

SUMMARY
We present a case of transient form of type 1 pseudohypoaldosteronism (S-PHA) in a 1.5-month-old male infant who presented with lethargy, failure to thrive, severe hyponatremia (Na=118 mmol/L), hypochloremia (Cl=93 mmol/L) and fever due to urinary tract infection. Potassium levels were normal. Markedly elevated serum aldosterone level and elevated serum renin confirmed the diagnosis of pseudohypoaldosteronism. Renal ultrasound showed grade III hydronephrosis on the left kidney while contrast-enhanced voiding urosonography excluded the existence of vesicoureteral reflux, which raised suspicion of obstructive uropathy at the level of vesicoureteral junction. Serum sodium normalized after several days of intravenous fluids and antibiotic therapy, after which oral supplementation of sodium was introduced. The levels of 17-hydroxyprogesterone, adrenocorticotropic hormone, cortisol and thyroid-stimulating hormone were normal. Functional magnetic resonance urography conducted at the age of 3 months confirmed the diagnosis of primary congenital obstructive megaureter and the infant was referred to a pediatric surgeon. Although a rare occurrence, S-PHA can be a potentially life-threatening condition in infants if not recognized and treated appropriately. Therefore, serum concentrations of electrolytes should be obtained in every child diagnosed with obstructive anomaly of the urinary tract and/or acute cystopyelonephritis. On the other hand, every child diagnosed with S-PHA should be evaluated for obstructive anomaly of the urinary tract.
Key words: Pseudohypoaldosteronism, Unilateral hydronephrosis, Obstructive megaureter
Introduction
Differential diagnosis in infants presenting with hyponatremia is broad. In co-occurrence with hyperkalemia, congenital adrenal hyperplasia (CAH) is usually suspected. However, the presence of hyponatremia in an infant with urinary tract infection (UTI) and/or urinary tract malformation (UTM) should raise suspicion of transient (secondary) form of type 1 pseudohypoaldosteronism (S-PHA) (1).
Pseudohypoaldosteronism is characterized by renal tubular resistance to aldosterone, resulting in potentially fatal electrolyte shifts, i.e., hyponatremia, hyperkalemia, metabolic acidosis, followed by elevated plasma renin and aldosterone levels. There are 2 types: pseudohypoaldosteronism type 1 (PHA-1), which can be primary (caused by mutation in mineralocorticoid receptor) or secondary (precipitated by UTI or UTM). Pseudohypoaldosteronism type 2 (PHA-2), or Gordon’s syndrome, is a rare inherited monogenic disorder characterized by hyperkalemia and hypertension (1, 2).
In case of S-PHA, electrolyte disturbances usually resolve with treatment of UTI and dehydration (3). In order to avoid unnecessary diagnostic procedures and raise awareness of this rare but potentially life-threatening condition, we present a case of transient PHA secondary to UTI in a 1.5-month-old male patient with unilateral hydronephrosis due to primary obstructive megaureter (POM) with ureterovesical junction obstruction (UVJO).
Case Report
A 1.5-month-old male infant was admitted to the Pediatric Department due to severe hyponatremia, UTI and failure to thrive. He initially presented to the Emergency Room with lethargy, fever, and poor weight gain (420 g from birth). He weighed 3730 g (9.c, -1.36 z-score), which was the same weight he had two weeks prior to admission during his first visit to pediatrician. He was born full-term by cesarean section, birth weight of 3380 g (49.c, -0.03 z-score) from noncomplicated pregnancy. The infant was restless at examination, had prolonged capillary refill and slow skin recoil. Physical examination showed normal genitalia without hyperpigmentation and no palpable abdominal mass. Initial laboratory testing revealed severe hyponatremia (Na 118 mmol/L; reference value 134-142 mmol/L) and hypochloremia (93 mmol/L; reference value 96-111 mmol/L) but surprisingly, without metabolic acidosis, hyperglycemia or hyperkalemia (potassium level at admission was 5.8 mmol/L; reference range 3-7 mmol/L). Urinalysis demonstrated pyuria and bacteriuria with slightly elevated C-reactive protein (22.9 mg/L), therefore empiric dual antibiotic treatment was introduced (ampicillin and gentamicin).
Serum sodium normalized after several days of intravenous fluids and antibiotic therapy, then oral supplementation of sodium was introduced initially at 3 mmol/kg with gradually lowering dosage until discharge. He was discharged with no oral sodium supplementation, his sodium levels remaining within the reference range. Initial abdominal ultrasound revealed grade 3 hydronephrosis on the left side. Urine culture subsequently grew Enterobacter spp., so antibiotic treatment was switched to ceftriaxone.
