

Abstract
Background
Inflammatory bowel disease [IBD] is a major debilitating disease. Recently, the gut microbiota has gained attention as an important factor involved in the pathophysiology of IBD. As a complement to the established bacterial ‘enterotypes’ associated with IBD, we focused here on viruses. We investigated the intestinal virome of IBD patients undergoing biological therapy for the presence of virome configurations associated with IBD, and to uncover how those configurations are associated with therapeutic success.
Methods
Viral-like particle enrichment followed by deep sequencing was performed on 432 faecal samples from 181 IBD patients starting biological therapy. Redundancy analysis and Dirichlet Multinomial Mixtures were applied to determine covariates of the virome composition and to condense the gut virota into ‘viral community types’, respectively.
Results
Patients were stratified based on unsupervised clustering into two viral community types. Community type CA showed a low α-diversity and a high relative abundance of Caudoviricetes [non-CrAss] phages and was associated with the dysbiotic Bact2-enterotype. Community type CrM showed a high α-diversity and a high relative abundance of Crassvirales and Malgrandaviricetes phages. During post-interventional analysis, endoscopic outcome was associated with gut virome composition. Remitting UC patients had a high percentage of community type CrM, a high Shannon diversity and a low lysogenic potential. Pre-interventional analyses also identified five novel phages associated with treatment success.
Conclusions
This study proposed two gut virome configurations that may be involved in the pathophysiology of IBD. Interestingly, those viral configurations are further associated with therapeutic success, suggesting a potential clinical relevance.
Keywords: IBD, virome, microbiome, community-typing, bacteriophages, metagenomics
1. Introduction
The gut microbiota is a complex ecosystem that consists of viruses, fungi, bacteria, archaea and protozoa. It can exert beneficial functions to the human host, such as protection against invading pathogens or production of essential vitamins. At times, this microbial ecosystem becomes disrupted, resulting in gut dysbiosis. Gut dysbiosis is associated with several diseases, one of which is inflammatory bowel disease [IBD].1 IBD is a group of chronic remitting diseases involving inflammation of the gut, and its two main phenotypes are ulcerative colitis [UC] and Crohn’s disease [CD]. Although the aetiology of IBD is unknown, the interplay between host genetic susceptibility, a mucosal immune response to the host microbiota and other environmental factors, has been suggested as a working hypothesis.2
In recent years, the bacterial component of the microbiota has been repeatedly associated with the pathology and activity of IBD.3–5 The gut microbiota of active IBD patients is characterized by a high abundance of Proteobacteria and a low abundance of Firmicutes, combined with a low bacterial α-diversity and cell count.2,6 Another frequently reported alteration is the reduction in anti-inflammatory bacteria, particularly butyrate-producing bacteria [e.g. Faecalibacterium prausnitzii].7 Community analysis can provide a more holistic view of the gut microbiota by collapsing the microbial variation into just a few categories. Enterotyping [or bacterial community typing] is such an analysis that can stratify patients based on their gut microbiota.8 Four enterotypes have been reported, named Bacteroides1 [Bact1], Bacteroides2 [Bact2], Prevotella [Prev] and Ruminococcus [Rum].6 One enterotype, Bact2, largely reflects the IBD-specific bacterial alterations, as described above.9 Up to 80% of IBD patients harbour the Bact2-enterotype, which is seen as a dysbiotic enterotype, while less than 15% of healthy individuals possess this enterotype.6,10
Despite the vast number of associations made with bacteria, little is known about the role of the viral component in the pathology and activity of IBD. A growing body of evidence suggests that disease pathology is associated with alterations in the gut virota as well.11–14 These alterations are largely characterized by a high abundance of members of the Caudovirales and a low abundance of Microviridae members.12,15 Other alterations show a highly lysogenic potential of the gut virota.11 In addition, diversity changes have also been reported, albeit in an inconsistent manner.11–14,16,17 One of the interesting viral groups is the recently identified CrAss-like phages, which belongs to the order Crassvirales. These viruses are a diverse group that are believed to be the most abundant viruses present in the human gut.18 In a recent study by Gulyaeva and colleagues, it was reported that Crassvirales are depleted in the gut microbiota of individuals with IBD.19 Other studies have suggested that some lytic phages [e.g. CrAss-like phage crAss001] could exist in symbiosis with their bacterial host, and even drive bacterial diversity through a process called phase variation.20 More specifically, phase variation allows the parallel multiplication of phages and their host by providing a balance between phage resistance and sensitivity. To date, no consistent associations have been made between the gut virota and disease activity. However, we anticipate that unravelling the full complexity of the human gut virome will deepen our understanding of complex human disease. Consequently, a viral counterpart of enterotyping [‘viral community-typing’] might improve this understanding and would allow stratification of individuals based on their gut virota. A first attempt was performed by Song and colleagues using published sequencing data of 2690 metagenomes, but was unable to describe the viral taxonomic composition of the community types due to a high number of unclassified viruses.21
In this study, we analysed faecal samples [n = 432] of a prospective cohort of active IBD patients [n = 181] starting biological therapies and investigated the factors shaping the gut virome composition. Furthermore, we investigated and found the presence of two gut-bound virome configurations that may be involved in the pathophysiology of IBD. Finally, those viral configurations were seen to be further associated with therapeutic success, suggesting a potential clinical impact.
2. Methods
Faecal samples were collected from a prospective IBD cohort [126 CD and 55 UC] based on sample availability and pairing [n = 432]. All IBD patients had active disease at baseline defined by endoscopy and were started on one of four approved biological therapies [infliximab, adalimumab, ustekinumab or vedolizumab]. Every patient provided a baseline and a follow-up [primary endpoint] sample. The NetoVIR protocol was used to prepare faecal samples for viral metagenomics, as described before [Extended Data Figure 2A].22 Further bioinformatic processing, viral community typing, diversity analysis and other details regarding the protocols are described in detail in the attachment [Extended Data Methods].
2.1. Ethics approval and consent to participate
The study was approved by the ethical commission of UZ Leuven [KU Leuven, reference number: S53684]. Participants provided signed informed consent to participate in the study. The design of the study was in accordance with the Declaration of Helsinki and Belgian privacy law.
