

Abstract
A significant fraction of non-small cell lung cancer (NSCLC) cases are due to oncogenic mutations in the tyrosine kinase domain of the epidermal growth factor receptor (EGFR). Anti-EGFR antibodies have shown limited clinical benefit for NSCLC, whereas tyrosine kinase inhibitors (TKIs) are effective, but resistance ultimately occurs. The current landscape suggests that alternative ligands that target wild-type and mutant EGFRs are desirable for targeted therapy or drug delivery development. Here we evaluate NSCLC targeting using an anti-EGFR aptamer (MinE07). We demonstrate that interaction sites of MinE07 overlap with clinically relevant antibodies targeting extracellular domain III and that MinE07 retains binding to EGFR harboring the most common oncogenic and resistance mutations. When MinE07 was linked to an anti-c-Met aptamer, the EGFR/c-Met bispecific aptamer (bsApt) showed superior labeling of NSCLC cells in vitro relative to monospecific aptamers. However, dual targeting in vivo did not improve the recognition of NSCLC xenografts compared to MinE07. Interestingly, biodistribution of Cy7-labeled bsApt differed significantly from Alexa Fluor 750-labeled bsApt. Overall, our findings demonstrate that aptamer formulations containing MinE07 can target ectopic lung cancer without additional stabilization or PEGylation and highlights the potential of MinE07 as a targeting reagent for the recognition of NSCLC harboring clinically relevant EGFRs.
Keywords: MT: Delivery Strategies, aptamers, non-small cell lung cancer, oligonucleotides, tumor cell-surface receptors, molecular engineering, biodistribution, targeted delivery, EGFR resistance mutations
Graphical abstract
Porciani and colleagues show that anti-EGFR aptamers are a feasible way to target EGFR-mutant NSCLC. These aptamers show promise as reagents for diagnostic and therapeutic development to address factors that contribute to poor survival rates in a subset of NSCLC patients, such as therapeutic resistance.
Introduction
Lung cancer is the leading cause of cancer-related deaths among both men and women worldwide, accounting for nearly 1.8 million deaths per year.1 Non-small cell lung cancer (NSCLC) is the most common subtype and includes ∼85% of all lung cancer diagnoses. Most NSCLC cases are attributed to adenocarcinoma (60%) and squamous cell carcinoma (30%).2 A significant number of lung adenocarcinoma cases are due to somatic, activating mutations within the tyrosine kinase domain (TKD) of the epidermal growth factor receptor (EGFR) gene that occur in 15%–20% of Caucasian and 40%–60% of Asian patients.3 Nearly 80%–90% of these EGFR genetic changes consist of a single-nucleotide missense mutation in exon 21 (L858R) or in-frame deletions of amino acids (aa) 746–749 within exon 19 (Ex19Del).4 These “classical” mutations act as oncogenic drivers, leading to a constitutively active EGFR that sustains cellular growth and survival signaling even in the absence of ligand stimulation.5 First-line treatment for EGFR-mutant NSCLC uses tyrosine kinase inhibitors (TKIs) that selectively inhibit EGFR kinase activity and downstream signaling.2 Unfortunately, TKI resistance eventually occurs within 12–18 months, and those patients show a 5-year survival rate that is <20%.6 The most recurring genetic mechanisms of resistance involve the acquisition of secondary (T790M, which accounts for ∼50% of resistance cases) and tertiary (C797S) mutations in the intracellular EGFR TKD, amplification of other cell-surface protein encoding oncogenes (e.g., MET), or mutations in downstream oncogenes (e.g., KRAS).3 More recently, wild-type (WT) EGFR amplification has been found in both lung adenocarcinoma and squamous cell carcinoma, where it is implicated in cancer pro-survival mechanisms.5,7 Genetic alterations in EGFR lead to NSCLC heterogeneity8 and resistance, which, in addition to delayed diagnoses,9 contribute significantly to poor survival. This underlines the importance of having a set of versatile ligands with different biological and physicochemical properties, such as antibodies (Abs) and aptamers, that target both WT and mutant EGFRs. In general, targeting of EGFR-mutant NSCLC is still a critical and unmet need of contemporary cancer research that requires the continued development of novel diagnostics and therapeutics to be used as an alternative or adjuvant to modern reagents.
Due to the heterogeneity of NSCLC, targeted cancer strategies must achieve specific targeting in mixed populations of closely related cells. Cells displaying a unique surface marker can be targeted with monoclonal Abs (mAbs), aptamers, peptides, or other classes of ligands, such as folic acid and transferrin.10,11 However, subsets of cells that do not express a given marker, or express it at low levels, evade targeted recognition.12 A validated strategy is to simultaneously target two or more markers that are expressed on the same tumor cell surface (called in cis binding).13,14 Dual targeting strategies performed with bispecific Abs (bsAbs) or bispecific chimeric antigen receptor (CAR) T cells have been shown to increase tumor selectivity.15,16,17,18 However, bsAbs have also displayed significant off-tumor toxicity.19,20,21 Engineering of mAbs has been performed to minimize their immunogenicity,22 but this process can be technically challenging, expensive, and time consuming.23,24
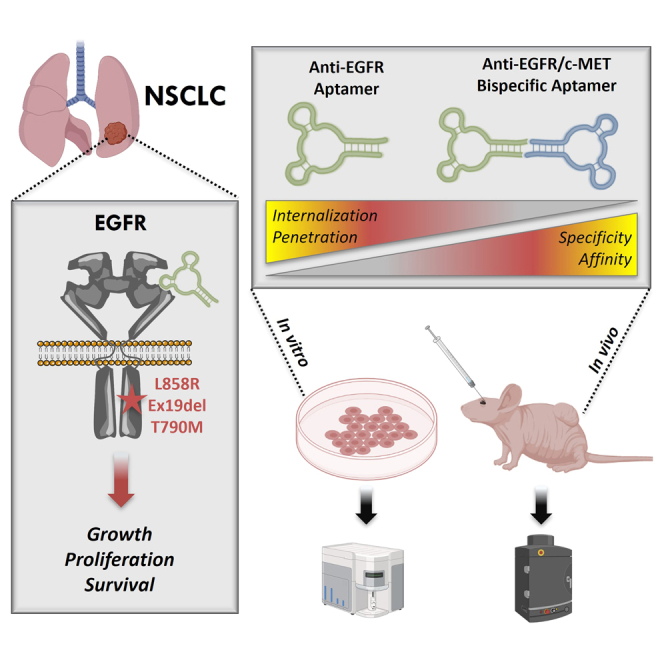
Oligonucleotide diagnostics and therapeutics have been indispensable in the biomedical sciences and in medicine, highlighted by being the topic of Noble Prize awardees and the clinical success of small interfering RNA (siRNA), antisense oligonucleotides (ASOs), and mRNA vaccines.25,26 Aptamer technology is still relatively underexplored compared with other types of RNA-based technologies. However, aptamers have various beneficial properties that make them suitable for new reagent evolution as they combine characteristics of small molecules and mAbs. Aptamers possess low immunogenicity as a foreign substance and do not activate immune effector cells unless they are engineered to act as immunomodulators.27,28 In addition, aptamer sequences are programmable making design and molecular engineering of bispecific aptamers (bsApts) or aptamer-based nanoarchitectures rapid and cost effective.29,30,31 Indeed, we and others have demonstrated that aptamers that target tumor cell-surface markers are valid tools to deliver several classes of molecular payloads in vitro31,32,33,34 and in vivo.35,36,37,38
Several RNA and DNA aptamers have been selected to bind WT EGFR.39,40,41 Fewer aptamers have been selected or utilized to target mutated EGFRs, such as EGFRvIII, a deletion variant (del6-273) commonly found on glioblastoma.42,43 Recently, a DNA aptamer (TuTu22) selected to bind WT EGFR41 was reported to label EGFR-mutant NSCLC H1975 cells,44 although we were unable to reproduce these results using our method of aptamer labeling (Figure S1). Aptamer data reproducibility has been a major bottleneck in the progression to and acceptance of aptamer technology in the clinic, as recently highlighted.45,46,47 Therefore, in addition to following the recently released minimum aptamer publication standards (MAPS),47 we utilized a 2′ fluoropyrimidine (FY) RNA anti-EGFR aptamer (MinE07) that was both rigorously selected and reproducible for our studies.
Aptamer MinE0748 binds WT EGFR with nanomolar affinity (Kd ∼1–2 nM) and displays antiproliferative effects in epidermoid cancer cells.39 MinE07 binds the extracellular domain of human EGFR in competition with epidermal growth factor (EGF) but also recognizes EGFRvIII (Kd ∼250 nM)49 and mouse EGFR (Kd ∼35 nM),39 albeit with lower affinity than recognition of WT EGFR. Over the past years, we and others have used MinE07 to target WT EGFR as a tool for super-resolution microscopy,50 in vivo imaging,49 and targeted drug delivery.51,52,53 However, using MinE07 to target tumors harboring clinically relevant EGFR mutations found in lung adenocarcinoma patients has not yet been demonstrated.
Here, we show that MinE07 binding to the extracellular domain of EGFR is retained even in the presence of EGFR TKD mutations associated with NSCLC. To improve recognition of NSCLC cells, we describe the engineering of a bsApt that dually targets EGFR and c-Met. We show that EGFR/c-Met bsApt significantly increases in vitro NSCLC cell labeling compared to monospecific anti-EGFR (MinE07) and anti-c-Met (CLN3) aptamers. We also demonstrate that MinE07 and EGFR/c-Met bsApt effectively target subcutaneous xenografts of human NSCLC cells. However, in contrast with our in vitro assessments, we found that increasing aptamer cell-targeting specificity did not significantly improve in vivo tumor targeting. We propose that these findings may be attributed to the heterogeneity of c-Met expression in vivo and to differences in endocytosis and tumor penetration between monospecific aptamer and EGFR/c-Met bsApt. Finally, we show that aptamer biodistribution can be affected by the choice of fluorophore used for biodistribution studies. Specifically, a near-infrared (NIR) fluorescent dye, Cy7, conjugated to our EGFR/c-Met bsApt inherently increased accumulation at liver and kidney tissues compared to a more hydrophilic NIR dye, Alexa Fluor 750. This warrants caution when interpreting biodistribution of dye-labeled oligonucleotides solely in terms of the targeting reagent without considering the dye.
Our findings establish monospecific aptamer and bsApt formulations containing MinE07 as potential reagents for future development of aptamer-based therapeutics and/or diagnostics for EGFR-mutant NSCLC. The proposed approaches are modular and can be fine-tuned and applied to other cancer types. Importantly, aptamer technology has not yet been widely accepted for clinical use; hence, future developments should strive for clinically useful reagents.54,55 The discontinuity between in vitro and in vivo studies suggests that translational in vitro models that better recapitulate three-dimensional tumor biology (e.g., organoids) should be used to validate aptamer technology. Further improvements to aptamer pharmacokinetic properties should be pursued to increase circulation half-life and decrease nuclease susceptibility, potentially by selecting and utilizing fully chemically modified aptamers.
Results
MinE07 aptamer binds cell-surface EGFRs harboring oncogenic and TKI-resistance mutations
To test whether MinE07 binds EGFR harboring mutations found in NSCLC patients, we performed flow cytometry binding assays. To facilitate aptamer conjugation to organic dyes, we used a modular design that was previously applied to several RNA and DNA aptamers,31,32 including MinE07,50 and is similar to constructs used extensively for targeted siRNA delivery.56 In this design, MinE07 sequence was extended at its 3′ end to include a short 21-nt oligonucleotide “tail” that can efficiently anneal with its complementary anti-tail bearing an organic dye (Figure S2). Far-red dyes (Cy5 or Atto647N) were used for flow cytometry and light microscopy assays, and NIR dyes (Cy7 or Alexa Fluor 750) were used for in vivo and ex vivo imaging.
Cell labeling with MinE07 was compared with that of control aptamer (mutMinE07), a highly mutated version of MinE07 with a different predicted secondary structure (Figure S2B). HEK293FT cells (low to no EGFR) were transiently transfected with WT EGFR or a panel of five mutant EGFRs (expression plasmid, pCMV EGFR_T2A_EGFP; Table S1; Figure S3A). We focused on EGFR harboring oncogenic classical mutations (L858R or exon19 deletions) that are present in 70%–90% of EGFR-mutant NSCLC cases, and on EGFR double mutants that include a secondary “gatekeeper” mutation (T790M) that drives resistance to first and second generations of EGFR TKIs in ∼50%–60% of patients.2,3 Each EGFR variant (WT and mutants) was co-expressed in HEK293FT cells with GFP to identify transfected cells and to normalize expression levels of WT and mutant EGFRs (see section “materials and methods”). Cy5-MinE07 targeted all five mutant EGFR-expressing HEK293FT cells, showing a superior labeling of GFP-positive HEK293FT cells relative to mutMinE07 (Figure 1A) and relative to non-transfected HEK293FT cells (Figure S3B). These results confirm that MinE07 recognized cell-surface EGFR expressed upon cell transfection. Among EGFR mutants, MinE07 showed the highest labeling of cells expressing L858R/T790M, followed by L858R, and then by cells transfected with EGFR exon19 deletions (del746-750; del747-751; del746-750/T790M) (Figure 1A). To rule out that such differences in cell labeling were due to changes in EGFR expression levels, we normalized mean fluorescence intensities (MFIs) of Cy5-MinE07 relative to MFIs measured for GFP, which is proportional to the EGFR level.57 Relative MFIs (expressed as MFICy5/MFIGFP) further confirmed that MinE07 effectively targets all EGFR mutants but indeed displays an increased labeling of cells expressing L858R/T790M (Figure S3C). These results illustrate that, when WT or mutant EGFRs are transiently transfected at similar levels in the same cell line (HEK293FT), MinE07 retained binding of EGFRs harboring intracellular oncogenic and resistance mutations. In these conditions, we also observed that MinE07 preferentially labels HEK293FT cells expressing EGFR L858R/T790M (and to a lesser extent L858R) compared with exon19 deletions, although it has not yet been determined whether such differences are due to an enhanced aptamer affinity of these mutant EGFRs or are cell line dependent.
Figure 1.

Anti-EGFR MinE07 aptamer binds cell-surface EGFRs harboring oncogenic and resistance mutations and competes with clinically relevant anti-EGFR Abs
(A) HEK293FT cells were transfected with an expression plasmid containing either WT or mutant EGFR and an EGFP reporter under control of the pCMV reporter (pCMV_EGFR_T2A_EGFP; Table S1). Transfected cells were incubated with 100 nM Cy5-labeled anti-EGFR aptamer (MinE07, green bar) or control aptamer (mutMinE07, gray bar) and cell labeling was analyzed via flow cytometry. Cy5 MFI of GFP+ cells is reported on the y axis. MinE07 variably labeled cells expressing both WT and mutant EGFR, relative to the control aptamer. Plotted values represent mean ± SD for n = 3 independent experiments. Statistical analysis was performed using a two-way ANOVA (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). (B) H1975 cells (NSCLC cell line harboring endogenous EGFR L858R/T790M) were incubated with 50 nM Cy5-labeled MinE07 in either PBS (no competition) or 10× (500 nM) competitor at 37°C, or at 4°C to mitigate receptor internalization, for 45 min. Cell labeling was defined via flow cytometry. MinE07 competed with mAbs cetuximab and nimotuzumab, suggesting a specific binding site within extracellular domain III of EGFR. Plotted values represent mean ± SD for n = 2 independent experiments. (C) Schematic of EGFR interactions with clinically relevant mAbs and MinE07, all of which are proposed to bind sub-domain III of EGFR ectodomain.
