

Abstract
Protein function annotation is one of the most important research topics for revealing the essence of life at molecular level in the post-genome era. Current research shows that integrating multisource data can effectively improve the performance of protein function prediction models. However, the heavy reliance on complex feature engineering and model integration methods limits the development of existing methods. Besides, models based on deep learning only use labeled data in a certain dataset to extract sequence features, thus ignoring a large amount of existing unlabeled sequence data. Here, we propose an end-to-end protein function annotation model named HNetGO, which innovatively uses heterogeneous network to integrate protein sequence similarity and protein–protein interaction network information and combines the pretraining model to extract the semantic features of the protein sequence. In addition, we design an attention-based graph neural network model, which can effectively extract node-level features from heterogeneous networks and predict protein function by measuring the similarity between protein nodes and gene ontology term nodes. Comparative experiments on the human dataset show that HNetGO achieves state-of-the-art performance on cellular component and molecular function branches.
Keywords: heterogeneous network, gene ontology, protein function annotation, graph neural network
INTRODUCTION
As the expression product of genes, protein forms the main material basis of life and plays a key role in life activity and function execution. Functional annotation of proteins is crucial to understanding life activity from the molecular level. Gene ontology (GO) [1] is a systematic method of annotating the properties of genes and gene products, which divides the function of proteins into three different sub-ontology: biological process (BP), cellular component (CC) and molecular function (MF). As shown in Figure 1, for each branch, GO is a direct acyclic graph, where each node has a unique label and refers to a specific term. The nodes with deeper hierarchies refer to a more detailed description of protein function. That means, when a protein is labeled with a specific term, it is also annotated by all ancestor nodes of the term, which is known as true path rule [2–4].
Figure 1.

A subgraph of GO. GO is a direct acyclic graph for each branch, where each node has a unique label and refers to a specific term.
Automatic protein function annotation aims to predict protein function through computational methods, which is more flexible and convenient than experimental methods and has important application prospects. In recent years, shallow machine learning and deep learning have been widely used in the field of bioinformatics, such as biological sequence analysis [5], protein structure prediction [6, 7] and medical image processing [8]. From the perspective of machine learning, protein function prediction is usually regarded as a multi-label classification problem. Methods based on shallow machine learning usually integrate the features extracted from multisource data to measure the similarity between proteins and functional terms and annotate similar functions for similar proteins. The most representative methods are GeneMANIA [9], MS-kNN [10] and NetGO [11]. GeneMANIA [9] is a semi-supervised algorithm based on network label propagation, which fuses heterogeneous networks into a network through linear regression and then makes functional predictions through Gauss label propagation algorithms. The MS-kNN [10] algorithm combines a variety of similarity measures to extract features from sequence similarity, protein–protein interaction (PPI) network and gene expression profile data, which is used by the kNN algorithm to predict protein function. Similarly, the NetGO [11] model predicts protein functions through a ranking framework based on ensemble learning, which comprehensively measures the similarity between proteins and GO terms and uses multiple sub-models to extract features from sequence and PPI network. Compared with MS-kNN only using sequence similarity, NetGO can effectively extract multiple features from the sequence through the sub-model, such as protein family and structural domain information, and thus achieves better performance. However, the reliance on complex feature engineering and model integration methods limits the development of such methods.
Deep learning methods can extract features from large-scale data in an end-to-end manner, leading to their increasing popularity in the field of automatic protein function annotation [12, 13]. Such models usually focus on extracting the features of protein sequence through deep learning networks such as convolution neural network (CNN) and recurrent neural network (RNN), and then integrate sequence similarity, PPI network and other data to improve model performance [14, 15]. In recent years, researchers have made a lot of efforts to develop deep learning models to predict protein function, among which the most representative methods are DeepGO [16] and DeepGOPlus [17], deepNF [18] and DeepMNE-CNN [19], as well as GONET [20] and DeepGOA [21, 22]. DeepGO [16] is one of the earliest models to annotate protein based on deep learning, which combines the sequence and PPI network features to predict protein function and achieve good results. DeepGOPlus [17] model is an improved version of the DeepGO, which does not rely on PPI network data and instead improves prediction performance by integrating sequence similarity information. DeepNF [18] extracts the high-order features of PPI network based on multimodal deep autoencoders, and then predicts protein function via support vector machine. DeepMNE-CNN [19] is an embedding-based function prediction method, which combines semi-supervised autoencoder and CNN to extract complex topological features of multi-networks. GONET [20] predicts protein function by integrating protein sequence and PPI network, which can effectively extract long-range features of protein sequences using RNN and achieves good performance in human and mouse datasets. DeepGOA [21] innovatively utilizes graph convolutional network to model the hierarchy structure of GO term network and annotate proteins by calculating the similarity between protein nodes and term nodes, which achieves good results in corn and human datasets. Based on deep learning algorithms, existing models can effectively extract features from protein sequences, while they still rely on artificially designed model integration or feature integration methods to process sequence similarity information [17, 23] and PPI data [16, 20], which are relatively shallow and inevitably cause information loss, and thus hinders the development of such methods [24].