3. Results
3.1. The gut virome is dominated by Caudoviricetes and Malgrandaviricetes phages in a multi-therapeutic IBD cohort
Faecal samples [n = 432] were collected from patients in a prospective multi-therapeutic IBD cohort [n = 181].23 Patients had either active ulcerative colitis [UC, n = 55] or active Crohn’s disease [CD, n = 126] and started biologicals as part of their medical care [51 infliximab, 29 adalimumab, 63 vedolizumab and 49 ustekinumab]. Patients were re-evaluated at the pre-defined primary endpoint [post-intervention] and classified as achieving remission or not [Extended Data Figure 1; Extended Data Methods; Supplementary Table S1]. For every included patient a baseline and primary endpoint faecal sample was analysed, and 50 CD patients contained and additional week 14 timepoint [Supplementary Table S1]. The gut virome was characterized using the NetoVIR protocol to isolate, enrich and sequence viruses in faecal samples [Extended Data Figure 2A]. Computational analyses on an input of 10.2 billion paired-end reads [1.52 TB, mean = 23.6 million reads per sample] was performed with the most up-to-date methodologies [Extended Data Figure 2B]. Most of the quality-controlled reads were found to be of viral origin [viral = 63.0%, bacterial = 32.3%, other = 3.0%, dark matter = 1.0%; Extended Data Figure 3]. Most of the identified viruses could reliably be classified at class-level taxonomy [classified = 93%, unclassified = 7%; Extended Data Figure 4; Supplementary Table S2]. The two most abundant viral classes were Caudoviricetes [58.1%, dsDNA tailed phages] and Malgrandaviricetes [37.7%, ssDNA circular phages], representing most quality-controlled reads [mean = 95.8%, range = 2.76–100% per sample]. The former viral class could be broken down into Caudoviricetes [non-CrAss] and Crassvirales phages, encompassing 41.9% and 16.2% of the quality-controlled reads, respectively [Supplementary Table S2].
3.2. The eukaryotic virome is small and is largely composed of plant viruses
In less than half of the samples eukaryotic viruses were detected [32.9%] which accounted for a minority of the quality-controlled viral reads [eukaryotic viruses = 0.2%, phages = 99.8%; Extended Data Figure 5; Supplementary Table S2]. Eukaryotic viruses could be grouped based on the host, which could be known [animal, plant or fungal viruses] or unknown [small circular viruses]. Most of the viruses detected in IBD patients belonged to the plant and fungal viral group, with only a few samples containing small circular viruses, or viruses potentially causing gastroenteritis. The two most prevalent viral species were pepper mild mottle virus [prevalence = 12.9%, genus Tobamovirus] and pepino mosaic virus [prevalence = 10.8%, genus Potexvirus], probably obtained via the patient’s diet [Supplementary Table S3].24 Given the low prevalence and abundance of eukaryotic viruses in adult faecal samples, we considered them to have limited importance for IBD patients.
3.3. The gut virota reveals the existence of two virome configurations in IBD patients
The gut microbiota is complex and variable, further complicating its thorough exploration. One approach is to condense the bacterial complexity into bacterial community types [‘enterotypes’]. By using Dirichlet Multinomial Mixture [DMM] modelling, bacterial research has consistently stratified large human gut microbiota studies into four enterotypes [Extended Data Methods].9,23,25 We used the same methodology and applied this for the first time to the gut virota. The gut virota observed in the IBD cohort consisted of no fewer than 874 genus-like groups [median = 26, range = 3–74 per individual, Extended Data Figure 6, Extended Data Methods]. Applying the DMM algorithm reduced the viral complexity and revealed the existence of two distinct clusters, or virome configurations [n = 363, genus-like group, Bray–Curtis dissimilarity; Figure 1A; Supplementary Table S4]. The groups showed a high probability of cluster assignment and were hereafter referred to as viral community types [median = 99.6%, Extended Data Figure 7; Supplementary Table S4].
Figure 1.

Viral community types to stratify individuals based on their virome composition. [A] Principal coordinates analysis [PCoA] of inter-individual differences of the gut virome composition [genus-like group, Bray–Curtis dissimilarity] of the IBD cohort [coloured by viral community types, n = 363] with shape representing categorical richness (median richness = 31, open circles < 31 [low richness], closed circles ≥ 31 [high richness], R2 = 0.0193, p = 0.001, Supplementary Table S8). [B] Boxplot showing relative abundance of major phage classes [≥1% of reads] stratified according to viral community type [n = 363, Mann–Whitney U, adj p < 0.05]. [C] Boxplot showing alpha-diversity metrics [observed richness and Shannon diversity] stratified according to viral community type [n = 363, Mann–Whitney U, adj p < 0.05]. [D, left] Violin plot showing comparisons of observed richness stratified according to phage lifestyle [lytic vs lysogenic] within each viral community type [comparison within viral community type CrM, n = 121, Mann–Whitney U, adj p < 0.05] and between viral community types [n = 363, Mann–Whitney U, adj p < 0.05]. [D, right] Barplot showing the distribution of phage lifestyles [lytic vs lysogenic] for major phage classes [≥1% of reads]. [E, left] Doughnut plot visualizing host prediction of phages on contig and read level [≥1% of reads]. [E, right] Boxplot visualizing host prediction of phages on read level [relative abundance] stratified according to viral community type [comparison Prevotella-infecting phages, n = 363, Mann–Whitney U, adj p < 0.05]. Adjustment for multiple testing [adj p] was performed using the Benjamini–Hochberg method. Significant associations [adj p < 0.05] are visualized by an asterisk [*]. Abbreviations: relative abundance [RA], community type Caudoviricetes [non-CrAss] [CA] and community type Crassvirales–Malgrandaviricetes [CrM].
To obtain insights into these viral community types, we investigated the virome compositional variation and found that viral community types were associated with distinct groups of viruses [Figure 1B]. The first viral community type [termed ‘CA’] revealed a high relative abundance of members of the Caudoviricetes [non-CrAss] [n = 363, Mann–Whitney U, r = 0.137, adj p = 0.0266; Supplementary Table S5], whereas the second [termed ‘CrM’] revealed a high relative abundance of members of the Crassvirales and Malgrandaviricetes [n = 363, Mann–Whitney U, adj p < 0.05]. Viral community types were characterized by a distinct α-diversity [Figure 1C]. Viral community type CrM had a higher viral richness [n = 363, Mann–Whitney U, r = 0.542, adj p < 2.2e-16] and Shannon diversity [n = 363, Mann–Whitney U, r = 0.384, adj p < 4.76e-10] compared to viral community type CA. We hypothesized that this diversity variation might be the result of a different phage lifestyle [lytic vs lysogenic], as determined by the presence or absence of lysogeny-specific proteins such as integrases [Supplementary Table S6].26 We found that viral community type CrM, but not CA, was associated with a richness expansion of lytic compared to lysogenic phages [n = 121, Mann–Whitney U, r = 0.308, adj p = 6.86e-06; Figure 1D]. In addition, only phages belonging to the classes Caudoviricetes and Crassvirales were predicted to be lysogenic in these samples (Caudoviricetes [non-CrAss] = 69.4%, Crassvirales = 10.8%, Malgrandaviricetes = 0%; Figure 1D). Overall, both community types showed a high relative abundance of lytic compared to lysogenic phages [Mann–Whitney U, adj p < 0.05; Supplementary Table S5], but only in viral community CrM did we observe a high relative abundance of lytic Caudoviricetes phages [CrAss + non-CrAss, n = 121, Mann–Whitney U, r = 0.307, adj p = 0.00360; Supplementary Table S5], suggesting that Caudoviricetes prophages were induced in patients with viral community type CA.
