

Abstract
The abnormal upregulation of programmed death ligand-1 (PD-L1) on tumor cells impedes T-cell mediated cytotoxicity through PD-1 engagement, and further exploring the mechanisms regulation of PD-L1 in cancers may enhance the clinical efficacy of PD-L1 blockade. Here, using single-guide RNAs (sgRNAs) screening system, we identify ubiquitin-specific processing protease 2 (USP2) as a novel regulator of PD-L1 stabilization for tumor immune evasion. USP2 directly interacts with and increases PD-L1 abundance in colorectal and prostate cancer cells. Our results show that Thr288, Arg292 and Asp293 at USP2 control its binding to PD-L1 through deconjugating the K48-linked polyubiquitination at lysine 270 of PD-L1. Depletion of USP2 causes endoplasmic reticulum (ER)-associated degradation of PD-L1, thus attenuates PD-L1/PD-1 interaction and sensitizes cancer cells to T cell-mediated killing. Meanwhile, USP2 ablation-induced PD-L1 clearance enhances antitumor immunity in mice via increasing CD8+ T cells infiltration and reducing immunosuppressive infiltration of myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), whereas PD-L1 overexpression reverses the tumor growth suppression by USP2 silencing. USP2-depletion combination with anti-PD-1 also exhibits a synergistic anti-tumor effect. Furthermore, analysis of clinical tissue samples indicates that USP2 is positively associated with PD-L1 expression in cancer. Collectively, our data reveal a crucial role of USP2 for controlling PD-L1 stabilization in tumor cells, and highlight USP2 as a potential therapeutic target for cancer immunotherapy.
Subject terms: Cancer microenvironment, Tumour-suppressor proteins, Cancer microenvironment
Introduction
Immune checkpoint therapies (ICTs) have achieved unprecedented clinical efficacy and reshaped the treatment landscape for multiple malignancies. Among the ICTs, blockade of programmed death-ligand1 (PD-L1) and its receptor programmed cell death 1 (PD-1) with antibodies can provide durable clinical responses and improve overall survival in various solid tumor [1, 2]. Despite these successes, ICTs are still limited by low clinical response rates, lack of durable remission and immune-related adverse events (irAE) [3]. As a membrane protein, PD-L1 is upregulated in many tumors responds to microenvironment stimuli and suppresses T cell function through PD-1 engagement [4]. PD-L1 is also responsible for the resistance to anti-PD-1/PD-L1 therapy via dynamically recycling in endosomes and trafficking to tumor cell membrane [5]. Therefore, it is necessary to explore the molecular mechanisms regulating PD-L1 level and provide hopeful strategies for tackling the challenges in ICTs.
Ubiquitination is a dynamic and reversible post-translational modifications (PTMs) that attaches various kinds of ubiquitin molecules to protein to control the fate of target protein or protein-protein interaction [6]. Accumulating evidences suggest that ubiquitination and deubiquitination play critical roles in the regulation of PD-L1 protein level and tumor cell immunosuppression [7, 8]. The ubiquitin E3 ligases cullin 3-SPOP and β-TrCP are proved to be involved in the polyubiquitination of PD-L1 and decreased PD-L1 protein pool through the proteasome degradation pathway [9, 10]. Meanwhile, the deubiquitinating enzymes (DUBs) cleaving polyubiquitin chains have also been reported to regulate PD-L1 level. A few members of DUBs have been identified to stable PD-L1 through their deubiquitinase activity. For example, COP9 signalosome 5 (CSN5) inhibits TNF-α-mediated ubiquitination and degradation of PD-L1 by removing K48-linked ubiquitination of PD-L1 [11]. OTUB1, another well-elucidated DUB, positively regulates PD-L1 stability and mediates cancer immune responses by removing K48-linked polyubiquitin chains from the PD-L1 intracellular domain [12]. Thus, targeting the ubiquitination/deubiquitinating of PD-L1 might be a plausible strategy to promote antitumor immunity. Despite the increasing awareness of the involvement of DUBs in regulating PD-L1 abundance, a systematic inquiry of other DUBs participation in PD-L1-mediated tumor immune escape has not been carried out.
In this study, we performed a screening system based on CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) to identify functional DUBs in the regulation of PD-L1 abundance. Our study revealed that USP2 specific interacts with and stabilizes PD-L1 in prostate and colorectal cancer cells, and USP2 is positively associated with PD-L1 expression in human colorectal cancer (CRC) tissue samples. Abrogation of USP2 resulted in increased ubiquitination and clearance of PD-L1 through the proteasome-dependent endoplasmic reticulum-associated degradation (ERAD) pathway. Depletion of USP2 not only sensitized cancer cells to T cell-mediated killing by downregulating cell surface PD-L1 level, but also enhanced antitumor immunity through regulating PD-L1 in mice. Our data provides new insight into USP2-mediated cancer immunosuppression via regulation of PD-L1 abundance, highlighting USP2 as a therapeutic target for PD-L1-dependent cancer immunotherapy.
Results
Identification of USP2 as a positive regulator of PD-L1
To explore the potential deubiquitinase (s) in PD-L1-mediated tumor immune escape, we screened the abilities of a panel of DUBs to regulate PD-L1 abundance by using a validated single-guide RNAs (sgRNAs) screening system (Fig. 1A). 180 sgRNAs targeting 90 DUB family genes were constructed into the CRISPRv2 lentiviral vectors expressing Cas9 [13]. RKO cells abundantly expressing endogenous PD-L1 were infected with lentiviruses delivering Cas9 and sgRNA to generate RKO sgDUB cells. The knockdown efficiency in RKO sgDUB cells was confirmed by real-time quantitative polymerase chain reaction (RT-qPCR) (Supplementary Fig. S1). We thus systematically analyzed the abilities of 90 DUBs to down-regulate the surface PD-L1 level by flow cytometry. Screening data were translated into a scatter diagram representing the relative change of PD-L1 responses to DUBs (Fig. 1B). Interestingly, we found ubiquitin specific peptidase 2 (USP2) significantly decreased the surface PD-L1 level in RKO cells (Fig. 1B). Additionally, COP9 signalosome 5 (CSN5) [11], USP7 [14], USP22 [15], and OTU domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) [12] were also identified as positive PD-L1 regulators, which were consistent with previous reports and validated the reliability of our screening data.
Fig. 1. USP2 positively maintains PD-L1 protein stability.

A Schematic diagram illustrating the screening of DUBs that regulate PD-L1 protein level. B Scatter diagram presentation of the influence of DUBs depletion by specific sgRNAs on PD-L1 abundance. C Immunoblotting examination of PD-L1 abundance in HEK293T cells transfected with increasing amounts of HA-USP2 (0.5, 1, 2 μg). D Immunoblotting analysis of the levels of PD-L1, USP2, PVR, CD47, HLA and Siglec-15 in PC3 cells infected with two distinct sgUSP2-encoding lentiviruses. E Immunoblotting analysis of the expression levels of PD-L1 and USP2 in RKO and Du145 cells infected with sgUSP2-encoding lentivirus. F Immunofluorescence approach determining the PD-L1 abundance in RKO cells infected with sgControl or sgUSP2-encoding lentivirus, respectively. Quantification of fluorescent intensity of PD-L1 was shown. Scale bars, 10 μm. G Flow cytometry analysis of the plasma membrane levels of PD-L1 in RKO cells infected with sgControl or sgUSP2-encoding lentivirus, respectively. Quantification of mean fluorescence intensity (MFI) of was shown. The experiments (C–G) were repeated three times independently with similar results. Data in (F, G) are shown as mean ± SD. ***p < 0.001, unpaired Student’s t test (F), one-way ANOVA test (G).
We next further investigated whether USP2 had regulatory effect on PD-L1. Overexpression of USP2 markedly increased PD-L1 abundance in a dose-dependent manner (Fig. 1C and Supplementary Fig. S2A). On the contrary, depletion of USP2 by specific sgRNAs dramatically decreased endogenous PD-L1 abundances in human prostate cancer PC3, Du145 cells, and CRC RKO, HCT116 cells (Fig. 1D, E and Supplementary Fig. S2B). The abundance of other immune inhibitory ligands, such as PVR, CD47, HLA and Siglec-15, were not changed in USP2-depletion or ectopic expression of USP2 tumor cells (Fig. 1D and Supplementary Fig. S2A, B), indicating that USP2 selectively regulates PD-L1 level in tumor cells. In addition, the positively regulation effect of USP2 on PD-L1 was also validated with similar results in other human tumor cell lines such as prostate cancer LNCaP cells, lung cancer H1299, A549 cells and breast cancer MDA-MB-231 cells (Supplementary Fig. S2A, C). Moreover, USP2-depletion-mediated PD-L1 reduction was also observed in primary human CRC cells (Supplementary Fig. S2D), which further validating our findings. The above data suggest a universal regulatory effect of USP2 on PD-L1 in different tumor cells. Consistently, taking an independent cellular immunofluorescence approach, we found depletion of USP2 by specific sgRNA resulted in a remarkable PD-L1 reduction (Fig. 1F and Supplementary Fig. S2E). Generally, PD-L1 exerts its function on the cancer cell membrane, we next probed whether USP2 affected plasma membrane level of PD-L1. Flow cytometry assays demonstrated that USP2 depletion significantly reduced the plasma membrane level of PD-L1 (Fig. 1G and Supplementary Fig. S2F, G). Together, these data suggest that USP2 is a positive PD-L1 regulator in tumor cells.
