

Significance
Subgenome dominance refers to gene fractionation and expression bias in different subgenomes of species experienced allopolyploidization events. Pieces of evidence showed that the differences in transposon density and resulted methylation levels were negatively associated with subgenome dominance. Therefore, many researchers suggested that transposons served to initiate subgenome dominance in allopolyploids. This study on allopolyploid progenies merging from Brassica rapa and Brassica oleracea provided a solid case arguing against the previous notion through investigating the transposon, methylation, and gene expression dynamics in this well-constructed system. Instead, we provide a “nuclear chimera” model to explain the subgenome difference in wide hybrid. The research highlights the complex nature of subgenome dominance and contributes significantly to our further understanding of polyploidized genome evolution.
Keywords: subgenome dominance, polyploidization, methylation, transposon
Abstract
Polyploidization is important to the evolution of plants. Subgenome dominance is a distinct phenomenon associated with most allopolyploids. A gene on the dominant subgenome tends to express to higher RNA levels in all organs as compared to the expression of its syntenic paralogue (homoeolog). The mechanism that underlies the formation of subgenome dominance remains unknown, but there is evidence for the involvement of transposon/DNA methylation density differences nearby the genes of parents as being causal. The subgenome with lower density of transposon and methylation near genes is positively associated with subgenome dominance. Here, we generated eight generations of allotetraploid progenies from the merging of parental genomes Brassica rapa and Brassica oleracea. We found that transposon/methylation density differ near genes between the parental (rapa:oleracea) existed in the wide hybrid, persisted in the neotetraploids (the synthetic Brassica napus), but these neotetraploids expressed no expected subgenome dominance. This absence of B. rapa vs. B. oleracea subgenome dominance is particularly significant because, while there is no negative relationship between transposon/methylation level and subgenome dominance in the neotetraploids, the more ancient parental subgenomes for all Brassica did show differences in transposon/methylation densities near genes and did express, in the same samples of cells, biased gene expression diagnostic of subgenome dominance. We conclude that subgenome differences in methylated transposon near genes are not sufficient to initiate the biased gene expressions defining subgenome dominance. Our result was unexpected, and we suggest a “nuclear chimera” model to explain our data.
Polyploidization is an important feature in all plant lineages (1–4). The repeated cycles of polyploidization, fractionation, and rediploidization helped engender the rich diversity of current conifers and flowering plants (5–7), and the genomic rearrangements happening during rediploidizations may help explain bursts of new species (2, 8). The divergence of multicopy genes generated by polyploidizations fueled trait innovations (9–11). As of 2022, about 50 independent polyploidization events have been located in plant lineages (1, 12, 13), while over 240 putative paleo- or meso-polyploidizations were inferred in the Viridiplantae (14). Among them, there are two types of polyploidizations based on the degree of genetic divergence of the progenitor genomes: allopolyploidization and autopolyploidization (15), where the inexact difference is assumed to be somewhere near the subspecies level of divergence. Allopolyploidization refers to the merging of two or more different progenitor genomes (16), while autopolyploidization is the duplication of very similar to identical genomes. When the postpolyploid contains two or more recognizable, more-or-less complete parental genomes, no matter the karyotype, these are called “subgenomes” (17, 18). Subgenome dominance is a conspicuous phenomenon that often accompanies allopolyploidization (19), but not autoploidization. Generally, subgenome dominance was observed and investigated in ancient polyploidized genomes, where asymmetric evolution of subgenomes is often obvious. Specifically, one subgenome was dominant. Dominant subgenomes retain more genes (biased fractionation/loss of a duplicate gene), and genes on the dominant homoeolog (syntenic paralog) tend to express to higher RNA levels than do their homoeologs on the submissive subgenome(s). Subgenome dominance is known to occur in Arabidopsis, maize, Brassicas, cotton, teff, and more (17, 20–25) and is thought to have enabled the domestication/breeding of agronomic traits in crops (11, 26, 27).
Although the origin of subgenome dominance is unknown, important clues came from studies in synthetic- and neo-allopolyploids. Subgenome dominance was observed in neo-polyploid cotton and synthetic tetraploid of Cucumis. In the analysis of cotton subgenomes A and D, Wendel and co-authors (28) found that genome-wide expression level dominance was biased toward the A subgenome in the haploid hybrid and the natural neo-allopolyploid cotton, whereas the direction was reversed in the synthesized allopolyploid, suggesting a combination of regulatory (cis and trans) and epigenetic interactions that accompanied the initial merger of two parental genomes. It was further found that the extent of homoeolog expression bias and expression level dominance increased over time, from the genome merger through the evolution at the polyploid level; this made for a complicated story. A more recent study investigated gene expression in the synthesized allopolyploid between Cucumis sativus and Cucumis hystrix (29). These researchers found that the subgenome of C. sativus was dominant over the subgenome of C. hystrix since the C. sativus subgenome had many more genes expressed to higher levels than their homoeologs in the C. hystrix subgenome. The dominance of the subgenome from the C. sativus parent emerged immediately at interspecific hybridization and was diminished as the new allotetraploid was stabilized during subsequent crossing. Similarly, Edger and colleagues studied the gene expression and genome methylation using the powerful monkeyflower (Mimulus) system (30): a natural less than 140-y-old allopolyploid (Mimulus peregrinus), a synthesized interspecies triploid hybrid (M. robertsii), a synthesized allopolyploid (M. peregrinus), and their progenitor species (M. guttatus and M. luteus). They found that the hybrid between the parental lines showed subgenome expression dominance immediately, as did the synthetic and the recent allotetraploid, and that subgenome dominance increased over generations, again suggesting that genome dominance “sets in” over time. They also found that the CHH methylation levels were reduced in regions near genes and within transposable elements (TEs) in the first-generation hybrid, intermediate in the synthesized allopolyploid, and were recovered and re-patterned differently between the dominant and recessive subgenomes in the natural allopolyploid. Since subgenome dominance begins at the first opportunity, the wide hybrid, the origin must involve some sort of compatibility aspect in the parent genomes.
Previous studies found structural variations (SVs) caused by homologous exchange (31–36), resulting in copy number difference of homoeologous fragments between subgenomes in the synthetic polyploids. These SVs have nothing to do with the subgenome dominance phenomenon since dominance is compared between two sets of unique homoeologs. However, the copy number variations of homoeologs in synthetic polyploids may cause false estimates of the expression of genes located at the corresponding variant site. Therefore, these SVs (37) are excluded from our subgenome dominance analysis.
Before this report, TEs and their methylation status were hypothesized to be involved in these parental compatibility differences. TEs are repeat sequences that clearly originated from autonomously replicating (selfish) DNA families and can accumulate to very high levels in particular gross chromosomal locations, like pericentromeres, but also exist near protein-coding DNA as well. In the Arabidopsis genome, the density of neighbor TEs was, but only when methylated, negatively associated with the RNA expression level of the associated gene (38). A trade-off effect was proposed to denote the relationship between the reduced transposition of methylated TEs (mTEs) and the reduced fitness caused by the suppressed expression of a neighbor gene, this suppression being a position effect. This association between small RNA-suppressed TEs—a surrogate for methylated transposons—and subgenome dominance was shown in Brassica rapa (39). TEs were also found to be significantly and negatively associated with subgenome dominance in the allopolyploidized genomes of maize and white lupin, etc. (26, 40, 41), with the dominant subgenome having many fewer TEs near genes than that for the other (submissive) subgenome(s). This negative association between a gene’s expression and its associated TE load was also observed in studies of methylation variations in the genome of autotetraploid rice (42). These findings suggested that TEs might play a role in the genomic interactions in the wide hybrid that eventually lead to the biased expression of homoeologous genes from different subgenomes and therefore might function in the original formation of subgenome dominance in allopolyploidized genomes. However strong the association between higher levels of methylated TEs near genes and submissiveness, associations do not prove causality, as we will demonstrate.
The Brassica genus is a particularly good system for studying the evolution of subgenome dominance. There are several Brassica genomes that have been sequenced, including ancient allohexaploid genomes of B. rapa, B. oleracea, and B. nigra, as well as the neo-allotetraploidized genomes of B. napus, B. juncea, and B. carinata (43–47). “Neo” for B. napus means that two sets of parental chromosomes are maintained and no rediploidization or chromosome number reduction has occurred. Genomes of these Brassica species are varied in TE content. For example, B. oleracea has a larger genome with more TEs as compared to B. rapa (44, 48).
We studied eight generations of progenies from the original hybridization of parental genomes B. rapa and B. oleracea and then analyzed the seedling leaves of the two parents, the hybrid F1, the tetraploid F2, as well as three individuals from each of the self-crossed generations F5 and F8. We used mRNA-seq and an assay for whole-genome methylation (bisulfite-seq). Since several organs were used in the original maize and B. rapa subgenome dominance proofs (17, 23), and all organ genomes expressed subgenome dominance to about the same level, our conclusions based on data from the seedling leaf are likely to apply over the entire plant.
