholar]
- [46].MacRitchie AN, Jun SS, Chen Z, German Z, Yuhanna IS, Sherman TS, et al. Estrogen upregulates endothelial nitric oxide synthase gene expression in fetal pulmonary artery endothelium. Circ Res 1997;81:355–62. [DOI] [PubMed] [Google Scholar]
- [47].Sumi D, Hayashi T, Jayachandran M, Iguchi A. Estrogen prevents destabilization of endothelial nitric oxide synthase mRNA induced by tumor necrosis factor alpha through estrogen receptor mediated system. Life Sci 2001;69:1651–60. [DOI] [PubMed] [Google Scholar]
- [48].Knot HJ, Lounsbury KM, Brayden JE, Nelson MT. Gender differences in coronary artery diameter reflect changes in both endothelial Ca2+ and ecNOS activity. Am J Physiol 1999;276:H961–9. [DOI] [PubMed] [Google Scholar]
- [49].Targeting Michel T. and translocation of endothelial nitric oxide synthase. Braz J Med Biol Res 1999;32:1361–6. [DOI] [PubMed] [Google Scholar]
- [50].Russell KS, Haynes MP, Sinha D, Clerisme E, Bender JR. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc Natl Acad Sci U S A 2000;97:5930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Guo X, Razandi M, Pedram A, Kassab G, Levin ER. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors alpha and beta. J Biol Chem 2005;280:19704–10. [DOI] [PubMed] [Google Scholar]
- [52].Caulin-Glaser T, Garcia-Cardena G, Sarrel P, Sessa WC, Bender JR. 17 beta-estradiol regulation of human endothelial cell basal nitric oxide release, independent of cytosolic Ca2+ mobilization. Circ Res 1997;81:885–92. [DOI] [PubMed] [Google Scholar]
- [53].Hisamoto K, Ohmichi M, Kurachi H, Hayakawa J, Kanda Y, Nishio Y, et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem 2001;276:3459–67. [DOI] [PubMed] [Google Scholar]
- [54].Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest 1999;103:401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, et al. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res 2000;87:677–82. [DOI] [PubMed] [Google Scholar]
- [56].Tan E, Gurjar MV, Sharma RV, Bhalla RC. Estrogen receptor-alpha gene transfer into bovine aortic endothelial cells induces eNOS gene expression and inhibits cell migration. Cardiovasc Res 1999;43:788–97. [DOI] [PubMed] [Google Scholar]
- [57].Widder J, Pelzer T, von Poser-Klein C, Hu K, Jazbutyte V, Fritzemeier KH, et al. Improvement of endothelial dysfunction by selective estrogen receptor-alpha stimulation in ovariectomized SHR. Hypertension 2003;42:991–6. [DOI] [PubMed] [Google Scholar]
- [58].Darblade B, Pendaries C, Krust A, Dupont S, Fouque MJ, Rami J, et al. Estradiol alters nitric oxide production in the mouse aorta through the alpha-, but not beta-, estrogen receptor. Circ Res 2002;90:413–9. [DOI] [PubMed] [Google Scholar]
- [59].Chambliss KL, Yuhanna IS, Anderson RG, Mendelsohn ME, Shaul PW. ERbeta has nongenomic action in caveolae. Mol Endocrinol 2002;16:938–46. [DOI] [PubMed] [Google Scholar]
- [60].Brandes RP, Mugge A. Gender differences in the generation of superoxide anions in the rat aorta. Life Sci 1997;60:391–6. [DOI] [PubMed] [Google Scholar]
- [61].Hernandez I, Delgado JL, Diaz J, Quesada T, Teruel MJ, Llanos MC, et al. 17beta-estradiol prevents oxidative stress and decreases blood pressure in ovariectomized rats. Am J Physiol Regul Integr Comp Physiol 2000;279:R1599–605. [DOI] [PubMed] [Google Scholar]
- [62].Wagner AH, Schroeter MR, Hecker M. 17beta-estradiol inhibition of NADPH oxidase expression in human endothelial cells. FASEB J 2001;15:2121–30. [DOI] [PubMed] [Google Scholar]
- [63].Barbacanne MA, Rami J, Michel JB, Souchard JP, Philippe M, Besombes JP, et al. Estradiol increases rat aorta endothelium-derived relaxing factor (EDRF) activity without changes in endothelial NO synthase gene expression: possible role of decreased endothelium-derived superoxide anion production. Cardiovasc Res 1999;41:672–81. [DOI] [PubMed] [Google Scholar]
- [64].Gragasin FS, Xu Y, Arenas IA, Kainth N, Davidge ST . Estrogen reduces angiotensin II-induced nitric oxide synthase and NAD(P)H oxidase expression in endothelial cells. Arterioscler Thromb Vasc Biol 2003;23:38–44. [DOI] [PubMed] [Google Scholar]
- [65].Calkin AC, Sudhir K, Honisett S, Williams MR, Dawood T, Komesaroff PA. Rapid potentiation of endothelium-dependent vasodilation by estradiol in postmenopausal women is mediated via cyclooxygenase 2. J Clin Endocrinol Metab 2002;87:5072–5. [DOI] [PubMed] [Google Scholar]
- [66].O’Sullivan MG, Goodrich JA, Adams MR. Increased prostacyclin synthesis by atherosclerotic arteries from estrogen-treated monkeys. Life Sci 2001;69:395–401. [DOI] [PubMed] [Google Scholar]
- [67].Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM, et al. COX-2-derived prostacyclin confers atheroprotection on female mice. Science 2004;306:1954–7. [DOI] [PubMed] [Google Scholar]
- [68].Jun SS, Chen Z, Pace MC, Shaul PW. Estrogen upregulates cyclooxygenase-1 gene expression in ovine fetal pulmonary artery endothelium. J Clin Invest 1998;102:176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sherman TS, Chambliss KL, Gibson LL, Pace MC, Mendelsohn ME, Pfister SL, et al. Estrogen acutely activates prostacyclin synthesis in ovine fetal pulmonary artery endothelium. Am J Respir Cell Mol Biol 2002;26:610–6. [DOI] [PubMed] [Google Scholar]
- [70].Jiang CW, Sarrel PM, Lindsay DC, Poole-Wilson PA, Collins P. Endothelium-independent relaxation of rabbit coronary artery by 17 beta-oestradiol in vitro. Br J Pharmacol 1991;104:1033–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Case J, Davison CA. Estrogen alters relative contributions of nitric oxide and cyclooxygenase products to endothelium-dependent vasodilation. J Pharmacol Exp Ther 1999;291:524–30. [PubMed] [Google Scholar]
- [72].Liu MY, Hattori Y, Fukao M, Sato A, Sakuma I, Kanno M. Alterations in EDHF-mediated hyperpolarization and relaxation in mesenteric arteries of female rats in long-term deficiency of oestrogen and during oestrus cycle. Br J Pharmacol 2001;132:1035–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Sakuma I, Liu MY, Sato A, Hayashi T, Iguchi A, Kitabatake A, et al. Endothelium-dependent hyperpolarization and relaxation in mesenteric arteries of middle-aged rats: influence of oestrogen. Br J Pharmacol 2002;135:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Webb CM, Ghatei MA, McNeill JG, Collins P. 17beta-estradiol decreases endothelin-1 levels in the coronary circulation of postmenopausal women with coronary artery disease. Circulation 2000;102:1617–22. [DOI] [PubMed] [Google Scholar]
