

Abstract
Regulating macrophage activation precisely is crucial in treating chronic inflammation in osteoarthritis (OA). However, the stable pro-inflammatory state and deep distribution of macrophages in vivo pose a great challenge to treatment. In this study, inspired by the innate immune, immune cell mobilized hydrogel microspheres were constructed by microfluidic methods and load chemokines, macrophage antibodies and engineered cell membrane vesicles (sEVs) via covalent and non-covalent junctions. The immune cell mobilized hydrogel microspheres, based on a mixture of streptavidin grafted hyaluronic acid methacrylate (HAMA-SA) and Chondroitin sulfate methacrylate (ChSMA) microspheres (HCM), can recruit, capture and reprogram proinflammatory macrophages in the joint cavity to improve the joint inflammatory microenvironment. In vitro experiments demonstrated that immune cell mobilized hydrogel microspheres had excellent macrophage recruitment, capture, and reprogramming abilities. Pro-inflammatory macrophages can be transformed into anti-inflammatory macrophages with an efficiency of 88.5 %. Animal experiments also revealed significant reduction in synovial inflammation and cartilage matrix degradation of OA. Therefore, the immune cell mobilized hydrogel microspheres may be an effective treatment of OA inflammation for the future.
Keywords: Engineered cell membrane vesicles, Hydrogel microspheres, Macrophage reprogram, Osteoarthritis
Graphical abstract
Inspired by the innate immune, hydrogel microspheres were constructed for mobilizing immune cell, which show excellent macrophage recruitment, capture, and reprogramming abilities. Pro-inflammatory macrophages can be transformed into anti-inflammatory macrophages with an efficiency of 88.5 %. Animal experiments also revealed significant reduction in synovial inflammation and cartilage matrix degradation of osteoarthritis via regulating mitochondrial function.
Highlights
-
•
Inflammatory cell recruitment offers a new approach to intraarticular drug delivery.
-
•
The immune cell mobilized hydrogel microspheres provide macrophage recruitment, capture, and reprogramming abilities.
-
•
Engineered cell membrane vesicles (sEVs) enhance macrophage reprogramming by repairing mitochondrial dysfunction.
1. Introduction
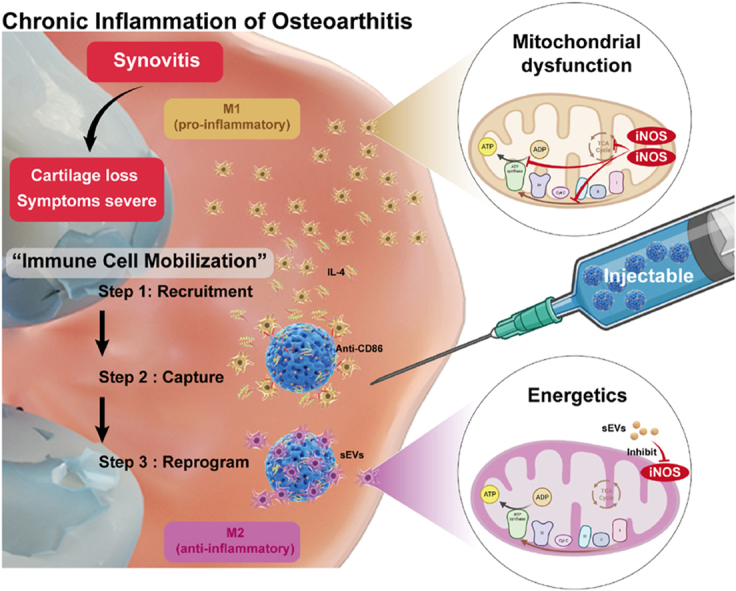
Osteoarthritis (OA) is a progressive joint disease affecting the entire joint [1,2]. Regrettably, chronic inflammation stands as a pivotal contributory element in the exacerbation of symptoms pertaining to OA, while the existing dearth of efficacious therapeutic interventions aimed at mitigating these symptoms persists [[3], [4], [5]]. Macrophages, which are the most abundant immune cells in the synovial membrane, play an essential role in controlling joint inflammation [6]. Macrophages that are activated can be categorized as either pro-inflammatory or anti-inflammatory state. Reprogramming pro-inflammatory into anti-inflammatory macrophages is crucial for treating synovial inflammation in OA [7,8]. Nevertheless, reprogramming pro-inflammatory macrophages into anti-inflammatory macrophages in vivo is not easy [9], due to the macrophages are distributed in various tissues within the joint - such as pro-inflammatory macrophages in deep synovial tissue and ligaments - which are difficult to reach an effective therapeutic concentration [10,11]. Especially after OA occurs, the difficulty of treating proinflammatory macrophages distributed in the deep synovial layer is further increased [12]. In cases of OA, the synovial membrane, composed of the lining layer and the sub-lining layer, undergoes a loss of integrity and leading to uneven fibrous proliferation [13]. Consequently, the challenge of drug penetration is significantly magnified [14]. Simultaneously, the sub-lining layer is accompanied by increased capillary permeability, which accelerates the excretion of drugs and reduces the duration of anti-inflammatory drugs in deep synovial tissue [15]. Therefore, the lack of a targeted treatment towards pro-inflammatory macrophage within the deep synovial layer remian a significant concern of utmost importance.
Drawing inspiration from the innate immune process, immune cell mobilization can recruit immune cells from bone marrow, spleen, and other immune organs into the bloodstream to respond to foreign pathogens [16,17]. Therefore, it makes more sense to recruit pro-inflammatory cells to the target site in a sequential manner than to simply drug disordered penetration. This method of targeting and centralizing treatment has been reported in some tumor studies, where the effect also suggests advantages in reducing drug diffusion loss and increasing lesion clearance range [[18], [19], [20]]. However, its effectiveness is unknown in the treatment of OA. The key to implementing immune cell mobilization strategy is the ability to recruit, capture, and reprogram pro-inflammatory macrophages into anti-inflammatory state. Macrophages possess strong chemotactic capabilities and can migrate to distant inflammatory sites by amoeboid movement under the action of chemokines [21,22]. Numerous drugs with macrophage chemotactic capabilities, such as interleukin4 (IL4), are currently undergoing preclinical experiments and have high biological safety, making them applicable to joint treatment [23]. Micron-sized hydrogel microspheres (HMs) can move freely in the joint cavity and do not diffuse out of the joint with the capillaries, making them ideal carriers for recruitment and capture [[24], [25], [26]]. Previous studies have used hydrogel microspheres loaded with cytokines to recruit and activate stem cells in joints [27]. Furthermore, surface modifications of hydrogel microspheres can enable them to load antibodies as “catchers” of pro-inflammatory macrophages. Therefore, the cell recruitment and capture of the immune cell mobilized hydrogel microsphere is theoretically feasible.
To achieve ideal macrophage reprogramming, it is essential not only anti-inflammatory induction but also the restoration of normal mitochondrial energy metabolism. The mitochondrial dysfunction induced by pro-inflammatory macrophage-derived iNOS significantly impairs the reprogramming plasticity [10,28]. Engineered cell membrane-derived EVs (mEVs) that deliver drugs to regulate organelle function are an attractive option due to their high biocompatibility and unique internal transport pathway, which greatly reduces cell toxicity and improves drug delivery efficiency within cells [[29], [30], [31], [32], [33]]. Jiang et al. reported that engineered macrophage membrane vesicles loaded with antioxidant nanoparticles successfully cleared mitochondrial reactive oxygen species, restoring the plasticity of pro-inflammatory macrophage reprogramming [34]. However, immunogenicity of macrophage membrane and nanoparticle degradation are potential negative factors. Synovial mesenchymal stem cells (SMSCs), as endogenous cells within the joint, have lower immunogenicity and have close membrane protein communication with macrophages [[35], [36], [37]]. Therefore, using engineered extracellular vesicles derived from SMSCs membranes (sEVs) to deliver soluble antioxidant drugs to regulate pro-inflammatory macrophage reprogramming may be safer and more efficient. However, directly injecting free EVs cannot remain in the joint for longtime [38,39]. Therefore, to better implement the strategy of immune cell mobilization to reprogram macrophages, hydrogel microspheres should not only release factors and capture macrophages but also load sEVs as a “drug release library” for reprogramming.
To investigate the therapeutic potential of immune cell mobilization strategy in reprogramming proinflammatory macrophages for OA management, this study introduces a novel approach utilizing hydrogel microspheres (HCM) fabricated through microfluidic techniques, incorporating streptavidin grafted hyaluronic acid methacrylate (HAMA-SA) and Chondroitin sulfate methacrylate (ChSMA). The ChSMA contained sulfonic acid groups, which combined with IL4 to recruit macrophages [40]. The grafted streptavidin protein (SA) could covalently bind to biotinylated antibody CD86 to capture pro-inflammatory macrophages [41]. Additionally, HAMA formed a tight connection with CD44 antigens of sEVs (Scheme 1) [42]. The immune cell mobilized HCM were constructed slowly released IL4 to enhance the migratory ability of pro-inflammatory macrophages, and captured them on the surface of microspheres by CD86 antibodies. Simultaneously, immune cell mobilized HCM regulated the mitochondrial energy metabolism of pro-inflammatory macrophages through sEVs and reprogrammed them into anti-inflammatory macrophages state in cooperation with IL4. Furthermore, immune cell mobilized HCM effectively reduced synovial inflammation and inhibited the progression of OA in rat models. Overall, due to the advantages mentioned above, the immune cell mobilization strategy is a suitable treatment option for inflammatory OA.
Scheme 1.

Schematic diagram of immune cell mobilized hydrogel microspheres preparation. (A) The extrusion method to prepare engineering cell membrane vesicles (sEVs) derived from synovial mesenchymal stem cells (SMSCs). (B) The microfluidic methods construct streptavidin grafted hyaluronic acid methacrylate (HAMA-SA) and Chondroitin sulfate methacrylate (ChSMA) microspheres (HCM). (C) sEVs, antibody of CD86 and IL4-loaded HCM (sEVs@IL4@HCM) are used for intra-articular injection to treat osteoarthritis. Schematic diagram of immune cell mobilization anti-inflammatory strategy: Healthy synovial tissue structure; pathological proliferation process under OA inflammatory microenvironment; drawbacks of traditional intra-articular administration; immune cell mobilization hydrogel microspheres (sEVs@IL4@HCM) recruitment, capture and reprogrammed intra-articular pro inflammation macrophages.