Furthermore, viral community types were associated with IBD subtype [proportion test, χ2 = 5.20 adj p = 0.0472; Supplementary Table S5] but not disease location [proportion test, χ2 = 1.04 adj p = 0.791; Supplementary Table S5], as characterized by a higher prevalence of viral community type CA in CD compared to UC patients [CD = 69.8%, UC = 57.9%, Supplementary Table S5].
3.4. Viral community type CA is mostly associated with the dysbiotic Bact2-enterotype
Next, we implemented in silico phage host prediction to determine the bacterial host. Most bacterial hosts were predicted at the phylum level [83.6%] but only a few hosts could be predicted at the genus level [27.7%; Extended Data Figure 8]. The majority of the host phyla were Bacteroidetes, Firmicutes and Proteobacteria, which were predicted to be infected preferentially by Crassvirales, Caudoviricetes and Malgrandaviricetes phages [Extended Data Figure 8A]. At the genus level, most of the predicted host genera were Bacteroides and Prevotella, which were preferentially infected by Caudoviricetes [non-CrAss] and Crassvirales phages, respectively [≥1% reads; Figure 1E; Extended Data Figure 8B]. Viral community type CrM had a high relative abundance of Prevotella-infecting phages compared to viral community type CA [n = 363, Mann–Whitney U, r = 0.235, adj p = 1.51e-05; Supplementary Table S5], but no significant differences were found in relative abundance of Bacteroides-infecting phages between community types [n = 363, Mann–Whitney U, r = 0.00530, adj p = 1.00; Supplementary Table S5].
The gut microbiota of IBD patients has been repeatedly associated with the dysbiotic Bact2-enterotype.9,23Extended Data Figure 9 shows a statistical correlation between the bacterial enterotypes [Bact1, Bact2, Prev and Rum] and the viral community types [Supplementary Table S7]. Viral community type CA was mostly associated with the Bact2-enterotype. This dysbiotic enterotype showed a higher prevalence in viral community type CA compared to viral community type CrM in UC [CA = 57.1%, CrM = 25.6%, n = 88, proportion test, χ2 = 7.55, adj p = 0.012; Supplementary Table S7] and in CD patients [CA = 79.6%, CrM = 38.3%, n = 207, proportion test, χ2 = 3.13, adj p = 8.96e-08]. Conversely, viral community type CrM was mostly associated with the Bact1-enterotype. This enterotype showed a higher prevalence in viral community CrM compared to viral community type CA in UC [CA = 16.3%, CrM = 48.7%, n = 88, proportion test, χ2 = 9.24, adj p = 9.44e-03] and CD patients [CA = 12.2%, CrM = 35.0%, n = 88, proportion test, χ2 = 1.30, adj p = 4.20e-04].
3.5. The gut virome composition is individual and is associated with disease location, the patient’s age and moisture content of the faecal samples
The factors shaping the gut virome composition were determined by distance-based redundancy analysis [dbRDA] in the IBD cohort [Supplementary Table S9]. Patient individuality was identified as the largest explanatory variable, thereby reinforcing the notion that the virome is highly individual-specific [n = 363, multivariate dbRDA, genus-like group, R2 = 75.8%, adj p = 0.001; Figure 2A]. Disease location was identified as the second largest explanatory variable of virome variation [n = 363, multivariate dbRDA, genus-like group, R2 = 1.40%, adj p = 0.001]. Next, patient age and moisture content of the stool also showed a limited contribution to the virome variation [n = 363, multivariate dbRDA, genus-like group, age R2 = 0.5%, moisture R2 = 0.3%, adj p < 0.05]. Remarkably, disease location [Montreal classification] stratifying patients according to ileal [L1CD], colonic [L2CD/UC] and ileocolonic [L3CD] phenotypes, revealed a higher explanatory power than simple diagnosis [UC/CD], and was observed in bacterial research as well [n = 363, univariate dbRDA, genus-like group, location R2 = 1.34%, diagnosis R2 = 0.47%, adj p < 0.05].23 Despite our expectations, the choice of biological therapy did not show an association [univariate or multivariate dbRDA] with the virome variation [n = 363, univariate dbRDA, genus-like group, therapy, adj p > 0.05 Supplementary Table S9].
Figure 2.