- [75].Akishita M, Kozaki K, Eto M, Yoshizumi M, Ishikawa M, Toba K, et al. Estrogen attenuates endothelin-1 production by bovine endothelial cells via estrogen receptor. Biochem Biophys Res Commun 1998;251:17–21. [DOI] [PubMed] [Google Scholar]
- [76].Tostes RC, David FL, Carvalho MH, Nigro D, Scivoletto R, Fortes ZB. Gender differences in vascular reactivity to endothelin-1 in deoxycorticosterone-salt hypertensive rats. J Cardiovasc Pharmacol 2000;36:S99–101. [DOI] [PubMed] [Google Scholar]
- [77].David FL, Carvalho MH, Cobra AL, Nigro D, Fortes ZB, Reboucas NA, et al. Ovarian hormones modulate endothelin-1 vascular reactivity and mRNA expression in DOCA-salt hypertensive rats. Hypertension 2001;38:692–6. [DOI] [PubMed] [Google Scholar]
- [78].Ba ZF, Lu A, Shimizu T, Szalay L, Schwacha MG, Rue LW, 3rd, et al. 17beta-Estradiol modulates vasoconstriction induced by endothelin-1 following trauma-hemorrhage. Am J Physiol Heart Circ Physiol 2007;292:H245–50. [DOI] [PubMed] [Google Scholar]
- [79].Morey AK, Razandi M, Pedram A, Hu RM, Prins BA, Levin ER. Oestrogen and progesterone inhibit the stimulated production of endothelin-1. Biochem J 1998;330 ( Pt 3):1097–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bilsel AS, Moini H, Tetik E, Aksungar F, Kaynak B, Ozer A. 17Beta-estradiol modulates endothelin-1 expression and release in human endothelial cells. Cardiovasc Res 2000;46:579–84. [DOI] [PubMed] [Google Scholar]
- [81].Dubey RK, Gillespie DG, Imthurn B, Rosselli M, Jackson EK, Keller PJ. Phytoestrogens inhibit growth and MAP kinase activity in human aortic smooth muscle cells. Hypertension 1999;33:177–82. [DOI] [PubMed] [Google Scholar]
- [82].Takeda-Matsubara Y, Nakagami H, Iwai M, Cui TX, Shiuchi T, Akishita M, et al. Estrogen activates phosphatases and antagonizes growth-promoting effect of angiotensin II. Hypertension 2002;39:41–5. [DOI] [PubMed] [Google Scholar]
- [83].Dubey RK, Gillespie DG, Mi Z, Rosselli M, Keller PJ, Jackson EK. Estradiol inhibits smooth muscle cell growth in part by activating the cAMP-adenosine pathway. Hypertension 2000;35:262–6. [DOI] [PubMed] [Google Scholar]
- [84].Crews JK, Khalil RA. Antagonistic effects of 17 beta-estradiol, progesterone, and testosterone on Ca2+ entry mechanisms of coronary vasoconstriction. Arterioscler Thromb Vasc Biol 1999;19:1034–40. [DOI] [PubMed] [Google Scholar]
- [85].Jiang C, Sarrel PM, Poole-Wilson PA, Collins P. Acute effect of 17 beta-estradiol on rabbit coronary artery contractile responses to endothelin-1. Am J Physiol 1992;263:H271–5. [DOI] [PubMed] [Google Scholar]
- [86].Kakucs R, Varbiro S, Nadasy GL, Monos E, Szekacs B. Acute, nongenomic vasodilatory action of estradiol is attenuated by chronic estradiol treatment. Exp Biol Med (Maywood) 2001;226:538–42. [DOI] [PubMed] [Google Scholar]
- [87].Murphy JG, Khalil RA. Gender-specific reduction in contractility and [Ca(2+)](i) in vascular smooth muscle cells of female rat. Am J Physiol Cell Physiol 2000;278:C834–44. [DOI] [PubMed] [Google Scholar]
- [88].Kanashiro CA, Khalil RA. Signal transduction by protein kinase C in mammalian cells. Clin Exp Pharmacol Physiol 1998;25:974–85. [DOI] [PubMed] [Google Scholar]
- [89].Johnson BD, Zheng W, Korach KS, Scheuer T, Catterall WA, Rubanyi GM. Increased expression of the cardiac L-type calcium channel in estrogen receptor-deficient mice. J Gen Physiol 1997;110:135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Bowles DK. Gender influences coronary L-type Ca(2+) current and adaptation to exercise training in miniature swine. J Appl Physiol (1985) 2001;91:2503–10. [DOI] [PubMed] [Google Scholar]
- [91].Murphy JG, Khalil RA. Decreased [Ca(2+)](i) during inhibition of coronary smooth muscle contraction by 17beta-estradiol, progesterone, and testosterone. J Pharmacol Exp Ther 1999;291:44–52. [PubMed] [Google Scholar]
- [92].White RE, Darkow DJ, Lang JL. Estrogen relaxes coronary arteries by opening BKCa channels through a cGMP-dependent mechanism. Circ Res 1995;77:936–42. [DOI] [PubMed] [Google Scholar]
- [93].Zhang F, Ram JL, Standley PR, Sowers JR. 17 beta-Estradiol attenuates voltage-dependent Ca2+ currents in A7r5 vascular smooth muscle cell line. Am J Physiol 1994;266:C975–80. [DOI] [PubMed] [Google Scholar]
- [94].Prakash YS, Togaibayeva AA, Kannan MS, Miller VM, Fitzpatrick LA, Sieck GC. Estrogen increases Ca2+ efflux from female porcine coronary arterial smooth muscle. Am J Physiol 1999;276:H926–34. [DOI] [PubMed] [Google Scholar]
- [95].Kanashiro CA, Khalil RA. Gender-related distinctions in protein kinase C activity in rat vascular smooth muscle. Am J Physiol Cell Physiol 2001;280:C34–45. [DOI] [PubMed] [Google Scholar]
- [96].Chrissobolis S, Budzyn K, Marley PD, Sobey CG. Evidence that estrogen suppresses rho-kinase function in the cerebral circulation in vivo. Stroke 2004;35:2200–5. [DOI] [PubMed] [Google Scholar]
- [97].Barrett-Connor E, Bush TL. Estrogen and coronary heart disease in women. JAMA 1991;265:1861–7. [PubMed] [Google Scholar]
- [98].O’Keefe JH Jr., Kim SC, Hall RR, Cochran VC, Lawhorn SL, McCallister BD Estrogen replacement therapy after coronary angioplasty in women. J Am Coll Cardiol 1997;29:1–5. [DOI] [PubMed] [Google Scholar]
- [99].Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, et al. Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). JAMA 2002;288:49–57. [DOI] [PubMed] [Google Scholar]
- [100].Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002;288:321–33. [DOI] [PubMed] [Google Scholar]
- [101].Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998;280:605–13. [DOI] [PubMed] [Google Scholar]
- [102].Smith NL, Heckbert SR, Lemaitre RN, Reiner AP, Lumley T, Weiss NS, et al. Esterified estrogens and conjugated equine estrogens and the risk of venous thrombosis. JAMA 2004;292:1581–7. [DOI] [PubMed] [Google Scholar]
- [103].Cushman M, Kuller LH, Prentice R, Rodabough RJ, Psaty BM, Stafford RS, et al. Estrogen plus progestin and risk of venous thrombosis. JAMA 2004;292:1573–80. [DOI] [PubMed] [Google Scholar]
- [104].Grodstein F, Manson JE, Stampfer MJ. Postmenopausal hormone use and secondary prevention of coronary events in the nurses’ health study. a prospective, observational study. Ann Intern Med 2001;135:1–8. [DOI] [PubMed] [Google Scholar]
- [105].Hendrix SL, Wassertheil-Smoller S, Johnson KC, Howard BV, Kooperberg C, Rossouw JE, et al. Effects of conjugated equine estrogen on stroke in the Women’s Health Initiative. Circulation 2006;113:2425–34. [DOI] [PubMed] [Google Scholar]