While using HEK293FT cells allowed for the rapid screening of MinE07 binding toward a panel of mutant EGFRs, this embryonic kidney cell line does not mimic the diversity of receptors and EGFR levels that are found on the surfaces of NSCLC cells. Thus, to determine whether MinE07 targets endogenously expressed EGFR mutants, we performed flow cytometry binding assays using two EGFR-mutant NSCLC cell lines: H820 (EGFR del746-750/T790M) and H1975 (EGFR L858R/T790M). A third NSCLC cell line (A549) that expressed WT EGFR was used as positive control. Atto647N-MinE07 specifically labeled all three NSCLC cell lines in a dose-dependent manner (Figure S4A). A549 cells emerged as the cell line with the highest expression of cell-surface EGFRs, equally followed by H820 and H1975 cells, which displayed comparable EGFR levels (Figure S4A). A similar trend of cell labeling (A549 >> H820 ≈ H1975) (Figures S4B and S4C) was obtained using an APC-labeled Ab (clone AY13) that targets the extracellular domain of EGFR but in a different site than MinE07 (Figure S5). These results further confirmed that MinE07 indeed labels NSCLC cells that express endogenous mutant EGFRs with primary oncogenic mutations (L858R or del746-750) and T790M resistance mutation.
MinE07 aptamer competes with anti-EGFR mAbs cetuximab and nimotuzumab
Mutations in the kinase domain are not expected to induce major changes in the extracellular region of EGFR, which contains four domains, I, II, III, and IV. MinE07 was previously shown to bind EGFRvIII and to compete with EGF,39 which preferentially recognizing domains I (aa 1–165) and III (aa 310–481), suggesting that major interactions between MinE07 and EGFR may involve domain III. To better define the MinE07 binding site, we performed cell binding competition assays with three anti-EGFR mAbs (nimotuzumab, cetuximab, and matuzumab) that target domain III in distinct epitopes.58,59,60 Nimotuzumab binds EGFR by engaging primarily aa 353 and a stretch of contiguous aa from 356 to 359 that are located toward the N-terminal end of domain III.61 All these residues are also found within the EGF binding pocket. Aa 353 is also recognized by cetuximab, which in turn shows critical interactions with aa 384, 408, 418, 440, 443, 465, 467, 468, 471, and 473.60,61 In contrast, matuzumab primarily binds an epitope loop on domain III spanning aa 454–464 that precedes the C-terminal end of domain III and does not block EGF binding as the other two anti-EGFR mAbs do.59
H1975 cells were incubated with Cy5-MinE07 (50 nM) in the absence or presence of 10-fold molar excess of anti-EGFR Ab. Competition between Cy5-MinE07 and a non-labeled version of MinE07 that lacks the tail sequence was also used as positive control (Figure 1B). Our results indicate that Cy5-MinE07 effectively labeled H1975 cells in the presence of matuzumab or a control immunoglobulin (Ig) G (anti-GAPDH mAb), but cell labeling markedly decreased when MinE07 was co-incubated with cetuximab or nimotuzumab (Figure 1B). Cell binding competition assays were repeated at 4°C to prevent EGFR internalization and to ensure that the reduction of Cy5 MFI was not caused by a decrease of cell-surface EGFRs upon Ab-mediated endocytosis. MinE07 again competed with cetuximab and nimotuzumab for binding to EGFR but not with matuzumab. These data illustrate that MinE07 interaction site overlaps with important residues involved in nimotuzumab and cetuximab recognition but is distinct from that of matuzumab (Figure 1C).
Next, we investigated whether MinE07 binding is affected by resistance mutations in the EGFR ectodomain that occur in colorectal cancer (CRC) in response to cetuximab therapy. EGFR domain III single point mutations (S464L, G465E, G465R, or S492R), which account for ∼20% of CRC patients with clinical resistance,62 were transfected into HEK293FT cells and labeled with Cy5-MinE07 as above. While MinE07 retained binding to each of the mutant EGFR at levels well above background, cell labeling was decreased approximately 50%–70% for HEK293FT cells expressing EGFR mutations at residues 464 and 465 but not at 492 (Figure S6). These results suggest that S464L, G465E, and G465R affect MinE07 binding properties to some extent but that they do not fully abolish its ability to label EGFR-expressing cells. From these results, it is unclear whether MinE07 establishes direct contacts with S464 or G465 or whether mutations of these residues drive conformational rearrangements in the MinE07 binding site that ultimately affect its affinity. Nevertheless, current efforts are ongoing to further define the EGFR-MinE07 interaction and provide a well-characterized RNA ligand to the list of available reagents for EGFR targeting.
Engineering of EGFR/c-Met bsApts
Targeting of two overexpressed cell-surface receptors on the same cells (in cis binding) is a proven strategy to increase tumor selectivity.15,17,18 To enhance targeting of NSCLC cells, we linked MinE07 to a second aptamer CLN0003 (hereinafter called CLN3) that binds c-Met, another tyrosine kinase receptor involved in lung cancer progression and drug resistance.3,63,64 CLN3 is a 50-nt DNA aptamer that has been widely used in vitro and in vivo by different groups and has been often reported with the name SL1.65,66,67,68 Monospecific CLN3 recognized endogenously expressed c-Met on the surface of NSCLC cells (blue bars in Figures 2B and S7A). EGFR/c-Met ratios for H820, H1975, and A549 cells as measured by MinE07 and CLN3 cell labeling were indistinguishable from ratios obtained from those measured employing commercially available APC-labeled mAbs against EGFR and c-Met (Figure S7). This result reinforces the precise receptor labeling and specificity of aptamers MinE07 and CLN3 and supports their use as valid Ab alternatives for diagnostic applications of cell phenotyping.
Figure 2.

Engineered EGFR/c-Met bsApts improve labeling of NSCLC cells in vitro compared to monospecific aptamers
(A) Schematics of EGFR/c-Met bsApt engineering composed of MinE07/CLN3 aptamers. All designs utilized a 21-nt complementary 3′ tail/anti-tail hybridization to anneal monospecific aptamers and yield bsApt complexes. In the three proposed bsApt designs (A–C), the structural variations are included in the CLN3 sequence between the 3′ end of the aptamer and anti-tail. Design A contains a short dinucleotide linker, design B has a flexible dTx15 linker, and design C possesses a structurally constrained stem-loop. (B) A panel of cell lines expressing moderate/high levels (H820, H1975, A549) or very low levels (MCF7) of endogenous EGFR (WT or mutant) and c-Met (WT) were incubated with 50 nM Atto647N-labeled aptamers. Flow cytometry was used to measure cell labeling. Atto647N MFI of cells is reported on the y axis. All designs (A–C) of EGFR/c-Met bsApt (yellow bars) increased labeling of NSCLC cell lines compared to monospecific aptamers (MinE07 and CLN3, green and blue bars, respectively), dual-aptamer complexes with specificity only for c-Met (CLN3 annealed to mutMinE07, blue/white bars), and monospecific control aptamer (mutMinE07, gray bars). (C) We titrated the amount of MinE07tail and tested bsApt labeling of H1975 cells (EGFR/c-Met ∼1) while keeping the concentration (50 nM) of the Atto647N-labeled CLN3 (design B) constant. Cell labeling was measured by flow cytometry and Atto647N MFI of cells is reported on the y axis. Again, bsApt complex (yellow bars) showed a superior cell labeling compared to monospecific aptamers (MinE07 and CLN3, green and blue bars, respectively), dual-aptamer complexes with specificity either for c-Met (CLN3 annealed to mutMinE07, blue/white bars) or EGFR (MinE07 annealed to mutCLN3, green/white bars), and monospecific control aptamers (mutMinE07, gray bar and mutCLN3, black bar). These results also showed that binding of H1975 cells was dependent upon the effective concentration of annealed bsApt complexes, as a reduction of cell staining was noted at 1:0.5 M ratio of CLN3:MinE07. (D) Labeling of H1975 and A549 cells was measured upon incubation with two different concentrations (25 and 50 nM) of Atto647N-EGFR/c-Met bsApt (yellow bars) or a cocktail of unligated aptamers (Atto647N-MinE07 and Atto647N-CLN3, dark gold bars). In the aptamer cocktail, we used a 50-nt CLN3 aptamer with no 3′-anti-tail sequence, such that it cannot anneal with MinE07tail. As shown in the schematic above the graphs, the actual concentration of Atto647N in the aptamer cocktail was double the concentration of Atto647N-labeled bsApt. So, when we tested 25 nM bsApt, the actual concentration of Atto647N in the cocktail was 50 nM (25 nM for each aptamer). A cocktail of unligated, control aptamers (mutCLN3 and mutMinE07, gray bars) was used to ensure that, even at higher concentration of Atto647N, the non-specific cell labeling was kept minimal. EGFR/c-Met bsApt showed a higher cell labeling at 25 nM relative to the aptamer cocktail, but at 50 nM they both had equivalent MFIs. Plotted values represent mean ± SD for n = 3 independent experiments. Statistical analysis was performed for each cell line using a one-way ANOVA test (∗p < 0.05; ∗∗p < 0.01).
We designed three EGFR/c-Met bsApts (termed A, B, and C) that anneal the 2′-FY RNA tail of MinE07 and a complementary DNA anti-tail at the 3′ end of CLN3 (Figure 2A). While MinE07tail was kept consistent for all three designs, subtle structural changes were introduced in the CLN3 module. In design A, the 50-nt CLN3 sequence was extended with a TA dinucleotide linker followed by 21-nt anti-tail. The annealed tail-anti-tail duplex in design A is expected to separate MinE07 and CLN3 by ∼60 Å (assuming ∼2.9 Å of helical rise per bp in A-form hybrid DNA:RNA duplexes69,70). Designs B and C introduce additional nucleotides to increase spatial separation between MinE07 and CLN3 or to alter their 3D orientation. Specifically, design B includes a flexible single-stranded linker composed of 15 dT residues (dTx15), adding ∼105 Å for a total estimated separation of ∼165 Å (calculation details in section “materials and methods”). Design C displays a more structurally constrained stem-loop that is not expected to increase aptamer separation significantly relative to design A because its length is comparable with that of a di- or tri-nucleotide linker (∼14–21 Å). However, the geometry of the stem could alter 3D orientation of CLN3 and thus affects the actual spatial distance between MinE07 and CLN3. In addition, this stem-loop can also increase steric hindrance and structural rigidity of CLN3, which could influence c-Met recognition. To minimize the potential impact of molecular-weight differences on bsApt-targeting properties, CLN3 B and C were designed with nearly identical sequence lengths (88 and 89 nt, respectively).
The three variants of CLN3anti-tail were directly labeled with Atto647 N at their 5′ ends prior to forming bsApt complexes. Annealing reactions between Atto647N-CLN3anti-tail (A–C) and MinE07tail used a 1:2 molar ratio (CLN3:MinE07) to ensure that all Atto647N-CLN3anti-tail was assembled into the complex. Aptamer assembly was determined by native polyacrylamide gel shift assay (Figure S8). All three designs yielded >90% of CLN3anti-tail assembled into bsApt complexes with MinE07tail.
Dual-aptamer recognition of EGFR and c-Met improves labeling of NSCLC cell lines
RNA tends to fold hierarchically, with local sequences dominating local structure.71 Nevertheless, individual modules can interfere with each other when they are assembled or co-transcribed as chimera.72 For these EGFR/c-Met bsApts, several lines of evidence suggest that both the MinE07 and CLN3 modules retain their individual abilities to bind their respective target receptors. First, cell labeling was determined by flow cytometry using non-saturating concentrations (25 and 50 nM) of Atto647N-EGFR/c-Met bsApts and compared with monospecific and mutant aptamer samples (Figures 2B, S9, and S10). All three bsApts (yellow bars) display dose-dependent labeling of NSCLC cell lines that overexpressed WT (A549 cells) or mutated EGFRs (H1975 and H820 cells). In each case, cell staining was enhanced relative to monospecific MinE07 and CLN3 aptamers (green and blue bars, respectively) (Figures 2B, S9, and S10). In contrast, this improved cell labeling was not observed for bsApts in which Atto647N-CLN3anti-tail was annealed to mutMinE07tail (a control sequence with reduced EGFR binding50). Second, MFI values were unchanged when the molar ratio of CLN3:MinE07 (design B) was 1:2, 1:1.5, or 1:1, but a reduction of cell labeling was noted at 1:0.5 (Figure 2C), when formation of the active bsApt complex was diminished. Third, when MinE07tail was annealed to mutCLN3anti-tail, which bears 11 point mutations relative to CLN3, its ability to label H1975 cells was reduced by approximately 50% relative to MinE07 linked to anti-tail. This was likely due to unwanted intramolecular aptamer interactions that alter MinE07 folding and binding of EGFR. In contrast, addition of mutMinE07tail to CLN3anti-tail only slightly reduced H1975 cell labeling relative to the presence of the tail alone, demonstrating that CLN3 folding and ability to interact with c-Met were minimally affected. These results confirm that dual receptor recognition is required for the observed enhancement of NSCLC cell targeting. Our data also suggest that any structural differences among designs A, B and C—such as aptamer separation, rigidity, and 3D orientation—have little to no effect on net cell labeling. Thus, all three bsApt designs can be considered as strong initial candidates.
Flow cytometry assays were also performed using MCF7 cells, a breast cancer cell line that expresses low levels of EGFR and c-Met (EGFRlow/c-Metlow) and that mimic the levels found on healthy epithelial cells (<104 receptor/cell).73,74 All aptamer samples show a reduced labeling of MCF7 cells (Figures 2B, S9, and S10) in agreement with a diminished expression of their target receptors. In addition, all three bsApts did not significantly increase MCF7 recognition compared to monospecific MinE07 or CLN3. These data suggest that, even when EGFR/c-Met bsApts are used for a dual targeting approach, their recognition of cells that expressed low levels of receptors can still be minimal and thus a targeted cell labeling could potentially be achieved even in more complex systems. Altogether, we demonstrated that EGFR/c-Met bsApts improve in vitro targeted labeling of cell lines that overexpressed their target receptors, as in the case of NSCLC cells. Although significant differences were not observed among the three designs of bsApts, we decided to focus on the most promising of the three (design B) and use it for our next sets of evaluations.