USP2 interacts with and stabilizes PD-L1
Having identification of the positive correlation between USP2 and PD-L1, we next investigate how USP2 impacted on PD-L1 abundance. Real-time PCR results revealed that depletion of USP2 did not affect the mRNA level of PD-L1 (Supplementary Fig. S3A). However, half-life analysis in the presence of protein translation inhibitor cycloheximide (CHX) indicated that depletion of USP2 significantly accelerated the degradation rate of PD-L1 protein (Fig. 2A). We thus speculated that USP2 might exert its function through directly interacting with PD-L1. Co-immunoprecipitation (IP) assay showed that USP2 could physically bind to exogenous PD-L1 in HCT116 ectopic PD-L1 cells (Fig. 2B) and endogenous PD-L1 in PC3 and RKO cells (Fig. 2C). In addition, in vitro pull-down assay also suggested the direct interaction between recombinant Flag-PD-L1 and His-USP2 proteins (Fig. 2D). Furthermore, endogenous USP2 and PD-L1 proteins formed a strong co-localization as observed by Duolink assay (Fig. 2E) and immunofluorescence assays (Supplementary Fig. S3B). Intriguingly, PD-L1 distributed in perinuclear region in sgControl RKO cells, while USP2 depletion greatly reduced the PD-L1 localization to the perinuclear region (Fig. 2F), further demonstrating USP2 might affect the intracellular distribution of PD-L1.
Fig. 2. USP2 specifically interacts with and stabilizes PD-L1.

A Half-life analysis of PD-L1 abundance in sgControl or sgUSP2 RKO cells for the indicated time periods in the presence of CHX (25 µg/mL). The PD-L1 level was normalized to GAPDH and quantification of PD-L1 intensity is shown on the right. Data are shown as mean ± SD of 3 independent experiments. **p < 0.01, two-way ANOVA test. B, C Co-immunoprecipitation assays determining the interactions between USP2 and exogenous PD-L1 (B) and endogenous PD-L1 (C), respectively, in the indicated cells. D In vitro pull-down assay showing the direct interaction between recombinant Flag-PD-L1 protein and His-USP2 protein. OVA protein was used as a negative control. E Duolink assay analysis of the interaction of endogenous USP2 and PD-L1 proteins in RKO cells. Red dots indicating the binding of two proteins. Nuclei were stained with DAPI. Scale bar, 10 μm. F Immunofluorescence assays detecting the co-localization of PD-L1 and USP2 in RKO cells infected with sgControl or sgUSP2-encoding lentivirus, respectively. The intensity profiles of USP2 and PD-L1 and quantification data are shown on the right panel. Scale bar, 10 μm. Data are shown as mean ± SD. ***p < 0.001, two-way ANOVA test. G Co-immunoprecipitation assay mapping of USP2 domains critical for PD-L1 binding. H Duolink assay analysis of the interaction between PD-L1 and full-length USP2, 1-258 aa USP2, 259-310 aa USP2 and 311-605 aa USP2 in RKO cells. Red dots indicating the binding of two proteins. Nuclei were stained with DAPI. Scale bar, 10 μm. I, J HEK293T cells were cotransfected HA-USP2 WT or HA-USP2 mutant with Myc-PD-L1 plasmids for 48 h, cell lysates were pull-down with Myc antibody; the precipitates were then immunoblotted by HA antibody. The experiments (A–J) were repeated three times independently with similar results.
We sought to identify which USP2 regions are critically required for its interaction with PD-L1. USP2 protein consists of an N-terminal domain, a ubiquitin binding (UB) domain, a QDS box domain, a metal binding domain and a His box domain (Supplementary Fig. S3C). Through domain deletion studies, we demonstrated that the N terminal, C-2, C-3 and C-4 domains in USP2 dramatically reduced the PD-L1 binding, while C-1 domain retained its association with PD-L1 (Fig. 2G), suggesting that UB domain (259-310 aa region) contributes to the USP2-PD-L1 interaction. This conclusion was further confirmed by the interaction between USP2 259-310 aa and endogenous PD-L1 as evidenced by duolink assay (Fig. 2H). To gain a better understanding of the interaction between USP2 and PD-L1, we searched for and predicted the binding sites between USP2 UB domain and PD-L1. Interestingly, UB domain in USP2 interaction with PD-L1 forms favorable hydrogen bonding sites with amino acids including Thr288, Glu290, Arg292, Asp293 and Gln297 (Supplementary Fig. S3D). To pinpoint the binding sites, we individually mutated these interacting residues in USP2 UB domain to alanine, resulting in USP2 T288A, E290A, R292A, D293A and Q297A mutants. Co-immunoprecipitation assay indicated that T288A, R292A and D293A mutants dramatically reduced the ability of USP2 binding with PD-L1 (Fig. 2I). Importantly, substitution of these three sites simultaneously (TRD-3A) led to a significantly reduction in the USP2-PD-L1 binding compared with the wild type (WT) PD-L1 (Fig. 2J). These data illustrated that Thr288, Arg292 and Asp293 at USP2 control its binding to PD-L1. On the other hand, PD-L1 is composed of an extracellular domain, a transmembrane domain and an intracellular domain (Supplementary Fig. S3E). Co-immunoprecipitation assay revealed that deletion of the intracellular (ΔICD) domain of PD-L1 disrupted its binding to USP2 (Supplementary Fig. S3F), indicating the intracellular region of PD-L1 was responsible for USP2 binding. Collectively, our findings show that USP2 positively regulates PD-L1 stability by direct interaction with intracellular domain of PD-L1.
USP2 maintains PD-L1 stability via modulating K48-linked polyubiquitin chains of PD-L1
As USP2 is a deubiquitinating enzyme, we next determined if USP2 regulates PD-L1 stabilization via deubiquitination. To this aim, we performed co-immunoprecipitation experiment to examine the level of PD-L1 ubiquitination in the presentence of proteasome inhibitor MG132. As expected, USP2 overexpression dramatically reduced PD-L1 poly-ubiquitination (Fig. 3A), while depletion of USP2 significantly elevated the poly-ubiquitination of PD-L1 in RKO cells (Fig. 3B) and primary human CRC cells (Supplementary Fig. S4A). Consistently, MG132-induced PD-L1 ubiquitination was largely abolished by overexpression of wild-type USP2, while catalytically inactive mutant USP2 C276A and binding deficient mutant USP2 TRD-3A failed to remove ubiquitination chains from PD-L1 (Fig. 3C and Supplementary Fig. S4B). In line with this, ectopic expression of wild-type USP2 caused an increase of PD-L1 abundance, but USP2 C276A mutant and USP2 TRD-3A mutant had no obvious effect (Fig. 3D and Supplementary Fig. S4C), indicating USP2 regulates PD-L1 abundance in a deubiquitylase activity-dependent manner.
Fig. 3. USP2 modulates K48-linked polyubiquitin chains of PD-L1.

A Immunoblotting analysis of the ubiquitination of endogenous PD-L1 in HCT116 cells ectopic expression of HA-USP2. B Immunoblotting analysis of the ubiquitination of endogenous PD-L1 in RKO cells infected with sgUSP2-encoding lentivirus. C Immunoblotting analysis of the ubiquitination of PD-L1 in USP2−/− RKO cells reconstituted with Myc-PD-L1 and HA-ubiquitin along with wild-type USP2, C276A or TRD-3A USP2 mutants. D Immunoblotting examination of the effect of wild-type, C276A or TRD-3A mutant USP2 on PD-L1 abundance in the indicated tumor cells. E, F Examination of the influence of USP2 on specific ubiquitination types of PD-L1. HEK293T cells were transfected with Myc-PD-L1 and His-USP2 along with indicated HA-Ubiquitin mutants. Following MG132 treatment, cell extracts were immunoprecipitated by Myc antibody and immunoblotted with HA antibody. WT, wild type; KR, K is mutated to R; K, K only. G The effect of endogenous PD-L1 ubiquitination by USP2 depletion in RKO cells was determined by immunoblotting analysis using the K48- or K63-ubiquitin linkage specific antibodies. H Immunoblotting analyzing the ubiquitination of ΔICD truncation or full-length of PD-L1 affected by USP2. HEK293T cells were transfected with His-USP2 and HA-Ubiquitin along with ΔICD truncation or full-length Myc-PD-L1. Following MG132 treatment, cell extracts were immunoprecipitated by PD-L1 antibody and immunoblotted with Ubiquitin antibody. I Effect of USP2 on the stability of PD-L1 and its mutants. HEK293T cells were transfected with the indicated plasmids for 24 h before immunoblotting analysis with the indicated antibodies. The experiments (A–I) were repeated at least two times independently with similar results.