Besides, even from the perspective of sequence feature extraction, these algorithms must train complex models on specific labeled datasets, resulting in their inability to use large-scale unlabeled sequence data [25–28]. Large-scale pretraining models can effectively alleviate this problem, such as the popular Bert [29] and XLNet [30] in the natural language field, which can train deep learning models through unsupervised learning on large amounts of unlabeled data, and then learn effective semantic representations of sentences. Inspired by such methods, researchers have proposed many pretrained methods [25, 31–34] to model protein sequences, among which the most representative models are ProtVec [31] and SeqVec [25] algorithm. The ProtVec model uses a method similar to the Subword-Embedding in fasttext [35] to obtain the representation of the sequence, which divides the full amino acid sequence into fixed-length substrings and represents each substring as a fixed dimensional vector through the word2vec [36] algorithm. Although the ProtVec model can effectively capture the local features of protein sequence, it ignores the context information of the amino acid sequence due to the context-independence of the word2vec model, and therefore cannot effectively extract the long-range relationship of protein sequence. To tackle this problem, the SeqVec model utilizes the bidirectional LSTM sequence model to capture the long-range association of protein sequence and generates an amino-acid-level embedding vector, which not only contains the semantic information of the amino acid itself, but also the information of its corresponding context [37–41].
In this paper, we propose an end-to-end protein function prediction model HNetGO to solve the problems mentioned above. Firstly, we utilize heterogeneous networks to integrate multisource data in an intuitive and effective way, which avoids the information loss caused by manual designed feature extraction methods to the greatest extent. Secondly, we use a pretraining model to extract protein-level sequence features, which can effectively capture functional-related semantic information within a single protein sequence. In the end, we design a link prediction model based on the attention mechanism to predict protein function. And comparative experiments on the human dataset show that HNetGO achieves state-of-the-art performance on CC and MF branches.
MATERIALS AND METHODS
Datasets acquisition and preprocessing
For our experiment, we downloaded human and mouse protein sequences and their corresponding experimentally verified GO annotation data from the UniProt [42] database, which contained 20 395 human protein sequences and 17 073 mouse protein sequences, respectively. And the PPI network data were downloaded from STRING [43] database (version 11). In addition, we downloaded the latest released GO data (releases/ 01 February 2021) from the official website. After that, we constructed a network of GO terms based on the information extracted from the file. It should be noted that each sub-ontology contains thousands of functional terms, while most of which have not appeared in our dataset. Therefore, we filter out terms with annotated proteins <300 in the BP branch, and the threshold for CC and MF branch is 100, which in turn resulted in unannotated proteins in the dataset. For these proteins, we keep them in the heterogeneous network to improve the connectivity of the network, but do not use them as part of the training set or test set. Table 1 shows some statistics of the dataset.
Table 1.
Dataset statistics
| Datasets | Terms (before filter) | Terms (after filter) | ||||
|---|---|---|---|---|---|---|
| BP | MF | CC | BP | MF | CC | |
| Human | 15 658 | 4803 | 1995 | 752 | 295 | 293 |
| Mouse | 15 838 | 4772 | 1985 | 666 | 262 | 291 |
BP, biological process; CC cellular component; MF molecular function.
Extract protein-level sequence features through pretrained model
The primary structure of protein refers to a one-dimensional sequence composed of 20 kinds of amino acids [44, 45], which can determine the secondary and tertiary structure of the protein, and thus can affect the protein functions [46]. Existing protein function prediction methods usually use one-hot encoding to represent amino acid sequences as a matrix or tensor, which can be used to extract protein-level semantic features through deep learning models. However, due to the inability to use large amount of existing unlabeled sequence data and the semantic independence of one-hot encoding, such methods cannot capture sufficient semantic features of protein sequences, resulting in their poor performance in predicting protein functions.
In this paper, we use the SeqVec, which is inspired by embeddings from language models (ELMO) model [47], to extract protein-level sequence features. ELMO, a powerful pretraining model for natural language sequence processing, can capture the contextual features of words and can generate different embedding vectors of the same word according to different contexts. Similarly, as shown in Figure 2, the SeqVec captures the long-range association of protein sequence and generates an amino-acid-level embedding vector, and then directly obtains an effective protein-level semantic representation through average aggregation. Experiments at the protein level [25, 48, 49] show that methods based on SeqVec features achieve similar results to the state-of-the-art model and have obvious superiority compared with embedding methods such as one-hot and ProtVec.
Figure 2.

Heterogeneous network construction process in HNetGO model. For protein sequence data, we first utilize the results of MSAs to construct a sequence similarity network, and then the pretrained model SeqVec is used to extract amino-acid-level features, which can be used to obtain protein-level features through average aggregation or principal component analysis. Next, a PPI network is built based on the interaction relationship, and a heterogeneous network is used to integrate all the information extracted from the original dataset.
Figure 3.

Framework of HNetGO model. In general, HNetGO model consists of three parts: a node-level mutual attention layer to learn the attention weights of all direct neighbors of the current node; a multi-head messaging layer enables different types of neighbors to deliver messages to the current node based on attention weights and a link prediction layer to predict protein function. In addition, HNetGO uses the cross-entropy loss function to optimize the model.
At the amino acid residue level, experiments [25] show that the performance of this model has some disadvantages compared with models that use protein evolutionary information. In fact, evolutionary information reflects the relationship between proteins, rather than the semantic information within a single protein sequence, so it is no wonder that such model designed to extract single sequence-level features cannot capture this information. To solve this problem, we explicitly integrate the sequence similarity information obtained from the multiple sequence alignments (MSAs) algorithm and the detailed approach is explained in the next section.
In terms of implementation, we use the SeqVec model deployed based on ELMO, which is about 360 MB in size and uses about 33 M sequence data in the UniRef50 [50] database for pretraining. In experiments, with a Tesla P40 graphics card, we can complete the training of 20 395 sequences with an average length of 555.53 in 40 min, which means that, on average, we can get sequence features of a protein in 0.11 s. For each sequence, we get a 1 × 1024-dimensional protein-level feature, and for the input protein that does not contain sequence data, we use a randomly generated vector as its sequence feature.