As shown in Figure 2 and as previously observed,75 when linking two targeting ligands, the assembled complex can display an improved binding of cells overexpressing their target receptors as a result of an avidity gain.76 Depending on the linker length and affinity of the monovalent ligands, the avidity effect can be due to either a “statistical rebinding” or “chelate” effect.77 Thus, we sought to determine cell labeling properties of our EGFR/c-Met bsApt in comparison with a cocktail of unligated MinE07 and CLN3 in which the separated monomers do not possess either statistical rebinding or chelate effect. When tested at 25 nM, our EGFR/c-Met bsApt (design B) had an improved labeling of H1975 and A549 cells relative to the aptamer cocktail (composed of 25 nM Atto647N-MinE07 and 25 nM Atto647N-CLN3) (Figure 2D). However, when we incubated 50 nM Atto647N-labeled bsApt with the cells, its MFI values were almost equivalent to those obtained upon incubation with the aptamer cocktail (50 nM Atto647N-MinE07 + 50 nM Atto647N-CLN3). This result suggests that, at very low concentrations (e.g., 25 nM), ligand avidity plays a major role in improving receptor recognition and, hence, labeling of target cells. Whether this avidity effect is driven by a statistical rebinding (i.e., occurrence of only one binding event, followed by ligand dissociation, and binding of the same or second receptor) or chelate effect (i.e., simultaneous binding of both receptors) is unclear. Nevertheless, considering the difference in aptamer affinities (MinE07 apparent Kd ∼20 nM50 vs. CLN3 apparent Kd ∼120 nM66), we can speculate that EGFR/c-Met bsApt first binds EGFR via MinE07, improving the local concentration of CLN3 on the cell surface and in turn promoting recognition of c-Met.
Overall, the flow cytometry assays of Figure 2 demonstrate that both MinE07 and CLN3 retain their ability to recognize their target receptors when assembled in a bsApt complex. The avidity gain of this EGFR/c-Met bsApt improves targeted cell labeling compared to MinE07 and CLN3, and compared to a cocktail of the two monovalent aptamers.
Internalization and surface retention onto NSCLC cells driven by opposing receptor dynamics
Several groups,35,37 including ours,31,32 have demonstrated that aptamer endocytosis is essential to enable delivery and accumulation of therapeutic or diagnostic payloads into target cancer cells. Aptamers can be internalized into cells via receptor-mediated endocytosis upon binding to their target receptors.78 To determine whether MinE07, CLN3, and EGFR/c-Met bsApt are effectively internalized into H1975 cells, cellular localizations of Atto647N-labeled aptamers were monitored by confocal microscopy on living cells that were stained with a membrane marker (CellBrite) and a nuclear label (Hoechst) (Figure 3). Only minimal background fluorescence was detected after 30-min incubation with a non-targeting control RNA aptamer (C36).45 MinE07 displays high intracellular accumulation with a punctate pattern that is consistent with its localization into endosomal vesicles, while CLN3 shows clear cell-surface staining (colocalization with CellBrite), along with some cytosolic puncta that could be due to endocytic recycling and/or plasma membrane turnover. Thus, on their own, these two aptamers favor opposing cell localization outcomes. EGFR/c-Met bsApt combined features from both aptamers and yielded both cell-surface staining and a punctate pattern in the cytosol. An increased fluorescence signal relative to monospecific MinE07 and CLN3 was also observed, consistent with the improved EGFR/c-Met bsApt binding to H1975 cells noted above.
Figure 3.

EGFR/c-Met bsApt stains the cell surface and is internalized into H1975 cells
Confocal microscopy images of H1975 cells stained upon incubation with 150 nM Atto647N-labeled aptamer samples: C36 (non-targeting control RNA aptamer), MinE07 (anti-EGFR aptamer), CLN3 (anti-c-Met aptamer), or EGFR/c-Met bsApt (bsApt). Cells were also stained with CellBrite Steady 488 (cell membrane stain) and Hoechst 33342 (nuclear marker stain). Representative images from one of n = 2 independent experiments are shown: Atto647N-aptamers (red, excitation 647 nm/emission 657–692 nm), cell membrane (green, excitation 488 nm/emission 521–551 nm), nuclei (blue, excitation 405 nm/emission 436–473 nm). Scale bars, 20 μm. Qualitatively, MinE07 was internalized after 45-min incubation at 37°C and found predominantly in the cytosol, as it did not show any colocalization with the cell membrane stain. While CLN3 internalized, it also co-localized with cell membrane stain. EGFR/c-Met bsApt showed both surface staining and fluorescent intracellular puncta, and its total cell staining was greater compared to monospecific aptamers (MinE07 and CLN3).
Internalization was independently evaluated by flow cytometry after first removing cell-surface membrane-bound aptamers by nuclease digestion using either a cocktail of RNases (RiboShredder) to target MinE07 or a deoxyribonuclease (DNase I) to target CLN3. In this assay, only internalized aptamers are protected from the nuclease digestion and contribute to the residual fluorescence measured by flow cytometry. Upon exposure to RiboShredder, MinE07 still retained ∼90% of cell labeling (Figure S11A), confirming its rapid internalization into H1975 cells. Cell staining by the DNA aptamer CLN3 was not affected by RiboShredder but, upon conjugation with MinE07, only ∼55% of EGFR/c-Met bsApt signal was resistant to the RNA aptamer digestion. Similarly, upon DNase I treatment, the residual fluorescence of CLN3 and EGFR/c-Met bsApt was reduced, showing ∼70% and ∼60% of residual cell labeling, respectively (Figure S11B). Exposure to DNase I increased cell staining for both of the RNA aptamers (MinE07 and non-binding control C36), likely due to removal of salmon sperm DNA, which had been included as a competitor/blocking agent to minimize non-specific binding and endocytosis.
Overall, our findings show that MinE07 and, to a lesser extent, EGFR/c-Met bsApt are internalized and show punctate intracellular staining in H1975 cells, whereas CLN3 predominantly localizes on the cell surface and displays a slower endocytic uptake than MinE07 (Figure 3). Mutant EGFRs harboring NSCLC oncogenic mutations are constitutively internalized via clathrin-mediated endocytosis even in the absence of ligand stimulation and show aberrant endosomal trafficking.79,80 Thus, we propose that intracellular uptake of MinE07 and EGFR/c-Met bsApt is predominantly driven by EGFR L858R/T790M as a result of its aberrant trafficking into the endocytic recycling compartment.
In vivo targeting of NSCLC harboring EGFR L858R/T790M
We next sought to determine whether MinE07 targets NSCLC harboring clinically relevant EGFR oncogenic and resistance mutations and to elucidate whether dual-aptamer specificity (EGFR/c-Met bsApt) improved tumor targeting in vivo. BALB/c nude mice were implanted with H1975 cells (EGFR L858R/T790M) to develop subcutaneous, ectopic xenograft tumors (see schematic in Figure S12). Approximately 3–4 weeks after cell engraftment, Cy7-labeled aptamer samples were injected retro-orbitally into blood circulation at a dose of 50 pmol/g, (1.0–1.5 nmol of aptamer per animal). To define aptamer-dye biodistribution and tumor-targeting properties, whole-body fluorescence images were acquired at different time points over the course of 48 h as indicated below for each experiment, followed by ex vivo fluorescence imaging of harvested tissues (liver, spleen, kidney, tumor, and muscle). H1975 cells were initially implanted subcutaneously into the right flank of each mouse (Figure 4A). However, the high level of Cy7 fluorescence in the kidneys hampered a clear evaluation of aptamer tumor targeting due to proximity to the kidneys. Nevertheless, Cy7 signal in the implanted tumors was still visible at 48 h post injection (hpi) in mice treated with MinE07 and EGFR/c-Met bsApt, whereas signal for C36 or vehicle was lower (orange arrows in Figure 4A).
Figure 4.

Cy7-labeled MinE07 and EGFR/c-Met bsApt accumulates primarily in tumor, kidney, and liver during in vivo biodistribution time course
H1975 cells were subcutaneously implanted into the flank (A) or shoulder (B) of 6- to 8-week-old nude mice and allowed to grow for 2–3 weeks until they reached ∼75–150 mm2. Yellow ovals highlight tumor location. Once tumors reached appropriate size, either PBS (vehicle, white) or 50 pmol/g (∼1-nmol bolus) of Cy7-labeled aptamers was injected into the retro-orbital (RO) sinus. (A) Injected aptamer samples were C36 (non-targeting aptamer, gray), monospecific MinE07 (anti-EGFR aptamer, green), or EGFR/c-Met bsApt (bsApt, yellow). (A) At 3, 6, 24, and 48 h post injection (hpi), mice were anesthetized with isoflurane and imaged using the IVIS Spectrum (excitation 710 nm/emission 760 nm). EGFR/c-Met bsApt and MinE07 localized more to the tumor at 48 hpi compared to controls. Earlier time points (3–24 hpi) showed fluorescence signal at the tumor for all aptamer samples, including non-binding control aptamer C36. (B) To elucidate whether aptamer size played a role in biodistribution and penetration into tumor tissues, we compared EGFR/c-Met bsApt (yellow) to monospecific aptamers (MinE07, green; CLN3, blue) that were annealed to a negative control aptamer (scDW4 or C36, gray) of similar size (∼30 kDa, as opposed to ∼7 kDa when annealed to tail or anti-tail oligos as shown in A). All dual-aptamer complexes in (A) range from ∼45 to ∼50 kDa. Mice were injected and imaged as in (A). Negative control aptamer (scDW4/C36) lost most tumor signal by 6 hpi, whereas monospecific anti-EGFR aptamer (MinE07/scDW4) did not lose the majority of tumor signal until 24 hpi. Monospecific anti-c-Met aptamer (CLN3/C36) and EGFR/c-Met bsApt still show Cy7 fluorescence signal in the tumors at 24 hpi, but a significant reduction occurred at 48 hpi. (A and B) Representative images of one mouse per condition from a single experiment are shown. Color scale of radiant efficiency in (A) is minimum = 2.5 × 107, maximum = 9.0 × 107. Color scale of radiant efficiency in (B) is minimum = 1.7 × 107, maximum = 2.5 × 108. Red arrows highlight signal retention in tumor at 48 h. Cohort of mice used in (A) was n = 2 mice for vehicle, n = 3 for C36 and EGFR/c-Met bsApt, and n = 4 for MinE07. Cohort of mice used in (B) was n = 2 mice for vehicle (not shown), C36/scDW4, MinE07/scDW4, and EGFR/c-Met bsApt; n = 3 for CLN3/C36.
To better define tumor targeting and Cy7 delivery with less interference from kidney accumulation, H1975 cells were implanted subcutaneously into the upper back/shoulder of the mice and lateral view images were acquired (Figure 4B). Early time points (3 and 6 hpi) showed no clear distinction in tumor targeting among the aptamer samples, potentially due to passive accumulation via the enhanced permeability and retention (EPR) effect due to leaky tumor vasculature.81 At 10 hpi, Cy7 fluorescence in the tumors persisted in mice treated with dual-aptamer complexes containing MinE07, CLN3, or both (EGFR/c-Met bsApt), while signal was reduced when Cy7 was delivered by a complex composed of two non-targeting aptamers (C36tail and scDW4anti-tail). Cy7 signal at the tumor persisted at 24 and 48 hpi when MinE07tail was linked to CLN3anti-tail (design B) to yield an EGFR/c-Met bsApt. In contrast, Cy7 signal at the tumor almost disappeared at 24 hpi for MinE07tail annealed to the control DNA (scDW4anti-tail), suggesting that H1975 tumor targeting by MinE07 diminished upon annealing with a less structured aptamer such as scDW4, similarly to what was observed in Figure 2C upon its annealing with mutCLN3.
Consistent with whole-body imaging above (Figure 4), analysis of Cy7 ex vivo fluorescence at 48 hpi showed the highest off-tumor levels in the kidneys (Figure 5A). To better define Cy7 delivery in the kidneys, we applied a color scale that reduced the overall fluorescence signal and avoided Cy7 saturation in kidney tissues (Figure S13). The highest Cy7 fluorescence in kidneys was detected when using MinE07 in the form of single, monospecific aptamer (i.e., MinE07tail linked to the small anti-tail) (Figure S13B). In contrast, when MinE07 formed the bsApt complex with CLN3, Cy7 off-tumor signal in the kidneys was reduced. This result indicates that, as the molecular weight of the aptamer formulation increased (from monospecific MinE07 ∼28 kDa to EGFR/c-Met bsApt ∼50 kDa), aptamers are less effectively removed from circulation through the renal glomerular filtration system (molecular-weight cutoff ∼45 kDa).
Figure 5.

Cy7-labeled MinE07 and EGFR/c-Met bsApt persist into H1975-derived tumor tissues at 48 hpi
Ex vivo imaging of major organs/tissues from cohort of mice described in Figure 4. At 48 hpi, organs were harvested and immediately imaged using the IVIS Spectrum (excitation 710 nm/emission 760 nm). (A) Representative ex vivo images of tissues are shown (from top to bottom): liver, kidney, spleen, tumor, muscle. (B) Ex vivo images of all H1975 tumors. Color scale of radiant efficiency in (A) and (B) is minimum = 9.9 × 106 maximum = 3.4 × 107. (C) Quantification of Cy7 fluorescence in all tumor tissues shown in (B). Average radiant efficiency ((p/s/cm2/sr)/(μW/cm2)) is plotted on the y axis and represent mean ± SD for n = 2–5 mice (two independent experiments). Monospecific MinE07 (anti-EGFR aptamer, green bar) and EGFR/c-Met bsApt (bsApt, yellow bar) showed significantly higher radiant efficiency compared to vehicle (PBS, light gray bar) and C36 (control aptamer, dark gray bar). Note that individual values of average radiant efficiency of C36 in (C) combine results obtained with C36 annealed to anti-tail (first three tumors from top) or scDW4 (last two bottom tumors). Scale bars represent relative radiant efficiency. Statistical analysis was performed using a one-way ANOVA test (∗p < 0.05, ∗∗p < 0.01).
Quantitative analysis of ectopic tumor targeting of all tumor tissues (Figures 5B and 5C) showed that Cy7 fluorescence accumulation in the H1975-derived tumor tissues was highest in mice injected with monomeric MinE07 or with EGFR/c-Met bsApt relative to mice treated with vehicle, a non-targeting aptamer (C36), or dual-aptamer complexes composed of C36 linked to scDW4 or CLN3 (Figure 5C). Importantly, monospecific MinE07 displayed significant tumor staining relative to C36 when annealed to the small anti-tail, accumulating to a similar magnitude as the EGFR/c-Met bsApt. However, the same monospecific MinE07 aptamer failed to retain tumor targeting at 48 hpi when linked to scDW4. These results are in contrast with the cell-based, in vitro comparative analysis above, where EGFR/c-Met bsApt showed a superior labeling of NSCLC cells relative to monospecific MinE07 and CLN3 (Figures 2 and 3). To better understand this discrepancy, we sought to determine whether antigen levels were altered in vivo. EGFR and c-Met expression levels were measured for H1975 cells recovered from ex vivo tumor tissue and this analysis revealed that CLN3 was downregulated in a small subset (n = 1 of 4) of animals, whereas EGFR was expressed at similar levels in all mice of that cohort (n = 4) (Figure S14). Thus, in tumors with low to no cell-surface expression of c-Met, only the MinE07 module of the EGFR/c-Met bsApt was still able to specifically recognize H1975 cells. The altered in vivo expression of c-Met may have played a role in the targeting of ectopic xenograft lung cancers, in conjunction with any differences in tumor penetration and endocytosis rate of MinE07 relative to EGFR/c-Met bsApt (see section “discussion” below).
