

Significance
Precursor tRNAs have flanking and intervening sequences removed by specific ribonucleases. We have previously demonstrated that transcription complexes of RNA polymerase III transcribe and process precursor tRNAs to mature forms. However, the control of processing of these extra sequences was unknown. Here, we show that RNase P cleaves the 5′ leader and then degrades it by its exoribonuclease subunit Rpp14. Biochemical and reverse genetic studies reveal that Rpp14 is required for removal of flanking and intervening sequences of precursor tRNAs by the tRNA splicing complex and RNase Z. RNase P coordinates the function of these complementary ribonucleases in transcription complexes consisting of two main modules, one for precursor tRNA polymerization and the other for processing and degradation of this transcript.
Keywords: RNA polymerase III, transcription complex, RNase P, tRNA, exoribonuclease
Abstract
Precursor tRNAs are transcribed with flanking and intervening sequences known to be processed by specific ribonucleases. Here, we show that transcription complexes of RNA polymerase III assembled on tRNA genes comprise RNase P that cleaves precursor tRNA and subsequently degrades the excised 5′ leader. Degradation is based on a 3′–5′ exoribonucleolytic activity carried out by the protein subunit Rpp14, as determined by biochemical and reverse genetic analyses. Neither reconstituted nor purified RNase P displays this magnesium ion-dependent, processive exoribonucleolytic activity. Markedly, knockdown of Rpp14 by RNA interference leads to a wide-ranging inhibition of cleavage of flanking and intervening sequences of various precursor tRNAs in extracts and cells. This study reveals that RNase P controls tRNA splicing complex and RNase Z for ordered maturation of nascent precursor tRNAs by transcription complexes.
The human genome has hundreds of transfer RNA (tRNA) genes mapped in several chromosomes. These small genes are transcribed by RNA polymerase (Pol) III, which generates precursor transcripts with flanking and intervening sequences shown to be removed by specific ribonucleases. Thus, the 5′ leader and 3′ trailer are excised by RNase P and RNase Z, respectively, whereas the intron is spliced out by the tRNA splicing complex (1–4). However, it remains unknown how these ribonucleases coordinate multistep processing with transcription of precursor tRNAs.
Human RNase P consists of H1 RNA and 10 protein subunits, termed Rpp14, Rpp20, Rpp21, Rpp25, Rpp29, Rpp30, Rpp38, Rpp40, Pop1, and Pop5 (5–10). These subunits form an interlocked ribonucleoprotein structure via extensive interactions (11). Except for Rpp14, the remaining protein subunits have homologs in yeast and Archaea (12, 13). In addition to its role in tRNA maturation, RNase P and its individual subunits serve unconventional tasks in chromatin structure and function (14). This ribonucleoprotein is implicated in transcription of small noncoding RNA genes by Pol III in cells and extracts (15, 16), but not in a defined reconstitution system (17). Genomic loci of transcriptionally active tRNA genes are bound by Rpp14, Rpp20, Rpp21, Rpp25, Rpp29, Rpp30, and Rpp40 (15, 16, 18). Since cleavage of precursor tRNA by RNase P is executed by H1 RNA (19, 20), the protein subunits seem to serve complementary tasks in transcription and maturation of this precursor. Here, we demonstrate that the Rpp14 subunit of RNase P found in transcription complexes of Pol III has a biologically relevant exoribonucleolytic activity that degrades the 5′ leader after its excision from precursor tRNA. Rpp14 is required by RNase P for controlling ordered cleavage of flanking and intervening sequences of nascent precursor tRNAs by the tRNA splicing complex and RNase Z preassembled in transcription complexes.
Results
RNase P Degrades the Excised 5′ Leader of Precursor tRNA.
Transcription complexes were assembled on a human tRNATyr gene (Fig. 1B) in S100 extract of HeLa cells. The resulting complexes were then purified by gel filtration chromatography using a Sephacryl S-500 HR column, which has a broad resolution range of 4 × 104 to 2 × 107. Transcription complexes proficient in synthesis of 32P-labeled precursor tRNATyr (GUA) were eluted in fractions F32 to F45 with peak activity seen in F38 to F40 (Fig. 1A, lanes 5 to 10). Extensive cleavage of the primary tRNATyr (Fig. 1B; 112-nt) by RNase P, as manifested in generation of the intermediate precursor tRNATyr, was evident in the same fractions (Fig. 1A, lanes 5 to 10; 96-nt band). This intermediate lacked the two flanking sequences, but retained the intron (Fig. 1B) (21), which was subsequently removed to produce tRNA (Fig. 1A, F38, 76-nt band). Fractions with the peak activities of RNase P and Pol III were enriched with their corresponding protein subunits (Fig. 1C, lanes 6 to 10; Dataset S2).
Fig. 1.