According to the conjugation style, eight types of lysine (K) ubiquitin linkage had been identified: K6, K11, K27, K29, K33, K48 and K63 [16]. We then sought to evaluate the influence of USP2 on specific ubiquitination modification of PD-L1. To this aim, we cotransfected PD-L1 with hemagglutinin (HA)-tagged ubiquitin mutants that contains only one of the seven lysine sites (K6, K11, K27, K29, K33, K48, and K63). Our co-immunoprecipitation assay revealed that USP2 dramatically removed K48-linked polyubiquitin chains of PD-L1 as well as moderate decreasing K27-linked polyubiquitin of PD-L1 (Fig. 3E). Consistently, when PD-L1 was cotransfected with HA-ubiquitin mutants in which only one lysine reside is mutated to arginine (KR), USP2 failed to remove K48R-linked PD-L1 polyubiquitination signals, whereas K27R displayed very marginal reduction in PD-L1 ubiquitination (Fig. 3F). These results indicate that USP2 predominantly deubiquitinates K48-linked polyubiquitin moieties from PD-L1, which is consistent with previous report that K48-linked ubiquitin chains is mainly related to proteasome degradation [17]. We thus further examined whether USP2 could remove K48-linked ubiquitin chains of PD-L1. Indeed, depletion of USP2 significantly increased the endogenous K48-linked, but not K63-linked ubiquitination of PD-L1 (Fig. 3G). Consistently, ectopic expression of USP2 greatly reduced the exogenous K48-linked ubiquitination of PD-L1 while had no effect on K63-linked ubiquitination of PD-L1 (Supplementary Fig. S4D). Given that USP2 associates with the ICD domain of PD-L1 (Supplementary Fig. S3F), we speculated that USP2 might remove the ubiquitin chains from the ICD region of PD-L1. As expected, ubiquitination assays indicated that compared with the full-length PD-L1, USP2 barely modified the ubiquitination of the PD-L1 ΔICD mutant (Fig. 3H).
We next sought to identify the residues of PD-L1 that are modified by USP2-mediated K48-linked polyubiquitination. To this aim, all of the 5 lysine residues (K263, K270, K271, K280, and K281) within the intracellular region of PD-L1were individually mutated to arginine and the effects of USP2 on PD-L1 mutants expression level were examined. We found that USP2 up-regulated the levels of wild-type PD-L1 as well as PD-L1 K263R, K271R, K280R and K281R, but had no effect on PD-L1 K270R (Fig. 3I). Additionally, USP2 failed to remove K48-linked polyubiquitination of PD-L1 K270R compared with WT PD-L1 (Supplementary Fig. S4E). All together, these findings suggest that USP2 modulates PD-L1 stability by eliminating the K48-linked polyubiquitination of the PD-L1 at lysine 270.
Depletion of USP2 induces an ERAD-dependent degradation of PD-L1
As depletion of USP2 markedly reduced the half-life of PD-L1 protein (Fig. 2A), we sought to investigate the underlying mechanism of USP2-depletion-mediated PD-L1 degradation. The degradation of mammalian membrane proteins is mainly through the following three pathways: proteasome-dependent ERAD (endoplasmic reticulum-associated degradation), ESCRT (endosomal sorting complexes required for transport and lysosome-dependent EGAD (Golgi-associated degradation) [18]. To explore which pathway contributes to USP2-depletion-induced PD-L1 degradation, USP2-depleted tumor cells were co-incubated with the proteasome inhibitor MG132, ERAD inhibitor Eer І, lysosomal inhibitor Bafilomycin A1 (Baf) or chloroquine (CQ), respectively. Immunoblotting results revealed that USP2-depletion-induced PD-L1 degradation was appreciably restored by MG132 (Fig. 4A and Supplementary Fig. S5A) and Eer I (Fig. 4B and Supplementary Fig. S5B) treatment in a time-dependent manner, while Baf and CQ treatment displayed no effect on PD-L1 abundance (Fig. 4C and Supplementary Fig. S5C, D). Furthermore, reconstitution of WT USP2 instead of USP2 C276A or USP2 TRD-3A mutant in USP2-/- RKO cells reversed USP2-depletion-induced PD-L1 degradation (Supplementary Fig. S5E). These findings suggest that USP2 regulates PD-L1 protein level through ERAD-dependent proteasome degradation pathway. As a membrane protein, PD-L1 goes through the endoplasmic reticulum (ER) and Golgi apparatus in sequence for maturation before it exposed to the cell surface [7], we thus presumed that silence of USP2 might lead to ER retention of PD-L1 for its ERAD degradation. Therefore, we examined whether USP2 may change the distribution of PD-L1 to this subcellular apparatus. As expected, immunofluorescence assay showed depletion of USP2 significantly reduced the distribution of PD-L1 to the ER membrane protein calreticulin, while MG132 treatment elevated PD-L1 accumulation in the ER (Fig. 4D). In line with this result, cell fractionation assay also demonstrated that depletion of USP2 indeed increased PD-L1 levels in the ER (Fig. 4E).
Fig. 4. USP2 depletion induced endoplasmic reticulum (ER)-associated degradation of PD-L1.

A–C Immunoblotting analysis of the effect of USP2 depletion on PD-L1 abundance in RKO cells in the absence or presence of proteasome inhibitor MG-132 (10 µM) (A), ERAD inhibitor Eer I (10 µM) (B), and lysosomal inhibitor Baf (100 nM) (C). D Immunofluorescence assays detecting the co-localization of PD-L1 and ER (Calreticulin) in RKO cells infected with sgControl or sgUSP2-encoding lentivirus, in the absence or presence of proteasome inhibitor MG132 (10 µM). The intensity profiles of PD-L1 and Calreticulin, and quantification data are shown in the right panel. Scale bar, 10 μm. Data are shown as mean ± SD. **p < 0.01, ***p < 0.001, one-way ANOVA test. E Immunoblotting examination of the abundance of PD-L1 and USP2 in different cell components of RKO by using antibodies against PD-L1 and USP2, the ER protein Calnexin, nuclear protein histone H3, as well as cytosolic protein GAPDH. F Immunoblotting evaluating the abundance of endogenous PD-L1 in PC3 cells transfected with USP2 siRNAs along with HRD1, XTP3B, and OS9 siRNAs as denoted. G Immunoblotting analysis of the abundance of endogenous PD-L1 in USP2-depleted RKO cells transfected with HRD1, XTP3B, and OS9 siRNAs. The experiments (A–G) were repeated three times independently with similar results.
ERAD-mediated protein degradation is major relied on HRD1 complex, which containing E3 ubiquitin ligase HRD1 and the lectin-like proteins OS9 and XTP3B [19, 20]. HRD1, OS9 and XTP3B have been reported to bind to PD-L1 and regulate its ERAD degradation [21]. To distinguish the role of HRD1, OS9 and XTP3B in USP2-depletion-mediated PD-L1 degradation, we knocked down the expressions of HRD1, OS9 and XTP3B in USP2-depleted tumor cells. As a result, USP2-depletion-mediated PD-L1 degradation could be restored by simultaneously silencing XTP3B and OS9 in PC3 cells (Fig. 4F). However, silencing HRD1 showed less effect on PD-L1 expression (Fig. 4F). The restore effect of silencing XTP3B and OS9 on PD-L1 expression was also observed in USP2-depleted RKO cells (Fig. 4G), indicating that OS9, XTP3B rather than HRD1 modulate USP2-depletion-mediated ERAD degradation of PD-L1. These data collectively suggest that depletion of USP2 triggers an OS9 and XTP3B-dependent ERAD degradation of PD-L1.
Depletion of USP2 promotes the cytotoxicity of T cell toward tumor cell
PD-L1 is known to interrupted T cell-mediated immune surveillance through binding with PD-1 [22]. As the above results showed USP2 stabilizes PD-L1 abundance and membrane distribution in cancer cells, we further evaluate the antitumor immunity of USP2. To validate whether USP2 depletion-mediated PD-L1 elimination could influence their ability to bind to PD-1, a recombinant human PD-1-Fc protein was incubated with USP2-depleted tumor cells. Both flow cytometry and immunofluorescence assays revealed that depletion of USP2 reduced PD-1 protein binding intensity to the tumor cell surface (Fig. 5A, B). Moreover, PD-1/PD-L1 blockage assay [23] also demonstrated that depletion of USP2 significantly induced transcriptional-mediated bioluminescent signal in both RKO and PC3 cells (Fig. 5C and Supplementary Fig. S6A), suggesting that depletion of USP2 interrupted PD-1/PD-L1 association thus repressed PD-L1’s checkpoint activity.
Fig. 5. Depletion of USP2 promotes the cytotoxicity of T cell toward tumor cell.