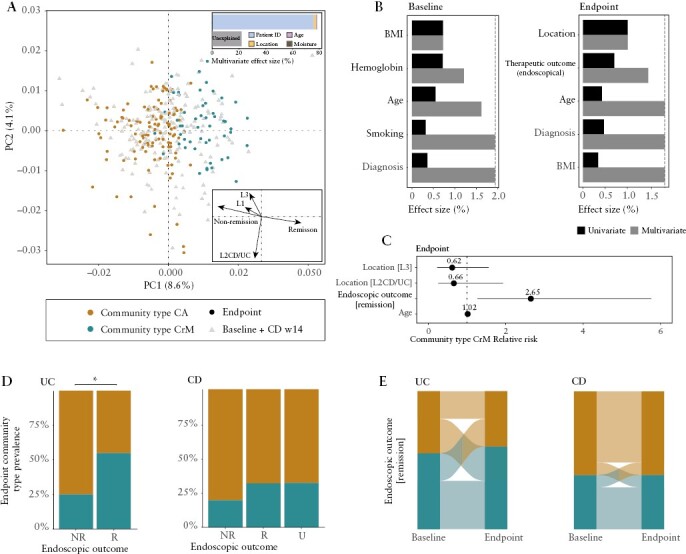
Virome covariates of the IBD cohort, and their association with viral community types. [A] Principal coordinate analysis of inter-individual differences of the gut virome composition [genus-like group, Bray–Curtis dissimilarity] of the IBD cohort [coloured by viral community types, n = 363] with shape representing sample time pointsa. Insert top: barplot representing significant covariates of virome composition of the IBD cohort, as identified in a multivariate model. The cumulative model [patient ID, age, location and moisture content] explains 78.0% of the virome variation [stepwise multivariate R2]. Arrows represent the effect sizes of a post hoc fit of significant virome covariates of post-intervention samples, as identified in a multivariate model in B [right]. [B] Metadata variables correlating [driving] significantly to virome variation in [left] pre-intervention and [right] post-intervention samples [dbRDA, genus-like group, Bray–Curtis dissimilarity]. Effect sizes of correlating metadata are calculated independently [univariate coloured in black, redundant covariates] or in a cumulative model [multivariate coloured in black, non-redundant covariates]. The purple dashed line represents the threshold for significant contribution to the multivariate model [metadata variables exceeding threshold coloured in black]. [C–E] Modelling the association between the prevalence of viral community type CrM and significant non-redundant covariates [location, therapeutic outcome as measured by endoscopy and age] of post-intervention samples [logistic regression model, n = 166]. [C] Relative risk ratio of IBD patients hosting community type CrM associated with significant non-redundant covariates of the virome composition in post-intervention samples (endoscopic outcome [remission], n = 166, RR = 2.65, adj p < 0.05). [D] Barplot showing viral community type [CA/CrM] prevalence in post-intervention samples stratified according to endoscopic outcome [R, NR, U] for UC [left] and CD [right] patients [comparison UC endoscopic outcome, n = 51, proportion test, adj p < 0.05]. [E] Alluvial diagram showing transitions of viral community types after successful intervention for UC [left] and CD [right] patients [logistic regression, n = 79 × 2, adj p > 0.05]. A total of 25.0% of UC patients and 9.09% of CD patients shift towards viral community type CrM, whilst only 20.0% of UC patients and 9.09% of CD patients shift towards viral community type CA. Adjustment for multiple testing [adj p] was performed using the Benjamini–Hochberg method. Significant associations [adj p < 0.05] are visualized with an asterisk [*]. Abbreviations: ulcerative colitis [UC], Crohn’s disease [CD], remission [R], non-remission [NR], unknown [U], pre-intervention [baseline], post-intervention [endpoint/primary endpoint], community type Caudoviricetes [non-CrAss] [CA] and community type Crassvirales–Malgrandaviricetes [CrM].
To further evaluate the factors shaping the gut virome composition in active IBD patients, we focused on baseline [pre-intervention] samples [Figure 2B; Supplementary Table S10]. We identified body-mass index [BMI], haemoglobin concentration, age and smoking behaviour as unique contributors to gut virome variation in baseline samples [n = 151, multivariate dbRDA, genus-like group, R2 = 1.94%, adj p = 0.050, full model]. We found that the patient’s BMI correlated with haemoglobin concentration [n = 151, ρ = 0.230, adj p = 0.00460; Supplementary Table S11] and reasoned that a low BMI and a low haemoglobin concentration [anaemia] probably expressed frailty of active IBD patients. In addition, we hypothesized that the gut virome might have a predictive capacity to determine therapeutic outcome [remission or non-remission at primary endpoint]. Unfortunately, none of the disease activity indices could be associated with virome composition at baseline; however, viral community types could be associated with endoscopic outcome [n = 151, adj p < 0.050; Supplementary Table S10]. Here, remitting patients harboured a 225% increased probability of hosting viral community type CrM, while simultaneously presenting a poor predictive power (univariate logistic regression, area under the curve [AUC] = 60.0, adj p = 0.0348; Supplementary Table S10), suggesting that community types had slight pharmacodynamic capabilities able to track patient outcomes.
3.6. The gut virome composition and community types are associated with endoscopic outcome in UC patients
To determine the effect of therapy outcome on the gut virome composition we investigated the association of all covariates on primary endpoint [post-intervention] samples in IBD patients [Figure 2B; Supplementary Table S12]. Disease location and the patient’s age were again identified as unique explanatory variables [n = 166, univariate dbRDA, genus-like group, Location R2 = 1.01%, Age R2 = 0.370%, adj p < 0.050]. Interestingly, therapeutic outcome appeared as a new covariate of the gut virome composition, as measured by the golden standard of endoscopic evaluation [n = 166, multivariate dbRDA, genus-like group, R2 = 0.460%, adj p = 0.0320]. On the other hand, other disease activity indices [clinical and biomarker evaluation] did not have a significant contribution to the virome variation. Moreover, no association was found between remission rates and disease location [n = 138, proportion test, χ2 = 7.47, r = 0.233, adj p = 0.0584; Supplementary Table S13], probably because of limited sample sizes.
By focusing on post-intervention samples, this study premiered endoscopic outcome as a significant covariate of the gut virome composition in IBD patients. We hypothesized that virome covariates might be associated with viral community types in post-intervention samples, and therefore modelled the association between viral community types and the three significant covariates of virome variation [n = 166, logistic regression; Figure 2C; Supplementary Table S14]. The disease location and patient’s age could not be associated with viral community types [n = 166, adj p > 0.05], while the endoscopic outcome could be associated with viral community types [n = 166, adj p = 0.0280]. Remitting patients had a 265% increased probability of hosting viral community type CrM (endoscopic remission relative risk [RR] = 2.65) and a 62% decreased probability of hosting viral community type CA [endoscopic non-remission RR = 0.38]. Stratification of endoscopic outcome for each IBD subtype demonstrated that UC but not CD patients had a higher prevalence of viral community type CrM when in remission [Figure 2D]. Viral community type CrM was found in 54.8% of the remitting and 25.0% of the non-remitting UC patients [n = 51, proportion test, χ2 = 4.41, r = 0.254, adj p = 0.0357; Supplementary Table S15]. Conversely, viral community type CrM was found in 31.7% of the remitting and 19.6% of the non-remitting CD patients [n = 115, proportion test, χ2 = 2.21, r = 0.136, adj p = 0.345; Supplementary Table S15]. Next, we evaluated the impact of treatment success [endoscopic outcome] on the gut virome, as determined by compositional changes between baseline and the primary endpoint [paired statistics, n = 2 × 103, Supplementary Table S9]. There were no changes in the gut virome composition over time associated with endoscopic remission, across all patients [paired dbRDA, n = 2 × 53, R2 = 0.869%, adj p = 0.688], or following stratification of the patients into corresponding UC and CD subtypes [Figure 2E; Supplementary Table S14].