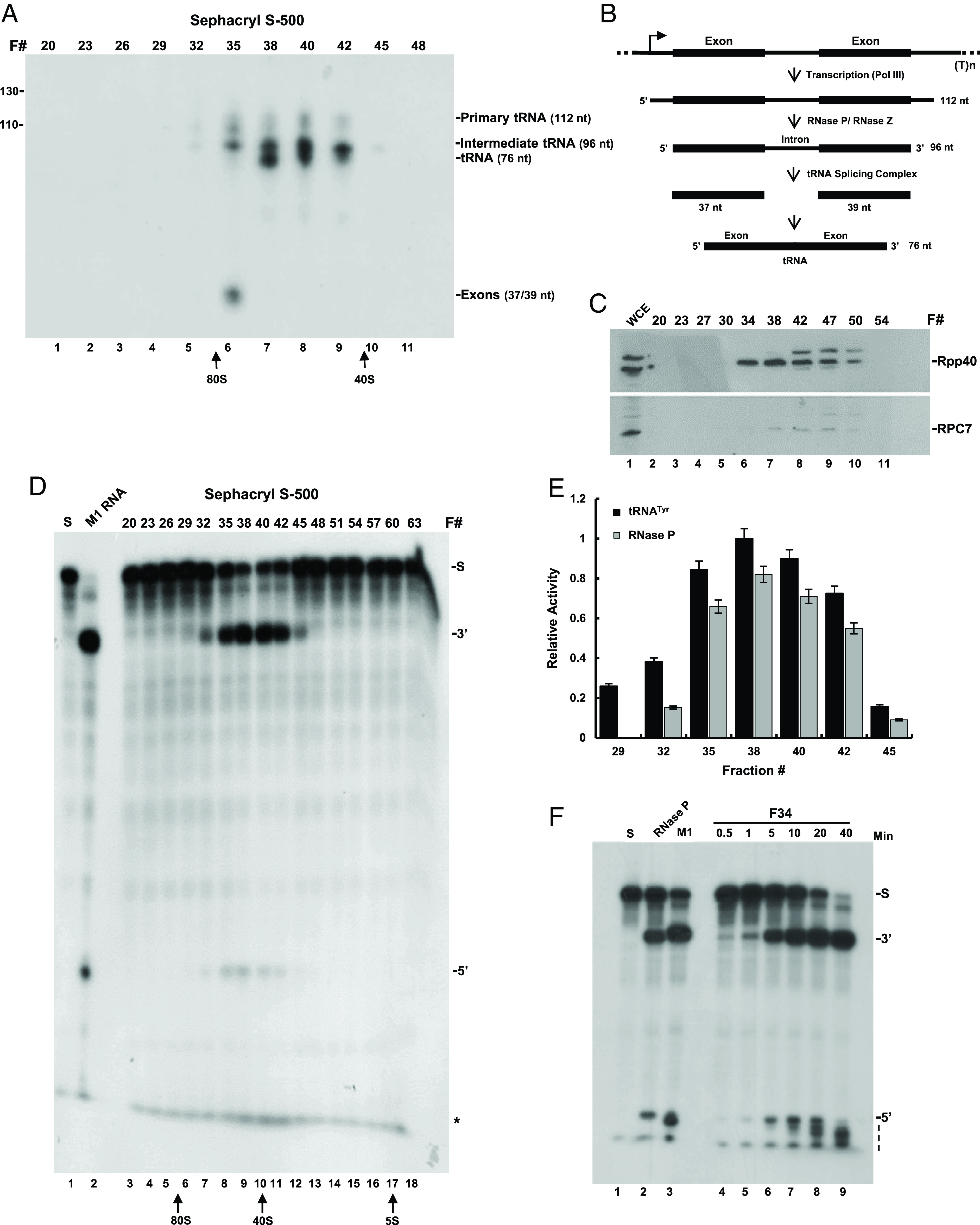
Transcription and cleavage of precursor tRNATyr by transcription complexes. (A) Transcription complexes were assembled on a human tRNATyr gene in S100 extract of HeLa cells and then were purified by gel filtration chromatography using a Sephacryl S-500 column (Materials and Methods). Eluted fractions were immediately tested for transcription of the tRNATyr gene in reactions containing 32P-UTP for 3 h. The resulted labeled RNAs were separated in a denaturing 8% polyacrylamide gel and visualized by autoradiography. The positions of the primary tRNATyr (112 nt), intermediate tRNATyr (96 nt) lacking the 5′ leader and 3′ trailer, tRNA (76 nt), and two excised exons (37/39 nt) bands are indicated. The positions of copurifying 40S and 80S ribosomal particles are pointed by arrows. (B) A scheme of the tRNATyr gene and processing of its primary transcript to mature forms. (C) Western blot analysis of Rpp40 and RPC7. Proteins in eluted fractions seen in A were analyzed by western blotting using antibodies against Rpp40 and RPC7. S100 cell extracts (WCE) were used as controls. The positions of the two proteins are shown. (D) Processing of an in vitro synthesized, internally 32P-labeled precursor tRNASer (S) by RNase P found in eluted fractions seen in A. The precursor tRNA (S), tRNA (3′), and 5′ leader (5′) were analyzed by electrophoresis in an 8% sequencing gel and visualized by autoradiography. The band seen in the gel front represents mononucleotides (asterisk) peaked in F35 to F42. (E) Quantitation of the intermediate 96-nt band (black bars) seen in A and 5′ leader band (gray bars) seen in D. Note the overlapping activities of the same RNase P, which acted on nascent precursor tRNATyr for generating the intermediate in A and synthetic precursor tRNASer in D. The error bars denote three independent experiments. (F) Kinetic analysis of cleavage (5′ band) and degradation (dashed line) of the 5′ leader of internally labeled precursor tRNASer by RNase P of fraction F34 in D. Reactions were in 1 × MRP/TNET buffer, containing 15 mM MgCl2. As controls, DEAE-purified HeLa RNase P (lane 2) and M1 RNA (lane 3) were checked for substrate cleavage. A typical one-nucleotide difference in 5′ leader sizes was produced by the two enzymes (lane 2 vs. 3).
The existence of RNase P in fractions F32 to F45 was corroborated by examining the ability of this endoribonuclease to cleave the 5′ leader of an in vitro-transcribed 32P-labeled precursor tRNASer in the presence of Mg2+ ions (SI Appendix, Fig. S1D, lanes 7 to 12, 5′ band). As expected, the peak activity was detected in fractions F38 to F40 (Fig. 1E), consistent with that exerted on nascent precursor tRNATyr in generating the 96-nt intermediate (Fig. 1A). Based on the size exclusion of cofractionating 40S and 80S ribosomal particles (Fig. 1A; arrows), which have molecular weights of ∼1.5 × 106 and ∼4.5 × 106, respectively, RNase P existed in transcription complexes with molecular weights of 1.5 to 3.5 × 106. The activity of RNase P was not detected in the latter fractions (Fig. 1D, lanes 13 to 18), which corresponded to eluates with low molecular weights (Fig. 1D; see 5S rRNA).
Copurification of RNase P and Pol III was also obtained when transcription complexes were preassembled on a human intron-containing tRNAArg (TCT) gene in S100 extracts of HEK293 cells, and purified by velocity sedimentation in a 15 to 35% glycerol gradient (Materials and Methods). Synthesis and processing of precursor tRNAArg peaked in fractions F14 to F16, as manifested in the production of mature tRNA (Fig. 2A, lanes 7 and 8; 76-nt band). These fractions contained protein subunits of copurifying RNase P and Pol III, as determined by mass spectrometry analysis (Dataset S3).
