

Abstract
Background and Aims
Exacerbated immune activation, intestinal dysbiosis and a disrupted intestinal barrier are common features among inflammatory bowel disease [IBD] patients. The polyamine spermidine, which is naturally present in all living organisms, is an integral component of the human diet, and exerts beneficial effects in human diseases. Here, we investigated whether spermidine treatment ameliorates intestinal inflammation and offers therapeutic potential for IBD treatment.
Methods
We assessed the effect of oral spermidine administration on colitis severity in the T cell transfer colitis model in Rag2−/− mice by endoscopy, histology and analysis of markers of molecular inflammation. The effects on the intestinal microbiome were determined by 16S rDNA sequencing of mouse faeces. The impact on intestinal barrier integrity was evaluated in co-cultures of patient-derived macrophages with intestinal epithelial cells.
Results
Spermidine administration protected mice from intestinal inflammation in a dose-dependent manner. While T helper cell subsets remained unaffected, spermidine promoted anti-inflammatory macrophages and prevented the microbiome shift from Firmicutes and Bacteroides to Proteobacteria, maintaining a healthy gut microbiome. Consistent with spermidine as a potent activator of the anti-inflammatory molecule protein tyrosine phosphatase non-receptor type 2 [PTPN2], its colitis-protective effect was dependent on PTPN2 in intestinal epithelial cells and in myeloid cells. The loss of PTPN2 in epithelial and myeloid cells, but not in T cells, abrogated the barrier-protective, anti-inflammatory effect of spermidine and prevented the anti-inflammatory polarization of macrophages.
Conclusion
Spermidine reduces intestinal inflammation by promoting anti-inflammatory macrophages, maintaining a healthy microbiome and preserving epithelial barrier integrity in a PTPN2-dependent manner.
Keywords: Spermidine, IBD, anti-inflammatory macrophages, dysbiosis, PTPN2
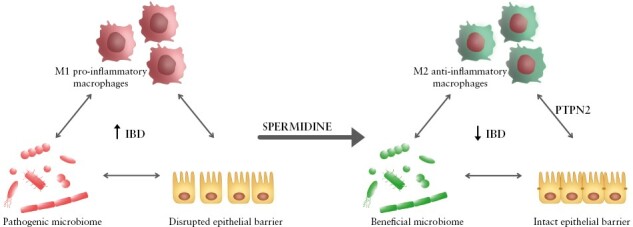
Graphical Abstract
Graphical Abstract.
1. Introduction
Spermidine is a polyamine naturally present in all living organisms. Levels of spermidine in the body depend on endogenous biosynthesis from putrescine and the conversion of spermine. Spermidine is also produced by intestinal microbes, and is an integral component of the human diet, with particularly high levels in soybeans, wheat germ and aged cheese.1–3 Physiologically, spermidine is essential for cell growth, survival and proliferation.1,2 It modulates and enhances autophagy and has been described to promote longevity.4 Furthermore, it has been shown that spermidine exerts anti-hypertensive properties in vivo,5 provides cardio- and neuroprotective effects, and stimulates anti-cancer immunosurveillance.2 Several studies have demonstrated that spermidine mediates anti-inflammatory effects, in particular in sepsis,6 lipopolysaccharide [LPS]-stimulated microglia,7 autoimmune encephalomyelitis,8 LPS-treated macrophages, LPS-treated zebrafish larvae,9 skin inflammation,10 interferon [IFN]-γ-stimulated epithelial cells,11 and in respiratory allergic inflammation,12, but its mode of action has not been fully elucidated. We have previously demonstrated protective effects of spermidine in a mouse model of acute dextran sodium sulphate [DSS]-induced colitis13 and demonstrated that the protein tyrosine phosphatase non-receptor type 2 [PTPN2] might be a key player in mediating the anti-inflammatory effects of spermidine.13,14 Notably, variants in the gene locus encoding PTPN2 confer an increased risk for the development of inflammatory bowel disease [IBD], while silencing PTPN2 in THP-1 monocytes and intestinal epithelial cells [IECs] abrogates the protective effects of spermidine. Nevertheless, the exact mechanisms by which spermidine exerts its anti-inflammatory effects in intestinal inflammation are still not understood, and it is unclear whether spermidine can prevent disease in more relevant, chronic and immune cell-mediated models of IBD.
IBD is a chronic, relapsing inflammatory disorder of the gastrointestinal tract, classified mainly into Crohn’s disease [CD] and ulcerative colitis [UC].15,16 The prevalence of IBD is increasing worldwide, especially in industrialized, developing countries, rendering IBD a global disease of the 21st century, affecting nearly 7 million people worldwide.17,18 Epithelial barrier impairment, excessive immune response and altered intestinal microbial composition are key events that drive IBD pathogenesis.19,20 Although great efforts have been made to understand the pathogenesis and the complexity of IBD over recent decades, there is still no cure for IBD. Thus, IBD poses a life-long burden on patients and often requires treatment for decades. Current therapeutic options, such as corticosteroids or biologicals, do not provide a causal treatment approach and are frequently not sustainably effective while exerting severe side effects. Surprisingly, targeting cytokines involved in the pathogenesis of IBD, such as IFN-γ, interleukin [IL]-17 or IL-13, failed to show significant clinical improvement in randomized, controlled clinical trials.21–23 On the other hand, most commonly used therapeutics such as anti-tumour necrosis factor [TNF]-α and anti-α4β7-integrin display great effectiveness in inducing remission, although efficacy varies significantly between IBD patients,24,25 and almost half of all patients eventually lose therapy response during the disease course.26,27 Therefore, it is undisputed that the development of safe and more effective therapeutic options is urgently needed.
Here we aimed to investigate the anti-inflammatory potential of spermidine in preclinical colitis models, explore its therapeutic potential and learn about spermidine’s mode of action as a potential novel therapeutic approach for the treatment of IBD.
2. Materials and Methods
2.1. Animal experiments
Wild-type C57BL/6J mice were obtained from Janvier, Rag2−/− mice were obtained initially from Taconic and bred in house, while Rag2−/− PTPN2fl/fl VilCre and Rag2−/− PTPN2fl/fl LysMCre mice were generated and bred in-house [described in detail in Supplementary methods]. For T cell transfer colitis, naïve T cells were isolated from the spleens of wild-type mice or in-house bred PTPN2fl/fl CD4Cre mice,28 enriched using a CD4+ T cell isolation kit [Stemcell Technologies] and sorted by fluorescence-activated cell sorting [FACS] [BD Aria III cell sorter, BD Bioscience] for CD4+CD62LhighCD44lowCD25− cells, which subsequently were injected intraperitoneally [0.5 × 106 cells per mouse] into Rag2−/− recipients. Chronic colitis was induced in C57BL/6J mice by administration of four cycles of 2% DSS [MP Biomedicals] in drinking water, each lasting for 7 days, followed by 10 days of recovery with drinking water. Sterile-filtered spermidine solution [Sigma Aldrich] was administrated via oral gavage [200 μL of a 44.7 mM solution, equivalent to the amount ingested when spermidine was supplemented at 3 mM in the drinking water; pharmacokinetics experiments] or supplied in drinking water [11.5 μM, 0.3 mM, 3 mM, 10 mM]. All studies were performed with 8–12-week-old mice receiving either standard chow or a low polyamine diet [M/R EXP AIN 93M, Granovit AG] as indicated.
2.2. Spermidine pharmacokinetics
Spermidine content was assessed in serum and faecal samples of C57BL/6J mice by ultra-performance liquid chromatography [UPLC] [as described in the Supplementary methods], as a collaborative effort at the Swiss Institute of Allergy and Asthma Research SIAF in Davos. Measurements were performed 15 min, 30 min, 1 h, 2 h, 4 h, 8 h and 24 h after a single administration of spermidine via oral gavage.
