

Summary
The organization of fear memory involves the participation of multiple brain regions. However, it is largely unknown how fear memory is formed, which circuit pathways are used for “printing” memory engrams across brain regions, and the role of identified brain circuits in memory retrieval. With advanced genetic methods, we combinatorially blocked presynaptic output and manipulated N-methyl-D-aspartate receptor (NMDAR) in the basolateral amygdala (BLA) and medial prefrontal cortex (mPFC) before and after cued fear conditioning. Further, we tagged fear-activated neurons during associative learning for optogenetic memory recall. We found that presynaptic mPFC and postsynaptic BLA NMDARs are required for fear memory formation, but not expression. Our results provide strong evidence that NMDAR-dependent synaptic plasticity drives multi-trace systems consolidation for the sequential printing of fear memory engrams from BLA to mPFC and, subsequently, to the other regions, for flexible memory retrieval.
Subject areas: Behavioral neuroscience, Cellular neuroscience
Graphical abstract
Highlights
-
•
We selectively manipulated NMDARs, synaptic transmission, and engram tagging
-
•
Cued memory is sequentially printed first from BLA to mPFC, then to other regions
-
•
mPFC pre- and BLA postsynaptic NMDARs are required for cued memory formation
-
•
Memory can be flexibly retrieved from BLA or mPFC neurons
Behavioral neuroscience; Cellular neuroscience
Introduction
Fear memories are vital for survival, are formed rapidly, but can last forever.1,2 They guide adaptive behavior and decision-making along the subconscious-conscious continuum.3,4,5 It is thought that functional interactions between distributed brain circuits across the different brain regions participate in generating fear memories with memory prints (or engrams).6,7,8 However, the underlying molecular and cellular mechanisms of systems consolidation and the organization of fear circuits remain largely elusive.9,10,11,12,13
In the hippocampal-cortical network for memory formation and storage, the “systems consolidation model” proposes a time-dependent functional reorganization of brain circuits, as memories are transferred over time from the hippocampus to the cortex for permanent storage.14 An alternative model, the “multi-trace theory,” suggests the simultaneous participation of multiple brain regions for memory formation and storage.15
The amygdala is a critical brain region for innate and conditioned fear expression in vertebrates.16,17,18,19,20 The basolateral amygdala (BLA) complex, including the lateral amygdala (LA) and the basal amygdala (BA), receives cortical and thalamic inputs for the multisensory integration of experiences.21,22,23,24 On the other hand, the central amygdala (CeA) acts as an output station to convert emotionally relevant sensory information into behavioral and physiological responses.17,18,25 Different brain regions, with a particular focus on the BLA, hippocampus, and cortex, have been shown to participate in the formation, storage, and retrieval of fear memories.26,27,28,29,30,31,32,33,34 The medial prefrontal cortex (mPFC) is hypothesized to provide top-down control over the amygdala to facilitate fear learning and modulate fear expression.35,36,37,38 Accordingly, multi-region optical brain imaging has revealed cellular activity in the mPFC, amygdala, and hippocampus during the contextual fear experience.26,28,29 In addition, the finding of mPFC engram inputs to BLA engram neurons39 further support the idea that the amygdala and mPFC neurons reciprocally interact in developing memory engrams in both regions at the time of memory formation. These studies support systems consolidation for the printing of fear engrams across brain networks for fear memory retrieval.27,28,29,31,40,41,42,43
We developed second generation genetic tools for the virus-delivered silencing of synaptic transmission (vINSIST-2). We used it to combinatorially block synaptic output from BLA and mPFC before and after cued-fear conditioning. Our results demonstrate that cued-fear memory is sequentially printed from BLA to mPFC, and subsequently to another brain region(s).