Fig. 2.

Transcription complexes contain a processive exoribonucleolytic activity that degrades the excised 5′ leader. (A) Transcription complexes were assembled on the human tRNAArg gene in S100 extracts of HEK293 cells and then were purified by velocity sedimentation using a 15 to 35% glycerol gradient (Materials and Methods). Derived fractions were immediately tested in transcription reactions containing 32P-UTP for 2 h. The products were analyzed in a denaturing 8% polyacrylamide sequencing gel. Positions of the primary tRNAArg (93 nt), intermediate tRNAArg (88 nt) lacking the 5′ leader and 3′ trailer, tRNA (76 nt), excised exons and intron are indicated. The asterisk points to a labeled transcript likely generated from inaccurate termination. (B) Fraction F14 in A was tested for site-specific cleavage of the 5′ leader of 32P-labeled precursor tRNASer and its subsequent, overlapping degradation in the presence of Mg2+ ions, for the indicated times. Cleavage products were then separated in 8% sequencing gel and visualized by autoradiography. The positions of precursor tRNASer (S) and 5′ leader (5′) are shown. The ladder is marked by a dashed line. The mononucleotide (Mono) band is indicated. The large gel was exposed to two x-ray films (arrowhead) for the same exposure time. Precursor tRNASer and 5′ leader (lane 1) were not degraded by M1 RNA (lane 2), recombinant Rpp14 (lane 3), or recombinant Rpp29 (lane 4) in the presence of Mg2+ ions. (C) The signal intensities of precursor tRNASer and excised 5′ leader bands seen in B were quantitated and plotted. The error bars are shown for at least three independent experiments.
Cleavage of precursor tRNASer by RNase P found in fractions F32-45 described above was accompanied by a degradation activity, as reflected by the increased label of the fuzzy band seen in the gel front (Fig. 1D, lanes 8 to 11; asterisk). This activity peaked with the endoribonucleolytic activity of RNase P in fraction F38 (Fig. 1E, F38). Kinetic analysis demonstrated that the degradation activity was exerted on the 5′ leader since this 28-nt sequence was digested to shorter fragments (Fig. 1F, lanes 4 to 9; 5′ and dashed line). A similar result was obtained when transcription complexes, purified by velocity sedimentation in glycerol gradient (Fig. 2A; F14), were checked for cleavage of a 5′-end 32P-labeled precursor tRNASer and degradation of the excised 5′ leader (Fig. 2B, lanes 5 to 10; dashed line). The 5′ leader was degraded by a processive exoribonucleolytic activity that proceeded in the 3′ to 5′ direction, generating a ladder of cleavage intermediates (Fig. 2B, lanes 5 to 10; dashed line). These intermediates were finally digested to mononucleotides seen in the gel front (Fig. 2B, lanes 5 to 10; Mono). The precursor tRNASer form was cleaved by RNase P, but without generating a ladder (Fig. 2B, S band). Hence, the detected mononucleotides were not produced by an exoribonucleolytic activity acting on precursor tRNA in the presence of Mg2+ ions (Fig. 2C) (see below). Degradation of the 5′ leader to mononucleotide was observed when Schizosaccharomyces pombe precursor tRNASer (SI Appendix, Fig. S1A, lanes 5 to 9 vs. 2; 5′ band) and Escherichia coli precursor tRNATyr (SI Appendix, Fig. S1A, lanes 10 to 14 vs. 4; 5′ band) were used as substrates. However, the addition of cold substrate to the cleavage reaction resulted in the appearance of the ladder due to the decrease in the excision and subsequent degradation of the 5′ leader to mononucleotides (SI Appendix, Fig. S1C vs. SI Appendix, Fig. S1B, lanes 6 to 10 vs. 1 to 5; 5′ band and dashed line) (Materials and Methods).
The overlap of 5′ leader cleavage and degradation (Fig. 2B, lanes 5 to 10; 5′ band vs. ladder) implies that the latter activity is associated with RNase P (see below).
Recombinant Rpp14, but not Pop5 and OIP2, Increases Degradation of the Excised 5′ Leader.
To assess whether one of the subunits of RNase P was responsible for the degradation activity described above, transcription complexes (Fig. 2A, F14) were examined for removal and degradation of the 5′ leader of precursor tRNASer in the presence and absence of recombinant Rpp14 (Fig. 3) and Pop5 (Fig. 4) proteins, which were purified to near homogeneity (SI Appendix, Fig. S2 A and B) (Materials and Methods). Rpp14 and its partner, OIP2 (EXOSC8), are parts of a putative exosome acting on the 3′ end of precursor tRNA in the absence of Mg2+ ions (22), whereas Pop5 has an overall structure similar to that of Rpp14 (11). The addition of recombinant Rpp14 increased the cleavage of the 5′ leader of a labeled precursor tRNASer, as well as its degradation to mononucleotides (Fig. 3A, lanes 10 to 15 vs. 4 to 9, and Fig. 3B). Recombinant Rpp14 acted via RNase P (Fig. 3A, lane 6 vs. 13; 5′ band, and Fig. 3C), as evidenced from the increase of the endonucleolytic cleavage of the 5′ leader (SI Appendix, Fig. S2C, lanes 3 to 10 vs. 2; 5′ band). Surprisingly, the addition of recombinant Pop5 also enhanced the cleavage of the 5′ leader and generation of mononucleotides in the presence of divalent ions (Fig. 4A, lanes 15 to 18 vs. lanes 5 to 8, Fig. 4 B and C). However, these ribonucleolytic activities coincided with a decline in precursor tRNASer (Fig. 4A, lanes 14 to 18 vs. lanes 4 to 8; S). Hence, Pop5 is an exoribonuclease, but it produces mononucleotides by acting on precursor tRNA (SI Appendix, Fig. S2C, lane 12 vs. 1, 11 and 13). The addition of recombinant Rpp25 showed no effect on precursor tRNA (SI Appendix, Fig. S3A, lanes 8 to 11 vs. 4 to 7; SI Appendix, Fig. S3 B and C).
Fig. 3.