3.6. Intestinal inflammation in UC patients is associated with a low viral diversity and a high lysogenic potential of Caudoviricetes
To investigate the effect of the endoscopic outcome on the major viral classes in more detail we focused on post-interventional samples of the IBD patients. The major viral classes of UC but not CD patients were characterized by a distinct α-diversity between remitting and non-remitting patients [Figure 3A and C; Supplementary Tables S16–S18]. Non-remitting UC patients revealed a low Shannon diversity of Caudoviricetes [non-CrAss] [n = 51, Mann–Whitney U, r = 0.372, adj p = 0.0143; Supplementary Table S16] and Malgrandaviricetes [n = 51, Mann–Whitney U, r = 0.329, adj p = 0.0378; Supplementary Table S16] phages compared to remitting UC patients. Other metrics [Pielou’s evenness and richness] were not seen to be affected by the endoscopic outcome. Conversely, none of the metrics [Shannon diversity, Pielou’s evenness and richness] were affected by the endoscopic outcome in CD patients [n = 94, Mann–Whitney U, adj p < 0.05; Supplementary Tables S16–S18]. Next, we hypothesized that therapeutic outcome might be associated with a changing phage lifestyle [Figure 3B and C]. A high relative abundance of lysogenic phages was observed in non-remitting compared to remitting UC patients [n = 51, Mann–Whitney U, r = 0.335, adj p = 0.0344], while no differences in phage lifestyle were found between non-remitting and remitting CD patients [n = 94, Mann–Whitney U, r = 0.219, adj p = 0.0677; Supplementary Table S19]. Moreover, the relative abundance of lysogenic phages in [non]-remitting UC patients was not associated with viral community types [n = 20, Mann–Whitney U, r = 0.127, adj p = 1.00; Supplementary Table S19]. Therefore, the expanded lysogenic potential of phages was thought to be associated with endoscopic outcome rather than viral community types. The high lysogenic potential in non-remitting UC patients appeared to be associated with the entire class of Caudoviricetes phages ([CrAss + non-CrAss], n = 51, Mann–Whitney U, r = 0.313, adj p = 0.0498; Supplementary Table S20). To evaluate the finding by Gulyaeva and colleagues that Crassvirales phages in the human gut were depleted in IBD patients, we also assessed the association between the prevalence of Crassvirales and endoscopic outcome.19 UC patients showed a prevalence of 77.4% in remission compared to 60.0% in non-remission [n = 51, proportion test, χ2 = 1.04, adj p = 0.617; Supplementary Table S21]. CD patients showed a prevalence of 71.9% in remission compared to 62.1% in non-remission [n = 94, proportion test, χ2 = 1.76, adj p = 0.419]. In addition, no associations were found between the relative abundance of Crassvirales and endoscopic outcome.
Figure 3.

Virome characteristics of post-intervention samples stratified according to endoscopic outcome. [A] Boxplot showing alpha-diversity metrics [observed richness and Shannon diversity] and evenness [Pielou’s evenness] for major phages classes [>1% reads] stratified according to endoscopic outcome [NR/R] for UC [left] and CD [right] patients (comparison Malgrandaviricetes and Caudoviricetes [non-CrAss] diversity in UC, Mann–Whitney U test, adj p < 0.05). [B] Boxplot showing relative abundance of lysogenic [top] and lysogenic Caudoviricetes phages stratified according to endoscopic outcome [NR/R] for UC [left] and CD [right] patients (comparison lysogenic Caudoviricetes [CrAss + non-CrAss]) phages in UC, Mann–Whitney U test, adj p < 0.05). [C] Graphical summary of virome characteristics detected in post-intervention samples. Adjustment for multiple testing [adj p] was performed using the Benjamini–Hochberg method. Significant associations [adj p < 0.05] are visualized with an asterisk [*]. Abbreviations: remission [R], non-remission [NR], community type Caudoviricetes [non-CrAss] [CA] and community type Crassvirales–Malgrandaviricetes [CrM].
3.7. Individual gut phages are associated with treatment success in IBD patients undergoing biological therapy
To investigate the capacity of individual phages to determine therapeutic success, we focused on pre-interventional samples of IBD patients. We argue that these phages should be [1] highly shared between patients and [2] differentiate between endoscopic treatment success [remission vs non-remission]. First, the 20 most prevalent viruses were determined for each IBD subtype and at least one of them was found in 97.7% of UC and 87.6% of CD patients [Extended Data Figure 10, Supplementary Table S22]. Second, phages with a capability to differentiate between endoscopic treatment success were identified for each IBD subtype (linear discriminant analysis score [log10] > 2, adj p < 0.05; Figure 4A; Supplementary Table S23]. Accordingly, five individual novel phages [both highly prevalent and differentially present] were found to meet these criteria [nUC = 44, nCD = 99; Figure 4B; Supplementary Tables S24 and S25]. Specifically, UC patients harboured two prevalent phages associated with endoscopic remission [CrAssella-R and Cripes-R] and one prevalent phage associated with endoscopic non-remission [Croccus-NR]. By contrast, CD patients harboured two prevalent phages associated with endoscopic remission [CrAssella-R and Croides-R] and one prevalent phage associated with endoscopic non-remission [Croides-NR]. CrAssella-R is a Crassvirales phage that was associated with treatment success and the only phage detected in both of the IBD subtypes. We argued that next-generation sequencing [NGS] methods, in combination with stringent bioinformatic criteria, might underestimate the presence and quantity of phages in IBD patients. Therefore, quantitative polymerase chain reaction [qPCR] was used to quantify and validate the association with endoscopic outcome of the five above-mentioned phages [Supplementary Table S26]. Consequently, two phages in UC [Cripes-R and Croccus-NR] revealed a higher qPCR positivity rate than NGS positivity rate [n = 44, qPCR = 79.5%/79.5%, NGS = 16.9%/18.2%, respectively; Figure 4C; Supplementary Table S27]. Three phages in CD [CrAssella-R, Croides-R and Croides-NR] revealed a higher qPCR positivity rate as well [n = 88, qPCR = 21.4%/52.4%/36.4%, NGS = 16.2%/21.2%/10.1%; Supplementary Table S27]. Only one phage [CrAssella-R] revealed a slightly lower qPCR positivity rate [n = 44, qPCR = 22.7%, NGS = 29.5%]. Next, the true predictive capacity (IBD predictive value [IPV]) of individual phages was determined based on the viral copy number [Supplementary Table S27]. Remitting IBD patients were predicted by a positive IPV [n = 82, IPV > 0, sensitivity = 0.67; Figure 4D; Supplementary Table S27] and non-remitting IBD patients were predicted by a negative IPV [n = 82, IPV < 0, specificity = 0.68]. No predictions were made for IBD patients with a neutral IPV [n = 46, IPV = 0]. Next, we modelled the association between prevalence of remission and IPV and found a predictive power of 72.8% [n = 82, logistic regression, AUC = 72.8%, adj p = 0.000394; Figure 4E; Supplementary Table S28]. Finally, we assessed a combination of IPV and viral community types and found that the latter variable does not significantly contribute to the model. In general, individual viruses revealed a capacity to determine therapeutic success in IBD patients, in which Crassvirales phages may play an important role.