- [106].Viscoli CM, Brass LM, Kernan WN, Sarrel PM, Suissa S, Horwitz RI. A clinical trial of estrogen-replacement therapy after ischemic stroke. N Engl J Med 2001;345:1243–9. [DOI] [PubMed] [Google Scholar]
- [107].Barrett-Connor E, Mosca L, Collins P, Geiger MJ, Grady D, Kornitzer M, et al. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med 2006;355:125–37. [DOI] [PubMed] [Google Scholar]
- [108].Barrett-Connor E, Grady D, Sashegyi A, Anderson PW, Cox DA, Hoszowski K, et al. Raloxifene and cardiovascular events in osteoporotic postmenopausal women: four-year results from the MORE (Multiple Outcomes of Raloxifene Evaluation) randomized trial. JAMA 2002;287:847–57. [DOI] [PubMed] [Google Scholar]
- [109].Douglas G, Cruz MN, Poston L, Gustafsson JA, Kublickiene K. Functional characterization and sex differences in small mesenteric arteries of the estrogen receptor-beta knockout mouse. Am J Physiol Regul Integr Comp Physiol 2008;294:R112–20. [DOI] [PubMed] [Google Scholar]
- [110].Pickar JH, Yeh IT, Wheeler JE, Cunnane MF, Speroff L. Endometrial effects of lower doses of conjugated equine estrogens and medroxyprogesterone acetate: two-year substudy results. Fertil Steril 2003;80:1234–40. [DOI] [PubMed] [Google Scholar]
- [111].Turgeon JL, Carr MC, Maki PM, Mendelsohn ME, Wise PM. Complex actions of sex steroids in adipose tissue, the cardiovascular system, and brain: Insights from basic science and clinical studies. Endocr Rev 2006;27:575–605. [DOI] [PubMed] [Google Scholar]
- [112].Gouva L, Tsatsoulis A. The role of estrogens in cardiovascular disease in the aftermath of clinical trials. Hormones (Athens) 2004;3:171–83. [DOI] [PubMed] [Google Scholar]
- [113].Ho JY, Chen MJ, Sheu WH, Yi YC, Tsai AC, Guu HF, et al. Differential effects of oral conjugated equine estrogen and transdermal estrogen on atherosclerotic vascular disease risk markers and endothelial function in healthy postmenopausal women. Hum Reprod 2006;21:2715–20. [DOI] [PubMed] [Google Scholar]
- [114].Zhu BT, Han GZ, Shim JY, Wen Y, Jiang XR. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 2006;147:4132–50. [DOI] [PubMed] [Google Scholar]
- [115].Alevizaki M, Saltiki K, Cimponeriu A, Kanakakis I, Xita N, Alevizaki CC, et al. Severity of cardiovascular disease in postmenopausal women: associations with common estrogen receptor alpha polymorphic variants. Eur J Endocrinol 2007;156:489–96. [DOI] [PubMed] [Google Scholar]
- [116].Emre A, Sahin S, Erzik C, Nurkalem Z, Oz D, Cirakoglu B, et al. Effect of hormone replacement therapy on plasma lipoproteins and apolipoproteins, endothelial function and myocardial perfusion in postmenopausal women with estrogen receptor-alpha IVS1–397 C/C genotype and established coronary artery disease. Cardiology 2006;106:44–50. [DOI] [PubMed] [Google Scholar]
- [117].Herrington DM, Howard TD, Hawkins GA, Reboussin DM, Xu J, Zheng SL, et al. Estrogen-receptor polymorphisms and effects of estrogen replacement on high-density lipoprotein cholesterol in women with coronary disease. N Engl J Med 2002;346:967–74. [DOI] [PubMed] [Google Scholar]
- [118].Post WS, Goldschmidt-Clermont PJ, Wilhide CC, Heldman AW, Sussman MS, Ouyang P, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res 1999;43:985–91. [DOI] [PubMed] [Google Scholar]
- [119].Wynne FL, Payne JA, Cain AE, Reckelhoff JF, Khalil RA. Age-related reduction in estrogen receptor-mediated mechanisms of vascular relaxation in female spontaneously hypertensive rats. Hypertension 2004;43:405–12. [DOI] [PubMed] [Google Scholar]
- [120].Kalesnykas G, Roschier U, Puolivali J, Wang J, Miettinen R. The effect of aging on the subcellular distribution of estrogen receptor-alpha in the cholinergic neurons of transgenic and wild-type mice. Eur J Neurosci 2005;21:1437–42. [DOI] [PubMed] [Google Scholar]
- [121].Carmel S. Health and Well-Being in Late Life: Gender Differences Worldwide. Front Med (Lausanne) 2019;6:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hagg S, Jylhava J. Sex differences in biological aging with a focus on human studies. Elife 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Wingrove CS, Garr E, Godsland IF, Stevenson JC. 17beta-oestradiol enhances release of matrix metalloproteinase-2 from human vascular smooth muscle cells. Biochim Biophys Acta 1998;1406:169–74. [DOI] [PubMed] [Google Scholar]
- [124].Lewandowski KC, Komorowski J, Mikhalidis DP, Bienkiewicz M, Tan BK, O’Callaghan CJ, et al. Effects of hormone replacement therapy type and route of administration on plasma matrix metalloproteinases and their tissue inhibitors in postmenopausal women. J Clin Endocrinol Metab 2006;91:3123–30. [DOI] [PubMed] [Google Scholar]
- [125].Hodis HN, Mack WJ, Azen SP, Lobo RA, Shoupe D, Mahrer PR, et al. Hormone therapy and the progression of coronary-artery atherosclerosis in postmenopausal women. N Engl J Med 2003;349:535–45. [DOI] [PubMed] [Google Scholar]
- [126].Miller VM, Black DM, Brinton EA, Budoff MJ, Cedars MI, Hodis HN, et al. Using basic science to design a clinical trial: baseline characteristics of women enrolled in the Kronos Early Estrogen Prevention Study (KEEPS). J Cardiovasc Transl Res 2009;2:228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Mikkola TS, Clarkson TB. Estrogen replacement therapy, atherosclerosis, and vascular function. Cardiovasc Res 2002;53:605–19. [DOI] [PubMed] [Google Scholar]
- [128].Mori T, Durand J, Chen Y, Thompson JA, Bakir S, Oparil S. Effects of short-term estrogen treatment on the neointimal response to balloon injury of rat carotid artery. Am J Cardiol 2000;85:1276–9. [DOI] [PubMed] [Google Scholar]
- [129].Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010;122:164–72. [DOI] [PubMed] [Google Scholar]
- [130].McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T, et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004;126:78S–92S. [DOI] [PubMed] [Google Scholar]
- [131].Barst RJ, Langleben D, Badesch D, Frost A, Lawrence EC, Shapiro S, et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol 2006;47:2049–56. [DOI] [PubMed] [Google Scholar]
- [132].Frost AE, Badesch DB, Barst RJ, Benza RL, Elliott CG, Farber HW, et al. The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US Contemporary Registries. Chest 2010;139:128–37. [DOI] [PubMed] [Google Scholar]
- [133].Patel KM, Lahm T, Crisostomo PR, Herring C, Markel T, Wang M, et al. The effects of endogenous sex hormones and acute hypoxia on vasoconstriction in isolated rat pulmonary artery rings. J Surg Res 2008;146:121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Smith AM, Jones RD, Channer KS. The influence of sex hormones on pulmonary vascular reactivity: possible vasodilator therapies for the treatment of pulmonary hypertension. Curr Vasc Pharmacol 2006;4:9–15. [DOI] [PubMed] [Google Scholar]