Choice of fluorophore affects in vivo aptamer biodistribution
Whole-animal and ex vivo imaging of dye-labeled probes such as aptamers and mAbs are often interpreted as representing the biodistribution propensities of the probe molecule itself. However, the dye moiety can contribute directly to biodistribution via its overall hydrophobicity and its impact on interactions with serum proteins and other molecules.82,83,84 Our biodistribution findings using Cy7-labeled aptamers appeared to be inconsistent with recent observations by Levy and colleagues45 where in vivo fluorescence of Alexa Fluor 750 (AF750)-labeled aptamers was largely undetectable by 24 h. Therefore, we directly compared biodistribution of EGFR/c-Met bsApt upon its conjugation with two NIR dyes with similar spectral properties but different physicochemical characteristics. Cy7 (excitation/emission, 743/767 nm) is highly hydrophobic and positively charged, while AF750 (excitation/emission, 749/775 nm) is hydrophilic and negatively charged. The EGFR/c-Met bsApt labeled H1975 cells with equivalent efficacy when conjugated to either dye, confirming that the choice of dye does not affect cell targeting in vitro (Figure S15). However, a clear distinction in the aptamer-dye biodistribution was apparent following systemic injection. Whole-body fluorescence imaging revealed that EGFR/c-Met bsApt conjugation to AF750 reduced non-specific interactions with and uptake within liver and kidneys relative to Cy7 conjugates. In addition, non-specific AF750 signal was rapidly cleared, allowing effective tumor targeting to be observed at 8 hpi with a strong fluorescence contrast that clearly delineates the ectopic lung cancer tissues (Figure 6A). In contrast, when the more hydrophobic Cy7 was used, fluorophore accumulation in the tumor persisted even at longer times (up to 48 h), but, as observed above (Figure 4), it also showed strong undesired kidney uptake (Figure 6). Ex vivo fluorescence imaging at 48 hpi further confirmed that Cy7 but not AF750 was retained in the kidney tissues (Figure 6B). When imaging only the tumor tissues, a fluorescence signal above the background was present at 48 h exclusively upon treatment with Cy7-labeled EGFR/c-Met bsApt (Figure 6C). It is important to consider that the signal from the fluorescent dye can persist even if it is separated from the probe by digestion or chemical cleavage. To assess the stability of the aptamers under in vivo-like conditions, aptamers were incubated in 55% serum at 37°C for various times and analyzed by native polyacrylamide gel and (RT)-qPCR (Figures S16A and S16B). These analyses showed that full-length, annealed MinE07 and the EGFR/c-Met bsApt were largely detectible at 10 h but mostly degraded by 24 h. Therefore, it is likely that the in vivo Cy7 signal observed at later time points (24 and 48 h; not observed with AF750) is either from tissue-accumulated dye (±aptamer) or partially digested circulating probe. Importantly, these results demonstrate that the conjugation to a small-molecule dye can affect observed (i.e., apparent) aptamer biodistribution and that hydrophobic dyes such as Cy7 can persist in certain tissues (kidneys, tumors) and contribute to the fluorescence signal even after digestion and clearance of the targeting aptamers.
Figure 6.

Fluorophore affects aptamer biodistribution in vivo
Cy7- or AF750-labeled EGFR/c-Met bsApt were retro-orbitally injected into mice bearing H1975 tumors on the upper back/shoulder. Yellow ovals highlight tumor location. (A) At 4, 8, 24, and 48 hpi, mice were anesthetized and imaged using the IVIS Spectrum using two tailored settings (see section “materials and methods” for further details). Representative images using the AF750 IVIS method (excitation 745 nm/emission 800 nm) are shown. EGFR/c-Met bsApt labeled with AF750 (a hydrophilic dye) clearly stained the tumor xenograft at 8 hpi with good contrast but showed a significant reduction at later time points, whereas the tumor staining from EGFR/c-Met bsApt labeled with Cy7 persisted out to 48 h. (B and C) Ex vivo imaging was performed at 48 hpi using the same conditions as in Figure 5: excitation 710 nm/emission 760 nm. Representative ex vivo images of major tissues collected (B) or tumors alone (C) from n = 1 independent experiment are shown. Cy7-labeled bsApt retained signal in kidney, liver, and in part tumor tissues out to 48 hpi. Color scale of radiant efficiency in (A) is minimum = 3.0 × 107 maximum = 4.0 × 108. Color scales in (B) and (C) are reported.
Discussion
Several solid cancers overexpress WT EGFR or harbor oncogenic EGFR mutations that drive tumor progression, as in the case of EGFR-mutant NSCLC. Targeting ligands that bind both WT and mutant EGFR is highly desirable to develop new diagnostics, therapeutics, or adjuvants to current treatments. Examples of ligands that target both WT and mutated EGFR are mAbs such as cetuximab, nimotuzumab, and matuzumab. However, unlike the notable clinical success for mAb treatment of CRC or head and neck cancer, anti-EGFR mAbs have shown marginal clinical benefit for the treatment of NSCLC.7 For instance, the phase III FLEX clinical trial demonstrated an improvement in overall survival for cetuximab combined with cisplatin and vinorelbine relative to chemotherapy alone (cisplatin plus vinorelbine), but the increased toxicity worsened the safety profile.85 In two recent case reports of patients with lung adenocarcinoma harboring oncogenic and acquired EGFR mutations, dual inhibition of EGFR by two different mechanisms using both TKIs (osimertinib or brigatinib) and an mAb (cetuximab) improved survival and showed tolerable toxicity.86,87 However, safety and efficacy of these approaches need to be further assessed in larger cohorts of NSCLC patients to determine applicability of combinatorial treatments. Indeed, a phase II clinical trial using EGFR-mutant NSCLC patients recently demonstrated that TKI (afatinib) in combination with cetuximab did not show significant clinical improvement and displayed a slight increase of toxicity relative to TKI alone.88
Aptamers have binding properties similar to mAbs but possess lower immunogenicity, and their synthesis uses a chemical technology with high batch fidelity that is not intrinsically prone to viral or bacterial contamination. These qualities make them ideal reagents to produce following the current Good Manufacturing Practice (cGMP) regulations. In this work we focused our attention on aptamer MinE07, a 2′-FY RNA aptamer that has been previously validated by our lab50 and others,45,49,52 which demonstrated high affinity and specificity for WT EGFR. Because MinE07 targets the extracellular domain of EGFR, we hypothesized that, like anti-EGFR mAbs, its recognition of EGFR would be less susceptible to oncogenic or acquired resistance mutations that occur in the intracellular TKD. Thus, we first determined that MinE07 binding was retained even when EGFR harbors clinically relevant mutations found in NSCLC patients (Figure 1A). Then, using competition assays, we found that MinE07 competes with cetuximab and nimotuzumab but not with matuzumab. Thus, we elucidated that important residues of the MinE07 binding site lie within domain III of the extracellular domain of EGFR (Figures 1B and 1C). Interaction of MinE07 with domain III is consistent with its ability to target EGFRvIII, an EGFR variant with deletions in domain I (aa 1–165) and II (aa 166–310) but not in domain III (aa 310–481).
MinE07 competition with cetuximab and nimotuzumab but not matuzumab is consistent with its ability to compete with EGF, as cetuximab and nimotuzumab do, and to inhibit ligand-dependent EGFR signaling.39,53 EGFR is present on the cell surface in two interchangeable conformations: a tethered, inactive conformation and an extended, active one (shown in Figure 2C). Oncogenic driver mutations (e.g., L858R) in the EGFR TKD found in NSCLC patients can promote an inside-out extracellular dimerization that activates EGFR even in the absence of ligand stimulation,89 ultimately driving canonical (EGF-like) or non-canonical signaling.7 Our experiments of single-molecule fluorescence tracking demonstrated that MinE07 still allows formation of ligand-independent EGFR dimers upon aptamer binding to EGFR L858R/T790M on the surface of H1975 cells (Video S1), consistent with our previous findings on HeLa cells.50 These preliminary findings suggest that MinE07 does not prevent ligand-independent EGFR dimerization, in contrast with cetuximab and nimotuzumab.90 However, whether these aptamer-bound EGFR dimers can still adopt the extended, active conformation is not yet clear. Importantly, our findings illustrate that, despite its competition with cetuximab and nimotuzumab, MinE07 interacts differently with EGFR. The nature of this RNA-protein interaction could potentially lead to different biological outcomes when using MinE07 to target distinct EGFR variants. Current studies in our lab are ongoing to clarify this point.
H1975 cells were incubated with 25 nM Atto647N-labeled MinE07 and single-molecule fluorescence imaging was performed. The video consists of a time-lapse sequence acquisition of diffraction-limited total internal reflection fluorescence (TIRF) images and shows extended colocalization of two individual EGFRs labeled with MinE07 on the basal cell membrane. A clear increase of fluorescence intensity is detected upon colocalization. The lifetime of the colocalization event is ∼1.9 s (21 frames), suggesting that this is not due to random molecular collisions or stochastic colocalization (lifetime <0.2 s). TIRF microscopy was performed on a Leica SR GSD 3D microscope using a 160× objective lens (HC PL APO 160× /1.43, oil immersion), a 500-mW 642-nm laser (MPBC, Montreal, Quebec, Canada), a 710/100-nm emission bandpass filter, and an EMCCD camera (iXon Ultra 897, Andor, UK). Time-lapse sequences were recorded as previously described in Delcanale et al. (https://doi.org/10.1002/anie.202004764). Camera exposure time, 50 ms; frame acquisition time (or total time per frame), 90 ms; laser intensity, 40% of maximum power; total frames in video S1, 78; video reproduction, 5 frames per second. Scale bar, 5 μm.
Because targeting of heterogeneous tumor cells is challenging, we sought to apply a dual targeting strategy to improve specificity toward EGFR-mutant NSCLC cells. Therefore, we used an approach of bottom-up assembly to enable MinE07 hybridization with a DNA aptamer (CLN3) that targets c-Met, a tyrosine kinase receptor overexpressed on several NSCLC cell lines. In vitro flow cytometry measurements of NSCLC cell labeling clearly demonstrated that EGFR/c-Met bsApt outperforms monospecific aptamers MinE07 and CLN3 (Figure 2). However, to our surprise, the same trend was not observed when testing these aptamers in preclinical mouse models of human lung cancer xenografts. Indeed, when labeled with Cy7, a hydrophobic NIR dye, MinE07 showed comparable ability with EGFR/c-Met bsApt in targeting subcutaneous H1975 cell-derived tumors in nude mice (Figures 5B and 5C). At least three key factors may have contributed to this disparity. First, we found that c-Met expression could be downregulated for a subset of tumors in vivo, thereby reducing the targeting ability of EGFR/c-Met bsApt. Second, the small size of MinE07 (∼28 kDa) compared with EGFR/c-Met bsApt (∼50 kDa) might facilitate its penetration and accumulation into the tumor tissues. Third, we found that MinE07 internalizes into H1975 cells more readily than EGFR/c-Met bsApt (Figures 3 and S11). This enhanced MinE07 endocytosis and recycling may lead to more efficient intracellular Cy7 delivery and net accumulation relative to EGFR/c-Met bsApt, despite the ability of the latter to target both EGFR and c-Met.
In this work, we also noted that recognition of NSCLC cells by their targeting aptamers relative to non-specific RNA sequences was remarkably lower in vivo than in vitro, making it more difficult to distinguish clearly among different aptamers. For example, MinE07-labeled H1975 cells in vitro ∼10-fold more than a control RNA (mutMinE07; Figures 2 and S4), and the ratio for EGFR/c-Met bsApt cell targeting was even higher (∼25-fold). In contrast, the fluorescence signal in ex vivo tumor tissues (IVIS) for MinE07 and EGFR/c-Met bsApt was only ∼1.5-fold higher than a control RNA (C36; Figure 5C). These data are consistent with previous findings in which signal of an anti-PSMA aptamer (monomeric A9g), in ex vivo tumor tissues, was ∼1.5- to 2-fold over background, and aptamer fluorescence relative to background was again significantly lower in vivo than in vitro.91 Clearly, the sensitivity of flow cytometry (single-cell level) is vastly different from in vivo and ex vivo fluorescence imaging of tissues or whole organisms. While aptamers usually bear a single fluorophore per molecule, adding more fluorophores per aptamer or using radionuclides in place of fluorescent probes might improve in vivo sensitivity. Nuances of tumor biology can also affect observed tumor targeting and biodistribution.92,93 In preliminary in vivo studies (not shown here), we noticed that increased tumor size, vasculature, and presence of necrotic tissues significantly increased the level of autofluorescence. Thus, to avoid misleading interpretation and minimize variation in fluorescence, only animals with similar tumor size (∼75–150 mm2) at the time of aptamer injection were used for the experiments described here.