A Flow cytometry detecting recombinant PD-1 Fc protein binding to RKO cells infected with sgControl or sgUSP2-encoding lentivirus. B Immunostaining of recombinant PD-1 Fc on RKO cells infected with sgControl or sgUSP2-encoding lentivirus. The nuclei were stained with DAPI. The intensity of green fluorescence indicates bound PD-1. Scale bar, 10 µm. C PD-1/PD-L1 blockade assay detecting the PD-1/PD-L1 interaction between Jurkat NFAT-luciferase reporter cells and RKO cells infected with sgControl or different sgUSP2-encoding lentivirus. Data are presented as fold induction over control. D Cell impedance assay analysis of cytotoxicity of human PBMCs toward RKO cells infected with sgControl or sgUSP2-encoding lentivirus. E Flow cytometry analysis of PBMCs-mediated killing of RKO cells infected with sgControl or sgUSP2-encoding lentivirus using Annexin V and propidium iodide (PI) double staining. Cells that in the late stage of apoptosis with Annexin V- and PI-positive were shown. F Immunoblotting analysis of cleaved caspase-3 levels in sgControl or sgUSP2 RKO cells after incubation with activated PBMCs. G sgControl or sgUSP2 RKO cells were co-incubated with activated PBMCs, the levels of GzmB and IFN-γ in CD8+ T cells were examined by flow cytometry. The experiments (A–G) were repeated three times independently with similar results. Data are shown as mean ± SD, **p < 0.01, ***p < 0.001, one-way ANOVA test (A, B, C, E, G) and two-way ANOVA test (D).
T-cell killing assays were also performed to further assess whether USP2 functionally influences the antitumor immunity by co-incubating tumor cells with activated human peripheral blood mononuclear cells (PBMCs). As a result, USP2 depletion rendered tumor cells more sensitive to PBMCs killing as determined by impedance assay (Fig. 5D). Moreover, depletion of USP2 markedly promoted PBMCs-mediated RKO (Fig. 5E) and PC3 (Supplementary Fig. S6B) tumor cell apoptosis and remarkably reduced the survival of tumor cells (Supplementary Fig. S6C). In addition, USP2-depleted tumor cells exhibited higher expression of cleaved caspase-3 further verified this result (Fig. 5F and Supplementary Fig. S6D). Moreover, reconstitution of WT USP2 instead of USP2 C276A or USP2 TRD-3A in USP2−/− RKO cells reversed USP2-depletion-mediated cytotoxicity of PBMCs toward RKO cell (Supplementary Fig. S6E). Granzyme B (GzmB) and IFN-γ are known to mediate the apoptotic effect of cytotoxic T lymphocytes [24, 25]. Importantly, increased GzmB and IFN-γ released from CD8+ T cells were observed in PBMCs incubation with USP2-depleted RKO cells than sgControl-treated cells (Fig. 5G), suggesting depletion of USP2 abolished the immunosuppressive function of PD-L1. Collectively, these results demonstrate that USP2 depletion-mediated PD-L1 destabilization effectively represses PD-1 binding and increases the cytotoxicity of T cells toward tumor cells.
USP2-silencing-mediated PD-L1 degradation promotes antitumor immunity
Next, we further elucidate the effect of USP2 depletion on PD-L1-dependent tumor immunosuppression in vivo. Endogenous USP2 in mouse MC38 cells were depleted by specific sgRNAs, and the rescued cells were generated by overexpressing mouse PD-L1 in MC38 or USP2-depleted MC38 cells. Both immunoblotting and flow cytometry assays demonstrated that USP2 depletion specifically reduced mouse PD-L1 abundance and surface PD-L1 level, while ectopic expression of PD-L1 elevated USP2-depletion-mediated PD-L1 reduction in MC38 (Fig. 6A, B) or RM1 cells (Supplementary Fig. S7A, B). Next, MC38 or RM1 cells with stably expressed sgControl, sgUSP2, PD-L1 and sgUSP2/PD-L1 were subcutaneously injected into the right armpits of C57BL/6 mice, respectively, and the tumor growths were monitored (Supplementary Fig. S8A). We found that overexpression of PD-L1 caused a substantial increase of tumor growth, whereas depletion of USP2 effectively alleviated tumor growth (Fig. 6C and Supplementary Fig. S7C), tumor volume (Fig. 6D and Supplementary Fig. S7D) and tumor weight (Supplementary Fig. S8B, C), and these suppressions could be rescued in sgUSP2+PD-L1 group. Moreover, IHC analysis of tumor tissues demonstrated that the PD-L1 abundance was dramatically reduced in sgUSP2 group, while the USP2-ablation-mediated PD-L1 reduction was recovered in the PD-L1 and sgUSP2+PD-L1 rescue groups (Fig. 6E and Supplementary Fig. S7E, upper panel). In contrast, USP2 depletion induced an elevation of the populations of tumor-infiltrating CD8+ T cells, while PD-L1 overexpression attenuated the infiltration of CD8+ T cells (Fig. 6E and Supplementary Fig. S7E, lower panel). IFN-γ and GzmB play a vital role in CD8+ T-cell-mediated antitumor immunity, tumor-infiltrating lymphocyte (TILs) profile analysis revealed that USP2 depletion led to a remarkable increase in IFN-γ and GzmB production by tumor-infiltrating CD8+ T cells, whereas PD-L1 rescue significantly reduced USP2 depletion-mediated IFN-γ and GzmB production (Fig. 6F, G and Supplementary Fig. S7F, G). Additional evaluation of the splenic lymphocytes showed no significant alterations in the frequency of CD8+ T cells and CD4+ T cells in each group (Supplementary Fig. S8D). These data demonstrate that depletion of USP2 specific activates cytotoxic CD8+ T cell in TILs.
Fig. 6. USP2-depletion-mediated PD-L1 degradation promotes antitumor immunity.

A, B Immunoblotting and flow cytometry examination of the PD-L1 abundances (A) and surface PD-L1 (B) in four different MC38 cell types (sgControl, sgUSP2, sgUSP2+PD-L1, PD-L1). C Photographs of mice tumors of each group (n = 6) at the end of the experiment. D Tumor growth curve of each group inoculated with indicated MC38 stable clones (n = 6). E Immunohistochemistry assay showing PD-L1 expression and CD8+ cell infiltration in indicated tumor tissues. Scale bars, 50 μm. Quantification of IHC staining is shown. F, G Flow cytometry analyzing the IFN-γ (F) and GzmB (G) levels in CD8+ T cells from sgControl, sgUSP2, sgUSP2+PD-L1, PD-L1 MC38 tumor tissues (n = 6). H, I Flow cytometry analyzing the populations of CD25+Foxp3+ Tregs cells (H) and CD11b+Gr1+ MDSCs cells (I) from sgControl, sgUSP2, sgUSP2+PD-L1, PD-L1 MC38 tumor tissues (n = 6). Data in (B), (D–I) are shown as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA test (B, E, F, G, H, I), two-way ANOVA test (D).
Myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) are the two major immunosuppressive immune cells within tumor microenvironment, which inhibits T-lymphocyte immunity and promotes tumor immune escape [23, 26]. TILs profile analysis indicated that USP2 depletion cause a remarkably decreased in the accumulation of CD11b+Gr1+ MDSCs and CD4+CD25+Foxp3+ Tregs cells within the tumor tissue, while PD-L1 rescue increased the accumulation of MDSCs and Tregs cells within the tumor tissue (Fig. 6H, I and Supplementary Fig. S7H, I), suggesting that USP2 depletion switches the tumor immune microenvironment from a suppressed state to an activated state. Collectively, these findings consistently suggest that disruption of USP2 remarkably attenuates tumor growth and enhances antitumor immunity through regulation of PD-L1 stability.
High expression of USP2 in colorectal cancer is negatively associated with anti-tumor immunity
To further investigate the pathological relevance of USP2 and PD-L1, we first evaluated the expression of USP2 protein in different types of tumor based on the Human Protein Atlas [27]. As shown in Fig. 7A, the expressions of USP2 protein in prostate cancer and colorectal cancer were significantly higher than that in other types of cancer, indicating that USP2 may be an oncoprotein. This finding was further validated by IHC staining for USP2 expression in tumor tissues using the human CRC tissue microarrays. Increased USP2 expression in tumor tissues was observed compared with normal tissues (Fig. 7B). Particularly, IHC staining assay demonstrated that the USP2 protein level was positively associated with PD-L1 abundance in CRC tumor tissues (Pearson r = 0.686, p < 0.0001) (Fig. 7C, D). Furthermore, overall survival analysis of patient samples in The Cancer Genome Atlas (TCGA) colon adenocarcinoma (COAD) database revealed that patients with high levels of USP2 were associated with poor clinical outcome (Supplementary Fig. S9A). Similar results were observed in patient samples treatment with anti-PD-1 or anti-PD-L1 (Supplementary Fig. S9B). To further investigate the role of USP2 in the immune microenvironment of human cancer, we analyzed the relationship between the USP2 expression level and infiltration of immunocytes in COAD or prostate adenocarcinoma (PRAD) through TIMER2.0 [28] based on TCGA database. We found that the high expression of USP2 was negatively correlated with intratumorally infiltration of CD8+ T cells, but positively correlated with intratumorally infiltration of Tregs in COAD or PRAD (Supplementary Fig. S9C), although the expression level of USP2 and the intratumorally infiltration of dendritic cells (DCs) (Supplementary Fig. S9D) and macrophages (Supplementary Fig. S9E) did not show a significant correlation. These results further confirmed that USP2 play a negative regulatory role in T cell immunity and inflammatory immunity. We then investigated whether depletion of USP2 combination with anti-PD-1 has a synergistic anti-tumor effect. Our results demonstrated that while USP2 depletion or anti-PD-1 alone both partially reduced mouse tumor burden from control, combination treatment of USP2 depletion with anti-PD-1 achieved the best tumor growth inhibition (Fig. 7E, F). Altogether, these findings suggest that USP2 maintenance of PD-L1 expression in tumor tissues and USP2 depletion can be combined with anti-PD-1 to improve its therapeutic effects.
