

Abstract
The respiratory syncytial virus polymerase complex, consisting of the polymerase (L) and phosphoprotein (P), catalyzes nucleotide polymerization, cap addition, and cap methylation via the RNA dependent RNA polymerase, capping, and Methyltransferase domains on L. Several nucleoside and non-nucleoside inhibitors have been reported to inhibit this polymerase complex, but the structural details of the exact inhibitor-polymerase interactions have been lacking. Here, we report a non-nucleoside inhibitor JNJ-8003 with sub-nanomolar inhibition potency in both antiviral and polymerase assays. Our 2.9 Å resolution cryo-EM structure revealed that JNJ-8003 binds to an induced-fit pocket on the capping domain, with multiple interactions consistent with its tight binding and resistance mutation profile. The minigenome and gel-based de novo RNA synthesis and primer extension assays demonstrated that JNJ-8003 inhibited nucleotide polymerization at the early stages of RNA transcription and replication. Our results support that JNJ-8003 binding modulates a functional interplay between the capping and RdRp domains, and this molecular insight could accelerate the design of broad-spectrum antiviral drugs.
Subject terms: Cryoelectron microscopy, Viral infection
Cryo-EM structure and inhibition assays reveal a non-nucleoside inhibitor of the respiratory syncytial virus polymerase complex that acts by binding to an induced-fit pocket in the capping domain.
Introduction
Respiratory syncytial virus (RSV) is a leading cause of infant hospitalization for acute lower respiratory tract infections1 and poses a major health burden in the elderly2, immunosuppressed patients3, and patients carrying comorbidities4,5. While next-generation antibodies and promising new vaccines under development offer possibilities for efficient and large-scale prevention, additional strategies are required for allowing pre-exposure prophylactic and post-exposure treatments. Several virus-entry inhibitors targeting the RSV fusion protein have demonstrated efficacy in proof-of-concept phase II human challenge and patient studies6–9, but it is unknown whether they alone will provide the best antiviral efficacy or combination with other therapies would be more suitable. Notably, the most advanced post-entry inhibitors target either the viral nucleoprotein10 or the viral polymerase11,12.
The RSV polymerase complex is composed of the large protein (L), a multidomain protein having RNA-dependent RNA polymerase (RdRp), polyribonucleotidyl transferase (PRNTase) or capping, and methyltransferase (MTase) activities, and four phosphoproteins (P) (Fig. 1a)13,14. The RdRp domain is essential for both transcription and replication of the viral RNA genome. The capping and the MTase domains contribute to the formation of capped and methylated mRNAs and have been functionally mapped to conserved regions V and VI of the L protein, respectively15–18. Co-transcriptional mRNA 5’ cap addition is a critical step in mRNA synthesis that allows for the production of full-length, functional mRNA transcripts in RSV and the closely related vesicular stomatitis virus (VSV)16,19,20. The intrusion (amino acid (a.a.) 1325–1354) and priming (a.a. 1261–1282) loops of the capping domain play critical roles in the capping activities21,22. P acts as a crucial component to control the efficiency of L, interacting with nucleocapsid, and guiding the newly synthesized RNA into the L active sites during transcription and replication processes (Fig. 1a)23–25.
Fig. 1. JNJ-8003 is a picomolar NNI of RSV L + P.

a Domain architecture of RSV L + P. Regions protected by JNJ-8003 binding in an HDX assay and resistance mutations are indicated on top and bottom of RSV L architecture, respectively. CD Connector domain. MT, Cap methyltransferase. NTD N-terminal domain. OD Oligomerization domain. CTD C-terminal domain. b Chemical structure and selected inhibition data of JNJ-8003 (Supplementary Table 1, 2). c Inhibition of RNA synthesis activity of the recombinant RSV L + P by JNJ-8003 in a biotinylated primer extension Flashplate assay. Data points from four technical replicates are shown. d Sequencing gel showing incorporation of a single nucleotide (lane 1 and 4) and multiple nucleotides (lane 2, 3, 5, 6, and 7) to a 4-mer RNA primer. 5 µM JNJ-8003 was used in the assay. ALS-8112-TP is a CTP analog that terminates elongation after its incorporation71. Data points and Gel image are in Supplementary Data 1, 2.
During the past decade, significant advancements have been made in comprehending the spatial organization of the RSV polymerase26,27. The two structures of the RSV L + P complex provide a clear picture of the central L- RdRp and capping domains, and L P interactions. In contrast to the structures of the VSV L, likely illustrating pre-initiation or initiation stages with a putative priming loop extending into the RdRp active site28,29, both RSV L structures are postulated to depict an elongation stage with the analogous priming loop being withdrawn from the polymerase catalytic center26,27.
Inhibiting the RSV polymerase activity is a promising strategy to discover anti-RSV antivirals, and several nucleoside analog inhibitors (NIs)30,31 and non-nucleoside inhibitors (NNIs)32–37 have been identified. Despite numerous studies exploring their precise modes of action, the discovery of RSV polymerase inhibitors has been lacking structural information revealing the exact drug-target interactions. Here, we document a RSV polymerase NNI, JNJ-8003, with its viral polymerase target, resistance profile, and inhibition characteristics. The cryo-EM structure of the RSV L + P complex with JNJ-8003 uncovered an induced fit binding pocket on the capping domain and provided insight into the mechanism of action (MOA) of this complex, which will guide future efforts in structural-based drug discovery.
Results
JNJ-8003 is a potent small-molecule inhibitor of the RSV polymerase
Approximately 5000 analogs were synthesized to optimize the pharmacological properties of a series that emerged from the initial screening campaign hit. This iterative optimization process involved a combination of phenotypic and target-based antiviral assessments, leading to the discovery of JNJ-8003 (Fig. 1b)38. JNJ-8003 exhibited significant inhibition against RSV A2 replication in a reporter assay (EC50: 0.78 nM; CC50: 27 µM), moderate inhibition against the closely related human metapneumovirus (HMPV, EC50: 88 nM), and negligible inhibition against the distantly related parainfluenza viruses (PIV-1 and PIV-3) and VSV (Supplementary Table 1, 2). JNJ-8003 inhibited the replication of RSV A and B clinical isolates and laboratory strains similarly by RT-qPCR (EC50: 0.18 nM and 0.10 nM, respectively; Supplementary Table 1). Inhibition of an RSV subgenomic replicon assay (EC50: 0.15 nM; CC50: 11 µM; Supplementary Table 1) indicated that JNJ-8003 possesses a post-entry mode of action. In biochemical assays, JNJ-8003 inhibited the RdRp activity in both Flashplate and gel-based 4-mer primer extension assays using the recombinant L and P viral proteins (IC50: 0.67 nM; Fig. 1c, d, and Supplementary Table 1), indicating that the RSV L + P polymerase complex is the primary target for JNJ-8003. In contrast, JNJ-8003 did not inhibit human DNA polymerases α, β, and γ, human mitochondrial RNA polymerase, and human RNA polymerase II (IC50: >100 µM; Supplementary Table 1).
JNJ-8003 inhibits viral RNA replication and transcription at the initiation and early elongation stage
RSV polymerase catalyzes de novo RNA synthesis for its genome replication and mRNA transcription39–41. RSV genome replication starts at the first nucleotide of its genome and anti-genome (+1 site at the 3’-end of the leader or trailer promoter sequence). RNA transcription starts at the third nucleotide of its genome (+3 site at the 3’-end of the leader sequence); after making a short RNA transcript, which is abortive, the polymerase scans to the first gene start (gs) signal to restart RNA transcription and makes the first capped mRNA39. The inhibitory effect of JNJ-8003 on RSV genome replication and mRNA transcription was confirmed by a cell-based minigenome assay, in which both the antigenome RNA and mRNA showed dose dependent reduction upon JNJ-8003 treatment (Supplementary Fig. 1a, b). Further analysis of these RNA molecules indicated that JNJ-8003 inhibited RNA synthesis both from the +1 (1U) and +3 (3C) initiation sites within the 44 nt leader (le) promoter region, suggesting it inhibits the initiation or early elongation of both RNA replication from the +1 site and RNA transcription from the +3 site in the le promoter (Supplementary Fig. 1c).