Results
Subgenome Dominance that Was Inherited from the Ancient PreBrassica Hexaploidization (generating β Homoeologs) Is Still Functioning in the Neotetraploids: The Control.
The B. rapa accession Chiifu and the B. oleracea accession JZS have high-quality genomes that were assembled by third-generation sequencing technologies (49, 50). That is, our parental genomes are sequenced. B. rapa was crossed with B. oleracea, and embryo rescue was conducted to obtain the hybrid F1. This F1 (single set of chromosomes from each parent) was then treated with colchicine to generate the F2 (the allotetraploid), which has two complete sets of homozygous chromosomes of both B. rapa and B. oleracea. The two parental genomes B. rapa and B. oleracea were merged as subgenomes after hybridization and tetraploidization and were then abbreviated rapa and oleracea, respectively. The syntenic paralogous gene pairs, those between the new subgenomes rapa and oleracea in the synthetic allotetraploid, are called “α homoeologs”, while the syntenic paralogs derived from the previous preBrassica hexaploidization are distributed among the three ancient subgenomes LF (the least fractionated subgenome), MF1 (more fractionated subgenome 1), and MF2 (more fractionated subgenome 2) (43). These more ancient duplicate genes are called “β homoeologs” (Fig. 1). F2 progeny were further self-crossed to produce tetraploid progenies from generations F3 to F8. We collected representative leaf samples from the two parents, the hybrid haploid F1, and the double haploid F2, as well as from three individuals from each of the self-crossed generations F5 and F8. Studying these inbred generations is important in case subgenome dominance “sets in over time.” mRNA levels expressed and the location of methyl groups were then determined from these samples (Materials and Methods). By design, we have RNA level and methylation data for leaves from the two parents, the hybrid F1, and the tetraploids F2, F5, and F8.
Fig. 1.

The schematic of the synthetic tetraploidization between B. rapa Chiifu and B. oleracea JZS. Parental chromosomes are color-coded. The blue-blue or red-red stars denote more ancient syntenic paralogous gene pairs/triplets; they are the β homoeologs (genes a1-a2-a3 or b1-b2-b3). The red-blue stars denote the new syntenic paralog pairs (genes a1-b1); these are the α homoeologs.
Data involving α and β homoeolog comparisons can seem complex. As Brassica species evolved from a common ancient hexaploidization event, many syntenic paralogous genes (homoeologs) were retained in the three resultant subgenomes LF, MF1, and MF2. As reported previously, subgenome LF showed expression dominance over the other two subgenomes in both B. rapa (43) and B. oleracea (44, 51). The method used to compare subgenomes was to tabulate winners in paired competitions (by greater or less, or by more than twofold differences) between an LF gene and its homoeolog MF (MF1 or MF2) genes. If this greater or less comparison were performed in, for example, B. rapa, α homoeologs would be competing, because α refers to the most recent polyploidy in this genome. If these same two genes were compared within a B. napus (AACC, red-blue of Fig. 1), where these two genes derived not from the most recent polyploidy, but the next most recent, then this would be a β homoeolog comparison within the genome of B. napus.
For our new allotetraploids, our new B. napus accessions, we asked whether or not the RNA-level data characterizing subgenome dominance, which exhibited in the parents, would still be exhibited in our new neotetraploids. We compared the expression of syntenic paralogous genes (LF vs. MFs) generated from the last polyploidization event, B. napus’ β homoeologs, in F1, F2, F5, and F8 to examine whether the LF subgenome still shows the expression dominance. Considering that these newly synthetic genomes contain two individual chromosome sets (rapa and oleracea), the comparisons were conducted in the parental genomes rapa and oleracea, separately. The results showed that the subgenome LF had more genes expressed to higher levels than the other subgenomes (Table 1 and SI Appendix, Table S1), which is similar to that found in B. rapa and B. oleracea (23, 44). For example, for all LF:MFs β homoeolog combinations in F2 rapa, there were 4,415 LF genes expressed to higher level as compared to 3,443 MFs genes expressed to higher level. Similarly, there were 4,354 LF genes expressed to higher level and 3,410 MFs genes expressed to higher level in F2 oleracea. We also performed stringent statistical tests and similar results were obtained (SI Appendix, Table S2 and Materials and Methods). Clearly, the β ancient hexaploidization shows subgenome dominance by these RNA-seq data comparisons. As a repeat experiment, we found these same results when analyzing published mRNA-seq datasets from a synthetic B. napus (SI Appendix, Table S3) (35).
Table 1.
The number of dominantly expressed genes of the β homoeologs among the three subgenomes LF, MF1, and MF2 in the parental genomes B. rapa or B. oleracea, as well as in the allotetraploid subgenomes rapa or oleracea of F1, F2, F5, and F8
| rapa | oleracea | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Samples | Comparison | Greater or less | P value** | Two folds | P value | Comparison | Greater or less | P value | Two folds | P value | |
| Parents | LF>MF1 | 2,451 | 4.57×10−22 | 1,696 | 1.67×10−20 | LF>MF1 | 2,320 | 1.67×10−15 | 1,657 | 2.23×10−21 | |
| LF<MF1 | 1,820 | 1,197 | LF<MF1 | 1,808 | 1,154 | ||||||
| LF>MF2 | 2,141 | 2.49×10−21 | 1,521 | 4.62×10−27 | LF>MF2 | 2,062 | 7.00×10−16 | 1,445 | 1.54×10−16 | ||
| LF<MF2 | 1,564 | 983 | LF<MF2 | 1,575 | 1,034 | ||||||
| MF1>MF2 | 1,320 | 3.70×10−02 | 912 | 2.08×10−01 | MF1>MF2 | 1,265 | 2.69×10−01 | 898 | 4.90×10−01 | ||
| MF1<MF2 | 1,214 | 858 | MF1<MF2 | 1,209 | 868 | ||||||
| F1 | LF>MF1 | 2,362 | 3.07×10−18 | 1,622 | 2.51×10−19 | LF>MF1 | 2,299 | 7.77×10−15 | 1,609 | 1.12×10−18 | |
| LF<MF1 | 1,800 | 1,149 | LF<MF1 | 1,801 | 1,146 | ||||||
| LF>MF2 | 2,051 | 7.09×10−17 | 1,427 | 3.23×10−21 | LF>MF2 | 2,041 | 9.68×10−15 | 1,420 | 2.39×10−15 | ||
| LF<MF2 | 1,550 | 965 | LF<MF2 | 1,575 | 1,028 | ||||||
| MF1>MF2 | 1,242 | 7.32×10−01 | 851 | 7.33×10−01 | MF1>MF2 | 1,253 | 2.66×10−01 | 876 | 3.10×10−01 | ||
| MF1<MF2 | 1,224 | 836 | MF1<MF2 | 1,197 | 833 | ||||||
| F2 | LF>MF1 | 2,355 | 4.85×10−14 | 1,653 | 4.69×10−13 | LF>MF1 | 2,312 | 1.22×10−14 | 1,642 | 3.40×10−18 | |
| LF<MF1 | 1,865 | 1,262 | LF<MF1 | 1,816 | 1,180 | ||||||
| LF>MF2 | 2,060 | 1.39×10−15 | 1,484 | 1.71×10−18 | LF>MF2 | 2,042 | 1.15×10−13 | 1,470 | 5.24×10−16 | ||
| LF<MF2 | 1,578 | 1,043 | LF<MF2 | 1,594 | 1,062 | ||||||
| MF1>MF2 | 1,276 | 2.62×10−01 | 885 | 5.18×10−01 | MF1>MF2 | 1,286 | 7.74×10−02 | 896 | 3.04×10−01 | ||
| MF1<MF2 | 1,219 | 857 | MF1<MF2 | 1,197 | 852 | ||||||
| F5A * | LF>MF1 | 2,113 | 2.20×10−01 | 1,512 | 1.91×10−01 | LF>MF1 | 2,644 | 1.66×10−80 | 2,097 | 1.69×10−90 | |
| LF<MF1 | 2,033 | 1,440 | LF<MF1 | 1,438 | 988 | ||||||
| LF>MF2 | 1,794 | 8.80×10−01 | 1,287 | 5.52×10−01 | LF>MF2 | 2,533 | 2.13×10−143 | 2,103 | 9.99×10−158 | ||
| LF<MF2 | 1,784 | 1,256 | LF<MF2 | 1,033 | 713 | ||||||
| MF1>MF2 | 1,205 | 2.51×10−01 | 840 | 2.79×10−01 | MF1>MF2 | 1,267 | 4.59×10−09 | 1,022 | 5.31×10−09 | ||
| MF1<MF2 | 1,263 | 886 | MF1<MF2 | 988 | 774 | ||||||
| F8A * | LF>MF1 | 2,502 | 1.77×10−57 | 2,012 | 1.68×10−70 | LF>MF1 | 2,004 | 9.62×10−01 | 1,492 | 2.51×10−01 | |
| LF<MF1 | 1,496 | 1,039 | LF<MF1 | 2,008 | 1,429 | ||||||
| LF>MF2 | 1,952 | 9.20×10−11 | 1,472 | 4.66×10−14 | LF>MF2 | 1,969 | 6.10×10−13 | 1,515 | 1.64×10−19 | ||
| LF<MF2 | 1,567 | 1,090 | LF<MF2 | 1,542 | 1,057 | ||||||
| MF1>MF2 | 1,005 | 1.44×10−14 | 744 | 6.49×10−16 | MF1>MF2 | 1,331 | 2.76×10−07 | 1,036 | 5.99×10−09 | ||
| MF1<MF2 | 1,381 | 1,090 | MF1<MF2 | 1,078 | 787 | ||||||
| ZS11 | LF>MF1 | 2,264 | 2.62×10−09 | 1,711 | 8.95×10−12 | LF>MF1 | 2,281 | 6.94×10−14 | 1,707 | 3.17×10−14 | |
| LF<MF1 | 1,880 | 1,334 | LF<MF1 | 1,802 | 1,291 | ||||||
| LF>MF2 | 2,066 | 2.08×10−17 | 1,540 | 1.09×10−17 | LF>MF2 | 2,046 | 3.88×10−16 | 1,536 | 1.79×10−19 | ||
| LF<MF2 | 1,555 | 1,100 | LF<MF2 | 1,557 | 1,075 | ||||||
| MF1>MF2 | 1,268 | 1.77×10−01 | 939 | 3.88×10−01 | MF1>MF2 | 1,259 | 2.26×10−01 | 925 | 6.57×10−01 | ||
| MF1<MF2 | 1,200 | 901 | MF1<MF2 | 1,198 | 905 | ||||||
*: Full datasets including that of F5B, F5C, F8B, and F8C are listed in SI Appendix, Table S1.