To reveal the systems consolidation mechanism at a molecular level, we removed N-methyl-D-aspartate receptor (NMDAR), an ionotropic voltage-gated glutamate receptor well-known to play a crucial role in synaptic plasticity, learning and memory, and oscillatory network activities.44,45,46 Previous studies reported that infusion of NMDAR specific antagonists specifically into the amygdala disrupted the acquisition, but not the expression of fear memory.47,48 However, there remains controversy with other studies that reported different results about the role of NMDAR function in fear expression.49,50 It is conceivable that the intra-amygdala injection of NMDAR antagonists blocks not only postsynaptic but also the presynaptic NMDARs, located both on local and distant inputs to the amygdala, including the mPFC afferents. In addition to the postsynaptic NMDARs, the presynaptic NMDARs appear to be crucial for neurotransmission, experience-dependent synaptic plasticity, and behavior.51,52,53,54,55 Along this line, the role of brain-derived neurotrophic factor (BDNF) in NMDAR-dependent synaptic plasticity and learning and memory processes is beginning to be elucidated.56 In support, presynaptic NMDAR of the cortico-striatal circuit is needed for long-term potentiation (LTP), and sustained elevated Ca2+ levels in the presynaptic terminals induced BDNF secretion for LTP.56,57,58 Notably, in the hippocampal circuit, the inhibition of the presynaptic NMDAR decreases glutamate release.59
We thus asked whether cued-fear memory engrams are printed from the BLA to the mPFC. We further asked whether the NMDAR-dependent synaptic plasticity along the mPFC-amygdala axis is required for cued-fear memory formation and retrieval. To address these questions, we established virus-based technologies to specifically target neurons of both BLA and mPFC in the mouse brain, to precisely block synaptic outputs (silencing of synaptic transmission) and remove NMDARs (Grin1 gene knockout) in these two regions in a combinatorial manner before and after fear conditioning. Moreover, we performed in vivo synaptic plasticity measurements between mPFC and BLA, simultaneous recordings of oscillatory network activities of amygdala and mPFC, and behaviorally related correlations in both control and BLA/mPFC specific double Grin1 gene knockout mice. Finally, we tagged engram neurons in BLA and mPFC and optogenetically tested their role in memory recall.
Our results demonstrate the sequential printing of cued-fear memory engrams from BLA to mPFC and, subsequently, to other brain regions requiring mPFC pre-to BLA postsynaptic NMDAR-dependent synaptic plasticity for cued-fear memory formation. By tagging experience-activated neurons and their optogenetic activation, we further demonstrate that memory retrieval can be achieved from BLA or mPFC.
Results
Advanced method for genetic silencing and un-silencing of synaptic transmission
To reveal the role of BLA and mPFC-specific synaptic output in fear memory formation and retrieval, we deployed virus-delivered inducible silencing of synaptic transmission (vINSIST),60 based on recombinant adeno-associated viruses (rAAVs) equipped with tetracycline (tet)-controlled genetic switches for conditional gene expression.61,62 We developed the next-generation advanced system with a destabilized tetanus toxin light chain (dsTeTxLC; vINSIST-2) (Dogbevia et al., article in the preparation) to block synaptic transmission by selectively cleaving synaptobrevin-2 (Syb-2). Our approach requires three rAAVs: (1) rAAV-PhSYN-rtTA, to express a reverse tet transactivator (rtTA) under the control of the human synapsin promoter (PhSYN); (2) rAAV-Ptetbi-dsTeTxLC, carrying a rtTA-dependent bidirectional tet promoter (Ptetbi) to express dsTeTxLC and (3) rAAV-PhSYN-tdTomato as a fluorescent tracer for validating injectioin sites (Figure 1A). The TeTxLC is therefore expressed under the control of the tet inducible system in the presence of the inducer, doxycycline (Dox), a hydrophobic derivative of tetracycline, that rapidly crosses the blood-brain-barrier.60,63
Figure 1.