To determine whether JNJ-8003 inhibits RNA synthesis at the early elongation steps, primed single nucleotide incorporation (SNI) assays with a set of short RNA primers complementary to the 3’end of an 11-mer leader (le-11) and 14-mer trailer (tr−14) promoter sequences were performed with the recombinant RSV L + P. JNJ-8003 inhibited nucleotide addition to a 2-, 4-, 5- or 6-mer primer complementary to le−11 (Fig. 2a) or tr-14 starting from the +1 site, representing primed RNA replication (Supplementary Fig. 2, 3). JNJ-8003 also inhibited SNI to a 2-, 3-, 4- and 5-mer primer complementary to le−11 starting from the +3 site representing primed RNA transcription (Fig. 2b). However, JNJ-8003 did not inhibit SNI to the 3-mer primer from the +1 site for both the le and tr promoter RNA, suggesting a distinctive SNI mechanism unaffected by JNJ-8003 (Fig. 2a, c, d, f, g). The IC50 values of JNJ-8003 in the SNI assays ranged from 17 to 32 nM, indicating JNJ-8003 can inhibit primed RNA elongation from as early as a dinucleotide primer for both +1 and +3 site-initiated RNA synthesis (Fig. 2f–h). De novo RNA synthesis assay from the tr−14 RNA template was performed to test if JNJ-8003 inhibits de novo dinucleotide formation. JNJ-8003 inhibited pppGpA dinucleotide formation initiated from the +3 site with an IC50 of 5.1 nM (Supplementary Fig. 4). The various IC50 values observed in these assays likely reflected the IC50 walls determined by the actual active enzyme concentrations but not the true potency of the compound. In sum, our current data supports the conclusion that JNJ-8003 inhibits RNA transcription as early as at the de novo dinucleotide formation step and RNA replication at the AC to ACG extension step. Since the assay development of terminal de novo formation of pppApC from the +1 site had not been successful in our hand yet, the direct effects of JNJ-8003 on de novo dinucleotide formation starting from the +1 site are unknown. Finally, JNJ-8003 had no impact on MTase activity, using a recombinant MTase-CTD fragment, or full-length RSV L + P protein (Supplementary Fig. 5).
Fig. 2. JNJ-8003 inhibits RNA synthesis at early elongation steps.

a Polyacrylamide gel showing single nucleotide incorporation (SNI) from a set of short primers with or without 5 µM JNJ-8003 with le−11 template (+1 site RNA synthesis). b Sequencing gel showing primer extension from a set of short primers with or without 5 µM JNJ-8003 with the le−11 RNA template (+3 site RNA synthesis). c Inhibition of SNI by JNJ-8003 at 5 µM to a set of primers with le−11 RNA template (+1 site RNA synthesis). Bars indicate the mean, and the data points from two independent experiments are shown. The gels are shown in Supplemental Data 1. d Inhibition of SNI by JNJ-8003 at 5 µM to a set of primers with tr−14 RNA template (+1 site RNA synthesis). e Inhibition of SNI by JNJ-8003 at 5 µM to a set of primers with le−11 RNA template (+3 site RNA synthesis). f–h Inhibition curves of JNJ-8003 on SNI with le−11 (+1 site), tr−14 (+1 site), and le−11 (+3 site synthesis), respectively. Data points from two independent experiments and fitted curves are shown. Gel images are in Supplementary Fig. 3 and Supplementary Data 3–10.
JNJ-8003 targets the capping domain of RSV L
JNJ-8003 stabilized the purified RSV L + P complex with an increase in melting temperature (Tm) of 6.0 °C in a thermal shift assay (Supplementary Fig. 6a, b). Surface plasmon resonance (SPR) demonstrated JNJ-8003 bound to the RSV L + P complex with a Kd of 0.84 nM, an on-rate of 2.16 µM−1 s−1, and a slow off-rate of 0.0000181 s−1. Escape mutations were selected at 1x EC90 concentrations of JNJ-8003 (Supplementary Table 3). Compared to Ribavirin, BI-D, or AZ-27, JNJ-8003 showed a distinctive resistance profile (Supplementary Table 3, 4)34,42,43. C1388G amino acid substitution appeared independently in the capping domain of RSV L (Fig. 1a, Supplementary Fig. 7, Supplementary Table 3, 4). Hydrogen-deuterium exchange (HDX) mass spectrometry analysis confirmed three regions in the capping domain showed reduced deuterium uptake in the presence of JNJ-8003 (Fig. 1a, Supplementary Fig. 8) suggesting JNJ-8003 binds within the capping domain.
Cryo-EM structure of RSV L + P complex with JNJ-8003 explains its potency
Single-particle cryo-electron microscopy (cryo-EM) analysis of RSV L + P in complex with JNJ-8003 resulted in a 3D reconstruction with a global resolution of 2.9 Å (Fig. 3a, Supplementary Fig. 9, Table 1). Similar to the Apo structure (PDBID: 6PZK), the L- RdRp and capping domains, and the P oligomerization domains (OD) and their partial C-terminal domains (CTD) were visible in the model. JNJ-8003 binding only induced subtle conformational changes of the L- RdRp and capping domains, with a root-mean-square deviation (RMSD) value of 0.41 Å. The priming loop shielded the conserved capping domain catalytic ‘HR’ motif (H1338, and R1339), leaving the nucleotide polymerization active-site wide open in an RNA elongation mode (Fig. 3b).
Fig. 3. Cryo-EM structure reveals the binding environment of JNJ-8003.

a Cryo-EM map of RSV polymerase complex with JNJ-8003. The RdRp, capping, and JNJ-8003 were colored in slate, green, and yellow, respectively. The EM density map display threshold was adjusted to enhance the visibility of JNJ-8003. b Close view of the priming loops and connecting regions from Apo or JNJ-8003 bound RSV (green) and VSV (cyan). Two additional putative docking sites (Site0 and Site1) were highlighted. The transitions of the priming loop and α54 were indicated using curve arrows. RSV L + P and JNJ-8003 were shown as cartoons, and sticks, respectively. c JNJ-8003 binding pocket. JNJ-8003 and residues within 4 Å were shown in sticks. d Two-dimensional interaction patterns of JNJ-8003 and RSV L were highlighted using dashed lines. One putative water molecule was shown as a red sphere. e The ligand binding pocket from the Apo RSV L. f Structural comparison at ligand binding pocket between JNJ-8003 bound and Apo RSV L. Rotamer transitions on H1338 and F1385 were highlighted using arrows. g Comparison of JNJ-8003 unbound NMR structures (pink and green) and the bound cryo-EM structure in the RSV L + P complex (yellow). Conformational transitions upon binding were highlighted using arrows. h, i Structural comparison at the ligand binding pockets from the Apo HMPV (H), and VSV (I). The side chains of key residues were shown in sticks.
Table 1.
Data collection, reconstruction, and model refinement statistics of cryo-EM structures.
| RSV L + P JNJ-8003EMD-29452, PDB: 8FU3 | |
|---|---|
| Data collection | |
| Microscope | Titan Krios |
| Voltage (keV) | 300 |
| Nominal magnification | 105000x |
| Exposure navigation | Image Shift |
| Electron exposure (e /Å2) | 52.66 |
| Total exposure time (sec) | 1.5 |
| Detector | K3 Summit |
| Pixel size (Å)* | 0.825 |
| Defocus range (µm) | −1.4 to −2.3 |
| Micrographs Used | 11,631 |
| Final Refined particles (no.) | 519,118 |
| Reconstruction | |
| Symmetry imposed | C1 |
| Resolution (global) | |
| FSC 0.143 | 2.88 Å |
| Applied B-factor (Å2) | −127 |
| Refinement | |
| Protein residues | 1657 |
| Ligand | 1 |
| Map Correlation Coefficient (Main chain) | 0.73 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.014 |
| Bond angles (°) | 1.01 |
| Ramachandran | |
| Outliers | 0.54% |
| Allowed | 1.57% |
| Favored | 97.89% |
| Rotamer outliers | 0.07% |
| MolProbity score | 0.93 |
| EMRinger score | 4.22 |
| Clashscore (all atoms) | 1.57 |
*Calibrated pixel size at the detector.
Non-protein density was observed at the capping domain and was modeled as JNJ-8003 (Fig. 3a, c). JNJ-8003 filled a mostly hydrophobic pocket of 397 Å3 volume. Noteworthy was the proximity of the ligand to its resistance substitution C1388G (Supplementary Fig. 9, Supplementary Table 3, 4). JNJ-8003 made multiple interactions with residues in this pocket. The cinnoline ring of JNJ-8003 made hydrogen-bond and π-stacking interactions with the guanidinium group of R1345. One weak van-der-Waals interaction was observed between the methoxy group of JNJ-8003 and S1266 from the priming loop. The amide group linker of JNJ-8003 intercalated with hydrogen bonds from the C = O of I1381 and NH of F1385. The (S)-OH group stereo-specifically interacted with the NH and C = O of I1381. The CF3 group remained solvent accessible. Both the cinnoline ring and CF3 groups had the potential to form van-der-Waals interactions with H1338 from the ‘HR’ motif in the capping active site. The OH group of the isopropanol substituent was in contact with the C = O of L1372 and could potentially form a hydrogen bond that forms essential interactions with the backbone groups of the protein. The fluoro-phenyl group interacted with F1385 with a T-shaped π-π stacking. WaterMap simulation predicted multiple water molecules around JNJ-8003, especially the one bridging the hydrogen-bond interaction between Q1386 and JNJ-8003 (Fig. 3d, Supplementary Fig. 10).