2. Results and discussion
2.1. Characterization of sEVs and protein profile
To prepare engineered extracellular vesicles derived from cell membranes of SMSCs, primary SMSCs were extracted from Sprague-Dawley (SD) rats (Fig. S1A) and their surface markers were confirmed through flow cytometry, showing over 90 % expression of CD44 antigen (Fig. S1B) [43]. Primary cells were amplified to the P3 generation, and the cell membrane was extracted to prepare engineered extracellular vesicles using the extrusion method. Transmission electron microscopy (TEM) indicated that sEVs had a relatively uniform and regular spherical vesicular structure (Fig. 1A). Analysis with nanoparticle tracking technology demonstrated that the particle size of sEVs was 139.8 ± 63.9 nm (Fig. 1B). The intracellular uptake of sEVs by macrophages was observed. Fluorescently-labeled sEVs (Dil, RFP) were successfully internalized into macrophages within 6 h, and with increased incubation time, more sEVs were absorbed by macrophages, indicating that sEVs could be efficiently taken up by macrophages (Fig. 1C, Fig. S2C). And quantitative results obtained by counting sEVs positive cells showed that 36.9 % of macrophages had internalized at 6 h, 94 % at 24 h, and 79.2 % at 48 h (Fig. S2 A,B). In previous reports, inhibition of iNOS (inducible nitric oxide synthase) expression was found to increase the plasticity of proinflammatory macrophages [10]. 1400 W is a safe and effective highly selective inhibitor of iNOS. We used 1400 W to enhance the ability of sEVs to reprogram macrophages. UV spectrophotometry experiments were conducted to detect the encapsulation efficiency of the lipophilic drug 1400 W in sEVs, which was found to be 84.9 % (Fig. S3). In summary, we successfully synthesized sEVs with similar characteristics to extracellular vesicles and successfully loaded small molecule drugs.
Fig. 1.

Characterization of sEVs, protein profiling, and biological functions. (A) Representative TEM image of sEVs. (B) Nanoparticle tracking analysis of sEVs size. (C) Dil-labeled sEVs uptake by RAWs. (D) Total protein distribution of cell membrane and sEVs, n = 3. (E) Subcellular localization of proteins in sEVs. (F) Cell immunofluorescence of iNOS and CD206 in RAWs and BMDMs; (G) The ratio of positive cell count to total cell count of RAWs and (H) BMDMs, data are expressed as mean ± standard deviation, n = 5, student-t test and One - way analysis of variance. (I) RT-PCR results of CD86, CD163, NOS2 and IL1β mRNA expressions in RAWs, data are expressed as mean ± standard deviation, n = 3, student-t test and One - way analysis of variance. (J) The mRNA expressions of CD86, CD163, NOS2 and IL1β in BMDMs, data are expressed as mean ± standard deviation, n = 3, student-t test and One - way analysis of variance. (** indicated p < 0.01, LPS group was pro-inflammatory control, while the IL4 group was anti-inflammatory control, respectively).
The comparative proteomics analysis was conducted on SMSCs cell membranes and sEVs to determine the loss of bioactive substances during the preparation of cell membrane-derived extracellular vesicles (Fig. S4). Through 4D label-free quantitative analysis, a total of 3338 proteins were quantified with a confidence level of more than 95 % in both protein samples. The relative abundance distribution of all identified proteins in a single sample is shown in the principal component analysis (PCA) (Fig. S5A), indicating differences in protein abundance between SMSCs cell membrane and sEVs groups, but not within groups. Hierarchical clustering analysis showed a high similarity in the overall distribution of the 3338 identified proteins in each group, as shown in the heatmap (Fig. 1D). Differentially expressed proteins (DEPs) between the two groups were determined as being up- or down-regulated by at least 2-fold with an FDR (False Discovery Rate) value of less than 0.05. A total of 387 proteins showed significant changes, with 232 proteins downregulated, as shown in the heatmap and volcano plot (Figs. S5B and C). To explore the impact of downregulated DEPs on biological functions, a GO (Gene Ontology) analysis was conducted. The top five enriched biological processes (Fig. S6A) included histone binding, low-density lipoprotein receptor binding, and protein tyrosine/serine/threonine phosphatase activity. The expression of these binding proteins may have decreased due to loss during the extrusion and centrifugation steps in the preparation of sEVs. KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis showed that both groups were enriched in unsaturated fatty acid biosynthesis, fatty acid metabolism, and glycosylphosphatidylinositol (GPI) anchor biosynthesis, suggesting no significant loss of lipid components during the preparation process (Fig. S6B). Furthermore, analyze the origin of the membrane based on protein subcellular localization analysis, the main proteins were classified into 5 categories: plasma membrane proteins, cytoskeleton proteins, extracellular proteins, mitochondrial proteins, and endoplasmic reticulum proteins. Plasma membrane proteins accounted for 47.6 %, while mitochondrial proteins accounted for 31.35 %. The heatmap showed the signature membrane proteins of SMSC-derived extracellular vesicles, mitochondrial oxidative phosphorylation-related proteins, macrophage communication-related proteins, and immune regulation-related proteins (Fig. 1E). Overall, these proteomic analysis results showed that sEVs prepared by the extrusion method retained the vast majority of bioactivity, providing strong evidence for drug delivery and mitochondrial function regulation to macrophages.
2.2. sEVs positively regulate pro-inflammatory macrophage reprogramming plasticity
Cell biocompatibility tests can be used to predict whether biomaterials pose potential risks to patients, so CCK-8 assay (Cell Counting Kit-8 assay) and live/dead staining were performed using different concentrations of sEVs to determine the safest application concentrations. In this study, RAW264.7 cells (RAWs) and the primary macrophages of bone marrow derived macrophages (BMDMs) were used. The live/dead staining results showed that the ratio of dead cells (red) were increased in 200 ng/ml and 1000 ng/ml of BMDMs (Fig. S7A). And lower live cells (green) counts both in RAWs and BMDMs (Figs. S7B and C). The CCK-8 results showed that when the concentrations of sEVs were greater than 1000 ng/mL, the cell survival rate decreased both in RAWs and BMDMs (Figs. S7D and E). Therefore, in the subsequent cell function experiments, concentrations of 100 ng/mL sEVs were chosen to maintain good cell proliferation ability.
Macrophages exhibit high sensitivity and plasticity to changes in the microenvironment, and also display a high degree of heterogeneity. However, research has shown that exposure to IL4, a cytokine that promotes anti-inflammatory state differentiation, cannot convert mouse and human macrophages pro-inflammatory to anti-inflammatory state, both in vitro and in vivo [6]. On the other hand, anti-inflammatory macrophages are more plastic and can be more easily repolarized to the pro-inflammatory macrophages. Menno et al. identified mitochondrial oxidative phosphorylation inhibition associated with pro-inflammatory as a factor that prevents anti-inflammatory reprogramming [10]. By inhibiting the production of nitric oxide in pro-inflammatory macrophages and improving the decline in mitochondrial function, macrophage reprogramming plasticity was enhanced. To investigate whether sEVs can enhance the reprogramming plasticity of macrophages, cell immunofluorescence was performed to examine the polarization marker protein of macrophages. As shown in Fig. 1F, the LPS group displayed pro-inflammatory activation of macrophages (control), while the IL4 group towards anti-inflammatory activation (positive control). Consistent with the existing literature, IL4 stimulation of pro-inflammatory macrophages to repolarize into anti-inflammatory state is extremely inefficient (Fig. 1F) [10]. Pro-inflammatory macrophages treated with sEVs and IL4 showed high efficiency in anti-inflammatory reprogramming, with anti-inflammatory state marker CD206 expression increasing 1.58 -fold in RAWs and 2.12 -fold in BMDMs compared to the LPS + IL4 group (Fig. 1G and H). However, treatment with sEVs lead to a considerable downregulation of iNOS expression, yet did not exhibit a noteworthy downregulation in the sEVs (empty) group, which unloaded 1400 W. In order to further confirm the influence of sEVs on macrophage reprogramming, RT-PCR was carried out to examine the expression levels of inflammatory factors CD86, CD163, NOS2 and IL-1β genes. The results demonstrated that the combination of sEVs and IL4 treatment had the strongest macrophage reprogramming ability, suggesting that the change in gene expression is consistent with alterations in protein expression (Fig. 1I and J). These findings suggest that sEVs positively regulate macrophage reprogramming plasticity, significantly enhancing the efficiency of IL4-induced anti-inflammatory reprogramming.
2.3. Characterization of immune cell mobilized hydrogel microspheres
An ideal immune cell mobilized hydrogel microsphere should not only have the characteristic of efficient loading and sustained release of sEVs, but also effectively chelate and deliver cytokines. As a component of articular cartilage, hyaluronic acid (HA) is widely used in the treatment of OA. Concurrently, CD44, which is highly expressed on sEVs, can tightly bind to hyaluronic acid. Chondroitin sulfate, a polysulfated glycosaminoglycan, is capable of forming strong interactions with cationic cytokines. By functioning as an intermediate linker, it facilitates the non-covalent binding of cytokines to the scaffold while also safeguarding them from denaturation. Both polymers were modified with methacryloyl groups, introducing polymerizable carbon-carbon double bonds. The effective grafting of methacrylate groups was verified by the 1H NMR analysis of HAMA and ChSMA. Methacrylic acid percentages were determined to be 53.43 % and 62.26 %, respectively. To further couple biotinylated antibody for pro-inflammatory macrophage capture, streptavidin-biotinylated protein was grafted onto HAMA via EDC/NHS amidation reaction, with a grafting rate of 21.11 % (Fig. 2A).
Fig. 2.

