

Summary
Maladaptive, non-resolving inflammation contributes to chronic inflammatory diseases such as atherosclerosis. Because macrophages remove necrotic cells, defective macrophage programs can promote chronic inflammation with persistent tissue injury. Here, we investigated the mechanisms sustaining vascular macrophages. Intravital imaging revealed a spatiotemporal macrophage niche across vascular beds alongside mural cells (MCs)—pericytes and smooth muscle cells. Single-cell transcriptomics, co-culture, and genetic deletion experiments revealed MC-derived expression of the chemokines CCL2 and MIF, which actively preserved macrophage survival and their homeostatic functions. In atherosclerosis, this positioned macrophages in viable plaque areas, away from the necrotic core, and maintained a homeostatic macrophage phenotype. Disruption of this MC-macrophage unit via MC-specific deletion of these chemokines triggered detrimental macrophage relocalizing, exacerbated plaque necrosis, inflammation, and atheroprogression. In line, CCL2 inhibition at advanced stages of atherosclerosis showed detrimental effects. This work presents a MC-driven safeguard toward maintaining the homeostatic vascular macrophage niche.
Keywords: macrophages, smooth muscle cells, pericytes, mural cells, chronic inflammation, chemokines, atherosclerosis, vascular macrophages, CCL2, MIF
Graphical abstract
Highlights
-
•
Mural cell-derived macrophage chemoattractants preserve the vascular macrophage niche
-
•
Interference with this axis results in repositioning and reduced macrophage coverage
-
•
This is accompanied by detrimental shifts in macrophage phenotype and function
-
•
Disruption of mural cell-macrophage communication exacerbates inflammation
Chronic inflammation causes a high burden of disability and mortality worldwide. Vascular macrophages are key orchestrators of homeostasis and vice versa of chronic inflammation. Pekayvaz et al. show how mural cells (the contractile cells of the vessel wall) actively sustain the vascular macrophage niche and keep it in a functional state.
Introduction
Macrophages (MΦs) are local immune cells that are present across organs and have multiple essential tasks in tissue homeostasis.1,2,3,4,5 Uncontrolled cell death, defective removal of necrotic cells, and dysregulated secretion of inflammatory mediators by MΦs result in an unresolvable inflammatory state.6,7,8 MΦs preferentially colonize distinct anatomical niches across tissues and frequently reside at the vascular and perivascular space.9,10,11
Vascular MΦs can reduce tissue fibrosis in the microvasculature9 and maintain the arterial tone in the macrovasculature.12 Furthermore, vascular MΦs are decisive orchestrators of vascular inflammation and atherosclerosis,6,7 which are key drivers of mortality and morbidity worldwide.7,13,14,15,16 Chemokine-mediated trafficking of monocytes across the bone marrow, blood, spleen, and finally the trans-endothelial recruitment of monocytes toward chronic inflammatory foci is well-described.13,17 However, what tunes mature MΦ biology after their recruitment to the subendothelial space—whether and how the anatomical and functional vascular MΦ niche is actively maintained—is poorly understood.
Below the endothelial barrier, vascular MΦs dwell within a microenvironment dominated by mural cells (MCs), comprising smooth muscle cells (SMCs) in the macrovasculature and pericytes (PCs) in the microvasculature. MCs themselves can express inflammatory mediators during acute inflammation.18,19,20 Consequently, we hypothesized that MCs actively communicate with MΦs to sustain the vascular MΦ niche and modulate vascular MΦ programs. Here, we provide evidence for a homeostatic MC-MΦ axis, which sustains a pro-resolving MΦ phenotype. We define how MC-derived chemokines generate a vascular niche, which counteracts chronic inflammation as seen in atherosclerosis. This has important implications for the development of anti-inflammatory therapies in chronic inflammatory diseases.
Results
Vascular MΦs reside in a dedicated niche along mural cells across vascular beds
MΦs localize in small and large blood vessels across different mammalian organs and tissues.9,10,11 To understand the behavior of vessel-associated MΦs in vivo, we employed different murine reporter strains. First, Cx3cr1Cre-ERT2; PC-G5-tdT (Cx3cr1-MΦCa-rep) mice were used to investigate the functional activation status of MΦs in vivo. The Cx3cr1-promotor is extensively characterized for MΦ imaging approaches.9,19,21,22,23 Cx3cr1Cre-ERT2 mice have been developed by the Jung lab and allow fate mapping of monocytes and tissue MΦs.21 PC-G5-tdT mice express the calcium indicator protein GCaMP5G variant, which allows dynamic monitoring of intracellular calcium levels, as well as constitutive tdTomato in a cre-dependent manner. GCAMP5 is a fusion protein consisting of GFP, calmodulin, and a peptide sequence of the myosin light chain kinase. Using this system, we analyzed functional MΦ activation by multi-photon imaging.24 The Cx3cr1-MΦCa-rep model indeed exhibited high specificity and sensitivity in tracing skin MΦs (Figures S1A–S1C), in line with previous work with Cx3cr1-CreERT2-mediated reporter systems.21,25,26 By tracing dynamic changes in MΦ positioning, morphology, and calcium-activity simultaneously in vivo, we observed that vascular MΦs respond to sterile microinjuries, reaching necrotic foci within minutes (Figure 1A; Video S1). Microinjuries occur on a frequent basis in homeostatic tissue, for example, by emerging apoptotic bodies. Therefore, the ability of MΦs to reach microinjuries is pivotal for preserving tissue homeostasis and preventing chronic unresolvable inflammation.23
Figure 1.

Mural cells sustain a vascular MΦ niche
(A) In vivo multi-photon imaging of Ca2+ signal and morphological changes of MΦs in Cx3cr1-MΦCa-rep mice in an environment of laser-induced microinjuries. MΦs are depicted in red; Ca2+ signal and vascular flow are depicted in green. MΦs have been rendered additionally below, with a pseudocolored depiction of the Ca2+ signal. Images are derived from Video S1.
(B) In vivo and ex vivo confocal and airy-scan imaging of MC-MΦ contacts across organs in MCRFP-rep; Cx3cr1-MΦGFP-rep mice, arrows depicting cell-cell contacts: top left: intravital imaging of the microvasculature in the mesentery, the dashed line is depicting MCs (scale bars, 5 μm); top middle: ex vivo imaging of the heart microvasculature (scale bars, 50 μm); top right: en face ex vivo imaging of the aortic atherosclerotic intima after 3 months of western-diet feeding (macrovasculature), the dashed line is subdividing the plaque core from the shoulder region (scale bars, 10 μm); bottom left: ex vivo imaging of the kidney microvasculature (scale bars, 50 μm), including higher magnification below (dashed line depicting MCs) (scale bars, 7 μm); bottom middle: ex vivo imaging of the lung microvasculature (scale bars, 20 μm), including higher magnification below (Cx3cr1hi CD68lo interstitial MΦs (iMΦs) in green, CD68hi Cx3cr1lo alveolar MΦs (aMΦs) in white, MCs in red) (dashed line depicting MCs) (scale bars, 5 μm); bottom right: ex vivo imaging of the stomach microvasculature (scale bars, 50 μm), including higher magnification below (dashed line depicting MCs) (scale bars, 10 μm). Interstitial MΦs are shown in green, and MCs are shown in red for all organs, with further subdifferentiation of MΦs in the lung (as depicted above).
(C) Analysis of the time until MΦs form their first dendrites (left) and time which MΦs require to reach injury (right), as the time in minutes after laser injury, in MΦGFP-rep mice treated locally (subcutaneously) and systemically with isotype or CCL2-neutralizing antibody (n = 17–37 individual cells analyzed from 3 to –4 mice/group).
(D) Reanalyzed single-cell RNA-seq data from human coronary arteries from Wirka et al., GEO: GSE131780. Uniform Manifold Approximation and Projection (UMAP) based dimensionality reduction of analyzed cells.
(E) Highly expressed cytokines and chemokines in human SMCs from coronary arteries analyzed from cells shown in (D) CCL2 is highlighted as the most prominently expressed chemokine.
(F) Percentage of peritoneal macrophage survival upon CCL2 stimulation at different time points under starvation stress conditions (n = 3 experiments).
(G) Quantification of CD68+ perivascular macrophage content in Ccl2MC+/+ and Ccl2MCΔ/Δ mice in percentage of total perivascular area (15 μm radius around the vessel) in the kidney (n = 5–6 mice/group).
(H) Quantification of cell proliferation as EdU+ cells relative to CD68+ area (as number of proliferating cells/μm2).
(I) Quantification of blood monocyte counts by automated blood counter (n = 5–6).
(J) Representative images from immunofluorescence staining of kidney sections in Ccl2MCΔ/Δ and Ccl2MC+/+ mice for ACTA2 (red), CD68 (green). Scale bars, 50 μm (left: Ccl2MC+/+; right: Ccl2MCΔ/Δ). (C, G, H, and I) Student’s t test was used. (F) Repeated measures two-way ANOVA was -808990139890500used. ∗ p < 0.05. Bar graphs show mean with SEM.
In vivo multiphoton microscopy of a Cx3cr1-MΦCa-rep mouse ear microvasculature. Laser microinjuries are labelled with broad arrows. Macrophages are shown by dashed lines. In the upper video, MΦs are depicted in red, Ca2+ activity in MΦs is depicted in green, vascular flow is depicted in green. In the lower video, the same segment is rendered in pseudocolor depicting the maximum detected Ca2+ intensity of the depicted MΦ in green (high Ca2+ activity) and blue (low Ca2+ activity). On the right, Ca2+ signals of MΦ 1 and MΦ 2 are aligned over time. Video is paused briefly at instants with high Ca2+ activity within either one of the MΦs. Pseudocolor scale, time scale and scale bar are included in the video.
We hypothesized that micro- and macro-vascular MCs control MΦ function. To trace MCs and MΦs by intravital imaging, NG2+ cells, encompassing PCs as well as SMCs throughout the arterial tree (excluding venular PCs), were labeled by the red fluorescent protein DsRed, as previously described.19,20,27 Cross-breeding with ApoE−/− mice28 allowed induction of atherosclerotic vessel disease in Ng2-DsRed; ApoE−/− (MCRFP-rep) mice. In addition, we used a constitutive GFP transgene under the control of the native Cx3cr1 promotor19,23—Cx3cr1GFP (Cx3cr1-MΦGFP-rep) mice. Histological ex vivo whole-mount confocal imaging confirmed CX3CR1+ cells to co-express F4/80 and CD11c (Figures S1B and S1C).
We examined MC-MΦ contacts in different micro- and macro-vascular beds using Ng2DsRed; Cx3cr1GFP; ApoE−/− mice (MCRFP-rep Cx3cr1-MΦGFP-rep), labeling MCs and mononuclear phagocytes.19,20,29,30 MΦs engaged in tight and frequent contacts with MCs in microvascular beds of different organs and in the atherosclerotic macrovasculature (Figure 1B). MCs highly express Ccl2 and Mif.19,31 We therefore investigated whether MC-derived CCL2 might actively preserve this pro-homeostatic role of MΦs in vivo. Indeed, disruption of CCL2 signaling in Cx3cr1-MΦGFP-rep mice impaired the ability of perivascular MΦs to reach microinjuries (Figure 1C; Figure S1D).
To augment our understanding of the broad cytokine and chemokine repertoire expressed by MCs, we reanalyzed single-cell RNA sequencing (scRNA-seq) data from human coronary vessels, particularly rich in a broad set of MC subsets, acquired by Wirka et al.32 (Figure 1D). CCL2 and MIF showed robust expression across MC clusters (Figure 1E). CCL19 also showed moderate expression across multiple MC subsets. The CXCL9-CXCR3 axis holds important roles for MΦ functions33; however, CXCL9 expression by MCs was comparably low (Figure S1E). In summary, vascular MΦs reside along MCs and react to sterile inflammatory stimuli supported by several MΦ active chemokines, including CCL2 and macrophage migration inhibitory factor (MIF).
MCs promote MΦ survival and sustain a homeostatic MΦ phenotype
Based on this MΦ-MC colocalization and high MC-derived expression of distinct MΦ active chemokines such as CCL2 and MIF, we hypothesized that these MΦ chemotactic proteins might act as niche-maintaining signals for vessel-associated MΦs. We found that addition of CCL2 to MΦs enhanced MΦ survival in vitro (Figure 1F). We next investigated whether MΦ origin dictated responses toward survival-enhancing chemokines and cytokines. However, survival upon macrophage colony-stimulating factor (M-CSF) or CCL2 and MIF stimulation was similar between embryonic or bone marrow-derived MΦs (Figure S1F). To allow a better understanding of the potential CCL2-mediated MC-MΦ interplay in vivo, we generated mice that lack MC-derived CCL2 signaling (Ng2cre; Ccl2fl/fl; ApoE−/−, referred to here as Ccl2MCΔ/Δ mice) to investigate phenotypic and functional changes in MΦs in the absence of MC-derived CCL2. Because the kidney and lung have dense microvascular beds rich in MCs, we focused on these tissues. In the kidney microvasculature of Ccl2MCΔ/Δ mice, perivascular MΦ coverage was reduced compared with controls (Figures 1G and 1J). This finding was not explained by changes in proliferation or blood monocyte counts (Figures 1H–1J and S1G). We also found a modest reduction in interstitial but not alveolar MΦ coverage in the lung (Figures S1H and S1I).
We next performed scRNA-seq of fluorescence-activated cell sorting (FACS)-enriched kidney and lung MΦs from Ccl2MCΔ/Δ mice and identified subclusters consistent with previous publications34,35 (Figures 2A, 2B, and S2A). The kidney and lung are profoundly traversed by a fine-meshed vascular bed; hence, MΦs hold frequent contacts to vascular MCs (compare Figures 1B and S2B). We investigated the expression of distinct marker genes that define perivascular MΦs, such as Adgre1, Cx3cr1, and Itgax.10,36 Within the kidney, particularly MΦ clusters 0 and 2 (further focused on below) showed high expressions of perivascular MΦ markers Adgre1, Cx3cr1, and Itgax, in contrast to MΦ clusters 1, 5, 8, 10, and 14. Cluster 3 showed high expression of Adgre1 and Cx3cr1 but low expression of Itgax. Within the lung, particularly clusters 1 and 3 showed high expression levels of CD64 (Fcgr1) as well as Adgre1 in combination with CD11b (Itgam) but low levels of CD11c (Itgax), associated with a perivascular MΦ localization10,36,37 (Figure S2C). MΦ/monocyte clusters underwent distinct shifts between Ccl2MCΔ/Δ and Ccl2MC+/+ mice (Figure S2D).
Figure 2.

MC-derived CCL2 sustains a homeostatic MΦ phenotype across the vascular tree
(A and B) UMAP based dimensionality reduction of single-cell RNA-seq of FACS-sort enriched CD45+ CD11bhi CD64hi F4/80hi cells in kidney (A) and lung (B) of Ccl2MCΔ/Δ and Ccl2MC+/+ mice (n = 4/group).
(C–G) Volcano and violin plots depicting selected significantly differentially regulated genes in (C) kidney resident MΦ cluster 2, (D) kidney resident MΦ cluster 0, (E) lung monocyte cluster 1, (F) lung alveolar MΦ cluster 2, (G) lung Folr2hi Mrc1hi interstitial MΦ cluster 3.
(H) Frequency of lung Zeb2hi interstitial MΦ cluster 4 cells among all analyzed cells.
(I) Significantly differentially regulated genes, associated with a functionally differentiated, efferocytotic MΦ phenotype in Ccl2MC+/+ and Ccl2MCΔ/Δ chimera mice. Low-input RNA-seq of FACS-sorted Cx3cr1+ MΦs from Ccl2MC+/+ or Ccl2MCΔ/Δ chimera mice with MCRFP-ep; Cx3cr1-MΦGFP-rep bone marrow after 20 weeks western diet (experimental setup further depicted in Figure S3) (n = 3–4 chimera mice). Student’s t test was used. Expression levels of depicted genes normalized to sample with highest expression (set as 1) across all samples. Bar graphs show mean with SEM. Violin plots with matching boxplot and mean expression. ∗ p < 0.05.
Perivascular MΦ/monocyte clusters in Ccl2MCΔ/Δ mice showed lower levels of key modulators of homeostasis/alternative MΦ activity, including Atf3,38 Dusp,39 Fos,40 or Fkbp5 (encoding FKBP51 protein),41 Ddit4,42 Cebpb,43 and Jun44 (Figures 2C–2G). In contrast, perivascular MΦ/monocyte clusters from Ccl2MCΔ/Δ mice had enriched transcripts associated with uncontrolled inflammatory responses such as Syk,45 Irf8,46 and Lst147 (Figures 2C–2G). An interstitial MΦ cluster with high Zebp2 expression, pivotal for the maintenance of a tissue-specific identity of MΦs,48 was reduced in lungs from Ccl2MCΔ/Δ mice (Figure 2H). In summary, the microvascular beds of CCL2 mutants displayed a decrease in MΦ survival and distinct shifts in MΦ transcriptome, consistent with a less homeostatic phenotype.
This observation raised the question of whether MC-derived inflammatory signatures shape MΦ activity in large vessels. To address this possibility, we examined the impact of macrovascular SMCs on intimal MΦ phenotype in atherosclerosis, a chronic inflammatory disease fundamentally affected by dysfunctional MΦ programs.13,49 To analyze transcriptomic changes in intimal macrovascular MΦs, we reconstituted Ccl2MCΔ/Δ mice with bone marrow from MCRFP-rep Cx3cr1-MΦGFP-rep donors. Chimeric mice allowed endogenous labeling and FACS-based isolation of intimal CX3CR1+ MΦs. Low-input RNA-seq revealed reduced expression of several efferocytosis-associated or M2-like MΦ defining genes such as Cd36,50,51 Folr2,52 Clec4e,53 and Pecam154 in MΦs from atherosclerotic Ccl2MCΔ/Δ mice (Figures 2I, S2E, and S2F). This could be recapitulated on a protein level after CCL2 stimulation of MΦs in vitro (Figure S2G). In addition, MΦ marker genes such as Adgre434 and Fabp455 were also reduced (Figures 2I and S2F). These findings underscore active maintenance of a differentiated and homeostatic MΦ phenotype by MC-derived CCL2 as determined on a transcriptomic level.
SMCs react to necrosis with intracellular Ca2+ bursts and show high CCL2 and MIF expression
We next dissected SMC heterogeneity and SMC-MΦ communication in atherosclerosis. We revisited scRNA-seq data from human coronary plaques and murine aortic roots from atherosclerotic SMClin mice.32 The UMAP algorithm clustered 6 different SMC subsets in human atherosclerosis. One cluster was distinct from the other SMCs and displayed a prominent chemokine-rich pattern, which we therefore termed chemotactic SMCs (cSMCs) (Figures 1D, 3A, and 3B). Comparative analysis identified Ccl2 as the most prominently expressed chemokine by cSMCs (Figure 3A). To reconcile the interplay between SMCs and MΦs, we mapped a chemokine—receptor interactome. CCL2 and MIF were highly expressed chemokines mediating the interplay between SMC and MΦ subsets, mainly involving the cSMC subset (Figures 3A–3C).
Figure 3.

Distinct chemotactic SMCs express high levels of MΦ chemoattractants, ameliorating atheroprogression
(A–C) Reanalyzed single-cell RNA-seq data from human coronary arteries from Wirka et al., GEO: GSE131780. (A) Violin plots (calculated on all cells expressing detectable baseline levels of the respective gene) of highly expressed cytokines and chemokines in chemotactic SMCs. Dots represent single cells, only cells exhibiting detectable expression of the particular gene are included (B) interactome depicting cell-cell interactions between MΦ and SMC subsets, prominent SMC → MΦ interactions are depicted in red. Intensity of red color depicts the respective portion of the CCL2-CCR2 axis for the concrete interaction (the darker the red color, the more the CCL2-CCR2 axis accounts for the respective inter-cluster interplay among all detected chemokine-receptor interactions). (C) Heatmap further unraveling SMC → MΦ chemokine:chemokine-receptor interactions. Blue box depicts interactions of chemotactic SMC subset, red box depicts CCL2-mediated interactions between SMC and MΦ subsets.
(D) Ccl2 and Mif expression in Ng2+ SMCs FACS-sorted from western-diet fed atherosclerotic MCRFP-rep mice compared to chow-diet fed non-atherosclerotic control mice. n = 3–4 mice per group.
(E) Representative images of BCA sections from Ccl2SMC +/+ and Ccl2SMCΔ/Δ littermates after 14 weeks of western diet stained for ACTA2 (green), LGALS3 (red), and Hoechst (blue). Scale bars, 100 μm.