Fig. 7. USP2 correlates with PD-L1 abundance in colorectal cancer.

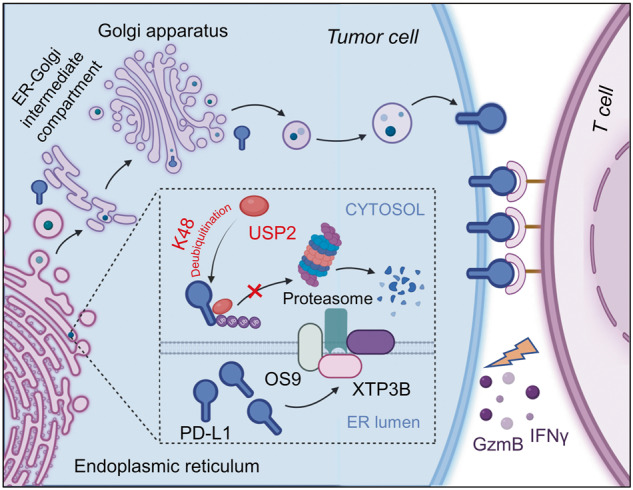
A Bioinformatic analysis of USP2 expression in in different types of tumors based on the Human Protein Atlas. B Representative IHC results of CRC tissue microarrays displaying USP2 expression in tumor tissues and normal tissues. Scale bars, 100 μm. Statistical result for USP2 expression was shown on the right. **p < 0.01 by two-sided Student’s t-test. C Representative IHC results of CRC tissue microarrays displaying the expression of USP2 and PD-L1 in tumor tissues of the same cases. Scale bars, 100 μm. D Statistical result for USP2 and PD-L1expression in C. The p value was determined by two-sided Pearson’s correlation (p < 0.0001). E, F C57BL/6 mice bearing MC38 (sgControl or sgUSP2) tumor were combination with anti-PD-1 (100 µg/mouse) treatment. Photographs of mice tumors of each group (n = 6) at the end of the experiment (E) and tumor growth curve of each group inoculated with indicated MC38 stable clones (F) were monitored. Data are shown as mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 by two-way ANOVA test. G Working model for regulation of PD-L1 stability and cancer cell immunosuppression by USP2. This figure was created with BioRender.com. USP2 interacts with and stabilizes PD-L1 via removing the K48-linked polyubiquitin chains of PD-L1. Depletion of USP2 triggers ERAD, which are recognized by OS9 and XTP3B, and eliminates PD-L1 by ubiquitin-proteasome pathway in the cytoplasm, thereby reprograms tumor microenvironment and sensitizes T cell antitumor immunity.
Discussion
In this study, we identified USP2, a member of the ubiquitin-specific proteases (USPs) subfamily of DUBs, as a novel regulator of PD-L1 stabilization for cancer cell evasion of immune surveillance. We found USP2 specifically interacts with PD-L1 and removes the K48-linked polyubiquitin chains of PD-L1, thus prevents its proteasomal degradation. USP2 depletion led to ERAD-dependent degradation of PD-L1, thereby functionally blocking PD-1 binding and enhancing T cell cytotoxicity as well as reprogramming the tumor microenvironment (Fig. 7G). Our findings suggest targeting USP2 may potentially improve PD-L1-dependent cancer immunotherapy.
So far, abundant studies have extensively described the mechanisms to control PD-L1 abundance, including genomic and epigenetic modifications, transcriptional and post-transcriptional regulations [1, 7, 29, 30]. However, the function of the ubiquitin proteasome system in controlling the PD-L1 stabilization is still not fully understood. Here, we systematically evaluated the effect of 90 DUBs on the expression of PD-L1 and revealed USP2 as a most significant PD-L1 positive regulator in tumor cells by using a validated single-guide RNAs (sgRNAs) screening system. In addition to USP2, several other DUBs with the moderate ability to regulate PD-L1 abundance, including CSN5, USP7, USP22, and OTUB1, were also confirmed in our screening assay. Our data are consistent with previous report that USP7 [14] and USP22 [15] influence PD-L1 abundance in gastric cancer and liver cancer, CSN5 [11] and OTUB1 [12] regulates PD-L1 in breast cancer through their deubiquitinase activity. Therefore, our and others’ findings indicate that in order to reinforce the tumor immunosuppressive microenvironment, some DUBs may exert functionally redundant role in maintenance of PD-L1 level in certain tumor types. Further studies are needed to explore whether USP2 will compete with other DUBs to regulate PD-L1 expression. Consistently, USP2 is highly expressed and participates in tumorigenesis in various types of malignant tumors, including prostate cancer [31], glioma [32], and triple triple-negative breast cancer [33]. Our results proved that USP2 specifically interacts with PD-L1 on the ICD region and regulates PD-L1 stability through its deubiquitination activity. Therefore, PD-L1 is identified as one of the targets for USP2-elicted responses. However, the upstream signaling pathway involved in USP2-modulated PD-L1 upregulation in tumor cells remains to be uncovered in further studies. A deeper exploration of the exact causes of USP2-mediated PD-L1 stabilization would improve the targeting specificity and efficiency of PD-L1-based immune checkpoint blockade therapy.
Diverse ubiquitin modifications of target protein induce various consequences in cells [34]. Among the different types of ubiquitin linkage, K48-linked and K63-linked polyubiquitination have been extensively studied [17]. It is well established that the K48-linked polyubiquitination generally triggers proteasome-dependent degradation of ligated protein, while K63-linked polyubiquitination modulates protein-protein interactions [17, 34]. USP2 has been reported to remove K48-linked polyubiquitination of STAT1 [35] and RIP1 [36]. In agreement, we found USP2 dramatically removed K48-linked polyubiquitin chains of PD-L1. Interestingly, moderate decrease of K27-linked polyubiquitin chains of PD-L1 was also observed in our in vitro ubiquitination assay. It has been reported that K27-linked polyubiquitin enhances protein-protein interactions, promotes proteasomal degradation, and provides binding platforms for DNA repair proteins [37]. We propose that K27-linked polyubiquitination of PD-L1 promotes its interaction with other unknown E3 ligase which triggering PD-L1 degradation, the disturbance of K27-linked ubiquitination of PD-L1 by USP2 therefore stabilize PD-L1 protein level. The role of K27-linked polyubiquitination in USP2-mediated PD-L1 degradation needs further investigation, other approaches such as “ubiquitinomics” may facilitates our understanding of the biological function of USP2 [38].
As a type I transmembrane protein, PD-L1 undergoes multiple trafficking and modification steps along the ER-Golgi-plasma membrane secretory pathway [21, 39, 40]. The modified proteins could be targeted for degradation at each step [18]. E3 ubiquitin ligase HRD1 and the lectin-like proteins OS9 and XTP3B have been reported to participate in the recognition and interaction with ERAD substrates for degradation [41, 42]. Specifically, OS9, XTP3B were indicated to control the ERAD of aberrant glycosylated-PD-L1 [21, 43]. In this work, our results revealed that depleting XTP3B or OS9 in USP2-depleted RKO cells could remarkably restore the abundance of PD-L1, however, silencing HRD1 seemed to have less effect on PD-L1 abundance. This finding suggests that XTP3B and OS9 instead of HRD1 drive the USP2-depletion-mediated ERAD degradation of PD-L1. Although a recent study has shown that HRD1 physically interact with PD-L1 and mediate its ERAD degradation [21], it is possible that other unknown ER-associated E3 ligase might involve in modulation of the ubiquitination and quantity of PD-L1. There are three transmembrane E3 ligase involved in ERAD: the canonical DOA10 and HRD1 as well as Asi [44, 45]. Further studies are warranted to validate whether DOA10 or Asi involved in USP2-abalation-mediated ERAD degradation of PD-L1 and that will unveil a novel mechanism for PD-L1 maturation.