- [135].Mueck AO, Seeger H. 2-Methoxyestradiol--biology and mechanism of action. Steroids 2010;75:625–31. [DOI] [PubMed] [Google Scholar]
- [136].Tofovic SP, Salah EM, Mady HH, Jackson EK, Melhem MF. Estradiol metabolites attenuate monocrotaline-induced pulmonary hypertension in rats. J Cardiovasc Pharmacol 2005;46:430–7. [DOI] [PubMed] [Google Scholar]
- [137].Tofovic SP, Jones TJ, Bilan VP, Jackson EK, Petrusevska G. Synergistic therapeutic effects of 2-methoxyestradiol with either sildenafil or bosentan on amelioration of monocrotaline-induced pulmonary hypertension and vascular remodeling. J Cardiovasc Pharmacol 2010;56:475–83. [DOI] [PubMed] [Google Scholar]
- [138].Vazquez F, Rodriguez-Manzaneque JC, Lydon JP, Edwards DP, O’Malley BW, Iruela-Arispe ML. Progesterone regulates proliferation of endothelial cells. J Biol Chem 1999;274:2185–92. [DOI] [PubMed] [Google Scholar]
- [139].Nisolle M, Gillerot S, Casanas-Roux F, Squifflet J, Berliere M, Donnez J. Immunohistochemical study of the proliferation index, oestrogen receptors and progesterone receptors A and B in leiomyomata and normal myometrium during the menstrual cycle and under gonadotrophin-releasing hormone agonist therapy. Hum Reprod 1999;14:2844–50. [DOI] [PubMed] [Google Scholar]
- [140].Molinari C, Battaglia A, Grossini E, Mary DA, Stoker JB, Surico N, et al. The effect of progesterone on coronary blood flow in anaesthesized pigs. Exp Physiol 2001;86:101–8. [DOI] [PubMed] [Google Scholar]
- [141].Rupnow HL, Phernetton TM, Shaw CE, Modrick ML, Bird IM, Magness RR. Endothelial vasodilator production by uterine and systemic arteries. VII. Estrogen and progesterone effects on eNOS. Am J Physiol Heart Circ Physiol 2001;280:H1699–705. [DOI] [PubMed] [Google Scholar]
- [142].Selles J, Polini N, Alvarez C, Massheimer V. Progesterone and 17 beta-estradiol acutely stimulate nitric oxide synthase activity in rat aorta and inhibit platelet aggregation. Life Sci 2001;69:815–27. [DOI] [PubMed] [Google Scholar]
- [143].Pieber D, Allport VC, Hills F, Johnson M, Bennett PR. Interactions between progesterone receptor isoforms in myometrial cells in human labour. Mol Hum Reprod 2001;7:875–9. [DOI] [PubMed] [Google Scholar]
- [144].Herkert O, Kuhl H, Busse R, Schini-Kerth VB. The progestin levonorgestrel induces endothelium-independent relaxation of rabbit jugular vein via inhibition of calcium entry and protein kinase C: role of cyclic AMP. Br J Pharmacol 2000;130:1911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Cox MW, Fu W, Chai H, Paladugu R, Lin PH, Lumsden AB, et al. Effects of progesterone and estrogen on endothelial dysfunction in porcine coronary arteries. J Surg Res 2005;124:104–11. [DOI] [PubMed] [Google Scholar]
- [146].Wassmann K, Wassmann S, Nickenig G. Progesterone antagonizes the vasoprotective effect of estrogen on antioxidant enzyme expression and function. Circ Res 2005;97:1046–54. [DOI] [PubMed] [Google Scholar]
- [147].Nickenig G, Strehlow K, Wassmann S, Baumer AT, Albory K, Sauer H, et al. Differential effects of estrogen and progesterone on AT(1) receptor gene expression in vascular smooth muscle cells. Circulation 2000;102:1828–33. [DOI] [PubMed] [Google Scholar]
- [148].Mueck AO, Seeger H. Progestogens and target tissues: vascular systems. Maturitas 2009;62:356–61. [DOI] [PubMed] [Google Scholar]
- [149].Seeger H, Mueck AO, Teichmann AT, Lippert TH. Effect of sequential estrogen/progestin treatment on biochemical vasoactive markers in postmenopausal women comparing oral and transdermal application. Clin Exp Obstet Gynecol 2000;27:17–20. [PubMed] [Google Scholar]
- [150].Viinikka L, Orpana A, Puolakka J, Pyorala T, Ylikorkala O. Different effects of oral and transdermal hormonal replacement on prostacyclin and thromboxane A2. Obstet Gynecol 1997;89:104–7. [DOI] [PubMed] [Google Scholar]
- [151].Preston RA, White WB, Pitt B, Bakris G, Norris PM, Hanes V. Effects of drospirenone/17-beta estradiol on blood pressure and potassium balance in hypertensive postmenopausal women. Am J Hypertens 2005;18:797–804. [DOI] [PubMed] [Google Scholar]
- [152].Coksuer H, Koplay M, Oghan F, Coksuer C, Keskin N, Ozveren O. Effects of estradiol-drospirenone hormone treatment on carotid artery intima-media thickness and vertigo/dizziness in postmenopausal women. Arch Gynecol Obstet 2011;283:1045–51. [DOI] [PubMed] [Google Scholar]
- [153].Koh KK, Jin DK, Yang SH, Lee SK, Hwang HY, Kang MH, et al. Vascular effects of synthetic or natural progestagen combined with conjugated equine estrogen in healthy postmenopausal women. Circulation 2001;103:1961–6. [DOI] [PubMed] [Google Scholar]
- [154].Clarkson TB, Anthony MS, Wagner JD. A comparison of tibolone and conjugated equine estrogens effects on coronary artery atherosclerosis and bone density of postmenopausal monkeys. J Clin Endocrinol Metab 2001;86:5396–404. [DOI] [PubMed] [Google Scholar]
- [155].Williams JK, Anthony MS, Honore EK, Herrington DM, Morgan TM, Register TC, et al. Regression of atherosclerosis in female monkeys. Arterioscler Thromb Vasc Biol 1995;15:827–36. [DOI] [PubMed] [Google Scholar]
- [156].Levine RL, Chen SJ, Durand J, Chen YF, Oparil S. Medroxyprogesterone attenuates estrogen-mediated inhibition of neointima formation after balloon injury of the rat carotid artery. Circulation 1996;94:2221–7. [DOI] [PubMed] [Google Scholar]
- [157].Reckelhoff JF, Yanes LL, Iliescu R, Fortepiani LA, Granger JP. Testosterone supplementation in aging men and women: possible impact on cardiovascular-renal disease. Am J Physiol Renal Physiol 2005;289:F941–8. [DOI] [PubMed] [Google Scholar]
- [158].Serock MR, Wells AK, Khalil RA. Modulators of vascular sex hormone receptors and their effects in estrogen-deficiency states associated with menopause. Recent Pat Cardiovasc Drug Discov 2008;3:165–86. [DOI] [PubMed] [Google Scholar]
- [159].Jiroutek MR, Chen MH, Johnston CC, Longcope C. Changes in reproductive hormones and sex hormone-binding globulin in a group of postmenopausal women measured over 10 years. Menopause 1998;5:90–4. [PubMed] [Google Scholar]
- [160].Higashiura K, Mathur RS, Halushka PV. Gender-related differences in androgen regulation of thromboxane A2 receptors in rat aortic smooth-muscle cells. J Cardiovasc Pharmacol 1997;29:311–5. [DOI] [PubMed] [Google Scholar]
- [161].Wynne FL, Khalil RA. Testosterone and coronary vascular tone: implications in coronary artery disease. J Endocrinol Invest 2003;26:181–6. [DOI] [PubMed] [Google Scholar]