(F and G) Morphometric analysis of plaque size (F) and vascular remodeling (G) from BCA sections at three consecutive locations from Ccl2SMC +/+ (n = 11) and Ccl2SMCΔ/Δ (n = 10) littermates.
(H and I) Quantification of ACTA2+ smooth muscle cell content as ACTA2+ area in percentage of total plaque area and percentage of 30 μm plaque surface area in valves (H) and in the BCA at three consecutive locations (I). (H and I) n = 10–11 mice per group.
(J and K) Analysis of intimal LGALS3+ area as percentage of plaque size in BCA sections at three consecutive locations (J) and in plaques from aortic valves (K) (n = 10–11 each).
(L) Schematic illustration of media and intima processing from aortae of Ccl2SMC+/+ and Ccl2SMCΔ/Δ littermates after 14 weeks of western diet (left). Heatmap displaying expression of differentially regulated genes in bulk RNA-seq of Ccl2SMC+/+ mice (n = 3) and Ccl2SMCΔ/Δ mice (n = 4). Rows represent individual replicates, differentially expressed genes are illustrated in columns (right).
(M) Volcano plots of intima/media RNA-seq showing differentially expressed genes in Ccl2SMC+/+ mice (n = 3) and Ccl2SMCΔ/Δ mice (n = 4), x-axis depicts Log2FC, y-axis depicts -Log10(adj. p-value). Data are shown as mean and SEM. (H and K), Student’s t test was used. (F, G, I, and J) Repeated measures two-way ANOVA or mixed-effects model was used. ∗p < 0.05; NS, not significant. Bar graphs show mean with SEM. Violin plots with matching boxplot and mean expression.
To understand whether chemokine expression in SMCs is actively calibrated, we analyzed whether SMCs respond to cell death. Incubation of cultured aortic SMCs, isolated from Myh11cre-ERT2; PC-G5-tdT mice (SMCCa2+-rep) with necrotic Jurkat cell supernatant induced a high-frequency burst in calcium activity, indicating an active response of SMCs to adjacent cell death (Video S2). Similar to other Myh11-driven models,56 SMCCa2+-rep mice also showed high recombination efficacy and specificity in the vasculature (Figure S3A).
In vitro epifluorescence microscopy of cultured SMCs from SMCCa2+ Rep mice coincubated either with living Jurkat cell supernatant or dead Jurkat cell supernatant. SMCs are depicted in red, Ca2+ activity is rendered and pseudocolored in green (high Ca2+ activity) and blue (low Ca2+ activity). Pseudocolor scale, time scale and scale bar are included in the video.
CCL2 and MIF also showed high protein expression in SMCs, as reported previously31,57 (Figure S3B). As CXCL12 and CCL19 also showed high expression in human SMCs, we further investigated whether these chemokines might also be induced in human coronary artery SMCs (HCASMCs). CCL19 and CXCL12 protein was low and showed no induction upon TLR stimulation (Figures S3C and S3D).
SMCs from MCRFP-rep mice showed an increase in Ccl2 expression in response to induction of atherosclerosis, Mif showed constant high expression (Figures 3D, S3E, and S3F). Along these lines, the expression of the MΦ active chemokine Ccl2 was mainly confined to distinct SMC populations, most prominently SMC 4 and cSMCs (Figure S3G).
Chemokine-driven SMC-MΦ axes dampen vascular inflammation in atherosclerosis
We next examined the relevance of the SMC-MΦ axis for MΦ function in the atherosclerotic plaque by conditionally deleting Ccl2 in MCs across the vascular tree (Ng2-cre; Ccl2fl/fl; ApoE−/− mice, referred to as Ccl2MCΔ/Δ and Myh11cre-ERT2; Ccl2fl/fl; ApoE−/− mice, referred to as Ccl2SMCΔ/Δ). Compared with (Cre−) Ccl2MC+/+ littermate controls, deletion of CCL2 in NG2+ SMCs exacerbated atherosclerosis progression without prompting systemic changes (Figures S3H–S3Q). In line, Myh11-dependent Ccl2 deletion increased the atherosclerotic plaque burden compared with Ccl2SMC+/+ littermate controls in the brachiocephalic artery (BCA) (Figures 3E–3G and S4A). Ccl2SMCΔ/Δ plaques further showed decreased α-SMA content in the fibrous cap without changes in overall plaque MΦ content, which was determined by LGAL-S3 (MAC2) staining as a frequently used and established marker for MΦs58,59 (Figures 3H–3K). Intima and media transcriptomics of Ccl2SMCΔ/Δ and Ccl2SMC+/+ littermate controls revealed an exacerbation of vascular inflammation in the absence of SMC-derived CCL2 (Figures 3L and 3M), indicating that SMC-derived CCL2 counteracts uncontrolled inflammation. Ccl2SMCΔ/Δ mutant mice did not display systemic or local differences in leukocyte trafficking or systemic lipid profiles. CCL2 deletion was confirmed in the vasculature of Ccl2SMCΔ/Δ mice and FACS-sorted tdTomato+ SMCs from Myh11cre-ERT2; Rosa26tdT; Cx3cr1GFP; Ccl2fl/fl; ApoE−/− (Ccl2SMCΔ/Δ; SMC-tdTlin; Cx3cr1-MΦGFP-rep) mice after tamoxifen injection in contrast to Ccl2fl/fl; ApoE−/− or Myh11cre-ERT2; ApoE−/− mice (Figures S4B–S4E).
Next, we tested whether the protective effect of chemokine-mediated intimal SMC-MΦ interplay is confined to CCL2-signaling or also pertains to other chemokines such as MIF (Figure 1E). We examined Ng2-cre+; Miffl/fl; ApoE−/− mice (MifMCΔ/Δ) and observed a drop in MΦ viability together with an increase in atherosclerotic lesion and necrotic core size upon western-diet feeding compared with Ng2-cre−; Miffl/fl; ApoE−/− (MifMC+/+) littermate controls. In line, interference with the SMC-MIF-MΦ signaling axis was accompanied by a reduced efferocytotic capacity of MΦs in vitro, providing an explanation for the exacerbated necrosis and atheroprogression in vivo (Figures S4F–S4R). We conclude that the release of CCL2 and MIF by SMCs jointly promotes MΦ homeostasis fostering inflammation resolution in atherosclerosis.
SMCs exert strong chemotactic programs on MΦs in atherosclerosis
To define the dynamics of the SMC-MΦ interplay in atherosclerotic lesions in vivo, we used Ng2DsRed; ApoE−/− mice (MCRFP-rep mice). In contrast to fate-mapping approaches,56 this direct reporter labeling focuses on differentiated MCs across the arterial vascular tree and does not allow labeling of SMC-progeny via fate-tracing19,20,29,30 (Figures S5A and S5B). For intravital imaging, direct fluorescent reporter systems are more suitable since they only label cells that express respective marker genes during the experimental imaging setup (Figures S5A–S5E). We therefore crossed Ng2DsRed; Lyz2eGFP; ApoE−/− (MCRFP-rep Lyz-MΦGFP-rep) mice and employed in vivo multi-photon imaging of the carotid artery. This approach revealed dynamic interactions between SMCs and MΦs in the carotid artery of MCRFP-rep Lyz-MΦGFP-rep mice, most prominently in the shoulder regions of the plaque (Figure 4A). Although some MΦs migrated in-between SMC interactions, others were sessile but dynamically interacted with SMC by extending protrusions toward the SMCs (Figures 4A–4F; Videos S3 and S4).
Figure 4.

SMCs exert chemotactic cues on plaque MΦs
(A–F) In vivo imaging of an atherosclerotic lesions within the carotid artery in atherosclerotic MCRFP-rep; Lyz-MΦGFP-rep mice after 14 weeks western diet by multi-photon microscopy. (A) Time-series with a focus on the shoulder region of the plaque, arrows depicting locally confined but dynamic protrusions formed by Lyz+ MΦs (green) toward SMCs (red). Images from Video S3. Scale bars, 20 μm. (B) In vivo imaging of Lyz+ MΦ-SMC contacts during Lyz+ MΦ migration within the intima. Rendered illustration of Lyz+ MΦs (green) migrating along SMCs (red), including exemplary migration tracks of 2 cells. Scale bars, 20 μm.
(C) Analysis of the duration of the interactions between SMCs (red) and Lyz+ MΦs (green). (D) Analysis of the displacement length during interaction and during free migration. (E) Velocity profile of cells 1 and 2 (labeled in the migration tracks above under B) over time: boxes indicate interactions; horizontal lines indicate mean velocity of the time period included. (F) Left: meandering index (track straightness) of Lyz+ MΦs during interaction with SMCs and during free migration without interaction. Right: displacement rate of Lyz+ MΦs during interaction with SMCs and during free migration without interaction with SMCs. (C–F) n = 46–52 cell tracks covering free migration (−) or subsequent SMC interaction (+) or vice versa from n = 3 mice, Mann-Whitney test used to compare groups, ∗∗∗p < 0.001, ∗∗p < 0.001.
(G) In vivo imaging of static SMC-Cx3cr1+ MΦ contacts in a MCRPF-rep; Cx3cr1-MΦGFP-rep mouse, arrow depicting a SMC embedded in two Cx3cr1+ MΦs. Scale bars, 20 μm.
(H) Ex vivo confocal imaging of cross-sections of atherosclerotic valves in MCRFP-rep; Cx3cr1-MΦGFP-rep mice after 12 weeks western diet, MΦs in green, SMCs in red, nuclei in blue, arrowheads depicting SMC-MΦ contacts. Scale bars, 40 μm.
(I and J) Ex vivo confocal imaging of SMClin; Cx3cr1-MΦGFP-rep mice after 22–24 weeks of western diet, (I) atherosclerotic valve cross-sections, MΦs in green, SMC and SMC-progeny in red, nuclei in blue, arrowheads depicting SMC-MΦ contacts, dashed line outlining SMC enveloping MΦ. Scale bars, 10 μm. (J) En face confocal z stacks of the atherosclerotic intima in SMC-tdTlin; Cx3cr1-MΦGFP-rep mouse, Cx3cr1+ MΦs in green, SMClin cells in red, blue arrowheads pointing toward SMC-MΦ contacts observable in the cross- and longitudinal-sections of the z stack, most frequent within the plaque surface. Scale bars, 50 μm (left), 5 μm (right) (K), left: representative immunohistochemical images of human aortic plaques stained for CD68 and α-SMA, scale bars, 300 μm, right: immunofluorescence staining for CD68 (red) and α-SMA (green) and with Hoechst (blue), dashed lines represent macrophage and SMCs, scale bars, 20 μm on the left and 10 μm on the right immunofluorescence image.
(L) Pearson correlation of the relative CD68+ area in fibrous cap environment (defined as the plaque area within the top 30% plaque surface) with, left: the relative necrotic core size, middle: the α-SMA content and right: the plaque vulnerability index (further elaborated in methods). Intermediate (n = 5) and advanced (n = 8) human plaques, graded accordingly by the pathology department, were included. Pearson r and two-tailed p value are included for every Pearson correlation.
(M) Summary illustration of the natural MΦ distribution within atherosclerotic lesions. MΦs mainly localize at areas of the plaque surface adjacent to SMCs in murine and in human atherosclerotic lesions. Bar graphs show mean with SEM.
In vivo microscopy of an atherosclerotic lesion in a MCRFP-rep Lyz-MΦGFP-rep carotid artery after 14 weeks Western diet. The atherosclerotic lesion is depicted with a continuous line, the box illustrates the shoulder region of the lesion and is further magnified in the middle and right video. The middle video is a 3D rendering of the indicated shoulder region, the video on the right depicts a higher magnification without 3D rendering of the shoulder region. Arrows indicate protrusions of MΦs towards SMCs that continuously form and regress during the video. SMCs are labelled in red, MΦs in green and second harmonic generation (SHG) in blue. Scalebar is 20μm.
In vivo microscopy of an atherosclerotic lesion in a MCRFP-rep Lyz-MΦGFP-rep carotid artery after 14 weeks Western diet. The lesion is depicted with a box. SMCs are labelled in red, MΦs in green and second harmonic generation (SHG) in blue. Scalebar is 50 μm.
An immunomodulatory, sessile CX3CR1hi MΦ subset was described in atherosclerosis.60 To dissect the interaction of this distinct CX3CR1hi MΦ-subset with SMCs, we generated Ng2DsRed; Cx3cr1GFP; ApoE−/− (MCRFP-rep Cx3cr1-MΦGFP-rep) mice. We observed multiple contacts between SMCs and CX3CR1hi MΦs, which were stable and enduring (Figure 4G). Ex vivo confocal imaging of atherosclerotic aortic valves recapitulated abundant cell-cell contacts between SMCs and CX3CR1+-MΦs at plaque surfaces (Figure 4H). However, since SMCs lose their classical SMC markers over the course of atheroprogression56 (compare Figure S5C), we also opted for a rigorous lineage tracing approach of SMCs. Therefore, we employed Myh11cre-ERT2; Rosa26tdT; Cx3cr1GFP; ApoE−/− (SMC-tdTlin Cx3cr1-MΦGFP-rep) mice. En-face confocal z stacks of lesions from SMC-tdTlin Cx3cr1-MΦGFP-rep mice revealed that almost every CX3CR1+-MΦ directly contacted SMCs and localized mostly at the plaque surface (Figures 4I and 4J). Consistently, MΦs and SMCs in human atherosclerotic lesions strongly colocalized, and MΦs were again most abundant at superficial plaque regions below the fibrous cap (Figure 4K). The MΦ content of SMC-rich superficial plaque regions did not correlate with overall α-SMA content or plaque vulnerability index. However, high MΦ content in proximity to fibrous cap SMCs was associated with smaller necrotic core size, suggesting beneficial consequences of the SMC-MΦ interplay (Figure 4L). Taken together, intimal MΦs abundantly interact with SMCs within the plaque surface (Figures 4J, 4K, and 4M). Therefore, we hypothesized that SMCs actively determine the distribution of MΦs within the plaque. In line with this conjecture, conditioned medium from resting and particularly from stimulated HCASMCs exerted chemotactic effects on monocytes, and this was mediated through CCL2 and MIF (Figures S5F–S5I). We ruled out any autocrine effects of CCL2 signaling on SMCs on a receptor level and functional level (Figures S5J–S5L). Therefore, SMCs have chemotactic properties on MΦs, and colocalization of SMCs and MΦs correlates with the plaque phenotype.
Fibrous cap SMCs preserve strategic MΦ positioning and secure pro-resolving MΦ functions in atherosclerosis
Single-cell in silico32 analysis of SMC heterogeneity recapitulated a distinct cSMC subset in murine aortic roots as described above in humans (Figure 5A). To achieve spatial resolution, we investigated the exact localization of cSMCs within atherosclerotic lesions. Marker gene analysis showed that murine cSMCs highly express Pdgfrb (Figure 5B). Spatial resolution in SMC-tdTlin Cx3cr1-MΦGFP-rep mice attributed high platelet-derived growth factor receptor-β (PDGFRβ) expression to fibrous cap SMCs. These PDGFRβhi cSMCs frequently interacted with MΦs (Figure 5C), supporting the concept that cSMCs actively maintain distinct intra-plaque MΦ positioning.
Figure 5.

SMCs within the fibrous cap preserve a strategic positioning of plaque MΦs and secure homeostatic MΦ functions
(A and B) Reanalyzed single-cell RNA-seq data from mouse aortic roots from atherosclerotic SMClin mice from Wirka et al., GEO: GSE131780. (A) UMAP based dimensionality reduction of analyzed cells (left), heatmap illustrating cytokine and chemokine expression of different SMC subsets (right). (B) Marker genes of SMC clusters illustrated in a heatmap, composed by ClustVis.
(C) Representative confocal image depicting the spatial distribution of the key cSMC marker PDGFRβ within an atherosclerotic valve in SMClin; Cx3cr1-MΦGFP-rep mice after 22–24 weeks of western diet, SMClin cells in red, MΦs in green, and PDGFRβ in white. Scale bars: 30 μm (left) and 15 μm (right images).
(D) Illustration of the experimental setup of the migration assay: macrophages undergo a migratory decision either moving toward the artificially composed SMC-rich fibrous cap below or residing at the artificially composed, necrotic cell rich, necrotic core. SMCs (representing the fibrous cap) are located in the lower chamber, whereas peritoneal macrophages have been attached on the transwell of the upper chamber. Necrotic Jurkat cells (representing the necrotic core) have been added to the upper chamber.
(E) Number of peritoneal MΦs from Lyz-MΦGFP-rep mice that transmigrated toward the lower chamber per field of view (FOV). Isotype or anti-CCL2 blocking antibody was simultaneously added to the lower chamber. MΦ numbers per FOV counted at 4 subsequent time points (n = 4 independent experiments).
(F) Distribution of macrophages as percentage of LGALS3+ area in 30 μm plaque surface area in percentage of total plaque LGALS3+ area at three subsequent locations (n = 10 each).
(G) Left: quantification of LGALS3+ surface macrophage content as relative LGALS3+ area in percentage of total plaque surface area (defined as the upper 30 μm of the plaque) from BCA sections at three consecutive locations (n = 10 each). Right: representative immunofluorescent images of BCA sections for ACTA2 (green), LGALS3 (red), and Hoechst (blue) with highlighted 30 μm plaque surface area from Ccl2SMC+/+ and Ccl2SMCΔ/Δ littermates after 14 weeks of western-diet feeding. Scale bars, 100 μm.
(H) Volcano plot depicting differentially regulated genes analyzed by RNA-seq of FACS-sort enriched peritoneal MΦs, coincubated either with live or dead Jurkat cell supernatant for 12 h.
(I) Quantification of peritoneal macrophages 12 h after addition of live or dead Jurkat cell supernatant (n = 6).
(J–L) Efferocytosis assay, analyzing the efferocytotic capacity of the MΦ population, isolated from Lyz-MΦGFP-rep mice. Apoptotic Jurkat cells were added for 1 h after 6 h incubation either with or without CCL2. (J) Quantification of MΦs with engulfed apoptotic cells upon presence or absence of CCL2 (n = 5 independent experiments). (K) Quantification of the total number of engulfed apoptotic cells upon CCL2 presence of absence. (L) Representative epifluorescence images of the efferocytosis assay with peritoneal macrophages (green) and apoptotic Jurkat cells (red), 1 h after Jurkat cell addition. Scale bars, 50 μm.
(M–O) Necrotic core analysis as total necrotic area in μm2 (M) and in percentage of plaque area (N), assessed with Masson Trichrom’s staining of valve sections, from Ccl2SMC+/+ (n = 9) and Ccl2SMCΔ/Δ (n = 10) littermates after 14 weeks of western diet. (O) Left: representative images of necrotic core content analyzed by Masson Trichrom’s staining of valve sections from Ccl2SMC+/+ and Ccl2SMCΔ/Δ littermates after 14 weeks of western diet. ∗ indicates necrotic areas. Scale bars, 100 μm. Right: representative images of immunofluorescence stainings of valve sections from Ccl2SMC+/+ and Ccl2SMCΔ/Δ littermates after 14 weeks of western diet for ACTA2 (green), LGALS3 (red), terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) (yellow), and DAPI (blue). Scale bars, 100 μm.
(P and Q) Quantification of cell apoptosis as total amount of TUNEL+ LGALS3+ Hoechst+ MΦs in plaque (P) and as total amount of TUNEL+ Hoechst+ apoptotic cells (Q) in Ccl2SMC+/+ (n = 9) and Ccl2SMCΔ/Δ (n = 10) individual littermates in total after 14 weeks of western diet, only including plaques at the proximal and intermediate BCA, without distal BCA areas with its early lesions.
(R) Quantification of valve atherosclerotic plaques for (left) total and relative TUNEL+ cells. Data are shown as mean and SEM.
(I, J, K, M, N, and R) Student’s t test was used for normally distributed data and Wilcoxon matched-pairs signed rank test for not normally distributed data. (E, F, G, P, and Q) Repeated measure two-way ANOVA or mixed-effects model was used. ∗p < 0.05; ∗∗ p < 0.01; NS, not significant. Bar graphs show mean with SEM.