USP2 is a multifunctional deubiquitinating enzyme that has been reported to participate in the carcinogenesis, rhythmic gene expression in the suprachiasmatic nucleus and liver, and regulation of energy metabolism by controlling hepatic gluconeogenesis [46]. Although the most documented topic associated with USP2 is tumorigenesis, the function and molecular mechanism of USP2 in tumor immunotherapy remains elusive. Our findings revealed USP2 level is positive correlated with PD-L1 expression in CRC tissues. In addition, silencing USP2 specifically attenuates the level of PD-L1 and the immunosuppressive ability of tumor cells by enhancing T cell cytotoxicity, thus promotes antitumor immunity. On the contrary, USP2 depletion has no impact on other immune inhibitory ligands’ expression on tumor cells. Given that USP2 KO mice exhibit normal growth and displayed no obvious phenotypic changes [47], our findings thus suggest USP2 is an ideal target to reduce PD-L1 level and potentiate the efficacy of immune checkpoint therapies.
To date, the function of USP2 in tumor microenvironment is rarely reported. In addition to demonstrating USP2 depletion activating CD8+ T cell for tumor eradication, our data also indicated that USP2 depletion activated T cell indirectly through alleviating M-MDSCs and Tregs accumulation in the tumor microenvironment. As the main subpopulations of immunosuppressive microenvironment, Tregs suppress the antitumor immune response driven by CD8+ T cells through producing inhibitory cytokines and consumption of IL-2 [48], and M-MDSCs inhibit tumor-reactive T and NK cells via upregulation of PD-L1 and arginase (ARG)-1 [49]. Although the precise functional relationships between USP2 and Tregs, MDSCs are unknown, our findings are consistent with other studies, showing that M-MDSCs and Tregs trigger immunosuppressive microenvironment and promote tumor progression [50]. It will be interesting to determine how USP2 depletion impairs the function of MDSCs and Tregs, thus expanding the T cell immune response.
In summary, our study unveils a novel regulation mechanism for PD-L1 maturation and establishes a critical association between USP2 and PD-L1-mediated tumor immune escape. We illustrate that USP2 specifically interacts with PD-L1 and maintains its stability via inhibition of ERAD pathway. Blockage of USP2 significantly degrades PD-L1, reprograms tumor microenvironment and sensitizes T-cell antitumor immunity. This work highlights USP2 as a promising and efficacious target to potentiate the efficacy of anti-PD-L1 immunotherapies.
Materials and methods
Cell culture
Human CRC RKO, HCT116 cells, NSCLC A549, H1299 cells, human prostate adenocarcinoma (PRAD) LNCaP, PC3 cells were obtained from Institute of Basic Medicine, Chinese Academy of Medical Sciences (Beijing, China). Mouse CRC MC38 cells and mouse PRAD RM1cells were purchased from Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Primary human CRC cells were kindly provided by Professor Guohui Wan (School of Pharmaceutical Sciences, Sun Yat-Sen University, Guangzhou, China). RKO, HCT116, PC3 cells and primary CRC cells were cultured in DMEM medium, and all the other cells were cultured in RPMI1640 medium (Gibico, Pittsburgh, PA, USA). The culture medium was supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were cultivated in a humidified atmosphere with 5% CO2 at 37 °C. The identities of cell lines were authenticated by short tandem repeat analysis. DAPI staining was used to confirm that no mycoplasma was contaminated in all cell lines. All study-used cell lines’ passages were limited in twenty after receipt or resuscitation.
Chemicals, antibodies, and plasmids
The reagents and commercial assay kits used in this study were listed in Supporting Information Table S1. The antibodies used for immunoblotting, flow cytometry, immunofluorescence and immunohistochemical are listed in Supporting Information Tables S2 and S3. C276A mutant of USP2 plasmid was kindly provided by Prof. Han Liu from Dalian Medical University (Dalian, China). Full-length, 1-258 aa, 259-605 aa, 311-605aa, 389-605aa, 485-605aa truncations of USP2, and ΔICD truncation of PD-L1 were cloned into the pcDNA3.0-HA vector. His-tagged USP2 was cloned into the pET28a vector. All constructs were confirmed by DNA sequencing. Site-mutant plasmids of USP2 (T288A, E290A, R292A, D293 A, Q297A and TRD-3A) and PD-L1 (K263R, K270R, K271R, K280R, and K281R) were obtained by total gene synthesis and confirmed by sequencing (DIA-UP Biotech, Beijing, China). Other plasmids were listed in Supporting Information Table S1.
CRISPR/Cas9-mediated knockout and RNA interference
CHOPCHOP (http://chopchop.cbu.uib.no/) was used to design guide RNAs (sgRNAs), which were then cloned into LentiCRISPRv2. HEK293T cells in 100 mm Petri dishes were co-transfected with 5 µg lentiviral plasmid, 5 µg psPAX2, and 1 µg VSVG. Lentivirus-containing media were gathered at 72 h after transfection and strained through a 0.45 µm filter. Target cells were infected with lentivirus for 24 h, and then treated with 2 μg/ml puromycin for 2 weeks before single cells were picked up. Cell clones were expanded in the presence of puromycin and positive knockout cells were identified by immunoblotting. sgRNAs target sequences were presented in Supporting Information Table S4.
siRNAs were designed on siDESIGN Center (https://horizondiscovery.com/) and cloned into the pSEB-HUS plasmid. For retrovirus production, the plasmids were transfected into HEK293T cells along with the plasmid pCL-Ampho. The supernatants containing retrovirus were collected and used to infect cells. The stably infected cells were selected and maintained using Blasticidin S (InvivoGen, USA). siRNAs for target genes were listed in Supporting Information Table S5.
Immunoblotting, co-immunoprecipitation, and immunofluorescence
Immunoblotting and co-immunoprecipitation were performed as described previously [23, 51]. For immunofluorescence, cells were planted in 8-well chamber slides (ThermoFisher Scientific) and incubated for 24 h. Cells were washed with PBS and fixed with 4% paraformaldehyde at room temperature for 15 min, and then permeabilized and blocked in 0.5% Triton X-100% and 1% BSA at room temperature for 1 h, followed by incubation with primary antibodies overnight at 4 °C. Cells were washed with PBS and then incubated with secondary antibodies at room temperature for 1 h. Nuclei were stained with DAPI (Cell Signaling Technology). The slides were scanned using Olympus confocal microscope. ImageJ v.1.53e was used to quantify fluorescence intensity and co-localization.
Quantitative real-time PCR (RT-qPCR)
Total RNA was extracted using RNeasy Kit (Qiagen) as manufacturer’s instructions. Reverse transcription was conducted using a SuperScript™ III CellsDirect™ cDNA Synthesis Kit (Invitrogen). RT-qPCR was carried out using TB Green® Premix Ex Taq™ (Tli RNaseH Plus) (Takara). Relative mRNA expression was determined by the ΔΔCt method and normalized to GAPDH. Primers were designed using the Primer3Plus online tool and listed in the Supporting Information Table S6.
Protein purification
Full length of human His-USP2 plasmid was transformed into pET28a vectors and expressed in Escherichia coli BL21 cells. The cells were grown at 37 °C until OD600 reached 0.6, and induced with 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 6 h at 30 °C. The cells were harvested by centrifugation at 4000 rpm for 15 min and resuspended in 20 mM Tris-HCl, 200 mM NaCl, pH 8.0 (buffer A) containing 1 μg/ml DNase I and 0.1 mg/ml lysozyme. After thawing, cells were ruptured by ultra-sonication, and the resulting lysate was cleared by centrifugation (75,000 g for 20 min). The supernatant was applied onto a nickel chelating affinity column equilibrated in buffer A and eluted by an imidazole gradient. This was followed by size-exclusion chromatography using a buffer of 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 1% glycerol on a Superdex 200 16/60 column (GE Healthcare). His-USP2-containing fractions were combined and concentrated for further use.
In vitro deubiquitination assay
For deubiquitination assay, HEK293T cells were co-transfected with Myc-PD-L1 and His-USP2 along with different HA-Ubiquitin mutants. Following MG132 treatment to increase the level of ubiquitinated PD-L1, cell extracts were immunoprecipitated by Myc antibody and protein A/G beads (B23201, Bimake), and immunoblotted with HA or ubiquitin antibodies.
Proximity ligation assay (Duolink assay)
In situ proximity ligation assay (Duolink® In Situ PLA® kit, Sigma Aldrich) was used to investigate the interactions between PD-L1 and USP2 according to the manufacturer’s protocol. Briefly, RKO cells were fixed with 4% paraformaldehyde for 20 min at room temperature and permeabilised with 0.5% Triton X-100/ PBS for 1 h. Cells were then incubated with Duolink blocking buffer for 30 min at room temperature, followed by incubation with diluted primary antibodies (USP2 1:200, PD-L1 1:200) at 4 °C overnight. After washing with PBS, cells were incubated with PLA probes for 1 h at 37 °C. The ligation solution was added to hybridize with the PLA probes and form a closed circle and then incubated for 30 min at 37 °C. Next, cells were incubated with fluorescently labelled oligonucleotides and polymerase at 37 °C for 100 min. lastly, nuclei were counterstained with DAPI. Fluorescence images were obtained under a confocal microscope (Olympus).
