

Summary
Genome-wide association studies (GWASs) have successfully identified 145 genomic regions that contribute to schizophrenia risk, but linkage disequilibrium makes it challenging to discern causal variants. We performed a massively parallel reporter assay (MPRA) on 5,173 fine-mapped schizophrenia GWAS variants in primary human neural progenitors and identified 439 variants with allelic regulatory effects (MPRA-positive variants). Transcription factor binding had modest predictive power, while fine-map posterior probability, enhancer overlap, and evolutionary conservation failed to predict MPRA-positive variants. Furthermore, 64% of MPRA-positive variants did not exhibit expressive quantitative trait loci signature, suggesting that MPRA could identify yet unexplored variants with regulatory potentials. To predict the combinatorial effect of MPRA-positive variants on gene regulation, we propose an accessibility-by-contact model that combines MPRA-measured allelic activity with neuronal chromatin architecture.
Keywords: MPRA, schizophrenia, GWAS, non-coding variants, accessibility-by-contact model, variant function, high-throughput reporter assay, gene regulation, chromatin architecture, eQTL
Graphical abstract
Highlights
-
•
MPRA functionally validates schizophrenia GWAS variants for differential allelic activity
-
•
An accessibility-by-contact model linked these variants to predicted gene expression
-
•
These genes include transcription factors, which can have broad gene expression impact
McAfee and Lee et al. utilized a massively parallel reporter assay to functionally validate schizophrenia-associated non-coding regulatory variants. Leveraging an accessibility-by-contact model, they linked functionally validated regulatory variants to their putative gene targets and predicted their gene expression.
Introduction
Schizophrenia is a polygenic neuropsychiatric disorder that affects about 24 million people worldwide.1 Heritability estimates of schizophrenia are 60%–80%, indicating a strong contribution of genetic variation to risk for the disorder.2 Common variation explains a significant portion of heritability (24% of single-nucleotide polymorphism [SNP] heritability), and the most recent genome-wide association study (GWAS) has identified 294 genome-wide significant (GWS) loci.3 However, it is challenging to understand the functional consequence of these GWS loci because (1) most reside in non-coding DNA with unknown functions, and (2) each GWS locus contains dozens of variants that show significant association due to linkage disequilibrium (LD).
Therefore, a critical step to bridging the gap between genetic loci and biological underpinning is to identify causal variants and delineate their functional impact. While computational fine-mapping approaches have been developed to predict putative causal variants,4 these methods merely narrow down the search space of causal variants by modeling their decay with LD rather than functionally validating variants. Moreover, different fine-mapping algorithms can provide different sets of fine-mapped variants.5 The general consensus in the field is that causal variants exert their function by altering gene expression. To accurately discern variants with gene-regulatory effects, experimental validation is pivotal.
Here, we employed a massively parallel reporter assay (MPRA) to experimentally verify the difference in allelic regulatory activity between protective and risk alleles of 5,173 schizophrenia-associated fine-mapped variants6 in the context of neurogenesis. MPRA provides a scalable genetic approach to characterize gene-regulatory effects of thousands of variants in a single experiment.7,8,9 We identified 439 MPRA-positive variants that showed allelic regulatory activity in human neural progenitors (HNPs). Pre-existing strategies to prioritize causal variants did not accurately identify MPRA-positive variants.
To link MPRA-positive variants to genes, we tried two different genomic approaches: expression quantitative trait loci (eQTLs) and chromatin interaction profiles (Hi-C). We found that eQTLs and Hi-C identify distinct sets of genes with different (epi)genomic properties. In particular, the Hi-C-based approach identified genes with functional annotation, higher selective constraints, and regulatory complexities, suggesting that chromatin architecture is instrumental in assigning GWAS variants to their cognate genes. Consequently, we propose an accessibility-by-contact model that supplements chromatin contexts to MPRA-measured allelic activity and demonstrate that this model can effectively translate variant function to targetable gene expression.
Results
MPRA on schizophrenia risk variants
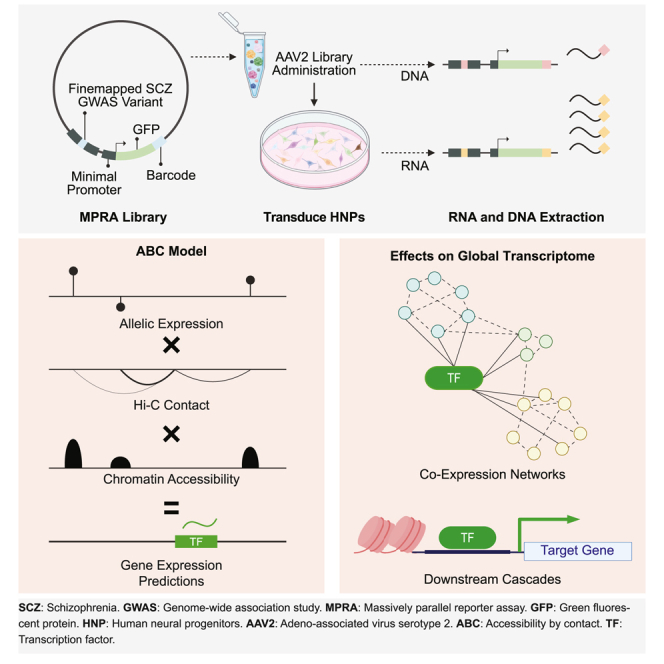
Because schizophrenia genetic risk factors are enriched in regulatory elements of the developing cortex,10,11,12,13 we conducted MPRA in HNPs that model human neural development14 (Figure 1A). To perform MPRA in HNPs, we built an adeno-associated virus-based MPRA vector (AAV-MPRA) that comprises a 150 base-pair (bp) target sequence with the variant in the center, a minimal promoter, green fluorescent protein (GFP), and a 20 bp unique barcode (Figure 1A and STAR Methods).
Figure 1.

MPRA on schizophrenia risk variants identifies functional regulatory variants
(A) We generated an MPRA library that contains 5,173 schizophrenia GWAS variants upstream to the promoter, reporter gene, and 20 bp barcode. This library was packaged into AAV, which was used to transduce HNPs. We compared barcode expression counts between risk and protective alleles to identify MPRA-positive variants.
(B) We display our MPRA results within a circular Manhattan plot. Red dotted line indicates FDR = 0.1.
(C) Volcano plot showing allelic regulatory activity of 5,173 fine-mapped variants.
(D) Out of 5,173 fine-mapped credible variants from 143 GWS loci, 439 variants exhibited allelic regulatory effects in HNPs covering 102 GWS loci (FDR < 0.1).
(E) Out of 102 GWS loci with regulatory activity, only 12 index variants showed allelic regulatory activity.
(F) MPRA dramatically reduced the number of causal variants per locus.
(G–I) GWAS association p values (G), GWAS odds ratio (OR, H), and fine-map posterior probabilities (PP, I) do not differ between MPRA-positive (MPRA-pos) and MPRA-negative (MPRA-neg) variants. p values were calculated by the two-sided Wilcoxon rank-sum test.
Using this AAV-MPRA backbone, we generated an AAV-MPRA library from a computationally predicted credible set of schizophrenia risk variants.6 We compiled 150 bp target sequences centered on 6,064 fine-mapped schizophrenia risk variants (Figures S1A and S1B). Among them, 470 target sequences that harbor either risk variants larger than 10 bp or recognition sites for restriction enzymes used in the cloning steps were removed (Figure S1A and STAR Methods). After filtering out low-quality and/or undetected variants, 5,173 variants (10,346 risk and protective alleles) were included in the final AAV-MPRA library that covers 143 out of 144 GWS loci (Figure S1A).
Because the size of variants (<10 bp) is smaller than the barcodes (20 bp), we reasoned that the effects of barcodes on GFP expression can be larger than allelic regulatory effects. To control for the potential effects of barcodes on GFP expression and to fully capture the small effect size of allelic regulatory activity, we mapped each allele to 185 barcodes on average (Figure S2A).
The resulting schizophrenia MPRA library was packaged into the AAV, which was administered to HNPs. Two weeks after administering the AAV-MPRA library to HNPs, RNA was extracted from the transduced cells and barcoded GFP expression was quantified by RNA sequencing (RNA-seq). To control for transduction efficiency and barcode dispersion during cloning, DNA barcode counts from the AAV-MPRA library were used for normalization (STAR Methods). The correlation coefficients across biological replicates ranged from 0.57 to 0.75 (Figure S2B).
To identify fine-mapped variants with allelic regulatory activity, RNA barcode counts for protective and risk alleles in a total of 10 biological replicates were compared against the corresponding viral DNA barcode counts using the mpra Bioconductor package (Figure 1B and STAR Methods). As a result, we identified 439 MPRA-positive variants that show allelic regulatory activity at a false discovery rate (FDR) threshold of 0.1 (Figures 1C and 1D; Table S1). We found that 102 out of 143 GWS loci contained at least one MPRA-positive variant (Figure 1D). Out of 102 GWS loci that harbor regulatory variants, index variants (variants with the strongest GWAS association statistics at given loci) of 12 loci showed regulatory activity (Figure 1E), suggesting that the most significant GWAS association cannot accurately predict functional variants.
MPRA not only refined the number of regulatory variants but also narrowed down the number of variants per locus (Figure 1F). On average, 36.2 variants per locus were identified via computational fine-mapping approaches. MPRA further pruned them to 4.3 variants per locus. Moreover, 18 out of 102 loci could be pinpointed to a single regulatory variant, demonstrating the power of MPRA in refining GWS loci.
We next evaluated whether association statistics from GWASs or computational fine-mapping may distinguish MPRA-positive variants from MPRA-negative variants (see STAR Methods for definition). MPRA-positive variants did not differ from MPRA-negative variants in their GWAS association statistics such as p values and effect sizes (Figures 1G and 1H). Similarly, fine-map posterior probabilities did not differ between MPRA-positive and -negative variants (Figure 1I). To further assess the predictive power of fine-mapping for variant regulatory function, we leveraged an independent MPRA dataset on eQTL variants.15 Consistent with our finding, the fine-map posterior probability demonstrated no significant difference between MPRA-positive and -negative variants in this independent dataset (Figure S2C). These results show that predictive models purely based on statistical associations do not accurately predict regulatory effects of variants.
Epigenetic properties of MPRA-positive variants
To further characterize MPRA-positive variants, we surveyed genomic annotations of MPRA-tested variants (Figure S3A). As expected, the majority of MPRA-tested variants were located in intergenic and intronic regions, with only a small proportion located in exons and promoters. We did not observe a clear distinction between MPRA-positive and -negative variants in their genomic annotation.
We next sought to characterize epigenetic properties of MPRA-positive variants. We first compared MPRA-positive variants with local (LD SNPs: non-fine-mapped SNPs within schizophrenia GWS loci) and genomic (random SNPs: SNPs that are matched for minor allele frequency and LD) background. MPRA-positive variants more frequently overlapped with brain (Figure 2A) and neuronal (Figures 2B and 2C) enhancers than the genomic background, but not the local background, corroborating the previous finding that schizophrenia genetic risk factors are enriched in brain and neuronal enhancers compared to the genomic background.11 Next, MPRA-positive variants were compared against MPRA-negative variants (Figures 2A–2C and S3B). MPRA-positive and -negative variants overlapped with H3K27ac peaks from the developing cortex16 at a similar proportion (Figure 2A), while MPRA-positive variants showed developmental stage-specific enrichment to assay for transposase-accessible chromatin sequencing (ATAC-seq) peaks during mid-gestation17 (Figure S3B). Moreover, MPRA-positive and -negative variants did not differ in their overlaps with H3K27ac peaks16 (Figure 2A) or DNase I hypersensitive site (DHS) peaks18 (Figure S3B) from the adult cortex, although MPRA-positive variants were more frequently located in DHS peaks from the cerebellum18 (Figure S3B). Because the cerebellum is neuron rich with less cellular heterogeneity than the cortex, we hypothesized that this enrichment could be due to neuronal enrichment of MPRA-positive variants. We therefore compared MPRA-positive and -negative variants with cell-type-specific enhancers. MPRA-positive and -negative variants did not significantly differ in their overlap with cell-type-specific H3K27ac peaks in the postnatal brain19 (Figure 2B). On the contrary, MPRA-positive variants were more frequently located in single-cell ATAC-seq peaks of early excitatory neurons and astrocytes/oligodendrocytes in the prenatal brain20 (Figure 2C). Enrichment of MPRA-positive variants in neuronal regulatory elements were further confirmed using ATAC-seq data from human induced pluripotent stem cell-derived neural progenitors and neurons21 (Figure S3B).