**: Binomial test.
The mTEs are negatively associated with the expression dominance of β homoeologs. Given that TEs—and especially those TE sequences near genes that were methylated and/or targeted by 24nt siRNAs—were reported to suppress those genes’ expression and be associated with subgenome dominance (26, 38, 39, 42), we investigated the genomic methylation variations among β homoeologs from the three subgenomes in each parent. Reads from bisulfite sequencing were mapped to the genome to determine the methylation status of each cytosine (C) (Materials and Methods and SI Appendix, Table S4). According to the flanking nucleotides of C, these methylation loci were separated into the three contexts: CpG, CHG, and CHH (52). Among them, take the genome of B. rapa as a represent, CpG is the largest group, comprising 46.38% of all the methylated C contexts, while 22.36% and 31.26% were for CHG and CHH, respectively. The Cs of CpG also showed a much higher methylation ratio (46.99%) than those of CHG (16.75%) or CHH (4.88%) (SI Appendix, Table S5), as estimated by a weighted methylation algorithm (53). We then analyzed the methylation of TEs and compared them among the three subgenomes in the two parental genomes. The results showed that the TEs had a high methylation level in all the three subgenomes (84.16–93.34%) (SI Appendix, Table S6). TEs that had a methylation ratio of more than 30% (weighted methylation at Cs of CpG) were further considered as mTEs (SI Appendix, Fig. S1). After that, we investigated the relationships between the level of gene expression and the density of mTEs in regions within the gene body (translation start to stop) plus 5-kb sequences in each of 5´ upstream and 3´ downstream regions (referred to as sequences around genes hereafter) (Materials and Methods). We compared the ratio of mTE sequences between the β homoeologs. These paired competitions might be called “methylation greater or less comparison”, and we found that there were more LF genes that had a lower density of mTE sequences than that of their MFs homoeologs (SI Appendix, Table S7). Similar results were found when using stringent statistical tests (Materials and Methods and SI Appendix, Table S8). Moreover, we separated each pair of the β homoeologs, based on their expression status, into one homoeolog group of dominantly expressed genes and the other group of submissively expressed ones. Comparison between the two groups of homoeologs showed that the dominantly expressed group had a significantly lower ratio of mTE sequences per gene than that of the submissively expressed group (Fig. 2 and SI Appendix, Fig. S2). These results were consistently observed in both genomes originated from B. rapa and B. oleracea in all these progenies after the distant hybridization, supporting the inheritance of the negative relationships of mTEs and gene expression among β homoeologs that retained from preBrassica ancient hexaploidization event into the synthetic tetraploids.
Fig. 2.

The density of methylated TE (mTE) sequences in genes and their flanking regions is negatively associated with the expression dominance of genes. The density of mTEs on average in and around two groups of genes in subgenome rapa from the synthetic tetraploid of F2: the dominant expression group in subgenome LF compared with the recessive expression group in MFs (MF1 and MF2) (A), the recessive expression group in subgenome LF compared with the dominant expression group in MFs (B). A similar plotting in subgenome oleracea from the synthetic tetraploid F2 (C and D). The red “d” means dominantly expressed genes.
The density of mTEs is positively associated with the methylation level of non-TE sequences around genes. To further investigate the potential mechanism underlying the mTEs-induced gene suppression, we separated genes into two groups, with one group having no mTEs in sequences around genes and the other group has more than 10% sequences that are mTEs. Methylation levels were then calculated in these non-TE sequences around each gene and further averaged and compared between the two gene groups. As shown in SI Appendix, Fig. S3 A and B, genes with mTEs around them had a much higher methylation ratio as compared to genes without mTEs, and that is true for both genomes of B. rapa and B. oleracea. We further separated genes into 11 groups based on the density of mTEs and investigated the distribution of the methylation level of these non-TE sequences around genes. The result showed a clear positive relationship between the level of mTEs and the level of methylation on non-TE sequences around genes (SI Appendix, Fig. S3C). Furthermore, we compared the relationship between the methylation level of non-TE sequences and the genes’ expression, and found that it showed a clear negative association (SI Appendix, Fig. S4). These results were also true in the two subgenomes rapa and oleracea in the synthetic tetraploids (SI Appendix, Fig. S5). These findings indicate that the mTEs-induced gene suppression may function through the methylation of nearby sequences, i.e., more mTEs—always highly methylated (>84% all subgenomes) (SI Appendix, Table S5)—resulted in the higher methylation ratio of non-TE sequences around genes and are thus thought to suppress the expression of the nearby genes.
It is clear that leaf nuclei of our tetraploids are competent to expresses subgenome dominance once such dominance has been established, even when established tens of millions of years ago. Thus, the existence of subgenome dominance among the β homoeologs (the derivatives of the Brassica hexaploidy) provides an important control for our experiments on the rapa-oleracea (α) homoeologs. To repeat, the α homoeologs and the β homoeologs are being compared for expression and methylation in the same cells of the synthetic B. napus seedling leaves.
Parental TE Density Bias Exists for the New Tetraploid but Subgenome Dominance Did Not Result.
Genome of B. oleracea has higher TE densities than does the genome of B. rapa, these being the parents of the synthetic allotetraploid. Considering that TEs, especially mTEs, were found to be associated with subgenome dominance in a previous report for B. rapa (39) and also in the β subgenomes in our synthetic allotetraploid, we then compared TE contents systematically between the two parents B. rapa and B. oleracea (the α subgenomes in the synthetic allotetraploid) using their high-quality genome sequences. We found that there were 285,935 and 515,612 copies of TEs, corresponding to 153.87 Mb (43.57% of genome size) and 294.96 Mb (52.56%) sequences in genomes of B. rapa and B. oleracea, respectively, showing significantly heavier TE load for the genome of B. oleracea (P value < 2.2 × 10−16). We further investigated the ratio of TEs in the flanking sequences of genes in the two parental genomes (Materials and Methods). As shown in Fig. 3A, a higher average density of TE sequences (TE sequence/all sequence examined) was found to be distributed around B. oleracea genes as compared to B. rapa genes. Since the high methylation levels were imposed on almost all TEs (SI Appendix, Table S6), the mTEs showed a similar distribution pattern as that of all TEs.
Fig. 3.

The differential TE loads around genes and the comparison of homoeologous gene expression between rapa and oleracea. (A) The differential TE sequence density in the flanking regions of genes located on parental genomes B. rapa and B. oleracea, with B. oleracea showing clearly higher TE density in 5′ and 3′ flanking regions, while a bit lower TE density in the gene body (introns). (B) The number of dominantly expressed genes from the two parental subgenomes, in which syntenic paralogs (α homoeologs) were compared (greater or less or eightfold winners between α homoeologs).
