

Abstract
We recently developed a screening system capable of identifying and evaluating inhibitors of the Hepatitis B virus (HBV) ribonuclease H (RNaseH), which is the only HBV enzyme not targeted by current anti-HBV therapies. Inhibiting the HBV RNaseH blocks synthesis of the positive-polarity DNA strand, causing early termination of negative-polarity DNA synthesis and accumulation of RNA:DNA heteroduplexes. We previously reported inhibition of HBV replication by N-hydroxyisoquinolinediones (HID) and N-hydroxypyridinediones (HPD) in human hepatoma cells. Here, we report results from our ongoing efforts to develop more potent anti-HBV RNaseH inhibitors in the HID/HPD compound classes. We synthesized and screened additional HIDs and HPDs for preferential suppression of positive-polarity DNA in cells replicating HBV. Three of seven new HIDs inhibited HBV replication, however, the therapeutic indexes (TI = CC50/EC50) did not improve over what we previously reported. All nine of the HPDs inhibited HBV replication with EC50s ranging from 110 nM to 4 μM. Cellular cytotoxicity was evaluated by four assays and CC50s ranged from 15 to > 100 μM. The best compounds have a calculated TI of >300, which is a 16-fold improvement over the primary HPD hit. These studies indicate that the HPD compound class holds potential for antiviral discovery.
Keywords: HBV, Ribonuclease H, N-hydroxyisoquinolinediones, N-hydroxypyridinediones, Antiviral, Structure-activity relationship
Introduction:
Hepatitis B virus (HBV) is a major global health problem. An estimated 257 million people are chronically infected worldwide, resulting in >880,000 deaths annually from HBV-associated liver diseases, including cirrhosis, hepatocellular carcinoma, and liver failure (WHO, 2017). HBV-associated health care costs are up to $39,898 per quality-adjusted life year (Xie et al., 2018). Available anti-HBV therapies are limited to two forms of the innate immunity cytokine interferon-α and six nucleoside/nucleotide analogs (NA) which block DNA chain elongation by the viral polymerase. These therapies significantly improve patient outcomes, and NA can suppress viremia by more than 4–5 log10 in the majority of patients (Cox and Tillmann, 2011; Kwon and Lok, 2011). However, HBV replication still persists in most patients, therefore these therapies must be continued indefinitely (Coffin et al., 2011; Zoulim, 2004). It is widely accepted that curing HBV will require new drugs against new targets capable of further suppressing HBV (Petersen et al., 2016). These new therapies will need to work together with other anti-HBV treatments, be well tolerated during prolonged use, and have a high genetic barrier to resistance.
We are exploring the HBV ribonuclease H (RNaseH) as an antiviral drug target. The RNaseH is one of two enzymatically active domains on the HBV polymerase that synthesizes the partially double-stranded DNA genome via reverse transcription. The reverse transcriptase (RT) domain of the polymerase protein copies the pregenomic RNA (pgRNA) template to form the minus-polarity DNA strand. The RNaseH recognizes RNA:DNA heteroduplexes that are formed during minus-polarity DNA synthesis and degrades the RNA strand (Seeger et al., 2013; Tavis and Badtke, 2009). The polymerase then synthesizes the positive-polarity DNA strand, but it typically arrests after making only ~50% of the plus-polarity DNA strand. Both enzymatic activities of the polymerase are required for synthesis of the HBV genome, yet the RNaseH is an unexploited drug target (Tavis and Lomonosova, 2015).
Blocking the HBV RNaseH activity prevents removal of the RNA strand from the minus–polarity DNA strand, resulting in an accumulation of RNA:DNA heteroduplexes (Hu et al., 2013). Failure to remove the pgRNA blocks synthesis of positive-polarity DNAs and causes premature arrest of minus-polarity DNA synthesis (Edwards et al., 2017; Gerelsaikhan et al., 1996; Hu et al., 2013; Tavis et al., 2013), with truncation of the minus-polarity DNAs possibly stemming from conformational constraints imposed on the viral DNA within capsids due to accumulation of the heteroduplexes. This prevents the formation of a complete viral genome, thus stopping both transmission of infectious viral particles and the intracellular “recycling” mechanism that replenishes the pool of covalently closed circular DNA (cccDNA), the template for all HBV RNAs.
We recently developed an HBV RNaseH screening pipeline (Cai et al., 2014; Edwards et al., 2017; Hu et al., 2013; Lomonosova et al., 2017a; Lomonosova et al., 2017b; Lu et al., 2015; Lu et al., 2016; Tavis et al., 2013; Tavis and Lomonosova, 2015). We identified >100 inhibitors primarily in three compound classes, the α-hydroxytropolones (αHT), N-hydroxyisoquinolinediones (HID), and N-hydroxypyridinediones (HPD), that suppress viral replication in cells by blocking the HBV RNaseH. These compounds preferentially suppress the plus-polarity DNA strands, induce truncation of the minus-polarity DNA strands, and cause accumulation of extensive RNA:DNA heteroduplexes in capsids as expected from their inhibition of the RNaseH. The best compounds have therapeutic indexes [cytotoxicity/efficacy (TI)] >200 (Lomonosova et al., 2017a). Representative HBV RNaseH inhibitors can work synergistically with approved and experimental therapies (Lomonosova et al., 2017b), inhibit multiple HBV isolates from genotypes B, C, and D equivalently (Lu et al., 2016), and suppress viral replication in a chimeric mouse model (Long et al., 2018). Therefore, the HBV RNaseH is a validated drug target.
We are pursuing a hit-to-lead medicinal chemistry campaign to optimize inhibitors of the HBV RNaseH. Here, we report the activity of seven new HIDs and nine new HPDs, with significant improvements in in vitro potency and cytotoxicity profiles for the HPD compounds.
2. Materials and methods
2.1. Compound acquisition and synthesis
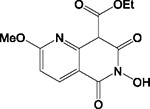
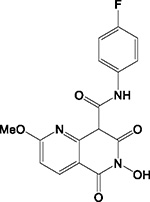
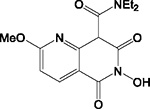
Compounds were synthesized by Arbutus Biopharma and all compounds were ≥95% pure. Compounds were dissolved in 100% DMSO and stored in amber tubes to protect them from light. All compounds were maintained in single-use aliquots and stored at −80°C until use. Compound structures and names are in Table 1.