To mimic the microanatomical distribution of SMCs and MΦs in vivo, we constructed a reductionist in vitro model in which MΦs were compelled to undergo a migratory decision either toward a layer of SMCs (representing the fibrous cap) or toward necrotic Jurkat cells (mimicking aspects of the necrotic core) (Figure 5D). Intact CCL2 signaling at the fibrous cap site attracted MΦs toward SMCs. Disruption of this axis resulted in decreased MΦ attraction to SMCs and increased migration to the necrotic core. This observation indicates that SMC-derived CCL2 competes with necrotic core damage-associated molecular patterns (DAMPs) for MΦ attraction (Figure 5E). We then defined the role of SMC-derived CCL2 on MΦ distribution in atherosclerotic lesions in vivo. Preferential MΦ positioning shifted from the plaque surface to the plaque core in Ccl2SMCΔ/Δ mice (Figures 5F and 5G). This indicates that SMC-derived CCL2 dictates MΦ positioning in atherosclerotic lesions and limits MΦ migration to the necrotic core.
We then tested whether spatiotemporal reorientation of MΦs toward the necrotic core in the absence of SMC-derived CCL2 promotes MΦ cell death and fosters necrotic core expansion. Coincubation of MΦs with necrotic cell supernatant triggered decreased MΦ survival, induction of apoptosis pathways, and a loss of MΦ markers (Adgre1, Fabp4, and Cxcr4) (Figures 5H, 5I, S6A, and S6B), resembling the phenotype of plaque MΦs from Ccl2MCΔ/Δ mice (Figures S2E–S2G). This suggests that some of the functional changes in MΦs observed in vivo result from spatiotemporal repositioning toward necrotic areas of the lesion in Ccl2MCΔ/Δ mice. However, we also considered potential direct effects of CCL2 on MΦs. CCL2 stimulation enhanced homeostatic functions of MΦs in vitro (Figures 5J–5L, S6C, and S6D). Consistent with this finding, in vivo analysis of advanced lesions in Ccl2SMCΔ/Δ mice revealed an increase in apoptosis and necrosis in advanced plaques. Local proliferation of MΦs or SMCs as well as SMC apoptosis did not change (Figures 5M–5R and S6E–S6I).
Next, we asked whether the chemokine-mediated SMC-MΦ axis relies on more than one chemokine, securing possible backup mechanisms. In SMC-MΦ coincubation systems, the co-inhibition of CCL2 and MIF reduced MΦ content. However, this was also the case at a similar magnitude when inhibiting CCL2 alone or MIF alone, emphasizing the non-redundant relevance of both chemokines for securing homeostatic SMC-MΦ communication (Figure S6J). In addition, direct physical SMC-MΦ interplay also strongly enhanced MΦ survival (Figure S6K). We however did not observe any differences in efferocytotic capacities in SMC-MΦ coculturing systems compared with MΦ monocultures (Figure S6L). This triggered the question, whether MΦ repositioning toward necrotic areas is the decisive mechanism or whether chemokine-mediated pro-survival cues play an additional, non-redundant role. We observed that the reduced MΦ survival in SMC-MΦ coculture systems after CCL2 inhibition can be reproduced in a necrotic environment, which per se reduces MΦ survival (as shown in Figure 5I) (Figure S6M). This indicates that the chemokine-mediated SMC-MΦ axis not only operates through spatiotemporally mediated functional changes in MΦs by retaining MΦs outside of necrotic areas but also by additional pro-survival effects, which are non-redundant to the chemotactic repositioning roles since they also enhance survival within necrotic areas (Figures 5I and S6M).
Although no direct effects on SMC proliferation in monocultures were observed, the chemokine-mediated SMC-MΦ axis enhanced SMC coverage in cocultures in vitro, explaining the observed plaque stabilization (Figure S6N). In contrast, the shear physical SMC-MΦ interplay did not influence SMC survival in vitro (Figure S6O). Therefore, we conclude that the spatiotemporal repositioning of MΦs together with direct functional effects of a lack of MC-derived CCL2 yields phenotypic changes in MΦs that exacerbate vascular inflammation.
Short-term pharmacological CCL2 inhibition promotes detrimental changes in advanced plaque phenotype
Our data suggest that a SMC-CCL2-MΦ axis operates in large vessels to sustain a protective MΦ phenotype by maintaining homeostatic MΦ programs and spatiotemporal distribution. However, prevailing concepts have considered CCL2 proatherogenic, predominantly by fueling vascular monocyte recruitment as observed by multiple experimental studies.61 These studies mainly focused on early atherogenesis,62,63,64 which is dominated by trans-endothelial monocyte recruitment.13 However, patients typically present with advanced atherosclerosis. Clinical studies that enrolled patients with advanced coronary artery disease undergoing percutaneous coronary intervention did not show a reduction in major adverse cardiac event rates following CCL2 inhibition.65 Hence, the effect of CCL2 inhibition in advanced atherosclerosis, when monocytic recruitment across the endothelium plays a less important role and MΦs mainly proliferate locally,66 remains unclear. Based on the protective SMC-CCL2-MΦ signaling, we hypothesized that systemic CCL2 inhibition in advanced atherosclerosis, after peaking early monocytic influx, could have detrimental consequences. When we performed antibody-mediated inhibition of CCL2 in advanced stages of atherosclerosis, we found marked indices of plaque destabilization compared with isotype-treated mice (Figures 6A–6D). Consistent with our data in SMC-specific CCL2 deficiency, we found a drop in fibrous cap coverage. However, there was no change in more steady plaque parameters (Figures 6E–6G) and no systemic changes (Figure 6I). Therefore, despite protective effects of CCL2 inhibition related to reduced monocyte recruitment in early atherosclerosis, in advanced lesions, CCL2 action appears to preserve the homeostatic SMC-MΦ axis within the vessel wall.
Figure 6.

Short-term CCL2 inhibition in advanced atherosclerosis triggers detrimental changes in plaque phenotype
(A) Acute pharmacological CCL2 inhibition in ApoE−/− mice after 6 months of western type diet. The anti-CCL2 or isotype control antibody was injected intravenous (i.v.) 2 weeks before sacrifice every 48 h (n = 7–8 / group).
(B) Quantification of fibrous cap coverage as continuity (percentage of fibrous cap covered plaque surface length relative to complete plaque surface length) at three subsequent BCA locations.
(C) Quantification of ACTA2+ area within plaque surface as % of plaque surface area (defined as the top 30 μm stripe of the lesion) at three subsequent BCA locations.
(D) Quantification of absolute ACTA2+ area in μm2 at three subsequent BCA locations.
(E) Quantification of macrophage area as LGALS3 area in μm2 at three subsequent BCA locations.
(F) Quantification of total plaque size as absolute plaque area in μm2 at three subsequent BCA locations.
(G) Quantification of cell apoptosis as total amount of TUNEL+ cells in plaque at three subsequent BCA locations.
(H) Representative images of BCA sections from ApoE−/− mice after 6 months of western diet stained for ACTA2 (green), LGALS3 (far red), TUNEL (red), and Hoechst (blue). Scale bars, 50 μm.
(I) Quantification of blood leukocytes, neutrophils, lymphocytes, monocytes, and plasma cholesterol (n = 7–8). (I) Student’s t test was used. (B–G) Repeated measures two-way ANOVA or mixed-effects model, with subsequent Šídák’s multiple comparisons test in (B)–(D), was used. ∗p < 0.05; ∗∗p < 0.01 NS, not significant. Bar graphs show mean with SEM.
Discussion
MCs and MΦs reside in close anatomical proximity along the vasculature and across tissues. Since MΦs preferably colonize vascular and perivascular “compartments” this vascular MΦ niche is gaining increasing attention.9,10 MΦ origin reaches beyond blood-borne monocytic populations to also comprise MΦ populations that are seeded perinatally.1,67 The exact origins and replacement of vascular MΦs in the micro- and macro-vasculature are established. In the uninflamed adventitial tissue of the macrovasculature, embryonic MΦs make up relevant proportions.68,69,70 The intimal resident MΦ population termed “MacAIR” is overtaken by monocytic MΦs during inflammation.11 In line, clonal hematopoiesis of indeterminant potential (CHIP) and the effects of environmental factors on atheroprogression via bone marrow activation and trans-endothelial recruitment are mainly mediated by increased recruitment of monocytic precursors to atherosclerotic lesions.58,71,72,73 Hence, although MΦ origins, modes of mobilization, and mechanisms of recruitment of monocytes are understood, the mechanisms of mature MΦ preservation within the vascular niche are undefined. In other words, how mature vascular MΦ biology and their distribution are actively tuned is unclear. Vascular MΦs dwell in an environment dominated by MCs, which express inflammatory mediators.18,19,20
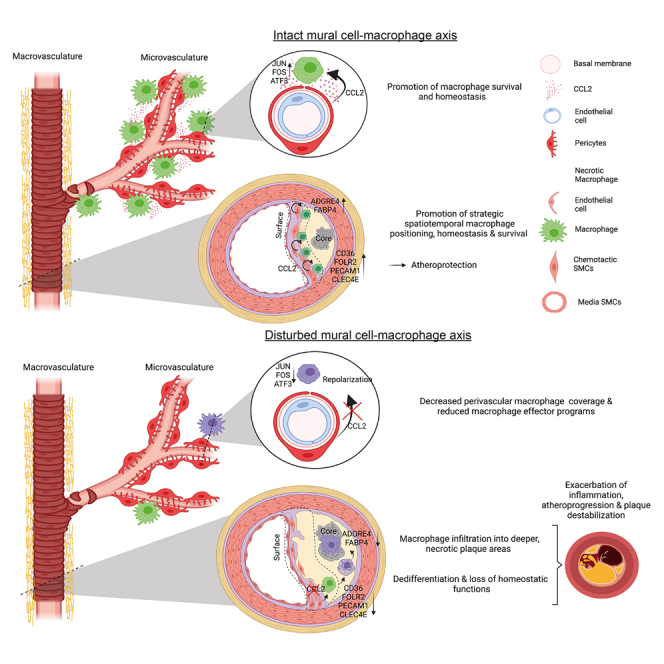
Our data provide evidence that MCs possess a fundamental role in sustaining MΦ homeostasis within this vascular niche. Beyond the traditionally well-studied bone marrow,74,75 blood,76 and endothelial-monocyte trafficking axes,13,64 we show that MCs critically determine MΦ function within the vessel wall, yet in an unexpected manner: MCs reacted to sterile inflammation by intracellular Ca2+ bursts and expressed high levels of MΦ chemotactic proteins, thereby imprinting a protective functional MΦ phenotype and spatial MΦ positioning, both independently securing vascular MΦ homeostasis. In addition, MΦ-chemotactic proteins CCL2 and MIF acted in a non-redundant fashion and were individually indispensable for actively maintaining MΦ homeostasis. Also, vice versa, these chemokine-mediated SMC-MΦ axes enhanced SMC survival, stabilizing the fibrous cap of atherosclerotic lesions. In the microvasculature, this chemokine-driven MC-MΦ axis also provided homeostatic cues for MΦ programming and enhanced MΦ survival. Disrupting this CCL2-mediated axis in the microvasculature led to decreased MΦ coverage and a transition to a less homeostatic MΦ phenotype at a transcriptomic level. In the atherosclerotic macrovasculature, maladaptive spatiotemporal repositioning of plaque MΦs in the absence of SMC-derived MΦ-chemotactic proteins resulted in a dedifferentiated transcriptional phenotype and impaired conduction of MΦ effector programs in vitro. These maladaptive processes culminated in exacerbated inflammation, plaque destabilization, and atheroprogression, shedding light on a homeostatic MC-MΦ checkpoint within the vascular niche.
Landmark studies in the 1990s linked the CCL2-CCR2 axis to chronic detrimental inflammation, providing pioneering evidence that inflammatory mediators play a crucial role in vascular disease.31,77 A myriad of studies show critical involvement of CCL2 in driving a plethora of chronic inflammatory diseases.62,74,77,78,79,80,81,82,83,84,85 CCL2 is strongly expressed by MCs.31 MIF is another proinflammatory chemoattractant, which is expressed by MCs86 and also associates with multiple chronic inflammatory diseases.87,88,89
These chemokines mainly affect chronic inflammatory responses by orchestrating excessive leukocyte trafficking: The CCL2-CCR2 axis is pivotal for monocyte mobilization from the bone marrow into the bloodstream,17 and in line, genetic CCR2 deletion results in reduced circulating monocyte counts and reduced chronic inflammatory disease burden.79 CCL2 is also centrally involved in monocyte trafficking from the bone marrow toward the spleen.90,91 In addition, systemic disruption of the CCL2-CCR2 axis strongly reduces trans-endothelial monocyte recruitment and tissue MΦ counts.64,92,93,94 In line, the atypical chemokine MIF holds similar functions for monocyte trafficking toward inflammatory foci.95
Most studies investigating cytokine and chemokine signaling in chronic inflammatory diseases have relied on germline genetic deletions or systemic pharmacological interventions, mainly interfering with immune-cell trafficking. However, how these chemokines influence mature MΦ functions, after their recruitment, remains unclear.
These findings now unravel that subendothelial canonical chemokine-driven MC-MΦ axes hold homeostatic effects on mature MΦs contrasting their detrimental effects on excessive monocyte recruitment. This MC-MΦ interplay is backed by several chemokines, most importantly CCL2, but also MIF and other MΦ active chemokines such as CCL1996 are highly expressed by vascular MCs. This counteracts uncontrolled, detrimental chronic inflammation. Along these lines, during chronic vascular inflammation, chemokines secreted by MCs are anatomically separated from endothelial-mediated (detrimental) leukocyte recruitment axes and mainly confined to functional and local spatiotemporal effects on tissue MΦs. In vitro studies suggest that canonical chemokines such as CCL2 can induce alternative MΦ activation states.97 Interestingly, one distinct SMC subset—we defined cSMCs—mainly accounted for this priming of MΦs. The Owens lab identifies that platelet-derived growth factor (PDGF)-PDGFR signaling is pivotal for SMC infestation into the fibrous cap.98 In line, our data showed that PDGFRβhi SMCs, which were mainly localized within the fibrous cap, were the SMC subtype that produced high amounts of MΦ chemotactic chemokines. These PDGFRβhi SMCs essentially kept MΦs within viable regions of the plaque (near the fibrous cap) and preserved a homeostatic MΦ phenotype. Previous studies prompted us to engage multiple inducible as well as non-inducible fate mapping and genetically deleted mice and conventional reporter systems to trace SMCs and their progeny.32,56,99,100 The Owen’s lab shows that a heterozygous depletion of MCP1 in MCs results in an increase in systemic circulating monocytes.101 In our model, we did not observe a monocytosis, neither in a Ng2-driven nor in a Myh11-driven conditional genetic deletion system. These discrepant results might be due to a lower cholesterol content in the western diet and a longer period of western-diet feeding in the experimental setup in the article by Owsiany et al.101
In advanced atherosclerosis, the role of continuous monocyte recruitment diminishes, and the significance of local MΦ proliferation for sustaining the plaque MΦ pool increases,66 hence reducing the relevance of endothelial-mediated leukocyte recruitment and confining possible chemokine-mediated effects to intra-plaque axes. Indeed, our data showed that short-term CCL2 inhibition, specifically in these advanced stages of atherosclerosis, triggers detrimental phenotypic changes in atherosclerotic lesions.
In summary, MCs exploit mediators, considered to be detrimental, to shape an immunomodulatory vascular MΦ niche. In contrast to the expected effects of chemokines in vascular inflammation, this MC-MΦ axis sustained homeostatic spatiotemporal positioning and a pro-resolving phenotype of MΦs in vivo. Genetic and pharmacologic disruption of this homeostatic niche resulted in exacerbated inflammation as shown for atherosclerosis. Therefore, our findings implicate that future studies are needed to recapitulate the roles of chemokines in inflammation in a source and stage-specific manner to differentiate between effects on leukocyte trafficking and effects on programming of mature leukocytes. Sole reliance on in vitro observations or on conventional genetic deletion experiments may mask compartment-specific immune-homeostatic effects of mediators which are deemed to be exclusively detrimental during chronic inflammation.
Together, this work identifies how the prominent vascular MΦ niche is pro-actively sustained in a homeostatic state by adjacent MCs, which dampens detrimental chronic inflammation.