The comparison of the ligand binding site from the JNJ-8003 bound and the Apo structures revealed notable side chain flips of F1385 and H1338 (Fig. 3c, e, f, Supplementary Movie 1). The side chains of F1385 and H1338 from the Apo structures produced steric clashes with the superimposed JNJ-8003 and had to reorganize to allow the observed binding mode. The solution NMR conformations of the free JNJ-8003 adopted mostly a V-shaped conformer (Fig. 3g, Supplementary Fig. 11, Supplementary Table 5, 6) with the pyridine-cinnoline rings spatially close with an angle of ~60°. This was substantially different from the extended pose of JNJ-8003 in the cryo-EM structure, indicating induced fits in both the RSV L capping domain and JNJ-8003 upon binding (Fig. 3f, g).
JNJ-8003 binding site explains its specificity
RSV and HMPV L proteins share 48% overall sequence identity, but the JNJ-8003 binding sites are highly conserved with an RMSD of 1.29 Å, and with I1383 and C1388 changed to Leucine and Alanine in HMPV L, respectively (Fig. 3c, h, Supplementary Fig. 12). In contrast, the homologous pocket is strikingly different in VSV L (Fig. 3c, i, Supplementary Fig. 12). VSV L hosts several residues with larger side chains within the corresponding JNJ-8003 binding site. In particular, S1221, I1368, N1369, I1381, I1383, and V1384 in RSV L are changed to V1103, M1257, R1258, F1265, F1267, and L1268 in VSV L, respectively. In addition, the R1233, and F1239 in the VSV L intrusion loop (R1345, and F1349 in RSV L, respectively) within the intrusion loop point away from the JNJ-8003 binding site (Fig. 3c, i, Supplementary Fig. 12). It is interesting to note that H1227 adopts a rotamer similar to the H1338 observed in the JNJ-8003 bound RSV L + P structure, suggesting that H1227 (H1338 in RSV L) may have various rotamers during capping process. These structural differences provide a molecular basis for why JNJ-8003 specifically inhibits RSV and HMPV, but not VSV, PIV-1, or −3 (Fig. 1b, Supplementary Fig. 12), and they further provide a rational for designing a new generation of broad-spectrum polymerase inhibitors targeting negative-sense single-stranded RNA (-ssRNA) viruses of members of the order Mononegavirales.
The JNJ-8003 binding pocket may participate in the capping activities
Mononegavirales polymerase adopts a distinctive mechanism for mRNA capping formation, as exemplified by the VSV polymerase. The mRNA capping involves the covalent attachment of the monophosphorylated 5’ terminus of the nascent transcript (pRNA) to a histidine side chain from the ‘HR’ motif (H1338 in RSV L) forming a covalent enzyme (L-pRNA). This is followed by the transfer of pRNA onto GDP (Gpp), ending with the GpppRNA capped product44,45 (Supplementary Fig. 13a). The development of the capping assays recapitulating these events using the recombinant RSV L + P complex was not successful yet, therefore the direct effect of JNJ-8003 on capping could not be tested. However, JNJ-8003 binding opens a ‘gate’ (H1338 and F1385), creating a novel connected cavity that could potentially be involved in capping (Fig. 4a). The guanosine diphosphate (GDP) guanine base can be docked to the left or right sides of the cavity with the phosphate group pointing towards the conserved ‘HR’ motif (Fig. 4b). Interestingly, GDP did not bind to the purified RSV L + P, suggesting GDP alone may not be sufficient to open the ‘gate’ for capping (Supplementary Fig. 13b). Further research involving structural studies on the RSV L + P complex with pre-mRNA, both in the presence and absence of GDP, and additional biochemical analyses are required to uncover the capping mechanism of RSV. Our structure sheds light on the plasticity of the capping domain that may be involved in modulating the capping activity and provides the molecular basis of an otherwise hidden druggable site for future inhibitor design.
Fig. 4. The JNJ-8003 binding pocket may be involved in RNA capping.
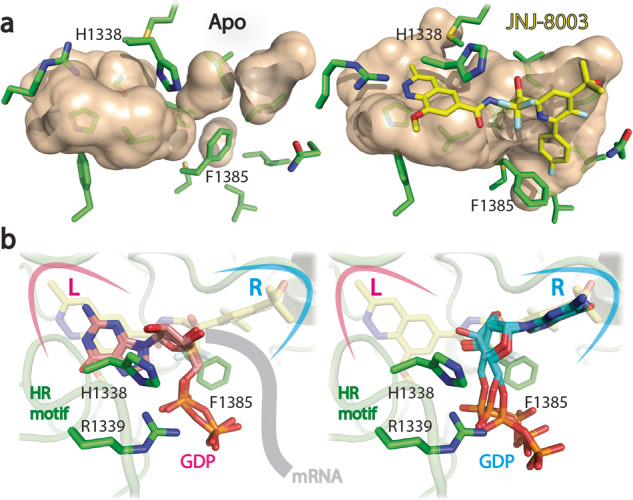
a Molecular surfaces of JNJ-8003 binding sites from Apo (left) and JNJ-8003 bound (right) structures. b Two different docking poses of GDP with the guanine nitrogenous base pointing to the left or right sides of the ligand binding site were shown in the left or right panels, respectively. JNJ-8003 (yellow) and GDP (pink or cyan) were shown as sticks.
Discussion
Here we employed multiple approaches to assess the mechanism of action of a RSV non-nucleoside inhibitor, JNJ-8003. The thermal shift assay and the SPR results demonstrated that JNJ-8003 binds tightly to the RSV L + P complex. Our structure showed JNJ-8003 binds at the capping domain forming multiple interactions with residues consistent with the resistant mutation profile. The inhibition of de novo RNA synthesis from a tr−14 RNA template indicated that JNJ-8003 inhibits de novo initiation of dinucleotide formation from the +3 site. The SNI primer extension assays showed JNJ-8003 can inhibit all primer extension events, except for ACG to ACGC or ACGA extension using the le−11 or tr−14 template respectively. This step may represent a distinctive step during the early elongation stage or the initiation to elongation transition stage, and further characterization of RNA synthesis at this stage is underway. Since the enzymatic activity of primer extension of the 6-mer primer was already low (Fig. 2a), suggesting RSV polymerase could not efficiently accept exogenous longer primer as substrate, the effect of JNJ-8003 on longer primer extension was not studied and its direct effect on productive RNA chain elongation in the middle or late stage of RNA synthesis is unknown. Inhibition of the de novo initiation and short primer extension activities by JNJ-8003 eventually leads to the inhibition of RNA genome replication and mRNA transcription in the minigenome assay and viral replication in the antiviral assays. Thus JNJ-8003 binding at the capping domain completely shut down the RdRp function of RSV L + P complex.
Several NIs and NNIs had been explored to inhibit the function of RSV polymerase (Supplementary Fig. 14a). While most of their binding sites on RSV L remained unknown, the locations of their resistance mutations had been mapped on RSV L and provided insight into their MOA (Supplementary Fig. 14b–d). Unlike VSV L + P, a structure of full-length RSV L is not currently available. However, during manuscript preparation, the availability of Alphafold246 provided a glimpse of a model of full-length RSV L, which will require validation using experimental cryo-EM structures in the future. The binding sites of YM-5340332, AZ-2733,34, PC78635, AVG-23336, and AstraZeneca inhibitor cpd 137 had been mapped to the connecting domain of an alphafold2 predicted model. Escape mutations in residues L1502, Y1631, and H1632 are clustered at the N-terminal boundary of the connecting domain of an alphafold2 predicted model (Supplementary Fig. 14d). Inhibitor binding did not interfere with the L P interaction but might disrupt the transition of the priming loop from preinitiation to elongation states36.
The binding site of BI-compound D (BI-D) was proposed to be in the capping domain evidenced by three resistance substitutions (I1381S, L1421F, or E1269D) in this domain (Supplementary Fig. 14c)47. In comparison to the resistance profile of JNJ-8003, BI-D was unlikely to target the JNJ-8003 binding site. Our structure revealed that the van-der-Waals interaction between C1388 and the fluorobenzene group of JNJ-8003 was disrupted by substitution with glycine, reducing the potency of JNJ-8003 by more than 200-fold (Supplementary Table 4). However, this substitution did not impact the inhibition potency of BI-D. Further, BI-D could not be docked into the JNJ-8003 pocket because of its larger size (Supplementary Fig. 14a, d). All these results suggested BI-D target a pocket near the JNJ-8003 binding site with a different MOA. Indeed, BI-D inhibited mRNA transcription differently from JNJ-8003. Short abortive RNA products (<50 nucleotides) were increased while full mRNA transcripts were inhibited in the presence of increased BI-D47. Later, it was found that BI-D increased RNA products initiated from the +3 site but had no significant effect on products from the +1 site, and it specifically reduced capped RNA formation21. Our polyacrylamide gel results indicated that JNJ-8003 inhibited primed RNA extension as early as from a 2-mer primer, during the early elongation stages of both transcription and replication (Fig. 2). It also was able to inhibit de novo dinucleotide initiation on the tr−14 template from the +3 position (Supplementary Fig. 4).