Table 1:
Activity profile of HID and HPD compounds
| Compound # | Compound structure | Derivative series (a) | Activity vs. HBV RNaseH (b) | Activity vs. huRNaseH1 (b) | Solubility (μM) | HBV replication inhibition (+) DNA (EC50, μM) | HBV replication inhibition (−) DNA (EC50, μM) | Cellular cytotoxicity (CC50, μM) (c) | TI | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular beacon | Oligonucleotide - directed cleavage | Molecular beacon | Oligonucleotide - directed cleavage | ||||||||
| A10 |

|
HID | - | - | - | ND (e) | >200 | - | - | >100 | N.A. (f) |
| A11 |
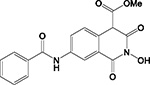
|
HID | IND (d) | - | + | ND | >200 | 3.3 ± 0.4 | 38 ± 9 | >100 | >30 |
| A12 |
|
HID | + | + | + | ND | >200 | - | - | 40 ± 8 | N.A. |
| A18 |
|
HID | - | + | - | ND | 50 | - | - | 24 ± 20 | N.A. |
| A19 |
|
HID | + | + | + | + | >200 | 3.8 ± 1.7 | >50 | 100 ± 0.2 | 26 |
| A20 |
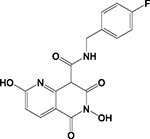
|
HID | - | + | - | ND | >200 | 2.4 ± 0.1 | >50 | 93 ± 9 | 39 |
| A21 |
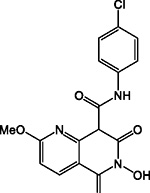
|
HID | - | + | - | ND | 50 | - | - | 18 ± 10 | N.A. |
| A13 |
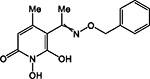
|
HPD | + | + | + | ND | >200 | 0.54 ± 0.3 | 56 ± 8 | 85 ± 15 | 157 |
| A14 |
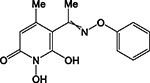
|
HPD | IND | + | IND | + | >200 | 2.1 ± 0.4 | 57 ± 4 | 81 ± 13 | 39 |
| A15 |

|
HPD | + | + | IND | - | >200 | 0.46 ± 0.1 | 9.8 ± 3 | 71 ± 17 | 154 |
| A16 |

|
HPD | + | + | + | ND | >200 | 4.0 ± 1.9 | >50 | 62 ± 15 | 16 |
| A17 |
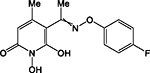
|
HPD | + | + | + | ND | >200 | 0.95 ± 0.2 | >50 | 15 ± 7 | 16 |
| A22 |

|
HPD | IND | + | + | ND | >200 | 0.32 ± 0.2 | 32 ± 17 | 90 ± 6 | 281 |
| A23 |

|
HPD | + | + | + | ND | >200 | 0.11 ± 0.01 | 10 ± 2 | 33 ± 8 | 300 |
| A24 |
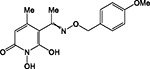
|
HPD | + | + | + | ND | >200 | 0.29 ± 0.1 | 31 ± 16 | 102 ± 3 | 352 |
| A25 |
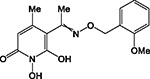
|
HPD | + | + | + | ND | >200 | 0.5 ± 0.1 | 30 ± 12 | 91 ± 16 | 182 |
N-hydroxyisoquinolinedione (HID) and N-hydroxypyridinediones (HPD)
(+) Detectable inhibition at or below 100 μM; (−) No inhibition detected at 100 μM
CellTiter 96™ AQueous Non-Radioactive Cell Proliferation Assay (MTS)
Indeterminante, (IND)
Not determined (ND)
Not applicable, (N.A.)
2.2. Expression and purification of HBV RNaseH and huRNaseH1
HBV RNaseH with an N-terminal SMT tag was expressed in Escherichia coli and purified by nickel affinity-chromatography as described previously (Villa et al., 2016). The human ribonuclease H1 (huRNaseH1) was expressed in Escherichia coli and purified by nickel affinity-chromatography as described previously (Villa et al., 2016).
2.3. Cells and cell culture
HepDES19 and HepDE19 cells are HepG2 (human hepatoblastoma) cell line derivatives stably transfected with an HBV genotype D genome under the control of a tetracycline-repressible promoter (Guo et al., 2007). The HepBHAe82 cell line is a HepG2 cell line derivative that has an in-frame HA epitope tag in the N-terminal coding sequence of HBV e antigen (HBeAg) in a transgene that does not disrupt any cis-elements critical for HBV replication, cccDNA transcription, and HA-HBeAg secretion (Cai et al., 2016). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) with 1 μg/mL tetracycline. Synchronized expression of HBV pgRNA was induced by removing tetracycline from the culture medium.
2.4. HBV q-PCR replication inhibition assay
HepDES19 or HepDE19 cells were seeded in 96-well plates at 4 × 104 cells per well in the absence of tetracycline. Test compounds in a final DMSO concentration of 1% were applied to cells 48 hours after induction of HBV replication and cells were incubated with compounds for 72 hours. Cells were washed in 200 μL of phosphate buffered saline (PBS) and lysed in 150 μL of core lysis buffer (10 mM Tris pH 7.4, 1% Tween20, 150 mM NaCl). Cells were incubated at room temperate on an orbital shaker at 350 rpm for 40 minutes. Cell lysate was transferred to a 96 well polymerase chain reaction (PCR) plate, and centrifuged at 3300 × g for 5 minutes. The supernatant (50 μL) from the cell lysate was transferred to a 96-well PCR plate and mixed with 20 units of micrococcal nuclease and 100 μM CaCl2. The lysate was incubated for 1 hour at 37°C, and then the nuclease was inactivated at 70°C for ten minutes. Qiagen protease (0.005 Anson units) was added to the lysate and the mixture was incubated overnight. Qiagen protease was then inactivated at 95°C for ten minutes.
The crude lysate was used as the template for strand-preferential quantitative polymerase chain reaction (q-PCR) analysis. Quantitative PCR was performed with 40 cycles of 95 °C for 15 s and 60 °C for 1 minute employing the Kappa Probe Force universal PCR master mix. The primers and probe (IDT Inc.) for the plus-polarity DNA strand were 5’CATGAACAAGAGATGATTAGGCAGAG3’, 5’GGAGGCTGTAGGCATAAATTGG3’, and 5’/56-FAM/CTGCGCACC/ZEN/AGCACCATGCA/3IABkFQ. The primers and probe for the minus-polarity DNA strand were 5’GCAGATGAGAAGGCACAGA3’, 5’CTTCTCCGTCTGCCGTT3’, and 5’/56-FAM/AGTCCGCGT/ZEN/AAAGAGAGGTGCG/3IABkFQ. EC50 values were calculated from the plus-polarity DNA data with GraphPad Prism using the three-parameter log(inhibitor)-versus-response algorithm with the bottom value set to zero.
2.5. Cytotoxicity assays
The effects of compounds on cell viability in HepDES19 or HepDE19 cells was assessed using the CellTiter 96™ AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) (MTS) as previously described (Edwards et al., 2017). Cells were seeded into a 96 well plate at 1 × 104 cells per well. The compounds were applied to cells 48 hours after the removal of tetracycline from the cell culture media and the HBV-expressing cells were incubated for 72 hours in the presence of compound containing media. Fifty percent cytotoxic concentration (CC50) values were calculated with GraphPad Prism using the three-parameter variable-response log(inhibitor)-versus-response algorithm with the bottom value set to zero.