Morphological characterization of immune cell mobilized hydrogel microspheres. (A) 1H NMR spectra of HAMA-SA, HAMA and ChSMA. (B) Size distribution of HCM. (C) Homogeneity of HCM. (D) Loading of Dil-labeled sEVs on HCM. (E) Loading of antibodies in HCM. (F) SEM observation of sEVs loaded on HCM. (G) In vitro degradation of HCM. (H) Encapsulation efficiency of IL4 and sEVs in HCM, data are expressed as mean ± standard deviation, n = 3, student-t test. (I) Release rate of IL4 in HM (HAMA hydrogel microspheres) and HCM, data are expressed as mean ± standard deviation, n = 3, student-t test. (J) Release rate of sEVs, data are expressed as mean ± standard deviation, n = 3, student-t test. (*p < 0.05, **p < 0.01).
In this study, hydrogel microspheres (HCM) were generated by photopolymerization of droplets produced through a microfluidic device. As shown by optical microscopy, the composite HCM made with microfluidics exhibited good dispersion, uniform size and shape (Fig. 2B). Smaller hydrogel microspheres are preferred for better injectability, however, too small a diameter may lead to poor diffusion within the joint. Usually, it is recommended to use hydrogel microspheres with diameters of 200–300 μm. In this study, monodisperse HCM with a diameter of 214.46 ± 14.68 μm was achieved by varying flow rates of the dispersed and continuous phases (Fig. 2C). Successful loading of DiI-labeled sEVs@HCM was confirmed by confocal laser scanning microscopy (CLSM) (Fig. 2D). SEM observation showed that HCM possessed a porous structure and also exhibited high loading efficiency of sEVs (Fig. 2F). To verify the uniformity of chondroitin sulfate grafting, element analysis and elemental mapping by energy dispersive spectroscopy (EDS) were performed, showing a uniform distribution of sulfur (Fig. S8). We assessed successful coupling of antibodies to the microsphere scaffold by incubating with fluorescent antibodies. Anti-SA (Green) and Anti-CD86 (Red) demonstrated successful antibody coupling (Fig. 2E). Understanding appropriate biodegradability of HCM is critical. If HCM degrades too quickly, drug release will not be completed. However, if HCM degrades too slowly, it may affect tissue remodeling in the joint microenvironment. As shown in Fig. 2G, hyaluronidase solution (1000 U/mL) were used to mimics the microenvironment in vivo, and the HCM underwent a two-stage degradation process, rapidly degrading in the initial 2 weeks before degrading slowly until almost completely degraded at 8 weeks.
Under the action of chemotactic factors, macrophages can migrate to distant inflammatory sites through amoeboid movement [21]. In this study, we aim to use IL4 to “mobilize” macrophages and make them traverse dense proliferating synovial tissue. Macrophage chemotactic factors are highly diverse, but mainly secreted by pro-inflammatory macrophages, with unknown safety when used in vivo and potential risks [20]. IL4, as the most commonly used cell factor for promoting anti-inflammatory activation in vitro, has also been shown to recruit pro-inflammatory macrophages [44]. In the HCM network, IL4 and sEVs are absorbed initially through swelling. Following this process, they interact with HCM through both covalent and non-covalent mechanisms. These interactions are mild in nature and are thought to have minimal impact on the stability of sEVs or IL4. In accordance with a specific ELISA kit, the encapsulation percentages of sEVs were determined to be 69.15 % and 75.02 % in HM (hyaluronic acid methacrylate hydrogel microspheres) and HCM, respectively (Fig. 2H). In addition, the encapsulation rates of IL4 in HM and HCM were 18.51 % and 51.02 %, respectively. The results show that the final load mass ratio of IL4 to microsphere in the HCM is 16.51 ng/mg, and in the HM is 5.99 ng/mg.
Macrophages can respond to concentration gradients of chemotactic factors (i.e., IL4) and migrate. As a result, it may be possible to establish a suitable concentration gradient around HCM by prolonging and regulating the release of IL4. In the subsequent drug release experiment, encapsulated IL4 showed a trend of sustained release from MGs, accumulating to 2267 pg/mL within 28 days (Fig. 2I). However, unlike IL4, sEVs were released at a uniform rate within 7 days, with a cumulative release of 188 ng/mL within 28 days (Fig. 2J). The sEVs release curve tended to be stable after 14 days, possibly due to rapid degradation of sEVs at room temperature. The drug release between IL4 and HCM can be explained by the protein isoelectric point (pI = 5.783) being positively charged at physiological pH, which can couple with negatively charged sulfate ions. In summary, our immune cell mobilized hydrogel microspheres can stably bind pro-inflammatory macrophage antibody CD86, and load sEVs and IL4 for sustained release. These features are of great significance for the synergistic work of macrophage recruitment, capture, and reprogramming in the joint.
2.4. Biocompatibility of the immune cell mobilized hydrogel microspheres
The in vitro cytotoxicity of immune cell mobilized hydrogel microspheres was evaluated by live/dead staining and Cell Counting Kit-8 (CCK8) assay. The grouping of experiments includes antibody-free hydrogel microspheres (HCM Anti-free), non-cytokine loaded hydrogel microspheres (HCM), IL4-loaded HCM (IL4@HCM), sEVs -loaded HCM (sEVs @HCM), IL4 and sEVs both loaded HCM (IL4@sEVs@HCM). For live/dead staining assay, RAWs were incubated with HCM (Anti-free), HCM, IL4@HCM, sEVs@HCM and IL4@sEVs@HCM for 3 day and then stained by a calcein–AM/PI kit. The images were shown in Figs. S9A and B; the live and dead cells were stained with green and red fluorescence, respectively. No marked difference was observed in different groups, indicating that microspheres were safe. In addition, the cytotoxicity of the microspheres was evaluated by CCK8 assay, and the results showed that all groups had safety biocompatibility (Fig. S9C).
2.5. Recruit and capture pro-inflammatory macrophages
Transwell device was employed to establish a co-culture system of cells and HCM in order to replicate the in vivo “amoeboid movement” of synovial macrophages that is triggered by chemotactic factors. Initially, hydrogel microspheres were positioned in the lower chamber, and pro-inflammatory activated RAWs in the upper chamber. The objective was to trigger IL4 release from the lower chamber to instigate macrophage migration towards it (Fig. 3A). We found that, after 24 h, the migrated macrophages in the IL4-loaded groups (IL4@HCM and IL4@sEVs@HCM) were significantly higher than other groups, with no statistical difference between IL4@HCM and IL4@sEVs@HCM (Fig. 3B). Furthermore, to simulate the process of macrophage migration through dense extracellular matrix in vivo, 0.1 wt% gelatin was coated on the Transwell upper chamber to observe whether the macrophages in the upper chamber could invade and migrate to the lower chamber. As shown in Fig. 3D, there were more macrophages in the IL4@sEVs@HCM and IL4@HCM groups than in other groups, indicating that IL4 can recruit macrophages to migrate to the joint cavity. The existence of cells that had migrated to the lower chamber following treatment with sEVs@HCM implies that sEVs might possess the ability to recruit macrophages.
Fig. 3.

In vitro simulation of immune cell mobilized hydrogel microspheres. (A) Schematic illustration of macrophage recruitment and capture in vitro. (B) Crystal violet staining in Transwell experiment, each group include: HCM(Anti-free) was unloaded CD86 antibody, sEVs@HCM(Anti-free), IL4@HCM(Anti-free), HCM, sEVs@ IL4@HCM. (C) cell counts of migration experiment (D) cell counts of invasion experiment, data are expressed as mean ± standard deviation, n = 3, One - way analysis of variance. (E) Immunofluorescence staining observation of cell quantity. (F) Alcian blue and nuclear fast red staining observation of cell quantity inside microspheres. (G) Quantification of immunofluorescence, data are expressed as mean ± standard deviation, n = 3, One - way analysis of variance. (H) Quantification of Alcian blue and nuclear fast red staining, data are expressed as mean ± standard deviation, n = 3, One - way analysis of variance. (*p < 0.05, **p < 0.01).
To further verify whether immune cell mobilized hydrogel microspheres have the ability to capture pro-inflammatory macrophages, polarized macrophages were co-cultured with HCM using a U-bottom low-adhesion culture plate, and the number of captured macrophages was observed by immunofluorescence. As shown in Fig. 3E, HCM and IL4@sEVs@HCM had a higher ability to capture macrophages than the other groups. Since HCM is composed of glycosaminoglycans and lacks RGD peptides required for cell adhesion, macrophage capture mainly relies on the coupling of CD86 antibodies. To verify the stability of macrophage capture, the microspheres with incubated macrophages were collected, shaken for 5 min and centrifugally filtered, and the number of captured cells in the microspheres was observed again (Fig. 3E). The number of cell captures in the HCM and IL4@sEVs@HCM groups was higher than in the other groups. The microsphere slice staining results also confirmed this trend (Fig. 3F, H). This indicates that the CD86 antibody significantly enhances the binding ability of the hydrogel microspheres to pro-inflammatory macrophages, which allows macrophages to stay in the high concentration of drugs for a longer period of time and provides a guarantee for reprogramming. Overall, the immune cell mobilized hydrogel microspheres enhance the migration ability of macrophages and capture them, providing spatial and temporal conditions for macrophage reprogramming.
2.6. The ability and mechanism of reprogramming pro-inflammatory into anti-inflammatory state
Firstly, the polarization state of RAWs adhered to microspheres was evaluated through immunofluorescence staining and flow cytometry. As show in Fig. 4A, macrophages expressing iNOS (green) and CD206 (red) were captured in different groups. A reduced cell count and a higher prevalence of the pro-inflammatory state were found in the HCM group. The IL4@HCM treatment group did not exhibit a noteworthy rise in anti-inflammatory macrophage proportion. However, an increased expression of CD206 was detected in the sEVs@HCM and IL4@sEVs@HCM groups. Importantly, in the IL4@sEVs@HCM group, the ratio of anti-inflammatory/pro-inflammatory was the highest (Fig. 4B). The expression of IL1 (proinflammatory cytokine secreted primarily by pro-inflammatory macrophage) and ARG1 (cytokine secreted primarily by anti-inflammatory macrophages) was consistent with polarization markers (Fig. 4C). In addition, we obtained consistent results with the immunofluorescence results by detecting the polarization markers CD86 and CD163 using flow cytometry (Fig. S10). The efficiency of anti-inflammatory differentiation in IL4@sEVs@HCM group reached 88.5 %.
Fig. 4.