- [162].Rubio-Gayosso I, Garcia-Ramirez O, Gutierrez-Serdan R, Guevara-Balcazar G, Munoz-Garcia O, Morato-Cartajena T, et al. Testosterone inhibits bradykinin-induced intracellular calcium kinetics in rat aortic endothelial cells in culture. Steroids 2002;67:393–7. [DOI] [PubMed] [Google Scholar]
- [163].Ba ZF, Wang P, Kuebler JF, Rue LW 3rd, Bland KI, Chaudry IH Flutamide induces relaxation in large and small blood vessels. Arch Surg 2002;137:1180–6. [DOI] [PubMed] [Google Scholar]
- [164].English KM, Steeds RP, Jones TH, Diver MJ, Channer KS. Low-dose transdermal testosterone therapy improves angina threshold in men with chronic stable angina: A randomized, double-blind, placebo-controlled study. Circulation 2000;102:1906–11. [DOI] [PubMed] [Google Scholar]
- [165].Khatibi A, Agardh CD, Shakir YA, Nerbrand C, Nyberg P, Lidfeldt J, et al. Could androgens protect middle-aged women from cardiovascular events? A population-based study of Swedish women: The Women’s Health in the Lund Area (WHILA) Study. Climacteric 2007;10:386–92. [DOI] [PubMed] [Google Scholar]
- [166].Rosano GM, Leonardo F, Pagnotta P, Pelliccia F, Panina G, Cerquetani E, et al. Acute anti-ischemic effect of testosterone in men with coronary artery disease. Circulation 1999;99:1666–70. [DOI] [PubMed] [Google Scholar]
- [167].Montalcini T, Gorgone G, Gazzaruso C, Sesti G, Perticone F, Pujia A. Endogenous testosterone and endothelial function in postmenopausal women. Coron Artery Dis 2007;18:9–13. [DOI] [PubMed] [Google Scholar]
- [168].Kloner RA, Carson C 3rd, Dobs A, Kopecky S, Mohler ER 3rd. Testosterone and Cardiovascular Disease. J Am Coll Cardiol 2016;67:545–57. [DOI] [PubMed] [Google Scholar]
- [169].Snyder PJ, Bhasin S, Cunningham GR, Matsumoto AM, Stephens-Shields AJ, Cauley JA, et al. Lessons From the Testosterone Trials. Endocr Rev 2018;39:369–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Yue P, Chatterjee K, Beale C, Poole-Wilson PA, Collins P. Testosterone relaxes rabbit coronary arteries and aorta. Circulation 1995;91:1154–60. [DOI] [PubMed] [Google Scholar]
- [171].Chou TM, Sudhir K, Hutchison SJ, Ko E, Amidon TM, Collins P, et al. Testosterone induces dilation of canine coronary conductance and resistance arteries in vivo. Circulation 1996;94:2614–9. [DOI] [PubMed] [Google Scholar]
- [172].Costarella CE, Stallone JN, Rutecki GW, Whittier FC. Testosterone causes direct relaxation of rat thoracic aorta. J Pharmacol Exp Ther 1996;277:34–9. [PubMed] [Google Scholar]
- [173].Honda H, Unemoto T, Kogo H. Different mechanisms for testosterone-induced relaxation of aorta between normotensive and spontaneously hypertensive rats. Hypertension 1999;34:1232–6. [DOI] [PubMed] [Google Scholar]
- [174].Somjen D, Kohen F, Jaffe A, Amir-Zaltsman Y, Knoll E, Stern N. Effects of gonadal steroids and their antagonists on DNA synthesis in human vascular cells. Hypertension 1998;32:39–45. [DOI] [PubMed] [Google Scholar]
- [175].Gonzales RJ, Ansar S, Duckles SP, Krause DN. Androgenic/estrogenic balance in the male rat cerebral circulation: metabolic enzymes and sex steroid receptors. J Cereb Blood Flow Metab 2007;27:1841–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Wild S, Pierpoint T, McKeigue P, Jacobs H. Cardiovascular disease in women with polycystic ovary syndrome at long-term follow-up: a retrospective cohort study. Clin Endocrinol (Oxf) 2000;52:595–600. [DOI] [PubMed] [Google Scholar]
- [177].Bairey Merz CN, Dember LM, Ingelfinger JR, Vinson A, Neugarten J, Sandberg KL, et al. Sex and the kidneys: current understanding and research opportunities. Nat Rev Nephrol 2019;15:776–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Gillams K, Juliebo-Jones P, Juliebo SO, Somani BK. Gender Differences in Kidney Stone Disease (KSD): Findings from a Systematic Review. Curr Urol Rep 2021;22:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Neugarten J, Golestaneh L. Sex Differences in Acute Kidney Injury. Semin Nephrol 2022;42:208–18. [DOI] [PubMed] [Google Scholar]
- [180].Ciarambino T, Crispino P, Giordano M. Gender and Renal Insufficiency: Opportunities for Their Therapeutic Management? Cells 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Hosszu A, Fekete A, Szabo AJ. Sex differences in renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 2020;319:F149–F54. [DOI] [PubMed] [Google Scholar]
- [182].Collins AJ, Foley RN, Chavers B, Gilbertson D, Herzog C, Johansen K, et al. ‘United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. Am J Kidney Dis 2012;59:A7, e1–420. [DOI] [PubMed] [Google Scholar]
- [183].Wyld MLR, Mata NL, Viecelli A, Swaminathan R, O’Sullivan KM, O’Lone E, et al. Sex-Based Differences in Risk Factors and Complications of Chronic Kidney Disease. Semin Nephrol 2022;42:153–69. [DOI] [PubMed] [Google Scholar]
- [184].Garcia GG, Iyengar A, Kaze F, Kierans C, Padilla-Altamira C, Luyckx VA. Sex and gender differences in chronic kidney disease and access to care around the globe. Semin Nephrol 2022;42:101–13. [DOI] [PubMed] [Google Scholar]
- [185].Carrero JJ, Hecking M, Chesnaye NC, Jager KJ. Sex and gender disparities in the epidemiology and outcomes of chronic kidney disease. Nat Rev Nephrol 2018;14:151–64. [DOI] [PubMed] [Google Scholar]
- [186].Swartling O, Rydell H, Stendahl M, Segelmark M, Trolle Lagerros Y, Evans M. CKD Progression and Mortality Among Men and Women: A Nationwide Study in Sweden. Am J Kidney Dis 2021;78:190–9 e1. [DOI] [PubMed] [Google Scholar]
- [187].Silbiger SR, Neugarten J. The role of gender in the progression of renal disease. Adv Ren Replace Ther 2003;10:3–14. [DOI] [PubMed] [Google Scholar]
- [188].Maric C, Sandberg K, Hinojosa-Laborde C. Glomerulosclerosis and tubulointerstitial fibrosis are attenuated with 17beta-estradiol in the aging Dahl salt sensitive rat. J Am Soc Nephrol 2004;15:1546–56. [DOI] [PubMed] [Google Scholar]
- [189].Zellweger R, Wichmann MW, Ayala A, Stein S, DeMaso CM, Chaudry IH. Females in proestrus state maintain splenic immune functions and tolerate sepsis better than males. Crit Care Med 1997;25:106–10. [DOI] [PubMed] [Google Scholar]
- [190].Acharya KD, Gao X, Bless EP, Chen J, Tetel MJ. Estradiol and high fat diet associate with changes in gut microbiota in female ob/ob mice. Sci Rep 2019;9:20192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Mueller S, Saunier K, Hanisch C, Norin E, Alm L, Midtvedt T, et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol 2006;72:1027–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity 2013;39:400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013;339:1084–8. [DOI] [PubMed] [Google Scholar]