The subgenome rapa that had lower load and density of TEs did not show subgenome dominance in the synthetic new allotetraploid B. napus. According to the hypothesis proposed in previous reports—including our own report—(26, 39), hypotheses based on strong associations but still associations only, the lower TE load in genome and lower TE density in the flanking regions of genes in B. rapa, as compared to B. oleracea, would predict that the rapa would dominate oleracea after the allotetraploidization. This prediction was falsified by the results of the following experiments. We identified 25,192 syntenic orthologs (α homoeologs in B. napus/newer syntenic paralogs in B. napus) between the two subgenomes rapa and oleracea (Materials and Methods) and compared the gene expression between these α homoeologs using the previously described methods, after removing genes that located at local genomic regions showing SVs in the synthetic tetraploids (37). These rapa:oleracea comparisons were performed for F1, F2, F5, and F8 (Fig. 3B and Table 2). Unexpectedly, we found that there are slightly more genes expressed to higher levels in oleracea, than their α homoeologs in rapa. For instance, there were 11,082 and 10,344 gene winners expressed higher in oleracea and rapa of F1, respectively. Among them, 545 expressed to more than eightfold more in oleracea than in rapa, while 425 expressed more than eightfold more in rapa than in oleracea (Table 2). This slight expression dominance in oleracea was also true for all the analyzed samples as F2, F5, and F8. We obtained the same results through statistical tests (SI Appendix, Table S9). These findings in the merged subgenomes rapa and oleracea showed a weak but stable dominance toward the higher TE load subgenome oleracea, rather than toward the subgenome with the lower TE load, subgenome rapa. A similar result was found using the public mRNA-seq data on other synthetic B. napus as aforementioned (SI Appendix, Table S10) (35).
Table 2.
The number of dominantly expressed genes of the α homoeologs between the two parental subgenomes rapa or oleracea in samples of F1, F2, F5, and F8
| Samples | Comparison* | Greater or less | P value** | Two folds | P value |
|---|---|---|---|---|---|
| F1 | Br>Bo | 10,344 | 4.77×10−07 | 3,597 | 2.09×10−16 |
| Br<Bo | 11,082 | 4,329 | |||
| F2 | Br>Bo | 10,246 | 1.61×10−17 | 3,815 | 4.51×10−32 |
| Br<Bo | 11,503 | 4,916 | |||
| F5A | Br>Bo | 6,291 | 5.63×10−81 | 2,477 | 2.61×10−96 |
| Br<Bo | 8,614 | 4,166 | |||
| F5B | Br>Bo | 8,509 | 2.18×10−43 | 3,488 | 1.05×10−58 |
| Br<Bo | 10,408 | 4,971 | |||
| F5C | Br>Bo | 7,895 | 1.40×10−06 | 3,110 | 8.82×10−15 |
| Br<Bo | 8,514 | 3,753 | |||
| F8A | Br>Bo | 8,436 | 2.03×10−43 | 3,701 | 2.51×10−58 |
| Br<Bo | 10,328 | 5,219 | |||
| F8B | Br>Bo | 7,712 | 3.78×10−16 | 3,536 | 2.80×10−22 |
| Br<Bo | 8,758 | 4,401 | |||
| F8C | Br>Bo | 8,173 | 2.74×10−09 | 3,884 | 2.52×10−13 |
| Br<Bo | 8,952 | 4,557 | |||
| ZS11 | Br>Bo | 9,817 | 3.80×10−56 | 5,413 | 8.51×10−48 |
| Br<Bo | 12,156 | 7,032 |
*: Br means rapa, Bo means oleracea.
**: Binomial test.
This result fails to support the previously hypothesized negative association relationships between subgenome dominance and TE load difference. These findings disconnect transposon densities near genes to subsequent subgenome dominance, and this casts doubt on the hypothesis that transposon density near genes in the parents somehow causes subgenome dominance following polyploidization, as will be discussed.
Sometime results are obtained that underscore just how much about comparative transposon patterning we do not know. Our transposon density comparisons between B. rapa and B. oleracea generated an unexpected result: Despite the much higher density of TEs/mTEs in the flanking regions of genes in B. oleracea than that in B. rapa, there were fewer TEs located in the gene body (introns) of B. oleracea than B. rapa, with 0.81 TEs per gene in B. rapa comparing to 0.61 TEs per gene in B. oleracea (Fig. 3A showing that the solid-lines of B. rapa in the gene body are higher than the corresponding dashed-lines of B. oleracea, which is reverse to that in the upstream and downstream regions). Fortunately, we have no way of knowing whether or not these differences in transposon patterning in the gene body, and the methylation consequences, has anything to do with subgenome dominance or lack thereof.
Overall Chromosomal DNA Methylation Levels Do Not Help Explain Subgenome Dominance in the Allotetraploids.
The overall methylation level differences between genomes B. rapa and B. oleracea and between subgenomes rapa and oleracea in the allotetraploid progenies were calculated, and we found a much higher methylation level in B. oleracea and oleracea than that in B. rapa and rapa, and this is true for all three contexts of methylation (SI Appendix, Table S5). For example, the genome-wide methylation ratio of CpG is 46.99% in B. rapa, while it is 69.70% in B. oleracea. The similar genome-wide methylation differences between the parental genomes and between the subgenomes in the progenies together indicated that the differential methylation pattern originated in the parental genomes, then inherited into the allotetraploid progenies.
The methylation level of genes in B. oleracea and oleracea was higher than that of B. rapa and rapa, respectively. We found that the average methylation level of CpG in the flanking regions of genes from parent genome B. oleracea is much higher than that from parent of B. rapa (Materials and Methods). For example, as shown in Fig. 4A, in the 2-kb region upstream of genes—protein-coding exons, the average methylation levels on non-TE sequences are ~35 and ~27% in B. oleracea and B. rapa, respectively. Similar to that of flanking sequences, both the exonic and intronic sequences of genes showed higher CpG methylation level in B. oleracea than those in B. rapa (Fig. 4A and SI Appendix, Table S11). Furthermore, we checked the methylation level of genes from the α subgenomes oleracea and rapa in the synthetic tetraploids. Both the flanking and gene body regions of oleracea showed a much higher methylation level than their homoeologs in rapa (Fig. 4B), which is consistent with the genome-wide methylation differences between the two parent genomes or parental subgenomes (SI Appendix, Tables S5 and S9). Additionally, the pattern found for CpG was also found for CHG and CHH contexts (SI Appendix, Tables S5 and S11). To summarize, B. oleracea and oleracea subgenomes are more highly methylated than B. rapa or rapa subgenomes. Take the CpG methylation for example, the difference is ~9 to ~22% (SI Appendix, Table S5). Most likely, these differences have nothing to do with subgenome dominance.
Fig. 4.
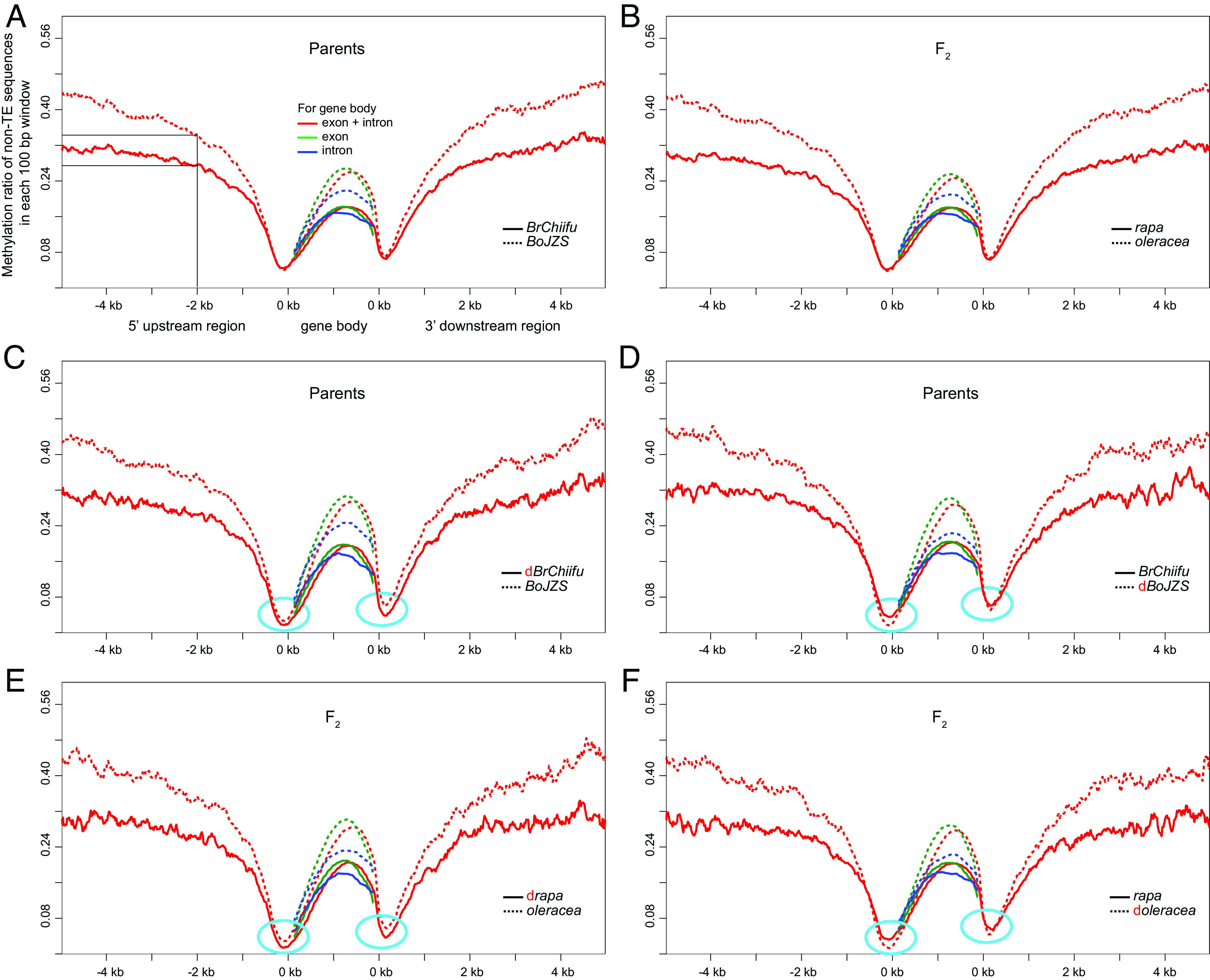
The methylation ratio on CpG loci of non-TE sequences in gene body (coding exons, introns) and their flanking regions. The CpG methylation ratio in average in and around all the syntenic orthologous genes between the two parents (A) and the α homoeologs between the two α subgenomes in the allotetraploid F2 (B). Syntenic orthologous α homoeologous gene pairs with one gene dominantly expressed over the other were further selected between the two parental genomes, then dominantly and recessively expressed orthologs/homoeologs were separated into two gene groups to be compared. (C and D) are the CpG methylation ratio in average in and around syntenic orthologs between the two parents, while (E and F) are the α homoeologs between the two parental subgenomes in F2. The red “d” denotes the dominantly expressed gene group. The small transcription start/stop regions of methylation levels that were negatively associated with the gene expression dominance between rapa and oleracea are highlighted in blue.