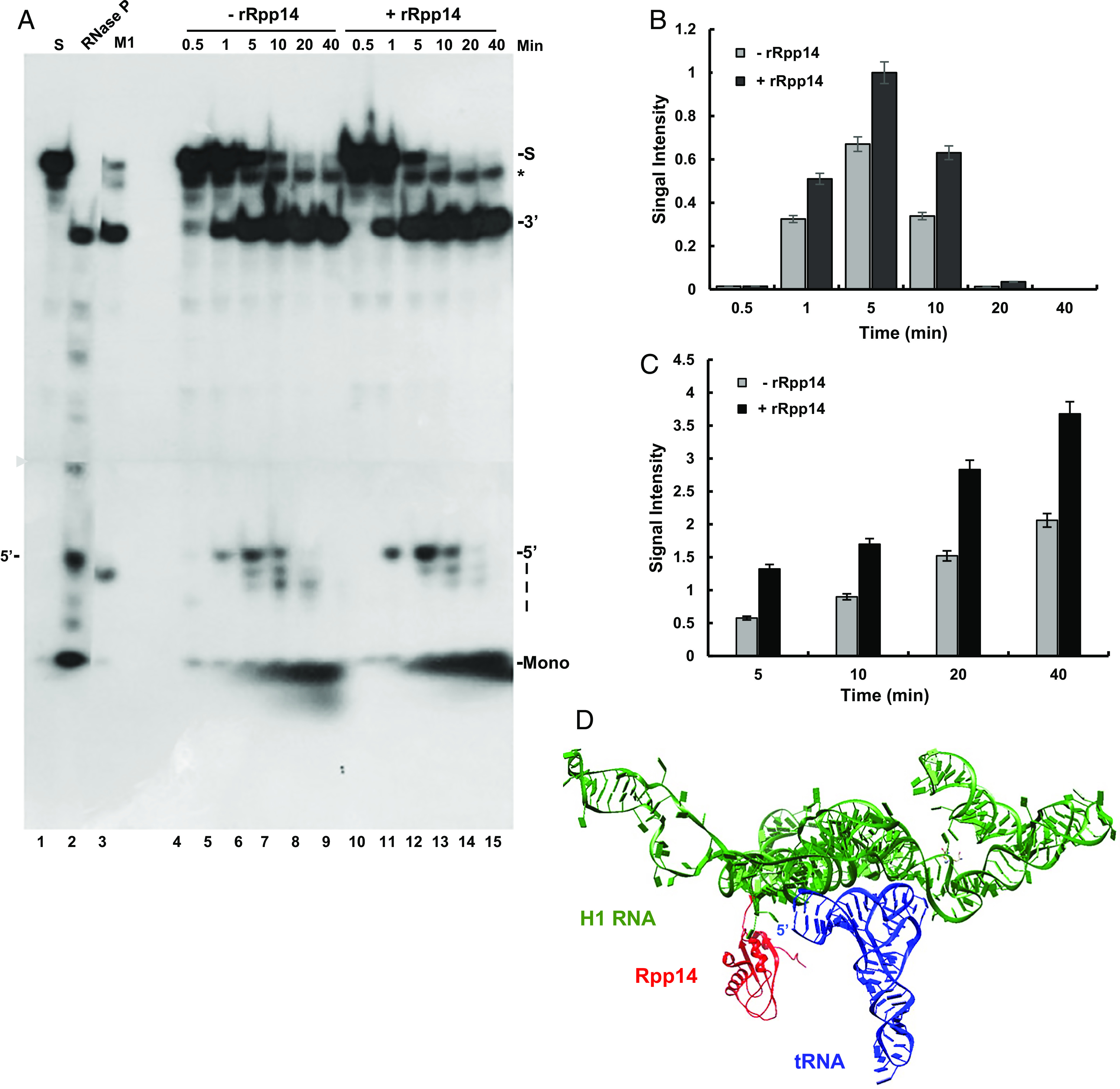
Recombinant Rpp14 increases cleavage and degradation of the 5′ leader by RNase P found in transcription complexes. (A) RNase P in fraction F14 described in Fig. 2B was tested for cleavage and degradation of the 5′ leader of an internally 32P-labeled precursor tRNASer for the indicated time points. Excess amounts of cold substrate were added to the reactions to slow down degradation of the 5′ leader. Cleavage products were analyzed in an 8% sequencing gel. The positions of the substrate (S), tRNA (3′), 5′ leader (28 nt), and mononucleotides (Mono) are shown. For cleavage controls, DEAE-purified HEK293 RNase P (lane 2) and M1 RNA (lane 3) were included (lane 1). The gel was exposed to two autoradiograms (arrowhead) for the same time. The asterisk points to a truncated substrate, as evident in lane 1. (B) The signal intensities of the 5′ leader bands seen in A were quantitated and plotted. (C) Bars depict quantitation of the peak signals of the mononucleotides in lanes 6 and 12 of the gel in A. The error bars in B and C are shown for at least three independent experiments. (D) Rpp14 position is close to the 5′ end of tRNA. Position of Rpp14 (red) relative to those of H1 RNA (green) and tRNA (blue) in RNase P, based on the cryo-EM structure of the holoenzyme (11). The other protein subunits were omitted for clarity. The distance of Rpp14 from the 5′ end of tRNA is ~71Å, which fits a 5′ leader of 21-nt in length. This length falls in the wide range of 5′ leader sizes, 2 to 40 nt, of human precursor tRNAs (1).
Fig. 4.

Recombinant Pop5 elicits degradation of precursor tRNA, but not excised 5′ leader, to mononucleotides by transcription complexes. (A) RNase P in transcription complexes eluted in fraction F39 of the gel filtration described in Fig. 1A were tested for cleavage and degradation of the 5′ leader of precursor tRNASer in the absence (lanes 4 to 8) or presence (lanes 14 to 18) of recombinant Pop5 protein. Protein alone was examined as a negative control (lanes 9 to 13). Excess amounts of cold precursor tRNASer was added to the reactions to slow down degradation of the 5′ leader. Cleavage products were analyzed in a sequencing gel and exposed to two x-ray films (arrowhead), as in Fig. 1A. (B) The signal intensities of the 5′ leader bands seen in A, lanes 4 to 8 and 14 to 18, were quantitated and plotted. Error bars are shown for at least three independent experiments. (C) The signal intensities of the mononucleotide bands in A, lanes 8, 13, and 18, were quantitated and plotted. Error bars are shown for at least three independent experiments.