Limitations of the study
Future studies are needed to further elaborate on the phenotypic shifts of vascular MΦs upon targeted interference with different MC functions. The modes of how MCs sense the microenvironment and how this subsequently influences MΦ phenotype and chronic inflammation require the attention of follow-up studies. Also, this study focuses on the MΦ chemoattractants CCL2 and MIF. However, it remains unclear whether this unexpected atheroprotective role of these chemokines specifically within the intimal niche can be extrapolated to other proinflammatory mediators that are known to be otherwise detrimental in chronic inflammatory settings. Lastly, these results underscore the need for cell-specific therapies when considering anti-inflammatory approaches. Hence, the development and translation of cell-specific delivery systems are urgently required to allow directed anti-inflammation solely at sites where respective mediators are considered detrimental.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CCL2 antibody (anti mouse) | BioXcell | #BE0185 |
| Isotype control antibody | BioXcell | #BE0091 |
| Anti-LGALS3 (anti mouse) | Cedarlane | #CL8942AP |
| Anti-ACTA2 (anti mouse) | Sigma | #F3777 |
| Anti-PDGFβR (anti mouse) | Abcam | #ab32570 |
| Anti-Ki-67 (anti mouse) | Abcam | #Ab15580 |
| Anti-CD68 (anti mouse) | ThermoFisher | #MCA1957 |
| Cy3-conjugated AffiniPure Goat Anti-Rat IgG (H+L) | Jackson ImmunoResearch | #112-165-071 |
| Cy5-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | #111-175-144 |
| Cy3-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | #111-165-003 |
| Anti-CD68 (anti human) | Dako | #M0876 |
| Cy3-conjugated AffiniPure Goat Anti-Mouse | Invitrogen | #A10521 |
| Fc-receptor blocking antibody | Ebioscience | # 14-0161-85 |
| Anti-CD45.2 (anti mouse) | BD Biosciences | #560694 |
| Anti-CD19 (anti mouse) | BioLegend | #152404 |
| Anti-CD3e (anti mouse) | BD Biosciences | #553062 |
| Anti-CD115 (anti mouse) | BioLegend | #15-0452-82 |
| Anti-Ly6C (anti mouse) | BioLegend | #128012 |
| Anti-Ly6G (anti mouse) | BioLegend | #127608 |
| Anti-CD45R (anti mouse) | ThermoFisher | #15-0452-82 |
| Fc-receptor blocking antibody | Ebioscience | #14-0161-86 |
| CD45+ magnetic selection beads (anti mouse) | Miltenyi Biotec | #130-052-301 |
| Anti-CD45 (anti mouse) | BD Biosciences | #557659 |
| Anti-CD11b (anti mouse) | Biolegend | #101228 |
| Anti-F4/80 (anti mouse) | Biolegend | #123110 |
| Anti-CD64 (anti mouse) | Biolegend | #139314 |
| MHCII (anti mouse) | ThermoFisher | #17-5321-82 |
| TotalSeq-B anti-mouse Hashtag antibodies no. 1-9 (anti mouse) | BioLegend | #B0301-B0309 |
| Anti-F4/80 Percp Cy5.5 (anti mouse) | BioLegend | #123128 |
| Anti-Ly6G BV711 (anti mouse) | BioLegend | #127643 |
| Anti-CD11b (anti mouse) | Biolegend | #101242 |
| Anti-Ly6C (anti mouse) | Biolegend | #128033 |
| Anti-CD14 (anti human) | Biolegend | #325619 |
| Anti-CD45 (anti human) | Miltenyi Biotec | #130-113-117 |
| Anti-CD14 Microbeads (anti human) | Miltenyi Biotec | #130-050-201 |
| Anti-CCL2 (anti mouse) | Invitrogen | #16-7096-85 |
| Isotype control | Invitrogen | #16-4888-85 |
| Anti-CCL2 (anti mouse) | Invitrogen | #16-7096-81 |
| Isotype control | Invitrogen | #16-4888-81 |
| Anti-CD16/32 antibody (anti mouse) | eBioscience | #15246827 |
| Anti-CD11b (anti mouse) | BioLegend | #101228 |
| Anti-CD64 (anti mouse) | BioLegend | #139304 |
| Anti-F4/80 (anti mouse) | BioLegend | #123132 |
| Anti-CD45 (anti mouse) | BD Biosciences | #557659 |
| Anti-CD45 (anti mouse) | BioLegend | #103132 |
| Anti-CD64 (anti mouse) | BioLegend | #139313 |
| Anti-F4/80 (anti mouse) | BioLegend | #123132 |
| Anti-CD11b (anti mouse) | Biolegend | #101241 |
| Anti-CD11c (anti mouse) | BioLegend | #117323 |
| Anti-Lyve-1 (anti mouse) | ThermoFisher | #50-0443-82 |
| Anti-CD11b (anti mouse) | BioLegend | #101236 |
| Anti-MHCII (anti mouse) | BioLegend | #107636 |
| Anti-CD206 (anti mouse) | BioLegend | #141721 |
| Anti-DC80 (anti mouse) | BioLegend | #104732 |
| Anti-CD204 (anti mouse) | BD Biosciences | #748088 |
| Anti-Tim4 (anti mouse) | BioLegend | #141721 |
| Anti-Mertk (anti mouse) | BioLegend | #151506 |
| Anti-CD64 (anti mouse) | BioLegend | #139320 |
| Anti-CCL2 (anti mouse) | Invitrogen | #16-7096-81 |
| Isotype control | Invitrogen | #16-4888-81 |
| Anti-CD11c (anti mouse) | BioLegend | #117318 |
| Anti-F4/80 (anti mouse) | BioLegend | #123128 |
| Anti-CD36 (anti mouse) | BD Biosciences | #56-0362-82 |
| Anti-CD68 (anti human) | Thermo | #4-0688-82 |
| Anti-aSMA (anti human) | Zytomed | #MOB001 |
| Anti-F4/80 (anti mouse) | eBioscience | #15287387 |
| Anti-CD11c (anti mouse) | ThermoFisher Scientific | #14-0114-82 |
| Goat anti rat Alexa Fluor 555 | Invitrogen | #A-21434 |
| Goat anti Armenian hamster Alexa Fluor 647 | Invitrogen | #A78967 |
| Anti-GAPDH antibody | Abcam | #9484 |
| Anti-ACTA2 biotin-conjugated antibody | Abcam | #ab125057 |
| Streptavidin AlexaFluor 647 | Biolegend | #405237 |
| Anti-MCP1 | Abcam | #ab25124 |
| Anti-MIF | Abcam | #ab7207 |
| Anti-CD31 | Abcam | # ab222783 |
| Anti-CD36 | Abcam | # ab124515 |
| Anti-FOLR2 | Abcam | # ab228643 |
| Chemicals, peptides, and recombinant proteins | ||
| Tamoxifen | Sigma-Aldrich | #10540-29-1 |
| Corn Oil | Sigma-Aldrich | # C8267 |
| Western Diet | Sniff EF | #D12079 |
| SMC medium | PELO Biotech | #PB-MH-200-2190 |
| RPMI1640 | Sigma Aldrich | R8758 |
| FBS | Biosell | #S 0613 |
| Penicillin-Streptomycin | Sigma Aldrich | #P4333 |
| b-Mercaptoethanol | Sigma Aldrich | #M3148 |
| b-Estradiol | Sigma Aldrich | #50-28-2 |
| M-CSF | Immunotools | #11343115 |
| Accutase | Sigma Aldrich | # A6964 |
| CellTrackerTM Red CMTPX | Invitrogen | # C34552 |
| CCL2 | Biolegend | # 571406 |
| MIF | Biolegend | # 599406 |
| Saponin | Roth | #4185.1 |
| PBS (10X) | Gibco | #70011-036 |
| PBS (1X) | Gibco | #14190-094 |
| Hoechst 33342 | Invitrogen | # H3570 |
| ApopTag® Red In Situ Apoptosis Detection Kit | Sigma-Aldrich | #S7165 |
| Oil-red-O solution | Sigma | #O1391 |
| Mayer’s hemalaum solution | Roth | #T865.2 |
| Masson Trichrome staining | ScyTek Laboratories | #TRM-1 |
| Sucrose | Sigma-Aldrich | #S1888 |
| Formaldehyde 4% | Microcos GmbH | #50-00-0 |
| Methylene-free formaldehyde solution | Thermos scientific | #28908 |
| Sudan III | Merck | #C.I. 26100 |
| BSA Albumin Fraction V ≥98 %, powdered | Carl Roth | #8076.1 |
| Sytox red dead cell staining | Invitrogen | # S34859 |
| collagenase I | Sigma-Aldrich | #C0130 |
| collagenase XI | Sigma-Aldrich | #C7657 |
| DNase I | Sigma-Aldrich | #D4527 |
| Hyaluronidase | Sigma-Aldrich | #H3506 |
| Sytox green | ThermoFisher | #S34860 |
| Collagenase type 2 | Worthington Biochemical | #LS004174 |
| Elastase | Worthington Biochemical | #LS002292 |
| Collagenase type XI | Sigma-Aldrich | #C7657 |
| DNase type 1 | Sigma-Aldrich | #H3506 |
| Collagenase type 1 | Sigma-Aldrich | #C0130 |
| Hyaluronidase | Sigma-Aldrich | #H3506 |
| Sytox orange | ThermoFisher | #S34861 |
| Sytox blue | ThermoFisher | #S34857 |
| Sytox red | ThermoFisher | #S34859 |
| Liberase | Roche | #5401127006 |
| DNase | Sigma-Aldrich | #D4527 |
| Hyaluronidase | Sigma-Aldrich | #H3506 |
| NEBNext Single Cell / Low Input RNA Kit for Illumina | New England Biolabs | #E6420S |
| Fluorescent dye-based Qubit® ds DNA HS Assay kit | Thermo Fisher Scientific | #Q33230 |
| RLT-Buffer | Qiagen | #79216 |
| b-Mercapto-Ethanol | Thermo | #Sigma M6250 |
| Proteinase K | Life Technologies | #AM2548 |
| DNase I | ThermoFisher | #EN0521 |
| Maxima H-enzyme | ThermoFisher | #EP0753 |
| Maxima H-Buffer | ThermoFisher | #EP0753 |
| dNTPs | ThermoFisher | #R0186 |
| Exonuclease I | New England Biolabs | #M0293L |
| 1X Kapa HS Ready Mix | Roche | #07958935001 |
| DNase/RNase-Free Distilled Water | ThermoFisher | #10977-049 |
| Quant-iT PicoGreen dsDNA Assay Kit | ThermoFisher | #P7581 |
| High-Sensitivity DNA Kit | Agilent | #5067-4627 |
| NEBNext Ultra II FS Library Preparation Kit | New England Biolabs | #E6177S |
| SPRI-select Beads | #B23317 | Beckman Coulter |
| Q5 Master Mix | New England Biolabs | M0544L |
| Ficoll® Paque Plus | Cytiva | #GE17-1440-02 |
| Chromium Next GEM Single Cell 3′reagent kit v3.1 | 10X Genomics | #CG000206 Rev D protocol with reagens |
| RNeasy Mini Kit | Qiagen | #74104 |
| TruSeq Stranded mRNA LP technology kit | Illumina | #20020594 |
| ISO-1 | Merck Millipore (Calbiochem) | #CAS 478336-92-4 |
| dsDNA | InvivoGen | #tlrl-ecdna |
| Non-oxidizable 3S-HMGB1 | HMGBiotech | #HM-132 |
| HSP60 | Enzo | #ADI-SPA-806-F |
| ODN2216 | InVivogen | #tlrl-2216-5 |
| RNeasy Micro Kit | Qiagen | #74004 |
| High Capacity cDNA synthesis kit | Applied Biosystems | #4368814 |
| DMEM VLE | Bio&Sell | #BS.FG1445 |
| Antibiotics-antimycotics | Thermo | #15240062 |
| LDL from human plasma | Invitrogen | #L3482 |
| eFluor proliferation dye | Invitrogen | #65-0840-85 |
| ISO-1 | MedChemExpress | #HY-16692 |
| DMSO | SigmaAldrich | #D2650-5X5ML |
| Fixable viability Dye eFluor780 | eBioscience | #64-0865 |
| Quantitect Primer assay | Qiagen | #249900 |
| Ssoadvanced SYBR green supermix | BioRad | #1725270 |
| EdU | Invitrogen | #A10044 |
| Protease 1 | Roche | #05266688001 |
| Smooth muscle cell growth medium | PromoCell | #C-22062 |
| TNFa | BioLegend | #570102 |
| NuPAGE 4-12% Bis-Tris Gel | Invitrogen | #NP0002 |
| Spectra Multicolor Broad Range Protein Ladder | ThermoFisher | #226634 |
| Recombinant mouse CCL2 | BioLegend | #578404 |
| Trypsin-EDTA (0,25%) | Gibco | #11560626 |
| Ripa Lysis and Exraction Buffer | ThermoFisher | #89900 |
| CellTracker Green | Invitrogen | #C2925 |
| Critical commercial assays | ||
| MIF ELISA | BioLegend | #444107 |
| CCL2 ELISA | IBL international | #BE45241 |
| Cholesterol Kit | Abcam | #Ab65390 |
| CCL19 ELISA | Sigma-Aldrich | RAB0052 |
| CXCL12/SDF-1 alpha | Bio-techne | #DSA00 |
| Peritoneal Macrophage isolation kit | Miltenyi Biotec | #130-110-434 |
| Experimental models: Cell lines | ||
| HCASMCs | PromoCell | #C-12511 |
| Primary murine smooth muscle cell | Cell Biologics | #C57-6080 |
| Jurkat cells | Merck | #88042803 |
| Murine SMCs | Cell Biologics | #PB-A57-6080 |
| Experimental models: Organisms/strains | ||
| ApoE-/- | The Jackson Laboratory | #002052 |
| Myh11cre-ERT2 | The Jackson Laboratory | #019079 |
| Rosa26tdTomato | The Jackson Laboratory | #007914 |
| Ng2DsRed | The Jackson Laboratory | #008241 |
| Ng2Cre | The Jackson Laboratory | #029926 |
| Ccl2RFP-fl/fl | The Jackson Laboratory | #016849 |
| PC-G5-tdT | The Jackson Laboratory | #024477 |
| Cx3cr1-CreER | The Jackson Laboratory | #021160 |
| Cx3cr1GFP/+ | The Jackson Laboratory | #005582 |
| LysM-eGFP | MGI | MGI ID: 2654931 |
| MIFfl/fl | Fingerle-Rowson et al.102 | PMID ID: 12878730 |
| Software and algorithms | ||
| Imaris Software | Bitplane | Version 8.4.1 |
| ZEN Software Black | Zeiss | Version 2.3 |
| ImageJ | FIJI | Version 2.9.0 |
| AxioVision | Zeiss | Version 4.8 |
| FlowJo | BD Biosciences | Version 10 |
| Fastqc | Andrews103 | Version 0.11.8 |
| Cutadapt | Martin104 | Version 1.12 |
| zUMI pipeline | Parekh et al.105 | Version 2.9.4.d |
| STAR | Dobin et al.106 | Version 2.7.3a |
| RSubread | Liao et al.107 | Version 1.32.4 |
| Hisat2 | Kim et al.108 | Version 2.1.0 |
| Feature Counts | Liao et al.109 | Version 1.6.3 |
| DESeq2 | Love et al.110 | Version 1.30.0 |
| DAVID | Huang et al.111,112 | Version 6.8 |
| ClustVis | Metsalu and Vilo113 | Online version |
| Seurat | Stuart et al.114 | Version 3.1.1 |
| SCTransform | Hafemeister and Satija115 | Version 0.2.0 |
| CellRanger | 10X Genomics | Version 6.0.2 |
| Seurat | Stuart et al.114 | Version 4.0.4 |
| SCTransform | Hafemeister and Satija115 | Version 0.3.2 |
| MAST | Finak et al.116 | Version 1.14.0 |
| QuPath | Bankhead et al.117 | Version 0.3.2 |
Resource availability
Lead contact
The lead contact is Dr. Kami Pekayvaz (kami.pekayvaz@med.uni-muenchen.de).
Materials availability
Materials are available upon request from the lead author. This study did not generate new unique reagents.
Experimental model and study participant details
Mice were used as an experimental research model. Publicly available human and mouse scRNA-seq datasets, human and mouse atherosclerotic plaques, and cells and cell lines were also analyzed. Further details are provided in the Method details.
Method details
Mice
ApoE-/- (Stock No.: 002052),28 Myh11cre-ERT2 (Stock No.: 019079),118 Rosa26tdTomato (Stock No.: 007914),119 Ng2DsRed (Stock No.: 008241),120 Ng2Cre (Stock No.: 029926)121 and Ccl2RFP-fl/fl (Stock No.: 016849),122 PC-G5-tdT (Stock No.: 024477),123 Cx3cr1-CreER (Stock No.: 021160)124 were obtained from Charles River and Jackson Laboratories. Previously described125,126 Cx3cr1GFP/+ and LyzeGFP/+ mice were employed. Ccl2RFP-fl/fl mice did not show RFP signal without further amplification and are hence referred to as Ccl2fl/fl. In Cx3cr1tm2.1(cre/ERT2)Litt mice the YFP signal was constant and very weak, and did not mask the alternating PC-G5 Ca2+ bursts. Miffl/fl mice were provided by Guenter Fingerle-Rowson and Richard Bucala and generated as described.102 Mice after the age of 8 weeks were included in the analysis. Both genders were used, except for Myh11-creERT2 driven reporter or deletion systems, where the Cre-recombinase is expressed on the y-chromosome only. Mice were kept at 55% humidity and 21 °C temperature with a 12h day-night rhythm. All animal studies were approved by the local legislation on protection of animals (Regierung von Oberbayern, Munich). Mouse strains have been summarized in Table S1.
Tamoxifen injection and western diet feeding
All transgenic mice were backcrossed to a C57BL/6 background for at least seven generations. In Myh11cre-ERT2; Ccl2fl/fl; ApoE-/- (Ccl2SMCΔ/Δ) mice and Myh11cre-ERT2; Ccl2+/+; ApoE-/- (Ccl2SMC+/+) littermate controls or Myh11cre-ERT2; Rosa26tdT; Cx3cr1GFP; ApoE-/- (SMC-tdTlin Cx3cr1-MΦGFP-rep) Cre-recombinase was activated in male mice with a series of 5 consecutive tamoxifen injections intraperitoneally daily at 2mg/day/mouse (Sigma-Aldrich 10540-29-1), dissolved in corn oil (Sigma-Aldrich C8267), at an age of 6 weeks. One week after the first Tamoxifen injection, mice were fed a Western diet (WD) containing 22% fat and 0.2% cholesterol (Sniff EF D12079) for the respective durations depicted in the figures. Myh11cre-ERT2; PC-G5-tdT mice (SMCCa2+-rep) and Cx3cr1Cre-ERT2; PC-G5-tdT (Cx3cr1-MΦCa-rep) mice were also induced with 5 consecutive Tamoxifen injections at the same dosage. Ca2+ reporter SMCs were isolated from the aorta as depicted below and cultured in SMC medium (PELOBiotech PB-MH-200-2190). Male Myh11cre-ERT2; CCL2fl/fl; ApoE-/- (Ccl2SMCΔ/Δ) were compared to male Myh11cre-ERT2; CCL2+/+; ApoE-/- (Ccl2 SMC+/+) littermate controls. Ccl2 SMCΔ/Δ or Ccl2 SMC+/+ littermate mice with a measured plasma LDL/VLDL-cholesterol level <250mg/dl and mice showing no atherosclerotic lesions neither in the BCA nor in the valves were excluded from further analyses. Ng2-cre+; Ccl2fl/fl; ApoE-/- mice (Ccl2MCΔ/Δ) mice and Ng2-cre-; Ccl2fl/fl; ApoE-/- mice (Ccl2MC+/+) were put on a Western diet (WD) beginning at 5 weeks of age for 14 weeks. Ng2-cre+; Miffl/fl; ApoE-/- mice (MifMCΔ/Δ) and Ng2-cre-; Miffl/fl; ApoE-/- mice (MifMC+/+) littermates were put on a WD beginning at 5 weeks of age for 8 weeks. Ng2-DsRed; ApoE-/- (MCRFP-rep), Ng2-DsRed; Cx3cr1-GFP; ApoE-/- (MCRFP-rep Cx3cr1-MΦGFP-rep) and Ng2-DsRed; Lyz-GFP; ApoE-/- (MCRFP-rep Lyz-MΦGFP-rep) mice were put on a WD beginning at 5 weeks of age for the amount of weeks respectively depicted in the figure captions.
Bone marrow transplant
1 x 107 bone marrow cells of donor Ng2DsRed; Cx3cr1GFP; ApoE-/- (MCRFP-rep Cx3cr1-MΦrep) mice were injected into irradiated (two times 6 Gγ with an interval of 4h) Ng2-cre-; Ccl2fl/fl; ApoE-/- mice (Ccl2MC+/+) and Ng2-cre+; Ccl2fl/fl; ApoE-/- mice (Ccl2MCΔ/Δ ). 4 weeks after transplantation mice were fed a WD for 20 weeks.
Intravital 2-photon microscopy
A TrimScope II (LaVision Biotech) equipped with a Chameleon laser (Coherent) and connected to an upright microscope with a 20× water immersion objective (Olympus) was used. Image stabilization, 3D rendering, and analysis was performed with Imaris.
In vivo atherosclerotic plaque imaging with single-cell resolution. Mice were anesthetized, the carotid artery was exposed and a custom build stage was placed. Imaging was performed with MCRFP-rep Cx3cr1-MΦGFP-rep and MCRFP-rep Lyz-MΦGFP-rep mice.
In vivo microscopy of the ear microvasculature. Mice were anesthetized, the ear was fixed on a custom-built-stage made of a plexiglass base with a round raised stage of resin where the ear can be pinned down. The ear was pinned down at the fringes and stretched across the resin stage with several pins, with minimal folds across its surface. Hair removal was not performed in order not to elicit any inflammatory response. The skin was scanned for an optimal fold-free and hairless spot that was well vascularized and contained an adequate number of MΦs before creating micro-sized laser injuries by prolonged, increased laser-power.
For CCL2 inhibition experiments, 200 μg of anti-CCL2 antibody (BioXcell #BE0185) or respective isotype control (BioXcell #BE0091) were injected i.v. twice every 24h for 48h, starting 72h before the experiment and once 30 minutes prior to imaging intravenously and also subcutaneously at the preparation site with an insulin syringe almost parallel to the ear surface, without injecting directly into the imaged area. Laser injuries were induced by elevated level of laser power (30% vs. 5% for imaging). Imaging was performed at the same spot over several hours. Micro laser injuries are circular and small in structure, and dendrites are motile and extend to several tens of micrometers. The time it took from laser injury onset for a tissue MΦs to form the first of possibly several dendrites is referred to as ‘time to dendrite formation’. The time it took one of several dendrites from formation/extension to reach a micro laser injury is termed as ‘time to reach injury’, ‘time to dendrite formation’ is the time it takes a cell to extend its first dendrite counted from the time the laser injury was created, ‘No. of dendrite forming MΦs‘ is the number of tissue MΦs that extended at least one dendrite in the field of view during the imaging period. When counting tissue MΦs in the field of view (FoV), only those with dendrites extending at least the length of the main cell body were included.