There was no negative relationship between levels of gene expression and methylation of sequences around genes in the α subgenomes of the synthetic allotetraploids. This result is now our new, expected result. Using a method similar to that used in our analysis of the β homoeologs, to separate and compare two groups of dominantly or recessively expressed α homoeologs, we found that the flanking sequences had a higher methylation level of CpG loci in gene groups from oleracea, regardless of whether or not they are the dominant or submissive gene group (Fig. 4 C–F and SI Appendix, Fig. S6). The results were consistent between the two parental subgenomes in F1, F2, F5, and F8. Both the CHG loci and CHH loci showed similar patterns. The findings indicate that the methylation variation in flanking sequences of genes is not associated with differential expression of α homoeologs generated from the synthetic Brassica allotetraploid. Said in another way, flanking methylation densities is not associated with expression differences between α homoeologs as BrLF vs. BoLF derived from the most recent neotetraploidy. This result is different from that of the β homoeologs retained from the ancient polyploidization of Brassica. These β homoeologs are exemplified by comparing BrLF and BoLF vs. BrMFs and BoMFs homoeologs, respectively. Intriguingly, the methylation level of the 100-bp sequences upstream and downstream of the translation start and stop regions do negatively correlate with the expression level of the paralogous gene groups (highlighted by light blue circles in Fig. 4 C–F), while the other parts of the gene body have no such relationship, as will be discussed.
DNA Methylation is Strongly Associated with TEs in the Parents and the Allotetraploids.
TEs from the parent B. oleracea showed a bit higher methylation level than that of B. rapa. Since TE loads showed a positive association with the levels of whole genome methylation and the methylation of non-TE sequences around genes, we further investigated the methylation variation of TEs in the parental genomes/subgenomes by calculating the average methylation level in TEs and their flanking sequences, using a method similar to those used in the analysis of genes (Materials and Methods). As shown in Fig. 5, the TE body, the element itself, is much more methylated than are its flanking sequences, as expected (38, 39). Moreover, TEs in the parent B. oleracea showed a bit stronger CpG methylation (~91%), by a factor of about ~4%, than that in B. rapa (~87%) (Fig. 5A). TE methylation difference between subgenomes oleracea and rapa in the synthetic progenies were also weighted toward the oleracea. Intriguingly, the difference of methylation level increased in the flanking sequences around TEs, and this increased methylation difference was not eliminated even that the distance to TEs extended to over 3 kb in both sides. For example, in the position of upstream 2 kb from the start of annotated TEs, the methylation of B. oleracea (~37%) is ~10% higher than that of the B. rapa (~27%), and the methylation difference is kept to the position of upstream/downstream 3 kb (Fig. 5A). This finding supports that TEs might suppress gene’s expression through the methylation of TEs themselves and non-TE sequences around genes (SI Appendix, Figs. S3 and S5). The similar result is also found for CHG and CHH contexts (Fig. 5 B and C). Additionally, the CHG methylation of TEs showed the strongest contrast between the parental genomes (Fig. 5B), and this contrast was largely reduced after the merging of the two genomes. For CHH methylation, the contrast between parental genomes was almost removed after their merging through hybridization and tetraploidization (Fig. 5C).
Fig. 5.

The methylation differences in TEs and their flanking regions between parental genomes and subgenomes in synthetic tetraploids. The methylation ratio average in and around TEs of the two parents and in the merged progenies F1, F2, F5, and F8 quantifying CpG loci (A), CHG loci (B), and the CHH loci (C).
The two parental genomes showed different TE methylation-induced gene suppression. Considering the importance of mTEs to both the whole genome and genes’ vicinity-region methylation, as well as the impact of nearby mTEs on gene expression, we compared the position effect of mTEs to gene expression differences between β homoeologs as well as between α homoeologs. The ratio of mTE nucleotides in sequences around genes was first calculated for each gene (Materials and Methods). When comparing the β homoeologs for each of the two parents’ genomes, we found that there were more genes in LF that had a lower density of mTE sequences around them, while more genes showed a higher density of mTE sequences in MFs (Fig. 6A). This difference between parents was inherited correspondingly in subgenomes rapa and oleracea into our synthetic allotetraploids (Fig. 6B). However, in comparison of these α homoeologs between rapa and oleracea, we found that α homoeologs from rapa had a much lower mTE density than that of their homoeologs from oleracea, regardless of whether the dominantly expressed genes are from subgenome rapa or oleracea (Fig. 6C). This result was further supported by the comparison of genes from rapa and oleracea that showed the same expression levels. We separated genes from rapa or oleracea of tetraploids based on their expression and compared the frequency distribution of the mTEs density of these genes under similar expression levels. Take F2 sample as example, Fig. 6D, genes from oleracea almost always had a much higher mTE density than genes with comparable expression from rapa in all these 11 expression levels analyzed. These results indicate that mTEs do show negative association with the expression of nearby β homoeologs in each of the two parents’ genomes even if the genes have different loads of mTEs (Fig. 6D). More importantly, genes in oleracea with much higher mTEs densities expresses about the same as those in rapa. This result will be discussed.
Fig. 6.

The difference in frequency distributions of methylated TE (mTE) sequences around genes. The frequency distribution of genes on the differential density of mTE sequences around genes (gene body plus 5-kb sequences in both 5′ upstream and 3′ downstream regions), syntenic paralogs among the three β subgenomes LF, MF1, and MF2 in B. rapa (A) and B. oleracea (B) were plotted separately. The frequency distribution of genes on the differential mTE density around genes, which are homoeologs between the α subgenomes rapa and oleracea (C); as well as all these genes that were separated by their expression levels, genes from subgenomes rapa and oleracea of F2 are plotted separately (D). The character “d” means the greater or less winners of gene expression between syntenic orthologs or homoeologs. TPM values estimate the level of gene expression.
Heritable Methylation Difference between Subgenomes.
The genome of B. napus cultivar Zhongshuang 11 (ZS) also showed higher methylation levels in subgenome Bo (B. oleracea subgenome in ZS) over subgenome Br (B. rapa subgenome in ZS). As aforementioned, parental genome methylation levels estimated using the leaf samples in B. oleracea is higher than in B. rapa and also higher in the synthetic allotetraploid lineage; this is true for eight inbreeding generations. The oilseed crop B. napus was naturally allotetraploidized, B. rapa + B. oleracea, five to ten thousand years ago (44). Being an annual plant, that implies tens of thousands of generations of divergence time. Do our results on a synthetic napus apply to the napus crop? We choose B. napus cultivar ZS as a material to investigate the subgenome features of naturally domesticated B. napus (54, 55). RNA-seq and bisulfite sequencing assays were then performed using ZS using the same Materials and Methods used in the work reported already. We found in this crop that the β subgenome LF kept dominance over MFs (Table 1). More importantly, there were a few more genes in subgenome Bo expressed to higher levels than their homoeologs in subgenome Br (Table 2), indicating the weak dominant role of Bo. Similar results were also obtained when using 12 public mRNA-seq datasets of natural B. napus in four previous studies (SI Appendix, Tables S3 and S10) (44, 54–56). Moreover, the higher methylation level of TEs and genes (SI Appendix, Table S11) and the whole genome sequences were also found in Bo than in Br (SI Appendix, Table S5). These results further supported the findings that the parent with the higher methylation level and TE density is not sufficient to identify the submissive subgenome and is not likely to be the “cause” of subgenome dominance.