Inducible silencing of the amygdala and prefrontal output
(A) Schematic showing the components of the second-generation virus-delivered INducible SIlencing of Synaptic Transmission (“vINSIST-2”) system and their operation. Abbreviations: Dox, doxycycline; dsTeTxLC, destabilized tetanus toxin light chain protein; PhSYN, human synapsin specific promoter; Ptetbi, bidirectional tet responder promoter; rAAV, recombinant adenovirus serotype 1/2; tdTOM, tdTomato red fluorescent protein; rtTA2-nM2, Dox-sensitive recombinant transactivator; Syb-2, synaptobrevin 2 vesicle protein.
(B) Silencing of BLA output by dsTeTxLC in dsTeTxLC-ONBLA mice before conditioning induces a significant decrease of freezing during the cued test (N = 7,8; two-way RM ANOVA; significant main effect of genotype F(1,13) = 4.98, p = 0.044; significant effect of cue/genotype interaction F(1,13) = 10.04, p = 0.007) ∗∗p < 0.01 by Bonferroni post-test. Scale bar, 1mm.
(C) Silencing of BLA output induced after fear conditioning does not affect freezing in the cued test (retrieval-2/dsTeTxLC-ONBLA compared to retrieval-1/dsTeTxLC-OFFBLA) (N = 10; two-way RM ANOVA; no significant effect of treatment F(1,18) = 0.099, p = 0.76).
(D) Silencing of mPFC output by constitutive TeTxLC expression has no significant effect on fear learning and expression (N = 11, 8; Freezing: two-way RM ANOVA, no significant effect of genotype F(1,17) = 0.96, p = 0.34).
(E) Silencing of mPFC outputs induced after fear conditioning has no significant effect on freezing in the cued test (retrieval-2/dsTeTxLC-ONmPFC compared to retrieval-1/dsTeTxLC-OFFmPFC) (N = 9; two-way RM ANOVA; no significant impact of treatment F(1,16) = 0.01, p = 0.91). Scale bar, 1mm. Data are presented as mean ± s.e.m.
Basolateral amygdala and medial prefrontal cortex synaptic outputs provide alternative pathways for fear memory expression
A group of mice was injected bilaterally in the BLA with the vINSIST-2 system; without and with Dox, the vINSIST-2 system is either OFF (dsTeTxLC-OFF) or ON (dsTeTxLC-ON), respectively. Control mice were similarly injected with rAAV expressing only a fluorescent protein and treated in a matched manner. In all cases, we used tdTOM expression to verify correct targeting (Figures 1B and 1D; right panels). Both control and dsTeTxLC-ON mice performed similarly in behavioral tests for exploration and innate anxiety, but dsTeTxLC-ON mice showed impairment in the passive avoidance test (Figure S1).
We used the delay cued-fear conditioning paradigm (Figure S2), in which a tone, the neutral conditioned stimulus (CS), is presented, and it co-terminates with a mild shock, an aversive unconditioned stimulus (US). After CS-US pairing, subjects (in our study mice) acquired the capacity to elicit a conditioned behavioral fear response (freezing) when the CS is presented alone.
The induced silencing of BLA synaptic output (dsTeTxLC-ONBLA) before fear conditioning was associated with a significant decrease in freezing measured in the cued retrieval test (Figure 1B). However, when silencing was induced only after fear conditioning, it didn’t interfere with the retrieval of the learned fear (Figure 1C). These results suggest that BLA output is necessary for fear acquisition but not for fear expression.