Figure 2.

Epigenetic characterization of MPRA-positive schizophrenia risk variants
(A–C) The proportion of epigenetic overlap of MPRA-positive (MPRA-pos) and MPRA-negative (MPRA-neg) variants, LD SNPs, and random SNPs to the adult and fetal brain enhancers (A), cell-type-specific enhancers in the adult brain (B), and cell-type-specific enhancers in the fetal brain (C). p values were calculated by one-sided Fisher’s exact test. Comparisons have been made between MPRA-positive variants and other sets of variants. Only significant p values are depicted. Neuro, neurons; Oligo, oligodendrocytes; Astro, astrocytes; Micro, microglia; RG, radial glia; IPC, intermediate progenitor cells; earlyEN, early excitatory neurons; ulEN, upper-layer excitatory neurons; dlEN, deep-layer excitatory neurons; MGE, medial ganglionic eminence; CGE, caudal ganglionic eminence; IN, inhibitory neurons.
(D) Proportion of MPRA-positive variants annotated by 15-core chromatin states.
(E) TFs whose motifs are predicted to be altered by MPRA-positive variants. TF enrichment was calculated by comparing TF binding motifs between MPRA-positive variants and LD SNPs. Each dot is color-coded based on the number of variants that are predicted to alter TF binding motifs, and the size of the dot represents the odds ratio. Dotted circles represent TFs that meet the FDR threshold (FDR < 0.1).
(F) Expression outcome of MPRA (measured by MPRA logFC) can be predicted by the combination of TF binding, activity, and expression (measured by corrected ΔSVM) for MPRA-positive variants, but not for MPRA-negative variants. r stands for Pearson’s correlation coefficient.
(G) Integration of MPRA and other functional genomic datasets unveils a causal variant (rs11062170), a trans regulator (RFX5), and a cell type (neuron) for the CACNA1C locus. The alternative allele (C) of rs11062170 breaks the binding motif of RFX5 and is correlated with lower expression of a reporter gene in MPRA.
Together, enhancer overlap could explain up to 31% of MPRA-positive variants, suggesting that variants do not always operate in the conventional way of promoting strong enhancer activity. To further investigate the epigenetic architecture of regulatory variants, we annotated MPRA-positive variants using 15-core chromatin states.22 Notably, 77% of MPRA-positive variants were annotated by chromatin states (Figure 2D), suggesting that variants can exert their regulatory effects through various epigenetic mechanisms. This result aligns with the previous finding that 27% and 74% of MPRA-positive variants can be functionally annotated by enhancers and chromatin states, respectively.23
It has been previously reported that schizophrenia GWAS signals are under strong selective pressure.6 We therefore explored the evolutionary conservation of variant-harboring regions (150 bp regions centered on each variant) for MPRA-positive, MPRA-negative, LD, and random SNPs. We employed evolutionary conservation scores calculated by comparative genomic analyses across 240 species.24 MPRA-positive variants showed elevated evolutionary constraints compared to random SNPs, albeit to a small degree (two-sided Wilcoxon rank-sum test, p = 0.018) (Figure S3C). On the contrary, evolutionary constraints did not differ between MPRA-positive and -negative variants (two-sided Wilcoxon rank-sum test, p = 0.46) or between MPRA-positive variants and LD SNPs (two-sided Wilcoxon rank-sum test, p = 0.86). Similar to this result, PhastCons scores, another metric for evolutionary conservation, did not differ between MPRA-positive variants and other sets of SNPs (Figure S3C).
Because we are using an episomal version of MPRA, the allelic regulatory activity is mainly driven by transcription factors (TFs). We used motifbreakR25 to identify TFs whose binding motifs are predicted to be disrupted or created by each set of variants (Table S2). We then identified TFs whose binding motifs are enriched in MPRA-positive variants compared to the local background (Figure 2E) or global background (Figure S4A). Top TFs enriched for MPRA-positive variants include the T cell factor/lymphoid enhancer factor (TCF/LEF) family (e.g., TCF7, TCF7L1, and LEF1), which mediates Wnt signaling pathways.26
To further address the relationship between TF binding and allelic regulatory activity, we leveraged a high-confidence delta support vector machine (SVM) framework that predicts variants with differential bindings to 94 TFs.27 Because TFs can act as activators or repressors, TF activity needs to be taken into account in translating TF binding to regulatory activity. Moreover, highly expressed TFs can have a larger impact on SNP-mediated regulatory activity than lowly expressed TFs. Consequently, we calculated corrected ΔSVM for each variant by combining preferential allelic binding (ΔSVM scores), expression levels, and activity (1 if a TF is an activator and −1 if a TF is a repressor) of TFs (see STAR Methods for the equation). Corrected ΔSVM scores were moderately correlated with allelic regulatory activity for MPRA-positive variants but not for MPRA-negative variants (Figures 2F and S4B). This result suggests that TFs are key drivers of allelic regulatory activity measured by MPRA. Given that SVM frameworks have been established for only 94 TFs, we expect that the allelic regulatory activity could be better modeled when we have a more complete understanding of TF-SNP interaction.
Because MPRA-positive variants were enriched in neuronal regulatory elements and TF binding occupancy showed moderate predictive power for nominating MPRA-positive variants, we also leveraged deep-learning-based sequence models such as SURF,28 DeepSEA,29 and Sei30 to identify any potential (epi)genomic features that distinguish MPRA-positive from -negative variants (Figure S4C). In all three models, MPRA-positive variants did not differ from MPRA-negative variants, again demonstrating that (epi)genomic properties alone cannot predict the regulatory function of variants.
Finally, in an example of tying together MPRA results and epigenetic profiles, we highlight an MPRA-positive variant, rs11062170, in the CACNA1C locus (Figure 2G). This variant is located within an H3K27ac peak for neurons,19 but not other brain cell types, alluding to the variant’s neuronal specificity. It is located within the intron of CACNA1C, a voltage-gated calcium-channel-encoding gene previously identified to be associated with schizophrenia.31 Allelic regulatory activity of rs11062170 measured by MPRA showed that the reference (protective) allele, G, induced significantly higher expression than the alternative (risk) allele, C. The alternative allele is predicted to break the binding motif of RFX5, alluding to the mechanism of action of lower expression for the alternative allele (Figure 2G). The allelic regulatory activity of rs11062170 was reproduced in our replication study (Figure S4D).
Cell-type specificity of MPRA results
The observed cell-type specificity of MPRA-positive variants (Figures 2B, 2C, and S3B) encouraged us to compare our results with previously published MPRA data obtained from K562 lymphoblast and SY5Y neuroblastoma cell lines.32 K562 lymphoblasts are a non-neuronal cell line, and SY5Y neuroblastomas were previously reported to display transcriptomic profiles that poorly resemble in vivo brain development compared with HNPs as used here.14 We therefore hypothesized that MPRA-positive variants identified from HNPs will be distinct from MPRA-positive variants from other cell lines. Out of 5,173 variants tested in our MPRA, only 565 variants were tested in K562 and SY5Y, due to the difference in SNP selection strategies (Figure S5A). We detected 49, 40, and 104 variants to have allelic regulatory activity in HNPs, SY5Y, and K562, respectively (FDR < 0.1 using 565 variants tested in both studies, Figure S5A). A minimal overlap of MPRA-positive variants was detected when comparing HNPs with SY5Y (4 variants, Figure S5B) and K562 (11 variants, Figure S5C).
Because this cell-type specificity could be caused by other contributing factors (e.g., batch effects, different experimental strategies, and statistical analysis), we introduced the same schizophrenia AAV-MPRA library (Figure S1A) to HEK293 cells (STAR Methods). Out of 5,137 variants tested in both HNPs and HEK293, 1,004 variants were MPRA positive in HEK293 (FDR < 0.1, Table S3). We found that 205 MPRA-positive variants were shared between HNPs and HEK293, again demonstrating cell-type specificity of schizophrenia risk variants (Figure S5D). The majority of MPRA-positive variants shared between HNPs and HEK293 showed the identical direction of effects (IDE) (Figure S5E).
MPRA identifies a different set of variants from eQTLs
eQTLs have become the primary genomic resource to functionally link GWASs to gene-expression measures.33 Since MPRA identifies allelic regulatory activity of variants as eQTLs do, we compared MPRA-positive variants with eQTLs detected in the adult prefrontal cortex.34 Notably, only 36% of MPRA-positive variants showed eQTL signals (Figure 3A). Among 157 variants with both MPRA allelic regulatory activity and eQTL signals, 130 variants (83%) exhibited the IDE (hereby referred to as IDE variants, Table S4), indicating a high level of concordance between MPRA and eQTLs when both signals are detected. Because HNPs better model developing brains than adult brains, we also compared MPRA-positive variants with eQTLs from the developing brain.35 Comparison with developing brain eQTLs gave similar findings, albeit to a lesser degree of overlap, which could be due to the low detection power of developing brain eQTLs from lower sample size (Figure S6). Because adult brain eQTLs (238,194 eQTLs associated with 32,944 genes) are better powered than developing brain eQTLs (7,962 eQTLs associated with 6,526 genes), we used adult brain eQTLs for the rest of the analysis.
Figure 3.

Comparison of MPRA results with adult brain eQTLs
(A) Nine percent of the variants tested in our MPRA were MPRA positive. Thirty-six percent of MPRA-positive variants overlapped with eQTLs. Within the 36% overlap with eQTLs, 83% of MPRA-positive variants showed IDE with the overlapping eQTL variants. Eighty-four percent of IDE variant-gene pairs were detected from the co-localization analysis between eQTLs and schizophrenia GWASs (Coloc).
(B) Seventy-eight schizophrenia GWS loci co-localize with eQTLs, providing 288 schizophrenia-associated eGenes (Coloc). Forty-two out of these 78 loci contain at least one MPRA-positive variant and are mapped to 161 eGenes (MPRA-Coloc). Thirty-six of MPRA-Coloc loci contain variants that have IDE between MPRA and eQTLs and are mapped to 132 eGenes (MPRA-Coloc-IDE).
(C) eQTLs for the CHRNA5 gene co-localize with a schizophrenia GWS locus on chromosome 15. Within this locus are two MPRA-positive variants. One of the MPRA-positive variants, rs8042059, shows IDE between MPRA and eQTLs that the alternative allele C is associated with downregulation of CHRNA5. MPRA p value was calculated by the mpra Bioconductor package, and p value for eQTL is from Wang et al.34
(D) MPRAnon-eQTL variants and MPRAeQTL variants differ in distance to the transcription start site (TSS). p values were calculated by two-sided Wilcoxon rank-sum test.
(E) MPRA non-eQTL variants more frequently overlap with neuronal enhancers compared to MPRAeQTL variants. p values were calculated by one-sided Fisher’s exact test.
Because eQTLs are affected by LD, simple genomic coordinate-level overlap between GWASs and eQTLs could lead to spurious overlap. Co-localization analysis has been implemented to evaluate whether GWASs and eQTLs are explained by a shared set of variants. To test how many IDE variants are also identified from the co-localization analysis, we compared IDE variants with co-localization between schizophrenia GWASs and adult brain eQTLs36 using the coloc package (STAR Methods). Because co-localization does not always indicate a specific variant, we tested how often eGenes (genes detected as having an associated eQTL) linked to IDE variants were also observed from co-localization analysis. From this analysis, 84% of IDE variants were linked to the same genes as predicted by co-localization analysis (Figure 3A).
Intersection of MPRA and eQTLs also pruned the gene list (Figure 3B). We initially detected 288 eGenes to be associated with schizophrenia by co-localization analysis, covering 78 loci.36 An orthogonal analysis of coordinate-level overlap between eQTLs and MPRA identified 269 eGenes covering 80 loci. We found that 161 eGenes were shared between co-localization and MPRA-eQTL overlap analysis. After pruning them further with the IDE between MPRA and eQTLs, 132 eGenes were detected, covering 36 loci.