Our results also bring attention to the importance of the choice of fluorescent dye in determining the outcomes of in vivo biodistribution studies. While several reports have highlighted that choice of dye, conjugation strategy, and degree of labeling can significantly affect in vivo biodistribution in the Ab and nanoparticle fields,82,83,84 a similar comparative analysis using aptamers labeled with different fluorophores has not yet been reported, to the best of our knowledge. Here, for the first time, we compared how two different NIR dyes (Cy7 and AF750) with similar spectral properties but distinct charge and hydrophobicity affect in vivo biodistribution of the same aptamer complex (EGFR/c-Met bsApt). For Cy7 and AF750, we used the same conjugation strategy (NHS ester labeling of 5′-NH2-CLN3) and high-pressure liquid chromatography (HPLC) purification, which ultimately provided aptamer-dye conjugates with the same degree of labeling (1:1 stoichiometry) and same ability to label H1975 in vitro (Figure S15). For the in vivo model, we observed that AF750-labeled EGFR/c-Met bsApt was cleared out from the circulation quite rapidly, whereas Cy7 contributed to an extended fluorescence signal in kidney and tumor tissues (Figure 6) even after the aptamer was likely degraded (Figure S16) or cleared from circulation. Our data are consistent with previous findings; indeed, persistence of fluorescence signal in the kidneys after >12 hpi were observed upon systematic injection of free Cy794 or several other aptamers68,91,95 that were labeled with other hydrophobic cyanine dyes (Cy5 or Cy5.5). Fluorescent signal recorded at longer time points is probably associated with free fluorophore or partially digested probe accumulating inside the cells. Upon aptamer digestion in the endo-lysosomal compartment, hydrophobic cyanine dyes, such as Cy7, could persist for several hours or days inside the cells, especially in highly vascularized tissues, such as tumors, or tissues that clear oligonucleotides from the circulation, such as the kidney or liver (e.g., Kupffer cells). In addition, it is also possible that, owing to its high hydrophobicity and/or positive charge, Cy7 might favor interactions with circulating serum proteins, such as albumin. Improved interactions with serum albumin were recently described for a DNA aptamer (Sgc8) that was chemically synthesized to incorporate seven hydrophobic residues (3,5-bis(trifluoromethyl)benzoyl) in its sequence.96 Similar to our Cy7-labeled aptamers, the modified Sgc8 aptamer showed a prolonged circulation time that favored fluorescence accumulation in kidney and tumor tissues at longer time points (up to 54 hpi). Accumulation in non-target tissues such as kidneys or liver could also reduce the effective concentration of circulating aptamer and the actual tumor-targeting capability of that given aptamer formulation. Indeed, when EGFR/c-Met bsApt was labeled with AF750, this reagent clearly defined the ectopic lung cancer tissues at 8 hpi with a stronger fluorescence contrast and minimal background signal in the nearby tissues compared with Cy7-labeled EGFR/c-Met bsApt (Figure 6A). The choice of the proper dye conjugate is critical when testing tumor targeting and biodistribution, especially as the observed signal is coming from the fluorophore (i.e., apparent biodistribution) rather than the oligonucleotide itself (i.e., true biodistribution, as in in situ hybridization). Based on our results, we believe that, for in vivo fluorescence imaging studies of aptamer biodistribution, the more hydrophilic AF750 is less likely to contribute to non-specific (i.e., non-aptamer-driven) signal from dye-protein interactions and thus may be preferred over Cy7. In conclusion, our in vivo and ex vivo data demonstrate that monospecific and bsApt formulations containing the 2′-FY-modified RNA aptamer, MinE07, can target ectopic EGFR-mutant lung cancer even without further stabilization or PEGylation. This outcome is quite remarkable considering that MinE07 also competes in vivo with endogenous EGF and not only is specific for human tumor cells but also cross reacts with the murine EGFR.39
We demonstrated that MinE07 recognizes WT and mutant EGFR on NSCLC cells, targets ectopic lung cancer, delivers small molecule dyes inside target cells, and can be easily engineered to yield effective bsApt constructs. Owing to these findings, MinE07 represents an attractive reagent for targeting NSCLC harboring clinically relevant EGFR mutations in studies of basic cancer research or for future development of aptamer-drug conjugates. That said, it is still important to address the pharmacokinetic properties that impede the clinical translation of aptamer technology, such as metabolic instability. Various groups have demonstrated the feasibility of alternative oligonucleotide delivery methods, such as intranasal drip or inhalation in the context of lung disease.97,98 Further developments in the field of local delivery (e.g., interventional radiology) may enable the use of aptamers with less extensive modifications, as these delivery methods can decrease aptamer exposure to serum nucleases before reaching their targets. Aptamer size, nucleotide sugar or backbone modifications, and conjugation methods also play key roles in aptamer pharmacokinetic properties. BsApts are larger and less likely to be quickly filtered through the kidney glomeruli compared to monospecific aptamers (Figure S13B), allowing for improved circulation times. Our aptamer constructs were designed using a hybridization method commonly used in the delivery of doxorubicin,32 in aptamer-siRNA conjugates (AsiC),56 and in immunomodulating bsApts.28 While hybridization is a relatively stable method of conjugation with respect to dissociation of the complex (calculated free energy of tail/anti-tail = −34 to −41 kcal/mol; Figure S16C), full-length MinE07 and EGFR/c-Met bsApt were still mostly degraded by 24 h under in vivo-like conditions (Figures S16A and S16B). This suggests additional sequence design and engineering methods should be explored to fine-tune aptamer pharmacokinetics. Chemical modifications that slow renal clearance (e.g., PEGylation), improve nuclease resistance (e.g., incorporation of 2′-OMe or addition of 3′ inverted dT), or enhance linker stability (e.g., circularization using DNA/RNA ligase or introduction of LNAs into the hybridized sequences) might extend its circulation half-life and tumor targeting properties, provided that tumor penetration, EGFR recognition, and rapid endocytosis are not drastically affected.14,55 In this regard and in light of current SELEX enzyme limitations,99 medicinal chemistry approaches that have been successfully applied to improve clinical success of other RNA-based oligonucleotide drugs (siRNAs and ASOs) offer a fantastic opportunity to academic labs and biotech companies that could apply similar strategies to unlock the therapeutic potential of aptamers.
Materials and methods
Reagents
DNA oligos were purchased from Integrated DNA Technologies (IDT, Coralville, IA). Stock solution of sheared salmon sperm DNA (sssDNA) at 10 mg/mL was purchased from Invitrogen (Waltham, MA). APC-labeled EGFR mAb (clone AY13, catalog #2364525) and isotype control (mouse IgG1, κ, clone MOPC-21, catalog #2600605) were purchased from Sony Biotechnology (San Jose, CA). APC-labeled anti-c-Met mAb (clone 271, catalog #10692-R271-A) was purchased from Sino Biological US (Houston, TX). EGFR mAbs (cetuximab, matuzumab, and nimotuzumab) were purchased from Novus Biotechnology (Centennial, CO). All other materials were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise noted.
Expression plasmids
pCMV_EGFR_EGFP was a purchased from Addgene (plasmid #32751, Cambridge, MA). All mutant EGFR constructs were generated using the Q5 site-directed mutagenesis kit (New England Biolabs, Ipswich, MA). To generate pCMV EGFR_T2A_EGFP (pBT002; Table S1), a DNA sequence encoding T2A (5′-GGCTCCGGCGAGGGCAGGGGAAGTCTTCTAACATGCGGGGACGTGGAGGAAAATCCCGGCCCA-3′) was inserted into pCMV_EGFR_EGFP using primer set 1 (Table S2). Plasmids pBT003, pBT004, pBT005, pMX016, pMX018, pMX019, and pMX020 (Table S1) were generated using primer sets 2, 3, 4, 6, 7, 8, and 9, respectively (Table S2). Plasmids pBT006 and pBT007 (Table S1) were generated using primer set 5 (Table S2).
Aptamer generation
All DNA and RNA oligonucleotides are reported in Tables S3 and S4. All oligos were resuspended in appropriate volume of Milli-Q water to reach a stock concentration of 100 μM. All 2′-FY-modified RNA aptamers included an extended 3′ tail sequence (5′-CGACGACGACGACGACGACGA-3′; Figure S2). For fluorescent imaging, complementary DNA anti-tail (5′-TCGTCGTCGTCGTCGTCGTCG-3′; Figure S2) or aptamer containing 3′ DNA anti-tail (e.g., CLN3) were purchased from IDT with a 5′C6 amino modifier. The 5′-amino-modified DNA oligos were conjugated to NHS ester-modified Atto647N, Cy5, Cy7, or AF750 and purified via HPLC to make 5′ dye-labeled DNA oligonucleotides (Figure S2; as described in next section). The 2′-FY-modified RNA aptamers were generated via in vitro run-off transcription (IVT). First, the DNA templates were PCR amplified with primers that appended a T7 promotor (Table S3). Then, 2′-FY-modified RNA aptamers were transcribed by overnight IVT at 37°C using recombinant mutant T7 RNA polymerase (Y639F), IVT buffer (50 mM Tris-HCl pH 7.5, 15 mM MgCl2, 5 mM DTT, 4% w/v PEG4000, and 2 mM spermidine), and 2 mM of each of ATP, GTP, 2′-fluoro modified CTP, and 2′-fluoro modified UTP (TriLink Biotechnologies, San Diego, CA, USA). All RNA aptamers were purified through denaturing polyacrylamide gel electrophoresis (PAGE) (0.75 mm, 6%–8% TBE-PAGE, 8 M urea) and bands corresponding to the expected product size were visualized by UV shadow, excised from the gel, and then eluted overnight while tumbling in 300 mM sodium acetate pH 5.4. Eluates were ethanol precipitated, resuspended in Milli-Q water, and stored at −20°C until further use.
Dye labeling and HPLC purification of DNA oligonucleotides
The 5′-amino-modified DNA oligonucleotides (aptamers or anti-tail) were labeled with NHS ester-modified Atto647N, Cy5, Cy7 (Lumiprobe) or AF750 (Thermo Scientific). The labeling reaction with Atto647N, Cy5, or Cy7 was performed as follows: 30 nmol of 5′-amino-modified DNA oligonucleotide were diluted in 0.2 M sodium bicarbonate buffer (pH 9) and mixed with 20-fold molar excess of dye in a final volume of 500 μL. The reaction mixture was kept overnight at 4°C. In the case of AF750, the labeling protocol included the following differences: the molar ratio between the 5′-amino-modified DNA oligo and NHS ester AF750 was 1:30, and MgCl2 (final concentration of 5 mM) was added to the labeling mix to reduce the electrostatic repulsion between the negative charges of the oligonucleotide and AF750. To remove excess fluorophore before analytical evaluation, oligonucleotides were ethanol precipitated and resuspended in a minimal volume of Milli-Q water (<100 μL).
Analytical evaluation of labeling reactions and purification of dye-labeled oligonucleotides were performed using reverse-phase HPLC (RP-HPLC). An Agilent 1100 series instrument with an Agilent Zorbax Eclipse XDB-C18 (4.6 × 150 mm2) was employed for RP-HPLC analysis. For all the analyses, column temperature was set at T = 50°C to partially denature aptamer secondary structures and improve sharpness and peak resolution. We used a flow rate of 1 mL/min with triethylammonium acetate (TEAA)/acetonitrile (ACN) buffer system (solvent A, 100 mM TEAA, pH 7; solvent B, 100% ACN). For Atto647N, Cy5, and Cy7 fluorophores, dye-labeled aptamers (retention time ∼18–20 min) were separated from the unlabeled fraction (retention time ∼8–9 min) and purified using a linear gradient from 5% to 30% ACN over 20 min. A further linear gradient was applied from 30% to 60% ACN over 5 min. The final step applied 60% ACN for 3 min to purge the column of unreacted fluorophore. For AF750 (more hydrophilic and negatively charged), we used a gradient protocol with a reduced slope to ensure good separation between the peaks of unreacted CLN3 and AF750-CLN3. Labeled CLN3 (design B) was separated (retention time ∼32 min) from the unlabeled fraction (retention time ∼27 min) and purified using a linear gradient from 5% to 20% ACN over 40 min. A further linear gradient was applied from 20% to 60% ACN over 5 min. The final step applied 60% ACN for 3 min to purge the column. Coupling efficiency of these labeling reactions strongly depends on the physicochemical properties of the dye and the aptamer.100,101 Cyanine and Atto dyes (more hydrophobic and positively charged) show a superior labeling efficiency compared with Alexa dyes (e.g., for a 20-nt DNA oligo, labeling efficiencies were >90% and ∼50%, respectively). Labeling efficiency is also affected by the oligonucleotide length (for 20- to 30-nt oligos, the coupling efficiency is >90%) and their purification method after synthesis (desalting vs. HPLC). Based on our experience, we recommend using HPLC-purified amino-modified DNA oligos from any commercial vendors when their length is >50 nt, as the labeling of these oligos with NHS ester dyes will improve significantly: e.g., for an 88-nt DNA oligo, labeling was ∼30% (for desalted-purified oligo) and ∼65% (for HPLC-purified oligo).
Purified dye-labeled oligonucleotides were concentrated and purified by the residual fraction of ACN using either spin desalting column (Amicon Ultra-15, 3,000 molecular-weight cutoff [MWCO]) or lyophilization. In the case of the latter, dry pellets were dissolved in 100 μL of Milli-Q water. Dye-to-aptamer ratio was evaluated from absorbance measured at 260 nm (for DNA oligo) and 646 nm (for Cy5 and Atto47N) or 750 nm (for Cy7 and AF750) on a NanoDrop 1000 spectrophotometer (Thermo Scientific). All dye-labeled oligonucleotides result in a dye-to-oligo stoichiometry of 1:1 within experimental error.
Aptamer folding and annealing with dye-labeled DNA oligonucleotide
All aptamers were folded and annealed to anti-tail as previously described.31,50 In brief, in vitro annealing reactions were prepared at room temperature in Dulbecco’s phosphate buffered saline (DPBS, pH 7.4) supplemented with 5 mM MgCl2 using a slight excess (1.5:1.0 to 2:1 M ratio) of RNA aptamer to the dye-labeled DNA anti-tail (monospecific aptamer; Figure S2) or DNA aptamer anti-tail (bsApt). For thermal renaturation, samples were transferred into a preheated aluminum insert within a dry heat block set to 90°C, where they were kept for 1–2 min to denature nucleic acid structures, and then the aluminum insert was removed from the block heater and placed on the workbench to cool slowly to below 37°C. Aptamer annealing efficiency was checked by electromobility shift assays (EMSAs) as previously described.31 In brief, ∼20 pmol of labeled aptamer was loaded and run on a native 8% polyacrylamide gel, stained with ethidium bromide, and analyzed using FIJI.102 Annealing efficiency is calculated using the limiting reagent (i.e., dye-labeled oligo) where the fluorescence of the complex is divided by the total fluorescence of the lane.
For in vitro applications, dye-conjugated aptamer samples were freshly prepared before each experiment at 5× concentration, unless otherwise noted. sssDNA was added to each sample at a final concentration of 1.0 mg/mL as a competitor for non-specific binding sites. MgCl2 was adjusted to maintain a final concentration of 5 mM Mg2+. For in vivo applications DNA oligos were conjugated to Cy7 or AF750 instead of Cy5 to enhance signal penetration through biological tissues. Dye-labeled aptamers or bsApts were prepared at a dose of 50–60 pmol per gram (mouse weight) in 100 μL of DPBS without MgCl2 the day before the experiment and stored at 4°C overnight. Aptamers were brought to room temperature for 15 min before injection.
Cell culture
HEK293FT were purchased from the American Type Culture Collection (ATCC, Manassas, VA). HeLa cells were gifted by Dr. Christian Lorson, University of Missouri-Columbia. A549 cells were gifted by Dr. Bumsuk Hahm, University of Missouri-Columbia. H1975 and H820 were gifted by Dr. Raghuraman Kannan, University of Missouri-Columbia. MCF7 cells were gifted by Dr. Thomas Quinn, University of Missouri-Columbia. HEK293FT, A549, and MCF7 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 2 mM L-glutamine, and 1× NEAAs. H1975 and H820 cells were cultured in HyClone RPMI 1640 supplemented with 10% FBS, 1 mM sodium pyruvate, 2 mM L-glutamine, and 1× NEAAs. All cell lines were maintained at 37°C in humidified incubator with 5% CO2 and passaged when ∼80% confluent. Cells were not passaged more than 20 times from thaw or past total passage number 30 (P30) to minimize genetic drift. Cells were consistently validated for EGFR and c-Met antigen presence using both Ab and aptamers staining. Consistency in cell morphology was checked prior to each passage.
Transient transfection
For flow cytometry, HEK293FT cells were seeded at 25,000–30,000 cells/well in a 48-well flat-bottom tissue culture plate 24 h prior to transfection. Transient transfection was performed using the jetPrime transfection kit (Polyplus, Illkirch, France) and 1 μg of plasmid DNA per milliliter of medium. Medium was replaced 5–6 h after transfection and cells were allowed to grow for an additional 24 h prior to use in binding assays, flow cytometry, and microscopy.