To investigate if mPFC output is also needed for fear conditioning, a group of mice was injected bilaterally in the mPFC to express TeTxLC under a constitutive pan-neuronal promoter (rAAV-PhSYN-TeTxLC-2A-mKO). TeTxLC expression induced a severe deficit in burrowing activity64 (Figure S3), which reflects a strong effect due to the diminished mPFC synaptic transmission. However, the same mice (TeTxLCmPFC) didn’t show a reduction of fear expression in the retrieval test (Figure 1D). To exclude that persistent TeTxLC expression in the mPFC may have activated a compensatory mechanism for fear expression later after learning, another group of mice was injected with vINSIST-2 system to block mPFC output after fear conditioning. The percentage of freezing measured in fear memory retrieval performed two days (dsTeTxLC-OFFmPFC) and four days (dsTeTxLC-ONmPFC) after fear conditioning didn’t differ (Figure 1E), in line with previous lesion studies.65,66
Fear memory is sequentially printed from the basolateral amygdala to the medial prefrontal cortex and then to other brain region(s)
To investigate whether the BLA and mPFC provide alternative pathways for memory retrieval, we permanently blocked the mPFC output (TeTxLCmPFC) and applied vINSIST-2 in the BLA for the inducible silencing of synaptic transmission (Figure 2A, left). After fear conditioning, we blocked BLA synaptic output (dsTeTxLC-ONBLA). We discovered that retrieval of fear memory was impaired (Figure 2A, right). This result indicates that fear memory is sequentially printed from the BLA to the mPFC, with no other alternative pathway. We next asked whether fear memory is printed from the mPFC to other regions to augment the potential pathways for dynamic fear memory retrieval. To test this hypothesis, we blocked output from both mPFC and BLA (dsTeTxLC-ONBLA-mPFC) after fear conditioning. We observed no attenuation of fear expression (Figure 2B), suggesting that the learned fear was printed from the mPFC to another brain region(s), offering alternative pathways for fear retrieval.
Figure 2.

Fear memory is distributed between the medial prefrontal cortex and the amygdala
(A) When mPFC outputs are inhibited since acquisition (TeTxLCmPFC), silencing of BLA output induced after conditioning (dsTeTxLC-ONBLA) has a significant effect on freezing in the cued test for memory recall (N = 8, two-way RM ANOVA; cue × treatment interaction, F(1,14) = 10.44, p = 0.006) ∗∗p < 0.01 by Bonferroni post-test.
(B) Silencing of both mPFC and BLA outputs after conditioning has no significant effect on freezing in the cued test (retrieval-2/dsTeTxLC-ONBLA-mPFCmPFC compared to retrieval-1/dsTeTxLC-OFFBLA-mPFCmPFC) (N = 6; two-way RM ANOVA; no significant effect of genotype F(1,10) = 0.02, p = 0.87). Data are presented as mean ± s.e.m. Scale bars, 1mm. All injection sites were validated by a tracer rAAV expressing tdTOM.
Recombinant adeno-associated viruses for inducible gene deletion
We developed a method called virus-delivered inducible gene knockout, or viKO, for conditional gene deletion.45,61 It requires two rAAVs: (1) rAAV-PhSYN-rtTA and (2) rAAV-Ptetbi-Cre/tdTOM, carrying the Ptetbi to simultaneously express Cre recombinase and tdTomato (tdTOM) genes (Figure 3A). While the Dox-induced expression of Cre recombinase allows for Cre/loxP mediated Grin1 gene deletion in Grin12lox mice,45 the co-expression of tdTOM allows for the direct visualization of targeted knockout neurons for the validation of proper targeting and expression analyses (Figures 3B, 3C, and S4).
Figure 3.

Role of NMDARs in fear memory conditioning
(A) rAAVs were bilaterally delivered into the BLA of Grin12lox mice to induce Cre/loxP mediated deletion of the Grin1 gene and the generation of BLA-specific Grin1 gene knockout mice (Grin1ΔBLA). Scale bar, 1 mm.
(B) tdTOM fluorescence and Cre immunostaining in the BLA are overlapping and evident in Dox-treated (Grin1ΔBLA) but not in untreated mice (Grin12lox mice, also called ControlBLA). Scale bar, 500 μM. Abbreviations: BA, basal amygdaloid nucleus; CpU, caudate-putamen; Cx, cortex; LA, lateral amygdaloid nucleus.
(C) in situ hybridization for Grin1 mRNA of coronal brain slices from ControlBLA and Grin1ΔBLA mice. Scale bar, 1 mm.
(D) Genetic deletion of Grin1 bilaterally in the BLA before fear conditioning had no effects on freezing during the cued test for memory recall in Grin1ΔBLA compared to ControlBLA mice (N = 8, 6; two-way RM ANOVA; no significant effect of genotype F(1,12) = 0.15; p = 0,7).