Yolk-sac- and bone-marrow-derived MΦs
Yolk sac (YS)- and bone marrow (BM)-derived Hoxb8 progenitors were generated as previously described.127 Progenitors were kept under proliferative conditions for the course of two months after establishing the cell line (RPMI1640 + 10% Foetal Bovine Serum [FBS] + 1% Penicillin-Streptomycin + 6% SCF-containing supernatant + 30 mM b-Mercaptoethanol + 1 mM b-estradiol [all from Sigma-Aldrich, except FBS from BioSell ®]). Differentiation to MΦs was induced by seeding the progenitors at a density of 3.0 x 10∧5 cells per 15 cm non-tissue treated dish in differentiation medium (RPMI1640 + 10% FBS + 1% Penicillin-Streptomycin + 6% SCF-containing supernatant + 30 mM b-Mercaptoethanol + 20 ng/mL M-CSF [ImmunoTools ®]). These conditions were kept for five days, with the medium being changed every second day. The MΦs were then washed with PBS, dissociated with Accutase (Sigma-Aldrich ®) for 10 min at 37°C and replated in non-tissue treated 12-well plates (1.75 x 10∧5 MΦs per well) in differentiation medium for another 24h. Afterwards, the Hoxb8 MΦs were stained with CellTracker™ Red CMTPX dye (Invitrogen ®) in RPMI 1640, according to the manufacturer’s instructions. After exchanging the dye solution with RPMI 1640 the cells were promptly imaged for a baseline count. Then, the MΦs were incubated with the designated cytokines/chemokines (50 ng/mL CCL2 [Biolegend ®] and/or 50 ng/mL MIF [Biolegend ®] and/or 20 ng/ml M-CSF [ImmunoTools] ®) in serum-free RPMI1640 and consecutively imaged at the indicated time-points. Three fields of vision were imaged per well, with each condition being done in triplicate per experiment. The number of cells on each field of vision was counted with CellProfiller™ software.128
Ex vivo analysis of the vasculature
After sacrificing the mice and perfusion with PBS/heparin (10:1), aortas were harvested. After fixation in 4% paraformaldehyde for 30 min the aortic valves, aortas, and brachiocephalic arteries were placed in 30% sucrose at 4°C overnight, embedded in optimal cutting temperature compound, frozen at −80°C, and then cut with a cryotome (CryoStar NX70 Kryostat; ThermoFisher Scientific) into 10 μm sections. The brachiocephalic artery was analyzed in a standardized manner and serially sectioned from the aortic arch to the right subclavian artery. Analyses were performed at a proximal location from the aortic arch, an intermediate, and a distal location in proximity to the right subclavian artery. Sections from the valve were fixed with 4% formaldehyde and blocked with 10% goat serum, 1% BSA and 0.5% saponine. Slides were stained with antibodies specific to LGALS3 (CL8942AP, Cedarlane), ACTA2 (F3777, Sigma), PDGFβR (ab32570, abcam,), Ki-67 (ab15580, abcam), CD68 (MCA1957). The respective secondary antibodies included Cy5-conjugated AffiniPure Goat Anti-Rat IgG (H+L) (#112-175-143, Jackson ImmunoResearch), Cy5-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) (#111-175-144, Jackson ImmunoResearch), Cy3-conjugated AffiniPure Goat Anti-Rat IgG (H+L) (#112-165-071, Jackson ImmunoResearch) Cy3-conjugated AffiniPure Goat Anti-Rabbit (#111-165-003, Jackson ImmunoResearch). Nuclei were stained with Hoechst 33342 (H3570). Immunofluorescence staining of human plaques was performed with mouse anti-human CD68 Clone PG-M1 40 (M0876, Dako), ACTA2 (F3777, Sigma) and secondary antibody Invitrogen® Cy3 goat anti mouse (A10521) after prior dewaxing and counterstained with DAPI. Apoptosis was assessed by TUNEL staining. (S7165, ApopTag® Red In Situ Apoptosis Detection Kit, Sigma-Aldrich). Plaque size was analysed in immunofluorescence images co-stained for LGALS3, ACTA2 and Hoechst. Oil-red staining was performed with Sigma O1391 Oil-red-O solution, counterstained with Mayer’s hemalaun solution (T865.2, Roth). Necrotic core formation, defined as area under the fibrous cap with loss of extracellular matrix and accumulation of cellular debris, was assessed by Masson Trichrome staining (ScyTek Laboratories, Trichrome Stain kit, modified Massons) or Hematoxylin and Eosin staining (HE) in Ccl2 SMCΔ/Δ, Ccl2 SMC+/+ mice or MifMCΔ/Δ, MifMC+/+ mice, respectively. Histological images were acquired using a Leica DM6B microscope. Immunofluorescence images were acquired using a Zeiss Axio imager microscope with an AxioCam. Confocal images were acquired with Zeiss Laser Scanning Microscope LSM 880 with an Airyscan module. Image analysis was performed in a standardized setting by colour thresholding with ZEN software or ImageJ. Cell counting was performed with a Zeiss AxioVision or a Leica DM6B microscope. Plaque quantification was performed in littermates. For en-Face ex vivo imaging of the aorta, harvested aortas from SMClin MΦrep mice were fixed in 4% PFA and subsequently left in 30% sucrose. overnight. Subsequently, the aorta was cut open longitudinally, embedded onto a slide and covered with a coverslip in PBS. Imaging was performed instantly with a Laser Scanning Microscope LSM 880 with an Airyscan module.
En face Sudan III staining
The ascending and descending aorta was fixed in 4% PFA overnight, washed with aqua bidest, post fixed 3 min in 75% ethanol and incubated for 30 min in Sudan III (Merck) with a subsequent washing step in aqua bidest. Perivascular fat tissue was removed thoroughly. Aortas were placed on a slide and covered with PBS under a coverslip. Images were acquired using a stereomicroscope (Zeiss) equipped with a Canon Powershot G5 camera.
Flow cytometry of blood, spleen, and bone marrow
Femurs and tibias were perfused with PBS to obtain bone marrow cells, which were filtered through a 40-μm mesh. The spleen was grinded through a 40 μm mesh to obtain single cell suspensions. Ammonium-chloride-potassium lysing buffer was applied twice on the blood samples and once on bone marrow/spleen samples to deplete red blood cells. Lysis was stopped with PBS and followed by a centrifugation step of 370 g for 7 min. Samples were resuspended, a purified Fc-receptor (CD16/32, FcγRIII/II) blocking antibody was added and incubated for 10 min (93, eBioscience®). Primary antibodies against CD45.2 (#560694, BD Biosciences®), CD19 (#152404, BioLegend®), CD3e (#553062, BD Biosciences®), CD115 (#135513, BioLegend®), Ly6C (#128012 BioLegend®), Ly6G(#127608, BioLegend®) or CD45R (#15-0452-82 ThermoFisher®) were used and incubated for 20 min. After a final centrifugation step cells were resuspended in PBS containing 0.5% BSA (FACS buffer). All steps were performed on ice or in a centrifuge at 4°C. In addition, Sytox red dead cell staining (S34859, Invitrogen®) was used for Ccl2SMCΔ/Δ and Ccl2SMC+/+ bone marrow, spleen and blood analyses. Cells were immediately analyzed by a BD LSR Fortessa ® cell analyser. Compensation was performed with beads. Quantification was performed with FlowJo V10®.
FACS of kidney and lung immune cells
After sacrificing the mice and perfusing with ice-cold PBS, kidneys and lungs were harvested, minced and resuspended in an enzyme digestion mix containing collagenase I (Sigma-Aldrich, C0130), collagenase XI (Sigma-Aldrich, C7657), DNase I (Sigma-Aldrich, D4527) and hyaluronidase (Sigma-Aldrich, H3506) and incubated for 30 minutes at 37°C on a shaker. Subsequently, the tissue was filtered across a 70μm filter and spun down at 350 g for 7 minutes at 4°C. The single cells were resuspended in 150 μl Fc-block (eBioscience, 14-0161-86). CD45+ magnetic selection beads (Miltenyi Biotec, 130-052-301) in FACS buffer (PBS with 0.5% BSA) were added and incubated on ice for 15 min. Then, 1ml FACS buffer were added, and the cells were spun down at 4°C, 300 g for 10min. Subsequently, magnetic bead separation with LS columns (Miltenyi Biotec®, 130-042-401) was performed according to the manufacturer’s instructions. After magnetic bead separation, cells were spun down at 4°C, 300 g, 10min and subsequently resuspended in the mastermix, containing CD45 APC-Cy7 (BD Biosciences®, 557659), CD11b Percp-Cy5.5 (Biolegend®, 101228), F4/80 PE (Biolegend®, 123110), CD64 PE-Cy7 (Biolegend®, 139314), MHCII APC (ThermoFisher®, 17-5321-82) and a respective TotalSeq™B anti mouse Hashtag antibody (i.e. Biolegend®, TotalSeq™-B0301 anti-mouse Hashtag 1; of this family, Hashtags 1, 2, 4, 5, 6, 7, 8, 9 were used). After 30 minutes incubation on ice and three subsequent washing steps, cells were resuspended in FACS Buffer with 2% FBS. Prior to FACS, Sytox green (ThermoFisher®, S34860), was added to the sample and CD11bhi CD64+ F4/80+ cells were enriched by FACS-sorting into FACS Buffer with 2% FBS. Subsequently the FACS Sorted cells were spun down, resuspended, counted and the cell concentration was adjusted to allow similar cell count yields across samples. Samples were subsequently pooled and proceeded with the single-cell RNA sequencing protocol described in detail below.
Digestion of the aorta
Aortas were harvested as described above and incubated in an enzyme mix containing collagenase type 2 (Worthington Biochemical® LS004174) and elastase (Worthington Biochemical® LS002292) in PBS containing Mg2+ and Ca2+ for 10 min at 37 °C in a thermo shaker with 400 rpm. Aortas were transferred to PBS without Mg++ and Ca++ on ice to stop the digestion. Adventitial tissue was subsequently removed from the rest of the aorta. The media and intima were transferred to a second enzyme mix containing collagenase type XI (Sigma-Aldrich® C7657), DNase type 1 (Sigma-Aldrich® D4527), collagenase type 1 (Sigma-Aldrich® C0130), hyaluronidase (Sigma-Aldrich® H3506) in PBS with Mg++ and Ca++ and incubated for 40 min at 37 °C in a shaker with 400 g. The cell suspension was filtered through a 70-μm mesh to obtain single cell suspensions. Cold PBS was added simultaneously to stop the enzymatic reactions. The cell suspension was centrifuged at 370 g for 7 min. Sytox blue, orange, or red dead cell staining (Invitrogen®) was added prior to analysis. Cell analysis and sorting was performed using a Beckman Coulter Astrios ® Cell Sorter. Subsequent analysis was performed by FlowJo V10®. Cell pellets from Ccl2SMCΔ/Δ and Ccl2SMC+/+ and Ccl2MCΔ/Δ and Ccl2MC+/+ aortas were shock-frozen on dry ice and subsequently processed for further bulk-RNA-seq or RT-PCR. Analysis of leukocyte subsets by flow cytometry of Ccl2SMCΔ/Δ mice was performed without excluding adventitial tissue. Therefore, the aorta (liberated of perivascular fat tissue) was minced in small pieces and placed in an enzyme mix of 4U/ml Liberase (TM research grade (ROCHE) Cat.: 5401127001), 120U/ml DNase (Sigma-Aldrich, D4527), 60U/ml Hyaluronidase (Sigma-Aldrich, H3506) and incubated at 37°C for 60 min. Subsequently, digested tissue was pushed through a 70μm filter and washed once. Subsequently the single-cell suspension was blocked with FC-block and subsequently incubated on ice with the antibody mix and prepared for flow cytometry as described above. The mentioned immune-cell subsets were gated by gating for live singlets expressing F4/80 (Cat.: 123128), Ly6G (Cat.: 127643), CD11b (Cat.: 101242), Ly6C (Cat.: 128033). FACS-sorting of Ccl2SMCΔ/Δ; SMC-tdTlin; Cx3cr1-MΦGFP-rep aortas was performed accordingly. TdTomato+ living singlets were FACS-sorted for downstream genetic deletion PCR.
Single-cell RNA sequencing
The Chromium Next GEM Single Cell 3’ reagent kit v3.1 (CG000206 Rev D) from 10X Genomics® protocol was used for FACS-sorted lung and kidney immune cell sequencing. To decrease batch-effect related artifacts, sample multiplexing with TotalSeqB™ anti-mouse Hashtag Antibodies, which were included into the FACS antibody mix, was performed. Up to eight samples were multiplexed into one library. A total of 1.26 x105 cells across runs were loaded for Gel Beads-in-emulsion (GEM) generation. According to the kit protocol first GEMs were generated, subsequently reverse transcription was performed, and cDNA was cleaned up, amplified, and size selected. After a quality control and quantification step, gene expression libraries and cell surface libraries were subsequently constructed. The libraries were sequenced with an Illumina NovaSeq by IMGM laboratories.
Bulk RNA sequencing of aortic intima/media
Total RNA from cell pellets of Ccl2SMCΔ/Δ and Ccl2SMC+/+ descending aortas was isolated using the RNeasy Mini Kit (Qiagen®) according to the manufacturer’s instructions including on-column DNase digestion. After quantification, purity and integrity control, the RNA library was prepared according to TruSeq® Stranded mRNA LP technology kit instructions.
Genetic deletion PCR for MIF and CCL2
Genetic deletion PCR was performed on frozen tissue from the aorta and brachiocephalic artery from Ccl2SMC+/+ and Ccl2SMCΔ/Δ mice and MifMC+/+ and MifMCΔ/Δ mice. In addition, tdTomato+ SMCs were FACS-sorted from atherosclerotic aortas from tamoxifen injected Myh11-creERT2; Rosa26-tdTomato; Cx3cr1-GFP; Ccl2fl/fl; ApoE-/- mice. The following primer sequences were employed for the respective genetic deletion PCR:
Ccl2-KO Fw: GGT CCC TGT CAT GCT TCT G
Ccl2-KO Rev: CAG GTT TCA GAG ACA TTG CTT C
MIF-KO Fw: AGG TTA GTC ACT CTA CTG GCC
MIF-KO Rev: GGC TCC TGG TCT CAG TCA GG
ITC Fw: ACA CAA CCC TTG CCT TCA CC
ITC Rev: GAA CTG GTG GCT CGG AAG AG
Low-input sequencing of FACS aortic MΦs
Aortic Cx3cr1+ MΦs from Ccl2MC+/+ or Ccl2MCΔ/Δ chimera mice with MCRFP-rep Cx3cr1-MΦGFP-rep bone marrow were directly sorted into NEBNext Cell Lysis Buffer. The RNA library was prepared with the NEBNext® Single Cell / Low Input RNA Kit for Illumina® (New England Biolabs), according to the manufacturers protocol. Quality control of RNA was performed by the 2100 Bioanalyzer (Agilent Technologies) and RNA was quantified using the highly sensitive fluorescent dye-based Qubit® ds DNA HS Assay kit (Thermo Fisher Scientific, according to the manufacturer’s protocol. All single libraries from both sample batches were pooled into a sequencing library with an equal DNA amount per sample and used for loading on the NextSeq® 500 sequencing system for cluster generation and sequencing. The library was sequenced on the next NextSeq® 500 sequencing system (Illumina®), performing one high output (HO) single-read 75 cycles (1x75bp SR) run, under the control of the NextSeq Control Software (NCS).
Prime-seq of peritoneal MΦs
An exact protocol for prime-seq129 is available at protocols.io (https://doi.org/10.17504/protocols.io.s9veh66). Briefly, of the MΦs sorted into RLT-Buffer (Qiagen®) with 1% Mercapto-Ethanol, 50 μL were further processed for library preparation. Proteinase K (AM2548, Life Technologies®) was used for lysate treatment, cleanup beads (GE65152105050250, Sigma-Aldrich®) (2:1 beads/sample ratio) were used for isolation, and DNase I (EN0521, Thermo Fisher®) for digestion. 30 units of Maxima H- enzyme (EP0753, Thermo Fisher®) were used for transcribing, 1x Maxima H- Buffer (EP0753, Thermo Fisher®), 1 mM each dNTPs (R0186, Thermo Fisher®), 1 μM template-switching oligo (IDT), 1 μM barcoded oligo-dT primers (IDT) in a 10 μL reaction volume at 42 °C for 90 minutes were included. Same cell-type samples were then pooled and cleaned using cleanup beads (1:1 beads/sample ratio). Remaining primers were digested with Exonuclease I (M0293L, NEB) following cleanup at 37 °C for 20 minutes followed by 80 °C for 10 minutes. The Exonuclease I digested samples were then again cleaned using cleanup beads (1:1 beads/sample ratio).
Using 1X KAPA HS Ready Mix (07958935001, Roche®) and 0.6 μM SINGV6 primer (IDT) in a 50 μL reaction synthesis of second strand and pre-amplification was performed. The PCR was cycled as follows: 98 °C for 3 minutes; 15 cycles of 98 °C for 15 s, 65 °C for 30 s, 72 °C for 4 minutes; and 72 °C for 10 minutes. Cleanup beads were used subsequently (0.8:1 beads/sample ratio) and then eluted in 10 μL of DNase/RNase-Free Distilled Water (10977-049, ThermoFisher®). Present cDNA was quantified by the Quant-iT PicoGreen dsDNA Assay Kit (P7581, Thermo Fisher®), the size distribution was qualified by the High-Sensitivity DNA Kit (5067-4627, Agilent®).
Following QC, cDNA was used to make libraries with the NEBNext Ultra II FS Library Preparation Kit (E6177S, NEB), with a five-fold lower reaction volume than the manufacturer’s instructions. The supplied Enzyme Mix and Reaction buffer was used for fragmentation in a 6 μL reaction. Adapter ligation was performed using the supplied Ligation Master Mix, Ligation Enhancer, in addition to a custom prime-seq Adapter (1.5 μM, IDT) in a reaction volume of 12.7 μL. Double-size selection using SPRI-select Beads (B23317, Beckman Coulter®), with 0.5 and 0.7 ratios, was performed following ligation. The samples were then amplified using a library PCR using Q5 Master Mix (M0544L, NEB), 1 μL i7 Index primer (Sigma-Aldrich®), and 1 μL i5 Index primer (IDT) using the following setup: 98 °C for 30 s; 13 cycles of 98 °C for 10 s, 65 °C for 1 m 15 s, 65 °C for 5 m; and 65 °C for 4 m. SPRI-select Beads were used for a final double size selection.
Concentration and quality was checked using a high-sensitivity DNA chip (Agilent Bioanalyzer), subsequently the libraries were 150 bp paired-end sequenced on a S4 or a S1 flow cell of a NovaSeq (Ilumina®).
The data was initially checked using fastqc (version 0.11.8103). Any regions on the 3′ end of the read where the sequence read into the poly-A tail were then removed by Cutadapt (version 1.12104). The zUMIs pipeline (version 2.9.4d105) was then used to filter the data, using a phred threshold of 20 for 4 bases for both the UMI and BC, to map the reads to the mouse genome with the Gencode annotation (v35) using STAR (version 2.7.3a),106 and count the reads using RSubread (version 1.32.4).107
Bioinformatic analysis
The paired-end sequencing data were aligned against the ensemble mus musculus release 97 (GRcm38). The alignment was performed using Hisat2108 (version 2.1.0). Genes were quantified using featureCounts109 (version 1.6.3) with parameters –primary -O -C -B (only primary alignments, assign reads to all overlapping features, not counting fragments having reads mapping to different chromosomes, only count fragments with both reads aligned). The minimal overlap between read and gene was set to 10bp (minOverlap) and at least 50% of all read bases must overlap the feature (fracOverlap). In the aortic intima/media bulk RNA-sequencing, one sample was excluded for its high mitochondrial fraction among the most frequently expressed genes.
Differential gene expression analysis
Differential read expression analysis for the low-input RNA-Seq data from FACS-Sorted MΦs from Ccl2MC+/+ and Ccl2MCΔ/Δ chimera mice, bulk RNA sequencing of Ccl2SMCΔ/Δ/Ccl2SMC+/+ aortic intima/media and the in vitro peritoneal MΦ - Jurkat cell coincubation RNA-Seq data was performed using the poreSTAT differential expression analysis pipeline (https://github.com/mjoppich/poreSTAT/, work in progress). Differential gene expression results used in this manuscript were received from calling DESeq2110 (version 1.30.0) with default parameters on either the acquired fragment counts (19083 data) or the read counts from the zUMIs pipeline.
Overrepresentation analysis
Overrepresentation analysis was performed using DAVID111,112 and the UP_KEYWORDS database.
ClustVis-based heatmaps
Respectively indicated heatmaps were created with ClustVis.113
scRNA-seq re-analysis
Data from Wirka et al.,32 Nature Medicine, 2019), GSE Accession number - GSE131780,32 were employed. The count matrices for both the human and mouse cells were downloaded and processed in R 3.5.3 using Seurat114 v3.1.1. The gene symbols of the mouse dataset were put to upper case. Subsequently, both count matrices were subset to only include common gene symbols. From the count matrices Seurat objects were created, filtering out genes expressed in less than 3 cells and cells with fewer than 200 genes expressed. After performing SCTransform115 (v0.2.0) on both the human and mouse object, we followed the SCTransform integration vignette with 3000 integration features.
After integration, it was found that all cells have less than 4000 expressed features and less than 15% mitochondrial content, which we deemed sufficiently to exclude doublets (usually filtering for 6000 features) and bad cells. Moreover, these results led us assume that Wirka et al.32 uploaded filtered expression matrices.
After integration, we identified cluster markers and assigned clusters and performed several visualizations on the SCT normalised values. The mean cluster gene expression values were also extracted for chemokine interactome analysis.
scRNA-seq (kidney, lung)
Data were obtained and processed using CellRanger v6.0.2 using the mm10 2020-A reference and the given barcodes for the hash-tag antibodies.
The subsequent analysis was performed in R 4.0.1 with Seurat130 v4.0.4 and sctransform115 v0.3.2. Because the data was split into M1 and M2 libraries, each subset matrix was loaded. Following Seurat object creation and calculation of mt- and ribosomal content, cells were filtered to contain more than 100 measured genes, but at most 6000, more than 500 measured UMIs per cell and at most 15% mt-content. Next, HTO classification had to take place. Because only 8 antibodies were used, the antibody expression matrix was subset to exclude Mtag3 and Mtag10 (these were not used). In general, we followed the demultiplexing with hashtag oligos vignette, and set the positive quantile to 0.99 for the HTODemux function. Both Seurat objects were subsequently filtered to only contain Singlets (removing Doublets and Negatives).