On day 5 of antibiotic treatment, he developed acalculous cholecystitis (elevated liver enzymes and bile acids) without signs of pseudocholelithiasis (Fig. 1), probably due to the iatrogenic effect of ceftriaxone, which led to switching antibiotic therapy to meropenem. Other frequent possible causes of liver lesion in early infancy (cytomegalovirus infection, cystic fibrosis, and alpha-1 antitrypsin deficiency) were all excluded and ursodeoxycholic acid was introduced into therapy.
Fig. 1.
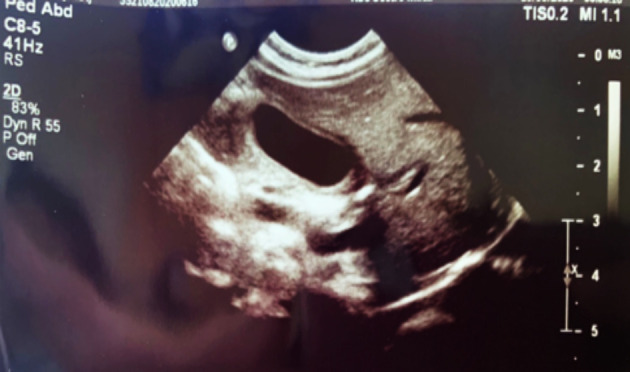
Thickened gallbladder wall as a sign of acalculous cholecystitis.
The diagnosis of PHA was confirmed when the serum aldosterone level obtained on admission was markedly elevated, >131.8 ng/dL (r.v. 7-39 ng/dL), with elevated serum renin, >330 ng/L (r.v. 23.5-370 ng/L). The levels of 17-hydroxyprogesterone, adrenocorticotropic hormone, cortisol and thyroid-stimulating hormone were all normal. No signs of neurological deficit were observed.
Renal ultrasound confirmed hydronephrosis on the left side while contrast-enhanced voiding urosonography excluded the existence of vesicoureteral reflux, which aroused suspicion of obstructive uropathy at the level of vesicoureteral junction, thus demanding further imaging.
Cardiology work-up included electrocardiography and heart ultrasound, which revealed no abnormalities.
Upon discharge, the patient continued receiving antibiotic prophylaxis with cephalexin. Ursodeoxycholic acid was gradually excluded upon normalization of liver enzymes. During follow up, he had no further UTIs, renin and aldosterone levels gradually decreased although, at the age of 3 months, they did not reach normal values (aldosterone at 3-month follow-up was still elevated at 108.3 ng/dL).
Functional magnetic resonance urography conducted at the age of 3 months confirmed the diagnosis of congenital POM and the infant was referred to a pediatric surgeon, who scheduled a surgery at the age of 1 year (Figs. 1-4).
Fig. 2.
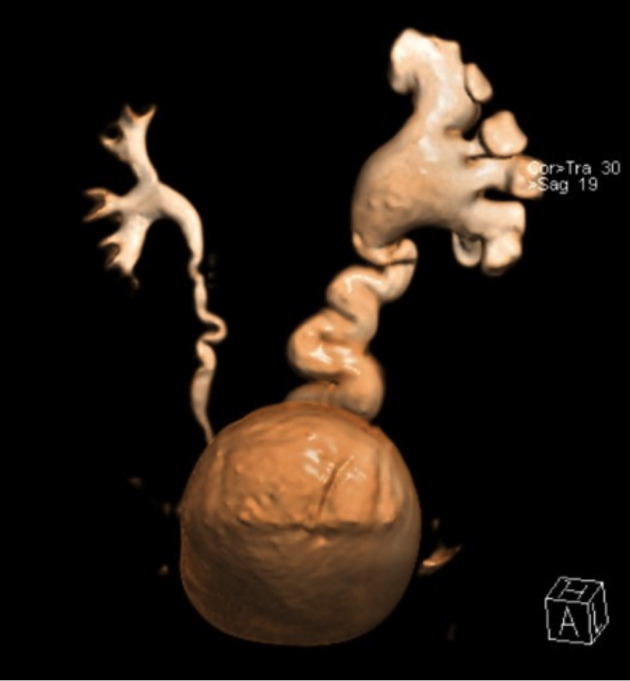
Magnetic resonance urography – volume rendering reconstruction: left-sided hydroureteronephrosis, primary obstructive megaureter.
Fig. 3.