2.3. Assessment of colitis and immune cell infiltrates
Colonoscopy was performed using a mouse endoscope [Karl Storz] and evaluated using the murine endoscopic index of colitis severity [MEICS] according to Becker et al.29 For histological assessment, paraffin-embedded, 5-μm sections of the most distal part of the colon were stained with haematoxylin-eosin [H&E] according to standard protocols and scored for epithelial damage and inflammatory infiltration as described.30 For the evaluation of immune cell infiltrates, colonic tissue was digested, and the immune cells obtained were stained for flow cytometry analysis [as indicated in the Supplementary methods]. Immunofluorescence staining was performed on paraffin-embedded colonic sections for CD3 and Ki67 evaluation [as described in the Supplementary methods].
2.4. Microbiota analysis
For 16S rDNA sequencing, DNA was extracted from the faeces and sequenced for the V4 region of the 16S rDNA by Microsynth AG. The QIIME2 pipeline was used for data analysis.31 After initial quality assessment, DAAD2 was used to merge the paired reads and denoise them, selecting a feature depth of 8000. The alpha rarefaction module in QIIME2 was used to ensure that sufficient depth was obtained to capture most of the features. Alpha and beta diversity were calculated using the core-metrics-phylogenetic module. For analysis of alpha diversity, the Faith’s phylogenetic matrix was used to compute the richness and incorporate the features’ phylogenetic relationships. For beta diversity, a weighted UniFrac distance matrix was used to quantify the dissimilarity between communities. Principal coordinate analysis [PCoA] was used for better visualization of beta diversity. The pre-trained Naïve Bayes silva-132-99-nb-classifier trained against Silva [release 132] full-length sequences was applied to explore the taxonomic composition. The taxa-bar plot module was used for visualization of the different taxonomic compositions.
2.5. Co-culture experiments
IEC–macrophage co-cultures were performed as described previously.32 In brief, 500 000 CaCo-2 cells wild-type for PTPN2 [WT] or knock-down [KD, generated as described32] were grown for 9 days on transwell inserts [Merck Millipore] to form a tight epithelial barrier. THP-1 monocytes or CD14+ monocytes, obtained from the blood of IBD patients from the Swiss Inflammatory Bowel Disease Cohort Study [SIBDCS], WT or single nucleotide polymorphism [SNP] rs1893217 for PTPN2 [isolated as described previously,32 patient characteristics are summarized in Supplementary Table S1], were cultured with macrophage colony stimulating factor [M-CSF, 20 ng/mL, Peprotech] for 8 days to differentiate into macrophages. CaCo-2-monolayers containing cell culture inserts were then transferred onto macrophage-containing wells. For the experiment, LPS [100 ng/mL, InvivoGen], spermidine [100 μM, Sigma Aldrich] or a combination of both was added to the apical side of the CaCo-2 monolayers, and transepithelial electrical resistance [TEER] and 4-kDa FITC-dextran [FD4] translocation were measured after 24 h as described previously32 and RNA was collected for gene expression analyses as described.32
2.6. RNA isolation and RT-PCR
Colon tissue was disrupted in Homogenization Solution [Promega] containing 1-thioglycerol [Promega] using a gentleMACS Octo Dissociator [Miltenyi Biotec]. Cells were washed twice with phosphate-buffered saline [PBS], and RNA isolation was performed using the Maxwell RSC simplyRNA Tissue Kit [Promega] and the Maxwell RSC Instrument [Promega] according to the manufacturer’s instructions. RNA concentration was determined by measuring absorbance at 260 nm using a microplate reader [Synergy H1, BioTek] and Gene5 V1.11 [BioTek] software. cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit [Thermo Fisher Scientific] following the manufacturer’s instructions. Real-time polymerase chain reaction [PCR] was performed using Taqman Gene Expression Assays and FAST qPCR MasterMix for Taqman Assays on a QuantStudio 6 System [all Thermo Fisher Scientific]. Each sample was measured in triplicate, mouse Actb was used as endogenous control and results were analysed by the ΔΔCT method.
2.7. Statistical analysis
All data are presented as means ± standard deviation [SD]. Statistical analyses were performed using GraphPad Prism 8 [GraphPad Software], with Student’s t-test or analysis of variance [ANOVA], followed by Tukey post hoc test or non-parametric Kruskal–Wallis test followed by multiple comparison test with Benjamini–Hochberg correction. Values of p below 0.05 were considered significant.
2.8. Ethical statement
All animal experiments were approved by the local animal welfare commission [Veterinary Office of the Canton Zurich, licence number ZH019/2018] and performed according to Swiss animal welfare legislation. All patients donating blood for isolation of peripheral blood mononuclear cells signed an informed consent prior to enrollment in the Swiss IBD cohort.
3. Results
3.1. Spermidine ameliorates experimental colitis in a dose-dependent manner
To assess whether spermidine treatment can ameliorate T cell-mediated colitis and to determine the most effective dosage, we supplemented drinking water with spermidine at concentrations of 3 mM, 0.3 mM and 11.5 μM based on previous reports13 and corresponding to concentrations used in dietary supplements,33 respectively. Colitis was induced in Rag2−/− mice by transfer of naïve T cells from WT mice. Supplementation of 3 mM and 0.3 mM spermidine in drinking water protected from T cell-induced colitis, as evidenced by reduced weight loss when compared to untreated mice or mice receiving 11.5 μM spermidine in drinking water [Figure 1A]. Administration of spermidine at 3 mM clearly reduced mucosal damage, as evidenced by mouse endoscopy and reduced MEICS scores [Figure 1B], as well as less severe signs of inflammation in H&E-stained sections of the terminal colon [Figure 1C]. In contrast, supplementation of 11.5 μM spermidine, which corresponds to the level recommended in food supplements for humans, did not affect colitis severity, while a concentration of 0.3 mM showed intermediate effects. Colon length was unaffected by any spermidine treatment [Figure 1D].
Figure 1.
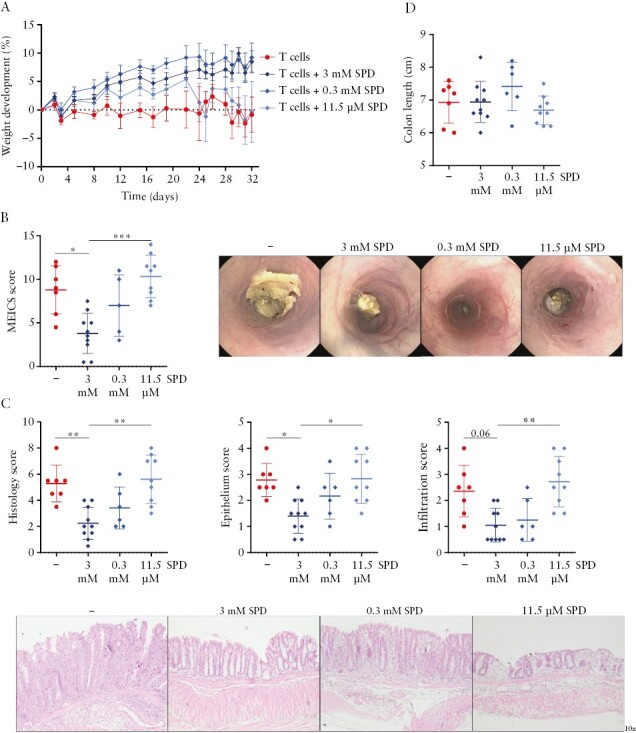
Administration of spermidine prevents colitis development in a dose-dependent manner. Splenic CD4+ CD62Lhi CD44low CD25− naïve T cells were injected intraperitoneally into Rag2−/− immunodeficient mice, and 3 mM, 0.3 mM or 11.5 μM spermidine [SPD] was administered in the drinking water from the day of the transfer. [A] Weight development, [B] MEICS score and representative images, [C] overall histology score, epithelium score, infiltration score and representative sections of H&E-stained distal colon tissue, and [D] colon length. *p < .05, **p < .01, ***p < .001.