Discussion
Subgenome dominance is one of the most distinct features associated with allopolyploidization (16, 17, 24). Natural paleo- or meso-polyploidizations occurred such a long time ago that it is impossible to reveal unequivocally how this subgenome dominance originated. Fortunately, we can observe this phenomenon in the current genomes recently or currently evolved on a background of ancient polyploidizations. Many studies investigated subgenome dominance using synthetic polyploid systems (28–30), and we have done the same. In this study, we systematically analyzed the expression and methylation variations of parental genomes (subgenomes) merged in synthetic allotetraploids and their progeny and found that the differences in TE load and/or methylation level near genes were not negatively associated with subgenome dominance. Since our allotetraploid is between different species of Brassica, our system is clearly “allo”. Thus, our results are contrary to previous suggestions, including our own (39). Differences in TE load and methylation between subgenomes are not sufficient to initiate or predict subgenome dominance.
Previous studies (26, 39–41) tried to address a classic chicken and egg problem: whether mTE load difference drives the formation of subgenome dominance, or—the other way around—does subgenome dominance cause the biased accumulation of mTEs near genes differently between subgenomes that are actually different for some other reason. Studies using the monkeyflower- and the Cucumis-based synthesized neopolyploidization genomes found subgenome dominance appeared immediately after the merging of two parental genomes that differed in mTE density near genes (29, 30), so the prevailing hypothesis fit these data well. It is not true for our synthetic Brassica polyploidy system.
We did not stop our analyses at the resynthesized B. rapa and B. oleracea hybrid and allotetraploid. Perhaps, as reviewed in the introductory paragraphs, subgenome dominance may need to set in over the generations or perhaps mTE densities near genes change over the generations. To address these possibilities, we generated self-cross progenies from F1 to F8, providing a good chance to systematically analyze the dynamic patterns of genome-wide expression and methylation and to compare them between subgenomes over a longer period. Subgenome dominance between the homoeologs of the two subgenomes never manifested. We conclude that differences in the TE densities near genes in homoeologous pairs are not sufficient to cause subgenome dominance. Our study included an important internal control. We observed subgenome dominance among the β subgenomes and also within each of the two parental genomes B. rapa and B. oleracea and in the subgenomes of our resynthesized allotetraploid. In other words, the α homoeologs—the most recent tetraploidy—show no subgenome dominance or even slight dominance toward the parental genome with significantly higher mTEs near genes, but, in the same cells, the more ancient β homoeologs do show subgenome dominance; gene expression is negatively associated with nearby mTEs, just as expected. It was formally possible that our resynthesized allopolyploid somehow lost the ability to express subgenome dominance. That explanation cannot be true because the β homoeologs express subgenome dominance as expected. We therefore conclude that allopolyploidization in our system simply did not initiate subgenome dominance, and progeny did not initiate subgenome dominance either because of some characteristic specific to the α duplication. This characteristic could be that the two subgenomes rapa and oleracea were regulated by independent systems of gene expression and genome methylation, even after tens of thousands years of co-evolution in natural B. napus. In a previous study, researchers found that chromosomes from different subgenomes in neo-allotetraploid cotton genome did not locate themselves randomly (57). Chromosomes that came from the same parent clustered together, with two subgenomes coordinated and located at different positions during meiosis (57). It supports the hypothesis that the two subgenomes may function independently, though they merged in one nucleus in the synthetic and natural tetraploids, and further suggests that subgenomes of neo-allopolyploids might exist as a “genome chimera”. The independent regulations of gene expression in genome chimera might be broken by a rediploidization process through genomic re-arrangements, and then the subgenome dominance might occur as what we observed among subgenomes of ancient polyploidization events (26, 39).
Our results suggest that some other “compatibility” or “interference” mechanism exists, not mTE density near genes, and is first realized in the wide-cross hybrid. This “mystery mechanism” is not known. However, speculations are always possible. There could be or have been species-level differences in the transcription efficiency that might counteract mTE transcriptional suppression. It is reasonable to think that genes may have evolved different efficiencies of transcription, or evolved new genome-wide increases in the expression level, in order to maintain basic biological activity to compensate for the bulk mTE-induced suppression, especially in those plant genomes with extremely heavy TE loads. This idea was supported by a previous study on the comparison of TEs and nearby gene expression between two genomes of Arabidopsis thaliana and Arabidopsis lyrata (58), though they have not investigated it in a synthetic tetraploid between A. thaliana and A. lyrata. Tests of this and other hypothetical mystery mechanisms that actually initiate subgenome dominance immediately, in the wide hybrid, must be left to the future.
Methylation differences between subgenomes happen even in the absence of subgenome dominance. Our data on the gene regions in our synthetic allotetraploid showed that when genes had mTEs in their flanking regions, then the methylation of the non-TE sequences around these genes becomes highly methylated. They were methylated to much higher levels than were gene regions that had no mTEs around them (SI Appendix, Figs. S3 and S5), suggesting that TEs may suppress their nearby genes even if several kb away. Methylation difference around TEs can be extended to over 3 kb away from TEs in both directions (Fig. 5). Comparing genes in our two different genomes and subgenomes, we found fewer TE insertions in the gene body (introns) of B. oleracea than of B. rapa, which is the opposite result to TE insertions in the flanking regions of these same genes (the 5′ upstream and 3′ downstream regions). It’s possible that the two genomes reflect different evolved strategies to cope with the impact of the TE insertions. As there are less TEs genome-wide in B. rapa than there are in B. oleracea, B. rapa may tolerate more TE insertions. However, considering that there were many more TEs insertions and higher methylation levels in the flanking regions of genes in B. oleracea genome, this genome should be under stronger selection against the TE insertions to its gene bodies than should B. rapa.
Additionally, we observed that the methylation level of the ~100-bp sequences around the gene translation start and stop regions showed negative associations to the expression level of genes chosen to represent the different α homoeologs (Fig. 3 C–F). This negative correlation could be explained if these sequences in the highly expressed genes are often kept in open chromatin status; open chromatin is generally less methylated than closed. In other words, the genes’ expression levels may not be regulated directly by the methylation levels of these regions (the ~100-bp sequences around the translation start/stop regions), but, rather, the methylation variations of these sequences near genes might be the results of differences to the genes’ expression activities.
Materials and Methods
Plant Materials and Sample Collection.
We selected B. rapa accession Chiifu (Chiifu-401-42) and B. oleracea accession JZS, whose high-quality genomes have been released (49, 50), as the diploid parents for our wide hybridization. After crossing the two parents, the hybrid immature embryo was rescued by culturing it on the nutrient medium to prevent the degradation caused by the reproductive barrier (59). The apical meristem of the F1 hybrid seedlings was further treated with 0.2% colchicine to synthesize the amphidiploids (F2). Sequential self-pollinations were performed to obtain the F3–F8 generations. For each generation, the seeds of individual plants were collected separately and were used for planting and next round self-crossing. The ~10 cm leaf organs were harvested from the eight-leaf stage seedlings of the diploid parents, hybrid F1, amphidiploid F2, the F5, and F8 progenies. Specifically, three randomly selected individual plants from each generation of F5 and F8 were used as biological replicates. All of the collected leaf samples were cut in half, flash-frozen in liquid nitrogen, and separately stored at −80 °C, one half for RNA sequencing and the other half for bisulfite sequencing.
Identification of Orthologous and Paralogous Genes.
The genome sequences of B. rapa Chiifu (v3) and B. oleracea JZS (v2), as well as corresponding gene annotations (v3.1 for B. rapa and v2 for B. oleracea) (49, 50) were downloaded from BRAD database (http://brassicadb.cn/#/Download/). The syntenic orthologous genes between B. rapa and B. oleracea genomes were identified using SynOrths (60) with default parameters (-m 20 -n 100 -r 0.2). These syntenic gene pairs were also considered as the syntenic paralogous gene pairs (rapa vs. oleracea) in the newly synthesized allotetraploid. For comparisons of paralogous genes generated by the common ancient hexaploidization, genes located in the subgenomes LF, MF1, and MF2 in B. rapa and B. oleracea were obtained based on a previous study (61). B. napus genome assembly and annotations were from variety Zhongshuang 11 (ZS); these were obtained from http://ocri-genomics.org/Brassia_napus_genome_ZS11/. The syntenic paralogous genes (α homoeologs) between rapa and oleracea subgenomes can be downloaded at http://www.bioinformaticslab.cn/files/subgenome_dominance/.
RNA Extraction, Sequencing, and Gene Expression Analysis.