Modeling evolutionary relationships with MSA
A pretrained model can effectively extract the semantic information contained in a single protein sequence, but theoretically it cannot capture the relationship between proteins. The evolutionary relationships of proteins are exactly a kind of relationship between proteins, which mainly refer to the homology of proteins and encode the information of biodiversity in the process of protein evolution. The amino acids in the protein sequence may mutate during the evolution process, causing the evolutionary tree to split. However, the protein sequence does not directly determine the function of protein, but indirectly affects the protein function through the protein structure, which means that the mutation of many amino acids may not cause the protein function to change [51]. Therefore, homologous proteins with different sequences tend to share similar structures and functions. Considering the significant sequence similarity between homologous proteins, protein sequence similarity, which can be easily obtained through MSAs, is suitable for inferring protein homolog. For example, some classic methods [52, 53] filter the results of MSAs according to a certain threshold and use these data to construct a protein similarity network, which is used by a subsequent clustering algorithm to infer protein homology.
Inspired by such methods, the HNetGO model uses diamond [54, 55], a fast and accurate MSA algorithm, to calculate sequence similarity between proteins, and selects the output with an e-value higher than 0.001 as the final alignment result, which is used to construct protein sequence similarity network. Compared with the manual integration approach used by the DeepGOPlus [17] model, our method can capture a wider range of local features through multi-layer aggregation operations between nodes on the sequence similarity network, whereas the manual integration method only considers the direct similarity between proteins.
PPI network
Existing studies [6, 56] have shown that deep learning models can predict the tertiary structure of proteins through sequences, so it is no wonder that pretraining models can extract structure-related local and global features contained in a single protein sequence. However, proteins usually do not perform their functions alone, but achieve specific functions through protein complexes formed by interacting with different proteins. This complex is the quaternary structure of proteins and reflects the association between proteins, which means that the semantic representation of a single protein sequence cannot express such type of relationship. PPI network is an undirected graph with proteins as nodes and protein interactions as edges, which can model the compound relationship between proteins, and thus can reflect the functional connection between proteins.
It should be noted that the protein ID used in the PPI data downloaded from the STRING database is not the same as the protein ID used in the sequence data. Therefore, we use the mapping file obtained from the Uniprot database to perform field alignment and data filtering on the PPI data. Table 2 shows some statistical information of the PPI network after processing.
Table 2.
PPI network statistics
| Datasets | Nodes | Edges | Average degree | Network density |
|---|---|---|---|---|
| Human | 18 560 | 11 098 152 | 597 | 0.064 |
| Mouse | 16 420 | 9 730 128 | 593 | 0.072 |
Heterogeneous information network
To avoid the use of manual design methods to converge the network, HNetGO uses a heterogeneous information network to model all the information extracted from the original data. The heterogeneous information network allows different types of edges and nodes to appear in the same network, and thus can model complex entity relationships. As shown in Figure 2, the heterogeneous network constructed in this paper consists of two types of nodes, namely protein and GO terms, and four types of edges between them, which can encode the hierarchical structure between terms, interactions and sequence similarity between proteins, and functional associations between proteins and terms. Formally speaking, we can define the heterogeneous graph as:
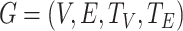 |
(1) |
and the corresponding node type mapping relationship is:
 |
(2) |
 |
(3) |
where  represents the collection of nodes,
represents the collection of nodes, 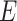 represents the collection of edges and each node
represents the collection of edges and each node 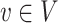 and each edge
and each edge 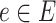 .
. 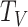 is the collection of all node types, including protein and term,
is the collection of all node types, including protein and term, 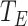 is a collection of all edge types, including four types of meta-relation, where
is a collection of all edge types, including four types of meta-relation, where  and
and  reflect the connection between proteins,
reflect the connection between proteins, 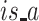 reflects the hierarchy structure between terms and
reflects the hierarchy structure between terms and  reflects the association between proteins and terms. Therefore, we can predict protein function by predicting the annotation relationship between protein nodes and term nodes in this heterogeneous network.
reflects the association between proteins and terms. Therefore, we can predict protein function by predicting the annotation relationship between protein nodes and term nodes in this heterogeneous network.
Model and implementation
Using heterogeneous network can greatly simplify data preprocessing and preserve more information extracted from original data, while at the same time it inevitably presents a great challenge to the design of prediction model. Inspired by the transformer-based deep learning model [57–59], we utilize a graph neural network based on attention mechanisms to learn embedding vector of nodes in heterogeneous networks, which are fed into a subsequent model for link prediction. As shown in Figure 3, our model is composed of three parts: a node-level mutual attention layer to learn the attention weights of all direct neighbors of the current node; a multi-head messaging layer enables different types of neighbors to deliver messages to the current node based on attention weights; a link prediction layer to predict protein function.
Node-level mutual attention
Similar to the Transformer model, for a given triplet  , we map the source node
, we map the source node 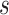 to a key vector, and the target node t to a query vector, and then calculate the contribution weight of the different source nodes to the target node through attention mechanism. If the input feature vector of the target node t is
to a key vector, and the target node t to a query vector, and then calculate the contribution weight of the different source nodes to the target node through attention mechanism. If the input feature vector of the target node t is 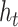 and the vector of the source node s is
and the vector of the source node s is 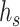 , the corresponding projected vector can be calculated as follows:
, the corresponding projected vector can be calculated as follows:
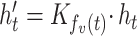 |
(4) |
 |
(5) |
where  and
and  are the projected vectors of target node and source node, respectively. And,
are the projected vectors of target node and source node, respectively. And, 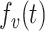 and
and  represent the type of node
represent the type of node 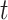 and
and 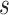 .