(E) Genetic deletion of Grin1 bilaterally in the mPFC before fear conditioning had no effects on freezing during the cued test in Grin1ΔmPFC compared to ControlmPFC mice (N = 8, 12; two-way RM ANOVA; no significant effect of genotype; F(1,18) = 0.004; p = 0.9).
(F) Genetic deletion of Grin1 bilaterally in both the BLA and mPFC before fear conditioning significantly reduced the percentage of freezing during the cued test in Grin1ΔBLA-mPFC compared to ControlBLA-mPFC mice (N = 8, 8; two-way RM ANOVA followed by Bonferroni post-test; significant main effect of genotype F(1,14) = 7.6; p < 0.001 during tone).
(G) Schematics reproducing the effects of targeted NMDAR removal in the BLA, in the mPFC, and in both areas.
(H) Schematic depicting in vivo electrodes implanted for stimulating neurons in the mPFC and recording in BLA. In the middle, examples of fEPSPs (averaged 5 times) were collected from representative ControlBLA-mPFC and Grin1ΔBLA-mPFC mice (n = 6 and n = 5, respectively). On the right, illustrated data correspond to fEPSPs evoked by the second pulse. While control mice presented a significant LTP (F(32,96) = 1.787, p = 0.016), Grin1ΔBLA-mPFC mice did not reach significant values (F(32,160) = 0.765, p = 0.812).
N-methyl-D-aspartate receptor-dependent synaptic plasticity along the basolateral amygdala-medial prefrontal cortex circuits is needed for cued-fear memory formation
To test if the genetic removal of BLA NMDAR would impair cued-fear memory acquisition, with our advanced genetic tools, we performed virus-delivered inducible gene knockout (viKO) on Grin12lox mice45 to delete the Grin1 gene that encodes for the obligatory NMDAR subunit GluN1.45 By stereotactic bilateral rAAV injections in the BLA of Grin12lox mice, we generated BLA-specific Grin1 knockouts (Grin1ΔBLA). Age-matched Grin12lox mice injected in the BLA with rAAV-PhSYN-tdTOM served as controls (ControlBLA). Molecular and electrophysiological analyses provided unequivocal evidence for physical and functional loss of GluN1 in the BLA; as expected, tdTOM fluorescence and Cre immunostaining were only evident in Dox-treated, but not in untreated mice (Figure 3B), and virus expression was largely restricted to the BLA. The loss of Grin1 mRNA signal and GluN1 protein, detected by in situ hybridization and immunostaining with a specific GluN1 antibody, respectively, was located at the virus injection site (Figure 3C and representative images Figures 3D–3F). In most AAV-targeted mice, the CeA was spared. However, Grin1 gene deletion was sometimes detectable in the ventral and dorsal endopiriform nuclei (Figure S4).
Electrophysiological studies performed in acute brain slices of mice tested in fear conditioning showed that in Grin1ΔBLA mice, the NMDAR component of the eEPSC current amplitudes (pA) was completely abolished and the NMDA/AMPA current ratio was highly reduced compared to controls (ControlBLA) (Figure S5).
Grin1 deletion in the BLA before conditioning didn’t affect the freezing response to the tone of Grin1ΔBLA mice compared to controls in the retrieval test performed two days after acquisition (Figures 3D and 3G). Not surprisingly, when the BLA Grin1 deletion was induced after conditioning, no differences in freezing were observed between groups during the second retrieval test (Figure S6A). ControlBLA and Grin1ΔBLA mice performed similarly in the open-field and visual association tests (Figure S7). These data suggest that Grin1 gene deletion in the BLA, both before and after fear conditioning, did not interfere with memory expression, indicating that BLA NMDAR is not required or, more likely, that NMDAR-dependent plasticity in the BLA can be compensated by other mechanisms, either locally or in different brain regions.