Reprogramming of pro-inflammatory macrophages and restoration of mitochondrial metabolism by immune cell mobilized hydrogel microspheres. (A) Immunofluorescence observation of pro-inflammatory/anti-inflammatory ratio (iNOS, CD206) and (IL1, ARG1). (B, C) Fluorescence quantification, data are expressed as mean ± standard deviation, n = 3, student-t test and One - way analysis of variance. (D) Polarization-related genes of macrophages, data are expressed as mean ± standard deviation, n = 3, student-t test. (E) GO enrichment pathways. (F) GSEA pathway analysis. (G) Changes in gene expression levels in glycolysis, TCA cycle and oxidative phosphorylation. (H) Expression levels of mitochondrial energy metabolism proteins, n = 3. (*p < 0.05, **p < 0.01).
To further investigate the efficiency and molecular mechanism of macrophage reprogramming after treatment with immune cell mobilized hydrogel microspheres, RNA-seq analysis was performed and correlation analysis was conducted between groups (Figs. S11A and B). A total of 2207 genes exhibited differential expression between the HCM group and the IL4@sEVs@HCM treatment group in this study. In addition, 1839 and 285 differentially expressed genes were observed between the IL4@HCM group and the IL4@sEVs@HCM group, and between the IL4@HCM group and the HCM group, respectively (Figs. S11C and D). Fig. 4D illustrates a decrease in the expression of pro-inflammatory macrophage-related genes and a corresponding increase in the expression of anti-inflammatory macrophage-related genes. GO enrichment analysis showed a high enrichment of mitochondrial-related pathways (Fig. 4E). The GSEA analysis found that there was a positive correlation between the IL4@sEVs@HCM group and the mitochondrial inner membrane and oxidative phosphorylation (Fig. 4F). Furthermore, RNA expression levels of glycolysis, tricarboxylic acid cycle, and mitochondrial oxidative phosphorylation were compared using a radar map (Fig. 4G). We found that the IL4@HCM and IL4@sEVs@HCM groups had higher levels of tricarboxylic acid cycle and oxidative phosphorylation. This suggests that macrophage reprogram to anti-inflammatory state and restored mitochondrial energy metabolic function. Mitochondrial dysfunction and oxidative stress are considered to be the main causes of the dysregulation of inflammatory response [3,10]. And mitochondria metabolic function was a vital source of dynamic signals that regulate macrophage biology to fine-tune immune responses [34]. Van den Bossche et al. showed that iNOS disrupt the oxygen-phosphorylated respiratory chain and are a key factor in the irreversibly functioning of pro-inflammatory macrophages, and that inhibition of iNOS expression by small molecule drugs improves mitochondrial function and reprogramming of anti-inflammatory macrophages [10]. Furthermore, we conducted a quantitative analysis of energy metabolic products in macrophages by employing liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis (Figs. S12 and 13). The IL4@sEVs@HCM group exhibited significant plasticity and successful anti-inflammatory macrophage reprogramming, as evidenced by its higher energy metabolism level and greater ATP production when contrasted with the other two groups (Fig. 4H).
2.7. In vivo retention time
Prolonging the retention time of sEVs within the joint cavity is of paramount importance to facilitating OA treatment. In this study, we used in vivo imaging systems (IVISs) to evaluate the retention time of the fluorescently labeled sEVs and sEVs loaded-microspheres (sEVs@HCM). sEVs and sEVs@HCM were injected into the right knee joint of the rat. The relative fluorescence intensity in the sEVs group declined rapidly and almost vanished on day 14 (Figs. S14A and C). The fluorescence intensity of the sEVs@HCM group existed for a longer time and diminished more slowly than sEVs group (Figs. S14B and C). The results suggested that HCM microspheres could provide excellent physical protection for the encapsulated sEVs, which was assumed to be essential for the reprogramming macrophages of OA in vivo.
2.8. Therapeutic effect of OA in vivo
The progression of osteoarthritis is usually manifested by narrowing of the joint space width (JSW), formation of bone spurs, and remodeling of subchondral bone in imaging studies [26]. Therefore, we conducted MicroCT scanning to analyze the imaging alterations in rat joints at 8 weeks post surgery (Fig. 5A). Fig. 5B reveals that a considerable decrease in JSW was observed in the PBS, HCM, IL4@HCM, sEVs@HCM, and IL4@sEVs@HCM groups compared to the sham group. The JSW in the PBS group was merely 0.2-fold compared to that of the sham group. Through quantitative measurement, it was found that in contrast to the PBS and HCM groups, the volume and mass of bone spurs in the IL4@sEVs@HCM group decreased. In this study, no significant bone spurs formation was observed in the sham group. However, intra-articular injection of IL4@sEVs@HCM did not significantly differ in subchondral bone density compared to the sham group, with a value of 1.10 ± 0.08 times, while the PBS group had a value of 1.66 ± 0.05 times (p < 0.01).
Fig. 5.

Imaging evaluation of immune cell mobilized hydrogel microspheres for the treatment of SD rat models with osteoarthritis. (A) Flow chart of animal experiments. (B) MicroCT experiment. (C) Quantitative analysis of (i) joint space, (ii) bone spur volume, (iii) bone spur bone mass, and (iv) subchondral bone density. (*p < 0.05, **p < 0.01; data are expressed as mean ± standard deviation, n = 5 independent experiments, student-t test and One - way analysis of variance).
In addition to radiographic assessments, we conducted histological analyses using hematoxylin and eosin (H&E) and Safranin O-fast green staining to identify changes in cartilage surface and synovium. According to the findings, the cartilage surface in the sham group exhibited a uniform and natural structure. Conversely, the joints in the PBS, HCM, and IL4@HCM groups demonstrated a remarkable decline in the cartilage matrix and substantial erosion of the cartilage surface. Conversely, in the IL4@sEVs@HCM treatment group, a reduction in the degenerative alterations was observed (Fig. 6A and B). At the same time, we scored the synovial tissue, and the scoring criteria are shown in (Table S1). In the groups loaded with sEVs, synovial inflammation was significantly inhibited compared to the groups without sEVs. The modified Mankin score was used to evaluate the severity of histological staining for cartilage damage, which was increased to varying degrees in the PBS, HCM, and IL4@HCM groups [27]. In addition, Type II collagen (COL2A1) expression was detected by immunohistochemistry. Compared with the sham group, the expression of aggregating proteoglycans in the cartilage of the PBS and HCM groups was significantly reduced, and moderately reduced in the IL4@HCM group (Fig. 6C).
Fig. 6.

Histological evaluation of immune cell mobilized hydrogel microspheres for the treatment of SD rat models with osteoarthritis. (A) HE staining. (B) Safranin O-Fast green staining. (C) Type II collagen immunohistochemical experiment. (D) Mankin score. (E) Immunohistochemical quantification. (F) Synovial histopathological score. (*p < 0.05, **p < 0.01; data are expressed as mean ± standard deviation, n = 5 independent experiments, student-t test and One - way analysis of variance).
Notably, an evaluation of the synovial macrophages reprogramming by immunofluorescence showed that the expression of iNOS in the IL4@sEVs@HCM group were significantly lower than those in the PBS and HCM groups (Fig. 7A,C). And the CD206 expression was an increased in the IL4@sEVs@HCM group, indicating that there was increased anti-inflammatory macrophages in these synovial tissues (Fig. 7A,C). Further, to observe the overall level of inflammation in the joint, we examined inflammatory cytokines matrix metalloproteinases 13 (MMP13) and a disintegrin and metalloproteinase thrombospondin 5 (ADAMTS5). MMP13 is low in normal human tissues, while the content of MMP13 is highly expressed in connective tissues in patients with osteoarthritis [45]. ADAMTS5 acts as a proteoglycanase that cleaves aggrecan, which is the main proteoglycan in cartilage, thus mediating cartilage damage in osteoarthritis [46]. In the IL4@sEVs@HCM and Sham group, MMP13 and ADAMTS5 were lower expression than other groups (Fig. 7B and C). This result provide further support that immune cell mobilized hydrogel microspheres can play an anti-inflammatory role in joint by reprogramming macrophages.
Fig. 7.

Immune cell mobilized hydrogel microspheres promoted macrophages reprogramming in vivo. (A) Images of immunofluorescence staining for iNOS and CD206. (B) Images of immunofluorescence staining for MMP13 and ADAMTS5. (C) And the quantification of immunofluorescence intensity of iNOS, CD206, MMP13 and ADAMTS5. (*p < 0.05, **p < 0.01; data are expressed as mean ± standard deviation, n = 5 independent experiments, student-t test and One - way analysis of variance).
In addition, rat organ staining showed that there was no significant difference between each group, proving that it had no potential in vivo toxicity (Fig. S15).
In summary, all these results indicate that immune cell mobilized hydrogel microspheres reduce inflammation and alleviate cartilage matrix degradation through efficient macrophage reprogramming, which has therapeutic potential for treating osteoarthritis.
3. Conclusion
In this study, we explored the advantages and feasibility of the immune cell mobilization strategy in the treatment of OA synovial inflammation. Injectable hydrogel microspheres prepared by microfluidic technology can efficiently load IL4 and CD86 antibodies, endowing them with excellent macrophage recruitment and capture capabilities, thereby achieving immune cell mobilize in the joint. In addition, sEVs were built based on the energy metabolism characteristics of pro-inflammatory macrophages, which endow the microspheres with the ability to improve mitochondrial function and efficiently reprogram macrophages. In summary, immune cell mobilized hydrogel microspheres provide a new approach for treating OA inflammation and resolving scattered macrophages, as well as provide reference value for subsequent research on EVs delivery.