. 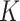 and
and 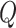 are type-specific transformation matrices related to node type which map different dimensions or different types of features into the same hidden semantic space and enable the model to calculate similarity score between any node pair. Then, the similarity score is calculated as follows:
are type-specific transformation matrices related to node type which map different dimensions or different types of features into the same hidden semantic space and enable the model to calculate similarity score between any node pair. Then, the similarity score is calculated as follows:
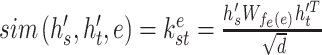 |
(6) |
where 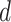 is the dimension of hidden space and
is the dimension of hidden space and 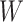 is a weight matrix associated with the type of edge
is a weight matrix associated with the type of edge 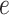 . They capture different semantic relationships formed by the same nodes over different type of edges. For example, two proteins with similar sequences may also interact with each other. By learning different weight matrices based on edge type, HNetGO model is able to extract different features between the same protein pairs. After calculating the similarity score of all neighbors of the target node
. They capture different semantic relationships formed by the same nodes over different type of edges. For example, two proteins with similar sequences may also interact with each other. By learning different weight matrices based on edge type, HNetGO model is able to extract different features between the same protein pairs. After calculating the similarity score of all neighbors of the target node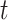 , the final attention weight can be obtained by normalizing the score through softmax function:
, the final attention weight can be obtained by normalizing the score through softmax function:
 |
(7) |
where 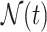 contains all neighbor nodes of the target node. If there is a node which is connected to the target node through different edges, we treat it as different neighbors during the calculation of attention weight. At the same time, for the same triple, the attention weight is not symmetrical for the source and target nodes, which means that their contributions to each other are different.
contains all neighbor nodes of the target node. If there is a node which is connected to the target node through different edges, we treat it as different neighbors during the calculation of attention weight. At the same time, for the same triple, the attention weight is not symmetrical for the source and target nodes, which means that their contributions to each other are different.
Multi-head attention and message passing
There are large differences between different types of nodes in heterogeneous networks, and even for the same type of nodes, their network characteristics, such as degree distribution, are usually extremely imbalance. To tackle this problem, we design a multi-head attention and message passing layer, which can extract the structure of heterogeneous network from different aspects. In particular, suppose we use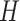 attention heads, then for a given triplet (s, e, t), the attention weight vector can be calculated by using:
attention heads, then for a given triplet (s, e, t), the attention weight vector can be calculated by using:
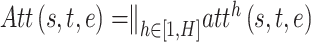 |
(8) |
Correspondingly, we design a multi-head message approach:
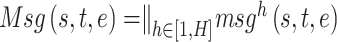 |
(9) |
 |
(10) |
where 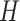 represents the number of attention heads,
represents the number of attention heads, 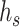 is the input feature of source node and
is the input feature of source node and 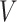 is a transformation matrix related to node type which can project
is a transformation matrix related to node type which can project 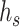 into a hidden space.
into a hidden space. 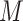 is an edge-type-specific matrix that allows the source node to deliver different messages to the destination node based on the edge type. Next, the embedding vector of target node
is an edge-type-specific matrix that allows the source node to deliver different messages to the destination node based on the edge type. Next, the embedding vector of target node 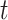 can be obtained by aggregating information from all of its neighbor nodes according to the corresponding attention weight vector:
can be obtained by aggregating information from all of its neighbor nodes according to the corresponding attention weight vector:
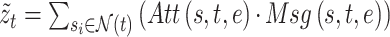 |
(11) |
Finally, we map the embedding vector back to the target node space and add residual connection to prevent network degradation:
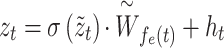 |
(12) |
where  is the parameter matrix of linear projection,
is the parameter matrix of linear projection,  is the original feature vector of node t, and
is the original feature vector of node t, and 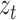 is the output embedding vector of the target node,
is the output embedding vector of the target node, 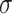 refers to sigmoid function.
refers to sigmoid function.
Protein-term link prediction
For any given protein node 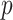 , as well as corresponding term node
, as well as corresponding term node 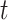 , we use the embedding vector obtained above to calculate a similarity score for this pair of nodes:
, we use the embedding vector obtained above to calculate a similarity score for this pair of nodes:
 |
(13) |
where 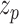 is the embedding vector of protein node,
is the embedding vector of protein node, 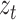 is the embedding vector of term node
is the embedding vector of term node  is a parameter matrix for training which maps embedding vector of protein node to the term space.
is a parameter matrix for training which maps embedding vector of protein node to the term space. 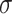 is the sigmoid function and converts the output value of the decoder into a probability value
is the sigmoid function and converts the output value of the decoder into a probability value 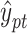 , which is between (0,1) and is regarded as the confidence value of the protein function prediction. Specifically, HNetGO regards node pairs with a
, which is between (0,1) and is regarded as the confidence value of the protein function prediction. Specifically, HNetGO regards node pairs with a 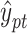 value >0.4 as positive examples of function prediction.
value >0.4 as positive examples of function prediction.
Finally, we use the cross-entropy loss function to optimize the model:
 |
(14) |
here  refers to the real annotation relationship between node p and t.
refers to the real annotation relationship between node p and t.
For any protein with unknown function, we use the following three steps to predict its function. First, extract sequence features of the protein through the SeqVec model. Then, use the Diamond algorithm to find the homologous protein node and add the new node to heterogeneous network according to the sequence similarity relationship. Finally, in the prediction stage, the HNetGO model only relies on the neighborhood information of the node, so the subgraph containing this protein node can be extracted through neighborhood sampling to perform function prediction.
Experiment and evaluation criterion
Evaluation criterion
As what was done in other works [20, 21, 60–63], we select area under the ROC curve (AUC), area under the precision-recall curve (AUPR) and Fmax to evaluate the performance of the model from different aspects [64, 65].