For both lung and kidney the following process was repeated. The so far processed M1 and M2 library was subset for only cells with hashtag relevant to lung or kidney, respectively. On these subsets SCTransform normalization was applied, regressing out mitochondrial and ribosomal contents. We selected 2000 integration features and ran the SCTransform115 integration workflow.
On the integrated object, PCA was performed with 50 PCs, which were also used for finding neighbors and subsequent UMAP calculation (using 50 neighbors). Clusters were determined at a resolution of 0.2. Upon normalizing the RNA assay, marker genes were determined for each cluster using the FindMarkers function with t-test. Differential expression analysis between cre+ and cre- cells was conducted using the FindMarkers function and with both MAST116 (v1.14.0) and t-test. Volcano plots in this manuscript show the results from the MAST-based analysis. Violin-Plots were enhanced to also show a box-plot and mean expression (black dot) using ggplot2 stat_summary.
Chemokine interactome analysis
Chemokine ligand-chemokine receptor interactions were collected from two different resources131 and https://www.rndsystems.com/pathways/chemokine-superfamily-pathway-human-mouse-lig-recept-interactions (year of access: 2021). Interactions are either classified as antagonist, agonist or undefined. In general, the steps described by Armingol et al.132 are followed for determining cell-cell communications. The experimental expression data (for each cluster) is read in and filtered to only contain genes from the above collection of chemokine interactors. For each ligand-receptor pair, and for each cluster-pair, the communication score is calculated. This communication score is the product of the ligand expression and the receptor expression (expression product).
This results in a data frame in which for each ligand-receptor pair in each cluster pair a score is associated. To determine the total communication between two clusters, all communication scores between these clusters are aggregated (sum).
In a second step, the data frame is arranged into matrix form, keeping only those clusters of interest (or all, if no filtering was requested). For highlighting specific interactions (e.g. CCL2->CCR2), the maximal interaction score among all cluster interactions for this specific interaction is determined. This value is then used to scale the single cluster interactions by the maximally observed interaction score (that is: the ratio of the interaction score divided by the maximal interaction score seen). This information is then used to plot a chord diagram (taken from https://github.com/tfardet/mpl_chord_diagram) showing LR-interactions between clusters.
In addition, the matrix plot shows the scaled (z-score) expression scores for all interactions in the selected clusters. In a filtered version, only interaction which have at least in one cluster pair a z-score > 1 are shown. Finally, the chemokines overview displays the ligand-receptor map in the lower left corner, and shows the expression values for the receptors in the selected clusters on top, and those for the ligands to the right. This visualization allows for a brief overview of ligand and receptor expressions, while also showing possible interactions. Source code for this analysis is available online https://github.com/mjoppich/chemokine_interactionmap. The specific MIF interactions with CXCR2, CXCR4 and CD44 were added to the ligand-receptor set.
Monocyte isolation
Blood was collected from healthy human donors into sodium citrate containing tubes. PBS-diluted blood was layered on 1.077 g/ml ficoll-paque plus (GE Healthcare®) and centrifuged 30 min at 450 g, RT. After discarding plasma, mononuclear cell layer was transferred in a fresh tube and washed in PBS+0.5% BSA+3mM sodium citrate buffer three times at 300 g and 200 g for 15 and 10 min respectively. Promptly, cell pellet was resuspended in the buffer and while keeping on ice, the cell viability was assessed by Trypan blue. Steps of magnetic labelling and bead separation were conducted according to manufacturer's instructions (Miltenyi Biotec®). Furthermore, the purity of enriched monocytes was confirmed by flow cytometry analysis with APC/Cy7-CD14 (Clone HCD14, Biolegend®) and FITC-CD45 (Clone5B1, Miltenyi Biotec®) to be >95%. To study the chemotaxis and to perform the RT-PCR experiments, CD14+ cells were isolated by CD14 micro beads.
Chemotaxis assay with human immune cells
Monocytes (1.8×105) were seeded in the upper chambers of cell culture inserts with 3 μm pore size (BD Biosciences®), while different media were added to the lower chambers. After 4 h, the numbers of migrated cells towards every medium in the lower chamber were counted with a cytometer. To analyse the effects of chemokines like CCL2 and MIF, the corresponding inhibitors, such as anti-CCL2 (10 μg/ml; R&D Systems®), ISO-1 (36 μg/ml; Merck®) or their controls were applied.
SMC isolation and culturing and calcium imaging
SMCs were isolated from Myh11cre-ERT2; PC-G5-tdT (SMCCa2+-rep) and CCL2MCΔ/Δ mice. For SMC isolation, aorta was harvested as described above, and the adventitial tissue was entirely removed under a stereomicroscope. The remaining aorta was subsequently minced with a scalpel and the shortly centrifuged aortic pieces were resuspended in collagenase II and elastase containing SMC medium (PELOBiotech® PB-MH-200-2190) and incubated in a shaker at 37°C for 45 min. Subsequently, SMC medium was added to dilute the enzyme mix and the suspension was centrifuged at 350 g for 7 min. at room temperature. Afterwards, supernatant was removed, and the washing step was repeated. The single cell suspension was then added to Gelatine pretreated wells in a 96-well plate. SMCs were cultivated with SMC medium (PELOBiotech® PB-MH-200-2190) for at least 1 week before they were split. Experiments were performed on IBIDI® μ-Slide 8 wells with SMCs that were split at least once after culturing. Imaging was performed with an Olympus® IX83 inverted microscope under 37°C and 5% CO2 incubation. Image visualization was performed with Imaris.
ELISA
The expression levels of chemokines, such as MIF, CCL2, CCL19 and CXCL12 were quantified by ELISA kits according to the manufacturer’s instructions (abcam®, ThermoFisher Scientific). Blood samples were collected from mice using heparin containing syringes. Blood samples were centrifuged for 20 min at 2000 g at 4 °C and frozen immediately at -80 °C. MIF (BioLegend®, #444107), CCL2 (IBL international®, BE45241) and cholesterol (abcam, ab65390) assays were performed according to the manufacturer’s instructions. Cell culture experiments. Primary human coronary artery smooth muscle cells (HCASMCs, PromoCell®) were seeded in 6-well plates (4x104 cells/well). When HCASMCs obtained 80% level of confluency, they were included for further downstream analyses. Subsequently, the cells were washed with PBS, thoroughly and incubated for further 24h with SMC growth medium. Collected media were used as conditioned media in our experiments. When HCASMCs obtained 80% level of confluency, for CCL19 and CXCL12 measurements, they were stimulated with dsDNA (50 μg/ml, InvivoGen®), non-oxidizable 3S-HMGB1 (1 μg/ml, HMGBiotech®), HSP60 (1 μg/ml, Enzo®), or ODN 2216 (35μg/ml) for 8h. Subsequently, the cells were washed thoroughly and incubated for further 24h with SMC growth medium. Collected media were used as conditioned media in our experiments. ELISAs (CCL19, Sigma-Aldrich®, RAB0052 Human and CXCL12/SDF-1 alpha, R&D® DSA00 Human) were performed according to manufacturers’ instructions.
Isolation of total RNA for RT-PCR
Total RNA was extracted from HCASMCs in different culture conditions and FACS-sorted Ng2+ SMCs using RNeasy Mini or Micro kits (Qiagen®). All binding, washing, and elution steps were performed by centrifugation in a benchtop microcentrifuge according to the manufacturer’s protocol. The purity (OD260/ OD280) and concentrations of isolated RNA were measured with a NanoDrop 2000 spectrophotometer (ThermoFisher Scientific®) and finally the RNA samples were stored at -80°C.
cDNA synthesis and quantitative RT-PCR
High Capacity cDNA synthesis kit (Applied Biosystems) was used for reverse transcription of total RNA to cDNA by Thermal cycler 2720 (Applied Biosystems). To examine quantitative RT-PCR, 2 μL of cDNA samples, 2 μL of primer (QuantiTect Primer Assay-Qiagen), 6 μL of nuclease free H2O, and 10 μL of 2X Ssoadvanced SYBR green supermix (Biorad) were precisely pipetted per reaction in a 96-well plate. It was analysed by MyiQ real time cycler (Biorad) using the following program: Denaturation step at 95°C for 3 min followed by 45 cycles of denaturation at 95°C for 15 sec, annealing at 55-65°C for 15 sec and extension at 72°C for 15 sec. An additional melting curve analysis was performed at the end of the PCR program to determine the specificity of the primers and analyse the PCR products. Finally, Ct values were mathematically calculated by comparative CT method (2−ΔΔCT) to estimate the gene expression level normalized to genes encoding B-actin, B2M, GAPDH or 18S rRNA.
MΦ survival assay
Murine peritoneal were isolated, counted and resuspended in a concentration of 5x105/mL in VLE DMEM (Bio&Sell) +10% FBS (Bio&Sell) + 1% antibiotics-antimycotics (Thermo Fisher Scientific). 5mL of this suspension were added to untreated 100mmx15mm petri dishes (Falcon®) and stored in the incubator overnight. On day 1, the cells were washed with sterile PBS and new medium was added. On day 2, after another washing step, the MΦs were counted under the microscope and stimulated with commercial CCL2 (200ng/mL) in serum starvation medium (VLE DMEM without supplements). After 6, 12 and 24h the peritoneal MΦs were counted.
MΦ migration assay
To perform this assay, a 24-well plate (TPP Techno Plastic Products AG) and FluoroBlok Inserts 8μm pore size (Falcon®) were used to create two compartments.
Primary murine smooth muscle cells (#C57-6080; Cell biologics) were seeded in the 24-well plate. When the SMCs obtained 80% level of confluency, they were included in experiments. Murine peritoneal MΦs were isolated, counted and resuspended in VLE DMEM. 100 μl of this suspension was added into the inlays and incubated for 2h. Cultured jurkat cells (#88042803; Merck) were centrifuged and resuspended in a concentration of 2.5x105/mL. Cells were brought into necrosis by alternately putting them into 60°C water bath and on ice. Shortly before coincubation, 20μg/mL Anti-CCL2 (16-7096-85; Invitrogen®) or Isotype control (16-4888-85; Invitrogen®) antibody was added to the SMCs. Subsequently, inlays with the attached peritoneal MΦs (upper chamber) were inserted into the 24-well plate containing the SMCs (lower chamber), mimicking the fibrous cap, and the the necrotic jurkat cells were added into the upper compartment to mimic the necrotic core. The MΦs that had migrated across the transwell were then imaged after 0, 1, 2 and 3h by an epifluroescence microscope. The analysis and counting of the MΦs was performed in a standardized manner with ImageJ software.
SMC migration (wound) assay
Migration capability of murine SMCs was assessed using a scratch wound assay. Cells were seeded into 8-well μ-slides IBIDI® after gelatin coating, at a concentration of 5×105 cells/ml and cultured in SMC medium (PELOBiotech PB-MH-200-2190) containing 10% FBS. Cells were incubated at 37°C and 5% CO2. Anti-CCL2 monoclonal antibodies (Ref. 16-7096-81, Invitrogen®) and IgG Isotype control (Ref. 16-4888-81; Invitrogen®) were added at a concentration of 20μg/ml prior to the wound generation. Wound area was measured at different time points and the generated images were analyzed using FIJI (ImageJ®).
SMC proliferation assay
Cells were seeded in a 6-well plate at a concentration of 5×105 cells/ml in SMC medium. Murine smooth muscle cells were placed in the incubator at 37°C and 5% CO2. Anti-CCL2 monoclonal antibodies (Ref. 16-7096-81, Invitrogen®) and IgG Isotype control (16-4888-81; Invitrogen®) were added at a concentration of 20μg/ml at the beginning of the experiment. Pictures were taken at different timepoints across the surface of the wells and the cells / field of view were counted using FIJI (ImageJ®).
Efferocytosis assay
Murine peritoneal MΦs were isolated, counted and resuspended in a concentration of 1x106/mL in VLE DMEM +10% FBS + 10% antibiotic-antimycotic. Next, 200 μl of this cell suspension were filled into uncoated IBIDI® μ-Slide 8 Wells and stored in the incubator overnight. On day 1, cells were washed and the medium was changed. On day 2, after another washing step, the MΦs were stimulated with commercial CCL2 (200ng/mL) in serum starvation medium or with Anti-CCL2 (16-7096-85; Invitrogen®) or Isotype control antibody (16-4888-85; Invitrogen®) in SMC supernatant. During the 6h incubation time, 10x106 Jurkat cells were counted, centrifuged, resuspended and then brought into apoptosis by putting them under the UV-light for 15 minutes. Next, Jurkat cells were placed in the incubator for about 3-4h. After this, the cells were stained with Cell proliferation dye efluor 670 (Invitrogen®) for 30 minutes, at 37°C. After the incubation step, 8-well chambers containing peritoneal MΦs were washed and 200μl of the apoptotic Jurkat cells were added to the MΦs. After 1h of incubation, the chambers were imaged by an epifluorescence microscope. Analysis and counting of the MΦs was performed in a standardized manner with ImageJ software. For the efferocytosis assays with SMC-MΦ coincubation and subsequent MIF inhibition, far-red labelled Cell Proliferation Dye eFluor™ 670 (65-0840-85, eBioscience ®) apoptotic Jurkat cells were added to the corresponding wells after 72h of incubation with MIF inhibitor 1600 μM (ISO-1 HY-16692, MedChemExpress ®) or DMSO only. Cells were incubated for 60 min prior to image acquisition. The resulting images were analysed in QuPath117 0.3.2 software. The phagocytosis index was calculated in the following way: Phagocytosis index = (total number of engulfed cells/number of MΦs containing engulfed cells) × (number of MΦs containing engulfed cells/total number of counted MΦs) × 100.
Cholesterol uptake
Murine peritoneal MΦs were isolated, counted and resuspended in a concentration of 1x106/mL in VLE DMEM +10% FBS + 10% antibiotic-antimycotic. 200 μl of this cell suspension were filled into uncoated 8-well chambers (IBIDI® GmbH) and stored in an incubator at 37°C and 5% CO2 overnight. On day 1, cells were washed, and the medium was changed. On day 2, after another washing step, the MΦs were stimulated with Anti-CCL2 or Isotype control antibody respectively in SMC supernatant for 6h. After the incubation step, the chambers were washed and 10μg/mL low Density lipoprotein from human plasma (L3482; Invitrogen®) in serum starvation medium was added. After another 18h of incubation, the chambers were quickly washed and imaged by an epifluorescence microscope. The analysis and counting of the MΦs was performed in a standardized manner with ImageJ software.
MΦ - Jurkat cell supernatant co-incubation
Jurkat cells were were brought to necrosis by alternately putting them into 60°C water bath and on ice and subsequently left in the incubator at 37°C, 5% CO2. After 24h, the dead cell and live cell suspensions were centrifuged, the supernatant was removed and stored at –20°C. Murine peritoneal MΦs were isolated and brought into culture as described earlier. MΦs were counted under the microscope before and every 12h after addition of the respective supernatant. To prepare the cells for FACS sorting, Petri dishes were washed, and the cells were detached by using a cell scraper (Sarstedt®). Cell suspension was subsequently centrifuged, resuspended and a purified Fc-receptor (CD16/32, FcγRIII/II; 93, eBioscience) blocking antibody was added and incubated for 10 min. Primary antibodies against CD11b (#101228; Biolegend®), CD64 (#139304; Biolegend®), F4/80 (#123132, Biolegend®) and a Fixable viability Dye eFluor 780 (#64-0865; eBioscience®) were added and incubated for 20 min. After a final centrifugation step cells were resuspended in a PBS containing 0.5% BSA (FACS buffer). All steps were performed on ice or in a centrifuge at 4°C. Cell analysis and subsequent sorting was performed using a Beckman Coulter Astrios Cell Sorter. PRIME-Seq was performed as depicted above to analyze MΦ phenotype.
Flow cytometry of lung and ear
After harvesting, the organs were prepared for flow cytometry as described before. The mastermix that was added to the cell suspensions after the centrifugation step contained the following antibodies: CD45 (#557659; BD Biosciences), CD45 (#103132, Biolegend®), CD64 (#139313; Biolegend®), F4/80 (#123132; Biolegend®), CD11b (#101241; Biolegend®), CD11c (#117323; Biolegend®), Lyve-1 (#50-0443-82; ThermoFisher), CD11b (#101236; Biolegend®), MHC II (#107636; Biolegend®), CD206 (#141721; Biolegend®), CD80 (#104732; Biolegend®), CD204 (#748088; BD Biosciences®), TIM4 (#742778; BD Biosciences®), Mertk (#151506; Biolegend®), CD64 (#139320; Biolegend®), CD11c (#117318; Biolegend®), F4/80 (#123128; Biolegend®), CD36 (#56-0362-82; BD Biosciences®). Sytox green (ThermoFisher®, S34860) was used as a viability dye prior to FACS. Cells were immediately analysed by a BD LSR Fortessa cell analyser®. Compensation was performed with beads. Quantification was performed with FlowJo V10®.
Analysis of the perivascular area in kidneys
EdU (5-ethynyl-2′-deoxyuridine) (#A10044; Invitrogen®) was injected i.p. to the mice in a concentration of 1mg in 150μl PBS every 48h (4 injections per mouse in total) before sacrifice. After organ harvesting, the kindeys were fixed in 4% PFA for 30 min and left in 30% sucrose at 4°C overnight and subsequently embedded in OCT medium. Kidneys were cut in 10μm slices as described before. The kidney was serially sectioned sagitally. Analyses were performed at the proximal part, the middle and the distal part of the sagittal sections. Slides were stained with antibodies specific to CD68 (MCA1957, Bio-Rad) and ACTA2 (F3777, Sigma®). The respective secondary antibody was Cy3-conjugated AffiniPure Goat Anti-Rat IgG (H+L) (#112-165-071, Jackson ImmunoResearch®). Nuclei were stained with Hoechst. EdU was stained according to the manufacturer’s protocol (#A10044; Invitrogen®). Histological images were acquired using a Leica DM6B microscope. Image analysis was performed in a standardized manner by colour thresholding with ImageJ software.
Human plaque analyses
Formalin-fixed paraffin-embedded (FFPE) human tissue samples of aortic cross-sections were retrieved from the archives of the Institute of Pathology of the LMU Munich with approval of the ethics committee of the LMU Munich University Hospital. For each sample, all FFPE blocks were examined in H&E and EvG stained and scanned slides. CD68 and SMA immunohistochemistry was performed following the automated, diagnostic standard protocol on BenchMark ULTRA IHC/ISH System (Roche®). In brief, for CD68 staining tissue was pre-digested with Protease 1 (Roche®) for 8 minutes. Mouse anti-human CD68 antibody (clone KP1, Thermo®) was diluted 1:100 and incubated at RT for 12 minutes. For SMA staining, mouse anti-human SMA antibody (clone 1A4, Zytomed®) was diluted 1:100 and incubated at RT for 28 minutes without any pre-treatment. Both primary antibodies were detected using the Ultraview (Roche®) system according to manufacturer protocol. Atherosclerosis was scored by an experienced surgical pathologist along a three-stage scoring-system: subtle, medium and pronounced atherosclerotic change. For plaque analysis, only plaques from cases showing medium and pronounced atherosclerosis were included (n=5 intermediate and n=8 advanced plaques were included) and isolated for further image analysis using the “cut“ function of ImageJ. Ulcerated plaques were excluded. Plaque analysis was performed by ImageJ. For every plaque, the total plaque area, the CD68+ area and the SMA+ area was determined. In addition, the necrotic core area was analyzed in the H&E stained images. Fibrous cap proximity was determined as the top 30% of the surface of each plaque diameter (calculated individually for every plaque diameter at the broadest point of the lesion). The plaque vulnerability index was calculated as VPI = (% Necrotic core area + % CD68 area) / (% SMA area), adapted from.133
Whole-mount ex vivo staining of the ear skin
The ear was fixed with PFA for 30 minutes and subsequently incubated in 2% FBS and 10% goat serum in PBS for 60 minutes at room temperature. Subsequently, the ear was incubated with rat monoclonal antibody anti mouse F4/80, Clone BM8, eBioscience and Armenian hamster anti mouse CD11c, Clone N418, Thermofisher Scientific. Subsequently, the tissue was incubated for 75 min at 37 °C on a Thermomixer (Thermomixer comfort, eppendorf, Hamburg). Afterwards, tissue was washed with 1 % Tween in PBS. Afterwards, the secondary antobodies (Goat anti rat AlexaFluor 555, Invitrogen ®, Thermofisher Scientific and Goat anti-armenian hamster AlexaFluor 647, abcam ®) were added and incubated again for 40 minutes at 37 °C. Afterwards, HOECHST was added for 30 minutes, washed twice with 1% Tween in PBS, mounted and subsequently stored at 4 °C until further imaging with a Laser Scanning Microscope LSM 880 with an Airyscan module.