4. Experimental section
4.1. sEVs fabrication
SD rats' synovial stromal cells were extracted according to previously reported methods, and the same age and sex (aged 8 weeks, male) were used to control the batch effect [35]. Subsequent to euthanizing the rat, after joint incision, the inner tissue is fully exposed and the superficial synovial tissue (lining layer) is carefully excised and subjected to an overnight digestion procedure utilizing 10 ng/mL collagenase type IV (Sigma-Aldrich, USA). After centrifugation, the supernatant was removed and the cells were cultured in DMEM/F12 (Gibco, USA) that contained 15 % FBS (Gibco, USA), leading to the generation of passage 0 (P0) synovial mesenchymal stem cells (SMSCs). The distinct procedures for the isolation of SMSCs membrane are outlined as follows: Once 90 % of cells reached P3, they were harvested using a cell scraper and underwent centrifugation at 300g for 5 min. The solution obtained from the centrifugation underwent elimination of its supernatant before being treated with membrane protein extraction buffer (Beyotime, China). The mixture was repeatedly blown and beaten in an ice bath for 15 min. The mixture underwent a series of repeated blowing and beating actions in an ice bath for 15 min. Subsequently, the resulting cellular lysate was centrifuged for 10 min at 4 °C and a speed of 700 g to yield a supernatant. This supernatant was then centrifuged at 13,000 g and 4 °C for 30 min, leading to the formation of sediment that was regarded as SMSCs membrane. sEVs carrying 1400 W (Sigma, USA) were prepared using a membrane extrusion method. In brief, the cell membrane sediment (Protein concentration: 1 mg/ml) was resuspended in PBS containing 1400 W (100 μM) and was extruded ten times through a 400 nm porous membrane (Polycarbonate membrane, Avanti, USA) with a liposome extruder (Avanti, USA), followed with ten cycles through 200 nm porous membrane (Polycarbonate membrane, Avanti, USA). Then, the sEVs were collected and stored at 4 °C. It is worth noting that the extrusion process should be slow and uniform to prevent the needle from clogging during injection.
4.2. Characterization of sEVs
To fully characterize the preparation of sEVs, transmission electron microscopy (TEM; HITACHI, HT7800, Japan) and nanoparticle tracking analysis (NTA; Particle Metrix, ZetaView, Germany) were used for morphology and particle size analysis. To confirm that sEVs can be internalized by macrophages, RAWs were incubated with Dil-labeled sEVs for a specified time, fixed with 4 % paraformaldehyde, stained with DAPI (Servicebio, China) for the cell nucleus, and stained with phalloidin (Beyotime, China) for the cell skeleton. The cells were then observed by fluorescence microscopy. The encapsulation rate of 1400 W was determined by ultraviolet spectrophotometry using the following formula: Encapsulation efficiency (%) = (successfully encapsulated drug weight/the initial feeding weight) × 100 %. Loading efficiency (%) = (successfully encapsulated drug weight/Total weight of the drug and carrier) × 100 %. The specific practice is to collect freshly prepared sEVs (Total drug concentration was 100 μM), and 10,000 g centrifugal to collect precipitation. Then the unloaded 1400 W in the supernatant and the concentration was measured by the standard curve method.
4.3. Surface marker proteins through a 4D-label free method
The main experimental procedures included protein extraction, quantification, detection, enzymatic digestion and desalination, fractionation separation, mass spectrometry detection, and data analysis. Protein extraction: The sEVs and cell membranes used for 4D-label free detection came from three different SD rats (aged 8 weeks, male). The sample was transferred it to a 1.5 ml centrifuge tube and lysed with DB lysis buffer (8 M Urea, 100 mM TEAB, pH 8.5), followed by 5 min of ultrasonication on ice. The lysate was centrifuged at 12,000 g for 15 min at 4 °C and the supernatant was reduced with 10 mM DTT for 1 h at 56 °C, and subsequently alkylated with sufficient iodoacetamide for 1 h at room temperature in the dark. Protein quantification: BSA standard protein solutions and sample solutions with different dilution multiples were added into 96-well plate to fill up the volume to 20 μL, respectively. Each gradient was repeated three times. The plate was added 180 μL G250 dye solution quickly and placed at room temperature for 5 min, the absorbance at 595 nm was detected. The standard curve was drawn with the absorbance of standard protein solution and the protein concentration of the sample was calculated. 20 μg of the protein sample was loaded to 12 % SDS-PAGE gel electrophoresis, wherein the concentrated gel was performed at 80 V for 20 min, and the separation gel was performed at 120 V for 90 min. The gel was stained by Coomassie brilliant blue R-250 and decolored until the bands were visualized clearly. Enzymatic digestion and desalination: Each protein sample was taken and the volume was made up to 100 μL with DB lysis buffer (8 M Urea, 100 mM TEAB, pH 8.5), trypsin and 100 mM TEAB buffer were added, sample was mixed and digested at 37 °C for 4 h. Then trypsin and CaCl2 were added digested overnight. Formic acid was mixed with digested sample, adjusted pH under 3, and centrifuged at 12,000 g for 5 min at room temperature. The supernatant was slowly loaded to the C18 desalting column, washed with washing buffer (0.1 % formic acid, 3 % acetonitrile) 3 times, then added elution buffer (0.1 % formic acid, 70 % acetonitrile). The eluents of each sample were collected and lyophilize. Fractionation separation: Mobile phase A (2 % acetonitrile, adjusted pH to 10.0 using ammonium hydroxide) and B (98 % acetonitrile, adjusted pH to 10.0 using ammonium hydroxide) were used to develop a gradient elution. The lyophilized powder was dissolved in solution A and centrifuged at 12,000 g for 10 min at room temperature. The sample was fractionated using a C18 column (Waters BEH C18, 4.6 × 250 mm, 5 μm) on a Rigol L3000 HPLC system, the column oven was set as 45 °C. The detail of elution gradient was shown in Table S2. The eluates were monitored at UV 214 nm, collected for a tube per minute and combined into 10 fractions finally. All fractions were dried under vacuum, and then, reconstituted in 0.1 % (v/v) formic acid (FA) in water. Mass spectrometry detection, and data analysis: A mass spectrometer (nanoElute, UHPLC + tims-pro2) and database (UniProt) were used, and protein profiling analysis was supported by LC-Bio laboratory (Hangzhou, China) in this study.
4.4. Synthesis of HAMA, HAMA-SA and ChSMA
HAMA and ChSMA were synthesized according to previously reported methods [26]. In brief, a reaction was initiated between 2 wt% HA (MW = 150 kDa; China) and ChSMA (MW = 85 kDa; China) with methacrylic anhydride (3-fold molar mass; Aladdin, China), utilizing deionized water at pH 8.0. After preparation, the solution was continuously stirred for 24 h at low temperatures below freezing. The purified product was obtained by dialysis against deionized water with a molecular weight cut-off (MWCO) of 3500 Da for a period of 3 days. The purified product was then freeze-dried. The obtained HAMA was dissolved in MES (pH = 6) buffer to obtain a 0.1 M HAMA solution. 10 mg of SA, 40 mg of EDC, and 60 mg of NHS were added sequentially, and the solution was reacted at 37 °C for 120 min, centrifuged, and the supernatant was collected. The product HAMA-SA was thoroughly dialyzed in water at 4 °C for 4 days using a dialysis bag (MWCO 3500Da) and subsequently freeze-dried. The grafting percentage of methacrylamide and SA was determined by 1H NMR (600 MHz, Bruker, Germany).
4.5. Characterization of hydrogel microspheres
Water-in-oil droplets were generated using microfluidic technology to prepare hydrogel microspheres [47]. For the water phase, HAMA-SA and ChSMA were mixed in a ratio of 4:1 (4 wt% HAMA-SA, 1 wt% ChSMA, and 0.5 wt% photosensitizer). For the oil phase, a blend of paraffin oil and 5 wt% Span 80 was prepared. Both the water phase and oil phase were injected through the inlet port of a microfluidic device, employing an appropriately adjusted flow rate ratio. The resultant droplets were frozen at - 40 °C prior to being cross-linked using UV light, which facilitated the formation of HCM. Following this, washing with ether was undertaken to remove paraffin oil and rinsing with PBS was repeated five times.
The characterization of microgels was performed using fluorescent microscopy (LSCM800, Zeiss, Germany) to determine the morphology and diameter of HCM. The surface morphology and microstructure of HCM that underwent freeze-drying were studied by conducting SEM analysis (FEI Sirion 200, USA), which enabled the identification of the element distribution. Loading of microspheres was characterized by LSCM upon co-incubation of DiI-labeled sEVs with HCM in the absence of light and at cold temperatures. In vitro validation was performed to determine the distribution of SA and the ability to chelate antibodies. A FITC-streptavidin antibody (Biolegend, USA), an anti-CD86 biotin antibody (Biolegend, USA), and an APC conjugated anti-mouse secondary antibody (Biolegend, USA) were utilized and subjected for night incubation under cooled darkness. Specifically, we collected 10 mg of freeze-dried hydrogel microspheres and used 1 ml of PBS (containing biotin antibodies at 100 μg/ml) swelling hydrogel microspheres. After the antibody incubation, 300 g centrifuged and precipitated washed with PBS. The samples underwent thrice washing with PBS and underwent observation via LSCM.
4.6. HCM degradation, encapsulation and release assay
To evaluate the degradation of HCM, a method previously described in literature was employed [27]. An amount of 80 mg empty HCM was packed into a dialysis bag (MWCO, 3500 Da) and immersed in a solution of 5 mL PBS containing 1000 U of HAase (Solarbio, China). The bag was positioned on a shaker at 37 °C and 50 g. At intervals of three days, the HAase solution was replenished and renewed. The residual weight of the sample was recorded at predetermined time points and compared to its initial weight.
To assess the encapsulation of IL4 (Novoprotein, China), 10 mg of freeze-dried HM and HCM were separately co-incubated with an IL4 solution (300 ng/mL IL4, pH = 7.4) overnight. The hydrogel microspheres (1 mg) were incubated and subsequently subjected to centrifugation to obtain unbound protein for calculation of loading efficiency. The above method was employed to acquire the hydrogel microspheres, which were placed in 1 mL of 0.1 % bovine serum albumin (BSA) PBS and kept at 37 °C within a cell culture incubator. The concentration of the protein released into the supernatant was evaluated at specific time points. ELISA (MULTI SCIENCES, China) was used to quantify protein concentration, and included blank wells, standard wells (50 μL of various concentrations of standard samples), and sample wells (40 μL of sample diluent + 10 μL of the experimental sample) as part of the experimental design. Further, enzyme-labeled reagent (100 μL) was added to all the wells except for blank ones, followed by incubation for 1 h at 37 °C in an enclosed plate. The liquid was then discarded, and the plate was washed using washing solution. Next, the plate was dried, and color development reagents A (50 μL) and B (50 μL) were added sequentially. The plate was left to incubate at 37 °C without illumination for 15 min before termination of color development, after which absorbance measurement was taken at a wavelength of 450 nm.