Figure 4.
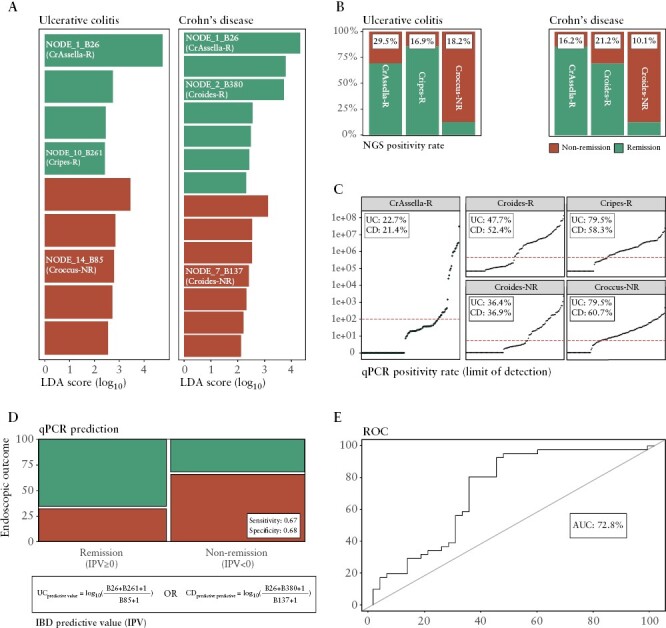
Predictive potential of the individual gut phages to determine therapeutic [endoscopic] outcome in IBD patients undergoing biological therapy. [A] Pre-interventional [baseline] differential abundance analysis of individual gut viruses to determine endoscopic outcome [non-remission/remission] by linear discriminant analysis [LDA] and effect size [Lefse] in IBD patients undergoing biological therapy (LDA score [log10] ≥ 2, n = 143, Mann–Whitney U test, adj p < 0.05). The NODE names of viruses with potentially predictive capacity for determining endoscopic outcome within UC [left, n = 44] or CD [right, n = 99] patients are coloured in white. [B] Barplot representing the prevalence of five phages with potential predictive capacity as stratified for endoscopic outcome for UC [left, three phages] and CD [right, three phages] patients. NODE_1_B26 is found to have predictive capacity for both IBD subtypes [bold]. NGS positivity rate [prevalence in baseline samples] is shown for each phage in the white boxes [n = 143]. [C] Scatterplot representing the number viral copies in baseline samples for each potential predictive phage [n = 128]. qPCR positivity rate [prevalence in baseline samples] is shown for each predictive phage in the white boxes [limit of detection ≥100 viral copies, represented by red dashed line] for IBD subtypes. [D, E] The predictive capacity of individual phages to determine endoscopic outcome is measured by the concept of IPV based on viral copy number. IPV is calculated by the natural logarithm of the remission predictive phages over the non-remission predictive phages for IBD subtypes [bottom formula, limit of quantification ≥500 viral copies]. [D] Mosaic plot displaying the distribution of [excluding samples with no predictions] the categorical variables endoscopic remission and qPCR prediction [based on IPV, sensitivity = 0.67, specificity = 0.68]. [E] Modelling the association between prevalence of remission and IPV [logistic regression, n = 88, RR = 1.32, adj p < 0.05]. ROC curve representing predictive potential of phages [n = 88, AUC = 72.8%]. Adjustment for multiple testing [adj p] was performed using the Benjamini–Hochberg method. Abbreviations: IBD predictive value [IPV], ulcerative colitis [UC], Crohn’s disease [CD], inflammatory bowel disease [IBD], next-generation sequencing [NGS] and receiver operating characteristic [ROC] curve.
4. Discussion
To date, this research represents one of the most comprehensive gut virome studies [using viral particle enrichment followed by deep sequencing] of IBD patients undergoing biological treatment. Here, we implemented the concept of viral community typing and found that patients could be stratified into two [CA/CrM] virome constellations [Figure 1A]. We hypothesize that patients harbouring viral community type CA exhibited a dysbalanced virome, characterized by a high abundance of Caudoviricetes [non-CrAss] and low viral diversity, which has been observed before [Figure 1B and C].12 The association with the dysbiotic Bact2-enterotype strengthens the belief that this constellation reflects the viral counterpart of gut dysbiosis [Extended Data Figure 9]. This association indicates that phages within viral community type CA may preferentially infect bacteria in the Bact2-enterotype. Conversely, patients harbouring viral community CrM might exhibit a more eubiotic-like virome characterized by a high abundance of members of the Crassvirales and Malgrandaviricetes, a high viral diversity, and a shift away from the dysbiotic Bact2-enterotype.
Furthermore, remitting UC patients are associated with viral community type CrM, whereas non-remitting UC patients are associated with viral community type CA, thereby reinforcing the notion that viral community types might reflect a potential dysbalanced or eubiotic-like state of the gut virome, and may therefore be clinically relevant [Figure 2B–D]. However, these findings could not be reproduced for CD patients, suggesting that these patients retain a dysbalanced state even in the case of remission or quiescent disease. Thus far, twin studies revealed a larger genetic impact in CD pathology and, in contrast, a larger non-genetic or environmental [e.g. virome] impact in UC pathology.27 These findings are corroborated in this study, further revealing a substantial role of the gut microbial ecosystem in UC pathology [Figure 3]. Consequently, thorough analysis of the gut inflammation in IBD revealed a decrease in viral diversity and an expansion of lysogenic phages. However, changes in viral diversity and phage lifestyle were not observed in CD pathology, thereby again confirming the different role of the virome regarding the IBD subtype. In addition, previous studies found a higher Caudoviricetes richness in IBD subtypes [Norman and colleagues13] and only in CD patients [Clooney and colleagues11], but no significant differences in virome richness or diversity across disease states. In our study, we did find a higher Caudoviricetes [non-CrAss] diversity in remitting UC but not remitting CD patients, and a higher prevalence of viral community type CrM. The different results obtained in different studies regarding richness and diversity might be largely due to methodological differences, specifically with respect to genome fragmentation bias. Corrections for this bias are necessary for accurate representation of viral richness and diversity, as described in detail previously.28 Furthermore, the lack of a healthy control cohort is a limitation in this study, but the results still show the presence of distinct viral constellations linked to endoscopic outcomes. Further research that includes a healthy matched control population could help to put our findings in a broader context.