Aptamer and Ab binding assay
A549, HeLa, H1975, H820, and MCF7 cells were seeded at 40,000–50,000 cells/well in a 48-well flat-bottom tissue culture plate 24 h before experiment. On the day of the assay, aptamers were folded and annealed as described above. APC-labeled EGFR and c-Met mAbs and isotype control were diluted to desired concentration in DPBS supplemented with 0.1% BSA. Cells were washed once with DPBS and then incubated in either aptamer or Ab binding solution for 45 min at 37°C and 5% CO2. After incubation, the solution was removed, cells were washed once with DPBS, and cells were lifted off with 1× TrypLE Express (Thermo Fisher, Waltham, MA) for <5 min at 37°C. TrypLE Express was diluted by the addition of complete medium, and cells were transferred to 1.5-mL Eppendorf tubes and gently centrifuged to pellet (5 min, 400 × g). Cells were then fixed in 4% paraformaldehyde (PFA) in DPBS in the dark for 5 min at 4°C before being pelleted (5 min, 400 × g) and resuspended in DPBS. Cells were kept in the dark at 4°C until analysis with flow cytometry. Transiently transfected 293FT cells were subjected to the same procedure but were not washed at either step due to their poor adherence to the plates.
Aptamer cocktail versus bsApt
To determine whether the avidity of the EGFR/c-Met bsApt complex improves cell labeling compared to a cocktail of monospecific anti-EGFR and c-Met aptamers, H1975 and A549 cells were incubated for 1 h at 37°C with either 25 nM Atto647N-labeled bsApt (design B) or a cocktail composed of 25 nM Atto647N-MinE07 and 25 nM Atto647N-CLN3 (50-nt sequence with no anti-tail). A second aptamer concentration was also tested; cells were incubated for 1 h at 37°C with either 50 nM Atto647N-labeled bsApt (design B) or a cocktail composed of 50 nM Atto647N-MinE07 and 50 nM Atto647N-CLN3. For both concentrations, a cocktail composed of two control aptamers (mutMinE07 and mutCLN3) was tested to define the non-specific uptake and background binding. After the incubation, the same protocol described above was applied prior to performing the flow cytometry analysis.
Competition assays
Binding assays in presence of competitor (aptamer or Ab) were done as previously described.50 In short, cells were seeded at 40,000–50,000 cells/well in a 48-well flat-bottom tissue culture plate 24 h before experiment. Cy5-conjugated aptamers were prepared as described above at 2× desired concentration. In separate tubes, aptamer competitor samples were annealed to free DNA anti-tail (unlabeled, no fluorophore) at 2× desired concentration (10-fold excess Cy5-apt). Alternatively, anti-EGFR mAbs (cetuximab, matuzimab, nimotuzimab) or control (anti-GAPDH, BioLegend, San Diego, CA) were prepared at 2× desired concentration (10-fold excess Cy5-apt) in DPBS supplemented with 0.1% BSA. Cy5-Apt samples and their corresponding competitors were combined and supplemented with 1.0 mg/mL sssDNA. H1975 or HeLa cells were incubated with these solutions as described above with one additional DPBS wash after removal of aptamer-Ab solution. Binding assays were done at 37°C, and at 4°C to minimize internalization.
Flow cytometry and analyses
Flow cytometry was performed on a BD Accuri C6 (BD Biosciences, San Jose, CA) or Attune NxT (Thermo Fisher Scientific, Waltham, MA) counting 10,000–30,000 live cell events. Fluorescence of far-red dyes (Cy5 or Atto647N) was excited using a 640-nm (Accuri) or 637-nm (Attune) laser and detected using a standard FL4 filter (675 ± 12.5 nm) (Accuri) or RL1 filter (670 ± 7 nm) (Attune). Flow cytometry data were then analyzed and processed using FlowJo Software (Treestar, Ashland, OR). Viable singlet cells were gated and analyzed for MFI. For 293FT transfection experiments, GFP fluorescence was excited using a 488-nm laser and detected using standard FL1 filter (530 ± 15 nm) (Accuri) or BL1 filter (530 ± 15 nm) (Attune). Viable singlet cells that were GFP+ were gated and Cy5 fluorescence was reported as either raw (Cy5 [GFP+]) or normalized to GFP fluorescence (Cy5 [GFP+]/GFP [GFP+]).
Confocal microscopy
H1975 cells were seeded at 50,000 cells/chamber in a Nunc Lab-Tek 4 Chamber Slide (Thermo Fisher) 24 h before experiment. Cells were stained with CellBrite Steady 488 Membrane Staining Kit (Biotium, Fremont, CA) for 1 h at 37°C and 5% CO2 and then washed twice with DPBS. Atto647N-labeled aptamers were prepared as described above to a final concentration of 150 nM and incubated with cells for 45 min at 37°C and 5% CO2. Cells were washed twice with DPBS, fixed in 4% PFA for 5 min at 4°C, and washed an additional three times with DPBS. Cells were counter-stained with 1 μg/mL Hoechst 33342 (Sigma Aldrich) for 15 min at room temperature, washed twice with DPBS, and stored in the dark in DPBS at 4°C until imaging. Confocal images were taken with a Lecia SP8X tandem scanning confocal microscope with a white-light laser using a 63× 1.2 NA water immersion objective over either a single or multiple z planes with a pinhole size of 1.00 (Leica Microsystems, Buffalo Grove, IL). Images were displayed as single xy planes. Images were rendered and analyzed using LASX (Leica Microsystems).
Aptamer internalization assay
Aptamer binding assays were done in a 48-well flat-bottom tissue culture plate as described above using a final aptamer concentration of 50 nM. After aptamer incubation, cells were treated with 4 U of DNase I (Invitrogen) or 3 U of RiboShredder (Epicentre Biotechnologies, Madison, WI) in 100 μL of DPBS for 30 min at 37°C. Then 400 μL of complete medium was added to dilute and stop the reaction. Supernatant was removed and cells were washed, lifted, and fixed as described above. To optimize nuclease conditions, various nuclease concentrations, treatment temperatures (22°C and 37°C), and incubation times (15 and 30 min) were assayed, and aptamer degradation (presence of a smear or multiple bands versus a single band) was accessed using native PAGE gels.
Subcutaneous xenograft of NSCLC cells
To develop cell-line-derived subcutaneous tumor models,103 H1975 cells were grown as described above in 15-cm dishes to ∼80% confluence. Cells were collected, washed twice with cold DPBS, and resuspended in ice-cold DPBS plus 50% extracellular matrix (ECM) at ∼5 million cells per 100 μL. Cells were kept on ice during injections to prevent solidification of ECM. To develop xenografts, BALB/c nude mice (Charles River, Wilmington, MA) were briefly anesthetized with isoflurane in an induction chamber (4%, 250 mL/s). Using a 28G 0.5″ 1-cc insulin syringe/needle, 5 × 106 cells were subcutaneously injected into the flank or shoulder of each mouse. Cells were allowed to engraft for 1 week and then mice were palpated every 3–4 days until tumors could be measured (>4 mm2), at which point tumors were measured using digital calipers at a similar interval until experimental start point (∼75–150 mm2).
Animal ethics statement
All animal procedures were conducted according to the NIH guidelines for the care and use of laboratory animals and were approved by the University of Missouri Institutional Animal Care and Use Committee.
In vivo and ex vivo imaging
Aptamers for in vivo experiments were folded and annealed as described above. Nude mice harboring subcutaneous tumors with sizes of ∼75–150 mm2 were briefly anesthetized with isoflurane in an induction chamber (4%, 250 mL/s). Using a 28G 0.5″ 1-cc insulin syringe/needle, 100 μL of aptamer solution (50 pmol/g) was injected into the retro-orbital (RO) sinus of the mice (time point = 0 h). RO injections104 were used to minimize pain and suffering. The NIR fluorescence imaging studies were conducted on an IVIS Spectrum imaging system (PerkinElmer)105 equipped with a cooled charge-coupled device (CCD) camera and a 150 W quartz halogen light source. At the indicated time points, four to five mice were simultaneously imaged under 1.5%–2.5% isoflurane anesthesia (nosecone) and maintained at a body temperature of 37°C during the imaging procedure, with the following settings: excitation, 710 nm; emission, 760 nm; binning, 8; exposure time, 1 s; F/stop, 2; lamp level, low. We used these parameters for the IVIS images included in Figure 4 (whole-body imaging), and Figures 5 and S13 (ex vivo imaging of tissues). The fluorescence intensity was measured as the average radiant efficiency with a unit of [p/s/cm2/sr]/(μW/cm2) using Living Image 4.4 (PerkinElmer).
To compare Cy7 and AF750, we performed a sequence acquisition using two tailored settings called “Cy7 custom method” (excitation, 710 nm; emission, 760 nm) and “AF750 IVIS method” (excitation, 745 nm; emission, 800 nm), which are the default IVIS settings for AF750. The other parameters used for both settings were as follows: Binning, 8; automatic exposure time; F/stop, 2; lamp level, high. The IVIS images included in Figure 6 were obtained using the AF750 IVIS method.
Mice were imaged in prone and right recumbent positions depending on tumor position. At 48 hpi, mice were humanely sacrificed (CO2 followed by cervical dislocation), and their vascular systems were flushed with DPBS prior to blood coagulation (<2 min after expiration). To flush the vascular system, a 50-mL syringe was filled with DPBS and equipped with a 25G butterfly needle. The mouse abdominal aorta was severed, and the needle was placed in the left ventricle. Then 30–50 mL of DPBS was injected until organs (specifically liver and kidney) were blanched. Tumor, liver, kidney, spleen, and muscle were harvested for endpoint IVIS imaging using the parameters reported above. Tumors were placed in 1.5-mL Eppendorf tubes, snap frozen in liquid nitrogen, and stored at −80°C for downstream analyses.
Ex vivo antigen expression
Snap-frozen tumor tissues were used for antigen expression analysis. Tumors were quickly thawed by the addition of 37°C medium (RPMI supplemented with 5% FBS). To digest and disaggregate into a single-cell suspension, tumor tissue was added to a 5-cm Petri dish containing 5 mL of medium supplemented with collagenase type I, cut and smashed into smaller pieces, and allowed to gently shake (80 rpm) for 30 min at 37°C. Note that no DNase was added to the digest to prevent aptamer or oligonucleotide digestion. After digestion, cell solution was filtered (40 μm) and washed with DPBS. Filtration was repeated two to four times to remove unwanted material. Cells were treated with red blood cell lysis solution for 5 min at room temperature, washed with DPBS, and resuspended in DPBS to a final concentration of 106 cells/mL in a single-cell suspension. Cells were then incubated with Fc block (BioLegend) for 15 min at room temperature, moved to a 96-well round-bottom plate (50,000–100,000 cells/well), washed with DPBS, and pre-blocked with 0.25% BSA, 1.0 mg/mL sssDNA, 1.0 mg/mL dextran sulfate, and 100 nM arbitrary 2′-FY RNA in DPBS with 5 mM MgCl2 for 20 min at room temperature. Cells were then incubated with Ab solutions for 25 min at room temperature, washed in DPBS, fixed in 4% PFA, and analyzed via flow cytometry as described above. In addition to antigen staining (anti-EGFR, anti-c-Met), cells were stained with anti-hCD71 (TfR, clone OKT9, BioLegend) to confirm that cells were of human origin and not from mouse.
Aptamer stability
To mimic in vivo-like conditions (1 nmol of injected material into ∼2 mL of circulating blood), MinE07 or EGFR/c-Met bsApt were folded and annealed as described above at 5 μM and diluted to 500 nM in 55% FBS in 1× DPBS. Aptamers were allowed to incubate for 0, 1, 3, 6, 10, 24, or 48 h at 37°C in 5% CO2 and then treated with 1 mg/mL proteinase K for 1 h at 37°C to stop nuclease activity and degrade serum proteins. For (RT)-qPCR, 1 μL of sample was diluted 1:100 in nuclease-free water, reverse transcribed (BST 3.0; New England Biolabs) for 30 min at 65°C (MinE07 primers only), and subjected to 40 rounds of qPCR (SYBR Green MasterMix; Bio-Rad, Hercules, CA). MinE07 was amplified using the same forward and reverse primers used for synthesis (Table S3). CLN3 was amplified using the following primers sequences: forward (5′-ATCAGGCTGGATGGTAGCTCGGTCG-3′) and reverse (5′-CGACGACGACGACGACGACGATAATC-3′). Analysis was done using CFX Maestro Software (Bio-Rad). For native polyacrylamide gel electrophoresis (TBM-PAGE), 25 μL of sample (∼12.5 pmol of starting material) was mixed with 10% glycerol and loaded onto 8% polyacrylamide gels. Gels were run at 15 W per gel for 2.5 h at 4°C, stained with ethidium bromide, imaged on the Typhoon (100-μm resolution; GE Healthcare, Marlborough, MA), and analyzed using FIJI.102 For doxorubicin (DOX) intercalation assays (i.e., quenching of fluorescent signal upon intercalation into CG base pairs present in the hybridized tail/anti-tail),32 2 μM DOX was added to the sample and an emission spectrum was acquired (excitation, 480 nm; emission, 500–700 nm) using the EnSpire Plate Reader (PerkinElmer). Area under the curve was used to determined quenching relative to doxorubicin in the presence of unannealed aptamers.
Data and code availability
Data are contained within the article or supplemental information. Unprocessed data and additional information regarding specific methods of interest to readers will be made available upon request.
Acknowledgments
This work was financially supported by the MU Life Sciences Center (LSC) Early Concept Grant (ECG) for Innovative Collaborative Research involving Post-Doctoral Researchers (principal investigator [PI], D.H.B-M.A.D,-D.P.), and the UM Research and Creative Works Strategic Investment Program grant (PI, D.H.B.). We would like to thank Melanie Alexis-Ruiz for her contribution in the development of expression plasmids. We acknowledge the VA Biomolecular Imaging Center resource jointly supported by the Harry S. Truman Veterans Memorial Hospital (Columbia, MO) and the University of Missouri.
Author contributions
B.J.T., conceptualization, methodology, investigation, visualization, and writing – review & editing; C.G., methodology and investigation. C.W., validation and investigation; M.B., validation and investigation; A.F.B., investigation; L.M., investigation; M.A.D., resources, supervision, and funding acquisition; D.H.B., project administration, writing – review & editing, supervision, and funding acquisition; D.P., conceptualization, methodology, investigation, visualization, writing – original draft, writing – review & editing, supervision, and funding acquisition.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.102046.
Contributor Information
Donald H. Burke, Email: burkedh@missouri.edu.
David Porciani, Email: dave.porciani@gmail.com.