AUC reflects the tradeoff between true positive rate (TPR) and false positive rate (FPR) of a model, whereas AUPR pays more attention to the tradeoff between TPR and positive predictive value. Fmax is an official evaluation criterion of the critical assessment of functional annotation [66], which measures the average accuracy and recall rate of the model. For a given threshold 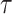 , the average precision (
, the average precision (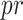 ), average recall rate (
), average recall rate (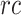 ) and Fmax on the test set are defined as follows:
) and Fmax on the test set are defined as follows:
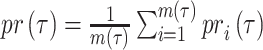 |
(15) |
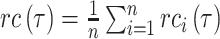 |
(16) |
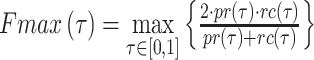 |
(17) |
where n is the total number of proteins in the dataset and 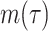 is the number of proteins that annotated with at least one term with the threshold
is the number of proteins that annotated with at least one term with the threshold 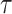 .
. 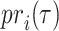 and
and  refer to the precision and recall rate of the i-th protein with the threshold τ and can be defined by the following formula:
refer to the precision and recall rate of the i-th protein with the threshold τ and can be defined by the following formula:
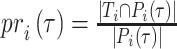 |
(18) |
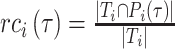 |
(19) |
where 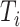 is the ground truth of protein i and
is the ground truth of protein i and 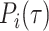 refers to the predicted label under specific threshold
refers to the predicted label under specific threshold 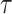 .
.
Experiments
To validate the rationality of data integration method and model design, we design different submodels and conduct experiments on mouse and human datasets. In total, we design three variants of HNetGO: HNetGO-PPI removes sequence similarity link from heterogeneous network, whereas HNetGO-SIM removes PPI link and HNetGO-RAN replace features extracted from the pretrained model with randomly generated vectors. In implementation, we take 5-fold cross-validation to reduce the experimental error and make full use of the dataset, which means that in each experiment, the training dataset contains 80% of the data, and the remaining 20% of the data is used as the test set. Specifically, the mouse dataset contains 16 420 protein nodes, of which 13 136 nodes are used as the training set, and the human dataset contains 18 560 protein nodes, of which 14 848 nodes are treated as training set.
As shown in Table 3, removing any part of a heterogeneous network will result in a performance degradation of the model, which precisely indicates that different types of relationships in the network contribute different information and also suggests that our model can effectively extract information encoded by different types of links. In particular, we can easily find that the removal of the PPI network has the greatest impact on the BP branch, which reflects the fact that both BP and PPI network focus on the functional interaction relationship between proteins. Besides, when replacing features extracted by pretrained model with randomly generated vectors, we observe not only a performance loss in the experiment, but also a decrease in convergence speed and stability, which indicates that pretrained models can effectively extract semantic information from a single protein sequence.
Table 3.
Results of ablation experiment
| Models | Human | Mouse | |||||
|---|---|---|---|---|---|---|---|
| F max | AUC | AUPR | F max | AUC | AUPR | ||
| BP | HNetGO | 0.561 | 0.909 | 0.625 | 0.543 | 0.890 | 0.579 |
| HNetGO-PPI | 0.448 | 0.879 | 0.450 | 0.422 | 0.838 | 0.419 | |
| HNetGO-SIM | 0.328 | 0.764 | 0.285 | 0.331 | 0.751 | 0.297 | |
| HNetGO-RAN | 0.404 | 0.819 | 0.389 | 0.393 | 0.806 | 0.371 | |
| CC | HNetGO | 0.748 | 0.971 | 0.812 | 0.742 | 0.969 | 0.808 |
| HNetGO-PPI | 0.498 | 0.891 | 0.494 | 0.537 | 0.893 | 0.531 | |
| HNetGO-SIM | 0.474 | 0.878 | 0.458 | 0.500 | 0.886 | 0.492 | |
| HNetGO-RAN | 0.441 | 0.854 | 0.425 | 0.432 | 0.867 | 0.414 | |
| MF | HNetGO | 0.697 | 0.959 | 0.771 | 0.674 | 0.953 | 0.743 |
| HNetGO-PPI | 0.566 | 0.892 | 0.584 | 0.551 | 0.888 | 0.563 | |
| HNetGO-SIM | 0.548 | 0.891 | 0.567 | 0.549 | 0.883 | 0.556 | |
| HNetGO-RAN | 0.589 | 0.911 | 0.615 | 0.573 | 0.904 | 0.579 | |
BP, biological process; CC, cellular component; MF, molecular function.
To further verify the performance of the model, we compared our model with several prevailing methods on the human protein dataset, including Naïve and BLAST [67], DeepGO [16] and DeepGOPlus [17], as well as GONET [20] and DeepGOA [21]. The results of comparative experiment are shown in Table 4.
Table 4.
Evaluation on human dataset with other models
| Methods | BP | CC | MF | ||||||
|---|---|---|---|---|---|---|---|---|---|
| F max | AUC | AUPR | F max | AUC | AUPR | F max | AUC | AUPR | |
| Naïve | 0.344 | 0.500 | 0.566 | 0.551 | 0.487 | 0.377 | 0.326 | 0.499 | 0.528 |
| BLAST | 0.339 | 0.577 | 0.489 | 0.441 | 0.563 | 0.269 | 0.411 | 0.623 | 0.461 |
| GONET | 0.612 | 0.934 | 0.581 | 0.718 | 0.972 | 0.780 | 0.646 | 0.973 | 0.709 |
| DeepGO | 0.327 | 0.639 | 0.571 | 0.589 | 0.695 | 0.448 | 0.404 | 0.760 | 0.625 |
| DeepGOPlus | 0.362 | 0.687 | 0.608 | 0.628 | 0.652 | 0.487 | 0.468 | 0.819 | 0.694 |
| DeepGOA | 0.385 | 0.698 | 0.622 | 0.629 | 0.757 | 0.500 | 0.477 | 0.820 | 0.710 |
| HNetGO | 0.561 | 0.909 | 0.625 | 0.748 | 0.971 | 0.812 | 0.697 | 0.959 | 0.771 |
BP, biological process; CC cellular component; MF molecular function.