3.2. Spermidine does not have adverse effects upon extended administration in mice
Next, we assessed potential adverse effects of prolonged spermidine administration by supplementing the drinking water of C57BL/6J mice with the most effective spermidine dose [3 mM, as determined in our previous experiment] or with even higher spermidine concentration [10 mM] over a period of 12 weeks. Serum levels of the liver markers alanine aminotransferase [ALT] and aspartate aminotransferase [AST], as well as the renal function marker creatinine [CRE], were not affected in mice after spermidine administration [Figure 2A]. Histological assessment of colon and kidney tissue showed no morphological abnormalities after extended spermidine administration [Figure 2B and C]. Furthermore, we observed no signs of enhanced proliferation or abnormal colon growth [Figure 2B]. Interestingly, mice fed with a low polyamine diet showed moderate lipid accumulation in the liver, an effect that was reverted upon spermidine supplementation [Figure 2D]. These data indicate that in mice, continuous administration of 3 mM spermidine in drinking water is safe and effective in preventing intestinal inflammation and does not lead to adverse effects upon extended administration.
Figure 2.
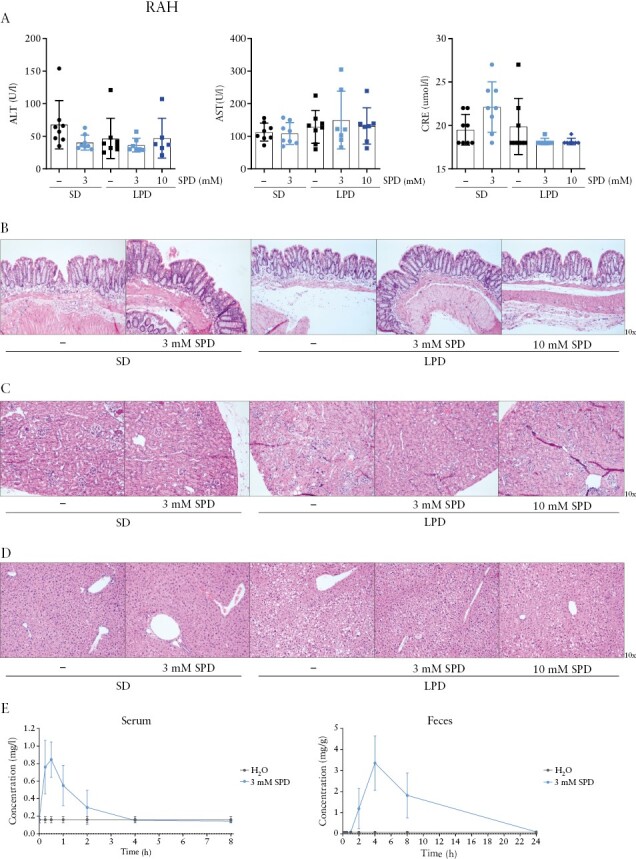
Safety of spermidine administration and spermidine pharmacokinetics. C57BL/6J mice received water or 3 mM or 10 mM spermidine [SPD] in the drinking water for 12 weeks and were fed with the standard diet [SD] or low polyamine diet [LPD]. [A] Serum levels of alanine aminotransferase [ALT], aspartate aminotransferase [AST], creatinine [CRE], and representative H&E-stained sections of [B] terminal colon, [C] kidney and [D] liver. C57BL/6J mice orally gavaged with 3 mM spermidine [SPD] and killed 15 min, 30 min, 1 h, 2 h, 4 h, 8 h and 24 h post-administration; [E] spermidine content was assessed using ultra-performance liquid chromatography [UPLC] of the serum and faeces.
When evaluating the pharmacokinetics of spermidine uptake, we observed that spermidine reached the highest serum concentration within 30 min after administration and returned to baseline levels within 4 h [Figure 2E], whereas the highest levels in faeces were observed 4 h post-administration, returning to baseline levels after 24 h [Figure 2E].
3.3. Spermidine has a prominent effect on myeloid cells and promotes anti-inflammatory [M2-like] macrophage differentiation
We next aimed to unravel the molecular mechanisms involved in mediating the anti-inflammatory effect of spermidine in the setting of experimental colitis. Thus, we repeated the transfer colitis experiment with the most effective spermidine dose [3 mM]. As before, spermidine treatment prevented disease-associated weight loss [Figure 3A] and colon shortening [Figure 3B]. Furthermore, spermidine administration had a drastic effect in ameliorating endoscopic and histologic colitis scores [Figure 3D]. In line with reduced immune cell infiltration into the colonic mucosa, myeloperoxidase activity within the colonic tissue was also significantly decreased, indicating lower infiltration/activation of myeloid cells [Figure 3E]. Confirming our safety studies described above, spermidine administration had no adverse effects in control animals that did not receive naïve T cells [Figure 3A–E]. These data confirm that spermidine alleviates experimental colitis and has no adverse effects in control animals.
Figure 3.
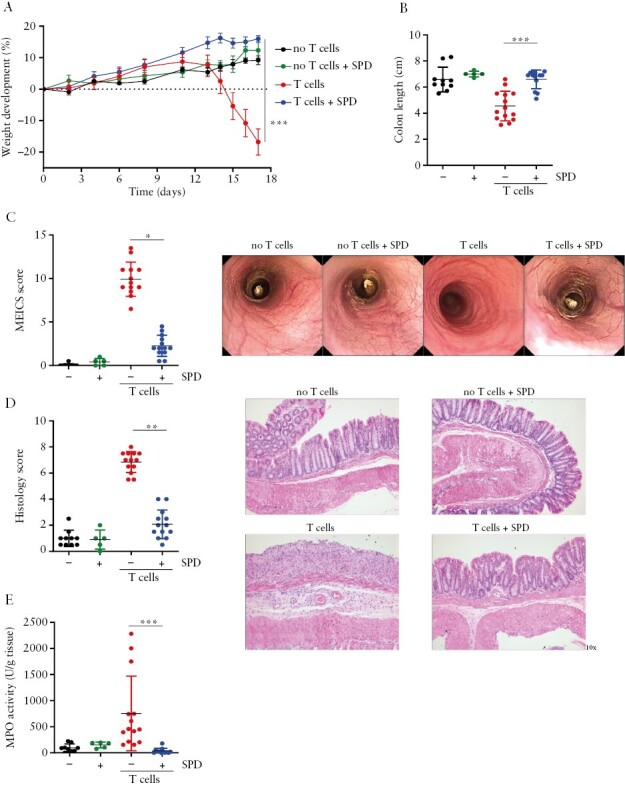
Spermidine treatment protects from T cell transfer colitis and has no effect on healthy mice. Rag2−/− immunodeficient mice were injected with saline intraperitoneally and received normal drinking water [no T cells] or drinking water supplemented with 3 mM spermidine [no T cells + SPD] or received splenic CD4+ CD62Lhi CD44low CD25− naïve T cells and normal drinking water [T cells] or drinking water supplemented with 3 mM spermidine [T cells + SPD]. Spermidine was administered in the drinking water from the day of T cell transfer. [A] Weight development, [B] colon length, [C] MEICS score and representative images, [D] histology score and representative sections of H&E-stained distal colon, and [E] myeloperoxidase activity [MPO] in the colonic tissue. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine the mode of action, we evaluated intestinal immune cell populations by flow cytometry analysis. We found that spermidine mediated a significant overall reduction of CD3+ T cells in the colonic lamina propria [Figure 4A], which might result from their reduced proliferation rate [Figure 4B]. However, no changes were observed in the proportion of CD4+ T helper [Th] subsets known to be involved in the pathogenesis of colitis, such as IFN-γ+ Th1, IL-17+ Th17, or double-positive IFN-γ+ IL-17+ T cells [Figure 4C, gating in Supplementary Figure S1A]. In contrast, noteworthy changes were observed within the myeloid cells: spermidine supplementation resulted in a significant reduction of infiltrating neutrophils, monocytes and, albeit not significant, dendritic cells, while the relative proportion of natural killer [NK] cells was elevated [Figure 4D, gating in strategy Supplementary Figure S1B]. Most notably, we observed a relative reduction of macrophages, especially pro-inflammatory, M1-like macrophages, while anti-inflammatory M2-like macrophages were increased in the spermidine-treated mice [Figure 4D]. In line with the flow cytometry data, mRNA expression of genes coding for the intestinal macrophage markers F4/80 and CD64 were decreased in the colonic tissue of spermidine-treated mice that received naïve T cells [Figure 4E]. Furthermore, the marked increase in mRNA expression of the pro-inflammatory macrophage marker gene coding for iNOS observed upon colitis induction was absent in mice receiving spermidine [Figure 4E]. In contrast, expression of the M2-specific marker Mrc1, which encodes CD206 protein, was increased upon spermidine supplementation. Taken together, this indicates that spermidine exerts its anti-inflammatory effects probably via modulating innate immune cells and in particular via acting on macrophages, while direct effects on T cells seemed to play a subordinate role. Following this observation, we further investigated the effect of spermidine on macrophage polarization in vitro. For this, we polarized bone marrow-derived macrophages [into classically activated [M1] and alternatively activated, anti-inflammatory [M2] macrophages using IFN-γ and LPS or IL-4 and IL-10, respectively. Spermidine was added during the polarization period. In line with our in vivo findings, spermidine administration significantly decreased mRNA expression of genes coding for MHCII, CD86 and iNOS in M1 macrophages and upregulated the expression of CD206. However, in M2 macrophages, there was no effect on the expression of MHCII and CD206 molecules [Figure 4F].