The capping domain plays an essential role in productive transcription and replication in addition to its capping activities21,22. Substitutions (P1261, W1262, G1264, T1267, E1269, N1335, and R1339) within the intrusion and priming loops of the capping domain were shown to cause defects in transcription and replication21,22. JNJ-8003 forms extensive interactions with L1337, H1338, R1345, and F1349 of the intrusion loop, potentially impacting the RdRp and capping activities (Figs. 3c, 5a). Moreover, the priming loop, which hosts the putative priming residues (P1261, and W1262) to stabilize the initiating NTPs during de novo initiation, undergoes large conformational changes from the pre-initiation/initiation state to the elongation state (Figs. 3b, 5b)22. Our results show that JNJ-8003 directly contacts S1266 of the priming loop with a van-der-Waals interaction (Fig. 5a), potentially reducing the mobility of the priming loop. The a.a. 660-688 is within the non-resolved regions of the RSV RdRp domain and does not have a direct interaction with JNJ-8003. An AlphaFold2-predicted RSV L model indicated that L659T660R661 sits at the top of an α helix, positioned next to the G810D811N812 active site, which may directly or allosterically coordinate with the priming loop during the pre-initiation/initiation and/or early elongation steps (Fig. 5b).
Fig. 5. Proposed mode of action of JNJ-8003.
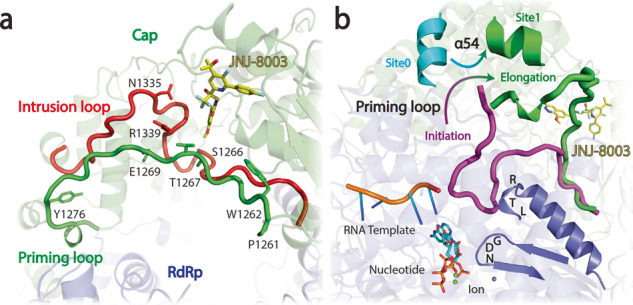
a Zoomed-in view of JNJ-8003 binding site with the priming and intrusion loops colored in red and green, respectively. Side chains of substitutions in the capping domain that cause defects in transcription and replication were shown. Side chain of S1266 forming direct interaction with JNJ-8003 was shown. b Overlay of RSV L derived from AlphaFold2 (Supplementary Fig. 14c), VSV L initiation complex72 models, and RSV L + P with JNJ-8003 bound structure. The priming loop was highlighted in magenta or green for pre-initiation/initiation or elongation states, respectively. The transition of the priming loop was indicated using curve arrow. RNA template, dinucleotides, and metal ions were shown in cartoons, sticks, and spheres, respectively. The L659T660R661 and G810D811N812 regions were highlighted. JNJ-8003 was shown in yellow sticks.
The binding of JNJ-8003 resulted in a novel pocket around the ‘HR’ motif that was not present in the Apo RSV, HMPV, VSV polymerase structures (Fig. 4). We hypothesize that this pocket may play a role in the capping process due to its location and size. The capping process needs meticulous coordination between newly synthesized uncapped mRNA and GDP near the ‘HR’ motif to ensure efficient mRNA capping (Supplementary Fig. 13a, b). The plasticity of the capping domain highlighted in our JNJ-8003 bound RSV L + P structure provides a rationale for developing a new generation of broad-spectrum polymerase inhibitors to combat Mononegavirales viruses.
Methods
Inhibition of rgRSV224 reporter RSV in HeLa cells
Black 384-well clear-bottom microtiter plates (Corning, Amsterdam, The Netherlands) were filled via acoustic drop ejection using the echo liquid handler (Labcyte, Sunnyvale, California). 200 nL of compound stock solutions (100% DMSO) were transferred to the assay plates. 9 serial 4-fold dilutions of compound were made. The assay was initiated by adding 20 µL of culture medium to each well (RPMI medium) without phenol red, 10% FBS-heat inactivated, 0.04% gentamycin (50 mg/mL) containing rgRSV22448 virus (multiplicity of infection (MOI) = 1). rgRSV224 virus is an engineered virus that includes an additional GFP gene48 and was in-licensed from the NIH (Bethesda, MD, USA). All addition steps are done by using a multidrop dispenser (Thermo Scientific, Erembodegem, Belgium). Finally, 20 µL of a HeLa cell suspension (3000 cells/well) were plated. Medium, virus- and mock-infected controls were included in each test. The wells contain 0.05% DMSO per volume. Cells were incubated at 37 °C in a 5% CO2 atmosphere. Three days post-virus exposure, viral replication was quantified by measuring GFP expression in the cells with an EnVision Multimode Plate Reader (Perkin Elmer, Zaventem, Belgium). The EC50 was defined as the 50% inhibitory concentration for GFP expression. In parallel, compounds were incubated for three days in a set of white 384-well microtiter plates (Corning) and the cytotoxicity of compounds in HeLa cells was determined by measuring the ATP content of the cells using the ATPlite kit (Perkin Elmer, Zaventem, Belgium) instructions and a ViewLux uHTS Microplate Imager apparatus (PerkinElmer, Zaventem, Belgium) according to the manufacturer’s instructions. The CC50 was defined as the 50% concentration for cytotoxicity.
Inhibition of RSV-A and B clinical isolates and laboratory strains
96 well blackview microtiter plates were filled with JNJ-8003 in quadruplicate in a 9 points 4-fold compound dilution starting (final volume of 50 µl) in RPMI-1640 supplemented with 10% fetal calf serum (FCS), 25 mM HEPES, 10 mM L-glutamine, and 0.02 µg/ml gentamicin. Then, 100 µL of a HeLa cell suspension (5 × 104 cells/mL) in culture medium was added to each well. Next, 50 µL of the appropriate virus dilution was added and cells were incubated at 37 °C in a 5% CO2 atmosphere. For each virus strain (Supplementary Table 7), virus stock dilutions with minimal intracellular cross point (Cp) values ranging from 19 to 28 and with minimal intra-group variation (Standard deviation [SD] <1) were selected for the antiviral assay. After 3 days of incubation, the supernatant was removed from wells. Plates were washed twice with ice-cold PBS (100 µL/well), and were then sealed and stored overnight at −80 °C. Next, 50 µL of lysis solution supplemented with DNAse was added to the plates and incubated for 5 min followed by the addition of 5 µL stop solution (Ambion Cell-to-CT bulk lysis reagent buffer). Plates were placed 2 minutes at room temperature and transferred thereafter on ice before being tested by RT-qPCR for positive strand RSV RNA.
In brief, cell lysate was added to RSV-A or RSV B or β-actin reverse primers (Supplementary Table 8) at 0.27 µM before initial denaturation (75 °C for 5 s). The product of the reaction was mixed with 10x PCR buffer BI (containing 15 mM MgCl2), MgCl2 (3.5 mM), dNTP (1 mM), RNase inhibitor (1 U/µL), and Expand Reverse Transcriptase (1 U/µL; Roche) for cDNA synthesis. Reverse transcription was performed at 42 °C for 30 min, followed by denaturation at 95 °C for 5 min. The PCR reaction was prepared by adding 2x master mix (Roche), RSV-A or RSV-B forward primer mixes at 0.3 µM, RSV-A or RSV-B reverse primers mix at 0.3 µM, RSV-A or RSV-B probes at 0.1 µM, β-actin forward primer at 0.3 µM, β-actin reverse primer at 0.3 µM, β-actin probe at 0.1 µM to the cDNA product (Supplementary Table 8). The reaction consisted of a denaturation step (95 °C for 600 s), 45 cycles of denaturation (95 °C for 15 s) and annealing/elongation (60 °C for 60 s), and a step of cooling (40 °C for 10 s).
Assuming that a change of 1 Cp value is equivalent to a 2-fold difference in original RNA content, EC50 value (inhibitor concentration leading to a 2-fold reduction of the viral RNA content as compared to the virus control) and EC90 value (inhibitor concentration leading to 10-fold reduction of the viral RNA content as compared to the virus control) were calculated from corresponding Cp values by graphic interpolation.