Molecular docking
The 3D structures of the USP2 (PDB ID: 3NHE) and PD-L1 (PDB ID: 6L8R) were downloaded from RCSB Protein Data Bank (https://www.rcsb.org/). Protein-protein docking between the USP2 and PD-L1 was simulated by ZDOCK with the USP2 selected as the receptor and PD-L1 as the ligand. The binding sites between USP2 and PD-L1 were predicted by Discovery Studio 4.5.
PD-L1/PD-1 interaction assay and T cell-mediated tumor cell-killing assay
To measure the PD-1 and PD-L1 protein interaction in vitro, cells were seeded on 6-well plates and then incubated with human recombinant PD-1 Fc protein and anti-human Alexa Fluor 488 dye conjugated [52]. DAPI was used to stain the cell nuclei. The green fluorescence signal was captured by a Zeiss Axio Vert A1 microscope (Carl Zeiss, Thornwood, NY, USA) or quantified by Novocyte flow cytometer with Novo Express software (Agilent). Human peripheral blood mononuclear cells (PBMCs) were obtained from SCHBIO (Shanghai, China). PBMCs-mediated tumor cell-killing assay was performed by the xCELLigence system (Agilent) [23]. Briefly, each well of E-plate 16 was added 50 μL of full medium for 30 min firstly. Additional 50 μL medium containing of 5 × 103 RKO or PC3 cells was added in plate and the final volume reached 100 μL. Each treatment was conducted two times. After 24 h treatment with the indicated conditions, PBMCs cells (activated by 100 ng/mL anti-CD3, 100 ng/mL anti-CD28 and 10 ng/mL IL-2) were incubated with tumor cells at the ratio of 10:1. The measurements for cell index values were performed by continuous impedance recordings every 10 min. The results were analyzed simultaneously by xCELLigence system (Agilent) with RTCA Software.
PD-L1/PD-1 blockade assay
Functional alterations in PD-1/PD-L1 interactions with USP2 depletion modulation were inspected with the PD-L1/PD-1 blockade assay kit (Promega, Madison, WI, USA) [23]. Parental cells or USP2-depleted RKO cells (1 × 104) were incubated with 1 × 104 Jurkat T cells (stably transfected with NFAT-luciferase reporter and human PD-1) for the indicated time. The NFAT-luciferase was activated through co-culturing of foresaid cells via TCR and MHC interaction. The NFAT-luciferase was downregulated by the interaction between PD-L1 and PD-1. Thus, the extent of decrease in luminescence is proportional to expression degree of PD-L1. Each well was added with Bio-Glo reagent (Promega) at the end of the experiment. After 5-minute incubation, the wells were measured by a LB942 multimode microplate reader (Berthold, Bad Wildbad, Germany) and the data were analyzed by ICE software.
ER and Golgi apparatus fractions enrichment
The extraction of ER and Golgi apparatus was performed by using Minute ER Enrichment Kit (ER-036, Invent) and Minute Golgi apparatus Enrichment Kit (GO-037, Invent), respectively. The experiments were performed according to the manufacturer’s instructions. All protein fractions were boiled with loading buffer and analyzed by immunoblotting.
Animal experiments
All procedures with C57BL/6 mice (6- to 8-week-old females, Beijing Vital River Laboratory, Beijing, China) were conducted under guidelines approved by the animal ethics committee of the Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences. All animals were handled following the ‘Principles for the Utilization and Care of Vertebrate Animals’ and the ‘Guide for the Care and Use of Laboratory Animals’. 1 × 106 mouse colorectal MC38 cells or prostate cancer RM1 cells (stable clones expressing sgControl, sgUSP2, PD-L1, sgUSP2+PD-L1) were injected subcutaneously into the right armpits of mice (mice were divided into four groups randomly, n = 6 for each group). For the sgUSP2 and anti-PD-1 combination assay, anti-PD-1 was administrated by i.p. (100 µg/mouse, once every other day) three times. Body weight and tumor volume were measured every 2 days using a vernier caliper and calculated with the formula π/6 × length × width2. Mice were sacrificed when the sizes of tumors reached 1500 mm3 or ulceration happened. The tumors were resected and weighed, and pretreated for further experiments.
Isolation and flow cytometry analysis of tumor-infiltrating lymphocytes [23, 51]
Tumors tissues in mice of each group were collected and divided into small pieces in culture medium. Then type 4 collagenase (1 mg/ml, Sigma-Aldrich) and DNase 1 (0.1 mg/ml, Sigma-Aldrich) were used to digest tissues’ pieces to signal cells for 1 h at 37 °C. After blocking with anti-CD16/CD32 antibodies, suspension cells were stained with fixable viability dye for 15 min at 4 °C. For T cell analysis, cells were stained with CD45, CD3, CD8, IFN-γ and GZMB. For Tregs analysis, cells were labeled with CD45, CD3, CD4, CD25 and Foxp3. For MDSCs analysis, cells were stained with CD45, CD11b, MHC II, F4/80, Gr-1. Stained cells were subsequently quantitatively analyzed by guava easyCyte flow cytometer with guavaSoft 3.1.
Immunohistochemical (IHC) staining and CRC tissue microarrays
IHC staining was conducted as described previously [23]. Briefly, tumor tissues were deparaffinized and rehydrated, the antigen retrieval was carried out in citrate antigen retrieval solution. After blocking with 3% H2O2 for 20 min and goat serum for 45 min, tissues samples were incubated with primary antibodies overnight at 4 °C, followed by incubation with a biotin-labeled secondary antibody and then co-cultured with an avidin-biotin-peroxidase complex. Amino-ethylcarbazole chromogen was used for visualization. CRC tissue microarray was purchased from Outdo Biotech (Shanghai, China) with informed patient consent provided. The tissue microarray involved 80 adjacent non-tumor samples and 81 Colorectal Adenocarcinoma samples. IHC staining was assessed using H score with the formula (weak)% × 1 + (moderate)% × 2 + (strong)% × 3, ranging from 0 to 300. The IHC staining intensity was ranked as 0 (negative), 1 (weak), 2 (moderate) and 3 (strong). Images were captured through an Axio Vert A1 microscope (Carl Zeiss).
Bioinformatics analysis
Three public web servers, the Human Protein Atlas (HPA, https://www.proteinatlas.org), the Tumor Immune Estimation Resource (TIMER, http://cistrome.shinyapps.io/timer/) and Kaplan-Meier plotter (KM-plotter, http://kmplot.com) were used to conduct bioinformatics analysis, respectively. The HPA was used to evaluate the expression of USP2 in different cancer types. KM-plotter was applied to explore cancer patient overall survival. The TIMER2.0 was used to analyze the correlation between the expression of USP2 in TCGA datasets and the intratumorally infiltration levels of CD8+ T cells, regulatory T (Treg) cells, macrophages and dendritic cells. One-way analysis of variance (ANOVA) was applied as the variable for calculating differential expression (Tumor or Normal). A p (disease state) <0.01 was defined as the differentially expressed genes.
Statistics
All statistical results are presented as mean ± SEM of three independent biological replicates. Data are analyzed using 2-tailed unpaired Student’s t test between two groups or one- or two-way ANOVA followed by Bonferroni’s post test between multiple groups. All graphs and statistical analyses were produced using GraphPad Prism 8 (GraphPad Software, La Jolla, CA). A p value of less than 0.05 was considered to be statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). For every figure, statistical tests are justified as appropriate.
Supplementary information
Acknowledgements
The authors sincerely thank Prof. Hong-bing Shu for providing HA-Ubiquitin KR plasmids, Prof. Ronggui Hu for providing HA-Ubiquitin K only plasmids, Prof. Han Liu for providing USP2 C276A plasmid, and Prof. Guohui Wan for providing primary human CRC cells. This study was supported by grants from National Natural Science Foundation of China (82273960, 81973366, 82273854, 82003792 and 82304512), CAMS Innovation Fund for Medical Sciences (2021-I2M-1-070), and Beijing Nova Program (20220484116). The funding sources had no involvements in study design, data collection, data analysis, data interpretation, manuscript preparation and submission.
Author contributions
HD overall designed, supervised and coordinated the study. ZK and XL performed most of the experiments. NZ, JD, MY, CS, YW and LL participated in the molecular and cellular biological experiments. DX, XZ performed molecular docking. YF and DS provided reagents and performed data analysis. HD supervised the study and interpreted results, wrote and revised the manuscript. All authors read and approved the manuscript.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All experiments using mice were approved by the animal ethics committee of the Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and complied with all relevant ethical guidelines.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Zean Kuang, Xiaojia Liu, Na Zhang.