Figure 4.
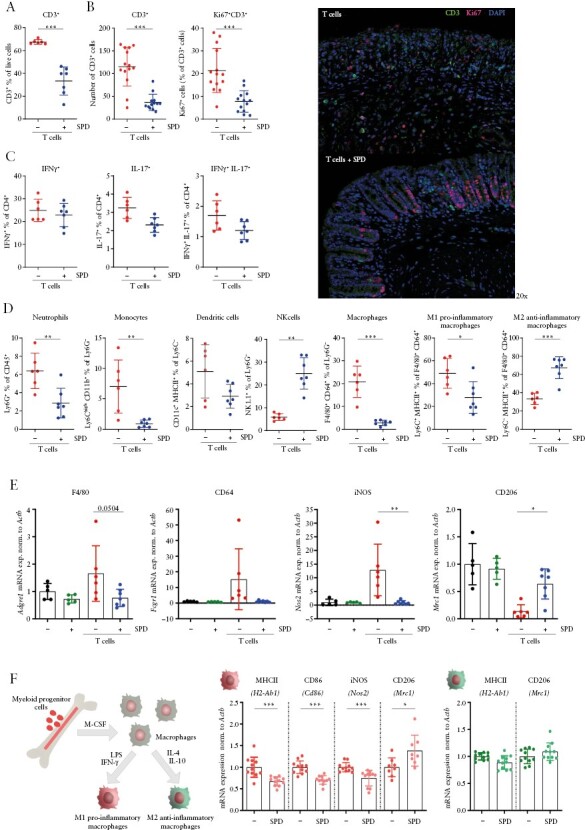
Spermidine promotes an M2-like anti-inflammatory macrophage phenotype. Mice were treated as in Figure 3, and colonic lamina propria immune cells from mice that received T cells transfer were analysed at the end of the experiment. [A] Relative abundance of CD3+ T cells measured by flow cytometry, [B] number of CD3+ T cells and Ki67+ CD3+ T cells assessed by immunofluorescence staining and representative images of the distal colon, [C] relative abundance of CD4+ T cells producing IFN-γ, IL-17 or both cytokines, and [D] myeloid cells measured by flow cytometry; [E] mRNA expression of macrophage-related genes within colonic tissue. Murine bone marrow-derived macrophages were polarized by stimulation with IFN-γ [100 ng/mL] and LPS [50 ng/mL] into M1-like pro-inflammatory type and IL-4 [50 ng/mL] and IL-10 [40 ng/mL] into M2-like anti-inflammatory type for 48 h in medium supplemented with or without spermidine [100 μM, SPD]; [F] mRNA expression of macrophage-related genes. *p < 0.05, **p < 0.01, ***p < 0.001.
3.4. Spermidine normalizes intestinal dysbiosis
Our data suggest a prominent role of macrophages in mediating the anti-inflammatory effect of spermidine in experimental colitis. Intestinal macrophages control bacterial infections by acting as the first line of defence against invasive bacterial species.34,35 In addition, spermidine might directly act on certain microbial species. Thus, next, we analysed the composition of the intestinal microbiota in our mice subjected to T cell transfer colitis. At the end of the experiment, we collected faecal samples of Rag−/− mice without naïve T cell transfer [no T cells], Rag−/− mice without naïve T cells supplemented with spermidine [no T cells + SPD], Rag−/− mice that received naïve T cells [T cells], and Rag−/− mice that received naïve T cells and were treated with spermidine [T cells + SPD]. Faecal DNA was sequenced for the V4 region of the 16S rDNA. Rarefaction reads based on Faith’s phylogenetic diversity [PD] indicated that sequencing depth was sufficient in each sample to capture alpha diversity [Figure 5A]. Samples from T cells mice showed significantly lower bacterial richness and phylogenetic diversity compared to T cells + SPD mice. Notably, T cells + SPD mice did not differ from the healthy controls [Figure 5A and B]. Moreover, spermidine alone [no T cells + SPD] did not alter alpha diversity in healthy mice. Evaluation of beta diversity based on weighted Unifrac PCoA revealed that samples from T cells mice were significantly different from the other three groups and that T cells + SPD mice clustered closer together with the healthy control groups [Figure 5C]. Together, our data demonstrated that spermidine administration prevented the microbiota shift observed in T cell transfer colitis.
Figure 5.
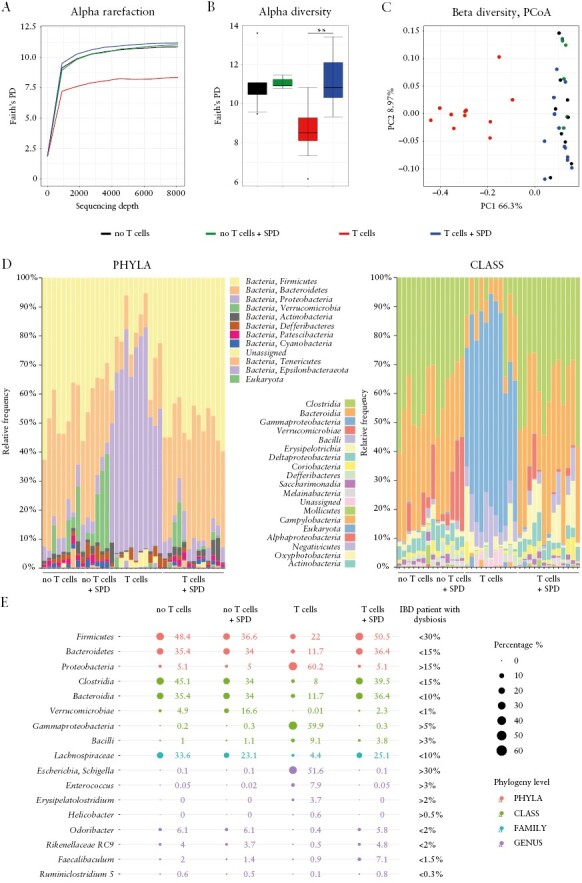
Administration of spermidine prevents intestinal dysbiosis. 16S rDNA sequencing data of faecal samples from mice subjected to T cell transfer colitis [as described in Figure 3]. [A] Rarefaction reads based on Faith’s phylogenetic diversity [PD], [B] alpha diversity, [C] beta diversity evaluation based on weighted Unifrac principal coordinate analysis, [D] taxonomic analysis of phyla and classes, and [E] bubble chart presenting various taxa known to be altered in IBD patients with dysbiosis and its corresponding levels measured in healthy mice [no T cells], healthy mice receiving spermidine [no T cells + SPD], transfer colitis mice [T cells] and transfer colitis mice receiving spermidine [T cells + SPD]. **p < 0.01.