- [194].Kozik AJ, Nakatsu CH, Chun H, Jones-Hall YL. Age, sex, and TNF associated differences in the gut microbiota of mice and their impact on acute TNBS colitis. Exp Mol Pathol 2017;103:311–9. [DOI] [PubMed] [Google Scholar]
- [195].Faith JJ, McNulty NP, Rey FE, Gordon JI. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 2011;333:101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Oustamanolakis P, Tack J. Dyspepsia: organic versus functional. J Clin Gastroenterol 2012;46:175–90. [DOI] [PubMed] [Google Scholar]
- [197].Narayanan SP, Anderson B, Bharucha AE. Sex- and Gender-Related Differences in Common Functional Gastroenterologic Disorders. Mayo Clin Proc 2021;96:1071–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Kim YS, Kim N. Functional Dyspepsia: A Narrative Review With a Focus on Sex-Gender Differences. J Neurogastroenterol Motil 2020;26:322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Kim YS, Kim N. Sex-Gender Differences in Irritable Bowel Syndrome. J Neurogastroenterol Motil 2018;24:544–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Drossman DA, Li Z, Andruzzi E, Temple RD, Talley NJ, Thompson WG, et al. U.S. householder survey of functional gastrointestinal disorders. Prevalence, sociodemography, and health impact. Dig Dis Sci 1993;38:1569–80. [DOI] [PubMed] [Google Scholar]
- [201].Jiang Y, Greenwood-Van Meerveld B, Johnson AC, Travagli RA. Role of estrogen and stress on the brain-gut axis. Am J Physiol Gastrointest Liver Physiol 2019;317:G203–G9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Brettle H, Tran V, Drummond GR, Franks AE, Petrovski S, Vinh A, et al. Sex hormones, intestinal inflammation, and the gut microbiome: Major influencers of the sexual dimorphisms in obesity. Front Immunol 2022;13:971048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Jelavic MM, Babic Z, Pintaric H. The importance of two metabolic syndrome diagnostic criteria and body fat distribution in predicting clinical severity and prognosis of acute myocardial infarction. Arch Med Sci 2017;13:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Ladeiras-Lopes R, Sampaio F, Bettencourt N, Fontes-Carvalho R, Ferreira N, Leite-Moreira A, et al. The Ratio Between Visceral and Subcutaneous Abdominal Fat Assessed by Computed Tomography Is an Independent Predictor of Mortality and Cardiac Events. Rev Esp Cardiol (Engl Ed) 2017;70:331–7. [DOI] [PubMed] [Google Scholar]
- [205].Pettersson US, Walden TB, Carlsson PO, Jansson L, Phillipson M. Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS One 2012;7:e46057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Christensen RH, von Scholten BJ, Hansen CS, Jensen MT, Vilsboll T, Rossing P, et al. Epicardial adipose tissue predicts incident cardiovascular disease and mortality in patients with type 2 diabetes. Cardiovasc Diabetol 2019;18:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Varlamov O, Bethea CL, Roberts CT Jr. Sex-specific differences in lipid and glucose metabolism. Front Endocrinol (Lausanne) 2014;5:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Nuutila P, Knuuti MJ, Maki M, Laine H, Ruotsalainen U, Teras M, et al. Gender and insulin sensitivity in the heart and in skeletal muscles. Studies using positron emission tomography. Diabetes 1995;44:31–6. [DOI] [PubMed] [Google Scholar]
- [209].Frias JP, Macaraeg GB, Ofrecio J, Yu JG, Olefsky JM, Kruszynska YT. Decreased susceptibility to fatty acid-induced peripheral tissue insulin resistance in women. Diabetes 2001;50:1344–50. [DOI] [PubMed] [Google Scholar]
- [210].Tramunt B, Smati S, Grandgeorge N, Lenfant F, Arnal JF, Montagner A, et al. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2020;63:453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Ostman J, Lonnberg G, Arnqvist HJ, Blohme G, Bolinder J, Ekbom Schnell A, et al. Gender differences and temporal variation in the incidence of type 1 diabetes: results of 8012 cases in the nationwide Diabetes Incidence Study in Sweden 1983–2002. J Intern Med 2008;263:386–94. [DOI] [PubMed] [Google Scholar]
- [212].Mauvais-Jarvis F Gender differences in glucose homeostasis and diabetes. Physiol Behav 2018;187:20–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Mendola ND, Chen TC, Gu Q, Eberhardt MS, Saydah S. Prevalence of Total, Diagnosed, and Undiagnosed Diabetes Among Adults: United States, 2013–2016. NCHS Data Brief 2018:1–8. [PubMed] [Google Scholar]
- [214].Lauretta R, Sansone M, Sansone A, Romanelli F, Appetecchia M. Gender in Endocrine Diseases: Role of Sex Gonadal Hormones. Int J Endocrinol 2018;2018:4847376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Gao X, Yamazaki Y, Tezuka Y, Omata K, Ono Y, Morimoto R, et al. Gender differences in human adrenal cortex and its disorders. Mol Cell Endocrinol 2021;526:111177. [DOI] [PubMed] [Google Scholar]
- [216].Liu X, Zhu X, Zeng M, Zhuang Y, Zhou Y, Zhang Z, et al. Gender-Specific Differences in Clinical Profile and Biochemical Parameters in Patients with Cushing’s Disease: A Single Center Experience. Int J Endocrinol 2015;2015:949620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Broersen LHA, van Haalen FM, Kienitz T, Biermasz NR, Strasburger CJ, Dekkers OM, et al. Sex Differences in Presentation but Not in Outcome for ACTH-Dependent Cushing’s Syndrome. Front Endocrinol (Lausanne) 2019;10:580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Zilio M, Barbot M, Ceccato F, Camozzi V, Bilora F, Casonato A, et al. Diagnosis and complications of Cushing’s disease: gender-related differences. Clin Endocrinol (Oxf) 2014;80:403–10. [DOI] [PubMed] [Google Scholar]
- [219].Pecori Giraldi F, Moro M, Cavagnini F. Gender-related differences in the presentation and course of Cushing’s disease. J Clin Endocrinol Metab 2003;88:1554–8. [DOI] [PubMed] [Google Scholar]
- [220].Bauer M, Glenn T, Pilhatsch M, Pfennig A, Whybrow PC. Gender differences in thyroid system function: relevance to bipolar disorder and its treatment. Bipolar Disord 2014;16:58–71. [DOI] [PubMed] [Google Scholar]
- [221].Mulder JE. Thyroid disease in women. Med Clin North Am 1998;82:103–25. [DOI] [PubMed] [Google Scholar]
- [222].Eckenrode HE, Carwie JC, Curtis LM. Does Gender Affirming Hormone Therapy Increase the Risk of Kidney Disease? Semin Nephrol 2022;42:151284. [DOI] [PubMed] [Google Scholar]
- [223].Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol 2016;16:626–38. [DOI] [PubMed] [Google Scholar]