Contributor Information
Yanchun Feng, Email: fyc@nifdc.org.cn.
Danqing Song, Email: songdanqingsdq@hotmail.com.
Hongbin Deng, Email: hdeng@imb.pumc.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-023-01219-9.
References
- 1.Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–52. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- 3.Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J, et al. The next decade of immune checkpoint therapy. Cancer Discov. 2021;11:838–57. doi: 10.1158/2159-8290.CD-20-1680. [DOI] [PubMed] [Google Scholar]
- 4.Vesely MD, Zhang T, Chen L. Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol. 2022;40:45–74. doi: 10.1146/annurev-immunol-070621-030155. [DOI] [PubMed] [Google Scholar]
- 5.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–23. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016;26:484–98. doi: 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu JM, Li CW, Lai YJ, Hung MC. Posttranslational modifications of PD-L1 and their applications in cancer therapy. Cancer Res. 2018;78:6349–53. doi: 10.1158/0008-5472.CAN-18-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Dang F, Ren J, Wei W. Biochemical aspects of PD-L1 regulation in cancer immunotherapy. Trends Biochem Sci. 2018;43:1014–32. doi: 10.1016/j.tibs.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–5. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–39. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu D, Xu R, Huang X, Tang Z, Tian Y, Zhang J, et al. Deubiquitinating enzyme OTUB1 promotes cancer cell immunosuppression via preventing ER-associated degradation of immune checkpoint protein PD-L1. Cell Death Differ. 2021;28:1773–89. doi: 10.1038/s41418-020-00700-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Q, Wu Y, Qin Y, Hu J, Xie W, Qin FX, et al. Broad and diverse mechanisms used by deubiquitinase family members in regulating the type I interferon signaling pathway during antiviral responses. Sci Adv. 2018;4:eaar2824. doi: 10.1126/sciadv.aar2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, Kang W, Li O, Qi F, Wang J, You Y, et al. Abrogation of USP7 is an alternative strategy to downregulate PD-L1 and sensitize gastric cancer cells to T cells killing. Acta Pharm Sin B. 2021;11:694–707. doi: 10.1016/j.apsb.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, et al. USP22 deubiquitinates CD274 to suppress anticancer immunity. Cancer Immunol Res. 2019;7:1580–90. doi: 10.1158/2326-6066.CIR-18-0910. [DOI] [PubMed] [Google Scholar]
- 16.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–29. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 17.Akutsu M, Dikic I, Bremm A. Ubiquitin chain diversity at a glance. J Cell Sci. 2016;129:875–80. doi: 10.1242/jcs.183954. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt O, Weyer Y, Baumann V, Widerin MA, Eising S, Angelova M, et al. Endosome and Golgi-associated degradation (EGAD) of membrane proteins regulates sphingolipid metabolism. EMBO J. 2019;38:e101433. doi: 10.15252/embj.2018101433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu C, Ng DT. Glycosylation-directed quality control of protein folding. Nat Rev Mol Cell Biol. 2015;16:742–52. doi: 10.1038/nrm4073. [DOI] [PubMed] [Google Scholar]
- 20.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–57. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–20.e7. doi: 10.1016/j.molcel.2018.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Liu X, Zhang N, Yin M, Dong J, Zeng Q, et al. Berberine diminishes cancer cell PD-L1 expression and facilitates antitumor immunity via inhibiting the deubiquitination activity of CSN5. Acta Pharm Sin B. 2020;10:2299–312. doi: 10.1016/j.apsb.2020.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. 2010;17:616–23. doi: 10.1038/cdd.2009.206. [DOI] [PubMed] [Google Scholar]
- 25.Kursunel MA, Esendagli G. The untold story of IFN-gamma in cancer biology. Cytokine Growth Factor Rev. 2016;31:73–81. doi: 10.1016/j.cytogfr.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7. doi: 10.1016/j.coi.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Thul PJ, Lindskog C. The human protein atlas: a spatial map of the human proteome. Protein Sci. 2018;27:233–44. doi: 10.1002/pro.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48:W509–W14. doi: 10.1093/nar/gkaa407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. 2017;549:106–10. doi: 10.1038/nature23669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, et al. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B. 2020;10:723–33. doi: 10.1016/j.apsb.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benassi B, Flavin R, Marchionni L, Zanata S, Pan Y, Chowdhury D, et al. MYC is activated by USP2a-mediated modulation of microRNAs in prostate cancer. Cancer Discov. 2012;2:236–47. doi: 10.1158/2159-8290.CD-11-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boustani MR, Khoshnood RJ, Nikpasand F, Taleshi Z, Ahmadi K, Yahaghi E, et al. Overexpression of ubiquitin-specific protease 2a (USP2a) and nuclear factor erythroid 2-related factor 2 (Nrf2) in human gliomas. J Neurol Sci. 2016;363:249–52. doi: 10.1016/j.jns.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 33.He J, Lee HJ, Saha S, Ruan D, Guo H, Chan CH. Inhibition of USP2 eliminates cancer stem cells and enhances TNBC responsiveness to chemotherapy. Cell Death Dis. 2019;10:285. doi: 10.1038/s41419-019-1512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clague MJ, Heride C, Urbe S. The demographics of the ubiquitin system. Trends Cell Biol. 2015;25:417–26. doi: 10.1016/j.tcb.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Ren Y, Zhao P, Liu J, Yuan Y, Cheng Q, Zuo Y, et al. Deubiquitinase USP2a sustains interferons antiviral activity by restricting ubiquitination of activated STAT1 in the nucleus. PLoS Pathog. 2016;12:e1005764. doi: 10.1371/journal.ppat.1005764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Augsten M, Bottcher A, Spanbroek R, Rubio I, Friedrich K. Graded inhibition of oncogenic Ras-signaling by multivalent Ras-binding domains. Cell Commun Signal. 2014;12:1. doi: 10.1186/1478-811X-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tracz M, Bialek W. Beyond K48 and K63: non-canonical protein ubiquitination. Cell Mol Biol Lett. 2021;26:1. doi: 10.1186/s11658-020-00245-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vere G, Kealy R, Kessler BM, Pinto-Fernandez A. Ubiquitomics: an overview and future. Biomolecules. 2020;10:1453. [DOI] [PMC free article] [PubMed]
- 39.Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–5. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chou CW, Yang RY, Chan LC, Li CF, Sun L, Lee HH, et al. The stabilization of PD-L1 by the endoplasmic reticulum stress protein GRP78 in triple-negative breast cancer. Am J Cancer Res. 2020;10:2621–34. [PMC free article] [PubMed] [Google Scholar]
- 41.Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J Cell Biol. 2010;188:223–35. doi: 10.1083/jcb.200910042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christianson JC, Shaler TA, Tyler RE, Kopito RR.OS-9. and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–82. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li C, Chi H, Deng S, Wang H, Yao H, Wang Y, et al. THADA drives Golgi residency and upregulation of PD-L1 in cancer cells and provides promising target for immunotherapy. J Immunother Cancer. 2021;9:e002443. [DOI] [PMC free article] [PubMed]
- 44.Khmelinskii A, Blaszczak E, Pantazopoulou M, Fischer B, Omnus DJ, Le Dez G, et al. Protein quality control at the inner nuclear membrane. Nature. 2014;516:410–3. doi: 10.1038/nature14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foresti O, Rodriguez-Vaello V, Funaya C, Carvalho P. Quality control of inner nuclear membrane proteins by the Asi complex. Science. 2014;346:751–5. doi: 10.1126/science.1255638. [DOI] [PubMed] [Google Scholar]
- 46.Kitamura H, Hashimoto M. USP2-related cellular signaling and consequent pathophysiological outcomes. Int J Mol Sci. 2021;22:1209. [DOI] [PMC free article] [PubMed]
- 47.Bedard N, Yang Y, Gregory M, Cyr DG, Suzuki J, Yu X, et al. Mice lacking the USP2 deubiquitinating enzyme have severe male subfertility associated with defects in fertilization and sperm motility. Biol Reprod. 2011;85:594–604. doi: 10.1095/biolreprod.110.088542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Itahashi K, Irie T, Nishikawa H. Regulatory T-cell development in the tumor microenvironment. Eur J Immunol. 2022;52:1216–27. doi: 10.1002/eji.202149358. [DOI] [PubMed] [Google Scholar]
- 49.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108–19. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019;120:16–25. doi: 10.1038/s41416-018-0333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Yin M, Dong J, Mao G, Min W, Kuang Z, et al. Tubeimoside-1 induces TFEB-dependent lysosomal degradation of PD-L1 and promotes antitumor immunity by targeting mTOR. Acta Pharm Sin B. 2021;11:3134–49. doi: 10.1016/j.apsb.2021.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang N, Dou Y, Liu L, Zhang X, Liu X, Zeng Q, et al. SA-49, a novel aloperine derivative, induces MITF-dependent lysosomal degradation of PD-L1. EBioMedicine. 2019;40:151–62. doi: 10.1016/j.ebiom.2019.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.