Minireplicon assay
The RSV subgenomic replicon system was licensed from Apath (Apath, NY, USA)49,50. The BHK-derived cell line (APC-126) containing the stable RSV A2 replicon was cultured in DMEM/Hams F-12 medium supplemented with 10% (v/v) FBS (Gibco), 1% (v/v) penicillin (10,000 units/mL) and streptomycin (10,000 µg/mL) solution (Gibco), 1% (v/v) MEM non-essential amino acids solution (Gibco), 5% (v/v) Tryptose Phosphate Broth solution (29.5 g/l) (Sigma-Aldrich) and 10 µg/ml of blasticidin solution (Invivogen). Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere. 5000 cells were plated in 96-well clear plates (Corning) in 100 µl/well of medium. On the following day, the tested compound was solubilized in 100% DMSO before being transferred in duplicate to the culture plate with 9 points 3-fold serial dilution starting at 10 µM for EC50 determination or 100 µM for CC50 determination (100 µl final volume; 1% DMSO final concentration). Cells were incubated with compounds for 3 days at 37 °C in a 5% CO2 atmosphere before measurement of the luciferase readout (Promega Renilla Glo assay, 100 µl/well of reagent). Cell viability (CC50 and CC90) was measured with a CellTiter-Glo cell proliferation assay (Promega, 100 µl/well of reagent). Results were expressed as EC50 and EC90 values as well as CC50 and CC90 values.
Resistance selection experiments
Resistance selection was performed on a recombinant RSV A2 strain harboring an enhanced green fluorescent protein (EGFP) reporter gene (rgRSV224)48, in HeLa cells (ATCC, Manassas, VA, USA), in triplicate, starting with a MOI of 0.01 and maintaining a compound pressure of 3 or 10 nM (1 or 3x EC90, respectively). Passages were performed when 90% of the culture appeared infected (GFP+). At each passage, culture supernatants were collected to inoculate a new culture flask. A fraction of the collected culture supernatant was RNA-sequenced by next generation sequencing (NGS). A cut-off value of 15% of the viral RNA population frequency, in comparison to the original sequence and non-treated control, was applied to eliminate background signals. Selected passages were also subjected to limiting dilution, to isolate single resistant mutants and evaluate shift in JNJ-8003 potency. Mutations confirmed by a > 10 shift in JNJ-8003 potency, or appearing in multiple replicates upon compound pressure, were considered as resistant mutations.
RSV minigenome assay
RSV transcription and RNA replication were analyzed using a replication-deficient minigenome, in which the minigenome template contained a single nucleotide substitution in the trailer region, limiting it to the antigenome step of RNA replication and uncoupling transcription from replication51. Minigenome transcription and replication were reconstituted intracellularly. BSR-T7 cells, a T7 RNA polymerase expressing cell line, were cultured in a 6-well dish and transfected with T7-driven plasmids that expressed the minigenome template (200 ng/ well), and the RSV proteins N (400 ng/ well), P (200 ng/ well), L (100 ng/ well) and M2-1 (100 ng/ well). Transfections were performed using Lipofectamine (Thermofisher) according to the manufacturer’s directions. The compound JNJ-8003 was diluted in DMSO and added to the media at the time of transfection at the concentrations indicated. DMSO was maintained at a consistent concentration in each transfection reaction. At 40–48 h post transfection, cells were harvested, and total intracellular RNA was purified using Trizol (Thermofisher) according to the manufacturer’s directions, except that following the isopropanol precipitation step, the RNA was subjected to an additional round of purification by phenol-chloroform extraction and ethanol precipitation. Input minigenome RNA, antigenome RNA, and mRNAs were detected by subjecting RNA to electrophoresis in 1.5% agarose-formaldehyde gels in MOPS buffer, followed by Northern blotting using 32P labeled riboprobes specific to negative sense minigenome (to detect the input RNA) and to positive sense antigenome and mRNAs. The same RNA samples were also analyzed by primer extension using 32P end-labeled primers corresponding in sequence to nucleotides 15–39 of the leader region (to detect RNA initiated from the +1U and +3 C sites on the promoter). Primer extension analysis to detect the RNAs initiated at +1U and +3 C was performed by reverse transcription at 37˚C using Sensiscript reverse transcriptase52. The +1U and +3 C primer extension products were migrated on 8% acrylamide gels containing 7 M urea gel in tris-borate-EDTA buffer. Quantification was performed by phosphorimage analysis. In each case, the signal from the -L lane was subtracted from the signal values for each of the other lanes, to account for background and RNA products generated by the RSV polymerase were normalized to the level of input minigenome RNA, which was used as a control for transfection efficiency.
Expression and purification of the RSV L + P complex
Codon-optimized Human RSV L protein (strain A2) with an N-terminal twin StrepTag and RSV P protein (strain A2) with a C-terminal 6x His-tag were subcloned into the pFastBac Dual vector, and the baculovirus was prepared using standard procedures. L + P complex was expressed in Sf9 cells by infecting with the recombinant baculoviruses at a multiplicity of infection (MOI) of 5 pfu/cell for 72 h post-infection. Cells were lysed by Dounce homogenization and passing through the microfluidizer in lysis buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM TCEP) supplemented with Complete EDTA-free Protease Inhibitor Cocktail (Roche) and 20 U/mL Benzonase (EMD Millipore). After clarification by high-speed centrifugation, the cell lysate was loaded onto a StrepTactin-XT column (Cytiva), and the bound protein was eluted using 50 mM biotin in Strep Buffer A (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM TCEP). The eluted L + P complex was pooled and diluted with half volume of heparin buffer A (50 mM Tris-HCl, 10% glycerol, and 1 mM TCEP, pH 8.0), and was further purified using a heparin column (Cytiva). The L + P complex was eluted from the heparin column using a NaCl gradient from 200-500 mM over 15 column volumes. The peak fractions containing pure L + P was pooled and supplemented with NaCl to bring the final NaCl concentration to 500 mM and concentrated with an Amicon Ultra 100 kDa cutoff centrifugal filter. The concentrated sample was loaded on to a size-exclusion column (Superpose 6 Increase 16/600, Cytiva) equilibrated in 50 mM HEPES, 500 mM NaCl, 5% glycerol, and 1 mM TCEP, pH 7.5. The fractions at the middle of the peak containing pure L + P complex were pool, concentrated to ~0.8 mg/ml, flash-frozen, and stored at −80 °C. The quality of purified proteins was analyzed by SDS-PAGE gel.
Biotinylated primer extension assay using recombinant RSV P + L
RNA polymerase reaction samples consisted of recombinant RSV L-P polymerase (3 nM) in a buffer containing Tris pH 7.5 (20 mM), KCl (10 mM), dithiothreitol (2 mM), Triton (0.01%), DMSO (10% v/v) with compound at appropriate dilution (10 points 1:5-fold serial dilution starting at 1 µM), and MgCl2 (6 mM). Reactions were started at 30 °C by adding an oligonucleotide template sequence derived from the RSV leader promoter (5’-UUUGUUCGCGU-3’, 0.2 μM) together with a biotinylated primer (5’-bi-ACGC, 4 μM) mixed with GTP (10 µM), ATP (10 µM) and [α-33P]CTP (0.025 µM, 3000 Ci/mmol) in a final volume of 10 μL. Reactions were stopped after 120 min by the addition of 90 µl EDTA (0.1 M). Samples were transferred to Flashplates and left 1 h at room temperature for incubation before being washed twice with 0.1% Tween 20, sealed (Topseal) and read on a Wallac MicroBeta TriLux Liquid Scintillation Counter.
Single and multiple nucleotide incorporation assay using recombinant RSV P + L
RNA polymerase reaction samples consisted of 0.2 μM recombinant RSV L-P polymerase, 0.2 μM of an RNA template (Le11 or Tr14), and 400 μM of a short primer (sequences as indicated in the figures), mixed in a buffer containing 50 mM Tris pH 7.4, 2 mM dithiothreitol and 6 mM MgCl2. For the assay in Fig. 1d, reactions were started by adding 100 nM [α-32P]GTP (3000 Ci/mmol) tracer and incubated at 30 °C for 10 min. Then 5% DMSO or 5 μM JNJ-8003 was added and incubated for 5 min before adding nucleotides that were incubated for another 15 min. For single nucleotide incorporation assays, the enzyme, RNA primer, and template were mixed with 5% DMSO or JNJ-8003 (at concentrations as indicated in figures) and incubated at 30 °C for 10 min and then the reactions were started by adding 100 nM [α-32P]NTP (3000 Ci/mmol) tracer and incubated at 30 °C for 30 min. Reactions were stopped by adding an equal volume of gel loading buffer containing 90% formamide, 50 mM EDTA, 0.05% Xylene Cyanol, and Bromophenol Blue. Samples were denatured at 95 °C for 5 min and run on a 22.5% polyacrylamide urea sequencing gel at 80 W. The gel was transferred onto a Whatman cellulose chromatography paper (Millipore Sigma) and dried on a gel dryer (Bio-Rad) for 1 h. The dried gel was exposed to a phosphor-screen, scanned on a Typhoon phosphorimager (GE Healthcare), and quantified using ImageQuant (GE Healthcare).