Naïve and BLAST are rule-based methods, which can directly annotate protein functions. Naïve is an intuitive method that annotates proteins according to the frequency of GO terms, and thus the algorithm predicts same annotations for all samples in the dataset. BLAST is a classic method based on protein sequence similarity, and as mentioned above, here we use Diamond to calculate sequence similarity between proteins. For other models, we explained in detail in the Introduction section, and it should be noted that GONET actually combines Prot2Vec and a well-designed deep learning model to extract protein sequence features. In detail, GONET first splices the amino acid-level vectors output by Prot2Vec into a matrix (which is a protein-level feature),then reduces the dimensionality of the matrix through a convolutional neural network and finally uses a RNN to extract the long-range connection of protein sequence. Besides, for DeepGO and DeepGOPlus, we predict the function of the protein based on the tools provided by the original author and calculate the prediction performance based on this result.
From Table 3, it is obvious that HNetGO achieves better performance on several evaluation metrics than other models, which indicates that the data integration method and model design of this paper are reasonable. However, despite the state-of-the-art performance achieved in AUPR, HNetGO performs relatively poorly on the AUC criterion, which means that the FPR of the predicted results of our model is slightly higher. This may be partly caused by the incompleteness of protein annotation dataset, which means that there may be some new functions added to a protein in the future dataset, whereas such functions in the prediction results will be regarded as negative examples under the current dataset. Therefore, we cannot determine whether the small decrease in the AUC reflects a decrease in the model’s predictive performance or a better generalization ability. In conclusion, compared with GONET, HNetGO obtains considerable performance, which suggests that it is feasible to replace complex models based on biological prior knowledge with pretrained models.
CONCLUSION AND DISCUSSION
In this paper, we propose an end-to-end, attention-based link prediction model named HNetGO to predict protein function, which can efficiently integrate protein sequence and interaction data through heterogeneous information networks. In particular, HNetGO innovatively utilizes heterogeneous information networks to model the complex relationship between proteins and GO terms and extract distributed embedding features of protein sequences based on the pretrained model. In addition, we introduced a powerful attention-based graph neural network to learn node embedding in heterogeneous networks.
At the same time, in general, amino acids only indirectly affect protein function through the structure of proteins, which determines that protein function prediction is a protein-level task, not an amino acid-level task. Therefore, it is an intuitive and effective choice to build functional prediction models based on protein-level pretrained features. And in this paper, we demonstrate that it is reasonable to replace complex models based on biological prior knowledge with pretrained models.
Finally, it should be noted that the GO database contains a large amount of information about genes and gene products, which means that the relationship between GO terms is very complex and each term has specific functional semantics and is dataset independent. However, due to the limitations of model complexity, we use only part of the association between GO terms, and the input feature of the term nodes is also randomly generated. In future work, we will try to design more complex models to fully mining the information in GO and explore the possibility of using pretrained language models to extract text semantic features of GO terms.
Additional Files
All additional files are available at: https://github.com/BIOGOHITSZ/HNetGO.
AUTHORS’ CONTRIBUTIONS
X.Z., Y.G. and F.Z. performed bioinformatics analysis, X.W., K.W., S.Q., B.L., Y.W., Y.H. and J.L. designed the study, and J.L. drafted the manuscript. All of the authors performed the analysis. J.L. conceived of the study, participated in its design and coordination and drafted the manuscript.
Acknowledgements
Nil.
Contributor Information
Xiaoshuai Zhang, School of Computer Science and Technology, Harbin Institute of Technology (Shenzhen), Shenzhen, Guangdong 518055, China.
Huannan Guo, General Hospital of Heilongjiang Province Land Reclamation Bureau, Harbin 150086, China.
Fan Zhang, Center NHC Key Laboratory of Cell Transplantation, The First Affiliated Hospital of Harbin Medical University, Harbin 150086, China.
Xuan Wang, School of Computer Science and Technology, Harbin Institute of Technology (Shenzhen), Shenzhen, Guangdong 518055, China.
Kaitao Wu, School of Computer Science and Technology, Harbin Institute of Technology (Shenzhen), Shenzhen, Guangdong 518055, China.
Shizheng Qiu, School of Computer Science and Technology, Harbin Institute of Technology, Harbin 150001, China.
Bo Liu, School of Computer Science and Technology, Harbin Institute of Technology, Harbin 150001, China.
Yadong Wang, School of Computer Science and Technology, Harbin Institute of Technology (Shenzhen), Shenzhen, Guangdong 518055, China; School of Computer Science and Technology, Harbin Institute of Technology, Harbin 150001, China.
Yang Hu, School of Computer Science and Technology, Harbin Institute of Technology, Harbin 150001, China.
Junyi Li, School of Computer Science and Technology, Harbin Institute of Technology (Shenzhen), Shenzhen, Guangdong 518055, China.
FUNDING
National Key Research Program (2021YFA0910700); Shenzhen Science and Technology University stable support program (GXWD20201230155427003-20200821222112001); National Natural Science Foundation of China (82003553); Guangdong Key Area Research Program (2020B0101380001); Shenzhen Science and Technology Program (JCYJ20200109113201726).