Considering the findings that the BLA and mPFC are reciprocally interconnected67 (Figure S8), and that the mPFC plays a crucial role in regulating fear-related behavior,35,36,37 we deleted the Grin1 gene in the mPFC (Grin1ΔmPFC mice) before fear conditioning. No differences in fear expression were observed between knockouts and controls (Figures 3E and 3G). Therefore, genetic removal of NMDAR, specifically in the BLA alone (Grin1ΔBLA) or in the mPFC alone (Grin1ΔmPFC), did not impair fear expression (Figures 3D and 3E).
Remarkably, simultaneous Grin1 gene deletion in the BLA and mPFC (Grin1ΔBLA-mPFC double knockout mice) before fear conditioning impaired memory expression in the retrieval test (Figures 3F and 3G). However, when we deleted NMDAR from both BLA and mPFC after fear conditioning, no differences in freezing were observed during the retrieval compared to the controls (Figure S6B). These results show that BLA-PFC NMDA receptors are needed for cued-fear memory acquisition, but not for its expression.
To investigate whether synaptic plasticity of the mPFC-BLA synapse is altered in the double Grin1 gene knockout (Grin1ΔBLA-mPFC), we performed in vivo electrophysiological LTP recordings in awake mice. High-frequency stimulation (HFS) of the mPFC region evoked significantly increased field excitatory postsynaptic potentials (fEPSPs) in BLA of control mice but not in Grin1ΔBLA-mPFC mice (Figure 3H). In light of the previous pharmacological NMDAR block in the BLA studies,47,48 our results suggest that both postsynaptic BLA NMDARs and presynaptic mPFC NMDARs are needed for cued-fear memory formation.
Perturbation of network oscillatory activities and coherence during behavior
Neural activity of the mPFC and the amygdala were examined by local field potential (LFP) recordings (Figures S9 and S10) in the double knockout Grin1ΔBLA-mPFC mice and controls during memory retrieval (Figure 3F). Spectral power was analyzed within selected bands (delta, 1.5–4 Hz; theta, 4–10 Hz; beta: 10–30 Hz; and gamma, 30–100 Hz) (Figures 4A and 4B) and represented in color-coded spectral power maps for visual display (Figures 4C and 4D). Compared to controls, LFPs recording in the amygdala of Grin1ΔBLA-mPFC mice revealed significant perturbation of all oscillation frequencies. During freezing, a reduction in theta, beta, and gamma bands was common in both genotypes. According to the previous finding,26,68 freezing in control mice was accompanied by an increase in the power spectra of the delta band, possibly playing an important role in long-range communication. However, unlike controls, Grin1ΔBLA-mPFC mice didn’t display such an increase.
Figure 4.

Spectral analyses of LFPs recorded in the amygdalar complex and mPFC of Grin1ΔBLA-mPFC and ControlBLA-mPFC mice
(A) Spectral power values during no freezing and freezing for the four selected bands in the amygdala of control (ControlBLA-mPFC) and double knockout mice (Grin1ΔBLA-mPFC).
(B) Spectral power values during no freezing and freezing for the four selected bands in the mPFC of control (ControlBLA-mPFC) and double knockout mice (Grin1ΔBLA-mPFC). ∗∗∗p < 0.001 and ∗∗p ≤ 0.01. Data are presented as mean ± s.e.m.
(C and D) Schematic diagrams representing the spectral power differences between Grin1ΔBLA-mPFC and ControlBLA-mPFC mice for the 4 selected bands in the BLA (C) and the mPFC (D). The color scale is illustrated in the middle.
(E) Time-frequency power analysis for delta, theta, and beta bands (0–40 Hz) of representative LFP signals recorded in the mPFC (left) and the amygdala (right) of ControlBLA-mPFC mice (upper panel) and Grin1ΔBLA-mPFC mice (lower panel) during a non-freezing period of 2 s (0–2 s) and a freezing period of 2 s (2–4 s).