We argue that a persistent inflamed state of the intestine will shape the gut virome by acting on different mechanisms. First, an increase in gut motility might lower microbial [and viral] diversity due to an increase in defecation frequency.29 Second, phage-mediated lysis might explain the observed expansion of lysogenic gut phages.30 Phage-mediated lysis describes a positive-feedback loop between phage induction and intestinal inflammation. Briefly, inflammation stimulates enterocytes to produce stressors [e.g. reactive oxygen species] activating a stress response in the host bacteria [‘SOS response’]. The stress response will trigger prophages to initiate the lytic lifecycle, leading to lysis of the bacterial host cell. An increased bacterial lysis will be accompanied by an increase of pathogen-associated molecular patterns [e.g. lipopolysaccharide, bacterial DNA] that stimulate pattern recognition receptors on enterocytes. These cells will produce more stressors and further promote prophage induction, thereby starting a positive feedback-loop and increasing the lysogenic potential under intestinal inflammation. Taken together, these mechanisms might explain the observed decrease in viral diversity and lysogenic expansion under inflammatory conditions [Figure 3].
Following viral study associated with gut inflammation, we also discovered five novel phages that were associated with treatment success, as confirmed by qPCR results [Figure 4]. One phage, a novel Crassvirales phage [CrAssella-R], showed an association with treatment success for both IBD subtypes. Shkoporov and colleagues described that long-term persistence of crAss0001 with its bacterial host could drive diversity [a hallmark of eubiosis] by a process called phase variation.20 We argue that biological treatment induces remission and that mechanisms such as phase variation could provide a path to maintain remission by stimulating eubiosis. Phages with predictive abilities might be those capable of driving diversity in combination with the respective host.
In conclusion, in this study we have shown that viral community types exist and allow the stratification of IBD patients based on a distinct viral composition in the gut, and could be used to better understand IBD subtypes, pathology and disease activity in the future.
Supplementary Material
Contributor Information
Daan Jansen, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute, Laboratory of Viral Metagenomics, Leuven, Belgium.
Gwen Falony, KU Leuven, Department of Microbiology Immunology and Transplantation, Rega Institute, Laboratory of Molecular Bacteriology, Leuven, Belgium; Center for Microbiology, VIB, B-3000 Leuven, Belgium.
Sara Vieira-Silva, KU Leuven, Department of Microbiology Immunology and Transplantation, Rega Institute, Laboratory of Molecular Bacteriology, Leuven, Belgium; Institute of Medical Microbiology and Hygiene, University Medical Center of the Johannes Gutenberg-University Mainz, Mainz, Germany; Institute of Molecular Biology (IMB), Mainz, Germany.
Ceren Simsek, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute, Laboratory of Viral Metagenomics, Leuven, Belgium.
Tine Marcelis, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute, Laboratory of Viral Metagenomics, Leuven, Belgium.
Clara Caenepeel, KU Leuven, Translational Research Center for Gastrointestinal Disorders (TARGID), University Hospitals Leuven, Leuven, Belgium.
Kathleen Machiels, KU Leuven, Translational Research Center for Gastrointestinal Disorders (TARGID), University Hospitals Leuven, Leuven, Belgium.
Jeroen Raes, KU Leuven, Department of Microbiology Immunology and Transplantation, Rega Institute, Laboratory of Molecular Bacteriology, Leuven, Belgium; Center for Microbiology, VIB, B-3000 Leuven, Belgium.
Séverine Vermeire, KU Leuven, Translational Research Center for Gastrointestinal Disorders (TARGID), University Hospitals Leuven, Leuven, Belgium.
Jelle Matthijnssens, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute, Laboratory of Viral Metagenomics, Leuven, Belgium.
Funding
This work was supported by the ‘Fonds Wetenschappelijk Onderzoek’ (Research foundation Flanders) with grant numbers [1S78021N,12M9118N].
Conflict of Interest
SV has received grants from AbbVie, J&J, Pfizer, Galapagos and Takeda. SV has received consulting and/or speaker fees from AbbVie, AbolerIS Pharma, AgomAb, Alimentiv, Arena Pharmaceuticals, AstraZeneca, Avaxia, BMS, Boehringer Ingelheim, Celgene, CVasThera, Dr Falk Pharma, Ferring, Galapagos, Genentech-Roche, Gilead, GSK, Hospira, Imidomics, Janssen, J&J, Lilly, Materia Prima, MiroBio, Morphic, MrMHealth, Mundipharma, MSD, Pfizer, Prodigest, Progenity, Prometheus, Robarts Clinical Trials, Second Genome, Shire, Surrozen, Takeda, Theravance, Tillots Pharma AG and Zealand Pharma. JR has received grants from the Colruyt group, Danone, DSM, MRM/Prodigest and Nestle. JR has received consulting and/or speaker fees from Biofortis, DSM, MRM/Prodigest, GSK, Janssen Pharmaceuticals and Metagenics. GF, SVS and JR report patents planned, issued or pending: (i) A new inflammation, low cell count enterotype (WO2019115755A1) and (ii) means and methods to diagnose gut flora dysbiosis and inflammation (WO2022073973A1). The other authors report no conflicts of interest. Readers are welcome to comment on the online version of the paper. Correspondence should be addressed to JM [jelle.matthijnssens@kuleuven.be].
Author Contributions
The study was conceived by JM, SV and JR. Experiments were designed by DJ, SV, JR and JM. Sampling was set up by CC, KM and SV. Experiments were performed by DJ, TM and CS. Bioinformatic and statistical analysis of the sequences reads was performed by DJ, SS and GF. DJ, CS, GF, SS and JM drafted the manuscript. All authors revised the article and approved the final version for publication.