Supplemental information
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Herbst R.S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 3.Leonetti A., Sharma S., Minari R., Perego P., Giovannetti E., Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer. 2019;121:725–737. doi: 10.1038/s41416-019-0573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumari N., Singh S., Haloi D., Mishra S.K., Krishnani N., Nath A., Neyaz Z. Epidermal Growth Factor Receptor Mutation Frequency in Squamous Cell Carcinoma and Its Diagnostic Performance in Cytological Samples: A Molecular and Immunohistochemical Study. World J. Oncol. 2019;10:142–150. doi: 10.14740/wjon1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigismund S., Avanzato D., Lanzetti L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018;12:3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin J.J., Cardarella S., Lydon C.A., Dahlberg S.E., Jackman D.M., Jänne P.A., Johnson B.E. Five-Year Survival in EGFR-Mutant Metastatic Lung Adenocarcinoma Treated with EGFR-TKIs. J. Thorac. Oncol. 2016;11:556–565. doi: 10.1016/j.jtho.2015.12.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas R., Weihua Z. Rethink of EGFR in Cancer With Its Kinase Independent Function on Board. Front. Oncol. 2019;9:800. doi: 10.3389/fonc.2019.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu F., Fan J., He Y., Xiong A., Yu J., Li Y., Zhang Y., Zhao W., Zhou F., Li W., et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021;12:2540. doi: 10.1038/s41467-021-22801-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellis P.M., Vandermeer R. Delays in the diagnosis of lung cancer. J. Thorac. Dis. 2011;3:183–188. doi: 10.3978/j.issn.2072-1439.2011.01.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen T.M. Ligand-targeted therapeutics in anticancer therapy. Nat. Rev. Cancer. 2002;2:750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- 11.Bajracharya R., Song J.G., Patil B.R., Lee S.H., Noh H.-M., Kim D.-H., Kim G.-L., Seo S.-H., Park J.-W., Jeong S.H., et al. Functional ligands for improving anticancer drug therapy: current status and applications to drug delivery systems. Drug Deliv. 2022;29:1959–1970. doi: 10.1080/10717544.2022.2089296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A., et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labrijn A.F., Janmaat M.L., Reichert J.M., Parren P.W.H.I. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019;18:585–608. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 14.Elskens J.P., Elskens J.M., Madder A. Chemical Modification of Aptamers for Increased Binding Affinity in Diagnostic Applications: Current Status and Future Prospects. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21124522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah N.N., Johnson B.D., Schneider D., Zhu F., Szabo A., Keever-Taylor C.A., Krueger W., Worden A.A., Kadan M.J., Yim S., et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat. Med. 2020;26:1569–1575. doi: 10.1038/s41591-020-1081-3. [DOI] [PubMed] [Google Scholar]
- 16.Yun J., Lee S.-H., Kim S.-Y., Jeong S.-Y., Kim J.-H., Pyo K.-H., Park C.-W., Heo S.G., Yun M.R., Lim S., et al. Antitumor Activity of Amivantamab (JNJ-61186372), an EGFR-MET Bispecific Antibody, in Diverse Models of EGFR Exon 20 Insertion-Driven NSCLC. Cancer Discov. 2020;10:1194–1209. doi: 10.1158/2159-8290.CD-20-0116. [DOI] [PubMed] [Google Scholar]
- 17.Castoldi R., Ecker V., Wiehle L., Majety M., Busl-Schuller R., Asmussen M., Nopora A., Jucknischke U., Osl F., Kobold S., et al. A novel bispecific EGFR/Met antibody blocks tumor-promoting phenotypic effects induced by resistance to EGFR inhibition and has potent antitumor activity. Oncogene. 2013;32:5593–5601. doi: 10.1038/onc.2013.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Z.-J., Wu Y., Hou W.-H., Wang Y.-X., Yuan Q.-Y., Wang H.-J., Yu M. A novel bispecific c-MET/PD-1 antibody with therapeutic potential in solid cancer. Oncotarget. 2017;8:29067–29079. doi: 10.18632/oncotarget.16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung C.H., Mirakhur B., Chan E., Le Q.-T., Berlin J., Morse M., Murphy B.A., Satinover S.M., Hosen J., Mauro D., et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N. Engl. J. Med. 2008;358:1109–1117. doi: 10.1056/NEJMoa074943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dalziel M., Beers S.A., Cragg M.S., Crispin M. Through the barricades: overcoming the barriers to effective antibody-based cancer therapeutics. Glycobiology. 2018;28:697–712. doi: 10.1093/glycob/cwy043. [DOI] [PubMed] [Google Scholar]
- 21.Pointreau Y., Commins S.P., Calais G., Watier H., Platts-Mills T.A.E. Fatal Infusion Reactions to Cetuximab: Role of Immunoglobulin E–Mediated Anaphylaxis. J. Clin. Oncol. 2012;30:334–335. doi: 10.1200/JCO.2011.38.4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lejeune M., Köse M.C., Duray E., Einsele H., Beguin Y., Caers J. Bispecific, T-Cell-Recruiting Antibodies in B-Cell Malignancies. Front. Immunol. 2020;11:762. doi: 10.3389/fimmu.2020.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng S., Moores S., Jarantow S., Pardinas J., Chiu M., Zhou H., Wang W. Cross-arm binding efficiency of an EGFR x c-Met bispecific antibody. mAbs. 2016;8:551–561. doi: 10.1080/19420862.2015.1136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park S., Shevlin E., Vedvyas Y., Zaman M., Park S., Hsu Y.-M.S., Min I.M., Jin M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-14749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khvorova A., Watts J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017;35:238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halloy F., Biscans A., Bujold K.E., Debacker A., Hill A.C., Lacroix A., Luige O., Strömberg R., Sundstrom L., Vogel J., Ghidini A. Innovative developments and emerging technologies in RNA therapeutics. RNA Biol. 2022;19:313–332. doi: 10.1080/15476286.2022.2027150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soldevilla M.M., Villanueva H., Pastor F. Aptamers: A Feasible Technology in Cancer Immunotherapy. J. Immunol. Res. 2016;2016 doi: 10.1155/2016/1083738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas B.J., Porciani D., Burke D.H. Cancer immunomodulation using bispecific aptamers. Mol. Ther. Nucleic Acids. 2022;27:894–915. doi: 10.1016/j.omtn.2022.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L., Liang H., Sun J., Liu Y., Li J., Li J., Li J., Yang H. Bispecific Aptamer Induced Artificial Protein-Pairing: A Strategy for Selective Inhibition of Receptor Function. J. Am. Chem. Soc. 2019;141:12673–12681. doi: 10.1021/jacs.9b05123. [DOI] [PubMed] [Google Scholar]
- 30.Zheng J., Zhao S., Yu X., Huang S., Liu H.Y. Simultaneous targeting of CD44 and EpCAM with a bispecific aptamer effectively inhibits intraperitoneal ovarian cancer growth. Theranostics. 2017;7:1373–1388. doi: 10.7150/thno.17826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porciani D., Cardwell L.N., Tawiah K.D., Alam K.K., Lange M.J., Daniels M.A., Burke D.H. Modular cell-internalizing aptamer nanostructure enables targeted delivery of large functional RNAs in cancer cell lines. Nat. Commun. 2018;9:2283. doi: 10.1038/s41467-018-04691-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porciani D., Tedeschi L., Marchetti L., Citti L., Piazza V., Beltram F., Signore G. Aptamer-Mediated Codelivery of Doxorubicin and NF-κB Decoy Enhances Chemosensitivity of Pancreatic Tumor Cells. Mol. Ther. Nucleic Acids. 2015;4:e235. doi: 10.1038/mtna.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou J., Rossi J.J. Cell-type-specific, Aptamer-functionalized Agents for Targeted Disease Therapy Molecular therapy. Nucleic acids. 2014;3:e169. doi: 10.1038/mtna.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu G., Zheng J., Song E., Donovan M., Zhang K., Liu C., Tan W. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. Proc. Natl. Acad. Sci. USA. 2013;110:7998–8003. doi: 10.1073/pnas.1220817110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McNamara J.O., Andrechek E.R., Wang Y., Viles K.D., Rempel R.E., Gilboa E., Sullenger B.A., Giangrande P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 36.Dassie J.P., Liu X.-Y., Thomas G.S., Whitaker R.M., Thiel K.W., Stockdale K.R., Meyerholz D.K., McCaffrey A.P., McNamara J.O., Giangrande P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Powell Gray B., Kelly L., Ahrens D.P., Barry A.P., Kratschmer C., Levy M., Sullenger B.A. Tunable cytotoxic aptamer–drug conjugates for the treatment of prostate cancer. Proc. Natl. Acad. Sci. USA. 2018;115:4761–4766. doi: 10.1073/pnas.1717705115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Binzel D.W., Shu Y., Li H., Sun M., Zhang Q., Shu D., Guo B., Guo P. Specific Delivery of MiRNA for High Efficient Inhibition of Prostate Cancer by RNA Nanotechnology. Mol. Ther. 2016;24:1267–1277. doi: 10.1038/mt.2016.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li N., Nguyen H.H., Byrom M., Ellington A.D. Inhibition of Cell Proliferation by an Anti-EGFR Aptamer. PLoS One. 2011;6 doi: 10.1371/journal.pone.0020299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esposito C.L., Passaro D., Longobardo I., Condorelli G., Marotta P., Affuso A., de Franciscis V., Cerchia L. A Neutralizing RNA Aptamer against EGFR Causes Selective Apoptotic Cell Death. PLoS One. 2011;6 doi: 10.1371/journal.pone.0024071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang D.-L., Song Y.-L., Zhu Z., Li X.-L., Zou Y., Yang H.-T., Wang J.-J., Yao P.-S., Pan R.-J., Yang C.J., Kang D.Z. Selection of DNA aptamers against epidermal growth factor receptor with high affinity and specificity. Biochem. Biophys. Res. Commun. 2014;453:681–685. doi: 10.1016/j.bbrc.2014.09.023. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X., Peng L., Liang Z., Kou Z., Chen Y., Shi G., Li X., Liang Y., Wang F., Shi Y. Effects of Aptamer to U87-EGFRvIII Cells on the Proliferation, Radiosensitivity, and Radiotherapy of Glioblastoma Cells. Mol. Ther. Nucleic Acids. 2018;10:438–449. doi: 10.1016/j.omtn.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y., Kuan C.-T., Mi J., Zhang X., Clary B.M., Bigner D.D., Sullenger B.A. Aptamers selected against the unglycosylated EGFRvIII ectodomain and delivered intracellularly reduce membrane-bound EGFRvIII and induce apoptosis. Biol. Chem. 2009;390:137–144. doi: 10.1515/BC.2009.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y., Dai X., Jiang S., Qahar M., Feng C., Guo D., Wang L., Ma S., Huang L. Targeted Co-Delivery of Gefitinib and Rapamycin by Aptamer-Modified Nanoparticles Overcomes EGFR-TKI Resistance in NSCLC via Promoting Autophagy. Int. J. Mol. Sci. 2022;23:8025. doi: 10.3390/ijms23148025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly L., Maier K.E., Yan A., Levy M. A comparative analysis of cell surface targeting aptamers. Nat. Commun. 2021;12:6275. doi: 10.1038/s41467-021-26463-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bottari F., Daems E., de Vries A.-M., Van Wielendaele P., Trashin S., Blust R., Sobott F., Madder A., Martins J.C., De Wael K. Do Aptamers Always Bind? The Need for a Multifaceted Analytical Approach When Demonstrating Binding Affinity between Aptamer and Low Molecular Weight Compounds. J. Am. Chem. Soc. 2020;142:19622–19630. doi: 10.1021/jacs.0c08691. [DOI] [PubMed] [Google Scholar]
- 47.McKeague M., Calzada V., Cerchia L., DeRosa M., Heemstra J.M., Janjic N., Johnson P.E., Kraus L., Limson J., Mayer G., et al. The minimum aptamer publication standards (MAPS guidelines) for de novo aptamer selection. OPEN ACCESS. 2022;6 [Google Scholar]
- 48.Avutu V. 2010. Avidity Effects of MinE07, an Anti-EGFR Aptamer, on Binding to A431 Cells. Doctoral dissertation. [Google Scholar]
- 49.Cheng S., Jacobson O., Zhu G., Chen Z., Liang S.H., Tian R., Yang Z., Niu G., Zhu X., Chen X. PET imaging of EGFR expression using an 18F-labeled RNA aptamer. Eur. J. Nucl. Med. Mol. Imag. 2019;46:948–956. doi: 10.1007/s00259-018-4105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delcanale P., Porciani D., Pujals S., Jurkevich A., Chetrusca A., Tawiah K.D., Burke D.H., Albertazzi L. Aptamers with Tunable Affinity Enable Single-Molecule Tracking and Localization of Membrane Receptors on Living Cancer Cells. Angew Chem. Int. Ed. Engl. 2020;59:18546–18555. doi: 10.1002/anie.202004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ray P., Cheek M.A., Sharaf M.L., Li N., Ellington A.D., Sullenger B.A., Shaw B.R., White R.R. Aptamer-Mediated Delivery of Chemotherapy to Pancreatic Cancer Cells. Nucleic Acid Therapeut. 2012;22:295–305. doi: 10.1089/nat.2012.0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kratschmer C., Levy M. Targeted Delivery of Auristatin-Modified Toxins to Pancreatic Cancer Using Aptamers. Mol. Ther. Nucleic Acids. 2018;10:227–236. doi: 10.1016/j.omtn.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mahajan U.M., Li Q., Alnatsha A., Maas J., Orth M., Maier S.H., Peterhansl J., Regel I., Sendler M., Wagh P.R., et al. Tumor-Specific Delivery of 5-Fluorouracil-Incorporated Epidermal Growth Factor Receptor-Targeted Aptamers as an Efficient Treatment in Pancreatic Ductal Adenocarcinoma Models. Gastroenterology. 2021;161:996–1010.e1. doi: 10.1053/j.gastro.2021.05.055. [DOI] [PubMed] [Google Scholar]
- 54.Zhou J., Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 2017;16:181–202. doi: 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ni S., Zhuo Z., Pan Y., Yu Y., Li F., Liu J., Wang L., Wu X., Li D., Wan Y., et al. Recent Progress in Aptamer Discoveries and Modifications for Therapeutic Applications. ACS Appl. Mater. Interfaces. 2021;13:9500–9519. doi: 10.1021/acsami.0c05750. [DOI] [PubMed] [Google Scholar]
- 56.Kruspe S., Giangrande P.H. Aptamer-siRNA Chimeras: Discovery, Progress, and Future Prospects. Biomedicines. 2017;5:45. doi: 10.3390/biomedicines5030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Z., Chen O., Wall J.B.J., Zheng M., Zhou Y., Wang L., Vaseghi H.R., Qian L., Liu J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017;7:2193. doi: 10.1038/s41598-017-02460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Talavera A., Friemann R., Gómez-Puerta S., Martinez-Fleites C., Garrido G., Rabasa A., López-Requena A., Pupo A., Johansen R.F., Sánchez O., et al. Nimotuzumab, an Antitumor Antibody that Targets the Epidermal Growth Factor Receptor, Blocks Ligand Binding while Permitting the Active Receptor Conformation. Cancer Res. 2009;69:5851–5859. doi: 10.1158/0008-5472.CAN-08-4518. [DOI] [PubMed] [Google Scholar]