To assess the encapsulation of sEVs, we separately co-incubated 1 mg of freeze-dried HM and HCM with freshly prepared sEVs solutions (Protein concentration: 377.6 ng/ml) overnight, followed by ELISA (Biosiy, China) measurement of the protein concentration of CD63 (one of the marker proteins of sEVs) in the original solution and supernatant to calculate the loading efficiency. The concentration of released CD63 protein was measured by ELISA at predetermined time points.
To assess the controlled release of sEVs in vivo, IVIS (Xenogen, USA) was utilized to evaluate the capacity of HCM to maintain controlled release of encapsulated sEVs. Firstly, the sEVs were labeled using Indocyanine Green (ICG, Merck, USA) according to a previous method [26], and loaded in HCM microspheres (sEVs@HCM). sEVs@HCM and solution that contained equal amounts of sEVs were injected into the knee joints of Rats (injection volume = 100 μL). The fluorescence intensity of the knee joint was detected using IVIS on day 0, 1, 2, 3, 7 and 14, respectively, with Ex/Em = 780/800 nm. The relative fluorescence intensities at different time points were recorded with the intensity on day 0 as reference.
4.7. Cell culture and macrophage reprogram assay
Throughout the study, RAW264.7 cells (RAWs) procured from ATCC were used. The primary macrophages were derived from bone marrow derived macrophages (BMDMs) from both femurs and tibias of C57BL/6 mice aged 6–8 weeks. The mice were killed by cervical vertebrae dislocated and placed in a culture bottle containing 75 % ethanol. After 5–10 min, the dried and sterilized scissors and tweezers were taken out, and the femur and tibia of the mice were removed. The bone was soaked in sterile PBS 3 times for 5 min each time, and the ends of the bone were cut with scissors. The bone cavity was rinsed with medium 3–5 times until the red marrow cavity cells were flushed out to white, the cells were blown 5–8 times with the flushing solution, and the bone marrow cell suspension was obtained. Then 500 g centrifuged for 5min and the supernatant was discarded. After the lysis of red cell lysate for 3min, the reaction was terminated by adding the MEM-α medium (Gibco, USA) with 10 % serum and 1 % penicillin streptomycin (Gibco, USA), centrifuged, and the supernatant was discarded. MEM-α medium containing 10 ng/ml M-CSF (Sigma, USA) was added. After 12–14 h, the unattached cells were collected and moved to the 6-well plate, and M-CSF was added to 30 ng/ml. The adherent cells were incubated in an incubator containing 5 % CO2 at a temperature of 37 °C for 3 days, and the supernatant was discarded.
For pro-inflammation macrophage activation, BMDMs were treated with IFNγ (20 ng/ml) and LPS (10 ng/ml) for 24 h. RAWs were treated with IFNγ (20 ng/ml) and LPS (100 ng/ml). For anti-inflammation macrophage activation, BMDMs and RAWs were treated with mouse IL-4 (10 ng/ml).
For macrophage reprogram, BMDMs and RAWs were primed with LPS and IFNγ. After 24 h, cells were washed and treated with the respective stimulus for another 48 h. Specifically, LPS group (positive control of activated pro-inflammation macrophage) and IL4 group (negative control of activated pro-inflammation macrophage) continued to be induced with LPS or IL4. Other groups, LPS was washed off and added with IL4, sEVs (unloaded 1400 W) and sEVs. LPS + IL4 group (IL4: 10 ng/ml), LPS + sEVs (sEVs unloaded 1400w, 100 ng/ml), LPS + sEVs (sEVs: 100 ng/ml) and LPS + IL4+sEVs (IL4: 10 ng/ml; sEVs: 100 ng/ml).
The reprogrammed macrophages were detected by immunofluorescent staining and RT-PCR. Firstly, the cells used to immunofluorescent staining were immobilized with paraformaldehyde for 30 min. Then washed with PBS for three times and treated with 0.2 % Triton X-100 (Solarbio, China) for 10 min 1 % BSA for protein blocking 1 h. The anti-iNOS (Abcam, USA) or anti-CD206 (Abcam, USA) antibody was added (1:100) and incubation overnight. The cells were subsequently treated with FITC or Cy3 Tyramide (Servicebio, China) for 30 min at 4 °C, and DAPI for 5 min at 4 °C. The cells observed under fluorescence microscope (Zeiss, Germany) and fluorescence quantification by ImageJ software subsequently.
To extract total RNA from RAWs and BMDMs, the Trizol method was adopted, followed by reverse transcription with a reagent kit (Takara, Japan). With primer sequences detailed in Table S3, RT-PCR was performed using the ABI 7300 Real-time PCR system (ABI, USA), and relative mRNA expression was calculated using the comparative cycle threshold (CT) approach. GAPDH serves as an internal reference gene. The experiments were repeated thrice.
4.8. Cytocompatibility of sEVs
Live/dead staining: RAWs and BMDMs were inoculated into 12-well plates at 1 × 105/well and treated with different concentration of sEVs (0, 10, 50, 100, 200 and 1000 ng/ml) for 3 days. Then the medium was discarded, and cells were washed with PBS three times. Live/dead staining reagent (Beyotime, China) was added for 20 min, and live cells (green) and dead cells (red) were then observed under a fluorescence microscope (Zeiss, Germany). The live cells were counted using ImageJ software subsequently. Cell count kit-8 assay (CCK8) was performed on 96-well plates inoculated with RAWs and BMDMs at 2 × 103/well. The different concentration of sEVs were added in different groups separately. On day 3, CCK8 reagent (Beyotime, China) was added and incubated for 2 h. The optical density (OD) values were then obtained at 450 nm using microplate reader (BioTek, USA).
4.9. Cytocompatibility of hydrogel microspheres
HCM(Anti-free), HCM, IL4@HCM, sEVs@HCM and IL4@sEVs@HCM microspheres were added to the low-adhesive U-bottomed plates in different groups. Then 2 × 103/well RAWs were inoculated. On day 3, the medium was discarded, and cells were washed with PBS three times. Then the Live/dead staining reagent (Beyotime, China) was added for 20 min, and observed under a fluorescence microscope (Zeiss, Germany). The CCK8 assay was performed as previously mentioned.
4.10. Recruitment and capture assay
To assess the impact of HCM on macrophage movement and mimic joint cell-cell interactions, migration assays were conducted utilizing the Transwell system (8 μm pore size, Corning, USA). Before performing the following experiments, we first activate macrophages, pro-inflammatory macrophages were induced by 100 ng/ml LPS (Sigma Aldrich, USA) and 20 ng/ml IFN-γ (Sigma Aldrich, USA) pretreatment for 24 h in RAWs.
Briefly, RAWs (1 × 105) were planted in the upper compartment where the bottom chamber was filled with culture medium incorporating 10 % serum and the corresponding hydrogel microspheres. The co-culture system was put in an incubator for 24 h. Afterward, with a cotton swab, gently wipe away the cells on the upper surface of the chamber, and the lower surface cells that adhered to the membrane were fixed and stained using a 0.5 % crystal violet solution (Macklin, China). The Transwell chamber was coated overnight at 4 °C using 0.1 wt% gelatin for the invasion experiment, with the RAWs being seeded the next day following the same migration assay procedures. Cell quantitative analysis was done by ImageJ software.
To determine the cell-capturing ability of HCM, RAWs were suspended and co-cultured with HCM in low-adhesive U-bottomed plates (7007, Corning, USA). After 24 h of culture, microspheres were polyformaldehyde-fixed and fluorescently stained with DAPI (Servicebio, China) to observe the captured macrophages under a fluorescence microscope. Meanwhile, another group of the captured cells on the microspheres were collected, agitated five times, centrifuged at 300 g, and filtered through a 70 μm cell strainer (Corning, USA). The captured cells on the residual microspheres were fixed with polyformaldehyde, stained with DAPI, and observed to determine the number of captured cells. To further count the internal cells of the microspheres, hydrogel microspheres were fixed by embedding in agarose, sectioned, and stained with Alcian blue and Nuclear fast red staining (Servicebio, China) to observe the number of macrophages.
4.11. Detection of macrophage reprogramming by hydrogel microspheres
Pro-inflammatory macrophages were induced by 100 ng/ml LPS (Sigma Aldrich, USA) and 20 ng/ml IFN-γ (Sigma Aldrich, USA) pretreatment for 24 h in RAWs. Subsequently, the cells were washed three times with PBS and treated with corresponding agents in fresh medium for an additional 2 days. The gathered cells underwent analysis with polarization markers using flow cytometry, and immunofluorescent staining.
Flow cytometry was performed based on previous reports [35]. Cells were harvested and suspended in 1 % bovine serum albumin-PBS buffer at a concentration of 1 × 106 cells/mL. Subsequently, 0.1 mL of the cell suspension was incubated with CD44 (Biolgend, USA), CD34 (Biosciences, USA), CD45 (Biosciences, USA), CD73 (Biolgend, USA), CD90 (Biolgend, USA) or CD105 (Biolgend, USA) conjugated antibodies in the dark at 4 °C for 30 min. After being washed with PBS three times, the labeled cells were resuspended in 0.2 mL of PBS and analyzed using Flowjo software.
Following cell collection, immunofluorescence staining was carried out. The cells were immobilized with paraformaldehyde at ambient temperature for half an hour, underwent treatment with 0.2 % Triton X-100 (Solarbio, China) for 10 min, and intermediately were prevented from non-specific binding with 1 % BSA for 45 min. Thereafter, anti-iNOS (Abcam, USA), anti-IL1 (Abcam, USA), anti-Arg1 (Abcam, USA), or anti-CD206 (Abcam, USA) antibody was added and left overnight for incubation. The cells were subsequently treated with FITC or Cy3 Tyramide (Servicebio, China) for 30 min at 4 °C, followed by imaging using a fluorescent microscopy, as per the instructions of TSA Fluorescent Double Stain Kit (Servicebio, China).