In an example of MPRA-eQTL IDE overlap, we highlight a schizophrenia GWS locus on chromosome 15 (Figure 3C). Two variants at this locus—rs11418931 and rs8042059—had significant MPRA allelic activity. Rs11418931 was missing in the eQTL analysis, while rs8042059 was detected as an eQTL for a nearby gene, CHRNA5. When comparing the directionality of the allelic expression of rs8042059, the reference (risk) A allele increased expression in comparison to the alternative (protective) C allele both in MPRA and eQTLs for CHRNA5.
Mostafavi et al. have recently postulated that eQTL studies and GWASs identify a different set of variants.37 In their analysis, variants detected in eQTL studies and GWASs differ by their distance to transcription start sites (TSSs) and regulatory architecture. To investigate whether MPRA could identify a distinct set of disease-associated variants that are not explained by variants detected as eQTLs, we characterized genomic and epigenomic properties of MPRA-positive variants with and without eQTL signature (hereafter referred to as MPRAeQTL variants and MPRAnon-eQTL variants, respectively).
MPRAnon-eQTL variants were more distal to the TSS than MPRAeQTL variants, hinting that MPRAnon-eQTL variants could be involved in distal regulatory relationships (Figure 3D). Because distal regulatory elements often encode enhancers, we next surveyed whether there is a difference between MPRAeQTL and MPRAnon-eQTL variants in their enhancer overlap. We found that a higher proportion of MPRAnon-eQTL variants (∼12%) overlapped with neuronal enhancers compared to MPRAeQTL variants (∼6%).19 Such a difference in enhancer overlap was not shown in other tested cell types (Figure 3E). Taken together, these results suggest that disease-associated variants may differ from variants detected as eQTLs, and MPRA could fill this gap by testing GWAS variant effects on gene regulation in a manner independent of issues related to eQTL study power.
Identification of schizophrenia candidate risk genes via long-range chromatin interactions
Because MPRA-positive variants exhibited epigenomic properties different from those of eQTLs (Figures 3D and 3E), we sought another method for assigning target genes for MPRA-positive variants. The previous finding that genes affected by GWAS variants show enhanced regulatory complexity37 prompted us to leverage long-range chromatin interaction datasets from the human brain. As MPRA-positive variants were preferentially located in enhancers of immature and mature neurons (Figures 2B, 2C, 3E, and S3B), we used chromatin loops in neural progenitors (germinal zone), immature postmitotic neurons (cortical plate), pediatric neurons, and adult neurons (Figure 4A and Table S5).19,38,39 Neuronal chromatin interaction datasets assigned 209 MPRA-positive variants to 272 protein-coding genes (hereby referred to as MPRAHi-C genes, Table S6). The resulting SNP-gene relationship was multivalent. On average, each SNP was mapped to 2.7 genes (Figure 4B), while each gene was mapped to 2.1 MPRA-positive SNPs (Figure 4C). Only 24 genes overlapped between the MPRAHi-C genes and the MPRAeQTL-IDE genes (Figure 4D), showing that the two datasets assign MPRA-positive variants to distinct sets of genes.
Figure 4.

Genes assigned to MPRA-positive variants using long-range interactome differ from eQTL-assigned genes
(A) Chromatin loops from neurons of four different developmental time points were used to map MPRA-positive variants to genes. GZ, germinal zone; CP, cortical plate; PN, pediatric neuron; AN, adult neuron.
(B) Distribution of the number of genes mapped per SNP. Red line denotes the mean.
(C) Distribution of the number of variants mapped per gene. Red line denotes the mean.
(D) Overlap between MPRAeQTL-IDE genes and MPRAHi-C genes.
(E) Gene Ontology (GO) analysis of MPRAHi-C genes indicates involvement of spliceosome and synaptic functions in schizophrenia etiology. Redundant GO terms were omitted (see Figure S7A for full GO terms).
(F) An example locus for CACNA1C shows that the MPRA-positive SNP rs11062170 physically interacts with CACNA1C promoter in adult neurons.
(G) Loss of function observed/expected upper-bound fraction (LOEUF) score distribution shows that MPRAHi-C genes are less tolerant to mutations compared to MPRAeQTL-IDE genes. p value was calculated by two-sided Wilcoxon rank-sum test.
(H) The number of promoter-anchored loops shows higher regulatory complexity for MPRAHi-C genes compared to MPRAeQTL-IDE genes. Loops from adult neurons were used. p value was calculated by two-sided Wilcoxon rank-sum test.
MPRAHi-C genes were enriched for Gene Ontology (GO) terms related to spliceosomes and synaptic functions (Figures 4E and S7A). Enrichment of MPRAHi-C genes in spliceosomes corroborates pervasive isoform-level dysregulation in schizophrenia brains.40 Furthermore, synaptic involvement of MPRAHi-C genes recapitulates a widely accepted notion that neurons are the central cell type for schizophrenia.11,41 Accordingly, MPRAHi-C genes showed elevated expression in neurons than non-neuronal cells in both the fetal (Figure S7B) and adult cortex (Figure S7C).
One of the genes that physically interacts with MPRA-positive variants was CACNA1C (Figure 4F). Chromatin interaction offers a complete mechanism of action for the CACNA1C locus (also depicted in Figure 2G): the MPRA-positive SNP rs11062170, located within a neuronal enhancer (Figure 2G), interacts with the promoter of CACNA1C in adult neurons (Figure 4F). The alternative (risk) allele C of rs11062170 disrupts RFX5 binding, which weakens the neuronal enhancer activity (Figure 2G). The weakened neuronal enhancer propagates to the decreased expression of CACNA1C via a neuronal chromatin loop.
Mostafavi et al. have shown that genes linked to variants detected in eQTL studies and GWASs differ by their functional annotation, mutational constraint, and regulatory complexity.37 In their study, all eGenes, regardless of disease association, were compared against genes proximal to GWAS variants, so it is unclear whether genes linked to GWAS variants also differed when different mapping strategies were used. Given the epigenetic difference between MPRAeQTL variants and MPRAnon-eQTL variants, we hypothesized that genes assigned to MPRA-positive variants via chromatin interactions (272 MPRAHi-C genes, Figure 4D) differ from those assigned by eQTLs (132 MPRAeQTL-IDE genes, Figures 3A and 4D).
Unlike MPRAHi-C genes that showed functional annotations related to synaptic biology (Figures 4E and S7A), MPRAeQTL-IDE genes were enriched for more generic cellular function (Figure S7D). Furthermore, MPRAHi-C genes exhibited higher mutational constraints than MPRAeQTL-IDE genes (Figure 4G), which is in line with the previous report that schizophrenia-associated common variation is enriched for mutation-intolerant genes.6 Finally, MPRAHi-C genes were engaged in more distal interaction than MPRAeQTL-IDE genes (Figures 4H and S7E), indicative of higher regulatory complexity. Taken together, these results suggest that gene assignment for GWAS variants may require an additional annotation strategy utilizing physical interactome, given the significant differences in properties of genes assigned by two different strategies (eQTLs vs. Hi-C).
Regulatory principles of multi-variant loci
Out of 102 GWAS loci with functional regulatory variants, only 18 loci were mapped to a single functional regulatory variant while 84 loci had more than one MPRA-positive variant. We explored regulatory relationships of multi-variant loci by mapping them to target genes with neuronal chromatin interactions (Figure 4A). Fifty-eight out of 84 multi-variant loci were mapped to genes, and 49 of them were mapped to more than one gene, indicating potential cases of pleiotropy. Adding to another layer of complexity, multiple variants often converged on a single gene. These results suggest that multi-variant loci are often engaged in a complex regulatory relationship that involves pleiotropy and convergence.
Multi-variant loci pose a challenge in translating variant effects to gene expression. Using 256 putative target genes of multi-variant loci, we sought to identify how variant effects can be aggregated to predict changes in gene expression using transcriptomic profiles of schizophrenia brain homogenates as a benchmark.40 Out of 256 putative targets, 192 genes showed detectable levels of expression in postmortem brains and were used for comparison between MPRA and schizophrenia postmortem expression. We recalibrated MPRA log fold change (logFC) values (alternative/reference allele) to reflect disease risk (risk/protective allele). Consequently, variants with positive logFC(risk/protective) values will increase gene expression, while those with negative logFC(risk/protective) values will decrease gene expression, in schizophrenia.
We first explored a simple additive model in which allelic activity of variants is added to predict gene expression (Figures 5A and 5B). Out of 192 genes compared between MPRA results and postmortem expression profiles, 107 genes (55.7%, permutation p = 0.03) showed IDE. Because variants with higher contact frequency may have larger impacts on gene expression, we next weighted allelic activity by chromatin contact frequency (hereby referred to as a contact model, Figure 5A). The number of genes with IDE grew from 107 to 109 (56.8%, permutation p = 0.0081) by the use of the contact model (Figure 5B). We next reasoned that variants within chromatin-accessible regions may have a larger impact on gene regulation. Because our episomal design measures allelic activity without taking chromatinization into account, we weighted allelic activity by chromatin accessibility and contact frequency (hereby referred to as an accessibility-by-contact [ABC] model, Figure 5A). With this model, 116 genes (60.4%, permutation p = 3 × 10−4) showed IDE with postmortem expression (Figure 5B). Applying the same model to MPRA-negative variants yielded 105 genes (54.7%, permutation p = 0.14) with IDE (Figure 5B). Remarkably, the ABC model outperformed transcriptome-wide association studies (TWASs) in predicting molecular pathology, as 371 out of 708 (52.4%) schizophrenia risk genes predicted by TWASs (TWAS FDR < 0.05) were in concordance with postmortem expression profiles.40
Figure 5.

Mapping multi-variant loci to genes
(A) Illustration of how the ABC model predicts gene expression outcome for multi-variant loci.
(B) The ABC model (allelic activity chromatin contact chromatin accessibility) outperforms additive (allelic activity) and contact (allelic activity chromatin contact) models in predicting gene-expression changes in schizophrenia postmortem brains. p values were calculated by permutation.
(C) GRIN2A locus is a multi-variant locus in which six variants are predicted to act together on GRIN2A regulation.
(D) The ABC model predicts GRIN2A to be downregulated, while additive and contact models predict GRIN2A to be upregulated in schizophrenia risk conditions.
(E) GRIN2A normalized gene expression in postmortem brain homogenates of neurotypical controls and individuals with schizophrenia (SCZ). FDR = 0.095.
(F) GRIN2A is downregulated in excitatory neurons from schizophrenia brain samples. Significant downregulation (padjusted < 0.05) is highlighted in blue.
The GRIN2A locus is an example in which additive and ABC models give opposite predictions (Figure 5C). In this locus, 6 MPRA-positive variants showed detectable levels of chromatin interactions with the GRIN2A promoter and were included in the model. An additive model suggested that the gene is upregulated (logFC = 0.17), while an ABC model predicted that the gene is downregulated (logFC contact accessibility = −2.72, Figure 5D). GRIN2A was modestly downregulated in schizophrenia postmortem brains (logFC = −0.036, Figure 5E), albeit with nominal significance (FDR = 0.095).40 Because this could be due to the cellular heterogeneity of the brain homogenate, we explored GRIN2A expression in a cell-type-specific fashion using single-cell RNA sequencing (scRNA-seq) data derived from schizophrenia postmortem brains.42 Consistent with the ABC model, GRIN2A was significantly downregulated in multiple excitatory neuronal subtypes of schizophrenia brains (Figure 5F). These results demonstrate that combining MPRA allelic activity with chromatin accessibility and contact frequency offers a framework to predict gene expression from MPRA-validated variant effects.
Linking variants to molecular pathology
To further investigate how combinatorial effects of the variants can be propagated to schizophrenia molecular pathology, we focused on two loci in which genes that encode transcriptional regulators are targeted by multiple MPRA-positive variants with the same direction of effects (Figure 6). This was based on two hypotheses. First, variants with the same directional effect may have a larger impact on the gene in aggregate. Second, the resulting dysregulation of transcriptional regulators may have a broader impact on the transcriptional landscape of schizophrenia.
Figure 6.

Combinatorial effects of variants on global transcriptome in schizophrenia
(A) Risk alleles of four MPRA-positive variants in the SETD8 locus downregulate the reporter gene compared with protective alleles.