Taxonomic analysis revealed that in healthy mice, Firmicutes and Bacteroides were the dominant phyla, with Clostridia and Bacteroidia being the most abundant classes. An increase in the relative abundance of Verrucomicrobia was the only change induced by spermidine in healthy control mice. In the T cells mice, Proteobacteria was the dominant phylum, whereas, on the class level, we observed a massive increase in the abundance of Gammaproteobacteria. Notably, T cells + SPD mice showed a similar microbiome composition as healthy mice [Figure 5D, Supplementary Figure S2A and B].
When comparing the microbiota composition in our mice with taxa known to be altered in IBD patients and which have been previously used to define intestinal dysbiosis,36–40 we found that the shift in intestinal microbiota induced by transfer of naïve T cells was similar to the alterations observed in IBD patients. This clearly underlines the relevance of this colitis model with regard to human IBD. Notably, the dysbiotic changes were almost completely reverted upon spermidine administration [Figure 5E], demonstrating a beneficial effect of spermidine on the intestinal microbiota in the setting of intestinal inflammation.
3.5. Pre-treatment with spermidine does not prevent colitis development
To determine whether prophylactic spermidine administration can prevent colitis development, we administrated 3 mM spermidine in drinking water for three different time periods: [1] starting on the day of naïve T cell transfer [treatment], [2] for 7 days prior to naïve T cell transfer only [pre-treatment] or [3] for 7 days prior to the transfer and throughout the experiment [continuous] [Supplementary Figure S3A]. The treatment group and the group receiving spermidine continuously were protected from colitis, as evidenced by reduced weight loss [Supplementary Figure S3B], lower endoscopic and histological colitis severity scores [Supplementary Figure S3C and D], while colon length was not affected [Supplementary Figure S3E]. In contrast, mice of the pre-treatment group did not show alleviation of colitis symptoms. To confirm the anti-inflammatory effect of spermidine in an independent colitis model, we applied the chronic DSS colitis model. Administration of 3 mM spermidine in drinking water throughout the experiment resulted in similar anti-inflammatory effects as observed in the T cell colitis model [Supplementary Figure S4].
3.6. The protective effect of spermidine depends on the presence of PTPN2 in intestinal epithelial cells and myeloid cells
Previous studies have demonstrated that spermidine activates PTPN2, which is at least partially responsible for its anti-inflammatory effects in vitro and in vivo.11,13,41 Therefore, we next investigated whether PTPN2 expression in CD4+ T cells, intestinal epithelial cells or myelomonocytic cells [including monocytes, macrophages and granulocytes] is required for the colitis-protecting effects of spermidine. For this, we induced T cell transfer colitis in mice either featuring a cell-specific deletion of PTPN2 in intestinal epithelial cells [Rag−/− PTPN2 VilCre mice], in myelomonocytic cells [Rag−/− PTPN2 LysMCre] or in T cells. In particular, we used the following four conditions: [i] WT naïve T cells transferred to Rag−/− mice [PTPN WT controls], [ii] PTPN2−/− naïve T cells into Rag−/− mice [Δ T cells], [iii] WT naïve T cells to Rag−/− PTPN2 VilCre mice [Δintestinal epithelial cells—ΔIEC], and [iv] WT naïve T cells to Rag−/− PTPN2 LysMCre mice [Δmonocytic myeloid cells—ΔMMCs] [Figure 6A]. PTPN2 deletion in the transferred T cells had no impact on the colitis-protective effect of spermidine, as mice receiving PTPN2 WT and PTPN2−/− T cells showed a comparable reduction of colitis severity upon spermidine administration [Figure 6B and C]. In contrast, deletion of PTPN2 in either IECs or MMCs abrogated the protective effect of spermidine [Figure 6B and C]. This clearly demonstrates the important cell-specific role of PTPN2 in mediating the anti-inflammatory effect of spermidine in colitis. When looking at the T cells within the colonic lamina propria, we found that spermidine administration did not have any impact on the abundance of CD3+ T cells in any of the PTPN2 settings [Figure 6D]. Spermidine reduced CD3+ T cells and induced more regulatory CD4+ T cells only in WT mice but not in the PTPN2 knockouts. There was no difference in the proportions of IFN-γ, and IL-17-producing CD4+ T cells in any of the spermidine-treated mice [Figure 6D]. This confirms that the main mode of action of spermidine does not seem to be via modulation of the T cell subsets.
Figure 6.

Expression of PTPN2 in intestinal epithelial cells and monocytic myeloid cells is essential for the colitis-protective effect of spermidine. Rag−/− mice received naïve CD4+ T cells expressing PTPN2 [PTPN2 WT], or naïve CD4+ T cells lacking PTPN2 [ΔT cells], Rag−/− mice lacking PTPN2 in IECs received naïve CD4+ T cells [ΔIEC], and Rag−/− mice lacking PTPN2 in MMC received naïve CD4+ T cells [ΔMMC]. Mice received drinking water without/with spermidine [3 mM, SPD] throughout the experiment. [A] Experimental set-up, [B] MEICS score and representative images, [C] histology score and representative sections of H&E-stained distal colon tissue, and [D] relative abundance of CD3+ T cells, regulatory CD4+ T cells, IFN-γ-producing CD4+ T cells and IL-17-producing CD4+ T cells measured by flow cytometry. *p < 0.05, **p < 0.01, ***p < 0.001.
3.7. Spermidine does not promote an anti-inflammatory phenotype in macrophages of IBD patients carrying PTPN2 SNP rs1893217
We have thus found that IECs and myeloid cells seem to play a crucial role in mediating the colitis-alleviating effect of spermidine. We have previously shown that macrophages promote intestinal epithelial barrier function in a PTPN2-dependent manner and that this effect is important in preventing intestinal inflammation.32 PTPN2-deficient macrophages, or macrophages on the other hand fail to mediate barrier protective effects and co-culture of IECs with PTPN2-deficient macrophages results in reduced TEER and increased permeability to FD4.32 Thus, to investigate the impact of spermidine on the interplay between macrophages, intestinal epithelial cells, bacteria-derived factors and PTPN2, we performed in vitro co-culture experiments. We co-cultured human CaCo-2 IECs with monocyte-derived macrophages from IBD patients either expressing PTPN2 [wild type, PTPN2 WT] or carrying the IBD-associated PTPN2 variant SNP rs1893217 [PTPN2 SNP] in the presence of spermidine and/or bacterial LPS [Figure 7A]. As observed previously, the barrier function of IECs co-cultured with PTPN2-deficient macrophages was reduced when compared to those co-cultured with WT macrophages, resulting in decreased TEER and elevated FD4 translocation [Figure 7B, Supplementary Figure S5B]. LPS treatment resulted in epithelial barrier defects, as observed by reduced TEER and increased FD4 translocation in CaCo-2 cells, regardless of the presence of the variant SNP rs1893217 in the macrophages. Notably, the addition of spermidine prevented epithelial barrier disruption. However, this was only observed when the epithelial cells were co-cultured with PTPN2 WT macrophages but not with PTPN2 SNP macrophages [Figure 7B, Supplementary Figure S5A]. Spermidine treatment alone had no impact on epithelial barrier integrity in any of the set-ups [Figure 7B, Supplementary Figure S5A]. Moreover, spermidine suppressed the LPS-induced increase of TNF and IL6 mRNA expression in epithelial cells only when co-cultured with PTPN2 WT macrophages [Figure 7C]. LPS treatment promoted IL10 expression in IECs, an effect on which spermidine had no impact regardless of the PTPN2 genotype in macrophages [Supplementary Figure S5B]. In PTPN2 WT macrophages, spermidine treatment reduced LPS-induced expression of CD86, TNF and IL6, all of which are genes associated with polarization into M1-like macrophages, while it increased the expression of the M2-type-associated genes CD163 and MRC1 [Figure 7D]. These effects were absent in PTPN2 SNP cells, indicating that expression of PTPN2 in macrophages is required to mediate the anti-inflammatory effects of spermidine. mRNA levels of genes coding for IL-10 and transforming growth factor [TGF]-β were not significantly affected by spermidine treatment in any of the co-culture setups [Supplementary Figure S5C].