- [224].Maleki Dana P, Sadoughi F, Hallajzadeh J, Asemi Z, Mansournia MA, Yousefi B, et al. An Insight into the Sex Differences in COVID-19 Patients: What are the Possible Causes? Prehosp Disaster Med 2020;35:438–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Roved J, Westerdahl H, Hasselquist D. Sex differences in immune responses: Hormonal effects, antagonistic selection, and evolutionary consequences. Horm Behav 2017;88:95–105. [DOI] [PubMed] [Google Scholar]
- [226].Jensen-Jarolim E, Untersmayr E. Gender-medicine aspects in allergology. Allergy 2008;63:610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Chowdhury NU, Guntur VP, Newcomb DC, Wechsler ME. Sex and gender in asthma. Eur Respir Rev 2021;30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Fuseini H, Newcomb DC. Mechanisms Driving Gender Differences in Asthma. Curr Allergy Asthma Rep 2017;17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Nguyen NT, Chinn J, De Ferrante M, Kirby KA, Hohmann SF, Amin A. Male gender is a predictor of higher mortality in hospitalized adults with COVID-19. PLoS One 2021;16:e0254066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].El Aidaoui K, Ait Benhamou R, Haoudar A, Ziati J, Kantri A, Agrad K, et al. Sex Differences in COVID-19 Outcomes. Cureus 2022;14:e25760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Pozzilli P, Lenzi A. Commentary: Testosterone, a key hormone in the context of COVID-19 pandemic. Metabolism 2020;108:154252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Kopel J, Perisetti A, Roghani A, Aziz M, Gajendran M, Goyal H. Racial and Gender-Based Differences in COVID-19. Front Public Health 2020;8:418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Stelzig KE, Canepa-Escaro F, Schiliro M, Berdnikovs S, Prakash YS, Chiarella SE. Estrogen regulates the expression of SARS-CoV-2 receptor ACE2 in differentiated airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 2020;318:L1280–L1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Liu J, Ji H, Zheng W, Wu X, Zhu JJ, Arnold AP, et al. Sex differences in renal angiotensin converting enzyme 2 (ACE2) activity are 17beta-oestradiol-dependent and sex chromosome-independent. Biol Sex Differ 2010;1:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Bangasser DA, Wiersielis KR. Sex differences in stress responses: a critical role for corticotropin-releasing factor. Hormones (Athens) 2018;17:5–13. [DOI] [PubMed] [Google Scholar]
- [236].Kessler RC, McGonagle KA, Swartz M, Blazer DG, Nelson CB. Sex and depression in the National Comorbidity Survey. I: Lifetime prevalence, chronicity and recurrence. J Affect Disord 1993;29:85–96. [DOI] [PubMed] [Google Scholar]
- [237].Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S, et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry 1994;51:8–19. [DOI] [PubMed] [Google Scholar]
- [238].Borrow AP, Handa RJ. Estrogen Receptors Modulation of Anxiety-Like Behavior. Vitam Horm 2017;103:27–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Choleris E, Galea LAM, Sohrabji F, Frick KM. Sex differences in the brain: Implications for behavioral and biomedical research. Neurosci Biobehav Rev 2018;85:126–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Dhingra H, Choudhari SG. Alzheimer’s Disease: Understanding Its Novel Drug Delivery Systems and Treatments. Cureus 2022;14:e31394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Hara Y, Waters EM, McEwen BS, Morrison JH. Estrogen Effects on Cognitive and Synaptic Health Over the Lifecourse. Physiol Rev 2015;95:785–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Vajaria R, Vasudevan N. Is the membrane estrogen receptor, GPER1, a promiscuous receptor that modulates nuclear estrogen receptor-mediated functions in the brain? Horm Behav 2018;104:165–72. [DOI] [PubMed] [Google Scholar]
- [243].Kuruca SE, Karadenizli S, Akgun-Dar K, Kapucu A, Kaptan Z, Uzum G. The effects of 17beta-estradiol on blood brain barrier integrity in the absence of the estrogen receptor alpha; an in-vitro model. Acta Histochem 2017;119:638–47. [DOI] [PubMed] [Google Scholar]
- [244].Tecalco-Cruz AC, Zepeda-Cervantes J, Ortega-Dominguez B. Estrogenic hormones receptors in Alzheimer’s disease. Mol Biol Rep 2021;48:7517–26. [DOI] [PubMed] [Google Scholar]
- [245].Scheyer O, Rahman A, Hristov H, Berkowitz C, Isaacson RS, Diaz Brinton R, et al. Female Sex and Alzheimer’s Risk: The Menopause Connection. J Prev Alzheimers Dis 2018;5:225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Schmidt R, Kienbacher E, Benke T, Dal-Bianco P, Delazer M, Ladurner G, et al. [Sex differences in Alzheimer’s disease]. Neuropsychiatr 2008;22:1–15. [PubMed] [Google Scholar]
- [247].Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s Disease-Associated beta-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018;100:1527–32. [DOI] [PubMed] [Google Scholar]
- [248].Samulowitz A, Gremyr I, Eriksson E, Hensing G. “Brave Men” and “Emotional Women”: A Theory-Guided Literature Review on Gender Bias in Health Care and Gendered Norms towards Patients with Chronic Pain. Pain Res Manag 2018;2018:6358624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Cabanero D, Villalba-Riquelme E, Fernandez-Ballester G, Fernandez-Carvajal A, Ferrer-Montiel A. ThermoTRP channels in pain sexual dimorphism: new insights for drug intervention. Pharmacol Ther 2022;240:108297. [DOI] [PubMed] [Google Scholar]
- [250].Hellstrom B, Anderberg UM. Pain perception across the menstrual cycle phases in women with chronic pain. Percept Mot Skills 2003;96:201–11. [DOI] [PubMed] [Google Scholar]
- [251].Seol SH, Chung G. Estrogen-dependent regulation of transient receptor potential vanilloid 1 (TRPV1) and P2X purinoceptor 3 (P2X3): Implication in burning mouth syndrome. J Dent Sci 2022;17:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Martinez-Lavin M Fibromyalgia in women: somatisation or stress-evoked, sex-dimorphic neuropathic pain? Clin Exp Rheumatol 2021;39:422–5. [PubMed] [Google Scholar]
- [253].Conversano C, Ciacchini R, Orru G, Bazzichi ML, Gemignani A, Miniati M. Gender differences on psychological factors in fibromyalgia: a systematic review on the male experience. Clin Exp Rheumatol 2021;39 Suppl 130:174–85. [DOI] [PubMed] [Google Scholar]
- [254].Yunus MB. Gender differences in fibromyalgia and other related syndromes. J Gend Specif Med 2002;5:42–7. [PubMed] [Google Scholar]
- [255].Wiesenfeld-Hallin Z Sex differences in pain perception. Gend Med 2005;2:137–45. [DOI] [PubMed] [Google Scholar]