4.12. Metabolites assay
This study includes three groups for metabolic analysis: HCM, IL4@HCM, and IL4@sEVs@HCM. Metabolite detection was performed using an LC-MS/MS detection platform. Each group included at least three samples, and each sample collected 1 × 107 cells, which were immediately snap-frozen in liquid nitrogen for storage. Subsequently, 300 μl of distilled water was supplemented to every sample. The samples were then pre-chilled in dry ice and underwent three cycles of freeze-thawing in liquid nitrogen. Protein concentration was determined by measuring the supernatant obtained from 50 μl of each sample. Subsequently, liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed. The data acquisition instrument system was composed of ultra-performance liquid chromatography (UPLC, Waters ACQUITY H-ClassD) and tandem mass spectrometry (tandem mass spectrometry, MS/MS, QTRAP® 6500+). A LC-BIO (Database) database was established utilizing standard substances, and the mass spectrum data were qualitatively examined. MultiQuant 3.0.3 software was used for processing the mass spectrum data.
4.13. RNA-seq assay
TRIzol (Thermo Fisher, USA) was used to isolate and purify total RNA from HCM [[48], [49], [50]], IL4@HCM, and IL4@sEVs@HCM. Sequencing was performed using Illumina NovaSeqTM 6000 (LC Bio Technology, Hangzhou, China) with standard operating procedures for paired-end sequencing, with a sequencing mode of PE150. The sequencing data was filtered to yield high-quality sequencing data (Clean Data) which underwent an analysis using R programming language. Differentially expressed genes (DEGs), with a p-value ≤0.05 and fold change ≥2, were identified. The identified DEGs were subjected to enrichment analyses for functional annotations in Gene Ontology (GO) and signal pathways in Kyoto Encyclopedia of Genes and Genomes (KEGG).
4.14. Rat model of osteoarthritis
The study involving animals was approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (Approval number: IACUC-CQMU-2023-0108). Male Sprague-Dawley (SD) rats, aged 12 weeks, were assigned randomly to two groups: sham group (n = 5) and osteoarthritis (OA) group (n = 25). The OA rats received medial meniscal transection of the knee joint, following anesthesia with 3 % pentobarbital sodium (40 mg/kg). Antibiotics (penicillin, 100,000 units of intramuscular injection per day) and painkillers (carprofen, 1 mg of jelly feeding per day) were used for 3 days after surgery to prevent postoperative infections and animal licking wounds. Thereafter twice-a-week exercise training was done. After one month, the OA group was further divided into five subgroups (n = 5 per group) and administered intra-articular injections of PBS, HCM, IL4@HCM, sEVs@HCM, or IL4@sEVs@HCM (injection volume = 100 μL; the ratio is 10 mg microspheres dissolved in 1 ml PBS). Repeat injections were administered weekly in the last 4 weeks up to euthanasia. At 8 weeks post-surgery, ex vivo micro-computed tomography analysis (SkyScan 1172, Belgium) was performed on the knee joints.
The knee joints were fixed in 4 % paraformaldehyde, decalcified, and embedded in paraffin for histological and immunohistochemical assessments. Subsequently, the samples were sectioned, and hematoxylin and eosin (H&E), as well as Safranin-O/Fast Green staining, were conducted on coronal sections for further histological analysis. An enhanced Mankin scoring system was employed by two assessors to assess the pathological state of knee joints [27]. The sections were subjected to immunohistochemical staining through overnight rabbit polyclonal anti-COL2A1 (Servicebio, China) antibody incubation at 4 °C, and followed by a secondary antibody treatment for 1 h 3,3′-diaminobenzidine (DAB) substrate was used to stain the paraffin sections. ImageJ software was employed to quantitatively determine the expression level of COL2A1. The immunofluorescence staining was used, including iNOS (Proteintech, China), CD206 (Proteintech, China), MMP13 (Abcam, USA) and ADAMTS5 (Abcam, USA). After overnight of antibody incubation, the sections were subsequently treated with FITC or Cy3 Tyramide (Servicebio, China) for 30 min at 4 °C, DAPI incubation 5 min, followed by imaging using a fluorescent microscopy.
4.15. Statistical analysis
Statistical analysis was carried out through the use of IBM SPSS Statistics version 26 (SPSS Inc, USA). The comparison of the experimental data between the two groups were performed using the student-t test. One- or two-way analysis of variance (ANOVA) was used to compare the experimental data between the multiple groups. The value of p < 0.05 and p < 0.01 was considered to indicate significant difference and extremely significant difference, respectively.
Ethics approval and consent to participate
All the animal experiments were approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (Approval number: IACUC-CQMU-2023-0108). All the authors were in compliance with all relevant ethical regulations.
CRediT authorship contribution statement
Pengcheng Xiao: Conceptualization, Methodology, Investigation, Data curation, Writing – original draft. Xiaoyu Han: Methodology, Investigation, Data curation, Writing – original draft. Yanran Huang: Methodology, Investigation, Data curation, Writing – original draft. Jianye Yang: Methodology, Investigation, Data curation. Li Chen: Methodology, Investigation, Data curation. Zhengwei Cai: Methodology, Investigation, Data curation. Ning Hu: Conceptualization, Supervision, Resources. Wenguo Cui: Conceptualization, Visualization, Supervision, Resources, Funding acquisition, Project administration, Writing – review & editing. Wei Huang: Conceptualization, Supervision, Resources, Funding acquisition, Project administration.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China-Joint Fund Project (U22A20284); the National Natural Science Foundation of China (81972069, 82202724); Doctoral Cultivating Project of the First Affiliated Hospital of Chongqing Medical University (CYYY-BSYJSCXXM-202227; 202204).
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2023.09.010.
Contributor Information
Ning Hu, Email: huncqjoint@yeah.net.
Wenguo Cui, Email: wgcui@sjtu.edu.cn.
Wei Huang, Email: huangw511@163.com, huangwei68@263.net.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Hunter D.J., Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393:1745–1759. doi: 10.1016/S0140-6736(19)30417-9. [DOI] [PubMed] [Google Scholar]
- 2.Li Y., Xie W., Xiao W., Dou D. Progress in osteoarthritis research by the national natural science foundation of China. Bone Res. 2022;10:41. doi: 10.1038/s41413-022-00207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson W.H., Lepus C.M., Wang Q., Raghu H., Mao R., Lindstrom T.M., Sokolove J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016;12:580–592. doi: 10.1038/nrrheum.2016.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez-Lopez E., Coras R., Torres A., Lane N.E., Guma M. Synovial inflammation in osteoarthritis progression. Nat. Rev. Rheumatol. 2022;18:258–275. doi: 10.1038/s41584-022-00749-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao Q., Wu X., Tao C., Gong W., Chen M., Qu M., Zhong Y., He T., Chen S., Xiao G. Osteoarthritis: pathogenic signaling pathways and therapeutic targets. Signal Transduct. Targeted Ther. 2023;8:56. doi: 10.1038/s41392-023-01330-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hannemann N., Apparailly F., Courties G. Synovial macrophages: from ordinary eaters to extraordinary multitaskers. Trends Immunol. 2021;42:368–371. doi: 10.1016/j.it.2021.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Mass E., Nimmerjahn F., Kierdorf K., Schlitzer A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat. Rev. Immunol. 2023:1–17. doi: 10.1038/s41577-023-00848-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park M.D., Silvin A., Ginhoux F., Merad M. Macrophages in health and disease. Cell. 2022;185:4259–4279. doi: 10.1016/j.cell.2022.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang D., Chai X.-Q., Hu S.-S., Pan F. Joint synovial macrophages as a potential target for intra-articular treatment of osteoarthritis-related pain. Osteoarthritis Cartilage. 2022;30:406–415. doi: 10.1016/j.joca.2021.11.014. [DOI] [PubMed] [Google Scholar]
- 10.Van den Bossche J., Baardman J., Otto N.A., van der Velden S., Neele A.E., van den Berg S.M., Luque-Martin R., Chen H.-J., Boshuizen M.C.S., Ahmed M., Hoeksema M.A., de Vos A.F., de Winther M.P.J. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016;17:684–696. doi: 10.1016/j.celrep.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 11.He L., Jhong J.-H., Chen Q., Huang K.-Y., Strittmatter K., Kreuzer J., DeRan M., Wu X., Lee T.-Y., Slavov N., Haas W., Marneros A.G. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knab K., Chambers D., Krönke G. Synovial macrophage and fibroblast heterogeneity in joint homeostasis and inflammation. Front. Med. 2022;9 doi: 10.3389/fmed.2022.862161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Culemann S., Grüneboom A., Nicolás-Ávila J.Á., Weidner D., Lämmle K.F., Rothe T., Quintana J.A., Kirchner P., Krljanac B., Eberhardt M., Ferrazzi F., Kretzschmar E., Schicht M., Fischer K., Gelse K., Faas M., Pfeifle R., Ackermann J.A., Pachowsky M., Renner N., Simon D., Haseloff R.F., Ekici A.B., Bäuerle T., Blasig I.E., Vera J., Voehringer D., Kleyer A., Paulsen F., Schett G., Hidalgo A., Krönke G. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature. 2019;572:670–675. doi: 10.1038/s41586-019-1471-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buckley C.D., Ospelt C., Gay S., Midwood K.S. Location, location, location: how the tissue microenvironment affects inflammation in RA. Nat. Rev. Rheumatol. 2021;17:195–212. doi: 10.1038/s41584-020-00570-2. [DOI] [PubMed] [Google Scholar]