In contrast to the above findings, the presence of recombinant Rpp14 and Pop5 in a reconstituted RNase P, consisting of H1 RNA and eight recombinant protein subunits, did not bring to degradation of the correctly cleaved 5′ leader of precursor tRNASer (Fig. 5A, lanes 2 and 5). Exclusion of the Mg2+ ions from the reconstitution reaction led to degradation of the substrate (Fig. 5A, lanes 2 and 5 vs. 3). Since neither the reconstituted nor purified RNase P eliminates the 5′ leader (Fig. 5A, lanes 2, 4, and 5, respectively), the context in which this enzyme occurs in transcription complexes is critical to degradation. Of note, except for Rpp14, the absence of the other recombinant proteins had only a minor effect on the reconstituted activity of RNase P (Fig. 5A, lane 11 vs. 6 to 13; 5′ band; Fig. 5 B and C), indicating that each protein is individually dispensable for substrate cleavage, which is based on H1 RNA (19, 20). Reconstitution required freshly prepared recombinant proteins, since these polypeptides become functionally impaired shortly after affinity purification and storage.
Fig. 5.

Reconstituted and purified RNase P do not degrade excised 5′ leader. (A) RNase P was reconstituted from in vitro–transcribed H1 RNA and eight recombinant proteins, Rpp14, Rpp20, Rpp21, Rpp25, Rpp29, Rpp30, Rpp40, and Pop5 (lanes 2 and 5). The resulted activity was determined by site-specific cleavage of the 5′ leader of a labeled precursor tRNASer. Reconstituted RNase P forms lacking one of the eight recombinant proteins were also examined under the same reaction conditions (lanes 6 to 13). Reconstituted RNase P degrades the precursor tRNA in the absence of Mg2+ ions (lane 3 vs. 2 and 5). (B) The signal intensities of the tRNA bands in A were quantitated and plotted. Error bars are shown for at least three independent experiments. (C) The signal intensities of the 5′ leader bands in A were quantitated and plotted. Error bars are shown for at least three independent experiments.
The results supported further assessment of Rpp14 as the exoribonuclease responsible for degradation of the 5′ leader by RNase P present in transcription complexes. The proximity of Rpp14 to the 5′ end of tRNA (Fig. 3D) seen in the cryo-EM structure of purified human RNase P holoenzyme (11) supports this view.
Exogenous Expression of Rpp14 Elicits Assembly of Atypical Transcription Complexes.
A Rpp14 protein fused to a myc-tag epitope was expressed in transiently transfected HeLa cells and S100 extracts were prepared for assembly and purification of transcription complexes, as described in Fig. 2A. Fractions derived from the glycerol gradient were then examined for cleavage and degradation of the 5′ leader of precursor tRNASer and compared to control activities exhibited by purified transcription complexes prepared from untransfected cells (Fig. 6A vs. Fig. 6B). Tagged Rpp14 was integrated into competent transcription complexes, but these were sedimented in fractions near the bottom of the gradient, when compared to the control complexes found in the upper fractions (Fig. 6 A and B, lanes 7 to 14), as expected (Fig. 2A). Apparently, expression of the tagged Rpp14 led to the assembly of large transcription complexes comprising RNase P. These atypical complexes exhibited increased endonucleolytic cleavage of the 5′ leader of precursor tRNASer, when compared to that displayed by the control complexes (Fig. 6A, lanes 11 to 14 vs. Fig. 6B, lanes 9 to 12; 5′ band). This effect, however, was not accompanied by an increase in 5′ leader degradation to mononucleotides (Fig. 6A, lanes 11 to 14 vs. Fig. 6B, lanes 9 to 12; Mono; Fig. 6C vs. Fig. 6D). Thereby, the tagged RNase P in the fractions was defective in 5′ leader degradation (see below), ruling out the possibility that the multiplexes were generated by aggregation of insoluble Rpp14.
Fig. 6.
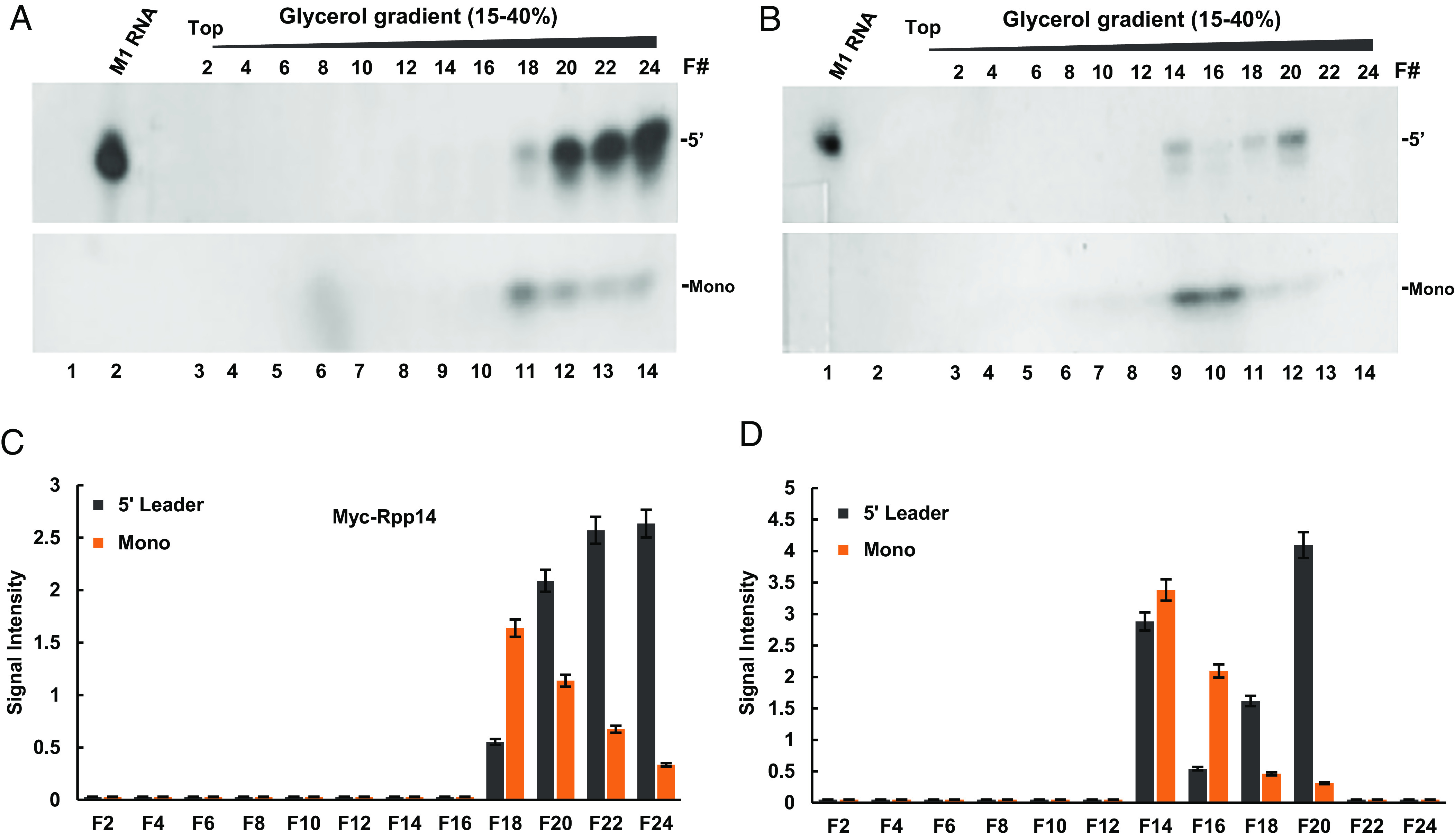