Active compounds were further evaluated in an expanded cytotoxicity panel. HepDES19 cells were seeded at 1 × 104 cells per well and HBV expression was synchronously induced. Cells were incubated in the presence of compound for 72 hours. Lysosomal function was tested using the neutral red (NR) uptake assay (Edwards et al., 2017; Repetto et al., 2008). Lactate dehydrogenase (LDH) release was measured using the LDH-cytotoxicity colorimetric assay kit (BioVision) as previously reported (Edwards et al., 2017). Fifty percent cytotoxic concentration (CC50) values were calculated with GraphPad Prism using the three-parameter variable-response log(inhibitor)-versus-response algorithm with the bottom value set to zero.
The effect of select compounds on cell viability in HepDE19 cells was assessed using replicate plates, with cells plated at a density of 5,000 cells/well and incubated for 4 days, to determine the ATP content as a measure of cell viability using the CellTiter-Glo™ reagent (CTG; Promega, Madison, WI) per manufacturer’s instructions. The plates were read using a Victor luminescence plate reader (PerkinElmer Model 1420 Multilabel counter) and the relative luminescence units (RLU) data generated from each well was calculated as percent inhibition of the untreated control wells and analyzed using XL-Fit module in Microsoft Excel to determine the CC50 (CTG) values using a four-parameter curve fitting algorithm.
The effect of select compounds on cell viability and proliferation in HepBHAe82 cells was assessed using replicate plates seeded at 10–20 % cell density that, after 4 days, were assayed for intracellular ATP content using the Cell-Titer Glo reagent (Promega, Madison, WI) per manufacturer’s instructions and plates were read using a Victor luminescence plate reader (Model 1420 Multilabel counter; PerkinElmer, Waltham, MA). Cell viability was calculated as a percentage of the untreated negative control wells. The relative luminescence units (RLU) data generated from each well was calculated as % inhibition of the untreated control wells and analyzed using XL-Fit module in Microsoft Excel to determine EC50 (HA-HBeAg AlphaLISA) and CC50 (CTG) values using a 4-parameter curve fitting algorithm.
2.6. RNaseH assays
Qualitative inhibition of the HBV RNaseH and huRNaseH1 in the biochemical assays was assessed using a molecular beacon assay as described previously (Edwards et al., 2017). HBV RNaseH (2.1 μg) was incubated with 25 nM of a RNA/DNA substrate, 50 mM HEPES pH 8.0, 100 mM NaCl, 2 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 0.05% Tween20, 5 units of RNaseOut, test compounds (0 to 500 μM), and 5% DMSO in a 100 μL reaction volume. The substrate was formed by annealing a complementary RNA oligonucleotide to a hairpin DNA oligonucleotide to hold the hairpin DNA oligonucleotide in an open conformation. The DNA oligonucleotide has a 5’ fluorescein reporter and a 3’ black hole quencher. When the RNA is degraded from the substrate the DNA oligonucleotide closes to form a hairpin structure, decreasing in signal intensity. Reactions were initiated with 5 mM MgCl2 and fluorescence was read using a Synergy HTS multi-mode plate reader at 37°C. All huRNaseH1 reactions were run with 0.3 μg huRNaseH1 and 100 nM of RNA:DNA substrate. All other reaction conditions were identical. Qualitative inhibition was defined as a dose-dependent reduction in the rate of substrate degradation over time compared to the vehicle-treated control.
Compounds were also analyzed in the oligonucleotide-directed RNA cleavage assay (Edwards et al., 2017; Hu et al., 2013; Tavis et al., 2013). 100 ng RNA was combined with 200 ng DNA oligonucleotide and the RNA:DNA substrate was incubated in the presence of the RNaseH enzyme and test compounds in 50 mM Tris (pH 7.5), 100 mM NaCl, 5 mM MgCl2, 2 mM TCEP, 0.05% Tween20, and 1% DMSO at 37°C for 60 minutes. The products were resolved by denaturing gel electrophoresis and RNA substrate and cleavage products were detected by staining with SYBR® gold according to the manufacturer’s instructions (Life Technologies, Carslbad, CA). Bands were imaged using a Typhoon Trio Variable Mode Imager (GE Healthcare, Marlborough,MA). Inhibition was defined as a dose-dependent reduction in the amount of substrate cleavage compared to the vehicle-treated control.
2.7. Synergy
Synergy between compounds was assessed using the Chou-Talalay method for HBV replication inhibition. HepDES19 cells were plated in 96 well plates at 4 × 104 cells per well and incubated in the absence of tetracycline for 48 hours prior to the addition of compound. Nine compound concentrations from 16- to 0.625-fold the EC50 of each compound (Table 1 and (Edwards et al., 2017)) were tested. All experiments were done with constant ratios of both compounds in the presence of 2% DMSO as described previously (Lomonosova et al., 2017b). Cell lysis and plus-polarity DNA quantification were done as described in section 2.4. The Chou-Talalay method was used to calculate the combination index (CI) to determine synergy (CI < 1), additive effects (CI = 1) or antagonism (CI > 1) using CompuSyn software from ComboSyn, Inc. (Chou, 2010). The weighted CI was calculated using the formula: (CI50 + 2CI75 + 3CI90 + 4CI95)/10 = CIwt (Lomonosova et al., 2017b).
2.8. Turbidimetric solubility assay
Compound solubility was assessed using a turbidimetric solubility assay in unmodified, phenol red-free DMEM media at pH 7.3. Compounds were diluted into DMEM (200 μM to 1.5 μM) in 1% DMSO. 100 μL of diluted compound was plated into a clear 96-well plate and the absorbance at 620 nm was read with a synergy HTX plate reader. The absorbance versus compound concentration was plotted and the inflection point was determined to be the point where the absorbance at 620 nm was more than 2-fold higher than background. Solubility limits are reported as the lower bound of the inflection point (Hoelke et al., 2009; Kerns et al., 2008).
2.9. bDNA quantitation of HBV rcDNA
HepDE19 (50,000 cells/well) were plated in 96 well collagen-coated tissue culture-treated microtiter plates in the presence of 1 μg/mL tetracycline and incubated in a humidified incubator at 37°C and 5% CO2 overnight. Next day, the cells were switched to fresh medium without tetracycline and incubated for 4 hrs at 37°C and 5% CO2. The cells were treated with fresh Tet-free medium with compounds at concentrations starting at 25 μM and a serial, half log, 8-point, titration series in duplicate. The final DMSO concentration in the assay was 0.5%. The plates were incubated for seven days in a humidified incubator at 37°C and 5% CO2. Following a seven day-incubation, the level of rcDNA present in the inhibitor-treated wells was measured using a Quantigene 2.0 bDNA assay kit (Affymetrix, Santa Clara, CA) with HBV specific custom probe set and manufacturer’s instructions. In order to differentiate HBV RNaseH inhibitors from other classes of HBV inhibitors (eg: capsid inhibitors), the assay method was configured by incorporating the quantitation of both the minus-strand and the plus-strand of HBV rcDNA levels using specifically designed HBV probe sets. This is based on the observation that inhibition of RNaseH enzyme activity results in a block of the synthesis of the positive-polarity strand synthesis by the viral polymerase that follows the synthesis of the minus-polarity strand of rcDNA. In this system, an RNaseH inhibitor would therefore show prominent inhibition of the plus-strand formation but minimal effect on the synthesis of the minus-strand of rcDNA. In contrast, treatment with HBV capsid inhibitors would reduce the production of both strands of HBV rcDNA in cells. Concurrently, the effect of compounds on HepDE19 cell viability was assessed as described in section 2.5. Data was analyzed using XL-Fit module in Microsoft Excel to determine EC50 and EC90 (bDNA) values using a 4-parameter curve fitting algorithm.