(B) SETD8 expression levels in excitatory neurons of postmortem schizophrenia brains. p values were obtained from Ruzicka et al.42
(C) The extent of dysregulation of SETD8 co-expressed genes in excitatory neurons of postmortem schizophrenia brains. Significant upregulation and downregulation (padjusted < 0.05) are highlighted in red and blue, respectively. Z scores were calculated from permutation.
(D) Genes upregulated by SETD8 knockdown were enriched for genes upregulated in excitatory neurons (Ex-L3 and Ex-L6b_SEMA3E) of postmortem schizophrenia brains. Significant enrichment (FDR < 0.05) for upregulated and downregulated genes in response to SETD8 perturbation is highlighted in red and blue, respectively. p values were calculated by two-sided Fisher’s exact test.
(E) Risk alleles of three MPRA-positive variants in the MEF2C locus upregulate the reporter gene compared with protective alleles.
(F) MEF2C expression levels in excitatory neurons of postmortem schizophrenia brains. p values were obtained from Ruzicka et al.42
(G) The extent of dysregulation of MEF2C co-expressed genes in excitatory neurons of postmortem schizophrenia brains. Significant upregulation and downregulation (padjusted < 0.05) are highlighted in red and blue, respectively. Z scores were calculated from permutation.
(H) Genes downregulated by Mef2c knockout were enriched for genes upregulated in excitatory neurons of postmortem schizophrenia brains. Significant enrichment (FDR < 0.05) for upregulated and downregulated genes in response to Mef2c perturbation is highlighted in red and blue, respectively. p values were calculated by two-sided Fisher’s exact test.
For example, a GWS locus in chromosome 12 has four MPRA-positive SNPs whose risk alleles indicate downregulation of SETD8 (Figure 6A). SETD8 encodes a lysine histone methyltransferase that represses downstream targets.43 Consistent with the MPRA results, SETD8 was downregulated in excitatory neurons of schizophrenia patients42 (Figure 6B). To understand how SETD8 downregulation translates to broader dysregulation in schizophrenia, we first queried genes that are co-expressed with SETD8 in excitatory neurons (STAR Methods). Similar to SETD8 downregulation in schizophrenia, its co-expressed genes were also downregulated in schizophrenia across multiple excitatory neuronal subtypes, except SEMA3E-expressing layer 6 excitatory neurons (Ex-L6b-SEMA3E, Figure 6C). Next, we evaluated genes that were differentially regulated in response to SETD8 perturbation (i.e., SETD8 knockdown [KD]).44 Genes upregulated upon SETD8 KD were significantly over-represented, with genes upregulated in schizophrenia in layer 3 excitatory neurons (Ex-L3) and Ex-L6b-SEMA3E (Figure 6D).
Another example is a locus in chromosome 5 that harbors three MPRA-positive SNPs which uniformly upregulate MEF2C with associated increased schizophrenia risk (Figure 6E). MEF2C encodes a TF, and mutations within this gene have been previously implicated in various psychiatric disorders.45 We found that both MEF2C and its co-expressed genes were significantly upregulated in excitatory neurons of schizophrenia patients42 (Figures 6F and 6G). We also searched for genes dysregulated upon Mef2c knockout (KO)45 and examined their expression profiles in schizophrenia postmortem brains. Downregulated genes in Mef2c KO brains significantly overlapped with genes upregulated in excitatory neurons of schizophrenia patients (Figure 6H). Notably, MEF2C co-expressed genes were enriched with TF regulatory networks with which MEF2C is affiliated46 (hypergeometric test p = 4.71 × 10−10, STAR Methods), suggesting that co-expressed genes are co-functional TF networks and may differ from the downstream targets.
Together, these results suggest how multiple variants with small effects can have an aggregated impact on gene regulation and how resulting gene dysregulation can be propagated to the widespread molecular pathology observed in schizophrenia40,42 via co-expressed TF networks and downstream targets.
Discussion
MPRA has demonstrated its ability to vastly narrow down GWAS variants to a list of functionally validated variants with differential allelic activity. We found 439 schizophrenia-associated variants with allelic regulatory effects within 102 GWS loci. Notably, MPRA-positive variants could not be solely distinguished from existing (epi)genomic features, highlighting the importance of experimental validation in addressing variant function.
The finding that 31% of MPRA-positive variants are located in enhancers could be due to a number of factors. First, our definition of an enhancer could be incomplete. Critically, there is a paucity of data on epigenomic states in a schizophrenia-relevant tissue. Given that the brain is an exceptionally heterogeneous organ, the existing enhancer definition can only provide a broad overview of stably open chromatin in the majority of cells. Therefore, MPRA-positive variants may be located in cell-type-specific enhancers that are yet to be identified. Second, regulatory variants may affect gene transcription through various epigenetic mechanisms, as evidenced by our finding that 77% of the MPRA-positive variants can be explained by chromatin states. Third, unlike previously conducted MPRA and self-transcribing active regulatory region sequencing (STARR-seq) studies that primarily examined regulatory elements identified by ATAC-seq and/or DHSs, our approach focuses on “allelic activity” rather than “enhancer activity” of variants. As a result, variants with differential allelic effects on gene expression could be captured even when they do not have strong enhancer activity. Fourth, we used an episomal version of MPRA that lacks epigenetic context (e.g., chromatinization). While enhancer activity is heavily dependent on chromatin accessibility, the episomal context of our MPRA enables characterization of variant function without an additional layer of chromatinization. For example, MPRA can be sensitive in identifying variants with regulatory activity that may be masked by closed chromatin in the baseline condition. These variants may only be functional under specific regulatory contexts (e.g., upon neuronal activity or cellular stress). While context-specific regulatory variants are difficult to detect via molecular assays in baseline conditions, their implications in disease association are emerging.47,48 We reason that MPRA could potentially identify regulatory effects of variants without the need of priming to make the chromatin accessible.
The pervasive standard in linking variants to gene expression is to leverage eQTL resources. However, a recent study suggested that variants detected in eQTL studies may capture a set of variants different from those of GWASs due to natural selection.37 In agreement with this, we found that 64% of MPRA-positive variants did not overlap with variants identified in adult brain eQTL studies. It is possible that the little overlap with eQTLs could be due to the differing cell type and developmental stage between eQTLs (heterogeneous adult brain homogenate) and MPRA (HNPs that model neural development) or the limited sample size of current eQTL studies. Well-powered cell-type-specific eQTLs (especially neuron-specific eQTLs) may be critical to filling this gap.
Despite the potential source of difference, we found that MPRAnon-eQTL variants showed epigenetic properties different from those of MPRAeQTL variants. In particular, MPRAnon-eQTL variants were more likely located in distal neuronal enhancers compared with MPRAeQTL variants. This prompted us to employ neuronal distal regulatory relationships to link MPRA-positive variants to their cognate genes. MPRAHi-C genes exhibited richer functional annotation and stronger selective constraints than MPRAeQTL-IDE genes. Moreover, MPRAHi-C genes were engaged in more distal regulatory interactions, which aligns with the reported enhancer redundancy of disease-associated mutation-intolerant genes.49 Collectively, our results suggest that unbiased characterization of GWAS variants via MPRA could identify functional regulatory variants under selective pressure that eQTLs may not be able to detect.
As chromatin architecture provides a complementary approach to annotate GWAS variants not cataloged by eQTLs, we explored how chromatin architecture can be integrated with allelic activity to predict gene expression from variant regulatory effects. We found that the ABC model outperformed a simple additive model in predicting the direction of gene-expression change. This model adds to the recently proposed activity-by-contact model that predicts the relationship between regulatory elements and genes.50 Current prediction accuracy of the ABC model was ∼60% when compared against the gene-expression profile from schizophrenia brain homogenates. Because neuronal chromatin accessibility51 and contact maps39 were used to translate the functional impact of MPRA-positive variants that are enriched in neuronal enhancers, we expect that the prediction accuracy could be further improved by the comparison with neuronal-specific transcriptomic signatures in schizophrenia.
We therefore leveraged scRNA-seq datasets obtained from the schizophrenia postmortem brain42 to study the functional impact of MPRA-positive variants within the neuronal context. In the examples in which MPRA-positive variants are predicted to act together to influence expression of transcriptional regulators (SETD8 and MEF2C), we showed that those transcriptional regulators were dysregulated in excitatory neurons of schizophrenia patients in the direction predicted by MPRA. Dysregulation of these transcriptional regulators were then propagated to shape the broader gene-expression landscape in schizophrenia through the co-expression networks and downstream cascades.
In conclusion, the combination of MPRA-measured allelic activity with chromatin architecture can complement the episomal design of MPRA that does not account for the endogenous genomic context and can provide a systematic framework to interpret variant effects on gene regulation, helping to shed light on the complex mechanisms underlying the molecular pathology of schizophrenia.
Limitations of the study
Despite its high-throughput ability to functionally validate the regulatory effect of genetic variants, MPRA lacks native local chromatin context due to its episomal design. Furthermore, MPRA does not identify putative target genes. To address this limitation, we link MPRA-positive variants to genes using endogenous genomic context. In addition, we compared MPRA-positive variants with (epi)genetic data primarily acquired from adult brains because of data availability and sample sizes. The observed low overlap in our MPRA conducted on HNPs may be attributed to differences in developmental stages and cell types.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Endura electrocompetent cells | Lucigen | cat#60242-1 |
| Chemicals, peptides, and recombinant proteins | ||
| Beta-Mercaptoethanol | Sigma-Aldrich | cat#60-24-2 |
| Poly-L-Ornithine | Sigma-Aldrich | cat#P3655-100MG |
| Fibronectin | Sigma-Aldrich | cat#F1141-5MG |
| Primocin | Invitrogen | cat#ant-pm-2 |
| BIT 9500 | STEMCELL | cat#09500 |
| glutamax | Fisher Scientific | cat#5112367 |
| Heparin | Sigma-Aldrich | cat#H3393-100KU |
| Epidermal growth factor (EGF) | PeproTech | cat#AF-100-15 |
| Fibroblast growth factors (FGF) | PeproTech | cat#AF-100-15 |
| Platelet-derived growth factor (PDGF) | PeproTech | cat#100-00AB |
| Leukemia inhibitory factor (LIF) | PeproTech | cat# 300-05 |
| DMEM | Gibco | cat#11995-065 |
| Fetal bovine serum | corning | cat#76418-024 |
| antibiotic-antimycotic | gibco | cat#15240062 |
| NEBNext 2X Q5 Hifi HS Mastermix | NEB | cat#M0453S |
| SpeI-HF | NEB | cat#R3133S |
| MluI-HF | NEB | cat#R3198S |
| rSAP | NEB | cat#M0371S |
| EcoR1-HF | NEB | cat#R3101S |
| T7 DNA ligase | NEB | cat#M0318S |
| Dynabeads MyOne Streptavidin C1 beads | Thermo Fisher | cat#65601 |
| KpnI-HF | NEB | cat#R3142S |
| XbaI | NEB | cat#R0145S |
| AMPure XP Beads | Beckman Coulter | cat#A63881 |
| Critical commercial assays | ||
| Qiagen Mini prep kit | Qiagen | cat#27106 |
| Qiagen Maxi prep kit | Qiagen | cat#12163 |
| Zymo DNA clean and concentrator-5 | Zymo | cat# D4014 |
| Zymo Gel DNA Recovery kit | Zymo | cat#D4007 |
| Zymo DNA clean and concentrator kit −25 | Zymo | cat#D4033 |
| Qiagen RNeasy kit | Qiagen | cat#74004 |
| SuperScript IV Reverse Transcriptase | Invitrogen | cat#18090050 |
| NucleoSpin virus kit | Macherey-Nagel | cat#740983.50 |
| Deposited data | ||
| Raw data | This paper | GEO: GSE211045 |
| Code for analysis | This paper | https://doi.org/10.5281/zenodo.8221493 |
| Experimental models: Cell lines | ||
| Human Neural Progenitor | Dr. Jason Stein’s lab, UNC-CH | Donor #54 |
| HEK293 | Dr. Jason Stein’s, UNC-CH | N/A |
| Recombinant DNA | ||
| pAAV-MPRA-MluI-SpeI-EcoRI | This paper | Addgene, cat#190196 |
| pLS-minP:Plasmid | Dr. Nadav Ahituv’s group | Addgene, cat#81225 |
| Software and algorithms | ||
| mpra | Myint et al.28 | https://bioconductor.org/packages/release/bioc/html/mpra.html |
| GARFIELD | Iotchkova et al.52 | https://www.bioconductor.org/packages/release/bioc/html/garfield.html |
| Annotatr | Bioconductor package | https://github.com/rcavalcante/annotatr |
| liftOver | Navarro et al.53 | http://genome.ucsc.edu/cgi-bin/hgLiftOver |
| motifbreakR | Bioconductor package | https://github.com/Simon-Coetzee/motifBreakR |
| SURF | Dong et al.28 | https://github.com/Boyle-Lab/RegulomeDB-TURF |
| DeepSEA | Zhou et al.29 | http://deepsea.princeton.edu/job/analysis/create/ |
| Sei | Chen et al.30 | https://github.com/FunctionLab/sei-framework |
| coloc | Giambartolomei et al.54 | https://github.com/chr1swallace/coloc |
| bedtools | Quinlan et al.55 | https://bedtools.readthedocs.io/en/latest/ |
| gprofiler2 | Kolberg et al.56 | https://biit.cs.ut.ee/gprofiler/gost |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hyejung Won (hyejung_won@med.unc.edu).