Figure 7.
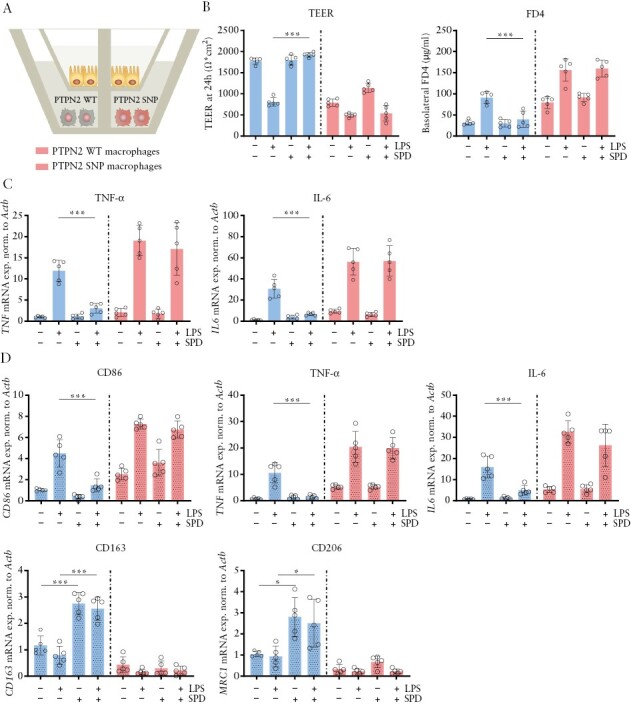
Spermidine treatment does not promote M2-like differentiation in macrophages from IBD patients carrying PTPN2 SNP rs1893217. Human CaCo-2 intestinal epithelial cells [IECs] were co-cultured with monocyte-derived macrophages from IBD patients wild type for PTPN2 [PTPN2 WT] or carrying the PTPN2 variant [PTPN2 SNP rs1893217] in the presence of LPS [100 ng/mL] and/or spermidine [SPD, 100 μM]. [A] Experimental set-up, [B] epithelial barrier function measured by TEER at 24 h and FD4 flux, [C] mRNA expression of TNF and IL6 in IECa, and [D] mRNA expression of CD86, TNF, IL6, CD163 and MRC1 in macrophages. **p < 0.01, ***p < 0.001.
3.8. Effects of spermidine on macrophage polarization depend on PTPN2 expression in IECs
Having shown the importance of the presence of PTPN2 in macrophages for epithelial barrier integrity and anti-inflammatory effects, we further investigated the impact of PTPN2 expression in IECs. Human THP-1 monocyte-derived macrophages were co-cultured with CaCo-2 cells expressing PTPN2-specific small hairpin [sh]RNA [PTPN2 KD—knock-down] or non-targeting control shRNA [PTPN2 WT] in the presence of LPS and/or spermidine [Supplementary Figure S6A]. Again, LPS induced a disruption of the epithelial barrier, as evidenced by a decreased TEER in both PTPN2 WT and PTPN2 KD cells. Notably, spermidine prevented a reduction of TEER and increased FD4 flux only in CaCo-2 cells expressing control shRNA but not in those expressing PTPN2-shRNA [Supplementary Figure S6B and C]. Moreover, LPS-induced expression of TNF was significantly decreased upon spermidine treatment in IECs, an effect independent of PTPN2. In contrast, the spermidine-mediated reduction of IL-6 was PTPN2-dependent [Supplementary Figure S6D]. There was no clear change in the expression of IL-10 in any of the settings, and spermidine itself did not have an impact on mRNA expression of TNF or IL6 genes [Supplementary Figure S6D]. When looking at THP-1-derived macrophages, we found that LPS promoted an increase in the mRNA expression levels of CD86 and a reduction of CD163 and MRC1, while addition of spermidine abrogated these effects [Supplementary Figure S6E]. Interestingly, spermidine alone enhanced the expression of CD163, MRC1, IL10 and TGFB1 [Supplementary Figure S6E], indicating a direct anti-inflammatory M2-like macrophage-promoting effect. Moreover, the LPS-mediated increased expression of TNF and IL6 was reverted upon spermidine supplementation [Supplementary Figure S6E]. Notably, all these effects in THP-1 macrophages required the expression of PTPN2 in epithelial cells.
4. Discussion
Our results indicate a strong anti-inflammatory and therapeutic potential of spermidine in the setting of intestinal inflammation. Anti-inflammatory effects of spermidine were also recently reported in the T cell transfer colitis model by Carriche et al.,42 DSS-induced colitis by Gobert et al.,43 and in DSS- and TNBS-induced colitis by Ma et al.44 Our current results show that the anti-inflammatory effect of spermidine is dose-dependent, and that the colitis-protective effect was not achieved in mice receiving spermidine doses equivalent to those currently proposed as dietary supplements for humans. These findings demonstrate that the dosage used in food supplements is not sufficient to reduce intestinal inflammation and that higher doses are required to achieve therapeutic effects. Interestingly, prolonged or high-dose administration of spermidine did not show any adverse effects in mice, indicating that administration of spermidine at a wide range of concentrations is safe. Schwarz et al. also confirmed the safety of spermidine administration by showing that no tumorigenesis or fibrosis was observed in mice receiving a spermidine‐rich plant extract and in a human study with compliance rates of 85%, indicating excellent tolerability.45
When exploring the effects of spermidine on the immune landscape in the intestine, we observed that treatment with spermidine reduced the abundance of CD3+ T cells, an effect probably due to their reduced proliferation upon spermidine administration. However, this effect seemed to encompass all Th cell subsets since we did not observe any differences in the proportions of Th subsets, i.e. Th1 and Th17 T cells, which are known drivers of IBD pathogenesis and are critical for the development of colitis in the adoptive T cell transfer model.46 Carriche et al. previously showed that spermidine does not affect IL-17-producing T cells in vitro or in vivo in the T cell transfer colitis model.42 Moreover, they reported that spermidine treatment increases the abundance of Foxp3+ regulatory T cells, which are known to promote homeostasis by inducing tolerance and by suppressing macrophages and dendritic cells. In line with this idea, we found increased numbers of these cells in spermidine-treated colitis mice. In addition, we saw prominent changes in myeloid cells, which have never been addressed in any previous study. In particular, we found that the relative abundance of M1-like pro-inflammatory macrophages was decreased, while M2-like anti-inflammatory macrophages were induced in spermidine-treated mice. This change correlated with decreased mRNA expression of the M1-associated gene coding for iNOS and upregulated expression of M2-specific CD206. Furthermore, in bone marrow-derived M1-polarized macrophages, the administration of spermidine decreased the mRNA expression of genes associated with a pro-inflammatory, M1-like phenotype, such as for MHCII, CD86 and iNOS, while increasing CD206, suggesting that spermidine directly affects macrophage polarization and promotes anti-inflammatory macrophages. This is in line with a report by Liu et al., showing that spermidine induces M2 polarization and that spermidine pre-treated bone marrow-derived macrophages ameliorate acute DSS-induced colitis.47
Macrophages play a key role in maintaining intestinal homeostasis and essentially interact with the intestinal microbiome. They play a crucial role in preventing the overgrowth of potentially pathogenic bacteria and intestinal dysbiosis, a key feature observed in IBD patients.35 Our 16S sequencing data showed that the microbiota in mice subjected to T cell transfer colitis closely resembled the dysbiotic changes described in IBD patients. This indicates that this model is well suited to mimic microbial changes observed in IBD and to investigate the effect of therapeutic agents on IBD-associated dysbiosis. Notably, spermidine administration protected mice from dysbiosis. It prevented the shift from a Clostridia- and Bacteroidia-dominated microbiome towards a microbiome with overgrowth of pathogenic Gammaproteobacteria, Escherichia and Shigella, as well as the genera Enterococcus and Helicobacter, which is often observed in IBD patients37 as well as in our T cell transfer colitis model. To our knowledge, our study for the first time provides evidence that spermidine beneficially affects the gut microbiome profile in the context of colitis. Yet, it remains unclear whether spermidine alleviates colitis by affecting the intestinal microbiome directly, or whether the maintenance of a beneficial microbiome is the result of the effects on immune cells and epithelial barrier integrity.