2.10. HepBHAe82 assay with cccDNA-dependent HA-HBeAg (e antigen) quantitation using AlphaLISA
The HepBHAe82 cell line is a HBV cell culture system is similar to the HepDES19 but has an in-frame HA epitope tag in the N-terminal coding sequence of HBeAg in the HBV transgene, without disrupting any cis-elements critical for HBV replication, cccDNA transcription, and HA-HBeAg secretion (Cai et al., 2016). In this cell culture system, the reporters are the precore RNA and its cognate protein product, the secreted HA-tagged HBV “e antigen” (HA-HBeAg) that is only formed upon the generation of cccDNA. An AlphaLISA assay for the detection of HA-tagged HBeAg with anti-HA antibody serving as a capture antibody and anti-HBeAg serving as detection antibody, specifically detects the cccDNA-dependent signal and eliminates the contaminating signal from HBV “core antigen” (HBcAg). The HepBHAe82 cell line exhibits high levels of cccDNA synthesis and HA-HBeAg production and secretion and provides a highly specific readout signal with low noise. To test the effect of compounds on cccDNA formation and expression, HepBHAe82 (50,000 cells/well) were plated in 96 well, tissue culture-treated, microtiter plates in DMEM/F12 medium supplemented with 10% FBS, 1% penicillin-streptomycin and tetracycline (1 μg/mL) and incubated in a humidified incubator at 37oC and 5% CO2 overnight. Next day, the cells were switched to fresh medium without tetracycline and treated with compounds at concentrations starting at 25 μM and a serial, ½ log, 8-point, titration series in duplicate. The final DMSO concentration in the assay was 0.5%. The plates were incubated for 9 days in a humidified incubator at 37oC and 5% CO2. On day 9, media was harvested and removed to a fresh plate, and level of HA-HBeAg was determined by AlphaLISA. Concurrently, the effect of inhibitor concentrations on cell viability and proliferation was assessed using the Cell-Titer Glo reagent (Promega, Madison, WI) as described in section 2.5.
2.11. Selectivity Assays
The selectivity assays for human immunodeficiency virus (HIV), human cytomegalovirus (HCMV), herpes simplex virus (HSV) and Eschericia coli tolC mutant were performed by Imquest Biosciences Inc. (Frederick, MD) using their standardized protocols under a service contract.
3. Results:
3.1. HBV replication inhibition and cytotoxicity
To evaluate the ability of compounds to inhibit HBV replication, HepDES19 cells, a human hepablastoma cell line (Guo et al., 2007), that carries a tetracycline-repressible HBV genomic expression vector were seeded onto a 96 well plate, HBV replication was synchronously induced by withdrawal of tetracycline, and compounds were incubated with the HBV-expressing cells for 72 hours. HBV nucleic acids were harvested and a strand-preferential q-PCR assay was done to quantify the amount of positive- and negative-polarity DNA strands because inhibiting the HBV RNaseH preferentially blocks synthesis of positive-polarity DNA. Three of the seven HIDs inhibited HBV replication, with effective concentration fifty percent (EC50) values from 2.4 to 3.5 μM (Table 1). All HPDs inhibited HBV replication, with EC50s from 0.11 to 4.0 μM (Table 1). The most effective compound was A23, an HPD, which has an EC50 of 0.11 ± 0.01 μM. All active compounds preferentially inhibited the HBV positive-polarity DNA strand and had little to no effect on accumulation of the minus-polarity DNA strand (Figure 1).
Figure 1. Representative replication inhibition efficacy and cytotoxicity assays.

Replication inhibition by representative HIDs and HPDs was measured against an HBV genotype D isolate in HepDES19 cells using a strand preferential q-PCR assay. EC50 values were calculated based on the decline of the positive-polarity DNA strand relative to the DMSO vehicle treated controls. Positive-polarity DNAs are in black; negative-polarity DNAs are in grey. EC50 values are the average of three experiments ± one standard deviation. Cytotoxicity was measured by MTS assay and CC50 values are the averages of three experiments ± one standard deviation.
To determine if efficacy of the compounds resulted from adverse effects on cellular health, we measured the 50 percent cytotoxicity concentration (CC50) values for the compounds in HepDES19 cells using an MTS assay that measures mitochondrial function over three days of compound exposure; cells were plated at a low enough density that they divided at least twice while in the presence of the compounds. CC50s ranged from 14.6 to >100 μM (Table 1, Figure 1). Eleven of the 16 compounds had CC50s over 50 μM (Table 1). Therapeutic index values (CC50MTS/EC50 = TI) ranged from 14 to 352 for the active compounds.
To expand assessment of cytotoxicity in HepDES19 cells, we evaluated all active compounds using a neutral red (NR) assay, which measures lysosomal function, and a lactate dehydrogenase (LDH) assay, which assesses cell membrane permeability. CC50 values for the NR assay ranged from 12 to >100 μM (Table 2), while for the LDH assay we observed values of 22 to >100 μM (Table 2). Select compounds were also evaluated for cytotoxicity in HepDE19 cells and HepBHAe82 cells using the CellTiter-Glo™ assay (CTG). CC50s in both cell lines ranged from 25 to >50 μM (Table 2), and there was a general concordance among all cytotoxicity assays.
Table 2:
Cytotxcicity analysis of selected compounds
| Compound # | HepDES19 | HepDE19 | HepBHAe82 | |||
|---|---|---|---|---|---|---|
| MTS (a) (μM) | NR (b) (μM) | LDH (c) (μM) | MTS (a) (μM) | CTG (d) (μM) | CTG (d) (μM) | |
| A11 | >100 | >100 | >100 | ND (e) | ND | ND |
| A13 | 85 ± 15 | 56 ± 11 | 90 ± 11 | ND | >25 | >25 |
| A14 | 81 ± 13 | 53 ± 8 | 77 ± 27 | ND | ND | ND |
| A15 | 71 ± 17 | 35 ± 15 | 96 ± 8 | ND | ~25 | >25 |
| A16 | 62 ± 15 | 52 ± 9 | 60 ± 12 | ND | ND | ND |
| A17 | 15 ± 7 | 17 ± 9 | 19 ± 13 | ND | ND | ND |
| A19 | 100 ± 0.2 | >100 | >100 | ND | >25 | >25 |
| A20 | 93 ± 9 | 42 ± 0.1 | 22 ± 8 | ND | ND | ND |
| A22 | 90 ± 6 | 63 ± 26 | 75 ± 13 | 71 ± 17 | ~25 | >25 |
| A23 | 33 ± 8 | 37 ± 15 | 62 ± 15 | 48 ± 20 | >25 | >25 |
| A24 | 102 ± 3 | 83 ± 30 | >100 | 97 ± 3 | >25 | >25 |
| A25 | 91 ± 16 | 90 ± 15 | 90 ± 1 | ND | ~25 | >25 |
CellTiter 96™ AQueous Non-Radioactive Cell Proliferation Assay (MTS) was used to measure mitochondrial function.