- 59.Schmiedel J., Blaukat A., Li S., Knöchel T., Ferguson K.M. Matuzumab Binding to EGFR Prevents the Conformational Rearrangement Required for Dimerization. Cancer Cell. 2008;13:365–373. doi: 10.1016/j.ccr.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li S., Schmitz K.R., Jeffrey P.D., Wiltzius J.J.W., Kussie P., Ferguson K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 61.Tundidor Y., García-Hernández C.P., Pupo A., Cabrera Infante Y., Rojas G. Delineating the functional map of the interaction between nimotuzumab and the epidermal growth factor receptor. mAbs. 2014;6:1013–1025. doi: 10.4161/mabs.28915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tintelnot J., Baum N., Schultheiß C., Braig F., Trentmann M., Finter J., Fumey W., Bannas P., Fehse B., Riecken K., et al. Nanobody Targeting of Epidermal Growth Factor Receptor (EGFR) Ectodomain Variants Overcomes Resistance to Therapeutic EGFR Antibodies. Mol. Cancer Therapeut. 2019;18:823–833. doi: 10.1158/1535-7163.MCT-18-0849. [DOI] [PubMed] [Google Scholar]
- 63.Tanizaki J., Okamoto I., Sakai K., Nakagawa K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br. J. Cancer. 2011;105:807–813. doi: 10.1038/bjc.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ortiz-Zapater E., Lee R.W., Owen W., Weitsman G., Fruhwirth G., Dunn R.G., Neat M.J., McCaughan F., Parker P., Ng T., Santis G. MET-EGFR dimerization in lung adenocarcinoma is dependent on EGFR mtations and altered by MET kinase inhibition. PLoS One. 2017;12 doi: 10.1371/journal.pone.0170798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boltz A., Piater B., Toleikis L., Guenther R., Kolmar H., Hock B. Bi-specific aptamers mediating tumor cell lysis. J. Biol. Chem. 2011;286:21896–21905. doi: 10.1074/jbc.M111.238261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ueki R., Sando S. A DNA aptamer to c-Met inhibits cancer cell migration. Chem. Commun. 2014;50:13131–13134. doi: 10.1039/c4cc06016d. [DOI] [PubMed] [Google Scholar]
- 67.Ueki R., Uchida S., Kanda N., Yamada N., Ueki A., Akiyama M., Toh K., Cabral H., Sando S. A chemically unmodified agonistic DNA with growth factor functionality for in vivo therapeutic application. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aay2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y., Gao H., Zhou W., Sun S., Zeng Y., Zhang H., Liang L., Xiao X., Song J., Ye M., et al. Targeting c-met receptor tyrosine kinase by the DNA aptamer SL1 as a potential novel therapeutic option for myeloma. J. Cell Mol. Med. 2018;22:5978–5990. doi: 10.1111/jcmm.13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiong Y., Sundaralingam M. Crystal structure and conformation of a DNA-RNA hybrid duplex with a polypurine RNA strand: d(TTCTTBr5CTTC)-r(GAAGAAGAA) Structure. 1998;6:1493–1501. doi: 10.1016/s0969-2126(98)00148-8. [DOI] [PubMed] [Google Scholar]
- 70.Conn G.L., Brown T., Leonard G.A. The crystal structure of the RNA/DNA hybrid r(GAAGAGAAGC). d(GCTTCTCTTC) shows significant differences to that found in solution. Nucleic Acids Res. 1999;27:555–561. doi: 10.1093/nar/27.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Woodson S.A. Compact intermediates in RNA folding. Annu. Rev. Biophys. 2010;39:61–77. doi: 10.1146/annurev.biophys.093008.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burke D.H., Willis J.H. Recombination, RNA evolution, and bifunctional RNA molecules isolated through Chimeric SELEX. RNA. 1998;4:1165–1175. doi: 10.1017/S1355838298980542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith L.M., Birrer M.J., Stampfer M.R., Brown P.H. Breast cancer cells have lower activating protein 1 transcription factor activity than normal mammary epithelial cells. Cancer Res. 1997;57:3046–3054. [PubMed] [Google Scholar]
- 74.Reilly R.M., Kiarash R., Sandhu J., Lee Y.W., Cameron R.G., Hendler A., Vallis K., Gariépy J. A comparison of EGF and MAb 528 labeled with 111In for imaging human breast cancer. J. Nucl. Med. 2000;41:903–911. [PubMed] [Google Scholar]
- 75.Rhoden J.J., Dyas G.L., Wroblewski V.J. A Modeling and Experimental Investigation of the Effects of Antigen Density, Binding Affinity, and Antigen Expression Ratio on Bispecific Antibody Binding to Cell Surface Targets. J. Biol. Chem. 2016;291:11337–11347. doi: 10.1074/jbc.M116.714287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kitov P.I., Bundle D.R. On the nature of the multivalency effect: a thermodynamic model. J. Am. Chem. Soc. 2003;125:16271–16284. doi: 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- 77.Dennis Wright B.S.P., Lynn Usher B.S.P. Multivalent Binding in the Design of Bioactive Compounds. COC. 2001;5:1107–1131. doi: 10.2174/1385272013374806. [DOI] [Google Scholar]
- 78.Tawiah K.D., Porciani D., Burke D.H. Toward the Selection of Cell Targeting Aptamers with Extended Biological Functionalities to Facilitate Endosomal Escape of Cargoes. Biomedicines. 2017;5 doi: 10.3390/biomedicines5030051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chung B.M., Raja S.M., Clubb R.J., Tu C., George M., Band V., Band H. Aberrant trafficking of NSCLC-associated EGFR mutants through the endocytic recycling pathway promotes interaction with Src. BMC Cell Biol. 2009;10:84. doi: 10.1186/1471-2121-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ménard L., Floc’h N., Martin M.J., Cross D.A.E. Reactivation of Mutant-EGFR Degradation through Clathrin Inhibition Overcomes Resistance to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2018;78:3267–3279. doi: 10.1158/0008-5472.CAN-17-2195. [DOI] [PubMed] [Google Scholar]
- 81.Danhier F., Feron O., Préat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Contr. Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 82.Debie P., Van Quathem J., Hansen I., Bala G., Massa S., Devoogdt N., Xavier C., Hernot S. Effect of Dye and Conjugation Chemistry on the Biodistribution Profile of Near-Infrared-Labeled Nanobodies as Tracers for Image-Guided Surgery. Mol. Pharm. 2017;14:1145–1153. doi: 10.1021/acs.molpharmaceut.6b01053. [DOI] [PubMed] [Google Scholar]
- 83.Cilliers C., Nessler I., Christodolu N., Thurber G.M. Tracking Antibody Distribution with Near-Infrared Fluorescent Dyes: Impact of Dye Structure and Degree of Labeling on Plasma Clearance. Mol. Pharm. 2017;14:1623–1633. doi: 10.1021/acs.molpharmaceut.6b01091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Álamo P., Pallarès V., Céspedes M.V., Falgàs A., Sanchez J.M., Serna N., Sánchez-García L., Voltà-Duràn E., Morris G.A., Sánchez-Chardi A., et al. Fluorescent Dye Labeling Changes the Biodistribution of Tumor-Targeted Nanoparticles. Pharmaceutics. 2020;12:1004. doi: 10.3390/pharmaceutics12111004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mazzarella L., Guida A., Curigliano G. Cetuximab for treating non-small cell lung cancer. Expet Opin. Biol. Ther. 2018;18:483–493. doi: 10.1080/14712598.2018.1452906. [DOI] [PubMed] [Google Scholar]
- 86.Wang X., Zhou L., Yin J.C., Wu X., Shao Y.W., Gao B. Lung Adenocarcinoma Harboring EGFR 19del/C797S/T790M Triple Mutations Responds to Brigatinib and Anti-EGFR Antibody Combination Therapy. J. Thorac. Oncol. 2019;14:e85–e88. doi: 10.1016/j.jtho.2019.01.015. [DOI] [PubMed] [Google Scholar]
- 87.Wang Y., Han R., Zhu M., He T., He Y. Case Report: Durable Response to the Combination of Brigatinib and Cetuximab Plus Icotinib in a NSCLC Patient Harboring EGFR L858R-T790M-cis-G796S and L718Q Resistance Mutations Following Progression With Osimertinib. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.875313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cortot A.B., Madroszyk A., Giroux-Leprieur E., Molinier O., Quoix E., Bérard H., Otto J., Rault I., Moro-Sibilot D., Raimbourg J., et al. First-Line Afatinib plus Cetuximab for EGFR-Mutant Non-Small Cell Lung Cancer: Results from the Randomized Phase II IFCT-1503 ACE-Lung Study. Clin. Cancer Res. 2021;27:4168–4176. doi: 10.1158/1078-0432.CCR-20-4604. [DOI] [PubMed] [Google Scholar]
- 89.Shan Y., Eastwood M.P., Zhang X., Kim E.T., Arkhipov A., Dror R.O., Jumper J., Kuriyan J., Shaw D.E. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 2012;149:860–870. doi: 10.1016/j.cell.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 90.Berger C., Krengel U., Stang E., Moreno E., Madshus I.H. Nimotuzumab and cetuximab block ligand-independent EGF receptor signaling efficiently at different concentrations. J. Immunother. 2011;34:550–555. doi: 10.1097/CJI.0b013e31822a5ca6. [DOI] [PubMed] [Google Scholar]
- 91.Omer M., Andersen V.L., Nielsen J.S., Wengel J., Kjems J. Improved Cancer Targeting by Multimerizing Aptamers on Nanoscaffolds. Mol. Ther. Nucleic Acids. 2020;22:994–1003. doi: 10.1016/j.omtn.2020.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Demos S.G., Gandour-Edwards R., Ramsamooj R., White R.d. Near-infrared autofluorescence imaging for detection of cancer. J. Biomed. Opt. 2004;9:587–592. doi: 10.1117/1.1688812. [DOI] [PubMed] [Google Scholar]
- 93.del Rosal B., Benayas A. Strategies to Overcome Autofluorescence in Nanoprobe-Driven In Vivo Fluorescence Imaging. Small Methods. 2018;2 doi: 10.1002/smtd.201800075. [DOI] [Google Scholar]
- 94.Goh W.J., Zou S., Ong W.Y., Torta F., Alexandra A.F., Schiffelers R.M., Storm G., Wang J.-W., Czarny B., Pastorin G. Bioinspired Cell-Derived Nanovesicles versus Exosomes as Drug Delivery Systems: a Cost-Effective Alternative. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-14725-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu X., Zhao Z., Bai H., Fu T., Yang C., Hu X., Liu Q., Champanhac C., Teng I.-T., Ye M., Tan W. DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition. Theranostics. 2015;5:985–994. doi: 10.7150/thno.11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang C., Zhao H., Sun Y., Wang C., Geng X., Wang R., Tang L., Han D., Liu J., Tan W. Programmable manipulation of oligonucleotide–albumin interaction for elongated circulation time. Nucleic Acids Res. 2022;50:3083–3095. doi: 10.1093/nar/gkac156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hariharan V.N., Shin M., Chang C.-W., O’Reilly D., Biscans A., Yamada K., Guo Z., Somasundaran M., Tang Q., Monopoli K., et al. Divalent siRNAs are bioavailable in the lung and efficiently block SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2219523120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ozeri-Galai E., Friedman L., Barchad-Avitzur O., Markovetz M.R., Boone W., Rouillard K.R., Stampfer C.D., Oren Y.S., Hill D.B., Kerem B., Hart G. Delivery Characterization of SPL84 Inhaled Antisense Oligonucleotide Drug for 3849 + 10 kb C- > T Cystic Fibrosis Patients. Nucleic Acid Therapeut. 2023 doi: 10.1089/nat.2023.0015. [DOI] [PubMed] [Google Scholar]
- 99.Freund N., Taylor A.I., Arangundy-Franklin S., Subramanian N., Peak-Chew S.-Y., Whitaker A.M., Freudenthal B.D., Abramov M., Herdewijn P., Holliger P. A two-residue nascent-strand steric gate controls synthesis of 2’-O-methyl- and 2’-O-(2-methoxyethyl)-RNA. Nat. Chem. 2023;15:91–100. doi: 10.1038/s41557-022-01050-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berlier J.E., Rothe A., Buller G., Bradford J., Gray D.R., Filanoski B.J., Telford W.G., Yue S., Liu J., Cheung C.-Y., et al. Quantitative Comparison of Long-wavelength Alexa Fluor Dyes to Cy Dyes: Fluorescence of the Dyes and Their Bioconjugates. J. Histochem. Cytochem. 2003;51:1699–1712. doi: 10.1177/002215540305101214. [DOI] [PubMed] [Google Scholar]
- 101.Gebhardt C., Lehmann M., Reif M.M., Zacharias M., Gemmecker G., Cordes T. Molecular and Spectroscopic Characterization of Green and Red Cyanine Fluorophores from the Alexa Fluor and AF Series. ChemPhysChem. 2021;22:1566–1583. doi: 10.1002/cphc.202000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stribbling S.M., Ryan A.J. The cell-line-derived subcutaneous tumor model in preclinical cancer research. Nat. Protoc. 2022;17:2108–2128. doi: 10.1038/s41596-022-00709-3. [DOI] [PubMed] [Google Scholar]
- 104.Yardeni T., Eckhaus M., Morris H.D., Huizing M., Hoogstraten-Miller S. Retro-orbital injections in mice. Lab. Anim. 2011;40:155–160. doi: 10.1038/laban0511-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Refaat A., Yap M.L., Pietersz G., Walsh A.P.G., Zeller J., del Rosal B., Wang X., Peter K. In vivo fluorescence imaging: success in preclinical imaging paves the way for clinical applications. J. Nanobiotechnol. 2022;20:450. doi: 10.1186/s12951-022-01648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
H1975 cells were incubated with 25 nM Atto647N-labeled MinE07 and single-molecule fluorescence imaging was performed. The video consists of a time-lapse sequence acquisition of diffraction-limited total internal reflection fluorescence (TIRF) images and shows extended colocalization of two individual EGFRs labeled with MinE07 on the basal cell membrane. A clear increase of fluorescence intensity is detected upon colocalization. The lifetime of the colocalization event is ∼1.9 s (21 frames), suggesting that this is not due to random molecular collisions or stochastic colocalization (lifetime <0.2 s). TIRF microscopy was performed on a Leica SR GSD 3D microscope using a 160× objective lens (HC PL APO 160× /1.43, oil immersion), a 500-mW 642-nm laser (MPBC, Montreal, Quebec, Canada), a 710/100-nm emission bandpass filter, and an EMCCD camera (iXon Ultra 897, Andor, UK). Time-lapse sequences were recorded as previously described in Delcanale et al. (https://doi.org/10.1002/anie.202004764). Camera exposure time, 50 ms; frame acquisition time (or total time per frame), 90 ms; laser intensity, 40% of maximum power; total frames in video S1, 78; video reproduction, 5 frames per second. Scale bar, 5 μm.
Data Availability Statement
Data are contained within the article or supplemental information. Unprocessed data and additional information regarding specific methods of interest to readers will be made available upon request.