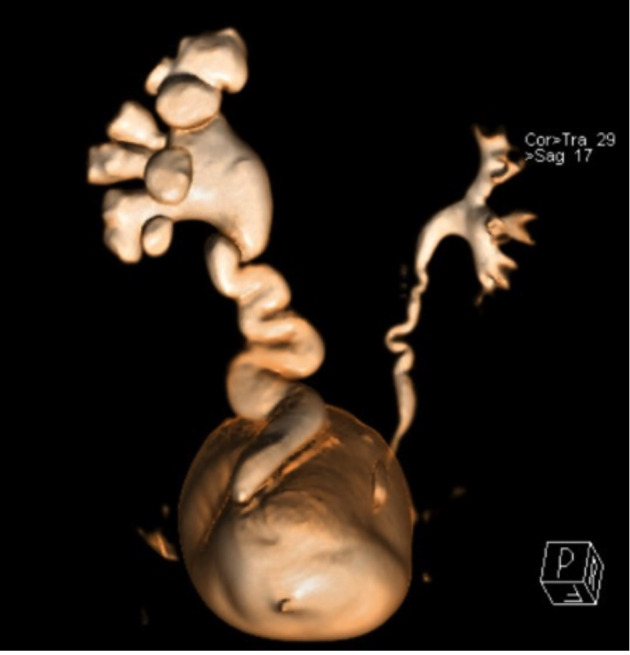
Magnetic resonance urography – volume rendering reconstruction: left-sided hydroureteronephrosis, dilated ureter with prevesical transition to aperistaltic normal caliper ureter, primary obstructive megaureter.
Fig. 4.
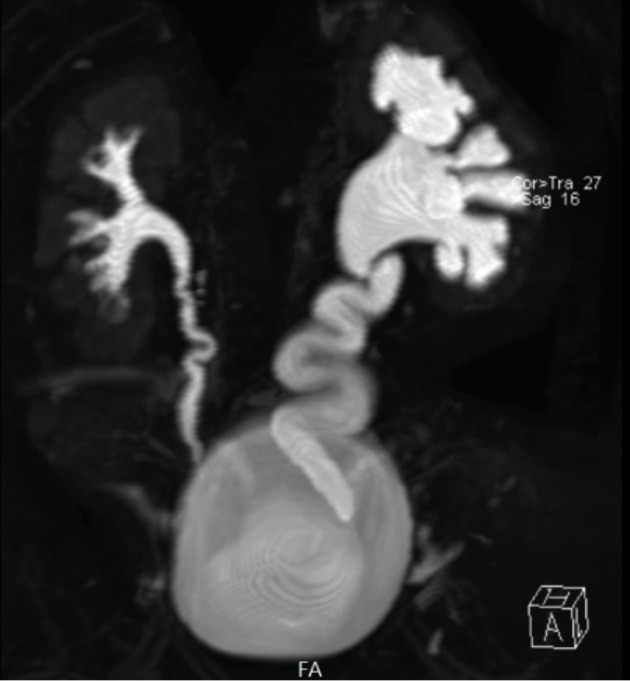
Magnetic resonance (MR) urography – coronal 3D T2-weighted fat-saturated FSE MR hydrogram: left-sided hydronephrosis, dilated ureter with prevesical transition to aperistaltic normal caliper ureter, parenchymal thinning.
Discussion
We present a patient who developed S-PHA secondary to UTI with unilateral hydronephrosis due to POM with UVJO. Eighteen more cases of S-PHA and POM have been described in the literature, 15 with associated UTI and 3 without associated UTI (2, 4, 5). Male gender and left-sided UTM in our patient confirm gender and left-side predominance of POM described in the literature (6). S-PHA mostly occurs in patients with UTI but around 10% of patients with S-PHA had UTM without proven UTI. In those patients, predominantly bilateral UTM was present. Furthermore, a retrospective study which included over 300 children with UTI showed that biochemical signs of aldosterone resistance were present in 3% of patients with electrolyte abnormalities occurring more often in younger infants (7). No connection was found between the type of UTM and the risk of developing S-PHA in the absence of UTI (2). Normalization of renin and aldosterone reportedly occurs after completion of treatment for UTI, which was not the case in our patient in the context of an underlying congenital abnormality of the kidney and urinary tract, indicative of ongoing distal tubular dysfunction.
It has been recognized that obstructive uropathy, which is most often associated with UTI, can lead to a salt-wasting syndrome resembling CAH by causing tubular resistance to aldosterone in neonates and young infants. The most prominent laboratory findings are hyponatremia, hyperkalemia and metabolic acidosis with elevations of plasma aldosterone concentration and plasma renin activity (8).
The signs and symptoms are usually nonspecific and include failure to thrive, vomiting, diarrhea and polyuria, which all lead to dehydration.
With hypernatremic dehydration being the most frequent type of dehydration in neonatal age due to poor feeding in the early days of life, hyponatremic dehydration should automatically arouse suspicion of endocrinological abnormalities, most commonly CAH. Contrary to CAH, in S-PHA the infant’s clinical status will not improve with stress-dose of corticosteroids but with volume resuscitation, sodium supplementation, and treatment of the underlying UTI.