Neutral red retention assay (NR) was used as a measure of lysosome function.
Lactate dehydrogenase release assay (LDH) was used as a measure of plasma membrane permeability.
CellTiter-Glo™ assay (CTG) was used as a measure of plasma membrane permeability.
Not determined (ND).
3.2. Biochemical screening against the HBV RNaseH
We evaluated all compounds for inhibition of the HBV RNaseH using a molecular beacon assay in which RNaseH activity causes a decline in fluorescence (Edwards et al., 2017); this assay is suitable for qualitative but not quantitative assessment of inhibition. Compounds were incubated with the HBV RNaseH plus a RNA:DNA heteroduplex substrate and the rate of substrate degradation was monitored, with inhibition being reported qualitatively as a dose-dependent reduction in the rate of substrate degradation. Of the seven HIDs evaluated, two inhibited the HBV RNaseH, four did not inhibit the enzyme, and one gave inconclusive results (Table 1, Figure 2). Of the nine HPDs, seven inhibited the HBV RNaseH and two gave inconclusive results (Table 1, Figure 2). Inconclusive results stemmed from interference with the fluorescence signal by the compounds or inhibition that lacked dose-responsiveness. Therefore, we evaluated inhibition by the HID/HPD compounds using a gel-based oligonucleotide-directed RNA cleavage assay that is specific for RNaseH activity compared to other RNase activities that could also be detected by the molecular beacon assay (Edwards et al., 2017). Compounds were incubated with the HBV RNaseH and an RNA:DNA heteroduplex substrate. The reaction products were resolved on a denaturing acrylamide gel and the amount of cleaved substrate was measured by imaging the resolved products using a Typhoon phosphorimager. Five of the HIDs inhibited the HBV RNaseH and all nine HPDs inhibited the HBV RNaseH in the oligonucleotide-directed RNA cleavage assay (Table 1, Figure 2b). Inhibition of the HBV RNaseH endonuclease activity in the oligonucleotide-directed RNaseH assay is best measured by observing the amount of substrate RNA remaining intact after the reaction relative to the uninhibited control (Figure 2b). This is because the HBV RNaseH has a 3’–5’ exonuclease activity that is stimulated by the endonuclease activity, and the RNaseH remains attached to the P1 product fragment and processively degrades it. (Villa et al., 2016). Both the endo- and exo-nuclease activities are inhibited by RNaseH inhibitors, resulting in stabilization of both the substrate and P1 products rather than just the substrate as would be the case if there were no exonucleolytic degradation (Figure 2b). In total, 14 of the 16 compounds inhibited the HBV RNaseH in one or both biochemical assays.
Figure 2. Effects of the compounds on purified HBV RNaseH.

(Panel A) HBV RNaseH inhibition was determined using a molecular beacon RNaseH assay in which quenching of fluorescence from the RNaseH substrate is measured following removal of the RNA strand by the RNaseH, causing the folding of the DNA strand into a hairpin. (Panel B) HBV RNaseH inhibition was determined using an oligonucleotide-directed RNA cleavage assay in which the cleavage of an RNA:DNA heteroduplex substrate was monitored by resolving the products on a gel and visualizing the stained gel using a Typhoon phosphorimager. Neg. control, reactions in which the DNA oligonucleotide was not homologous to the RNA; EDTA control, reactions containing 10 mM EDTA to prevent catalysis.
3.3. huRNaseH1 inhibition
All compounds were counter-screened using the molecular beacon assay for activity against the human ribonuclease H1 (huRNaseH1), which is a potential off-target cellular enzyme. Three of seven HIDs and seven of nine HPDs inhibited the huRNaseH1 in the molecular beacon assay. Two compounds were inconclusive and were therefore evaluated using the oligonucleotide-directed RNA cleavage assay, where both compounds inhibited the huRNaseH1 (Table 1).
3.4. bDNA quantitation of HBV rcDNA
Based on the results obtained in the strand-preferential q-PCR assay for the HPD compounds, one HID (A19) and six HPDs (A13, A15, A22, A23, A24, A25) compounds were further evaluated for inhibition of HBV replication in HepDE19 cells using a branched chain DNA (bDNA) hybridization assay. HepDE19 cells were plated in tetracycline containing DMEM on 96 well collagen-coated plates. 24 hours later, HBV replication was synchronously induced by the removal of tetracycline. Four hours after induction of HBV replication, test compounds were applied and incubated in the presence of HBV replicating cells for seven days. A bDNA assay kit with specific HBV primers and probes was used to quantify HBV rcDNA levels. All compounds tested inhibited HBV replication with EC50s from 3.4 to 15 μM (Table 3).
Table 3:
Expanded activity profile of selected RNaseH inhibitors
| Compound | EC50 | CC50 | q-PCR TI HepDES19 | q-PCR TI HepDE19 | bDNA TI HepDE19 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| q-PCR HepDES19 (μM) | q-PCR HepDE19 (μM) | bDNA_rcDNA HepDE19 (μM) | MTS HepDES19 (μM) | MTS HepDE19 (μM) | CTG HepDE19 (μM) | |||||||
| (+) strand | (−) strand | (+) strand | (−) strand | (+) strand | (−) strand | |||||||
| HID | ||||||||||||
| A19 | 3.8 ± 1.7 | ND (a) | ND | 7.6 ± 4.7 | >25 | 100 ± 0.2 | ND | >25 | 26 | NA | >3.2 | |
| HPD | ||||||||||||
| A13 | 0.54 ± 0.3 | 56 ± 8 | ND | ND | 10.7 ± 2.2 | >25 | 85 ± 15 | ND | >25 | 157 | NA | >2.3 |
| A15 | 0.46 ± 0.1 | 9.8 ± 3 | ND | ND | 4.4 ± 2.7 | ~25 | 71 ± 17 | ND | ~25 | 154 | NA | ~5.7 |
| A22 | 0.32 ± 0.2 | 32 ± 17 | 0.33 ± 0.2 | 11± 7 | 3.4 ± 2.5 | >25 | 90 ± 6 | 71 ± 17 | >25 | 281 | 215 | >7.3 |
| A23 | 0.11 ± 0.01 | 10 ± 2 | 0.1 ± 0.03 | 6.7 ± 4 | 5.2 ± 1.6 | >25 | 33 ± 8 | 48 ± 20 | ~25 | 300 | 480 | ~4.8 |
| A24 | 0.29 ± 0.1 | 31 ± 16 | 0.4 ± 0.01 | 32 ± 10 | 7.1 | >25 | 102 ± 3 | 97 ± 3 | >25 | 352 | 243 | >3.5 |
| A25 | 0.5 ± 0.1 | 30 ± 12 | ND | ND | 3.7 ± 2.3 | >25 | 91 ± 16 | ND | ~25 | 182 | NA | ~6.7 |
Not determined (ND)
3.5. HepBHAe82 assay with cccDNA-dependent HA-HBeAg (e antigen) quantitation using AlphaLISA
Select compounds were further evaluated for the ability to inhibit cccDNA formation and expression. HepBHAe82 cells were seeded into 96 well plates and 24 hours later tetracycline was removed to synchronously induce HBV replication. Test compounds were applied and incubated in the presence of HBV replicating cells for nine days. Media was harvested on day nine and transferred to a fresh plate.