Data Availability
Metadata can be found in Supplementary Table S1. The raw sequence data have been deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA804384. Sequences [predictive markers] have been deposited in GenBank under the following accession numbers: ON493177–ON493181. The ViPER [Virome Paired-End Reads pipeline] script was used to process raw paired-end reads and is publicly available at https://github.com/Matthijnssenslab/ViPER. All the data required to reproduce virome analyses will be made available at https://github.com/Matthijnssenslab/IBDVirome.
References
- 1. Wang B, Yao M, Lv L, Ling Z, Li L.. The human microbiota in health and disease. Engineering 2017;3:71–82. [Google Scholar]
- 2. Glassner KL, Abraham BP, Quigley EMM.. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol 2020;145:16–27. [DOI] [PubMed] [Google Scholar]
- 3. Öhman L, Lasson A, Strömbeck A, et al.. Fecal microbiota dynamics during disease activity and remission in newly diagnosed and established ulcerative colitis. Sci Reports 2021;11:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tedjo DI, Smolinska A, Savelkoul PH, et al.. The fecal microbiota as a biomarker for disease activity in Crohn’s disease. Sci Reports 2016;6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prosberg M, Bendtsen F, Vind I, Petersen AM, Gluud LL.. The association between the gut microbiota and the inflammatory bowel disease activity: a systematic review and meta-analysis. Scand J Gastroenterol 2016;51:1407–15. doi: 10.1080/0036552120161216587 [DOI] [PubMed] [Google Scholar]
- 6. Vieira-Silva S, Sabino J, Valles-Colomer M, et al. Quantitative microbiome profiling disentangles inflammation- and bile duct obstruction-associated microbiota alterations across PSC/IBD diagnoses. Nat Microbiol 2019;4:1826–31. [DOI] [PubMed] [Google Scholar]
- 7. Sokol H, Pigneur B, Watterlot L, et al.. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci 2008;105:16731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Costea PI, Hildebrand F, Manimozhiyan A, et al.. Enterotypes in the landscape of gut microbial community composition. Nat Microbiol 2017;3:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vandeputte D, Kathagen G, D’Hoe K, et al.. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017;551:507–11. [DOI] [PubMed] [Google Scholar]
- 10. Libby P. Statin drugs might boost healthy gut microbes. Nature 2020;581:263–4. [DOI] [PubMed] [Google Scholar]
- 11. Clooney AG, Sutton TDS, Shkoporov AN, et al. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe 2019;26:764–778.e5. [DOI] [PubMed] [Google Scholar]
- 12. Zuo T, Lu XJ, Zhang Y, et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019;68:1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Norman JM, Handley SA, Baldridge MT, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015;160:447–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liang G, Cobián-Güemes AG, Albenberg L, Bushman F.. The gut virome in inflammatory bowel diseases. Curr Opin Virol 2021;51:190–8. [DOI] [PubMed] [Google Scholar]
- 15. Liang G, Conrad MA, Kelsen JR, et al.. Dynamics of the stool virome in very early-onset inflammatory bowel disease. J Crohns Colitis 2020;14:1600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pérez-Brocal V, García-López R, Vázquez-Castellanos JF, et al.. Study of the viral and microbial communities associated with Crohn’s disease: a metagenomic approach. Clin Transl Gastroenterol 2013;4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernandes MA, Verstraete SG, Phan T, et al. Enteric virome and bacterial microbiota in children with ulcerative colitis and Crohn disease. J Pediatr Gastroenterol Nutr 2019;68:30–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yutin N, Benler S, Shmakov SA, et al. Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features. Nat Commun 2021;12:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gulyaeva A, Garmaeva S, Ruigrok RAAA, et al. Discovery, diversity, and functional associations of crAss-like phages in human gut metagenomes from four Dutch cohorts. Cell Rep 2022;38:110204. [DOI] [PubMed] [Google Scholar]
- 20. Shkoporov AN, Khokhlova EV, Stephens N, et al. Long-term persistence of crAss-like phage crAss001 is associated with phase variation in Bacteroides intestinalis. BMC Biol 2021;19:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Song L, Zhang L, Fang X.. Characterizing enterotypes in human metagenomics: a viral perspective. Front Microbiol 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Conceição-Neto N, Zeller M, Lefrère H, et al. Modular approach to customise sample preparation procedures for viral metagenomics: a reproducible protocol for virome analysis. Sci Rep 2015;12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caenepeel C, Falony G, Machiels K, et al. Dysbiosis-associated stool features improve prediction of response to biological therapy (anti-TNF, alpha, anti-integrin and anti-interleukin 12/23) in inflammatory bowel disease. Gastroenterology 2022. [DOI] [PubMed] [Google Scholar]
- 24. Schwarz D, Beuch U, Bandte M, Fakhro A, Büttner C, Obermeier C.. Spread and interaction of Pepino mosaic virus (PepMV) and Pythium aphanidermatum in a closed nutrient solution recirculation system: effects on tomato growth and yield. Plant Pathol 2010;59:443–52. [Google Scholar]
- 25. Vieira-Silva S, Falony G, Belda E, et al.. Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature 2020;581:310–5. [DOI] [PubMed] [Google Scholar]
- 26. Court DL, Oppenheim AB, Adhya SL.. A new look at bacteriophage λ genetic networks. J Bacteriol 2007;189:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Loddo I, Romano C.. Inflammatory bowel disease: Genetics, epigenetics, and pathogenesis. Front Immunol 2015;6:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jansen D, Matthijnssens J.. The emerging role of the gut virome in health and inflammatory bowel disease: challenges, covariates and a viral imbalance. Viruses 2023;15:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bassotti G, Antonelli E, Villanacci V, et al. Abnormal gut motility in inflammatory bowel disease: an update. Tech Coloproctol 2020;24:275–282. [DOI] [PubMed] [Google Scholar]
- 30. Zuppi M, Hendrickson HL, O’Sullivan JM, Vatanen T.. Phages in the gut ecosystem. Front Cell Infect Microbiol 2022;11:1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metadata can be found in Supplementary Table S1. The raw sequence data have been deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA804384. Sequences [predictive markers] have been deposited in GenBank under the following accession numbers: ON493177–ON493181. The ViPER [Virome Paired-End Reads pipeline] script was used to process raw paired-end reads and is publicly available at https://github.com/Matthijnssenslab/ViPER. All the data required to reproduce virome analyses will be made available at https://github.com/Matthijnssenslab/IBDVirome.