Materials availability
The MPRA backbone generated in this study, pAAV-MPRA-MluI-SpeI-EcoRI, has been deposited to Addgene (Addgene number: 190196).
Experimental model and subject details
Acquisition, generation, and culture of HNP have been previously described.57 Donor number 54 (genetically male) was used for all experiments. Briefly, 6-well plates were coated with Poly-L-Ornithine (10 μg/mL; Sigma-Aldrich, cat#P3655-100MG) and fibronectin (5 μg/mL; Sigma-Aldrich, cat#F1141-5MG). HNPs were plated at 400K cells/well in a 6-well plate. The cells were plated in Neurobasal A media (Thermo Fisher, cat#10888022) supplemented with primocin (100 μg/mL; Invitrogen, cat#ant-pm-2), BIT 9500 (10%; STEMCELL, cat#09500), glutamax 100X (1%; Fisher Scientific, cat#5112367), heparin (1 μg/mL; Sigma-Aldrich, cat#H3393-100KU), and growth factors: EGF (20 μg/mL; PeproTech, cat#AF-100-15), FGF (20 μg/mL; PeproTech, cat#AF-100-15), PDGF (20 ng/mL; PeproTech, cat#100-00AB), and LIF (2 ng/mL; PeproTech, cat# 300-05).
HEK293 cells (genetically female) were plated at a density of 1 million cells/well in a 6-well plate and fed with media consisting of DMEM (gibco, cat#11995-065), 10% fetal bovine serum (corning, cat#76418-024), and antibiotic-antimycotic (gibco, cat#15240062). All cells were grown in an incubator at 37°C with 5% carbon dioxide.
Method details
Variant selection
FINEMAP58 was applied to 144 schizophrenia GWS loci (excluding the MHC locus) from Pardinas et al.6 A set of fine-mapped variants that can explain a given GWS loci with 95% probability for containing causal configuration was selected as previously described.59 In total, we identified 6,064 fine-mapped variants for 144 schizophrenia GWS loci. A 150bp sequence flanking each variant was then selected to be inserted to the MPRA library. For indels, we used the same sized fragment (150bp) centered to the variant. We found that 150bp flanking sequences of 470 variants out of 6,064 fine-mapped variants contained sequences for restriction enzymes (MluI, SpeI, KpnI, XbaI) used for molecular cloning. These variants were excluded, resulting in 5,594 variants that were tested via our MPRA framework.
Creating variant oligo library
The 202 bp library oligos that contain schizophrenia risk variants were synthesized by Agilent and amplified using NEBNext 2X Q5 Hifi HS Mastermix (NEB, cat#M0453S; primers: MPRA-chipprimer-R and MPRA-chipprimer-F). Primer information is available in Table S7.
We then used a pair of primers, one with the 20bp random barcode and SpeI restriction site (MPRA-BC_Primer_R) and the other with the MluI restriction site (MPRA-BC_Primer_F) to add random barcodes and restriction sites to the library oligos via PCR (NEBNext 2X Q5 Hifi HS Mastermix). The resulting library was digested with SpeI-HF (NEB, cat#R3133S) and MluI-HF (NEB, cat#R3198S) for 1 h at 37°C, followed by rSAP treatment (NEB, cat#M0371S) for 1 h at 37°C and heat inactivation for 5 min at 65°C. After digestion, the library was cleaned up using Zymo DNA clean and concentrator kit −25 (Zymo, cat#D4033).
Engineering of AAV-MPRA backbone
We obtained the AAV backbone plasmid (pAAV-hSyn-EGFP) from the UNC vector core (https://www.addgene.org/50465/). We digested pAAV-hSyn-EGFP using MluI-HF and EcoR1-HF (NEB, cat#R3101S), and ligated in an oligo that contains the sequences for MluI, SpeI, and EcoRI restriction sites using T7 DNA ligase (NEB, cat#M0318S). The ligated plasmid was transformed into Endura electrocompetent cells (Lucigen, cat#60242-1) via electroporation and grown in ampicillin LB overnight at 30°C. The cells were mini prepped with Qiagen Mini prep kit (Qiagen, cat#27106) resulting in the AAV backbone that harbors the multicloning site of MluI-SpeI-EcoR1 (hereby referred to as AAV-MluI-SpeI-EcoR1).
Inserting variant oligo library into AAV-MPRA backbone
The AAV-MluI-SpeI-EcoR1 plasmid and the variant library were digested with SpeI-HF, MluI-HF, and rSAP for 3 h at 37°C, and heat inactivated for 20 min at 80°C. The digested plasmid (∼4kb) was run through a 1% agarose gel and gel extracted using Zymo Gel DNA Recovery kit (Zymo, cat#D4007). The digested library was cleaned up using Dynabeads MyOne Streptavidin C1 beads (Thermo Fisher, cat#65601). The digested library and plasmid were ligated together using T7 DNA ligase at room temperature for 30 min using a 1:3 ratio (plasmid:library). The ligated product was cleaned up using Zymo DNA clean and concentrator-5 (Zymo, cat#11-303C), and transfected into Endura electrocompetent cells via electroporation, and plated on 10 cm circular LB agar plates with ampicillin. The plates were grown overnight at 30°C. The colonies were scraped and grown in 2 L of LB with ampicillin for 7 h at 37°C. The resulting plasmid was maxi prepped using Qiagen Maxi prep kit (Qiagen, cat#12163) resulting in the AAV library that contains variant-barcode combinations (hereby referred to as an AAV-variant-barcode library).
Barcode mapping
The variant and barcode region of the AAV-variant-barcode library was PCR amplified using NEBNext 2X Q5 Hifi HS Mastermix with primers that contain Illumina P5 and P7 adapters (Bcmap_P5_AAV_R and Bcmap_P7_AAV_F). The PCR product was cleaned up using Zymo DNA clean and concentrator-5. The resulting library was sequenced using custom sequencing primers (BCmap_R1Seq_AAV_R and BCmap_R2Seq_AAV_F) via Novaseq 6000 SP (2x250bp) by the UNC High-Throughput Sequencing Facility (HTSF). Barcodes were assigned to each variant using the custom code available in the github repository (https://github.com/kiminsigne-ucla/bc_map).
Adding in minimal promoter and GFP
We obtained pLS-minP, a plasmid that contains a minimal promoter (minP) and GFP (minP-GFP), from Dr. Nadav Ahituv’s group (https://www.addgene.org/81225/). The minP-GFP fragment was amplified from the plasmid via PCR using NEBNext 2X Q5 Hifi HS Mastermix and cleaned up using Zymo DNA clean and concentrator-5 (primers: minP-GFP-F and minP-GFP-R). The minP-GFP fragment and AAV-variant-barcode library were both digested with KpnI-HF (NEB, cat#R3142S) and rSAP for 3 h at 37°C, which was followed by heat inactivation for 10 min at 65°C. Both of these products were then gel extracted from a 0.8% agarose gel using Zymo Gel DNA Recovery kit. The gel extracted products were then digested with XbaI (NEB, cat#R0145S) and rSAP for 3 h at 37°C, and then for 10 min at 65°C for heat inactivation. The digested products were cleaned up using Zymo DNA clean and concentrator-5.
The digested minP-GFP and AAV-variant-barcode library plasmid were ligated together using T7 DNA ligase. The ligation mix was incubated at room temperature for 30 min, then cleaned up using Zymo DNA clean and concentrator-5. The ligation mix was transformed into Endura electrocompetent cells, which were then plated on 10 cm circular LB agar plates with ampicillin, resulting in the AAV-variant-minP-GFP-barcode library. The AAV-variant-minP-GFP-barcode library was grown in 2 L of LB with ampicillin, and maxi prepped using Qiagen Maxi prep kit.
The UNC vector core packaged the AAV-variant-minP-GFP-barcode library into AAV serotype 2 (AAV2). The resulting virus had the titer of 7x1012 viral particles/uL.
Administration of AAV-MPRA to HNPs
HNPs were plated and fed as described in EXPERIMENTAL MODEL. The day after plating, each well was transduced with the AAV-MPRA library at 7,000 multiplicity of infection (MOI). One well per plate was a no-virus control to monitor general cell health. After the AAV-MPRA library was added to the cells, the plates were spun in a centrifuge for 5 min at 37 °C at 1000 rcf. Cells were half-fed with 2X growth factors every other day for two weeks after transduction. RNA was extracted from each well two weeks after transduction. To enhance detectability of transduced cells, we pooled 3 wells for one replicate, resulting in 1.2 million cells per replicate.
Administration of AAV-MPRA to HEK293s
HEK293 cells were plated and fed as described in EXPERIMENTAL MODEL. The day after plating the cells, each well was transduced with the AAV-MPRA library at 10,000 MOI. After the AAV-MPRA library was added to the cells, plates were spun in a centrifuge for 5 min at 37 °C at 1000 rcf. The cells were half-fed 48hrs after transduction. The RNA was extracted from each well 72 h after transduction. We pooled 3 wells per replicate, resulting in 3 million cells per replicate.
Processing RNA and DNA for sequencing
RNA was extracted from each well using Qiagen RNeasy kit (Qiagen, cat#74004), using 10 μL of -Mercaptoethanol (Sigma-Aldrich, cat#60-24-2) per 1 mL of Qiagen RLT buffer. The columns were treated with DNase (Qiagen, cat#79256). cDNA was generated from the extracted RNA by SuperScript IV Reverse Transcriptase (Invitrogen, cat#18090050) using a primer that targets downstream of the barcodes (Lib_Hand_RT_AAV).
To acquire an initial input of DNA put into the cells, DNA was extracted from the AAV2 virus which contained the AAV-MPRA library using a NucleoSpin virus kit (Macherey-Nagel, cat#740983.50).
Amplification of RNA-seq libraries
DNA extracted from the AAV-MPRA library and cDNA from each transduced well were amplified via PCR using NEBNext 2X Q5 Hifi HS Mastermix (primers for DNA: Lib_Hand_RT_AAV and Lib_Seq_GFP_AAV_R; primers for cDNA: Lib_Hand_AAV and Lib_Seq_GFP_AAV_R). The samples were cleaned up using Zymo DNA clean and concentrator-5. This was followed by the second amplification step to add on sequencing adaptors and unique Illumina indices (primers: P5_Seq_GFP_AAV_F and P7_Ind_#_Han). Again, NEBNext 2X Q5 Hifi HS Mastermix was used for amplification. The resulting libraries were cleaned up using 0.75X ampure beads (Beckman Coulter, cat#A63881) and sequenced by UNC HTSF via Novaseq 6000 SP (1x35bp), with custom primers that capture the barcode sequence and sequencing index (read 1 primer: Exp_R1_seq_P_AAV, index primer: Exp_Ind_seq_P_AAV).