The mRNA of leaf samples was extracted using the Dynabeads mRNA DIRECT Kit (Invitrogen), and the library was then constructed using the VAHTS mRNA-seq v2 Library Prep Kit for Illumina. The Illumina NovaSeq platform was used to sequence the libraries. The quality of the resultant 150-bp paired-end RNA-seq reads was assessed by FastQC (available at https://qubeshub.org/resources/fastqc), and low-quality bases/reads were trimmed or filtered out using Trimmomatic (62) with default parameters (ILLUMINACLIP:adapter:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36). For the RNA-seq data derived from the parent plants (Chiifu and JZS), respective reference genomes (B. rapa and B. oleracea) were used directly as targets for read alignment, whereas for the RNA-seq data derived from the progenies, the individual genomes of Chiifu and JZS were merged as the reference genome (B. rapa + B. oleracea). The ZS genome was used as reference genome for RNA-seq data of ZS. RNA-seq fragments were aligned to the reference genomes using Hisat2 (63) with parameters “--max-intronlen 20000 -k 5”, and the fragment counts for each annotated gene were calculated by FeatureCounts (64) with parameters “-p -D 1000 -Q 5”. We evaluated the effect of possible cross-mapping reads between genomes of B. rapa and B. oleracea on the quantification of gene expression in our synthetic tetraploids. In detail, fragments with multiple alignments locating in two parents’ genomes (B. rapa and B. oleracea) were extracted from the alignment files. These extracted fragments were then assigned to B. rapa genome with other alignments in B. oleracea masked, and the resultant fragment-assigned SAM files were subjected to FeatureCounts to determine whether these fragments were able to be mapped onto a certain gene. Only those successfully mapped to genes were retained. We also performed the same analyses on the B. oleracea alignments with B. rapa alignments masked. Based on these data, if one fragment was mapped to a syntenic gene pair between B. rapa and B. oleracea in the two quantification processes, it was considered as a cross-mapping fragment. Finally, less than 1% (0.63–0.82%) of the aligned fragments were found to be identified as cross-mapping ones, which did not interfere with our quantification of gene expression. For these limited number of cross-mapped fragments, we assigned them equally to genes in each syntenic pair. The additional fragment numbers were then added to the unique fragment numbers of each gene to calculate the TPM (transcripts per million) value.
Comparison of Expression Levels of Gene Pairs.
Genes were sorted according to their expression levels evaluated by TPM. To determine the dominantly expressed homoeolog for a given gene pair (homoeolog pairs), their TPM values were compared: Greater or less was recorded, and the relative expression fold—dominant over submissive—was calculated. We also used the non-parametric statistical test of the Mann-Whitney-U test (65) in the Python package SciPy to identify the significance of differentially expressed gene pairs from a subset of genes. Gene pairs with P value < 0.05 were considered significantly differentially expressed.
Whole-Genome Bisulfite Sequencing and Data Analysis.
Genomic DNA of collected leaf samples was extracted using the New Plant DNA Kit (TIANGEN). During the construction of sequencing libraries, the DNA was treated with sodium bisulfite using the EZ DNA Methylation-GoldTM Kit to convert the unmethylated cytosines to uracil, while the methylated cytosines stayed unchanged. The bisulfite-converted libraries were sequenced on Illumina NovaSeq 6000 platform at Berry Genomics (Beijing), and 150-bp paired-end reads were generated. The strategy for choosing the reference genome for different materials is the same as the one described in the RNA-seq analysis methods. The Bismark tool (66) was used for processing the bisulfite sequencing data with default parameters (--bowtie2 --score-min L,0,-0.2 --no-discordant --maxins 500 --dovetail --no-mixed --ignore-quals). Specifically, both the sequencing reads and the genomes were transformed into bisulfite-converted versions before read aligning. To estimate the sodium bisulfite non-conversion rate, the lambda genome (NC_001416 in GenBank) was included in the reference genome as a control. Bowtie embedded in Bismark was responsible for aligning the reads onto genomes, and only the uniquely mapped reads were retained for downstream analyses (Supplementary SI Appendix, Table S4). The functions “deduplicate_bismark” and “bismark_methylation_extractor” in Bismark were used to remove the PCR duplicates and to identify the methylated loci, respectively. The MethylExtract tool (67) was used to calculate the conversion rate using the reads aligned to the lambda sequences with parameters “flagW=99,147 flagC=83,163.” Due to the high conversion rate estimated in our samples (>99.5%), the methylation results from Bismark were directly used for downstream analyses. Considering that both the CpG and CHG are symmetric methylations, the read counts (both the methylated and unmethylated reads) of adjacent cytosine and guanine in CpG and CHG loci were merged. The weighted methylation level of a given region was calculated—the summed number of reads supporting the methylated cytosine loci was divided by the summed number of all reads mapped in this region—following the method reported previously (53). To compare the methylation levels between the synthetic tetraploid genome and B. napus ZS, the methylation data of ZS were also aligned to the merged genome of B. rapa and B. oleracea, and similar analyses were conducted to determine the DNA methylation level.
Determination of Repeat Content Around Genes.
The repeat annotation of B. rapa and B. oleracea were obtained using tools of RepeatModuler and RepeatMasker (version 4.0.3) (68). The gff files of repeat annotation are freely available through http://www.bioinformaticslab.cn/files/subgenome_dominance/repeatsGff/. The ratio of repeat DNA around genes was calculated following the method described in our previous study (40). Specifically, upstream and downstream 5-kb sequences around the genes (translation start to stop) were defined as the flanking sequences. Flanking sequences were hard-masked as “N” when they started to overlap with the coding regions of adjacent genes. A 100-bp sliding window with step set as 10 bp was used to screen these flanking sequences to determine the ratio of TE sequences in each window. While for the gene body regions, whole gene body/whole exons/whole introns of each gene (transcript with the longest coding sequences was chosen as the “representative” gene model) were separated into 20 bins with an equal length, then submitted to the window screening and calculation. For a given gene set of interest, the resulting ratio for each window or bin was averaged in these genes. The custom scripts were freely available through http://www.bioinformaticslab.cn/files/subgenome_dominance/code/.
Determination of DNA Methylation Level Around Genes and TEs.
The method used to calculate the average DNA methylation level in the flanking regions of genes, and TEs is similar to that used to calculate the repeat content in the flanking regions of genes as described above. The weighted methylation level (53) was determined in each 100-bp sliding window. DNA methylation levels around the TEs were also calculated in the same way, except for the TE body, which was divided into ten equal-length bins and subjected to DNA methylation calculation.
Comparison of mTE Densities between Gene Pairs.
TEs with a weighted methylation ratio >30% were considered as mTEs. The densities of mTEs in sequences around genes were compared between gene pairs using a similar principle as in the comparison of expression levels as described above. Specifically, the Cochran–Mantel–Haenszel test (69) in R package mantelhaen was used to identify gene pairs with statistically different mTE densities (P value < 0.05 as cutoff).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC grants 31972411, 31722048, and 31630068), Central Public-interest Scientific Institution Basal Research Fund (No. Y2022PT23), Innovation Program of the Chinese Academy of Agricultural Sciences, and Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture and Rural Affairs, China. M.F. was supported by NIFA, U.S. Department of Agriculture via UC-Berkeley.
Author contributions
F.C. and X.W. designed research; Y.C., Y.Y., J.W., J.L., X.L., X.Z., Y.Z., Z.G., Lei Zhang, and S.C. performed research; F.C., K.Z., Lingkui Zhang, and J.R. analyzed data; and F.C., M.F., and K.Z. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Michael Freeling, Email: freeling@berkeley.edu.
Xiaowu Wang, Email: wangxiaowu@caas.cn.
Feng Cheng, Email: chengfeng@caas.cn.