It is worth mentioning that spermidine has enzyme-modulating functions and has been demonstrated to directly activate PTPN2.41,48 PTPN2 regulates intestinal epithelial barrier and immune cell functions49,50in vitro and in vivo,51,52 and PTPN2 dysfunction has been linked to an elevated risk of developing IBD.53 We have previously shown that in THP-1 monocytes, the anti-inflammatory effects of spermidine were PTPN2-dependent,13 and Penrose et al. demonstrated that spermidine protects from IFN-γ-induced epithelial barrier defects in a PTPN2-dependent manner.11 Here we demonstrate that the lack of PTPN2 in CD4+ T cells does not affect the colitis-protective properties of spermidine in the T cell transfer model, while the presence of PTPN2 in intestinal epithelial cells and monocytic myeloid cells was critical for disease prevention. These findings clearly demonstrate a significant role of PTPN2 for the anti-inflammatory effects of spermidine in the setting of intestinal inflammation.
Our co-culture experiments further confirmed the importance of a normal interaction between intestinal epithelial cells and macrophages and the significance of PTPN2 for the anti-inflammatory effects of spermidine. The bacterial cell wall component LPS reduced epithelial barrier integrity and promoted the expression of TNF and IL-6 in epithelial cells and macrophages co-cultured with LPS-treated epithelial cells, leading to their polarization into a pro-inflammatory phenotype. Spermidine treatment prevented epithelial barrier disruption, reversed the LPS-mediated expression of pro-inflammatory cytokines in IECs and macrophages, and promoted an anti-inflammatory, M2-like macrophage phenotype. However, these effects were only present when PTPN2 was functional in epithelial cells and macrophages, clearly demonstrating the PTPN2 dependence of the beneficial effects of spermidine on barrier integrity. This suggests that spermidine-induced PTPN2 activation can prevent barrier defects induced by microbial products. It has been demonstrated that PTPN2 negatively regulates pro-inflammatory signalling cascades induced by IL-6, and thus its activation might promote a non-inflammatory environment upon spermidine addition. Together with the beneficial effect of spermidine on the intestinal microbiota, the PTPN2-dependent anti-inflammatory effects seem to be required to mediate its colitis-protective effects.
In conclusion, our data confirm the colitis-protective and anti-inflammatory properties of spermidine and demonstrate that these effects are mediated by PTPN2 in intestinal epithelial cells and monocytic myeloid cells. Our data further suggest that spermidine acts in multiple ways by promoting anti-inflammatory macrophages, maintaining a healthy gut microbiome, and preserving epithelial barrier integrity. These three components heavily influence each other and collectively maintain intestinal homeostasis. By positively influencing these factors in the absence of overt negative effects, spermidine administration might represent a promising novel therapeutic strategy for the treatment of IBD.
Supplementary Material
Acknowledgments
Flow cytometry data were recorded on the instruments of the Flow Cytometry Facility of the University of Zurich.
Contributor Information
Anna Niechcial, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Marlene Schwarzfischer, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Marcin Wawrzyniak, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Kirstin Atrott, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Andrea Laimbacher, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Yasser Morsy, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Egle Katkeviciute, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Janine Häfliger, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Patrick Westermann, Swiss Institute of Allergy and Asthma Research (SIAF), University of Zurich, Davos, Switzerland.
Cezmi A Akdis, Swiss Institute of Allergy and Asthma Research (SIAF), University of Zurich, Davos, Switzerland.
Michael Scharl, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Marianne R Spalinger, Department of Gastroenterology and Hepatology, University Hospital Zurich, University of Zurich, Zurich, Switzerland.
Funding
This research was supported by grants from the Stiftung Experimentelle Biomedizin to M.S., the Swiss National Science Foundation [Grant Nos.: 314730-166381, 320030-184753, 320030E_190969 to M.S.] and a Litwin IBD Pioneer Award from the Crohn’s and Colitis Foundation [CCF] to M.S.
Conflict of Interest
The authors have declared no conflicts of interest.
Author Contributions
A.N.: data acquisition, analysis, interpretation, drafting the manuscript; Ma.Sc., M.W., K.A., A.L., E.K., J.H.: data acquisition and analysis; Y.M.: processing and analysis of 16S rDNA data; P.W., C.A.A.: UPLC analysis and data interpretation; M.S.: conceived the study, study design, supervision, data interpretation, funding; M.R.S., study design, supervision, data interpretation. All authors were involved in writing the manuscript and approved its final version prior to submission.
Data Availability
The data underlying this article are available in the article and in its online Supplementary Material. Sequencing data underlying this article will be made available upon acceptance of the manuscript for publication.