Quantification and statistical analysis
Quality check and barcode aggregation
Using the barcode-variant relationship decoded from the barcode mapping step, RNA and DNA barcodes from RNA- and DNA-sequencing were mapped back to their corresponding variants. We counted the number of barcodes mapped to each variant and found that each variant was mapped to ∼200 barcodes on average. Next, we aggregated the RNA and DNA barcode counts for each variant. Because different combinations of barcodes could be introduced to different biological replicates, using the same DNA counts measured from the AAV-MPRA library for all biological replicates could lead to incorrect normalization. To mitigate this, if a given RNA barcode was missing in one biological replicate, that barcode was not counted in aggregating DNA counts for that replicate. This way, even when the DNA from the AAV-MPRA library was used, each biological replicate could have different DNA barcode counts guided by RNA barcodes. For example, if barcodes 1, 2, and 3 were mapped to the variant1, and barcode 2 was missing from the RNA barcode count, we would simply sum up the counts for barcodes 1 and 3 for both DNA and RNA. In contrast, if none of the barcodes were measured, that corresponding variant will have NA count. After merging both DNA and RNA counts by those criteria, we discarded any variants that had more than eight NAs across ten replicates.
Identification of MPRA-positive variants
Using the aggregated DNA and RNA counts, we used mpra (version 1.18.0) Bioconductor package to calculate differential allelic regulatory activity.60 We used mpralm() function which uses the linear model to measure differential regulatory activity between two alleles with following parameters:
mpra_lm_object <- mpralm(object = mpra_set, design = design_matrix, aggregate = "none", normalize = T, block = samples, model_type = "corr_groups")
Here, mpra_set refers to the mpra object created by MPRAset() function consisting DNA and RNA counts and design_matrix refers to the matrix that specifies the reference and alternative allele status of the corresponding DNA/RNA counts. For our code we used 1) aggregate = “none” since we aggregated our barcodes before running mpralm and 2) normalize = T as DNA and RNA counts were not pre-normalized. Lastly, we named our replicates with the samples variable and used model_type = “corr_groups'' for paired mixed-model fit.
The resulting mpra_lm_object provides summary statistics (e.g., logFC, average expression, t-statistics, P-value, adjusted P-values, and B-statistics) of each variant (Table S1). We defined MPRA-positive variants as variants that show statistical RNA count difference between reference and alternative allele at FDR <0.1, while defining MPRA-negative variants as variants with no significant allelic regulatory activity at nominal p > 0.1.
Measuring reproducibility
We applied mpralm normalization method to 10 biological replicates to scale all replicates to have a common size of 10 million reads. We then used corrplot R package’s corrplot.mixed(upper = "number", lower = "square", col.lim = c(0.5,0.8)) function to compare the RNA/DNA count ratio between biological replicates (Figure S2).
Circular Manhattan plots
Circular Manhattan plots (Figures 1B and S1) for fine-mapped and MPRA variants were created by using CMplot R package.61 For fine-mapped GWAS variants, CMplot( …, type = “p”, plot.type = “c”, threshold = 5e-8) was used. For our MPRA variants, we used threshold = 0.1 and all other parameters were identical.
LD SNPs and random SNPs
To define the local background, LD SNPs were selected. LD (or a schizophrenia GWS locus) was defined as a region that encompasses SNPs with r2 > 0.6 to the index SNP. All SNPs within 144 schizophrenia GWS loci with nominal association (p < 0.001) were selected. Fine-mapped variants were then extracted from these variants, leaving non-fine-mapped SNPs with nominal association within LD.
To define the global background, random SNPs with matched minor allele frequency (MAF) and LD were selected. For each fine-mapped SNP, we randomly selected 10 SNPs within the same chromosome that have matching ( 10%) MAF and the number of SNPs in LD (defined as r2 > 0.1). If less than 10 SNPs were identified for a given SNP, we selected all SNPs matched with MAF and LD. MAF and the number of LD buddies for genome-wide SNPs were obtained from garfield Bioconductor package62 (version 1.28.0).
Genomic annotation
Using the annotatr Bioconductor package (version 1.22.0), MPRA-positive and MPRA-negative variants were mapped to its corresponding genomic annotations. After labeling MPRA-positive and MPRA-negative variants to its corresponding genomic annotations, we noticed that some of the variants are overlapping with multiple annotations which leads to overrepresentation of certain variants. To mediate this issue, we prioritized certain annotations (i.e., exons/UTRs (can be duplicated) > promoters > 1kb–5kb from promoter > introns > intergenic) so that each SNP is mapped to a single genomic annotation.
Epigenetic annotation
We used 1) H3K27ac peaks from the fetal and adult dorsolateral prefrontal cortex (DLPFC) as fetal and adult brain enhancers, respectively,16 2) H3K27ac peaks from sorted brain cells as cell type-specific adult brain enhancers,19 and 3) single-cell ATAC-seq peaks from the fetal brain as cell type-specific fetal brain enhancers20 to compare the epigenetic differences of MPRA-positive, MPRA-negative, LD, and random SNPs. We overlapped MPRA-positive, MPRA-negative, LD, and random SNPs with each enhancer set using findOverlaps() function in GenomicRanges Bioconductor package. Additionally, we used 1) ATAC-seq peaks from developing human telencephalon,17 2) DNAse peaks from different regions of the adult brain18 and 3) ATAC-seq peaks from hiPSC-derived neural progenitor cells (NPCs) and differentiated neurons21 to overlap MPRA-positive and MPRA-negative variants using findOverlaps() function. Finally, we leveraged the 15-state chromHMM model in brain cell and tissue types (neural progenitors, neurons, fetal brain, and adult brain)22 to define chromatin states of the MPRA-positive variants. TssA, TssAFlnk, TxFlnk, and TssBiv were grouped into TSS; Tx and TxWk were grouped into Transcribed; EnhG, Enh, EnhBiv were grouped into Enhancers; ZNF/Rpts, Het, BivFlnk, ReprPC, and ReprPCWk were grouped into repressors.
The overlap proportion was calculated by dividing the number of overlapped variants by the original number of variants (e.g., 4 out of 10 variants within the variant set A overlapped with enhancer set B gives 40% overlap). To compare the overlap between two SNP categories, Fisher’s exact test with the contingency table below was used.
| The number of MPRA-positive variants overlapping with epigenetic region A | The number of MPRA-positive variants not overlapping with epigenetic region A |
|---|---|
| The number of MPRA-negative/LD/random overlapping with epigenetic region A | The number of MPRA-negative/LD/random overlapping with epigenetic region A |
Evolutionary conservation
From the Zoonomia consortium, we obtained human phyloP scores predicted from the comparative genomic analysis of 240 mammalian species.24 Since phyloP scores were available in hg38, we converted them to hg19 using liftOver52 (version 1.04.00). As we used 150 bp sequences centered (76th position) on each variant for our MPRA experiment, we calculated average phyloP scores for 150 bp sequences flanking the variants of interest. Average phyloP scores were calculated for MPRA-positive, MPRA-negative, LD, and random SNPs and compared against each other using Wilcoxon rank-sum test. To ensure that this finding is not dependent on the size of the window used, we also used different window sizes (e.g., 100bp, 200bp, and 300bp centered on each variant), but the choice of window sizes did not change the results.
Similar to phyloP scores, average phastCons scores of the same sequences were obtained using the Bioconductor package phastCons100way.UCSC.hg19.53
TF motif analysis
To observe TF motif altering properties of MPRA-positive variants, we used motifbreakR Bioconductor package25 (version 2.10.2). Following the motifbreakR vignette, we subsetted the TF motif database by Hsapiens and excluded stamlabs since they are not annotated. This database included TF motif data from cisbp_1.02, HOCOMOCOv10, HOCOMOCOv11, hDPI, JASPAR_2014, JASPAR_CORE, jaspar2016, jaspar2018, jolma2013, SwissRegulon, and UniPROBE. Then we ran motifbreakR( …, filterp = TRUE, method = “ic”, threshold = 1e-4) and filtered the result by effect = “strong” to observe strong TF motif alterations only.
TF enrichment analysis
To calculate the TF enrichment for MPRA-positive variants, we also ran motifbreakR on LD and random SNPs. Then we compared the number of TF motif alterations between MPRA-positive and LD/random SNPs and calculated statistical significance by Fisher’s exact test with the contingency table of.
| The number of MPRA-positive variants altering TF motif 1 | The number of MPRA-positive variants not altering TF motif 1 |
|---|---|
| The number of LD/random SNPs altering TF motif 1 | The number of LD/random SNPs not altering TF motif 1 |
Calculation of corrected SVM scores
To predict the impact of TFs on SNP-mediated regulatory activity, we calculated corrected delta support vector machine ( SVM) scores with the following formula for each variant.
SVM scores for each TF-SNP pair were obtained from Yan et al.27; expression levels of TFs in HNPs have been obtained from Aygün et al.57; information about whether TFs are activators or repressors has been obtained from Savitskaya.63 For TFs that are predicted to act as both activators and repressors, we assumed that they mainly act as an activator.
The resulting corrected SVM scores were compared against MPRA logFC values at a variant level. Pearson’s correlation coefficients between corrected SVM scores and MPRA logFC were calculated for MPRA-positive and MPRA-negative variants. We then randomly sampled corrected SVM scores and MPRA logFC values for MPRA-negative variants for 1,000 times to calculate the permuted distribution of Pearson’s correlation coefficient. The observed Pearson’s correlation coefficient for MPRA-positive variants was compared against the permuted distribution to calculate the permuted P-value.
Deep learning-based sequence models
We leveraged SURF,28 DeepSEA,29 and Sei30 models to unbiasedly identify (epi)genomic features that can distinguish MPRA-positive from -negative variants. The SURF model predicts the generic regulatory function of SNPs in the range of [0, 1].28 A higher value indicates the more likely an SNP would function as a regulatory variant. The DeepSEA model is a deep learning model that predicts genomic variant effects on a wide range of regulatory features.29 We used its functional significance in the range of [0, 1] which is meant to be a general functionality score, not specific to a particular purpose. The Sei model is the successor of DeepSEA with larger model architecture to enable it to predict more (epi)genomic assays simultaneously (900 vs. 21,000).30 We used its maximum absolute difference prediction as the indicator of variant function.
eQTL overlap
eQTL datasets from the adult DLPFC (n = 1,387) and fetal cortices (n = 201) were obtained from Wang et al.34 and Walker et al.,35 respectively. We overlapped our MPRA-positive variants with brain eQTL resources by matching variant information (i.e., chromosome, position, rsid). One discrepancy that we found was that our data contained SNPs in chromosome X, whereas both eQTLs lacked SNPs in sex chromosomes.
Colocalization analysis between adult DLPFC eQTLs and schizophrenia GWAS was obtained from Liu et al.36 Same analytic pipeline was used to perform colocalization analysis between developing brain eQTLs and schizophrenia GWAS. Briefly, we intersected developing brain eQTLs with schizophrenia GWS loci using findOverlap() function in GenomicRanges Bioconductor package. We then performed colocalization analysis between schizophrenia GWAS and eQTLs using the default setting of coloc R package64 (version 5.1.0.1). We selected loci and eGenes with colocalization posterior probability greater than 0.6 (H4 PP > 0.6) to compare against MPRA-positive variants.
For the variant level overlap analysis, the proportion of eQTL overlap was calculated by dividing the number of MPRA-positive variants that overlapped with eQTLs (i.e., matching rsid, chr, and pos) by the total number of MPRA-positive variants. Then, the proportion of IDE overlap was calculated by dividing the number of MPRA-positive-eQTL overlapped variants that has any IDE variant-gene pairs (i.e., MPRA log2FC > 0 & eQTL beta >0 and vice versa) by the number of MPRA-positive-eQTL overlapped variants. Lastly, we overlapped our IDE variants’ genomic coordinates to the colocalized GWS loci using findOverlap() function and calculated the overlap by dividing the number of IDE variants that overlapped to the colocalized locus by the number of IDE variants. For each overlap, the number of genes and loci was counted as well.
TSS distance analysis
Using the Gencode v19 promoter definition,54 we employed bedtools65 (version 2.29) closest function to calculate the distance to the nearest promoters for MPRAnon-eQTL and MPRAeQTL variants. Then Wilcoxon rank-sum test was used to calculate the statistical significance between two distributions.