Data, Materials, and Software Availability
All sequence data generated in this study have been deposited at the BIG data (https://ngdc.cncb.ac.cn/gsub/) with BioProject accession number PRJCA012535 (70). The files of syntenic gene pairs and TE annotations, as well as custom scripts used in this work are freely available through the website (https://www.bioinformaticslab.cn/files/subgenome_dominance/) (71). All other data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Van de Peer Y., Mizrachi E., Marchal K., The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Soltis P. S., Marchant D. B., Van de Peer Y., Soltis D. E., Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 35, 119–125 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Ruprecht C., et al. , Revisiting ancestral polyploidy in plants. Sci. Adv. 3, e1603195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soltis D. E., Visger C. J., Soltis P. S., The polyploidy revolution then.and now: Stebbins revisited. Am. J. Bot. 101, 1057–1078 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Soltis D. E., Visger C. J., Marchant D. B., Soltis P. S., Polyploidy: Pitfalls and paths to a paradigm. Am. J. Bot. 103, 1146 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Zhuang W., et al. , The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 51, 865–876 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wendel J. F., The wondrous cycles of polyploidy in plants. Am. J. Bot. 102, 1753–1756 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Schranz M. E., Mohammadin S., Edger P. P., Ancient whole genome duplications, novelty and diversification: The WGD radiation lag-time model. Curr. Opin. Plant Biol. 15, 147–153 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Salmanminkov A., Sabath N., Mayrose I., Whole-genome duplication as a key factor in crop domestication. Nat. Plants 2, 16115 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Pophaly S. D., Tellier A., Population level purifying selection and gene expression shape subgenome evolution in maize. Mol. Biol. Evol. 32, 3226–3235 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Hughes T. E., Langdale J. A., Kelly S., The impact of widespread regulatory neofunctionalization on homeolog gene evolution following whole-genome duplication in maize. Genome Res. 24, 1348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanneste K., Baele G., Maere S., Van de Peer Y., Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous-Paleogene boundary. Genome Res. 24, 1334–1347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng F., et al. , Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 4, 258–268 (2018). [DOI] [PubMed] [Google Scholar]
- 14.One Thousand Plant Transcriptomes Initiative, One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garsmeur O., et al. , Two evolutionarily distinct classes of paleopolyploidy. Mol. Biol. Evol. 31, 448–454 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Steige K. A., Slotte T., Genomic legacies of the progenitors and the evolutionary consequences of allopolyploidy. Curr. Opin. Plant Biol. 30, 88–93 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Schnable J. C., Springer N. M., Freeling M., Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. U.S.A. 108, 4069–4074 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glover N. M., Redestig H., Dessimoz C., Homoeologs: What are they and how do we infer them? Trends Plant Sci. 21, 609–621 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bird K. A., VanBuren R., Puzey J. R., Edger P. P., The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytol. 220, 87–93 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Wang J., et al. , Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172, 507–517 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodhouse M. R., et al. , Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol. 8, e1000409 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renny-Byfield S., Gong L., Gallagher J. P., Wendel J. F., Persistence of subgenomes in paleopolyploid cotton after 60 my of evolution. Mol. Biol. Evol. 32, 1063–1071 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Cheng F., et al. , Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS ONE 7, e36442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murat F., et al. , Shared subgenome dominance following polyploidization explains grass genome evolutionary plasticity from a seven protochromosome ancestor with 16K protogenes. Genome Biol. Evol. 6, 12–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.VanBuren R., et al. , Exceptional subgenome stability and functional divergence in the allotetraploid Ethiopian cereal teff. Nat. Commun. 11, 884 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renny-Byfield S., Rodgers-Melnick E., Ross-Ibarra J., Gene fractionation and function in the ancient subgenomes of maize. Mol. Biol. Evol. 34, 1825–1832 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Wang M., et al. , Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 49, 579–587 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Yoo M. J., Szadkowski E., Wendel J. F., Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity (Edinb) 110, 171–180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu X., et al. , Whole-genome sequence of synthesized allopolyploids in cucumis reveals insights into the genome evolution of allopolyploidization. Adv. Sci. (Weinh) 8, 2004222 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edger P. P., et al. , Subgenome dominance in an interspecific hybrid, synthetic allopolyploid, and a 140-year-old naturally established neo-allopolyploid monkeyflower. Plant Cell 29, 2150–2167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaeta R. T., Pires J. C., Iniguez-Luy F., Leon E., Osborn T. C., Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 19, 3403–3417 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong Z., Gaeta R. T., Pires J. C., Homoeologous shuffling and chromosome compensation maintain genome balance in resynthesized allopolyploid Brassica napus. Proc. Natl. Acad. Sci. U.S.A. 108, 7908–7913 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferreira de Carvalho J., et al. , Untangling structural factors driving genome stabilization in nascent Brassica napus allopolyploids. New Phytol. 230, 2072–2084 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Hurgobin B., et al. , Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 16, 1265–1274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bird K. A., et al. , Replaying the evolutionary tape to investigate subgenome dominance in allopolyploid Brassica napus. New Phytol. 230, 354–371 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mason A. S., Wendel J. F., Homoeologous exchanges, segmental allopolyploidy, and polyploid genome evolution. Front. Genet. 11, 1014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang K., et al. , Variations in homoeologous dosage and epigenomics mark the early evolution of synthetic Brassica tetraploids. bioRxiv [Preprint] (2023). 10.1101/2023.06.27.543697 (Accessed 27 June 2023). [DOI]
- 38.Hollister J. D., Gaut B. S., Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 19, 1419–1428 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woodhouse M. R., et al. , Origin, inheritance, and gene regulatory consequences of genome dominance in polyploids. Proc. Natl. Acad. Sci. U.S.A. 111, 5283–5288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng F., et al. , Epigenetic regulation of subgenome dominance following whole genome triplication in Brassica rapa. New Phytol. 211, 288–299 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Xu W., et al. , The genome evolution and low-phosphorus adaptation in white lupin. Nat. Commun. 11, 1069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J., et al. , Autotetraploid rice methylome analysis reveals methylation variation of transposable elements and their effects on gene expression. Proc. Natl. Acad. Sci. U.S.A. 112, E7022–E7029 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X., et al. , The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Chalhoub B., et al. , Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Yang J., et al. , The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48, 1225–1232 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Song X., et al. , Brassica carinata genome characterization clarifies U’s triangle model of evolution and polyploidy in Brassica. Plant Physiol. 186, 388–406 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dönmez A. A., Aydın Z. U., Wang X., Wild Brassica and its close relatives in Turkey. Hortic. Plant J. 7, 97–107 (2021). [Google Scholar]
- 48.Zhao M., et al. , Shifts in the evolutionary rate and intensity of purifying selection between two Brassica genomes revealed by analyses of orthologous transposons and relics of a whole genome triplication. Plant J. 76, 211–222 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Zhang L., et al. , Improved Brassica rapa reference genome by single-molecule sequencing and chromosome conformation capture technologies. Hortic. Res. 5, 50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai X., et al. , Improved Brassica oleracea JZS assembly reveals significant changing of LTR-RT dynamics in different morphotypes. Theor. Appl. Genet. 133, 3187–3199 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Parkin I. A., et al. , Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15, R77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H., Lang Z., Zhu J. K., Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Schultz M. D., Schmitz R. J., Ecker J. R., “Leveling” the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 28, 583–585 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun F., et al. , The high-quality genome of Brassica napus cultivar “ZS11” reveals the introgression history in semi-winter morphotype. Plant J. 92, 452–468 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Chen X., et al. , A high-quality Brassica napus genome reveals expansion of transposable elements, subgenome evolution and disease resistance. Plant Biotechnol. J. 19, 615–630 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Q., et al. , Asymmetric epigenome maps of subgenomes reveal imbalanced transcription and distinct evolutionary trends in Brassica napus. Mol. Plant 14, 604–619 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Han J., et al. , A and D genomes spatial separation at somatic metaphase in tetraploid cotton: Evidence for genomic disposition in a polyploid plant. Plant J. 84, 1167–1177 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Hollister J. D., et al. , Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. U.S.A. 108, 2322–2327 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wen J., et al. , Improving ovary and embryo culture techniques for efficient resynthesis of Brassica napus from reciprocal crosses between yellow-seeded diploids B. rapa and B. oleracea. Euphytica 162, 81–89 (2008). [Google Scholar]
- 60.Cheng F., Wu J., Fang L., Wang X., Syntenic gene analysis between Brassica rapa and other Brassicaceae species. Front. Plant Sci. 3, 198 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng F., et al. , Deciphering the diploid ancestral genome of the Mesohexaploid Brassica rapa. Plant Cell 25, 1541–1554 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim D., Paggi J. M., Park C., Bennett C., Salzberg S. L., Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Vengatesan K., et al. , Performance analysis of gene expression data using Mann-Whitney U test. Lect. Notes Electr. Eng. 442, 701–709 (2018). [Google Scholar]
- 66.Krueger F., Andrews S. R., Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barturen G., Rueda A., Oliver J. L., Hackenberg M., MethylExtract: High-Quality methylation maps and SNV calling from whole genome bisulfite sequencing data. F1000Res 2, 217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tarailo-Graovac M., Chen N., “Chapter 4, Unit 4: Using RepeatMasker to identify repetitive elements in genomic sequences” in Current Protocols in Bioinformatics (Wiley, 2009), p. 10. [DOI] [PubMed] [Google Scholar]
- 69.Lu K., Multiple imputation score tests and an application to Cochran–Mantel–Haenszel statistics. Stat. Med. 39, 4025–4036 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Zhang K., et al. , Data for “The lack of negative association between TE load and subgenome dominance in synthesized Brassica allotetraploids” BIG data. https://ngdc.cncb.ac.cn/gsub/. Deposited 9 September 2023. [DOI] [PMC free article] [PubMed]
- 71.Zhang K., et al. , Data for “The lack of negative association between TE load and subgenome dominance in synthesized Brassica allotetraploids” Bioinformaticslab. http://www.bioinformaticslab.cn/files/subgenome_dominance/. Deposited 9 September 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All sequence data generated in this study have been deposited at the BIG data (https://ngdc.cncb.ac.cn/gsub/) with BioProject accession number PRJCA012535 (70). The files of syntenic gene pairs and TE annotations, as well as custom scripts used in this work are freely available through the website (https://www.bioinformaticslab.cn/files/subgenome_dominance/) (71). All other data are included in the article and/or SI Appendix.