In this cell culture system the levels of cccDNA are detected by measuring the secreted HA-HBeAg that is only formed upon generation of cccDNA. An AlphaLISA assay for the detection of HA-tagged HBeAg with anti-HA antibody serving as a capture antibody and anti-HBeAg serving as detection antibody was used. All seven compounds inhibited cccDNA formation and expression with EC50s ranging from 0.43 to 1.5 μM (Table 4).
Table 4:
Inhibition of cccDNA by selected compounds
| Compound | HepBHAe82_ AlphaLISA_eAg | HepBHAe82_ Cytotoxicity |
|---|---|---|
| EC50 (μM) | CC50 (μM) | |
| HID | ||
| A19 | 1.3 ± 0.39 | >25 |
| HPD | ||
| A13 | 1.5 ± 0.94 | >25 |
| A15 | 0.76 ± 0.71 | >25 |
| A22 | 0.56 ± 0.34 | >25 |
| A23 | 0.56 ± 0.2 | >25 |
| A24 | 0.43 ± 0.11 | >25 |
| A25 | 0.65 ± 0.51 | >25 |
3.6. Synergy
New anti-HBV drugs are likely to be used in combination with other drugs. Therefore, we determined if an HPD (A23) and the approved nucleoside analog Lamivudine act synergistically in inhibiting HBV replication. Three independent synergy experiments were performed in which the amount of DNA accumulation was measured using our strand preferential q-PCR assay. The Chou-Talalay method was used to calculate the combination indexes (CI), and weighted CI (CIwt) values are reported in (Table 5). All CI values were well below 1, indicating that Lamivudine and A23 synergistically suppressed HBV replication (Table 5). We also determined if two RNaseH inhibitors from HID (#86) and HPD (A23) compound classes can act synergistically with one another in inhibiting HBV replication as was recently shown for α-hydroxytropolone (αHT) and an HIDs (Lomonosova et al. 2017). As expected, both compounds preferentially inhibited the positive-polarity DNA strand and had little to no effect on the minus-polarity DNA. The CIwt values were below 1, indicating that #86 and A23 act synergistically against HBV replication (Table 5).
Table 5:
(a) Combination index (CI)
| Compound Combination | CI values at various inhibition levels: (a) | Weighted CI values (a) | |||
|---|---|---|---|---|---|
| 50% | 75% | 90% | 95% | ||
| A23 + Lam | 0.21 ± 0.25 | 0.20 ± 0.11 | 0.22 ± 0.13 | 0.24 ± 0.14 | 0.21 |
| A23 + #86 | 0.50 ± 0.33 | 0.46 ± 0.32 | 0.43 ± 0.31 | 0.41 ± 0.31 | 0.46 |
Combination index (CI)
3.7. Compound solubility limits at pH 7.3
Solubility of all compounds was evaluated using a turbidimetric solubility assay (Hoelke et al., 2009; Kerns et al., 2008). Compounds were diluted from 200 to 1.5 μM in DMEM in 1% DMSO to mimic the assay conditions of the HBV replication inhibition assay. Most compounds were soluble in DMEM up to 200 μM, except two HIDs, A18 and A21, which were soluble up to 50 μM (Table 1).
3.8. Selectivity assays
Selectivity assays evaluating inhibition of HIV, HCMV, HSV-1, and E. coli tolC mutant were performed by Imquest Biosciences Inc. (Frederick, MD) for three representative compounds. No substantial evidence of activity was observed against any of the pathogens tested (Table 6).
Table 6:
Selectivity profile of selected compounds
| Compound | HIV | HSV1 | CMV | E. coli tolC | |||
|---|---|---|---|---|---|---|---|
| EC50 (μM) | CC50 (μM) | EC50 (μM) | CC50 (μM) | EC50 (μM) | CC50 (μM) | MIC (μM) | |
| A15 | >34.4 | 34.4 | >20.2 | 20.2 | >39 | 39 | >50 |
| A22 | >50 | >50 | >40.9 | 40.9 | >50 | >50 | >50 |
| A25 | >50 | >50 | >43.3 | 43.3 | >50 | >50 | >50 |
Discussion and Summary:
We previously reported that 11 HIDs and a structurally related HPD inhibited HBV replication with low- to sub-micromolar efficacies, with TI values of 2.4 to 71 (Edwards et al., 2017). Therefore, we synthesized 16 new compounds, seven HIDs and nine HPDs, to examine the anti-HBV potential of these scaffolds.
HIDs:
We explored modifications to the HID scaffold, primarily at the R5 and R6 positions based on our preliminary SAR (Edwards et al., 2017), to determine which modifications to the scaffold improved potency, specificity, and reduce cytotoxicity. Of the seven HIDs screened, only three were active against HBV replication, with EC50 values between 2.4 to 3.8 μM (Table 1). CC50s for all HIDs evaluated ranged from 14 to >100 μM, but the three active compounds were relatively nontoxic with CC50s above 90 μM (Table 1). Two of the previously reported HIDs have TI >40, with the best inhibitor (#86) having a TI of 71 (Edwards et al., 2017). None of the new HIDs had a TI >40, indicating that modifications to the HIDs did not improve the in vitro potency or cytotoxicity profiles of this class of compounds.
One HID (A19) was further evaluated for inhibition of HBV replication in HepDE19 cells using a bDNA hybridization assay. Consistent with data from the strand-preferential q-PCR assay, compound A19 inhibited HBV replication with an EC50 of 7.6 μM. While the EC50 was approximately 2-fold higher in the bDNA hybridization assay compared to the q-PCR assay, both assays reported only modest inhibition of HBV replication by A19.
We previously reported our biochemical HBV RNaseH inhibition assays consistently underreports RNaseH activity for a number of HID inhibitors of HBV replication (Edwards et al., 2017). Therefore, biochemical analysis of the HIDs were limited to qualitative assessment. Five of the seven HIDs inhibited HBV RNaseH in one or both biochemical assays employed (Table 1). Coupled with the preferential suppression of the positive-polarity DNA strand synthesis in the replication inhibition assays, this indicates that the HIDs inhibit HBV replication by inhibiting HBV RNaseH in culture.
Solubility issues were encountered with A18 and A21. Therefore, the solubility profiles of all the HIDs were evaluated in unmodified DMEM media under conditions that mimic our replication inhibition assay. Compounds A18 and A21 were insoluble in DMEM media above 50 μM (Table 1) which may help explain the large standard deviation in assays with them. However, the other HIDs were soluble above 200 μM. Therefore, inactivity in the replication inhibition assay of the compounds A10 and A12 is not due to lack of solubility.