Assigning genes to MPRA-positive variants using Hi-C data
To assign genes to MPRA-positive variants using long-range interactome, first we filtered the Hi-C loops from the four datasets (GZ, CP, PN, AN) that interact with Gencode v19 promoters (hereafter referred to as promoter-anchored loops). Then we overlapped 439 MPRA-positive SNP coordinates with the other end of the promoter-anchored loops (the non-promoter anchor) to identify variants that interact with promoters through loops. SNP-gene pairs obtained this way were filtered for protein-coding genes with HUGO Gene Nomenclature Committee (HGNC) symbols, resulting in a total 272 genes (MPRAHi-C genes). To visualize the loci of MPRAHi-C genes (variants, genes, Hi-C loops), plotgardener Bioconductor package was used.55 When loops were plotted, we only visualized the midpoint of each loop’s end for simplicity.
Gene ontology
For gene ontology (GO) analysis, we used gprofiler2 R package66 (version 3.4.2). GO terms with term size between 5 and 1000 were filtered, resulting in 26 terms (FDR<0.1). To reduce redundant GO terms, REVIGO web interface was used (http://revigo.irb.hr/).
LOEUF score
A LOEUF score for each gene was obtained from Karczewski et al.56 LOEUF scores for MPRAeQTL-IDE genes were compared against MPRAHi-C genes. Statistical significance of the difference in LOEUF scores between two gene sets was calculated by the Wilcoxon rank-sum test.
Regulatory complexity
To analyze regulatory complexity, we counted the number of loops anchored at promoters of MPRAeQTL-IDE, MPRAeQTL-IDE protein-coding, and MPRAHi-C genes. Because eQTLs from the adult DLPFC were used to identify MPRAeQTL-IDE and MPRAeQTL-IDE protein-coding genes, we used loops from the adult neuronal Hi-C dataset.39 Loops that overlap with the promoter of each gene were selected and counted. Kolmogorov-Smirnov test was used to compare the difference in the number of promoter-anchored loops between MPRAeQTL-IDE and MPRAHi-C genes.
Cell type-specific gene expression in fetal and adult prefrontal cortex
In order to visualize cell type-specific gene expression, we used the single-cell gene expression matrix from the fetal67 and adult PFC.34 Gene expression matrix was filtered for MPRAHi-C genes. Then scaled, average expression across all genes was calculated for each cell type as previously described.11
Adding the chromatin context to allelic activity within multi-variant loci
Multi-variant loci were defined as GWS loci that have more than one MPRA-positive SNP detected. We identified 256 MPRAHi-C genes that were mapped to the multi-variant loci. To understand how these genes were expressed in schizophrenia, we used transcriptomic signature from postmortem adult brains with schizophrenia (hereby referred to as RNA-seq data).40 MPRAHi-C genes whose expression was not detected in RNA-seq data (due to their low expression level) were discarded, leaving 192 genes to compare between MPRA and RNA-seq. Because MPRA logFC values were initially calculated to compare the ratio between alternative and reference alleles, we converted them to compare the ratio between risk and protective alleles. The resulting logFC(risk/protective) values encode disease risk: whether the variant will up- or down-regulate the target gene in schizophrenia. We then aggregated variant-level logFC(risk/protective) values to cognate genes using the following three strategies.
1) Additive model: For each MPRAHi-C gene, we aggregated logFC(risk/protective) values of all MPRA-positive variants within the GWS locus that were assigned to the gene via Hi-C loops. Using all variants within the GWS locus (regardless of showing chromatin interactions with the gene) gave a similar result.
2) Contact model: For each MPRAHi-C gene, we used all MPRA-positive variants within the locus, because each variant is weighted by contact frequency. We weighted logFC(risk/protective) values with log(normalized contact frequency) between the variant and gene promoter using contact maps of adult neurons.39 For a gene with multiple promoters, we used the maximum normalized contact frequency.
3) Accessibility by contact model: For each MPRAHi-C gene, we used all MPRA-positive variants within the locus, because each variant is weighted by contact frequency and chromatin accessibility. We weighted logFC(risk/protective) with log(normalized contact frequency) between the variant and gene promoter and average chromatin accessibility of the 150bp element flanking the variant. Contact maps of adult neurons39 and chromatin accessibility from the Brain Open Chromatin Atlas51 were used to extract contact frequency and chromatin accessibility, respectively. For a gene with multiple promoters, we used the average value of log(normalized contact frequency) × chromatin accessibility.
We then compared predicted gene expression with RNA-seq logFC values. We did not stratify genes with significant differential expression for this comparison because the effect sizes of common variants are small, which may not necessarily yield significant differential expression in idiopathic schizophrenia. Accordingly, we measured the percentage of genes that show the same direction of effects (e.g., up- or down-regulation) between predicted gene expression and RNA-seq logFC.
Because the third model (accessibility by contact model) outperformed other models, we used the same model to calculate predicted gene expression from MPRA-negative variants as a control. In addition, we randomly sampled logFC(risk/protective) values for MPRA-positive and -negative variants for 10,000 times to calculate permuted predicted gene expression. The percentage of genes that show the same direction of effects between permuted predicted gene expression and RNA-seq logFC was compared against what was predicted from MPRA-positive variants to calculate the permutation P-value.
Single-cell RNA-seq from schizophrenia postmortem brains
We surveyed cell type-specific gene expression patterns of the targeted genes (e.g., GRIN2A, SETD8, MEF2C) from single-cell (sc)RNA-seq datasets of schizophrenia postmortem brains.42 Gene co-expression analysis was performed by estimating Pearson’s correlation coefficient between the expression profile of a given target gene and that of all other genes in the same dataset.42 Genes with positive correlation values supported by evidence at Bonferroni-corrected p value <0.01 were considered as co-expressed. Correlation analyses were performed using pseudobulk gene expression profiles with normalized log-transformed expression values reported in Ruzicka et al.42 Analyses were performed independently for each subpopulation of excitatory neurons. To estimate the degree to which co-expressed genes tend to be dysregulated in schizophrenia, average differential scores for co-expressed genes were contrasted with random expectation by computing expected values for 10,000 randomly resampled and equally-sized gene sets. Differential scores were estimated using the -log10 adjusted p values signed by the directionality in fold-change from expression level comparisons between schizophrenia and control subjects42 (multi-cohort meta-analysis). Z-scores were used to measure deviation from random expectation. We then identified downstream target genes of SETD8 and MEF2C by querying genes that are perturbed by SETD8 knockdown (KD) in medulloblastoma44 and Mef2c knockout (KO) in the mouse cortex,45 respectively. Significance of overlap between these downstream targets and genes dysregulated in each excitatory neuronal subtypes of schizophrenia postmortem brains42 was calculated using a Fisher’s exact test. Significance of overrepresentation was plotted, while significance of underrepresentation was omitted.
TF pathway overlap analysis
To understand the functional properties of MEF2C co-expressed genes, we compared them with the MEF2C harboring TF network that is involved in differentiating human embryonic stem cells to brain cell types.46 Because TF networks only contain TFs, we first filtered the MEF2C co-expressed genes by TFs. Moreover, combinatorial TF analysis result was partitioned by brain cell types (i.e., Astrocytes, Excitatory neurons, Ganglion cells, Granule neurons, Inhibitory interneurons, Inhibitory neurons, Oligodendrocytes, and Schwann cells).46 We then compared the two gene lists (MEF2C co-expressed TFs vs. MEF2C harboring TF network) using the phyper( …, lower.tail = F) function in R.
Acknowledgments
We thank members of the Won lab for helpful discussions and comments about this paper and Dr. Patrick Sullivan at the University of North Carolina for his advice on evolutionary conservation analysis. This research was supported by the PsychENCODE consortium (R01MH122509, H.W. and J.L.S.), the IGVF consortium (UM1HG012003, H.W. and M.I.L.), the National Institute of General Medical Sciences (5T32GM067553, S.L; 5T32GM135128, J.C.M. and M.B.), the National Institute on Aging (R01AG066871, H.W. and D.H.P.), the National Institute of Child Health and Human Development (5T32HD040127, J.L.), the NIH New Innovator Award from the National Institute of Mental Health (DP2MH122403, H.W.), and the NARSAD Young Investigator Award from the Brain and Behavior Research Foundation (H.W.).
Author contributions
H.W., J.D., and S.K. designed schizophrenia MPRA libraries. J.C.M. created the AAV-MPRA vector and generated MPRA libraries. J.C.M. performed MPRA with help from J.L.B. O.K. helped J.C.M. with HNP culture. K.I. created the script to map barcodes to variants. S.L. conducted the bioinformatic analyses of the resulting MPRA datasets. N.Z. and A.P.B. compared MPRA-positive and -negative variants using deep-learning-based models. J.L. identified target genes of MPRA-positive variants using Hi-C datasets. W.B.R. and J.D.-V. integrated MPRA results with schizophrenia scRNA-seq datasets. M.L.B., D.H.P., and M.I.L. helped establish statistical analytic pipelines. O.K. and J.L.S. optimized transduction protocols for HNPs. H.W., S.L., J.C.M., and J.L. generated figures. H.W., J.C.M., S.L., J.L., and J.L.B. co-wrote the manuscript, which was subsequently reviewed and edited by the rest of the authors.
Declaration of interests
The authors declare no competing interests.
Published: September 13, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100404.
Supplemental information
Data and code availability
Sequencing data are available via the Gene Expression Omnibus. Custom codes used to generate our SCZ MPRA results are available on our GitHub page (https://github.com/thewonlab/schizophrenia-MPRA) and Zenodo.
GEO accession number and Zenodo DOIs are listed in the key resources table.