Exogenous expression of Rpp14 enhances cleavage, but not degradation, of the 5′ leader by RNase P existing in transcription complexes. (A and B) A myc-tagged Rpp14 was expressed in transfected HeLa cells for 42 h and S100 extract was prepared. Transcription complexes assembled on a tRNAArg gene in this extract (A) and control extract (B), prepared from untransfected cells, were purified by velocity sedimentation in 15 to 35% glycerol gradients. Selected fractions derived from the two gradients were then checked for RNase P activity and cleavage products were analyzed in a denaturing 8% polyacrylamide gels, which were exposed to x-ray autoradiography. The 5′ leader (Upper) and mononucleotide (Lower) bands are shown. A sign of a sellotape is seen in the gel right side. (C) The 5′ leader and mononucleotides bands seen in A were quantified and plotted. Error bars are shown for at least three independent cleavage assays. (D) The 5′ leader and mononucleotides bands seen in B were quantified and plotted. Error bars are shown for at least three independent assays.
Knockdown of Rpp14 Leads to a General Inhibition of Cleavage of Nascent Precursor tRNAs.
Attempts to investigate Rpp14 by knocking out its respective gene using standard and inducible CRISPR-Cas9 techniques were unsuccessful, as the resulting cell clones survived for a few passages in culture. Therefore, Rpp14 mRNA was targeted in HEK293 cells by the use of three shRNAs directed against the coding region and 3′-UTR (Materials and Methods) (23). The shRNAs were expressed by minigenes cloned in psi-U6 vector. S20 extracts and total RNA were prepared from transfected cells at 24, 48, and 72 h. Knockdown was confirmed by semiquantitative RT-PCR using specific primers that amplified the open reading frame sequence of the Rpp14 mRNA (Fig. 7A, lane 2 vs. 3 to 5, lane 6 vs. 7 to 9 and lane 10 vs. 11 to 13, and Fig. 7C). A reduction of ~ 90% in the steady-state level of the Rpp14 mRNA was obtained in 48 h posttransfection (Fig. 7A, lanes 7 to 9 vs. 6, and Fig. 7C), which was followed by a recovery in 72 h (Fig. 7A, lanes 11 to 13 vs. 7 to 9). The steady-state levels of GAPDH mRNA remained largely unchanged (Fig. 7B, lanes 2 to 13). Repeated attempts to demonstrate Rpp14 knockdown at the protein level by western blot analysis using commercially available antibodies were unsuccessful. Strikingly, transcription of the tRNAArg gene in S20 extracts prepared from cells with Rpp14 knockdown produced a large amount of precursor tRNAArg, when compared to that produced by the control extracts (Fig. 7D, lane 2 vs. 3 to 5, 6 vs. 7 to 9, and 10 vs. 11 to 13; Fig. 7H). By contrast, production of the mature tRNAArg sharply declined at 24 and 48 h (Fig. 7D, lanes 2 vs. 3 to 5, and 6 vs. 7 to 9; 75-bp band; Fig. 7I), but reestablished at 72 h (Fig. 7D, lane 10 vs. 11 to 13; Fig. 7I) owing to the transient knockdown. Similarly, examination of transcription of a human tRNALys gene revealed synthesis of the precursor tRNALys (UUU) containing a long 5′ leader of 23-nt in length during Rpp14 knockdown (Fig. 7E, lane 6 vs. 7 to 9; Fig. 7J). Unabated transcription of the tRNAiMet gene was also evident, as manifested in the continuous synthesis of unprocessed precursor tRNAiMet (Fig. 7G, lane 2 vs. 3 to 5, lane 6 vs. 7 to 9 and lane 10 vs. 11 to 13; Fig. 7L). Transcription of a human tRNAHis (GTG) gene, which codes for a primary transcript with a long 3′ trailer of 38-nt in length, was unaffected too (Fig. 7F, lane 2 vs. 3 to 5, 6 vs. 7 to 9 and 10 vs. 11 to 13; Fig. 7K). These precursor tRNAs were generated from their respective cloned genes, as exemplified for the precursor tRNAiMet (Fig. 7M, lane 2 vs. 3 and 4). Moreover, the cells with Rpp14 knockdown had high steady-state levels of endogenous precursor tRNAArg, precursor tRNALys, and precursor tRNAHis, as verified by RT-PCR analysis of total RNA (Fig. 7 N–P, lane 2 vs. 3 to 5; Fig. 7 Q–S).
Fig. 7.