- 15.Perretti M., Cooper D., Dalli J., Norling L.V. Immune resolution mechanisms in inflammatory arthritis. Nat. Rev. Rheumatol. 2017;13:87–99. doi: 10.1038/nrrheum.2016.193. [DOI] [PubMed] [Google Scholar]
- 16.Moral J.A., Leung J., Rojas L.A., Ruan J., Zhao J., Sethna Z., Ramnarain A., Gasmi B., Gururajan M., Redmond D., Askan G., Bhanot U., Elyada E., Park Y., Tuveson D.A., Gönen M., Leach S.D., Wolchok J.D., DeMatteo R.P., Merghoub T., Balachandran V.P. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature. 2020;579:130–135. doi: 10.1038/s41586-020-2015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaplin D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010;125:S3–S23. doi: 10.1016/j.jaci.2009.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maier-Hauff K., Ulrich F., Nestler D., Niehoff H., Wust P., Thiesen B., Orawa H., Budach V., Jordan A. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J. Neuro Oncol. 2011;103:317–324. doi: 10.1007/s11060-010-0389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spoială A., Ilie C.-I., Motelica L., Ficai D., Semenescu A., Oprea O.-C., Ficai A. Smart magnetic drug delivery systems for the treatment of cancer. Nanomaterials. 2023;13:876. doi: 10.3390/nano13050876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang J., Mei J., Ma Y., Jiang S., Zhang J., Yi S., Feng C., Liu Y., Liu Y. Tumor hijacks macrophages and microbiota through extracellular vesicles. Explorations. 2022;2 doi: 10.1002/EXP.20210144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qu F., Guilak F., Mauck R.L. Cell migration: implications for repair and regeneration in joint disease. Nat. Rev. Rheumatol. 2019;15:167–179. doi: 10.1038/s41584-018-0151-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao W.-J., Liu J.-X., Liu M.-N., Yao Y.-D., Liu Z.-Q., Liu L., He H.-H., Zhou H. Macrophage 3D migration: a potential therapeutic target for inflammation and deleterious progression in diseases. Pharmacol. Res. 2021;167 doi: 10.1016/j.phrs.2021.105563. [DOI] [PubMed] [Google Scholar]
- 23.Jayme T.S., Leung G., Wang A., Workentine M.L., Rajeev S., Shute A., Callejas B.E., Mancini N., Beck P.L., Panaccione R., McKay D.M. Human interleukin-4-treated regulatory macrophages promote epithelial wound healing and reduce colitis in a mouse model. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aba4376. eaba4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao Z., Wang Z., Li G., Cai Z., Wu J., Wang L., Deng L., Cai M., Cui W. Injectable microfluidic hydrogel microspheres for cell and drug delivery. Adv. Funct. Mater. 2021;31 doi: 10.1002/adfm.202103339. [DOI] [Google Scholar]
- 25.Lin F., Wang Z., Xiang L., Deng L., Cui W. Charge‐guided micro/nano‐hydrogel microsphere for penetrating cartilage matrix. Adv. Funct. Mater. 2021 doi: 10.1002/adfm.202107678. [DOI] [Google Scholar]
- 26.Lei Y., Wang Y., Shen J., Cai Z., Zhao C., Chen H., Luo X., Hu N., Cui W., Huang W. Injectable hydrogel microspheres with self-renewable hydration layers alleviate osteoarthritis. Sci. Adv. 2022;8 doi: 10.1126/sciadv.abl6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei Y., Wang Y., Shen J., Cai Z., Zeng Y., Zhao P., Liao J., Lian C., Hu N., Luo X., Cui W., Huang W. Stem cell‐recruiting injectable microgels for repairing osteoarthritis. Adv. Funct. Mater. 2021 doi: 10.1002/adfm.202105084. [DOI] [Google Scholar]
- 28.Sridharan R., Cavanagh B., Cameron A.R., Kelly D.J., O'Brien F.J. Material stiffness influences the polarization state, function and migration mode of macrophages. Acta Biomater. 2019;89:47–59. doi: 10.1016/j.actbio.2019.02.048. [DOI] [PubMed] [Google Scholar]
- 29.Herrmann I.K., Wood M.J.A., Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021;16:748–759. doi: 10.1038/s41565-021-00931-2. [DOI] [PubMed] [Google Scholar]
- 30.EL Andaloussi S., Mäger I., Breakefield X.O., Wood M.J.A. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 31.Ikeda G., Santoso M.R., Tada Y., Li A.M., Vaskova E., Jung J.-H., O'Brien C., Egan E., Ye J., Yang P.C. Mitochondria-rich extracellular vesicles from autologous stem cell-derived cardiomyocytes restore energetics of ischemic myocardium. J. Am. Coll. Cardiol. 2021;77:1073–1088. doi: 10.1016/j.jacc.2020.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y.-J., Wu J.-Y., Liu J., Xu W., Qiu X., Huang S., Hu X.-B., Xiang D.-X. Artificial exosomes for translational nanomedicine. J. Nanobiotechnol. 2021;19:242. doi: 10.1186/s12951-021-00986-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu C., Ju D., Zhang X. Cell membrane-derived vesicle: a novel vehicle for cancer immunotherapy. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.923598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L., Chen X., Cai P., Sun H., Shen S., Guo B., Jiang Q. Reprogramming mitochondrial metabolism in synovial macrophages of early osteoarthritis by a camouflaged meta‐defensome. Adv. Mater. 2022;34 doi: 10.1002/adma.202202715. [DOI] [PubMed] [Google Scholar]
- 35.Xiao P., Zhu Z., Du C., Zeng Y., Liao J., Cheng Q., Chen H., Zhao C., Huang W. Silencing Smad7 potentiates BMP2-induced chondrogenic differentiation and inhibits endochondral ossification in human synovial-derived mesenchymal stromal cells. Stem Cell Res. Ther. 2021;12:132. doi: 10.1186/s13287-021-02202-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang F., Wei K., Slowikowski K., Fonseka C.Y., Rao D.A., Kelly S., Goodman S.M., Tabechian D., Hughes L.B., Salomon-Escoto K., Watts G.F.M., Jonsson A.H., Rangel-Moreno J., Meednu N., Rozo C., Apruzzese W., Eisenhaure T.M., Lieb D.J., Boyle D.L., Mandelin A.M., Accelerating Medicines Partnership Rheumatoid Arthritis and Systemic Lupus Erythematosus (AMP RA/SLE) Consortium. Boyce B.F., DiCarlo E., Gravallese E.M., Gregersen P.K., Moreland L., Firestein G.S., Hacohen N., Nusbaum C., Lederer J.A., Perlman H., Pitzalis C., Filer A., Holers V.M., Bykerk V.P., Donlin L.T., Anolik J.H., Brenner M.B., Raychaudhuri S. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019;20:928–942. doi: 10.1038/s41590-019-0378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin T., Liu Y., Ji W., Zhuang J., Chen X., Gong B., Chu J., Liang W., Gao J., Yin Y. Engineered mesenchymal stem cell-derived extracellular vesicles: a state-of-the-art multifunctional weapon against Alzheimer's disease. Theranostics. 2023;13:1264–1285. doi: 10.7150/thno.81860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siefen T., Bjerregaard S., Borglin C., Lamprecht A. Assessment of joint pharmacokinetics and consequences for the intraarticular delivery of biologics. J. Contr. Release. 2022;348:745–759. doi: 10.1016/j.jconrel.2022.06.015. [DOI] [PubMed] [Google Scholar]
- 39.Cao Y., Ma Y., Tao Y., Lin W., Wang P. Intra-articular drug delivery for osteoarthritis treatment. Pharmaceutics. 2021;13:2166. doi: 10.3390/pharmaceutics13122166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He Y., Sun M., Wang J., Yang X., Lin C., Ge L., Ying C., Xu K., Liu A., Wu L. Chondroitin sulfate microspheres anchored with drug-loaded liposomes play a dual antioxidant role in the treatment of osteoarthritis. Acta Biomater. 2022;151:512–527. doi: 10.1016/j.actbio.2022.07.052. [DOI] [PubMed] [Google Scholar]
- 41.Lim K.H., Huang H., Pralle A., Park S. Stable, high-affinity streptavidin monomer for protein labeling and monovalent biotin detection. Biotechnol. Bioeng. 2013;110:57–67. doi: 10.1002/bit.24605. [DOI] [PubMed] [Google Scholar]
- 42.Weng X., Maxwell-Warburton S., Hasib A., Ma L., Kang L. The membrane receptor CD44: novel insights into metabolism. Trends Endocrinol Metab. 2022;33:318–332. doi: 10.1016/j.tem.2022.02.002. [DOI] [PubMed] [Google Scholar]
- 43.McGonagle D., Baboolal T.G., Jones E. Native joint-resident mesenchymal stem cells for cartilage repair in osteoarthritis. Nat. Rev. Rheumatol. 2017;13:719–730. doi: 10.1038/nrrheum.2017.182. [DOI] [PubMed] [Google Scholar]
- 44.Heeb L.E.M., Egholm C., Boyman O. Evolution and function of interleukin-4 receptor signaling in adaptive immunity and neutrophils. Gene Immun. 2020;21:143–149. doi: 10.1038/s41435-020-0095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grillet B., Pereira R.V.S., Van Damme J., Abu El-Asrar A., Proost P., Opdenakker G. Matrix metalloproteinases in arthritis: towards precision medicine. Nat. Rev. Rheumatol. 2023;19:363–377. doi: 10.1038/s41584-023-00966-w. [DOI] [PubMed] [Google Scholar]
- 46.Zhao S.S., Karhunen V., Morris A.P., Gill D. ADAMTS5 as a therapeutic target for osteoarthritis: mendelian randomisation study. Ann. Rheum. Dis. 2022;81:903–904. doi: 10.1136/annrheumdis-2021-222007. [DOI] [PubMed] [Google Scholar]
- 47.Chen Z., Lv Z., Zhang Z., Weitz D.A., Zhang H., Zhang Y., Cui W. Advanced microfluidic devices for fabricating multi‐structural hydrogel microsphere. Explorations. 2021;1 doi: 10.1002/EXP.20210036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mao J., Saiding Q., Qian S., Liu Z., Zhao B., Zhao Q., Lu B., Mao X., Zhang L., Zhang Y., Sun X., Cui W. Reprogramming stem cells in regenerative medicine. Smart Medicine. 2022;1 doi: 10.1002/SMMD.20220005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z., Zhou Y., Li T., Zhang J., Tian H. Stimuli‐responsive hydrogels: fabrication and biomedical applications. View. 2022;3 doi: 10.1002/VIW.20200112. [DOI] [Google Scholar]
- 50.Cheng W., Zhang J., Liu J., Yu Z. Granular hydrogels for 3D bioprinting applications. View. 2020;1 doi: 10.1002/VIW.20200060. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.