De novo RNA synthesis assay using recombinant RSV P + L
The reactions were performed as described by Cressey et al.22. The reactions contained 10 nM RSV L/P, 2 μM RNA template (Tr14), 500 μM GTP, 10 μM ATP, 170 nM [α-33P]ATP (3000 Ci/mmol), and 5% DMSO or JNJ-8003 at various concentrations, in a buffer consisted of 50 mM Tris pH7.4, 8 mM MgCl2, 6 mM dithiothreitol, and 10% glycerol. The reactions were incubated at 30 °C for 1 h and then stopped by adding gel loading buffer containing 90% formamide, 50 mM EDTA, 0.05% Xylene Cyanol and Bromophenol Blue. Samples were denatured at 95 °C for 5 min and run on a 22.5% polyacrylamide urea sequencing gel at 80 W with radiolabeled RNA markers generated via the T7 DNA dependent RNA polymerase (DdRp). The gel was dried, exposed to a phosphor-screen, scanned on a Typhoon scanner (GE Healthcare), and quantified using ImageQuant (GE Healthcare). Bands were identified as extension products or initiation products by the 11-nucleotide pppGAGAAAAAAAG and 2-nucleotide pppGA RNA markers, respectively. Radiolabeled RNA markers corresponding to pppGA, pppGAGAAAAAAAG, and intermediates were generated utilizing a reaction by T7 DNA-dependent RNA polymerase (AM2718, Ambion)53. DNA oligos: 5’-TAATACGACTCACTATA-3’ (T7 promoter primer) and 5’-ACTTTTTTTCTCATATAGTGAGTCGTATTA-3’ (T7 promoter primer + target sequence template) were synthesized at IDT. Due to a mechanistic idiosyncrasy in T7 DdRp, an additional Adenosine residue was placed between the T7 promoter and the target sequence in the template. Each 40 µl reaction contained 5 mM ATP, 5 mM CTP, 5 mM GTP, 100 nM annealed DNA primer and template, 20 U T7 DdRp, 1x T7 DdRp Buffer (40 mM Tris pH 7.8, 20 mM NaCl, 6 mM MgCl2, 2 mM Spermidine HCl, 10 mM DTT), and 20 µCi of [α-33P]ATP. Reactions were incubated at 30 °C for 5 h. Then 1 µl of reconstituted DNase I (Ambion) was added and incubated for an additional 10 min at 37 °C. This reaction was quenched with the addition of 60 µl gel loading buffer.
NMR conformational analysis
All NMR spectra were recorded at 298 K on a Bruker 500 MHz instrument equipped with a 5 mm room temperature SmartProbe. Chemical shifts (δ values) are given in parts per million (ppm) and referenced to the DMSO (2.50 ppm) residual signal. For the structural assignment of the molecule the following spectra were acquired in DMSO-d6: 1D 1H, 2D COSY, 13C-HSQC, 13C-HMBC, and 2D EASY-ROESY (mixing time 300 ms; relaxation delay 5 s) using the standard pulse sequences available in TopSpin (v. 4.1.0, Bruker GmbH). NMR data was processed and analyzed using MNova (v. 14.2.1, Mestrelab Research S.L.). 2D cross-peaks from the 2D EASY-ROESY spectrum were integrated and converted into distances using the Stereofitter plug-in (v. 1.1.1) embedded in MNova, where the PANIC method is used to normalize intensities relative to the diagonal peaks54, and correction factors applied to compensate for the number of spins in each environment (corrected integral).
The exploration of the conformational landscape of the molecule was carried out using Maestro (v. 12.6, Schrödinger, LLC) using a mixed torsional/low-mode sampling method with the OPLS3e force field. The energy threshold was set to 15 Kcal/mol and the cutoff for maximum atom deviation to 0.5 Å, generating 638 conformers. Using the Stereofitter tool, this set of conformers was fitted against the NMR constraints to yield solutions representing the poses that give the best agreement with the experimental data.
Grid preparation and data acquisition
The purified RSV L + P complex was diluted 1:1 with the buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 1 mM TCEP) containing 50 µM JNJ-8003 and incubated on ice for one hour prior to freezing grids. 3.5 µL of the RSV L + P JNJ-8003 complex was applied to the plasma-cleaned (Gatan Solarus) Quantifoil 1.2/1.3 holey gold grid and subsequently vitrified using a Vitrobot Mark IV (FEI Company). Grids were loaded into a Titan Krios transmission electron microscope (ThermoFisher Scientific) with a post-column Gatan Image Filter (GIF) operating in a nanoprobe at 300 keV with a Gatan K3 Summit direct electron detector and an energy filter slit width of 20 eV. Images were recorded with Leginon in counting mode with a pixel size of 0.832 Å and a nominal defocus range of −1.4 to −2.3 μm. Images were recorded with a 1.5 s exposure and 40 ms subframes (38 total frames) corresponding to a total dose of ~52 electrons per Å2. All details corresponding to individual datasets are summarized in Table 1.
Electron microscopy data processing
A total of 11631 dose-fractioned movies were gain-corrected, and beam-induced motion correction using MotionCor255 with the dose-weighting option. The particles were automatically picked from the dose-weighted, motion-corrected average images using Relion 3.056. CTF parameters were determined by Gctf57. A total of ~0.8 million particles were then extracted using Relion 3.0 with a box size of 280 pixels. The 2D and 3D classifications and refinements were performed with Relion 3.0 using the binned datasets. Two rounds of 2D classification and one round of 3D classification were performed to select the homogenous particles. One set of 519118 unbinned homogenous particles were re-extracted and then submitted to 3D auto-refinement without symmetry imposed. Per-particle CTF estimation was refined using the CTF refinement within Relion and followed by one round of 3D auto-refinement with a soft binary mask. CryoDRGN58 was performed using the parameters from the last iteration of the 3D auto-refinement. Further analysis via CryoDRGN, or signal subtraction & refinement was not able to trace the signals corresponding to the CD, MT, and CTD domains indicating those are highly mobile, probably due to the flexible loops flanked on both sides of the CD domain as indicated from the HDX results. Bulky densities corresponding to region 660-688 were observed on top of the palm close to the active site GD811N, highlighting its dynamic nature. 3D classifications and 3D refinements were started from a 60 Å low-pass filtered version of an ab initio map generated with Relion 3.0. All resolutions were estimated by applying a soft mask around the protein complex density and based on the gold-standard (two halves of data refined independently) FSC = 0.143 criterion. Prior to visualization, all density maps sharpened by applying different negative temperature factors using automated procedures, along with the half maps, were used for model building. Local resolution was determined using ResMap59 (Supplementary Fig. 9).
Model building and refinement
The initial template of the RSV L + P complex was derived from a homology-based model calculated by SWISS-MODEL60. The model was docked into the EM density map using Chimera61 and followed by manual adjustment using COOT62. Note that the EM density around the P regions was poor relative to other parts of the model. The P regions were modeled using the unsharpened maps together with the deepEMhancer63 maps that were calculated with the half maps from the focus refinements. Each model was independently subjected to global refinement and minimization in real space using the module phenix.real_space_refine in PHENIX64 against separate EM half-maps with default parameters. The model was refined into a working half-map, and improvement of the model was monitored using the free half map. Model geometry was further improved using Rosetta65. The geometry parameters of the final models were validated in COOT and using MolProbity66 and EMRinger67. These refinements were performed iteratively until no further improvements were observed. The final refinement statistics were provided in Table 1 Model overfitting was evaluated through its refinement against one cryo-EM half map. FSC curves were calculated between the resulting model and the working half map as well as between the resulting model and the free half and full maps for cross-validation (Supplementary Fig. 9). Figures were produced using PyMOL68 and Chimera.
WaterMap
WaterMap computes within a workflow of restrained molecular dynamics simulations, solvent clustering, and statistical thermodynamic analysis, various properties of water molecules, including location, occupancy, enthalpy, entropy, and free energy69,70. 2 ns explicit-solvent MD simulation of the holo protein-ligand complex, and the holo protein unbound ligand were performed using the program Desmond of the Schrodinger Maestro 2021-1 suite. The OPLS4 force field was used for protein and ligand atoms together with the TIP4P water model. In the truncated protein set-up mode, only residues within 10 Å of the ligand were simulated as restrained flexible residues. The coordinates of the protein were restrained with a 5.0 kcal/mol/Å2 harmonic potential applied to the initial positions of the heavy atoms, which ensured convergence of the water sampling around the protein conformation of interest. The protein structure of the L was prepared with the Schrodinger Maestro 2021-1 Protein Preparation Wizard; missing loop regions with less than 25 residues were modeled using the program Prime and free N- and C-termini were caped for the simulations.
GDP docking
GDP docking was performed in Glide SP of the Schrödinger Maestro 2021-1 suite using the JNJ-8003 structure as a reference. The ligand box of 20 Å edge length was centered on JNJ-8003.