Rpp14 knockdown leads to a wide-ranging inhibition of processing of flanking and intervening sequences of nascent precursor tRNAs. (A) Rpp14 mRNA was targeted for destruction in HEK293 cells (lanes 2, 6, and 10) by the use of shRNA A (lanes 3, 7, and 11), shRNA B (lanes 4, 8, and 12) or shRNA C (lanes 5, 9, and 13) for the indicated time points. shRNA A and B were directed against the coding region of Rpp14, whereas shRNA Ctargeted the 3′-UTR (Materials and Methods). S20 extracts and total RNAs were then prepared from transfected and control cells. The steady-state levels of Rpp14 mRNA were determined by semiquantitative RT-PCR at 24, 48, and 72 h. The PCR product, 321 bp in size, was visualized by electrophoresis in 2% agarose gel. The identity of the product was confirmed by direct DNA sequencing. Lanes 1 and 14 depict DNA size markers. (B) The steady-state levels of GAPDH mRNA were determined by RT-PCR and used as internal controls and for standardization. (C) The PCR products of Rpp14 seen in A were quantitated and plotted. (D–G) S20 extracts of control and transfected cells described in A were examined for transcription of tRNAArg (D), tRNALys (E), tRNAHis (F), and tRNAiMet (G). In D, the lower band (75 bp) denotes the mature tRNAArg, whereas the larger one (88 bp) represents intron-containing precursor tRNAArg. The sizes of the PCR products are indicated and their identities were confirmed by direct DNA sequencing. Lanes 1 and 14 have DNA size markers. Schematics of precursor tRNAArg, tRNALys, and tRNAHis are shown below. Precursor tRNAHis has long 5′ leader and 3′ trailer sequences enabling the detection of the primary transcript using corresponding primers. The lower band is unknown. Error bars designate the SD of three independent experiments. (H and I) The PCR products that correspond to precursor tRNAArg and tRNAArg seen in D were quantitated and plotted. The error bars denote the SD of three independent experiments. (J–L) The PCR bands of tRNAHis, tRNALys, and tRNAiMet seen in E–G were quantitated and plotted. (M) To confirm the specificity of the RT-PCR in detecting in vitro transcribed tRNAs, but not the endogenous tRNA counterparts, the tRNAiMet gene was tested in control transcription reaction (lane 2) or reactions lacking extract (lane 3) or plasmid (lane 4) for 3 h. After extraction of nucleic acids, precipitation, and treatment with DNase I (Materials and Methods), total RNA was reverse transcribed and then analyzed for tRNAiMet, as in G. The PCR product was seen only in lane 2, which represents the nascent tRNAiMet generated by in vitro transcription. (N–P) RT-PCR analysis of tRNAArg, tRNALys, and tRNAHis in total RNA extracted from cells with knockdown of Rpp14 or control (lanes 3 to 5, 7 to 9, and 11 to 13 vs. 2, 6, and 10). (Q–S) PCR bands of tRNAArg, tRNALys, and tRNAHis seen in N–P were quantitated and plotted.
The results uncover that Rpp14 knockdown leads to inhibition of cleavage of flanking and intervening sequences of various precursor tRNAs in cells and extracts.
Discussion
We have shown that human RNase P exhibits two consecutive ribonucleolytic activities by which this holoenzyme cleaves precursor tRNA and subsequently degrades the excised 5′ leader. These activities are interdependent and coordinated by RNase P present in transcription complexes. The 5′ leader sequence is removed from precursor tRNA before it is degraded by a processive 3′ to 5′ exoribonucleolytic activity displayed by the Rpp14 subunit. It remains unknown, however, if this exoribonuclease subunit recognizes and degrades 5′ leader sequences of all precursor tRNAs from diverse organisms (SI Appendix, Fig. S1). The two overlapping ribonucleolytic activities of RNase P require cooperation of Rpp14 with H1 RNA, which is supported by the proximity of this subunit, as part of the Pop5-Rpp14-(Rpp30)2-Rpp40 subcomplex (11), to the catalytic core of H1 RNA (Fig. 3D). Noticeably, highly purified and reconstituted RNase P possess Rpp14, but these catalytic ribonucleoproteins do not degrade excised 5′ leaders (Fig. 5A). Accordingly, Rpp14 displays its 3′ to 5′ exoribonuclease activity in a context-dependent manner, as a subunit of RNase P existing in transcription complexes. Since the 5′ leader of nascent precursor tRNA (Fig. 1A) or in vitro-transcribed precursor tRNA (Fig. 1D) is eliminated by RNase P in transcription complexes, the function of Rpp14 as an exoribonuclease subunit is independent of transcription. Though, precursor tRNAs are synthesized in transcription factories in the cell (24), and hence, the cleavage and degradation of the 5′ leader of precursor tRNA by RNase P should be linked to transcription. The assembly of multiplexes of transcription complexes that are defective in degradation of the 5′ leader (Fig. 6) supports a direct role of Rpp14 in the latter activity. Further study is required to assess whether multiplexes are formed in cells expressing high levels of Rpp14.
Rpp14 has an interacting partner, OIP2, which has been shown to degrade precursor tRNAs in the absence of divalent ions in vitro (22). Our study, however, does not support a collaboration of these two exoribonucleases in excision and elimination of the 5′ leader (Figs. 1 and 2). Therefore, the exosome, which contains OIP2 (EXOSC8), is not involved in the degradation of the leader by transcription complexes. Instead, it is RNase P that eliminates this sequence, a task that renders this catalytic ribonucleoprotein a major degradation machinery, when considering the large number of precursor tRNAs transcribed in the cell and their corresponding 5′ leaders are excised and degraded. In addition to Rpp14, the related Pop5 also has an exoribonuclease activity, but this highly conserved protein subunit (7) degrades precursor tRNA. RNase P contains putative ribonucleases that form the Pop5-Rpp14-(Rpp30)2-Rpp40 subcomplex, shown to bind the catalytic domain of H1 RNA, a ribonuclease by itself.