- [256].Becker JB, McClellan ML, Reed BG. Sex differences, gender and addiction. J Neurosci Res 2017;95:136–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Nicolas C, Zlebnik NE, Farokhnia M, Leggio L, Ikemoto S, Shaham Y. Sex Differences in Opioid and Psychostimulant Craving and Relapse: A Critical Review. Pharmacol Rev 2022;74:119–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Quigley JA, Logsdon MK, Turner CA, Gonzalez IL, Leonardo NB, Becker JB. Sex differences in vulnerability to addiction. Neuropharmacology 2021;187:108491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].White AM. Gender Differences in the Epidemiology of Alcohol Use and Related Harms in the United States. Alcohol Res 2020;40:01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Kezer CA, Simonetto DA, Shah VH. Sex Differences in Alcohol Consumption and Alcohol-Associated Liver Disease. Mayo Clin Proc 2021;96:1006–16. [DOI] [PubMed] [Google Scholar]
- [261].Becker JB. Sex differences in addiction. Dialogues Clin Neurosci 2016;18:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Shah DA, Khalil RA. Bioactive factors in uteroplacental and systemic circulation link placental ischemia to generalized vascular dysfunction in hypertensive pregnancy and preeclampsia. Biochem Pharmacol 2015;95:211–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Uzan J, Carbonnel M, Piconne O, Asmar R, Ayoubi JM. Pre-eclampsia: pathophysiology, diagnosis, and management. Vasc Health Risk Manag 2011;7:467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Poon LC, Shennan A, Hyett JA, Kapur A, Hadar E, Divakar H, et al. The International Federation of Gynecology and Obstetrics (FIGO) initiative on pre-eclampsia: A pragmatic guide for first-trimester screening and prevention. Int J Gynaecol Obstet 2019;145 Suppl 1:1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Brown MA, Magee LA, Kenny LC, Karumanchi SA, McCarthy FP, Saito S, et al. The hypertensive disorders of pregnancy: ISSHP classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens 2018;13:291–310. [DOI] [PubMed] [Google Scholar]
- [266].Podymow T, August P. Hypertension in pregnancy. Adv Chronic Kidney Dis 2007;14:178–90. [DOI] [PubMed] [Google Scholar]
- [267].Roberts JM, Gammill HS. Preeclampsia: recent insights. Hypertension 2005;46:1243–9. [DOI] [PubMed] [Google Scholar]
- [268].Weinstein L Syndrome of hemolysis, elevated liver enzymes, and low platelet count: a severe consequence of hypertension in pregnancy. Am J Obstet Gynecol 1982;142:159–67. [DOI] [PubMed] [Google Scholar]
- [269].Qu H, Khalil RA. Vascular mechanisms and molecular targets in hypertensive pregnancy and preeclampsia. Am J Physiol Heart Circ Physiol 2020;319:H661–H81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Coats LE, Davis GK, Newsome AD, Ojeda NB, Alexander BT. Low Birth Weight, Blood Pressure and Renal Susceptibility. Curr Hypertens Rep 2019;21:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Sandovici I, Fernandez-Twinn DS, Hufnagel A, Constancia M, Ozanne SE. Sex differences in the intergenerational inheritance of metabolic traits. Nat Metab 2022;4:507–23. [DOI] [PubMed] [Google Scholar]
- [272].Dearden L, Bouret SG, Ozanne SE. Sex and gender differences in developmental programming of metabolism. Mol Metab 2018;15:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Sutherland S, Brunwasser SM. Sex Differences in Vulnerability to Prenatal Stress: a Review of the Recent Literature. Curr Psychiatry Rep 2018;20:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Bukowski R, Smith GC, Malone FD, Ball RH, Nyberg DA, Comstock CH, et al. Human sexual size dimorphism in early pregnancy. Am J Epidemiol 2007;165:1216–8. [DOI] [PubMed] [Google Scholar]
- [275].Galjaard S, Ameye L, Lees CC, Pexsters A, Bourne T, Timmerman D, et al. Sex differences in fetal growth and immediate birth outcomes in a low-risk Caucasian population. Biol Sex Differ 2019;10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Goldenberg RL, Andrews WW, Faye-Petersen OM, Goepfert AR, Cliver SP, Hauth JC. The Alabama Preterm Birth Study: intrauterine infection and placental histologic findings in preterm births of males and females less than 32 weeks. Am J Obstet Gynecol 2006;195:1533–7. [DOI] [PubMed] [Google Scholar]
- [277].Challis J, Newnham J, Petraglia F, Yeganegi M, Bocking A. Fetal sex and preterm birth. Placenta 2013;34:95–9. [DOI] [PubMed] [Google Scholar]
- [278].Bale TL. The placenta and neurodevelopment: sex differences in prenatal vulnerability. Dialogues Clin Neurosci 2016;18:459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Grether JK, Nelson KB, Walsh E, Willoughby RE, Redline RW. Intrauterine exposure to infection and risk of cerebral palsy in very preterm infants. Arch Pediatr Adolesc Med 2003;157:26–32. [DOI] [PubMed] [Google Scholar]
- [280].Arnold ML, Saijo K. Estrogen Receptor beta as a Candidate Regulator of Sex Differences in the Maternal Immune Activation Model of ASD. Front Mol Neurosci 2021;14:717411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Inkster AM, Fernandez-Boyano I, Robinson WP. Sex Differences Are Here to Stay: Relevance to Prenatal Care. J Clin Med 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Branstrom R, Andreasson S. Regional differences in alcohol consumption, alcohol addiction and drug use among Swedish adults. Scand J Public Health 2008;36:493–503. [DOI] [PubMed] [Google Scholar]
- [283].Buford TW. Hypertension and aging. Ageing Res Rev 2016;26:96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics−-2015 update: a report from the American Heart Association. Circulation 2015;131:e29–322. [DOI] [PubMed] [Google Scholar]
- [285].Zucker DR, Griffith JL, Beshansky JR, Selker HP. Presentations of acute myocardial infarction in men and women. J Gen Intern Med 1997;12:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Rexrode KM, Madsen TE, Yu AYX, Carcel C, Lichtman JH, Miller EC. The Impact of Sex and Gender on Stroke. Circ Res 2022;130:512–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Kovesdy CP. Epidemiology of chronic kidney disease: an update 2022. Kidney Int Suppl (2011) 2022;12:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Barbour KE, Helmick CG, Boring M, Brady TJ. Vital Signs: Prevalence of Doctor-Diagnosed Arthritis and Arthritis-Attributable Activity Limitation - United States, 2013–2015. MMWR Morb Mortal Wkly Rep 2017;66:246–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2019;18:88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]