Statistics and reproducibility
Graphs were prepared using GraphPad Prism 9.0. Descriptive statistics were used to summarize the data, including mean, standard deviation, and standard error, as indicated in relevant figure legends and tables. Nonlinear regression analysis was used to derive IC50 values by fitting inhibition data to a four-parameter logistic equation (Hill equation). All graphs either show the mean as a bar in the bar graphs or show fitted-curves and individual data points in dot plot graphs; refer to the legends for the number of independent or technical replicates performed. Independent replicates of experiments were performed on different days or at different times of the same day or with separately prepared materials. The source data for the graphs presented is included in Supplementary Data 1–13.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
Janssen virology and medicinal chemistry teams collaboratively lead to JNJ-8003 discovery. Structural predictions were performed using an internal deployment of AlphaFold2, which was setup and deployed by the In Silico Discovery & External Innovation (ISDEI) team of Johnson & Johnson Innovative Medicine. We thank Dr. Stephen Mason for his critical review of the manuscript and Dr. Patrick Byrne at the University of Texas at Austin for the discussion on the RSV L + P Apo structure. The cryo-EM data were collected at NanoImaging Services (San Diego, CA).
Author contributions
Conceptualization: P.R., D.R., S.S. and Z.J. Investigation and visualization: X.Y., Z.J., P.A., B.B., I.B., B.A., A.F., E.J., S.A., A.S., P.S-O., A.B., R.J.C, R.M., S.D.B., N.Y., N.V. Writing – Original Draft: X.Y., P.R., E.J. and Z.J. Writing – Reviewing & Editing: X.Y., P.A., B.B., I.B., B.A., A.F., E.J., S.A., A.B., R.J.C, R.M., M.P., Q.P., S.D.B., E.P., N.Y., N.V., H.V.V., T.J., S.M., F.H., J.S.M., E.D., R.F., S.G., D.R., S.S., P.R., and Z.J. Supervision, H.V.V., T.H.M.J., S.M., F.H., J.M., E.D., R.F., S.G., D.R., P.A., S.S., P.R., and Z.J.
Peer review
Peer review information
This manuscript has been previously reviewed at another Nature Portfolio journal. The manuscript was considered suitable for publication without further review at Communications Biology.
Data availability
The map was deposited in the electron microscopy data bank (EMDB) with ID EMD-29452, and the atomic model in the protein data bank (PDB) with ID 8FU3. The source data for graphs can be found in Supplementary Data 1–13. The original gel images for Fig. 2f–h are in Supplemental Fig. 3 and Supplemental Data 1. All other data that support the findings in this study are available from the corresponding authors upon reasonable request.
Competing interests
The authors declare no competing interests. N.Y., S.D.B., M.P., N.V., A.F., I.B., B.A., Z.J., A.B., A.P., P.A. S.A. X.Y. R.J.C S.G., E.J. S.S., T.J., P.R., N.Y., D.R., and F.H. are employees and possible shareholders of Johnson & Johnson Innovative Medicine. R.F., P.S-O., E.D. and J.S.M. received research funding from Johnson & Johnson Innovative Medicine. During the manuscript preparation, R.F. has a sponsored research agreement with Merck & Co. B.B. is a graduate student from Antwerp University and has received funding from Johnson & Johnson Innovative Medicine and from the agency Flanders Innovation and Entrepreneurship (VLAIO) for this work.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Xiaodi Yu, Pravien Abeywickrema.
These authors jointly supervised this work: Xiaodi Yu, Zhinan Jin.
Contributor Information
Xiaodi Yu, Email: xyu6@its.jnj.com.
Zhinan Jin, Email: zjin14@its.jnj.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-023-05451-4.
References
- 1.Li Y, et al. National burden estimates of hospitalisations for acute lower respiratory infections due to respiratory syncytial virus in young children in 2019 among 58 countries: a modelling study. Lancet Respir. Med. 2021;9:175–185. doi: 10.1016/S2213-2600(20)30322-2. [DOI] [PubMed] [Google Scholar]
- 2.Shi T, et al. Global disease burden estimates of respiratory syncytial virus-associated acute respiratory infection in older adults in 2015: a systematic review and meta-analysis. J. Infect. Dis. 2020;222:S577–S583. doi: 10.1093/infdis/jiz059. [DOI] [PubMed] [Google Scholar]
- 3.Chatzis O, et al. Burden of severe RSV disease among immunocompromised children and adults: a 10 year retrospective study. BMC Infect. Dis. 2018;18:111. doi: 10.1186/s12879-018-3002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaw PS, et al. Acute lower respiratory infections associated with respiratory syncytial virus in children with underlying congenital heart disease: systematic review and meta-analysis. J. Infect. Dis. 2020;222:S613–S619. doi: 10.1093/infdis/jiz150. [DOI] [PubMed] [Google Scholar]
- 5.Chaw PS, et al. Respiratory syncytial virus-associated acute lower respiratory infections in children with bronchopulmonary dysplasia: systematic review and meta-analysis. J. Infect. Dis. 2020;222:S620–S627. doi: 10.1093/infdis/jiz492. [DOI] [PubMed] [Google Scholar]
- 6.DeVincenzo JP, et al. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N. Engl. J. Med. 2014;371:711–722. doi: 10.1056/NEJMoa1401184. [DOI] [PubMed] [Google Scholar]
- 7.Korell J, Green B, DeVincenzo J, Huntjens D. A human challenge model for respiratory syncytial virus kinetics, the pharmacological effect of a novel fusion inhibitor, and the modelling of symptoms scores. Eur. J. Pharm. Sci. 2017;109S:S154–S160. doi: 10.1016/j.ejps.2017.05.070. [DOI] [PubMed] [Google Scholar]
- 8.Stevens M, et al. Antiviral activity of oral JNJ-53718678 in healthy adult volunteers challenged with respiratory syncytial virus: a placebo-controlled study. J. Infect. Dis. 2018;218:748–756. doi: 10.1093/infdis/jiy227. [DOI] [PubMed] [Google Scholar]
- 9.DeVincenzo, J. et al. A Randomized, Placebo-Controlled, Respiratory Syncytial Virus Human Challenge Study of the Antiviral Efficacy, Safety, and Pharmacokinetics of RV521, an Inhibitor of the RSV-F Protein. Antimicrob. Agents Chemother.64, 10.1128/AAC.01884-19 (2020). [DOI] [PMC free article] [PubMed]
- 10.Kazmierski, W. M. & ebrary Inc. xiii, 438 p. (Wiley,, Hoboken, N.J., 2011).