References
- 1. Lenis YY, Elmetwally MA, Maldonado-Estrada JG, Bazer FW.. Physiological importance of polyamines. Zygote 2017;25:244–55. [DOI] [PubMed] [Google Scholar]
- 2. Madeo F, Eisenberg T, Pietrocola F, Kroemer G.. Spermidine in health and disease. Science 2018;359:eaan2788. [DOI] [PubMed] [Google Scholar]
- 3. Madeo F, Hofer SJ, Pendl T, et al. Nutritional aspects of spermidine. Annu Rev Nutr 2020;40:135–59. [DOI] [PubMed] [Google Scholar]
- 4. Eisenberg T, Knauer H, Schauer A, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 2009;11:1305–14. [DOI] [PubMed] [Google Scholar]
- 5. Eisenberg T, Abdellatif M, Zimmermann A, et al. Dietary spermidine for lowering high blood pressure. Autophagy 2017;13:767–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu S, Ashok M, Li J, et al. Spermine protects mice against lethal sepsis partly by attenuating surrogate inflammatory markers. Mol Med 2009;15:275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Choi YH, Park HY.. Anti-inflammatory effects of spermidine in lipopolysaccharide-stimulated BV2 microglial cells. J Biomed Sci 2012;19:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Q, Zheng C, Cao J, et al. Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages. Cell Death Differ 2016;23:1850–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jeong JW, Cha HJ, Han MH, et al. Spermidine protects against oxidative stress in inflammation models using macrophages and zebrafish. Biomol Ther [Seoul] 2018;26:146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paul S, Kang SC.. Natural polyamine inhibits mouse skin inflammation and macrophage activation. Inflamm Res 2013;62:681–8. [DOI] [PubMed] [Google Scholar]
- 11. Penrose HM, Marchelletta RR, Krishnan M, McCole DF.. Spermidine stimulates T cell protein-tyrosine phosphatase-mediated protection of intestinal epithelial barrier function. J Biol Chem 2013;288:32651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wawrzyniak M, Groeger D, Frei R, et al. Spermidine and spermine exert protective effects within the lung. Pharmacol Res Perspect 2021;9:e00837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moron B, Spalinger M, Kasper S, et al. Activation of protein tyrosine phosphatase non-receptor type 2 by spermidine exerts anti-inflammatory effects in human THP-1 monocytes and in a mouse model of acute colitis. PLoS One 2013;8:e73703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Niechcial A, Butter M, Manz S, et al. Presence of PTPN2 SNP rs1893217 enhances the anti-inflammatory effect of spermidine. Inflamm Bowel Dis 2020;26:1038–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xavier RJ, Podolsky DK.. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007;448:427–34. [DOI] [PubMed] [Google Scholar]
- 16. Yadav V, Varum F, Bravo R, Furrer E, Bojic D, Basit AW.. Inflammatory bowel disease: exploring gut pathophysiology for novel therapeutic targets. Transl Res 2016;176:38–68. [DOI] [PubMed] [Google Scholar]
- 17. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 2017;390:2769–78. [DOI] [PubMed] [Google Scholar]
- 18. Collaborators GBDIBD. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol 2020;5:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ananthakrishnan AN, Bernstein CN, Iliopoulos D, et al. Environmental triggers in IBD: a review of progress and evidence. Nat Rev Gastroenterol Hepatol 2018;15:39–49. [DOI] [PubMed] [Google Scholar]
- 20. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res 2019;2019:7247238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reinisch W, Hommes DW, Van Assche G, et al. A dose escalating, placebo controlled, double blind, single dose and multidose, safety and tolerability study of fontolizumab, a humanised anti-interferon gamma antibody, in patients with moderate to severe Crohn’s disease. Gut 2006;55:1138–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hueber W, Sands BE, Lewitzky S, et al. ; Secukinumab in Crohn’s Disease Study Group. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012;61:1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Danese S, Rudzinski J, Brandt W, et al. Tralokinumab for moderate-to-severe UC: a randomised, double-blind, placebo-controlled, phase IIa study. Gut 2015;64:243–9. [DOI] [PubMed] [Google Scholar]
- 24. De Souza HSP, Fiocchi C, Iliopoulos D.. The IBD interactome: an integrated view of aetiology, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol 2017;14:739–49. [DOI] [PubMed] [Google Scholar]
- 25. De Mattos BR, Garcia MP, Nogueira JB, et al. Inflammatory bowel disease: an overview of immune mechanisms and biological treatments. Mediators Inflamm 2015;2015:493012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chudy-Onwugaje KO, Christian KE, Farraye FA, Cross RK.. A state-of-the-art review of new and emerging therapies for the treatment of IBD. Inflamm Bowel Dis 2019;25:820–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hazel K, O’Connor A.. Emerging treatments for inflammatory bowel disease. Ther Adv Chronic Dis 2020;11:2040622319899297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spalinger MR, Kasper S, Chassard C, et al. PTPN2 controls differentiation of CD4+ T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol 2015;8:918–29. [DOI] [PubMed] [Google Scholar]
- 29. Becker C, Fantini MC, Neurath MF.. High resolution colonoscopy in live mice. Nat Protoc 2006;1:2900–4. [DOI] [PubMed] [Google Scholar]
- 30. Obermeier F, Kojouharoff G, Hans W, Schölmerich J, Gross V, Falk W.. Interferon-gamma (IFN-gamma)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin Exp Immunol 1999;116:238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 2019;37:852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spalinger MR, Sayoc-Becerra A, Santos AN, et al. PTPN2 regulates interactions between macrophages and intestinal epithelial cells to promote intestinal barrier function. Gastroenterology 2020;159:1763–1777.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Information about commercially available spermidine offered by Longevity Labs. https://www.spermidinelife.com/.
- 34. Rubio CA, Schmidt PT.. Severe defects in the macrophage barrier to gut microflora in inflammatory bowel disease and colon cancer. Anticancer Res 2018;38:3811–5. [DOI] [PubMed] [Google Scholar]
- 35. Wang J, Chen WD, Wang YD.. The relationship between gut microbiota and inflammatory diseases: the role of macrophages. Front Microbiol 2020;11:1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arumugam M, Raes J, Pelletier E, et al. ; MetaHIT Consortium. Enterotypes of the human gut microbiome. Nature 2011;473:174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsuoka K, Kanai T.. The gut microbiota and inflammatory bowel disease. Semin Immunopathol 2015;37:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lloyd-Price J, Arze C, Ananthakrishnan AN, et al. ; IBDMDB Investigators. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019;569:655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alam MT, Amos GCA, Murphy ARJ, Murch S, Wellington EMH, Arasaradnam RP.. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog 2020;12:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu S, Zhao W, Lan P, Mou X.. The microbiome in inflammatory bowel diseases: from pathogenesis to therapy. Protein Cell 2021;12:331–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mattila E, Marttila H, Sahlberg N, et al. Inhibition of receptor tyrosine kinase signalling by small molecule agonist of T-cell protein tyrosine phosphatase. BMC Cancer 2010;10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carriche GM, Almeida L, Stuve P, et al. Regulating T-cell differentiation through the polyamine spermidine. J Allergy Clin Immunol 2021;147:335–348.e11. [DOI] [PubMed] [Google Scholar]
- 43. Gobert AP, Latour YL, Asim M, et al. Protective role of spermidine in colitis and colon carcinogenesis. Gastroenterology 2022;162:813–827.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ma L, Ni L, Yang T, et al. Preventive and therapeutic spermidine treatment attenuates acute colitis in mice. J Agric Food Chem 2021;69:1864–76. [DOI] [PubMed] [Google Scholar]
- 45. Schwarz C, Stekovic S, Wirth M, et al. Safety and tolerability of spermidine supplementation in mice and older adults with subjective cognitive decline. Aging [Albany NY] 2018;10:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Imam T, Park S, Kaplan MH, Olson MR.. Effector T helper cell subsets in inflammatory bowel diseases. Front Immunol 2018;9:1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu R, Li X, Ma H, et al. Spermidine endows macrophages anti-inflammatory properties by inducing mitochondrial superoxide-dependent AMPK activation, Hif-1alpha upregulation and autophagy. Free Radic Biol Med 2020;161:339–50. [DOI] [PubMed] [Google Scholar]
- 48. Ylilauri M, Mattila E, Nurminen EM, et al. Molecular mechanism of T-cell protein tyrosine phosphatase (TCPTP) activation by mitoxantrone. Biochim Biophys Acta 2013;1834:1988–97. [DOI] [PubMed] [Google Scholar]
- 49. Scharl M, Paul G, Weber A, et al. Protection of epithelial barrier function by the Crohn’s disease associated gene protein tyrosine phosphatase n2. Gastroenterology 2009;137:2030–2040.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McCole DF. Regulation of epithelial barrier function by the inflammatory bowel disease candidate gene, PTPN2. Ann N Y Acad Sci 2012;1257:108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Scharl M, Hruz P, McCole DF.. Protein tyrosine phosphatase non-receptor Type 2 regulates IFN-gamma-induced cytokine signaling in THP-1 monocytes. Inflamm Bowel Dis 2010;16:2055–64. [DOI] [PubMed] [Google Scholar]
- 52. Spalinger MR, Manzini R, Hering L, et al. PTPN2 regulates inflammasome activation and controls onset of intestinal inflammation and colon cancer. Cell Rep 2018;22:1835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Parkes M, Barrett JC, Prescott NJ, et al. ; Wellcome Trust Case Control Consortium. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet 2007;39:830–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online Supplementary Material. Sequencing data underlying this article will be made available upon acceptance of the manuscript for publication.