HPDs:
In previous studies, the HPD compound #208 was active against HBV replication with an EC50 of 0.69 μM, a CC50 of 15 μM, and a TI of 22 (Edwards et al., 2017). This compound showed the best combination of potency and TI in the HID/HPD compound classes, and therefore it was the starting point for exploring the anti-HBV potential of the HPDs.
The EC50 values for the nine new HPD compounds by the three-day q-PCR replication inhibition assay ranged from 0.11 to 4 μM, and the CC50s ranged from 15 to >100 μM, resulting in TIs from 14 to 352 (Table 1). The best HPD compounds, A22, A23, and A24, were significantly more potent than the hit compound #208 (EC50 = 0.69μM) and also less cytotoxic than #208 (CC50 = 15 μM), resulting in a 16-fold improvement of the calculated TI for A23. Similar to data for the HIDs (Edwards et al., 2017), there was a general concordance among the various cytotoxicity assays employed.
Inhibition of HBV cccDNA formation and expression is considered essential for the elimination of HBV because the cccDNA is the persistent form of the viral genome. Therefore, we evaluated six HPDs with EC50s from 0.11 to 0.54 μM and TIs between 154 to 352 for inhibition of cccDNA formation and expression in the HepBHAe82 cell line that expresses the HBV e antigen only from the cccDNA. EC50s in these assays ranged from 1.5 to 0.43 μM. These results indicate that these compounds have a moderate effect on cccDNA formation and/or expression, which would be the predicted result of blocking the recycling mechanism that replenishes the cccDNA pool in the nucleus by inhibiting genome maturation via RNaseH inhibition. Explicit evaluations of the effects of RNaseH inhbiitors on cccDNA formaton will be conducted during subsequent optimization of the HPDs.
Due to the results from the strand-preferential q-PCR assay, six HPDs were tested for inhibition of HBV replication in HepDE19 cells using a bDNA assay. The EC50s for the six compounds tested ranged from 3.4 to 10.7 μM. It is unclear why the potencies in this assay were lower than the strand-preferential q-PCR assay. The possibility that the discrepancy could have resulted from differences in the HepDES19 cells used for the q-PCR assay and HepDE19 cells used for the bDNA assay was tested by screening A22, A23, and A24 for HBV replication inhibition in HepDE19 cells using the q-PCR assay. EC50s were consistent in the HepDES19 and HepDE19 cells (Table 3), precluding cell line differences as the cause for the discrepancy. Other technical explanations for the discrepancy would require either a selective under-detection of the plus-polarity DNA strand relative to the minus-polarity strand for those two compounds by the q-PCR assay, or over-detection of the plus-polarity strand by the bDNA hybridization assay, potentially due to hybridization with incompletely degraded HBV RNA and/or HBV DNA from the transcriptional template in the cells. We consider it unlikely that there was a selective under-detection of the positive polarity DNA in the q-PCR based assay because these compounds were tested more than a dozen times and results were consistent. As none of these technical explanations is probable and as HPD inhibitors such as A23 and A25 could inhibit cccDNA formation in the HepBHAe82 assay with EC50s ≈ 0.5 μM, we feel that the most plausible explanation is that compounds were less potent in the bDNA hybridization assay because the cells were incubated with the compounds for seven days versus three days for the q-PCR assay. Over this time some compound degradation may have occurred, resulting in resumption of reverse transcription. Such resurgence of viral replicaton late in the 7-day assay would have had a lesser effect on the cccDNA-dependent assay because cccDNA formation is slow and requires prior formation of capsid-associated HBV DNA (Guo et al., 2007).
All HPD compounds were active against the HBV RNaseH in the biochemical assays (Table 1, Figure 2). However, only qualitative assessment of the biochemical assayswas possilbe. This is likely due to the known insensitivity of the biochemical assay, possibly because the recombinant RNaseH is only a fragment of the full HBV polymerase (Villa et al., 2016) and may not fully mimic the native enzyme.
Selectivity of the HPDs for the HBV RNaseH was evaluated by counter-screening them against the huRNaseH1 because huRNaseH1 is the cellular enzyme whose catalytic activity most nearly resembles that of the HBV RNaseH. Although these enzymes have similar catalytic function, they share only 19% amino acid identity in global alignments, with only two of the DEDD residues aligning. Eight of the nine HPDs inhibited huRNaseH1, indicating that selectivity of the HPDs needs to be improved. However, the qualitative nature of the assays precludes identifying moderate selectivity if it were present. Interestingly, A15 inhibited the HBV RNaseH in both the biochemical and cell based assays but it did not inhibit the huRNaseH1. These data indicate that it may be possible to develop an HBV RNaseH inhibitor that minimizes undesired effects on huRNaseH1.
Further selectivity studies were done against HIV, HSV-1, HCMV, and E. coli tolC for three representative HPDs. No appreciable activity was observed against any of these pathogens. The failure to inhibit the HIV with these RNaseH inhibibitors may stem from the large genetic distance between the HIV and HBV RNaseHs, which share only 23% amino acid identity (Tavis et al., 2013), or possibly due to different interactions of the compounds with the lymphocytic cells in wich HIV is studied versus the hepatic cells used for HBV. Regardless, these data indicate that it is possible to develop a highly specific HBV RNaseH inhibitor.
Curing HBV will require new drugs against new targets capable to further suppressing HBV, and these drugs will likely be used in combination with other drugs. Therefore, we evaluated the effects on HBV replication of combining an HPD (A23) with Lamivudine, an approved NA drug that targets the HBV reverse transcriptase active site. A23 and Lamivudine were strongly synergistic, with a CIwt of 0.21 (Table 5). Therefore, blocking both enzymatic activities of the multifunctional HBV polymerase resulted in much stronger suppression of viral replication than blocking either activity alone.
We also evaluated the effects of combining two RNaseH inhibitors with each other, the HID #86 and the HPD A23, on HBV replication. The HID and HPD compound classes share a common pharmacophore, so we hypothesized that combing them would be antagonistic. Surprisingly, we found that combing A23 and #86 synergistically suppressed HBV replication, with a CIwt of 0.46 (Table 5). We previously showed that combining an αHT and HID synergistically suppresses HBV replication (Lomonosova et al., 2017b). However, these two compound classes share different core pharmacophores, which suggests that these compound classes may not interact with RNaseH identically. Our data indicate that the HID and HPD compounds also likely do not inhibit the enzyme in identical manners despite the shared pharmacophore. We see three possibilities for how HIDs and HPDs can synergize. First, both inhibitors could bind simultaneously in a cooperative manner. Second, the compounds could bind to the RNaseH during different stages of the RNaseH catalytic cycle and therefore not compete for the same protein conformation or complex. Third, the compounds may also contact the RNaseH substrate in the active site, and the local sequence of the substrate could modulate binding of the HIDs and HPDs differently. Regardless, these data indicate that RNaseH inhibitors could be used in combination with other anti-HBV therapies in the future.