References
- 1. Consortium GO . The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res 2019;47(D1):D330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Valentini G. True path rule hierarchical ensembles for genome-wide gene function prediction. IEEE/ACM Trans Comput Biol Bioinform 2010;8(3):832–47. [DOI] [PubMed] [Google Scholar]
- 3. Abbass J, Nebel J-C. Rosetta and the journey to predict proteins' structures, 20 years on. Curr Bioinform 2020;15(6):611–28. [Google Scholar]
- 4. Cheng L, Hu Y, Sun J, et al. DincRNA: a comprehensive web-based bioinformatics toolkit for exploring disease associations and ncRNA function. Bioinformatics 2018;34(11):1953–6. [DOI] [PubMed] [Google Scholar]
- 5. Jurtz VI, Johansen AR, Nielsen M, et al. An introduction to deep learning on biological sequence data: examples and solutions. Bioinformatics 2017;33(22):3685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature 2020;577(7792):706–10. [DOI] [PubMed] [Google Scholar]
- 7. Hu Y, Qiu S, Cheng L. Integration of multiple-omics data to analyze the population-specific differences for coronary artery disease. Comput Math Methods Med 2021;2021:7036592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Razzak MI, Naz S, Zaib A. Deep learning for medical image processing: overview, challenges and the future. In: Dey N, Ashour AS, Borra S (eds). Classification in BioApps, Cham, Switzerland: Springer International Publishing AG, 2018;323–50. [Google Scholar]
- 9. Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 2010;38(suppl_2):W214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lan L, Djuric N, Guo Y, et al. MS-kNN: protein function prediction by integrating multiple data sources. BMC Bioinformatics 2013;14(Suppl 3):S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. You R, Yao S, Xiong Y, et al. NetGO: improving large-scale protein function prediction with massive network information. Nucleic Acids Res 2019;47(W1):W379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao T, Hu Y, Peng J, et al. DeepLGP: a novel deep learning method for prioritizing lncRNA target genes. Bioinformatics 2020;36(16):4466–72. [DOI] [PubMed] [Google Scholar]
- 13. Cheng L. Computational and biological methods for gene therapy. Curr Gene Ther 2019;19(4):210. [DOI] [PubMed] [Google Scholar]
- 14. Mosharaf MP, Hassan MM, Ahmed FF, et al. Computational prediction of protein ubiquitination sites mapping on Arabidopsis thaliana. Comput Biol Chem 2020;85:107238. [DOI] [PubMed] [Google Scholar]
- 15. Zhu H, Du X, Yao Y. ConvsPPIS: identifying protein-protein interaction sites by an ensemble convolutional neural network with feature graph. Curr Bioinform 2020;15(4):368–78. [Google Scholar]
- 16. Kulmanov M, Khan MA, Hoehndorf R, et al. DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics 2018;34(4):660–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kulmanov M, Hoehndorf R. DeepGOPlus: improved protein function prediction from sequence. Bioinformatics 2020;36(2):422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gligorijević V, Barot M, Bonneau R. deepNF: deep network fusion for protein function prediction. Bioinformatics 2018;34(22):3873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peng J, Xue H, Wei Z, et al. Integrating multi-network topology for gene function prediction using deep neural networks. Brief Bioinform 2021;22(2):2096–105. [DOI] [PubMed] [Google Scholar]
- 20. Li J, Wang L, Zhang X, et al. GONET: a deep network to annotate proteins via recurrent convolution networks. In: 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Seoul, Korea, p.29–34. IEEE, New York, NY, USA. [Google Scholar]
- 21. Zhou G, Wang J, Zhang X, Yu G. Deepgoa: predicting gene ontology annotations of proteins via graph convolutional network. In: 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, p.1836–41. IEEE, New York, NY, USA. [Google Scholar]
- 22. Lv Z, Ao C, Zou Q. Protein function prediction: from traditional classifier to deep learning. Proteomics 2019;19(14):1900119. [DOI] [PubMed] [Google Scholar]
- 23. Cao Y, Shen Y. TALE: transformer-based protein function annotation with joint sequence-label embedding. Bioinformatics 2021;37(18):2825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yan N, Lv Z, Hong W, et al. Editorial: feature representation and learning methods with applications in protein secondary structure. Front Bioeng Biotechnol 2021;9(822):748722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heinzinger M, Elnaggar A, Wang Y, et al. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinformatics 2019;20(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lv Z, Wang P, Zou Q, et al. Identification of sub-Golgi protein localization by use of deep representation learning features. Bioinformatics 2020;36(24):5600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lv Z, Cui F, Zou Q, et al. Anticancer peptides prediction with deep representation learning features. Brief Bioinform 2021;22:bbab1008. [DOI] [PubMed] [Google Scholar]
- 28. Cheng L, Shi H, Wang Z, et al. IntNetLncSim: an integrative network analysis method to infer human lncRNA functional similarity. Oncotarget 2016;7(30):47864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Devlin J, Chang MW, Lee K, et al. BERT: Pre-Training of Deep Bidirectional Transformers for Language Understanding, Stroudsburg, PA, USA: Association for Computational Linguistics, 2018.
- 30. Yang Z, Dai Z, Yang Y, et al. XLNet: Generalized Autoregressive Pretraining for Language Understanding, New York, NY, USA: Curran Associates Inc., 2019.