References
- 1.McCutcheon R.A., Reis Marques T., Howes O.D. Schizophrenia-An Overview. JAMA Psychiatr. 2020;77:201–210. doi: 10.1001/jamapsychiatry.2019.3360. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan P.F., Kendler K.S., Neale M.C. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatr. 2003;60:1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 3.Ripke S., Walters J.T., O’Donovan M.C., O’Donovan M.C. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv. 2020 doi: 10.1101/2020.09.12.20192922. Preprint at. [DOI] [Google Scholar]
- 4.Schaid D.J., Chen W., Larson N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018;19:491–504. doi: 10.1038/s41576-018-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mah W., Won H. The three-dimensional landscape of the genome in human brain tissue unveils regulatory mechanisms leading to schizophrenia risk. Schizophr. Res. 2020;217:17–25. doi: 10.1016/j.schres.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pardiñas A.F., Holmans P., Pocklington A.J., Escott-Price V., Ripke S., Carrera N., Legge S.E., Bishop S., Cameron D., Hamshere M.L., et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018;50:381–389. doi: 10.1038/s41588-018-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tewhey R., Kotliar D., Park D.S., Liu B., Winnicki S., Reilly S.K., Andersen K.G., Mikkelsen T.S., Lander E.S., Schaffner S.F., Sabeti P.C. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell. 2016;165:1519–1529. doi: 10.1016/j.cell.2016.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulvey B., Lagunas T., Dougherty J.D. Massively parallel reporter assays: defining functional psychiatric genetic variants across biological contexts. Biol. Psychiatr. 2021;89:76–89. doi: 10.1016/j.biopsych.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McAfee J.C., Bell J.L., Krupa O., Matoba N., Stein J.L., Won H. Focus on your locus with a massively parallel reporter assay. J. Neurodev. Disord. 2022;14:50. doi: 10.1186/s11689-022-09461-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de la Torre-Ubieta L., Stein J.L., Won H., Opland C.K., Liang D., Lu D., Geschwind D.H. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell. 2018;172:289–304.e18. doi: 10.1016/j.cell.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sey N.Y.A., Hu B., Mah W., Fauni H., McAfee J.C., Rajarajan P., Brennand K.J., Akbarian S., Won H. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci. 2020;23:583–593. doi: 10.1038/s41593-020-0603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spiess K., Won H. Regulatory landscape in brain development and disease. Curr. Opin. Genet. Dev. 2020;65:53–60. doi: 10.1016/j.gde.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pratt B.M., Won H. Advances in profiling chromatin architecture shed light on the regulatory dynamics underlying brain disorders. Semin. Cell Dev. Biol. 2022;121:153–160. doi: 10.1016/j.semcdb.2021.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stein J.L., de la Torre-Ubieta L., Tian Y., Parikshak N.N., Hernández I.A., Marchetto M.C., Baker D.K., Lu D., Hinman C.R., Lowe J.K., et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron. 2014;83:69–86. doi: 10.1016/j.neuron.2014.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abell N.S., DeGorter M.K., Gloudemans M.J., Greenwald E., Smith K.S., He Z., Montgomery S.B. Multiple causal variants underlie genetic associations in humans. Science. 2022;375:1247–1254. doi: 10.1126/science.abj5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M., Santpere G., Imamura Kawasawa Y., Evgrafov O.V., Gulden F.O., Pochareddy S., Sunkin S.M., Li Z., Shin Y., Zhu Y., et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362 doi: 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markenscoff-Papadimitriou E., Whalen S., Przytycki P., Thomas R., Binyameen F., Nowakowski T.J., Kriegstein A.R., Sanders S.J., State M.W., Pollard K.S., Rubenstein J.L. A chromatin accessibility atlas of the developing human telencephalon. Cell. 2020;182:754–769.e18. doi: 10.1016/j.cell.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B., et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nott A., Holtman I.R., Coufal N.G., Schlachetzki J.C.M., Yu M., Hu R., Han C.Z., Pena M., Xiao J., Wu Y., et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019;366:1134–1139. doi: 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziffra R.S., Kim C.N., Ross J.M., Wilfert A., Turner T.N., Haeussler M., Casella A.M., Przytycki P.F., Keough K.C., Shin D., et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature. 2021;598:205–213. doi: 10.1038/s41586-021-03209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S., Zhang H., Zhou Y., Qiao M., Zhao S., Kozlova A., Shi J., Sanders A.R., Wang G., Luo K., et al. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science. 2020;369:561–565. doi: 10.1126/science.aay3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roadmap Epigenomics Consortium. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper Y.A., Teyssier N., Dräger N.M., Guo Q., Davis J.E., Sattler S.M., Yang Z., Patel A., Wu S., Kosuri S., et al. Functional regulatory variants implicate distinct transcriptional networks in dementia. Science. 2022;377:eabi8654. doi: 10.1126/science.abi8654. [DOI] [PubMed] [Google Scholar]
- 24.Zoonomia Consortium A comparative genomics multitool for scientific discovery and conservation. Nature. 2020;587:240–245. doi: 10.1038/s41586-020-2876-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coetzee S.G., Coetzee G.A., Hazelett D.J. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015;31:3847–3849. doi: 10.1093/bioinformatics/btv470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cadigan K.M., Waterman M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harbor Perspect. Biol. 2012;4 doi: 10.1101/cshperspect.a007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan J., Qiu Y., Ribeiro Dos Santos A.M., Yin Y., Li Y.E., Vinckier N., Nariai N., Benaglio P., Raman A., Li X., et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature. 2021;591:147–151. doi: 10.1038/s41586-021-03211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong S., Boyle A.P. Predicting functional variants in enhancer and promoter elements using RegulomeDB. Hum. Mutat. 2019;40:1292–1298. doi: 10.1002/humu.23791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou J., Troyanskaya O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015;12:931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen K.M., Wong A.K., Troyanskaya O.G., Zhou J. A sequence-based global map of regulatory activity for deciphering human genetics. Nat. Genet. 2022;54:940–949. doi: 10.1038/s41588-022-01102-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roussos P., Mitchell A.C., Voloudakis G., Fullard J.F., Pothula V.M., Tsang J., Stahl E.A., Georgakopoulos A., Ruderfer D.M., Charney A., et al. A role for noncoding variation in schizophrenia. Cell Rep. 2014;9:1417–1429. doi: 10.1016/j.celrep.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myint L., Wang R., Boukas L., Hansen K.D., Goff L.A., Avramopoulos D. A screen of 1,049 schizophrenia and 30 Alzheimer’s-associated variants for regulatory potential. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2020;183:61–73. doi: 10.1002/ajmg.b.32761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang D., Liu S., Warrell J., Won H., Shi X., Navarro F.C.P., Clarke D., Gu M., Emani P., Yang Y.T., et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362 doi: 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker R.L., Ramaswami G., Hartl C., Mancuso N., Gandal M.J., de la Torre-Ubieta L., Pasaniuc B., Stein J.L., Geschwind D.H. Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell. 2019;179:750–771.e22. doi: 10.1016/j.cell.2019.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S., Won H., Clarke D., Matoba N., Khullar S., Mu Y., Wang D., Gerstein M. Illuminating links between cis-regulators and trans-acting variants in the human prefrontal cortex. bioRxiv. 2021 doi: 10.1101/2021.09.07.459322. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mostafavi H., Spence J.P., Naqvi S., Pritchard J.K. Limited overlap of eQTLs and GWAS hits due to systematic differences in discovery. bioRxiv. 2022 doi: 10.1101/2022.05.07.491045. Preprint at. [DOI] [Google Scholar]
- 38.Won H., de la Torre-Ubieta L., Stein J.L., Parikshak N.N., Huang J., Opland C.K., Gandal M.J., Sutton G.J., Hormozdiari F., Lu D., et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538:523–527. doi: 10.1038/nature19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu B., Won H., Mah W., Park R.B., Kassim B., Spiess K., Kozlenkov A., Crowley C.A., Pochareddy S., et al. PsychENCODE Consortium Neuronal and glial 3D chromatin architecture informs the cellular etiology of brain disorders. Nat. Commun. 2021;12:3968. doi: 10.1038/s41467-021-24243-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gandal M.J., Zhang P., Hadjimichael E., Walker R.L., Chen C., Liu S., Won H., van Bakel H., Varghese M., Wang Y., et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018;362 doi: 10.1126/science.aat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skene N.G., Bryois J., Bakken T.E., Breen G., Crowley J.J., Gaspar H.A., Giusti-Rodriguez P., Hodge R.D., Miller J.A., Muñoz-Manchado A.B., et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018;50:825–833. doi: 10.1038/s41588-018-0129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruzicka W.B., Mohammadi S., Davila-Velderrain J., Subburaju S., Tso D.R., Hourihan M., Kellis M. Single-cell dissection of schizophrenia reveals neurodevelopmental-synaptic axis and transcriptional resilience. medRxiv. 2020 doi: 10.1101/2020.11.06.20225342. Preprint at. [DOI] [Google Scholar]
- 43.Milite C., Feoli A., Viviano M., Rescigno D., Cianciulli A., Balzano A.L., Mai A., Castellano S., Sbardella G. The emerging role of lysine methyltransferase SETD8 in human diseases. Clin. Epigenet. 2016;8:102. doi: 10.1186/s13148-016-0268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Veschi V., Liu Z., Voss T.C., Ozbun L., Gryder B., Yan C., Hu Y., Ma A., Jin J., Mazur S.J., et al. Epigenetic siRNA and Chemical Screens Identify SETD8 Inhibition as a Therapeutic Strategy for p53 Activation in High-Risk Neuroblastoma. Cancer Cell. 2017;31:50–63. doi: 10.1016/j.ccell.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harrington A.J., Raissi A., Rajkovich K., Berto S., Kumar J., Molinaro G., Raduazzo J., Guo Y., Loerwald K., Konopka G., et al. MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife. 2016;5 doi: 10.7554/eLife.20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joung J., Ma S., Tay T., Geiger-Schuller K.R., Kirchgatterer P.C., Verdine V.K., Guo B., Arias-Garcia M.A., Allen W.E., Singh A., et al. A transcription factor atlas of directed differentiation. Cell. 2023;186:209–229.e26. doi: 10.1016/j.cell.2022.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alasoo K., Rodrigues J., Mukhopadhyay S., Knights A.J., Mann A.L., Kundu K., HIPSCI Consortium. Hale C., Dougan G., Gaffney D.J. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat. Genet. 2018;50:424–431. doi: 10.1038/s41588-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Umans B.D., Battle A., Gilad Y. Where Are the Disease-Associated eQTLs? Trends Genet. 2021;37:109–124. doi: 10.1016/j.tig.2020.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X., Goldstein D.B. Enhancer domains predict gene pathogenicity and inform gene discovery in complex disease. Am. J. Hum. Genet. 2020;106:215–233. doi: 10.1016/j.ajhg.2020.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fulco C.P., Nasser J., Jones T.R., Munson G., Bergman D.T., Subramanian V., Grossman S.R., Anyoha R., Doughty B.R., Patwardhan T.A., et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019;51:1664–1669. doi: 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fullard J.F., Hauberg M.E., Bendl J., Egervari G., Cirnaru M.-D., Reach S.M., Motl J., Ehrlich M.E., Hurd Y.L., Roussos P. An atlas of chromatin accessibility in the adult human brain. Genome Res. 2018;28:1243–1252. doi: 10.1101/gr.232488.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Navarro Gonzalez J., Zweig A.S., Speir M.L., Schmelter D., Rosenbloom K.R., Raney B.J., Powell C.C., Nassar L.R., Maulding N.D., Lee C.M., et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021;49:D1046–D1057. doi: 10.1093/nar/gkaa1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frankish A., Diekhans M., Jungreis I., Lagarde J., Loveland J.E., Mudge J.M., Sisu C., Wright J.C., Armstrong J., Barnes I., et al. GENCODE 2021. Nucleic Acids Res. 2021;49:D916–D923. doi: 10.1093/nar/gkaa1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kramer N.E., Davis E.S., Wenger C.D., Deoudes E.M., Parker S.M., Love M.I., Phanstiel D.H. Plotgardener: cultivating precise multi-panel figures in R. Bioinformatics. 2022;38:2042–2045. doi: 10.1093/bioinformatics/btac057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aygün N., Elwell A.L., Liang D., Lafferty M.J., Cheek K.E., Courtney K.P., Mory J., Hadden-Ford E., Krupa O., de la Torre-Ubieta L., et al. Brain-trait-associated variants impact cell-type-specific gene regulation during neurogenesis. Am. J. Hum. Genet. 2021;108:1647–1668. doi: 10.1016/j.ajhg.2021.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benner C., Spencer C.C.A., Havulinna A.S., Salomaa V., Ripatti S., Pirinen M. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016;32:1493–1501. doi: 10.1093/bioinformatics/btw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schork A.J., Won H., Appadurai V., Nudel R., Gandal M., Delaneau O., Revsbech Christiansen M., Hougaard D.M., Bækved-Hansen M., Bybjerg-Grauholm J., et al. A genome-wide association study of shared risk across psychiatric disorders implicates gene regulation during fetal neurodevelopment. Nat. Neurosci. 2019;22:353–361. doi: 10.1038/s41593-018-0320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Myint L., Avramopoulos D.G., Goff L.A., Hansen K.D. Linear models enable powerful differential activity analysis in massively parallel reporter assays. BMC Genom. 2019;20:209. doi: 10.1186/s12864-019-5556-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yin L., Zhang H., Tang Z., Xu J., Yin D., Zhang Z., Yuan X., Zhu M., Zhao S., Li X., Liu X. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Dev. Reprod. Biol. 2021;19:619–628. doi: 10.1016/j.gpb.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iotchkova V., Ritchie G.R., Geihs M., Morganella S., Min J.L., Walter K., Timpson N., Dunham I., Birney E., Soranzo N., et al. GARFIELD - GWAS Analysis of Regulatory or Functional Information Enrichment with LD correction. bioRxiv. 2016 doi: 10.1101/085738. Preprint at. [DOI] [Google Scholar]
- 63.Savitskaya A. Florida Atlantic University; 2010. Activators and Repressors of Transcription: Using Bioinformatics Approaches to Analyze and Group Human Transcription Factors. [Google Scholar]
- 64.Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kolberg L., Raudvere U., Kuzmin I., Vilo J., Peterson H. gprofiler2 -- an R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Res. 2020;9 doi: 10.12688/f1000research.24956.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nowakowski T.J., Bhaduri A., Pollen A.A., Alvarado B., Mostajo-Radji M.A., Di Lullo E., Haeussler M., Sandoval-Espinosa C., Liu S.J., Velmeshev D., et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science. 2017;358:1318–1323. doi: 10.1126/science.aap8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are available via the Gene Expression Omnibus. Custom codes used to generate our SCZ MPRA results are available on our GitHub page (https://github.com/thewonlab/schizophrenia-MPRA) and Zenodo.
GEO accession number and Zenodo DOIs are listed in the key resources table.