This study indicates that the HPD compound class appears a better candidate for anti-HBV RNaseH inhibitor optimization than the HIDs. By synthesizing only nine new HPDs we achieved a 16-fold improvement over our initial hit compound, whereas no improvement was seen with seven new HIDs. Future efforts will explore additional modifications to the HPD scaffold in an effort to develop improved inbhitory compounds.
Summary:
We evaluated 16 novel compounds, seven HIDs and nine HPD compounds for activity against HBV replication in culture. Only three of the seven HIDs were active against HBV replication, and none of these compounds had a TI better than the best prior HID (#86, TI = 71). In comparison, synthesis of only nine new HPDs resulted in a more than 16-fold improvement in the TI over the best previous HPD (#208, TI = 22) (Edwards et al., 2017). We were able to achieve greater gains in potency and TI with the HPDs compared to the HIDs. Future efforts will focus on further improving the HPD scaffold with better TIs and improved selectivity over huRNaseH1, with the goal of developing a novel HBV RNaseH drug that can be used in combination with other inhibitors to provide better treatment to the millions of chronically infected patients.
Supplementary Material
Acknowldgements:
We thank Reese Foster for technical support and Maureen Donlin for assistance with the genetic alignments.
Financial support:
This research was funded by corporate funds from Arbutus Biopharma and NIH grant R21 AI124672 to JET.
Footnotes
Competing Intrests: None
References:
- Cai CW, Lomonosova E, Moran EA, Cheng X, Patel KB, Bailly F, Cotelle P, Meyers MJ, Tavis JE, 2014. Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antiviral Res 108, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Wang X, Yan R, Mao R, Liu Y, Ji C, Cuconati A, Guo H, 2016. Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged HBeAg for screening of cccDNA modulators. Antiviral research 132, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, 2010. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Coffin CS, Mulrooney-Cousins PM, Peters MG, van MG, Roberts JP, Michalak TI, Terrault NA, 2011. Molecular characterization of intrahepatic and extrahepatic hepatitis B virus (HBV) reservoirs in patients on suppressive antiviral therapy. J.Viral Hepat. 18, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox N, Tillmann H, 2011. Emerging pipeline drugs for hepatitis B infection. Expert.Opin.Emerg.Drugs 16, 713–729. [DOI] [PubMed] [Google Scholar]
- Edwards TC, Lomonosova E, Patel JA, Li Q, Villa JA, Gupta AK, Morrison LA, Bailly F, Cotelle P, Giannakopoulou E, 2017. Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles. Antiviral Research 143, 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerelsaikhan T, Tavis JE, Bruss V, 1996. Hepatitis B Virus Nucleocapsid Envelopment Does Not Occur without Genomic DNA Synthesis. J.Virol. 70, 4269–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT, 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81, 12472–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelke B, Gieringer S, Arlt M, Saal C, 2009. Comparison of nephelometric, UV-spectroscopic, and HPLC methods for high-throughput determination of aqueous drug solubility in microtiter plates. Analytical chemistry 81, 3165–3172. [DOI] [PubMed] [Google Scholar]
- Hu Y, Cheng X, Cao F, Huang A, Tavis JE, 2013. beta-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res 99, 221–229. [DOI] [PubMed] [Google Scholar]
- Kerns EH, Di L, Carter GT, 2008. In vitro solubility assays in drug discovery. Current drug metabolism 9, 879–885. [DOI] [PubMed] [Google Scholar]
- Kwon H, Lok AS, 2011. Hepatitis B therapy. Nat.Rev.Gastroenterol.Hepatol. 8, 275–284. [DOI] [PubMed] [Google Scholar]
- Lomonosova E, Daw J, Garimallaprabhakaran AK, Agyemang NB, Ashani Y, Murelli RP, Tavis JE, 2017a. Efficacy and cytotoxicity in cell culture of novel α-hydroxytropolone inhibitors of hepatitis B virus ribonuclease H. Antiviral Research 144, 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonosova E, Zlotnick A, Tavis JE, 2017b. Synergistic Interactions between Hepatitis B Virus RNase H Antagonists and Other Inhibitors. Antimicrobial agents and chemotherapy 61, e02441–02416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KR, Lomonosova E, Li Q, Ponzar NL, Villa JA, Touchette E, Rapp S, Liley RM, Murelli RP, Grigoryan A, 2018. Efficacy of hepatitis B virus ribonuclease H inhibitors, a new class of replication antagonists, in FRG human liver chimeric mice. Antiviral research 149, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Lomonosova E, Cheng X, Moran EA, Meyers MJ, Le Grice SF, Thomas CJ, Jiang JK, Meck C, Hirsch DR, D’Erasmo MP, Suyabatmaz DM, Murelli RP, Tavis JE, 2015. Hydroxylated Tropolones Inhibit Hepatitis B Virus Replication by Blocking the Viral Ribonuclease H Activity. Antimicrob Agents Chemother 59, 1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Villa JA, Donlin MJ, Edwards TC, Cheng X, Heier RF, Meyers MJ, Tavis JE, 2016. Hepatitis B virus genetic diversity has minimal impact on sensitivity of the viral ribonuclease H to inhibitors. Antiviral research 135, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen J, Thompson AJ, Levrero M, 2016. Aiming for cure in HBV and HDV infection. Journal of hepatology 65, 835–848. [DOI] [PubMed] [Google Scholar]
- Repetto G, del Peso A, Zurita JL, 2008. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nature protocols 3, 1125–1131. [DOI] [PubMed] [Google Scholar]
- Seeger C, Zoulim F, Mason WS, 2013. Hepadnaviruses, in: Knipe DM., Howley PM. (Eds.), Fields Virology, 6 ed. Lippincott Williams & Wilkins, Philadelphia PA, pp. 2185–2221. [Google Scholar]
- Tavis JE, Badtke MP, 2009. Hepadnaviral Genomic Replication, in: Cameron, C.E., G”tte, M., Raney, K.D. (Eds.), Viral Genome Replication. Springer Science+Business Media, LLC, New York, pp. 129–143. [Google Scholar]
- Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG, 2013. The hepatitis B virus ribonuclease h is sensitive to inhibitors of the human immunodeficiency virus ribonuclease h and integrase enzymes. PLoS pathogens 9, e1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Lomonosova E, 2015. The Hepatitis B Virus Ribonuclease H as a Drug Target. Antiviral Res. 118, 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa JA, Pike DP, Patel KB, Lomonosova E, Lu G, Abdulqader R, Tavis JE, 2016. Purification and enzymatic characterization of the hepatitis B virus ribonuclease H, a new target for antiviral inhibitors. Antiviral Research 132, 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, 2017. Hepatitis B vaccines: WHO position paper – July 2017. Weekly Epidemiological Record 92, 369–392.28685564 [Google Scholar]
- Xie L, Yin J, Xia R, Zhuang G, 2018. Cost-effectiveness of antiviral treatment after resection in hepatitis B virus–related hepatocellular carcinoma patients with compensated cirrhosis. Hepatology. [DOI] [PubMed] [Google Scholar]
- Zoulim F, 2004. Antiviral therapy of chronic hepatitis B: can we clear the virus and prevent drug resistance? Antivir.Chem.Chemother. 15, 299–305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.