- 31. Asgari E, Mofrad MR. Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One 2015;10(11):e0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alley EC, Khimulya G, Biswas S, et al. Unified rational protein engineering with sequence-based deep representation learning. Nat Methods 2019;16(12):1315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rives A, Meier J, Sercu T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci U S A 2021;118(15):e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Elnaggar A, Heinzinger M, Dallago C, et al. ProtTrans: towards cracking the language of Life's code through self-supervised deep learning and high performance computing. arXiv preprint arXiv:200706225 2020. [Google Scholar]
- 35. Joulin A, Grave E, Bojanowski P, et al. Bag of tricks for efficient text classification. arXiv preprint arXiv:160701759 2016. [Google Scholar]
- 36. Mikolov T, Chen K, Corrado G, et al. Efficient estimation of word representations in vector space. arXiv preprint arXiv:13013781 2013. [Google Scholar]
- 37. Di L, He Y, Lu Y. Deep novo a plus: improving the deep learning model for de novo peptide sequencing with additional ion types and validation set. Curr Bioinform 2020;15(8):949–54. [Google Scholar]
- 38. Long H, Sun Z, Li M, et al. Predicting protein phosphorylation sites based on deep learning. Curr Bioinform 2020;15(4):300–8. [Google Scholar]
- 39. Zhang T, Wei X, Li Z, et al. Natural scene nutrition information acquisition and analysis based on deep learning. Curr Bioinform 2020;15(7):662–70. [Google Scholar]
- 40. Zhang Y, Yan J, Chen S, et al. Review of the applications of deep learning in bioinformatics. Curr Bioinform 2020;15(8):898–911. [Google Scholar]
- 41. Ahmad F, Farooq A, Khan MUG. Deep learning model for pathogen classification using feature fusion and data augmentation. Curr Bioinform 2021;16(3):466–83. [Google Scholar]
- 42. Consortium U . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47(D1):D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47(D1):D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Charoenkwan P, Nantasenamat C, Hasan MM, et al. BERT4Bitter: a bidirectional encoder representations from transformers (BERT)-based model for improving the prediction of bitter peptides. Bioinformatics 2021;37(17):2556–62. [DOI] [PubMed] [Google Scholar]
- 45. Hasan MM, Schaduangrat N, Basith S, et al. HLPpred-fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 2020;36(11):3350–6. [DOI] [PubMed] [Google Scholar]
- 46. Anfinsen CB. Principles that govern the folding of protein chains. Science 1973;181(4096):223–30. [DOI] [PubMed] [Google Scholar]
- 47. Peters ME, Neumann M, Iyyer M, et al. Deep contextualized word representations. arXiv preprint arXiv:180205365 2018. [Google Scholar]
- 48. Littmann M, Heinzinger M, Dallago C, et al. Embeddings from deep learning transfer GO annotations beyond homology. Sci Rep 2021, 11(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Villegas-Morcillo A, Makrodimitris S, van Ham RC, et al. Unsupervised protein embeddings outperform hand-crafted sequence and structure features at predicting molecular function. Bioinformatics 2021;37(2):162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suzek BE, Wang Y, Huang H, et al. Consortium U: UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015;31(6):926–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rao R, Bhattacharya N, Thomas N, et al. Evaluating protein transfer learning with tape. Adv Neural Inf Process Syst 2019;32:9689. [PMC free article] [PubMed] [Google Scholar]
- 52. Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 2002;30(7):1575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Azad A, Pavlopoulos GA, Ouzounis CA, et al. HipMCL: a high-performance parallel implementation of the Markov clustering algorithm for large-scale networks. Nucleic Acids Res 2018;46(6):e33–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015;12(1):59–60. [DOI] [PubMed] [Google Scholar]
- 55. Buchfink B, Reuter K, Drost H-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 2021;18(4):366–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Billings WM, Hedelius B, Millecam T, et al. ProSPr: democratized implementation of alphafold protein distance prediction network. BioRxiv 2019;830273. [Google Scholar]
- 57. Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. arXiv preprint arXiv:170603762 2017. [Google Scholar]
- 58. Veličković P, Cucurull G, Casanova A, et al. Graph attention networks. arXiv preprint arXiv:171010903 2017. [Google Scholar]
- 59. Hu Z, Dong Y, Wang K, Sun Y. Heterogeneous graph transformer. In: Proceedings of The Web Conference, Taipei, Taiwan, 2020, p.2704–10. Association for Computing Machinery, New York, NY, USA.
- 60. Cai Y, Wang J, Deng L. SDN2GO: an integrated deep learning model for protein function prediction. Front Bioeng Biotechnol 2020;8:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wei L, He W, Malik A, et al. Computational prediction and interpretation of cell-specific replication origin sites from multiple eukaryotes by exploiting stacking framework. Brief Bioinform 2021;22(4):bbaa275. [DOI] [PubMed] [Google Scholar]
- 62. Hasan MM, Alam MA, Shoombuatong W, et al. NeuroPred-FRL: an interpretable prediction model for identifying neuropeptide using feature representation learning. Brief Bioinform 2021;22(6):bbab167. [DOI] [PubMed] [Google Scholar]
- 63. Charoenkwan P, Chiangjong W, Nantasenamat C, et al. StackIL6: a stacking ensemble model for improving the prediction of IL-6 inducing peptides. Brief Bioinform 2021;22:bbab172. [DOI] [PubMed] [Google Scholar]
- 64. Zhao TY, Wang DH, Hu Y, et al. Identifying Alzheimer's disease-related miRNA based on semi-clustering. Curr Gene Ther 2019;19(4):216–23. [DOI] [PubMed] [Google Scholar]
- 65. Zhuang H, Zhang Y, Yang S, et al. A Mendelian randomization study on infant length and type 2 diabetes mellitus risk. Curr Gene Ther 2019;19(4):224–31. [DOI] [PubMed] [Google Scholar]
- 66. Zhou N, Jiang Y, Bergquist TR, et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol 2019;20(1):1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25(17):3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]