- 11.DeVincenzo J, et al. Safety and anti-viral effects of nebulized PC786 in a respiratory syncytial virus challenge study. J. Infect. Dis. 2020 doi: 10.1093/infdis/jiaa716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeVincenzo JP, et al. Activity of oral ALS-008176 in a respiratory syncytial virus challenge study. N. Engl. J. Med. 2015;373:2048–2058. doi: 10.1056/NEJMoa1413275. [DOI] [PubMed] [Google Scholar]
- 13.Grosfeld H, Hill MG, Collins PL. RNA replication by respiratory syncytial virus (RSV) is directed by the N, P, and L proteins; transcription also occurs under these conditions but requires RSV superinfection for efficient synthesis of full-length mRNA. J. Virol. 1995;69:5677–5686. doi: 10.1128/jvi.69.9.5677-5686.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu Q, Hardy RW, Wertz GW. Functional cDNA clones of the human respiratory syncytial (RS) virus N, P, and L proteins support replication of RS virus genomic RNA analogs and define minimal trans-acting requirements for RNA replication. J. Virol. 1995;69:2412–2419. doi: 10.1128/jvi.69.4.2412-2419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogino, T. & Green, T. J. Transcriptional control and mRNA Capping by the GDP polyribonucleotidyltransferase domain of the rabies virus large protein. Viruses11, 10.3390/v11060504 (2019). [DOI] [PMC free article] [PubMed]
- 16.Li J, Rahmeh A, Morelli M, Whelan SP. A conserved motif in region v of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J. Virol. 2008;82:775–784. doi: 10.1128/JVI.02107-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogino T, Banerjee AK. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol. Cell. 2007;25:85–97. doi: 10.1016/j.molcel.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 18.Ogino, M., Ito, N., Sugiyama, M. & Ogino, T. The rabies virus L protein catalyzes mRNA capping with GDP polyribonucleotidyltransferase activity. Viruses8, 10.3390/v8050144 (2016). [DOI] [PMC free article] [PubMed]
- 19.Tekes G, Rahmeh AA, Whelan SP. A freeze frame view of vesicular stomatitis virus transcription defines a minimal length of RNA for 5’ processing. PLoS Pathog. 2011;7:e1002073. doi: 10.1371/journal.ppat.1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogino T. Capping of vesicular stomatitis virus pre-mRNA is required for accurate selection of transcription stop-start sites and virus propagation. Nucleic Acids Res. 2014;42:12112–12125. doi: 10.1093/nar/gku901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braun MR, et al. RNA elongation by respiratory syncytial virus polymerase is calibrated by conserved region V. PLoS Pathog. 2017;13:e1006803. doi: 10.1371/journal.ppat.1006803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cressey TN, et al. Distinctive features of the respiratory syncytial virus priming loop compared to other non-segmented negative strand RNA viruses. PLoS Pathog. 2022;18:e1010451. doi: 10.1371/journal.ppat.1010451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahmeh AA, et al. Critical phosphoprotein elements that regulate polymerase architecture and function in vesicular stomatitis virus. Proc. Natl Acad. Sci. USA. 2012;109:14628–14633. doi: 10.1073/pnas.1209147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sourimant J, et al. Fine mapping and characterization of the L-polymerase-binding domain of the respiratory syncytial virus phosphoprotein. J. Virol. 2015;89:4421–4433. doi: 10.1128/JVI.03619-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galloux M, et al. Characterization of a viral phosphoprotein binding site on the surface of the respiratory syncytial nucleoprotein. J. Virol. 2012;86:8375–8387. doi: 10.1128/JVI.00058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao D, et al. Cryo-EM structure of the respiratory syncytial virus RNA polymerase. Nat. Commun. 2020;11:368. doi: 10.1038/s41467-019-14246-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilman MSA, et al. Structure of the respiratory syncytial virus polymerase complex. Cell. 2019;179:193–204.e114. doi: 10.1016/j.cell.2019.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang B, et al. Structure of the L protein of vesicular stomatitis virus from electron cryomicroscopy. Cell. 2015;162:314–327. doi: 10.1016/j.cell.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenni S, et al. Structure of the vesicular stomatitis virus L protein in complex with its phosphoprotein cofactor. Cell Rep. 2020;30:53–60.e55. doi: 10.1016/j.celrep.2019.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang G, et al. Discovery of 4’-chloromethyl-2’-deoxy-3’,5’-di-O-isobutyryl-2’-fluorocytidine (ALS-8176), a first-in-class RSV polymerase inhibitor for treatment of human respiratory syncytial virus infection. J. Med. Chem. 2015;58:1862–1878. doi: 10.1021/jm5017279. [DOI] [PubMed] [Google Scholar]
- 31.Sidwell RW, Robins RK, Hillyard IW. Ribavirin: an antiviral agent. Pharm. Ther. 1979;6:123–146. doi: 10.1016/0163-7258(79)90058-5. [DOI] [PubMed] [Google Scholar]
- 32.Sudo K, et al. YM-53403, a unique anti-respiratory syncytial virus agent with a novel mechanism of action. Antivir. Res. 2005;65:125–131. doi: 10.1016/j.antiviral.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Noton SL, et al. Respiratory syncytial virus inhibitor AZ-27 differentially inhibits different polymerase activities at the promoter. J. Virol. 2015;89:7786–7798. doi: 10.1128/JVI.00530-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiong-Yip CL, et al. Characterization of a respiratory syncytial virus L protein inhibitor. Antimicrob. Agents Chemother. 2014;58:3867–3873. doi: 10.1128/AAC.02540-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coates, M. et al. Preclinical characterization of PC786, an inhaled small-molecule respiratory syncytial virus L protein polymerase inhibitor. Antimicrob. Agents Chemother.61, 10.1128/AAC.00737-17 (2017). [DOI] [PMC free article] [PubMed]
- 36.Sourimant J, et al. Orally efficacious lead of the AVG inhibitor series targeting a dynamic interface in the respiratory syncytial virus polymerase. Sci. Adv. 2022;8:eabo2236. doi: 10.1126/sciadv.abo2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laganas VA, et al. Characterization of novel respiratory syncytial virus inhibitors identified by high throughput screen. Antivir. Res. 2015;115:71–74. doi: 10.1016/j.antiviral.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Grosse, S. et al. RSV INHIBITING 3-SUBSTITUTED QUINOLINE AND CINNOLINE DERIVATIVES (2021).
- 39.Noton SL, Fearns R. Initiation and regulation of paramyxovirus transcription and replication. Virology. 2015;479-480:545–554. doi: 10.1016/j.virol.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao, D. et al. In Vitro primer-based RNA elongation and promoter fine mapping of the respiratory syncytial virus. J. Virol.95, 10.1128/jvi.01897-20 (2020). [DOI] [PMC free article] [PubMed]
- 41.Cressey TN, Noton SL, Nagendra K, Braun MR, Fearns R. Mechanism for de novo initiation at two sites in the respiratory syncytial virus promoter. Nucleic Acids Res. 2018;46:6785–6796. doi: 10.1093/nar/gky480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCormick JB, et al. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986;314:20–26. doi: 10.1056/NEJM198601023140104. [DOI] [PubMed] [Google Scholar]
- 43.Mason SW, et al. Polyadenylation-dependent screening assay for respiratory syncytial virus RNA transcriptase activity and identification of an inhibitor. Nucleic Acids Res. 2004;32:4758–4767. doi: 10.1093/nar/gkh809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogino T, Yadav SP, Banerjee AK. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc. Natl Acad. Sci. USA. 2010;107:3463–3468. doi: 10.1073/pnas.0913083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neubauer J, Ogino M, Green TJ, Ogino T. Signature motifs of GDP polyribonucleotidyltransferase, a non-segmented negative strand RNA viral mRNA capping enzyme, domain in the L protein are required for covalent enzyme-pRNA intermediate formation. Nucleic Acids Res. 2016;44:330–341. doi: 10.1093/nar/gkv1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jumper J, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liuzzi M, et al. Inhibitors of respiratory syncytial virus replication target cotranscriptional mRNA guanylylation by viral RNA-dependent RNA polymerase. J. Virol. 2005;79:13105–13115. doi: 10.1128/JVI.79.20.13105-13115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hallak LK, Spillmann D, Collins PL, Peeples ME. Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J. Virol. 2000;74:10508–10513. doi: 10.1128/JVI.74.22.10508-10513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malykhina O, Yednak MA, Collins PL, Olivo PD, Peeples ME. A respiratory syncytial virus replicon that is noncytotoxic and capable of long-term foreign gene expression. J. Virol. 2011;85:4792–4801. doi: 10.1128/JVI.02399-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tiong-Yip CL, et al. Development of a high-throughput replicon assay for the identification of respiratory syncytial virus inhibitors. Antivir. Res. 2014;101:75–81. doi: 10.1016/j.antiviral.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 51.Tremaglio CZ, Noton SL, Deflube LR, Fearns R. Respiratory syncytial virus polymerase can initiate transcription from position 3 of the leader promoter. J. Virol. 2013;87:3196–3207. doi: 10.1128/JVI.02862-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noton SL, Fearns R. The first two nucleotides of the respiratory syncytial virus antigenome RNA replication product can be selected independently of the promoter terminus. RNA. 2011;17:1895–1906. doi: 10.1261/rna.2813411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 54.Hu H, Krishnamurthy K. Revisiting the initial rate approximation in kinetic NOE measurements. J. Magn. Reson. 2006;182:173–177. doi: 10.1016/j.jmr.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 55.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife7, 10.7554/eLife.42166 (2018). [DOI] [PMC free article] [PubMed]
- 57.Zhang K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhong ED, Bepler T, Berger B, Davis JH. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods. 2021;18:176–185. doi: 10.1038/s41592-020-01049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat. Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waterhouse A, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 62.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanchez-Garcia R, et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 2021;4:874. doi: 10.1038/s42003-021-02399-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Afonine PV, et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D. Struct. Biol. 2018;74:531–544. doi: 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang, R. Y. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife5, 10.7554/eLife.17219 (2016). [DOI] [PMC free article] [PubMed]
- 66.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barad BA, et al. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods. 2015;12:943–946. doi: 10.1038/nmeth.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
- 69.Abel R, Young T, Farid R, Berne BJ, Friesner RA. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc. 2008;130:2817–2831. doi: 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Young T, Abel R, Kim B, Berne BJ, Friesner RA. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl Acad. Sci. USA. 2007;104:808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deval J, et al. Molecular basis for the selective inhibition of respiratory syncytial virus RNA polymerase by 2’-Fluoro-4’-Chloromethyl-Cytidine triphosphate. PLoS Pathog. 2015;11:e1004995. doi: 10.1371/journal.ppat.1004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ogino M, Gupta N, Green TJ, Ogino T. A dual-functional priming-capping loop of rhabdoviral RNA polymerases directs terminal de novo initiation and capping intermediate formation. Nucleic Acids Res. 2019;47:299–309. doi: 10.1093/nar/gky1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The map was deposited in the electron microscopy data bank (EMDB) with ID EMD-29452, and the atomic model in the protein data bank (PDB) with ID 8FU3. The source data for graphs can be found in Supplementary Data 1–13. The original gel images for Fig. 2f–h are in Supplemental Fig. 3 and Supplemental Data 1. All other data that support the findings in this study are available from the corresponding authors upon reasonable request.