Western blot
270.000 HCASMC (human coronary artery smooth muscle cells) were seeded in a 6-Well chamber and left at 37°C and 5% CO2 to settle in Smooth Muscle Cell Growth Medium, #C-22062, PromoCell ®. At 80% confluency, HCASMS were stimulated with TNFa, #570102, Biolegend ®, 100ng/ml or control. Subsequently, cells were incubated for 8h in the incubator. Afterwards, cells were washed with HBSS 3x, trypsinized, neutralized with TNS and centrifuged at 200xg. After one more washing step, the cell pellet was frozen at -80°C. For Western Blot analyses, cells were lysed in RIPA-buffer instantly after thawing. For Western Blot, NuPAGE 4-12% Bis-Tris Gel, #NP0321, Invitrogen was used as a Gel, NUPAGE MES SDS RUNN.B.500ML, #NP0002, Invitrogen as a buffer, ThermoFisher ®, #26634, Spectra Multicolor Broad Range Protein Ladder 2x250μl as a Marker, 5% Milk powder in TBS-T was used for blocking. Anti-GAPDH (Abcam 9484), Anti-MIF (Abcam 7207), Anti-MCP1 (Abcam 25124) showing cross species reactivity were used.
Myh11-CreERT2; PCG5-tdT tissue staining
n=3 Myh11-Cre-ERT2; PC-G5-tdT mice were injected with tamoxifen 2mg/day/mouse (Sigma-Aldrich ®, 10540-29-1) in 5 consecutive days, mice rested for another 5 days prior to organ harvest. Sections from valve, brachiocephalic artery, and aortic arch were fixed with 4% methanol-free formaldehyde and blocked with 10% goat serum, 1% BSA and 0.5% saponin. Slides were stained with ACTA2 (ab125057, abcam ®) biotin-conjugated antibody and the corresponding streptavidin AlexaFluor 647 conjugated (405237, BioLegend).
In vitro MΦ CCL2 stimulation and Western blot
Peritoneal MΦs were isolated from C57BL/6 mice using the Mouse peritoneal MΦ Isolation Kit (Milteny Biotec ®) according to the manufacturer’s instructions. Following isolation, MΦs were resuspended in RPMI media containing penicillin and streptomycin and allowed to settle on uncoated 6 well plates for two hours. Cells were washed once with fresh RPMI and then incubated with either phosphate buffered saline (PBS) or recombinant mouse CCL2 (Biolegend ®, 200 ng/ml) for 24 hours at 37°C, 5% CO2. Hereafter, cells were washed once with PBS and then dissociated from the plate using Trypsin-EDTA (0.25%, Gibco ®). Cells were spun down at 4°C, 400 G, for 7 min and the resulting pellets were snap-frozen using dry ice. Western blotting was performed as previously described.134 Briefly, pellets were lysed in the presence of proteinase inhibitors using RIPA Lysis and Extraction Buffer (ThermoFisher ®), followed by protein quantification. Per sample, the same amount of protein was loaded per lane. Levels of CD31 (abcam ®, ab222783), CD36 (abcam ®, ab124515) and FOLR2 (abcam ®, ab228643) as well as beta-actin, which was used as housekeepking gene, were identified using primary antibodies and visualized using horse raddish peroxidase (HRP)-coupled secondary antibodies and chemiluminescent agents. Intensities were assessed through densitometry, which was performed using the Fiji ImageJ gel analysis tool.
SMC-MΦ coculture
MΦs were isolated intraperitoneally from C57BL/6 mice. Isolated cells were MACS sorted (130-110-434, Miltenyi Biotec) to ensure macrophage purity. Murine SMCs (PB-A57-6080, CellBiologics) or C57BL/6 macrophages were labelled with CellTracker Green (CMFDA) (C2925, Invitrogen) prior to seeding. All cells were seeded at timepoint 0h containing 104 SMCs or 5∗104 MΦs per well. Cells were cultured for 96h. All cells were cultured in SMC full Medium (PELOBiotech® PB-MH-200-2190).
SMC and MΦ coincubation
MΦs were isolated intraperitoneally from C57BL/6 mice. Isolated cells were MACS sorted for MΦs (130-110-434, Miltenyi Biotec ®). Murine SMCs were labelled with CellTracker red (CMTPX) (C34552, Invitrogen ®), and MΦs were labelled with CellTracker Green (CMFDA) (C2925, Invitrogen ®) prior to culture. Labelled cells were cultured in SMC full Medium (PELOBiotech® PB-MH-200-2190); measurements were taken over the course of 72 hours. MIF inhibitor 1600 μM (ISO-1 HY-16692, MedChemExpress), CCL2 specific monoclonal antibody 20 μg/ml (MCP-1 16-7096-85, eBioscience) and IgG isotype control 20 μg/ml (16-4888-85, Invitrogen ®) reagents were supplemented to the different conditions as described. Image analysis was carried out in FIJI (ImageJ).
Acute short-term pharmacological CCL2 inhibition
ApoE-/- mice were fed a Western diet for 6 months. Anti-CCL2 (Bio X Sell®; BE0185) or Isotype control antibody (Bio X Sell®; BE0091) were injected i.v. in a concentration of 200 μg in 150 μl PBS every 48h (6 injections in total per mouse). In addition, EdU was injected i.p. as described previously. The aortic valves and brachiocephalic arteries were harvested, embedded and sectioned as described before. The sections were stained with the following primary antibodies: ACTA2 (F3777, Sigma®) and LGALS3 (CL8942AP, Cedarlane®). Apoptosis staining was analysed by TUNEL staining. (S7165, ApopTag® Red In Situ Apoptosis Detection Kit, Sigma-Aldrich®). Nuclei were stained with Hoechst. Histological images were acquired using a Leica DM6B microscope. Image analysis was performed in a standardized manner by colour thresholding with ImageJ software.
Quantification and statistical analysis
All data are shown as mean and s.e.m., unless indicated otherwise. Values were tested for normal distribution and t-test (two-tailed if not specified otherwise) or Mann-Whitney test were performed to compare groups as indicated, if not specified otherwise. Graphs tested for statistical significance without a depicted p-value showed no significant difference. Comparisons between more than two groups were made using ANOVA followed by LSD-post hoc-test, unless further specified. If not further specified, repeated measures in individual samples across two groups were compared by repeated measures two-way ANOVA or mixed-effects model with Geisser-Greenhouse correction was used. A value of P<0.05 was considered significant.
Acknowledgments
We thank Nicole Blount, Beate Jantz, and Sebastian Helmer for excellent technical assistance and Marco E. Bianchi (San Raffaele University and Scientific Institute, Milan, Italy) for providing HMGB1 redox forms. The graphical abstract was created with biorender.com.
This study was supported by the Deutsche Forschungsgemeinschaft—collaborative research center 1123 project A07 (K.S. and C.S.), A01, A10 (C.W.), B06 (S.M., L.N.), Z02 (R.Z., W.E.); collaborative research center 914 project B02 (K.S. and S.M.); collaborative research center 359 project A03 (K.S.) and B09 (C.S.); the DFG Clinician Scientist Program PRIME (413635475) (K.P.); LMUexcellent (K.S., K.P.); the DZHK (K.S., K.P., and S.M.); the European Research Council (ERC) (T-MEMORE); grant agreement no. 947611 (K.S.); the Deutsche Herzstiftung e.V. (K.P., K.S.); the Else Kröner-Fresenius-Stiftung (K.P.); and the Fondation Leducq (S.M.). P.T. receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892 and 1R01HL163099-01), the RRM Charitable Fund, and the Simard Fund.
Author contributions
Conception, experimental design, project administration, and supervision, K.P. and K.S.; methodology, investigation, and formal analysis, K.P., C.G., P.H., A.E., M.J., A.J., F.M., V.P., L.E., M. Knott, M. Kirschner, R.C., A.B., A.T., B.K., M.L., G.F.-R., R.B., K.S., L.T., R.K., and F.D.; visualization: K.P., C.G., M.J., and F.M.; writing original draft, K.P. and K.S.; writing & editing, all authors; funding acquisition, K.P., W.E., R.Z., C.S., C.W., P.L., S.M., and K.S.
Declaration of interests
P.L. is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Moderna, Norvo Nordisk, Novartis, Pfizer, and Sanofi-Regeneron. P.L. is a member of the scientific advisory board for Amgen, Caristo Diagnostics, Cartesian Therapeutics, CSL Behring, DalCor Pharmaceuticals, Eulicid Bioimaging, Kancera, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, Dewpoint, Plaque Tec, PlaqueTec, TenSixteen Bio, Soley Thereapeutics, and XBiotech, Inc.
P.L.'s laboratory has received research funding in the last 2 years from Novartis, Novo Nordisk, and Genentech. P.L. is on the Board of Directors of XBiotech, Inc. P.L. has a financial interest in Xbiotech (a company developing therapeutic human antibodies), in TenSixteen Bio (a company targeting somatic mosaicism and clonal hematopoiesis of indeterminate potential [CHIP] to discover and develop novel therapeutics to treat age-related diseases), and in Soley Therapeutics (a biotechnology company that is combining artificial intelligence with molecular and cellular response detection for discovering and developing new drugs, currently focusing on cancer therapeutics). P.L.'s interests were reviewed and are managed by Brigham and Women’s Hospital and Mass General Brigham in accordance with their conflict-of-interest policies.
Published: August 30, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2023.08.002.
Contributor Information
Kami Pekayvaz, Email: kami.pekayvaz@med.uni-muenchen.de.
Konstantin Stark, Email: konstantin.stark@med.uni-muenchen.de.
Supplemental information
Data and code availability
Count data for RNA-seq experiments will be made available (Zenodo: https://doi.org/10.5281/zenodo.8065731) after publication. Other data is available upon request from the corresponding authors. All original code has been deposited at Zenodo and will be publicly available.
References
- 1.Blériot C., Chakarov S., Ginhoux F. Determinants of resident tissue macrophage identity and function. Immunity. 2020;52:957–970. doi: 10.1016/j.immuni.2020.05.014. [DOI] [PubMed] [Google Scholar]
- 2.Guilliams M., Thierry G.R., Bonnardel J., Bajenoff M. Establishment and maintenance of the macrophage niche. Immunity. 2020;52:434–451. doi: 10.1016/j.immuni.2020.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Park M.D., Silvin A., Ginhoux F., Merad M. Macrophages in health and disease. Cell. 2022;185:4259–4279. doi: 10.1016/j.cell.2022.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koelwyn G.J., Corr E.M., Erbay E., Moore K.J. Regulation of macrophage immunometabolism in atherosclerosis. Nat. Immunol. 2018;19:526–537. doi: 10.1038/s41590-018-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wynn T.A., Vannella K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore K.J., Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524–533. doi: 10.1038/s41586-021-03392-8. [DOI] [PubMed] [Google Scholar]
- 8.Newton K., Dixit V.M., Kayagaki N. Dying cells fan the flames of inflammation. Science. 2021;374:1076–1080. doi: 10.1126/science.abi5934. [DOI] [PubMed] [Google Scholar]
- 9.Chakarov S., Lim H.Y., Tan L., Lim S.Y., See P., Lum J., Zhang X.M., Foo S., Nakamizo S., Duan K., et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019;363 doi: 10.1126/science.aau0964. [DOI] [PubMed] [Google Scholar]
- 10.Lapenna A., De Palma M., Lewis C.E. Perivascular macrophages in health and disease. Nat. Rev. Immunol. 2018;18:689–702. doi: 10.1038/s41577-018-0056-9. [DOI] [PubMed] [Google Scholar]
- 11.Williams J.W., Zaitsev K., Kim K.W., Ivanov S., Saunders B.T., Schrank P.R., Kim K., Elvington A., Kim S.H., Tucker C.G., et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 2020;21:1194–1204. doi: 10.1038/s41590-020-0768-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim H.Y., Lim S.Y., Tan C.K., Thiam C.H., Goh C.C., Carbajo D., Chew S.H.S., See P., Chakarov S., Wang X.N., et al. Hyaluronan receptor LYVE-1-Expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity. 2018;49:326–341.e7. doi: 10.1016/j.immuni.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Weber C., Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 14.Tabas I., Glass C.K. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Björkegren J.L.M., Lusis A.J. Atherosclerosis: recent developments. Cell. 2022;185:1630–1645. doi: 10.1016/j.cell.2022.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams J.W., Huang L.H., Randolph G.J. Cytokine circuits in cardiovascular disease. Immunity. 2019;50:941–954. doi: 10.1016/j.immuni.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi C., Pamer E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proebstl D., Voisin M.B., Woodfin A., Whiteford J., D’Acquisto F., Jones G.E., Rowe D., Nourshargh S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J. Exp. Med. 2012;209:1219–1234. doi: 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stark K., Eckart A., Haidari S., Tirniceriu A., Lorenz M., von Brühl M.L., Gärtner F., Khandoga A.G., Legate K.R., Pless R., et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and 'instruct' them with pattern-recognition and motility programs. Nat. Immunol. 2013;14:41–51. doi: 10.1038/ni.2477. [DOI] [PubMed] [Google Scholar]
- 20.Stark K., Pekayvaz K., Massberg S. Role of pericytes in vascular immunosurveillance. Front. Biosci. (Landmark Ed.) 2018;23:767–781. doi: 10.2741/4615. [DOI] [PubMed] [Google Scholar]
- 21.Yona S., Kim K.W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgess M., Wicks K., Gardasevic M., Mace K.A. Cx3CR1 expression identifies distinct macrophage populations that contribute differentially to inflammation and repair. Immunohorizons. 2019;3:262–273. doi: 10.4049/immunohorizons.1900038. [DOI] [PubMed] [Google Scholar]
- 23.Uderhardt S., Martins A.J., Tsang J.S., Lämmermann T., Germain R.N. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell. 2019;177:541–555.e17. doi: 10.1016/j.cell.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehari F.T., Miller M., Pick R., Bader A., Pekayvaz K., Napoli M., Uhl B., Reichel C.A., Sperandio M., Walzog B., et al. Intravital calcium imaging in myeloid leukocytes identifies calcium frequency spectra as indicators of functional states. Sci. Signal. 2022;15 doi: 10.1126/scisignal.abe6909. [DOI] [PubMed] [Google Scholar]
- 25.Ensan S., Li A., Besla R., Degousee N., Cosme J., Roufaiel M., Shikatani E.A., El-Maklizi M., Williams J.W., Robins L., et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat. Immunol. 2016;17:159–168. doi: 10.1038/ni.3343. [DOI] [PubMed] [Google Scholar]
- 26.Hoyer F.F., Naxerova K., Schloss M.J., Hulsmans M., Nair A.V., Dutta P., Calcagno D.M., Herisson F., Anzai A., Sun Y., et al. Tissue-specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity. 2019;51:899–914.e7. doi: 10.1016/j.immuni.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunisaki Y., Bruns I., Scheiermann C., Ahmed J., Pinho S., Zhang D., Mizoguchi T., Wei Q., Lucas D., Ito K., et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–643. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piedrahita J.A., Zhang S.H., Hagaman J.R., Oliver P.M., Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc. Natl. Acad. Sci. USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.She Z.G., Chang Y., Pang H.B., Han W., Chen H.Z., Smith J.W., Stallcup W.B. NG2 proteoglycan ablation reduces foam cell formation and atherogenesis via decreased low-density lipoprotein retention by synthetic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2016;36:49–59. doi: 10.1161/ATVBAHA.115.306074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanlandewijck M., He L., Mäe M.A., Andrae J., Ando K., Del Gaudio F., Nahar K., Lebouvier T., Laviña B., Gouveia L., et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554:475–480. doi: 10.1038/nature25739. [DOI] [PubMed] [Google Scholar]
- 31.Wang J.M., Sica A., Peri G., Walter S., Padura I.M., Libby P., Ceska M., Lindley I., Colotta F., Mantovani A. Expression of monocyte chemotactic protein and interleukin-8 by cytokine-activated human vascular smooth muscle cells. Arterioscler. Thromb. 1991;11:1166–1174. doi: 10.1161/01.atv.11.5.1166. [DOI] [PubMed] [Google Scholar]
- 32.Wirka R.C., Wagh D., Paik D.T., Pjanic M., Nguyen T., Miller C.L., Kundu R., Nagao M., Coller J., Koyano T.K., et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019;25:1280–1289. doi: 10.1038/s41591-019-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hasegawa T., Venkata Suresh V., Yahata Y., Nakano M., Suzuki S., Suzuki S., Yamada S., Kitaura H., Mizoguchi I., Noiri Y., et al. Inhibition of the CXCL9-CXCR3 axis suppresses the progression of experimental apical periodontitis by blocking macrophage migration and activation. Sci. Rep. 2021;11:2613. doi: 10.1038/s41598-021-82167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimmerman K.A., Bentley M.R., Lever J.M., Li Z., Crossman D.K., Song C.J., Liu S., Crowley M.R., George J.F., Mrug M., et al. Single-cell RNA sequencing identifies candidate renal resident macrophage gene expression signatures across species. J. Am. Soc. Nephrol. 2019;30:767–781. doi: 10.1681/ASN.2018090931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schyns J., Bai Q., Ruscitti C., Radermecker C., De Schepper S., Chakarov S., Farnir F., Pirottin D., Ginhoux F., Boeckxstaens G., et al. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat. Commun. 2019;10:3964. doi: 10.1038/s41467-019-11843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stamatiades E.G., Tremblay M.E., Bohm M., Crozet L., Bisht K., Kao D., Coelho C., Fan X., Yewdell W.T., Davidson A., et al. Immune monitoring of trans-endothelial transport by kidney-resident macrophages. Cell. 2016;166:991–1003. doi: 10.1016/j.cell.2016.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Byrne A.J., Maher T.M., Lloyd C.M. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol. Med. 2016;22:303–316. doi: 10.1016/j.molmed.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Sha H., Zhang D., Zhang Y., Wen Y., Wang Y. ATF3 promotes migration and M1/M2 polarization of macrophages by activating tenascin-C via Wnt/β-catenin pathway. Mol. Med. Rep. 2017;16:3641–3647. doi: 10.3892/mmr.2017.6992. [DOI] [PubMed] [Google Scholar]
- 39.Hammer M., Mages J., Dietrich H., Servatius A., Howells N., Cato A.C., Lang R. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J. Exp. Med. 2006;203:15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ray N., Kuwahara M., Takada Y., Maruyama K., Kawaguchi T., Tsubone H., Ishikawa H., Matsuo K. c-Fos suppresses systemic inflammatory response to endotoxin. Int. Immunol. 2006;18:671–677. doi: 10.1093/intimm/dxl004. [DOI] [PubMed] [Google Scholar]
- 41.Troiani T., Giunta E.F., Tufano M., Vigorito V., Arrigo P.D., Argenziano G., Ciardiello F., Romano M.F., Romano S. Alternative macrophage polarisation associated with resistance to anti-PD1 blockade is possibly supported by the splicing of FKBP51 immunophilin in melanoma patients. Br. J. Cancer. 2020;122:1782–1790. doi: 10.1038/s41416-020-0840-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ip W.K.E., Hoshi N., Shouval D.S., Snapper S., Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356:513–519. doi: 10.1126/science.aal3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruffell D., Mourkioti F., Gambardella A., Kirstetter P., Lopez R.G., Rosenthal N., Nerlov C. A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA. 2009;106:17475–17480. doi: 10.1073/pnas.0908641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fontana M.F., Baccarella A., Pancholi N., Pufall M.A., Herbert D.R., Kim C.C. JUNB is a key transcriptional modulator of macrophage activation. J. Immunol. 2015;194:177–186. doi: 10.4049/jimmunol.1401595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yi Y.S., Son Y.J., Ryou C., Sung G.H., Kim J.H., Cho J.Y. Functional roles of Syk in macrophage-mediated inflammatory responses. Mediators Inflamm. 2014;2014:270302. doi: 10.1155/2014/270302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langlais D., Barreiro L.B., Gros P. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med. 2016;213:585–603. doi: 10.1084/jem.20151764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fabisik M., Tureckova J., Pavliuchenko N., Kralova J., Balounova J., Vicikova K., Skopcova T., Spoutil F., Pokorna J., Angelisova P., et al. Regulation of inflammatory response by transmembrane adaptor protein LST1. Front. Immunol. 2021;12:618332. doi: 10.3389/fimmu.2021.618332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scott C.L., T'Jonck W., Martens L., Todorov H., Sichien D., Soen B., Bonnardel J., De Prijck S., Vandamme N., Cannoodt R., et al. The transcription factor ZEB2 is required to maintain the tissue-specific identities of macrophages. Immunity. 2018;49:312–325.e5. doi: 10.1016/j.immuni.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doran A.C., Yurdagul A., Tabas I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020;20:254–267. doi: 10.1038/s41577-019-0240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferracini M., Rios F.J., Pecenin M., Jancar S. Clearance of apoptotic cells by macrophages induces regulatory phenotype and involves stimulation of CD36 and platelet-activating factor receptor. Mediators Inflamm. 2013;2013:950273. doi: 10.1155/2013/950273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orecchioni M., Ghosheh Y., Pramod A.B., Ley K. Macrophage Polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS–) vs. Alternatively Activated Macrophages. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puig-Kröger A., Sierra-Filardi E., Domínguez-Soto A., Samaniego R., Corcuera M.T., Gómez-Aguado F., Ratnam M., Sánchez-Mateos P., Corbí A.L. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009;69:9395–9403. doi: 10.1158/0008-5472.CAN-09-2050. [DOI] [PubMed] [Google Scholar]