The present study reveals that RNase P functions in transcription complexes that comprise the tRNA splicing complex and RNase Z. These three complementary ribonucleases form an intricate ribonuclease module, in which RNase P controls the other two ribonucleases for ordered cleavage of flanking and intervening sequences of nascent precursor tRNA. This module constitutes a counterpart to the adjoining Pol module, which is made of Pol III and its core and auxiliary transcription factors. In view of the primordial evolutionary origins of H1 RNA and tRNA and their incomparable coevolution as enzyme and substrate, the present study of Pol III transcription complexes will help in elucidating the origin of short RNA polymerization by exploring the opposite reaction, RNA degradation, carried out by these versatile complexes.
Materials and Methods
Cell cultures and transfection, RNA interference, S20 and S100 extract preparation, tRNA gene constructs, in vitro transcription of 32P-labeled precursor tRNAs, RT-PCR, purification of transcription complexes by sedimentation in glycerol gradient, gel filtration chromatography using Sephacryl S-500 columns, velocity sedimentation analysis in glycerol gradient, and enzymatic assay of RNase P were essentially done, as previously described (15, 25, 26). For specific details, see SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Acknowledgments
We thank Manoj Komar for help with Fig. 3D. This research was supported by the Israel Science Foundation (grants #538/21 and #1205/17) and United States–Israel Binational Science Foundation (grant #2015/157).
Author contributions
N.O., D.M., and N.J. designed research; N.O. and D.M. performed research; D.M., A.R., and N.J. analyzed data; and N.J. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Gogakos T., et al. , Characterizing expression and processing of precursor and mature human tRNAs by hydro-tRNAseq and PAR-CLIP. Cell Rep. 20, 1463–1475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Popow J., et al. , HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science 331, 760–764 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Trotta C. R., et al. , The yeast tRNA splicing endonuclease: A tetrameric enzyme with two active site subunits homologous to the archaeal tRNA endonucleases. Cell 89, 849–858 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Turowski T. W., Tollervey D., Transcription by RNA polymerase III: Insights into mechanism and regulation. Biochem. Soc. Trans. 44, 1367–1375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eder P. S., Kekuda R., Stolc V., Altman S., Characterization of two scleroderma autoimmune antigens that copurify with human ribonuclease P. Proc. Natl. Acad. Sci. U.S.A. 94, 1101–1106 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarrous N., Altman S., Human ribonuclease P. Methods Enzymol. 342, 93–100 (2001). [DOI] [PubMed] [Google Scholar]
- 7.van Eenennaam H., Lugtenberg D., Vogelzangs J. H. P., van Venrooij W. J., Pruijn G. J. M., hPop5, a protein subunit of the human RNase MRP and RNase P endoribonucleases. J. Biol. Chem. 276, 31635–31641 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Guerrier-Takada C., Eder P. S., Gopalan V., Altman S., Purification and characterization of Rpp25, an RNA-binding protein subunit of human ribonuclease P. RNA 8, 290–295 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lygerou Z., Allmang C., Tollervey D., Séraphin B., Accurate processing of a eukaryotic precursor ribosomal RNA by ribonuclease MRP in vitro. Science 272, 268–270 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Lygerou Z., Pluk H., van Venrooij W. J., Séraphin B., hPop1: An autoantigenic protein subunit shared by the human RNase P and RNase MRP ribonucleoproteins. EMBO J. 15, 5936–5948 (1996). [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J., et al. , Cryo-EM structure of the human ribonuclease P holoenzyme. Cell 175, 1393–1404.e11 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Chamberlain J. R., Lee Y., Lane W. S., Engelke D. R., Purification and characterization of the nuclear RNase P holoenzyme complex reveals extensive subunit overlap with RNase MRP. Genes Dev. 12, 1678–1690 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall T. A., Brown J. W., Archaeal RNase P has multiple protein subunits homologous to eukaryotic nuclear RNase P proteins. RNA 8, 296–306 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarrous N., Roles of RNase P and its subunits. Trends Genet. 33, 594–603 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Reiner R., Ben-Asouli Y., Krilovetzky I., Jarrous N., A role for the catalytic ribonucleoprotein RNase P in RNA polymerase III transcription. Genes Dev. 20, 1621–1635 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serruya R., et al. , Human RNase P ribonucleoprotein is required for formation of initiation complexes of RNA polymerase III. Nucleic Acids Res. 43, 5442–5450 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrari R., Dieci G., The transcription reinitiation properties of RNA polymerase III in the absence of transcription factors. Cell. Mol. Biol. Lett. 13, 112–118 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reiner R., Krasnov-Yoeli N., Dehtiar Y., Jarrous N., Function and assembly of a chromatin-associated RNase P that is required for efficient transcription by RNA polymerase I. PLoS One 3, e4072 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kikovska E., Svärd S. G., Kirsebom L. A., Eukaryotic RNase P RNA mediates cleavage in the absence of protein. Proc. Natl. Acad. Sci. U.S.A. 104, 2062–2067 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mann H., Ben-Asouli Y., Schein A., Moussa S., Jarrous N., Eukaryotic RNase P: Role of RNA and protein subunits of a primordial catalytic ribonucleoprotein in RNA-based catalysis. Mol. Cell 12, 925–935 (2003). [DOI] [PubMed] [Google Scholar]
- 21.van Tol H., Stange N., Gross H. J., Beier H., A human and a plant intron-containing tRNATyr gene are both transcribed in a HeLa cell extract but spliced along different pathways. EMBO J. 6, 35–41 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang T., Altman S., A protein subunit of human RNase P, Rpp14, and its interacting partner, OIP2, have 3’–>5’ exoribonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 99, 5295–5300 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarrous N., Eder P. S., Wesolowski D., Altman S., Rpp14 and Rpp29, two protein subunits of human ribonuclease P. RNA 5, 153–157 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pombo A., Regional specialization in human nuclei: Visualization of discrete sites of transcription by RNA polymerase III. EMBO J. 18, 2241–2253 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiner R., Krasnov-Yoeli N., Dehtiar Y., Jarrous N., Function and assembly of a chromatin-associated RNase P that is required for efficient transcription by RNA polymerase I. PLoS One 3, e4072 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramanathan A., et al. , A mutation in POLR3E impairs antiviral immune response and RNA polymerase III. Proc. Natl. Acad. Sci. U.S.A. 117, 22113–22121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Data Availability Statement
All study data are included in the article and/or supporting information.