Breast-fed infants (like our patient) are more prone to hyponatremia since breast milk has low sodium content in order to protect against hypernatremic dehydration. However, in infants losing sodium, it predisposes them to hyponatremic dehydration since sodium loss leads to contraction of circulatory volume with activation of the renin-angiotensin system and antidiuretic hormone. With resistance to aldosterone, the release of antidiuretic hormone further exacerbates hyponatremia (3).
Megaureter in children is ureter that exceeds 7 mm in diameter (9). It can occur due to various causes and abnormalities, and the exact etiology needs to be recognized in order to treat it correctly. POM is caused by intrinsic ureteral factors involving the ureterovesical junction, either mechanical (stricture, ureterocele, tumor) or functional (adynamic ureteral segment) (10). In comparison to non-obstructive forms of megaureter, it is less likely that POM will resolve spontaneously (6). According to the British Association of Pediatric Urologists consensus from 2014, surgical correction of POM is recommended for symptomatic patients (febrile UTI, pain) and asymptomatic patients with differential renal function below 40% or progressive hydronephrosis. In other cases, initial conservative management (annual ultrasound, prophylactic antibiotic therapy) is suggested (10).
Conclusion
Although a rare occurrence, S-PHA can be a potentially life-threatening condition in infants if not recognized and treated appropriately. Therefore, serum concentrations of electrolytes should be obtained in every child diagnosed with obstructive anomaly of the urinary tract and/or acute cystopyelonephritis. On the other hand, every child diagnosed with S-PHA should be evaluated for obstructive anomaly of the urinary tract. For the time being, no such recommendations are part of the current guidelines for diagnosis and treatment of UTI.
References
- 1.White PC. Adrenocortical insufficiency. In: Kliegman RM, St. Geme J, editors. Nelson Textbook of Pediatrics. 21st edn., Vol 2. Philadelphia: Elsevier, 2019; p. 2969. [Google Scholar]
- 2.Delforge X, Kongolo G, Cauliez A, Braun K, Haraux E, Buisson P. Transient pseudohypoaldosteronism: a potentially severe condition affecting infants with urinary tract malformation. J Pediatr Urol. 2019;15(3):265.E1-265.E7. doi: 10.1016/j.jpurol.2019.03.002. Epub 2019 Mar 9. PMID: 30962012. 10.1016/j.jpurol.2019.03.002 [DOI] [PubMed]
- 3.Abraham MB, Larkins N, Choong CS, Shetty VB. Transient pseudohypoaldosteronism in infancy secondary to urinary tract infection. J Paediatr Child Health. 2017;53(5):458–63. 10.1111/jpc.13481 [DOI] [PubMed] [Google Scholar]
- 4.Makino Y, Kanematsu A, Imamura M, Yoshimura K, Nishiyama H, Yorifuji T, et al. Pseudohypoaldosteronism type 1 in an infant with bilateral primary obstructive megaureter: a case report. Hinyokika Kiyo. 2011;57(4):193–7. 10.1681/ASN.2005111188 [DOI] [PubMed] [Google Scholar]
- 5.Belot A, Ranchin B, Fichtner C, Pujo L, Rossier BC, Liutkus A. Pseudohypoaldosteronisms, report on a 10 patient series. Nephrol Dial Transplant. 2008;23(5):1636–41. 10.1093/ndt/gfm862 [DOI] [PubMed] [Google Scholar]
- 6.Gimpel C, Masioniene L, Djakovic N, et al. Complications and long-term outcome of primary obstructive megaureter in childhood. Pediatr Nephrol. 2010;25:1679–86. 10.1007/s00467-010-1523-0 [DOI] [PubMed] [Google Scholar]
- 7.Sperl W, Guggenbichler JP, Warter T. Changes in electrolyte and acid-base equilibrium in children with acute urinary tract infections. Padiatr Padol. 1988;23:121–8. [in German] [PubMed] [Google Scholar]
- 8.Bogdanović R, Stajić N, Putnik J, et al. Transient type 1 pseudo-hypoaldosteronism: report on an eight-patient series and literature review. Pediatr Nephrol. 2009. November;24(11):2167–75. 10.1007/s00467-009-1285-8 [DOI] [PubMed] [Google Scholar]
- 9.Farrugia MK, Hitchcock R, Radford A, et al. British Association of Paediatric Urologists consensus statement on the management of the primary obstructive megaureter. J Pediatr Urol. 2014;10:26–33. Epub 2013 Oct 16. 10.1016/j.jpurol.2013.09.018 [DOI] [PubMed] [Google Scholar]
- 10.Lockhart JL, Singer AM, Glenn JF. Congenital megaureter. J Urol. 1979;122(3):310–4. 10.1016/S0022-5347(17)56380-6 [DOI] [PubMed] [Google Scholar]