- 53.Clément M., Basatemur G., Masters L., Baker L., Bruneval P., Iwawaki T., Kneilling M., Yamasaki S., Goodall J., Mallat Z. Necrotic cell sensor Clec4e promotes a proatherogenic macrophage phenotype through activation of the unfolded protein response. Circulation. 2016;134:1039–1051. doi: 10.1161/CIRCULATIONAHA.116.022668. [DOI] [PubMed] [Google Scholar]
- 54.Vernon-Wilson E.F., Auradé F., Brown S.B. CD31 promotes beta1 integrin-dependent engulfment of apoptotic Jurkat T lymphocytes opsonized for phagocytosis by fibronectin. J. Leukoc. Biol. 2006;79:1260–1267. doi: 10.1189/jlb.1005571. [DOI] [PubMed] [Google Scholar]
- 55.Furuhashi M., Tuncman G., Görgün C.Z., Makowski L., Atsumi G., Vaillancourt E., Kono K., Babaev V.R., Fazio S., Linton M.F., et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447:959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shankman L.S., Gomez D., Cherepanova O.A., Salmon M., Alencar G.F., Haskins R.M., Swiatlowska P., Newman A.A., Greene E.S., Straub A.C., et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015;21:628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miyoshi T., Tian J., Matsumoto A.H., Shi W. Differential response of vascular smooth muscle cells to oxidized LDL in mouse strains with different atherosclerosis susceptibility. Atherosclerosis. 2006;189:99–105. doi: 10.1016/j.atherosclerosis.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 58.Fidler T.P., Xue C., Yalcinkaya M., Hardaway B., Abramowicz S., Xiao T., Liu W., Thomas D.G., Hajebrahimi M.A., Pircher J., et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592:296–301. doi: 10.1038/s41586-021-03341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marx C., Novotny J., Salbeck D., Zellner K.R., Nicolai L., Pekayvaz K., Kilani B., Stockhausen S., Bürgener N., Kupka D., et al. Eosinophil-platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood. 2019;134:1859–1872. doi: 10.1182/blood.2019000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McArdle S., Buscher K., Ghosheh Y., Pramod A.B., Miller J., Winkels H., Wolf D., Ley K. Migratory and dancing macrophage subsets in atherosclerotic lesions. Circ. Res. 2019;125:1038–1051. doi: 10.1161/CIRCRESAHA.119.315175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Živković L., Asare Y., Bernhagen J., Dichgans M., Georgakis M.K. 2021. CCL2/CCR2 inhibition in atherosclerosis: a meta-analysis of preclinical studies. Preprint at bioRxiv. [DOI] [Google Scholar]
- 62.Lutgens E., Faber B., Schapira K., Evelo C.T., van Haaften R., Heeneman S., Cleutjens K.B., Bijnens A.P., Beckers L., Porter J.G., et al. Gene profiling in atherosclerosis reveals a key role for small inducible cytokines: validation using a novel monocyte chemoattractant protein monoclonal antibody. Circulation. 2005;111:3443–3452. doi: 10.1161/CIRCULATIONAHA.104.510073. [DOI] [PubMed] [Google Scholar]
- 63.Cynis H., Hoffmann T., Friedrich D., Kehlen A., Gans K., Kleinschmidt M., Rahfeld J.U., Wolf R., Wermann M., Stephan A., et al. The isoenzyme of glutaminyl cyclase is an important regulator of monocyte infiltration under inflammatory conditions. EMBO Mol. Med. 2011;3:545–558. doi: 10.1002/emmm.201100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winter C., Silvestre-Roig C., Ortega-Gomez A., Lemnitzer P., Poelman H., Schumski A., Winter J., Drechsler M., de Jong R., Immler R., et al. Chrono-pharmacological targeting of the CCL2-CCR2 axis ameliorates atherosclerosis. Cell Metab. 2018;28:175–182.e5. doi: 10.1016/j.cmet.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 65.Colombo A., Basavarajaiah S., Limbruno U., Picchi A., Lettieri C., Valgimigli M., Sciahbasi A., Prati F., Calabresi M., Pierucci D., et al. A double-blind randomised study to evaluate the efficacy and safety of Bindarit in preventing coronary stent restenosis. EuroIntervention. 2016;12 doi: 10.4244/EIJY15M12_03. e1385–e1394. [DOI] [PubMed] [Google Scholar]
- 66.Robbins C.S., Hilgendorf I., Weber G.F., Theurl I., Iwamoto Y., Figueiredo J.L., Gorbatov R., Sukhova G.K., Gerhardt L.M., Smyth D., et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013;19:1166–1172. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gomez Perdiguero E., Klapproth K., Schulz C., Busch K., Azzoni E., Crozet L., Garner H., Trouillet C., de Bruijn M.F., Geissmann F., et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weinberger T., Esfandyari D., Messerer D., Percin G., Schleifer C., Thaler R., Liu L., Stremmel C., Schneider V., Vagnozzi R.J., et al. Ontogeny of arterial macrophages defines their functions in homeostasis and inflammation. Nat. Commun. 2020;11:4549. doi: 10.1038/s41467-020-18287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schulz C., Gomez Perdiguero E., Chorro L., Szabo-Rogers H., Cagnard N., Kierdorf K., Prinz M., Wu B., Jacobsen S.E., Pollard J.W., et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 70.Stremmel C., Stark K., Schulz C. Heterogeneity of macrophages in atherosclerosis. Thromb. Haemost. 2019;119:1237–1246. doi: 10.1055/s-0039-1692665. [DOI] [PubMed] [Google Scholar]
- 71.McAlpine C.S., Kiss M.G., Rattik S., He S., Vassalli A., Valet C., Anzai A., Chan C.T., Mindur J.E., Kahles F., et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature. 2019;566:383–387. doi: 10.1038/s41586-019-0948-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schumski A., Ortega-Gómez A., Wichapong K., Winter C., Lemnitzer P., Viola J.R., Pinilla-Vera M., Folco E., Solis-Mezarino V., Völker-Albert M., et al. Endotoxinemia accelerates atherosclerosis through electrostatic charge-mediated monocyte adhesion. Circulation. 2021;143:254–266. doi: 10.1161/CIRCULATIONAHA.120.046677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fuster J.J., MacLauchlan S., Zuriaga M.A., Polackal M.N., Ostriker A.C., Chakraborty R., Wu C.L., Sano S., Muralidharan S., Rius C., et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jordan S., Tung N., Casanova-Acebes M., Chang C., Cantoni C., Zhang D., Wirtz T.H., Naik S., Rose S.A., Brocker C.N., et al. Dietary intake regulates the circulating inflammatory monocyte pool. Cell. 2019;178:1102–1114.e17. doi: 10.1016/j.cell.2019.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Serbina N.V., Pamer E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 76.Georgakis M.K., Malik R., Björkbacka H., Pana T.A., Demissie S., Ayers C., Elhadad M.A., Fornage M., Beiser A.S., Benjamin E.J., et al. Circulating monocyte chemoattractant Protein-1 and risk of stroke: meta-analysis of population-based studies involving 17 180 individuals. Circ. Res. 2019;125:773–782. doi: 10.1161/CIRCRESAHA.119.315380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boring L., Gosling J., Cleary M., Charo I.F. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 78.Aiello R.J., Bourassa P.A., Lindsey S., Weng W., Natoli E., Rollins B.J., Milos P.M. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1999;19:1518–1525. doi: 10.1161/01.atv.19.6.1518. [DOI] [PubMed] [Google Scholar]
- 79.Combadière C., Potteaux S., Rodero M., Simon T., Pezard A., Esposito B., Merval R., Proudfoot A., Tedgui A., Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 80.Dawson T.C., Kuziel W.A., Osahar T.A., Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–211. doi: 10.1016/s0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 81.Gu L., Okada Y., Clinton S.K., Gerard C., Sukhova G.K., Libby P., Rollins B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 82.He J., Song Y., Li G., Xiao P., Liu Y., Xue Y., Cao Q., Tu X., Pan T., Jiang Z., et al. Fbxw7 increases CCL2/7 in CX3CR1hi macrophages to promote intestinal inflammation. J. Clin. Invest. 2019;129:3877–3893. doi: 10.1172/JCI123374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Inoue S., Egashira K., Ni W., Kitamoto S., Usui M., Otani K., Ishibashi M., Hiasa K., Nishida K., Takeshita A. Anti-monocyte chemoattractant protein-1 gene therapy limits progression and destabilization of established atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2002;106:2700–2706. doi: 10.1161/01.cir.0000038140.80105.ad. [DOI] [PubMed] [Google Scholar]
- 84.Lim S.Y., Yuzhalin A.E., Gordon-Weeks A.N., Muschel R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. 2016;7:28697–28710. doi: 10.18632/oncotarget.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rose C.E., Jr., Sung S.S., Fu S.M. Significant involvement of CCL2 (MCP-1) in inflammatory disorders of the lung. Microcirculation. 2003;10:273–288. doi: 10.1038/sj.mn.7800193. [DOI] [PubMed] [Google Scholar]
- 86.Burger-Kentischer A., Goebel H., Seiler R., Fraedrich G., Schaefer H.E., Dimmeler S., Kleemann R., Bernhagen J., Ihling C. Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation. 2002;105:1561–1566. doi: 10.1161/01.cir.0000012942.49244.82. [DOI] [PubMed] [Google Scholar]
- 87.Kang I., Bucala R. The immunobiology of MIF: function, genetics and prospects for precision medicine. Nat. Rev. Rheumatol. 2019;15:427–437. doi: 10.1038/s41584-019-0238-2. [DOI] [PubMed] [Google Scholar]
- 88.Bernhagen J., Krohn R., Lue H., Gregory J.L., Zernecke A., Koenen R.R., Dewor M., Georgiev I., Schober A., Leng L., et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007;13:587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 89.Pan J.H., Sukhova G.K., Yang J.T., Wang B., Xie T., Fu H., Zhang Y., Satoskar A.R., David J.R., Metz C.N., et al. Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2004;109:3149–3153. doi: 10.1161/01.CIR.0000134704.84454.D2. [DOI] [PubMed] [Google Scholar]
- 90.Sponaas A.-M., Freitas do Rosario A.P., Voisine C., Mastelic B., Thompson J., Koernig S., Jarra W., Renia L., Mauduit M., Potocnik A.J., et al. Migrating monocytes recruited to the spleen play an important role in control of blood stage malaria. Blood. 2009;114:5522–5531. doi: 10.1182/blood-2009-04-217489. [DOI] [PubMed] [Google Scholar]
- 91.Bronte V., Pittet M.J. The spleen in local and systemic regulation of immunity. Immunity. 2013;39:806–818. doi: 10.1016/j.immuni.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kurihara T., Warr G., Loy J., Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kuziel W.A., Morgan S.J., Dawson T.C., Griffin S., Smithies O., Ley K., Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl. Acad. Sci. USA. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gschwandtner M., Derler R., Midwood K.S. More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front. Immunol. 2019;10:2759. doi: 10.3389/fimmu.2019.02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hoffmann A., Zwißler L.C., El Bounkari O., Bernhagen J. Studying the pro-migratory effects of MIF. Methods Mol. Biol. 2020;2080:1–18. doi: 10.1007/978-1-4939-9936-1_1. [DOI] [PubMed] [Google Scholar]
- 96.Xuan W., Qu Q., Zheng B., Xiong S., Fan G.H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J. Leukoc. Biol. 2015;97:61–69. doi: 10.1189/jlb.1A0314-170R. [DOI] [PubMed] [Google Scholar]
- 97.Sierra-Filardi E., Nieto C., Domínguez-Soto A., Barroso R., Sánchez-Mateos P., Puig-Kroger A., López-Bravo M., Joven J., Ardavín C., Rodríguez-Fernández J.L., et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J. Immunol. 2014;192:3858–3867. doi: 10.4049/jimmunol.1302821. [DOI] [PubMed] [Google Scholar]
- 98.Newman A.A.C., Serbulea V., Baylis R.A., Shankman L.S., Bradley X., Alencar G.F., Owsiany K., Deaton R.A., Karnewar S., Shamsuzzaman S., et al. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by PDGFRβ and bioenergetic mechanisms. Nat. Metab. 2021;3:166–181. doi: 10.1038/s42255-020-00338-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alencar G.F., Owsiany K.M., Karnewar S., Sukhavasi K., Mocci G., Nguyen A.T., Williams C.M., Shamsuzzaman S., Mokry M., Henderson C.A., et al. Stem cell pluripotency genes Klf4 and Oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation. 2020;142:2045–2059. doi: 10.1161/CIRCULATIONAHA.120.046672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cherepanova O.A., Gomez D., Shankman L.S., Swiatlowska P., Williams J., Sarmento O.F., Alencar G.F., Hess D.L., Bevard M.H., Greene E.S., et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat. Med. 2016;22:657–665. doi: 10.1038/nm.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Owsiany K.M., Deaton R.A., Soohoo K.G., Tram Nguyen A., Owens G.K. Dichotomous roles of smooth muscle cell-derived MCP1 (monocyte chemoattractant protein 1) in development of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2022;42:942–956. doi: 10.1161/ATVBAHA.122.317882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fingerle-Rowson G., Petrenko O., Metz C.N., Forsthuber T.G., Mitchell R., Huss R., Moll U., Müller W., Bucala R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc. Natl. Acad. Sci. USA. 2003;100:9354–9359. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Andrews S. Babraham Bioinformatics; 2010. FastQC: a quality control tool for high throughput sequence data.http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- 104.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. j. 2011;17:3. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 105.Parekh S., Ziegenhain C., Vieth B., Enard W., Hellmann I. zUMIs - a fast and flexible pipeline to process RNA sequencing data with UMIs. GigaScience. 2018;7 doi: 10.1093/gigascience/giy059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 110.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 113.Metsalu T., Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015;43:W566–W570. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bankhead P., Loughrey M.B., Fernández J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G., et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7:16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wirth A., Benyó Z., Lukasova M., Leutgeb B., Wettschureck N., Gorbey S., Orsy P., Horváth B., Maser-Gluth C., Greiner E., et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 119.Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R., et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu X., Bergles D.E., Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]
- 121.LeBleu V.S., Taduri G., O'Connell J., Teng Y., Cooke V.G., Woda C., Sugimoto H., Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shi C., Jia T., Mendez-Ferrer S., Hohl T.M., Serbina N.V., LiPuma L., Leiner I., Li M.O., Frenette P.S., Pamer E.G. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34:590–601. doi: 10.1016/j.immuni.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gee J.M., Smith N.A., Fernandez F.R., Economo M.N., Brunert D., Rothermel M., Morris S.C., Talbot A., Palumbos S., Ichida J.M., et al. Imaging activity in neurons and glia with a Polr2a-based and cre-dependent GCaMP5G-IRES-tdTomato reporter mouse. Neuron. 2014;83:1058–1072. doi: 10.1016/j.neuron.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Littman D.R. An inducible Cre recombinase driven by Cx3cr1. MGI Direct Data Submission J:190965. 2013 https://www.informatics.jax.org/allele/reference/J:190965 [Google Scholar]
- 125.Jung S., Aliberti J., Graemmel P., Sunshine M.J., Kreutzberg G.W., Sher A., Littman D.R. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000;20:4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Faust N., Varas F., Kelly L.M., Heck S., Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96:719–726. [PubMed] [Google Scholar]
- 127.Elhag S., Stremmel C., Zehrer A., Plocke J., Hennel R., Keuper M., Knabe C., Winterhalter J., Gölling V., Tomas L., et al. Differences in cell-intrinsic inflammatory programs of yolk sac and bone marrow macrophages. Cells. 2021;10:3564. doi: 10.3390/cells10123564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stirling D.R., Swain-Bowden M.J., Lucas A.M., Carpenter A.E., Cimini B.A., Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics. 2021;22:433. doi: 10.1186/s12859-021-04344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Janjic A., Wange L.E., Bagnoli J.W., Geuder J., Nguyen P., Richter D., Vieth B., Vick B., Jeremias I., Ziegenhain C., et al. Prime-seq, efficient and powerful bulk RNA-sequencing. Genome Biol. 2022;23:88. doi: 10.1186/s13059-022-02660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., III, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bhusal R.P., Eaton J.R.O., Chowdhury S.T., Power C.A., Proudfoot A.E.I., Stone M.J., Bhattacharya S. Evasins: tick salivary proteins that inhibit mammalian chemokines. Trends Biochem. Sci. 2020;45:108–122. doi: 10.1016/j.tibs.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Armingol E., Officer A., Harismendy O., Lewis N.E. Deciphering cell–cell interactions and communication from gene expression. Nat. Rev. Genet. 2021;22:71–88. doi: 10.1038/s41576-020-00292-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Silvestre-Roig C., Braster Q., Wichapong K., Lee E.Y., Teulon J.M., Berrebeh N., Winter J., Adrover J.M., Santos G.S., Froese A., et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. 2019;569:236–240. doi: 10.1038/s41586-019-1167-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kaiser R.W.J., Ignarski M., Van Nostrand E.L., Frese C.K., Jain M., Cukoski S., Heinen H., Schaechter M., Seufert L., Bunte K., et al. A protein-RNA interaction atlas of the ribosome biogenesis factor AATF. Sci. Rep. 2019;9:11071. doi: 10.1038/s41598-019-47552-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vivo multiphoton microscopy of a Cx3cr1-MΦCa-rep mouse ear microvasculature. Laser microinjuries are labelled with broad arrows. Macrophages are shown by dashed lines. In the upper video, MΦs are depicted in red, Ca2+ activity in MΦs is depicted in green, vascular flow is depicted in green. In the lower video, the same segment is rendered in pseudocolor depicting the maximum detected Ca2+ intensity of the depicted MΦ in green (high Ca2+ activity) and blue (low Ca2+ activity). On the right, Ca2+ signals of MΦ 1 and MΦ 2 are aligned over time. Video is paused briefly at instants with high Ca2+ activity within either one of the MΦs. Pseudocolor scale, time scale and scale bar are included in the video.
In vitro epifluorescence microscopy of cultured SMCs from SMCCa2+ Rep mice coincubated either with living Jurkat cell supernatant or dead Jurkat cell supernatant. SMCs are depicted in red, Ca2+ activity is rendered and pseudocolored in green (high Ca2+ activity) and blue (low Ca2+ activity). Pseudocolor scale, time scale and scale bar are included in the video.
In vivo microscopy of an atherosclerotic lesion in a MCRFP-rep Lyz-MΦGFP-rep carotid artery after 14 weeks Western diet. The atherosclerotic lesion is depicted with a continuous line, the box illustrates the shoulder region of the lesion and is further magnified in the middle and right video. The middle video is a 3D rendering of the indicated shoulder region, the video on the right depicts a higher magnification without 3D rendering of the shoulder region. Arrows indicate protrusions of MΦs towards SMCs that continuously form and regress during the video. SMCs are labelled in red, MΦs in green and second harmonic generation (SHG) in blue. Scalebar is 20μm.
In vivo microscopy of an atherosclerotic lesion in a MCRFP-rep Lyz-MΦGFP-rep carotid artery after 14 weeks Western diet. The lesion is depicted with a box. SMCs are labelled in red, MΦs in green and second harmonic generation (SHG) in blue. Scalebar is 50 μm.
Data Availability Statement
Count data for RNA-seq experiments will be made available (Zenodo: https://doi.org/10.5281/zenodo.8065731) after publication. Other data is available upon request from the corresponding authors. All original code has been deposited at Zenodo and will be publicly available.