(F) Time-frequency power analysis for the gamma band (60–100 Hz) of representative LFP signals recorded in the mPFC (left) and the amygdala (right) of ControlBLA-mPFC mice (upper panel) and Grin1ΔBLA-mPFC mice (lower panel) during a non-freezing period of 2 s (0–2 s) and a freezing period of 2 s (2–4 s). The color scale is illustrated at the bottom right.
(G) Coherence analysis between mPFC and amygdala during no freezing and freezing behavior. Coherence spectra of Grin1ΔBLA-mPFC and ControlBLA-mPFC mice during freezing (dark colors) and no freezing (light colors) behaviors (N = 100 epochs of 1 s per behavior). The dashed lines correspond to 8 Hz and 14 Hz. Mean ± s.e.m. of coherence values in the 8–14 Hz band.
(H) Grin1ΔBLA-mPFC mice presented more significant coherence at the indicated band during the non-freezing period (N = 5, 5; ∗p < 0.05).
In the mPFC, Grin1ΔBLA-mPFC mice showed significant increases in the delta, theta, and beta bands when the CS evoked freezing, similar to other studies.56 Gamma band was elevated both during no freezing and freezing; however, compared to control mice, it was decreased during freezing.
Interestingly, Grin1ΔBLA-mPFC mice showed a significantly high beta band, which further increased upon tone presentation, in an opposite way to what was observed for control mice. Moreover, while control mice did not present any significant change in the delta band during freezing compared to non-freezing periods, Grin1ΔBLA-mPFC mice showed a significant increase. Altogether, these opposite power trends in different frequency bands between Grin1ΔBLA-mPFC and control mice might be relevant for the observed NMDAR-dependent deficits in fear conditioning, supporting the hypothesis that neural oscillatory activity in distinct frequency bands serves to link neural signals in multisensory processing.69,70
To better understand the role of mPFC and BLA during memory recall, we measured their functional connectivity by LFP coherence analysis. Grin1ΔBLA-mPFC mice presented a larger coherence value at the 8–14 Hz band (with a peak at 11.5 Hz) during non-freezing periods than during freezing (Videos S1 and S2), indicating a possible deficit in communication between the mPFC and the amygdala during fear memory expression (Figure 4E).
Fear memory engrams are located in both basolateral amygdala and medial prefrontal cortex
For the tagging of engram cells activated by a learning experience, we deployed a synthetic activity-dependent labeling method based on the promoter elements of c-fos, a gene well-known able to integrate neuronal activity.71 Our virus-delivered genetic activity-induced tagging of cell ensembles, or “vGATE” allows for Dox-controlled, activity-dependent rtTA expression,72 which activates the expression of responder genes under Ptetbi, such as Cre and a fluorescent protein. With the use of a flip-excision (FLEX) construct, the Cre recombinase enables the expression of Channelrhodopsin (hChR2)73 in the vGATE-tagged neurons (Figure 5A). We bilaterally injected the vGATE system and implanted optic fibers for the blue light (BL) optical stimulation of hChR2 expressed in the tagged cells, either in the BLA or in the mPFC of two groups of mice (Figures 5B and 5C). After cued fear conditioning, mice were tested for memory recall by optogenetic BL stimulation of hChR2-expressing cells in BLA or mPFC, respectively (Video S3). We found that the engram cells represented 16–18% of the cell population in the vGATE-infected regions (Figure S11; Table S1). Of note, activating vGATE-labeled engram cells in either the BLA or mPFC 10 days after conditioning was sufficient to recall fear memory (Figures 5B and 5C).
Figure 5.

Optogenetic reactivation of mPFC and BLA engram cells
(A) A schematic of the vGATE method (virus-delivered Genetic Activity-induced Tagging of Ensembles).
(B) Blue light stimulation of the BLA (BL+, 3mW) allowed the recruitment of the same ensemble of cells activated during conditioning. It produced significant freezing (vGATE-ONBLA), comparable to the fear-conditioned response observed in the cued test (vGATE-OFFBLA) for memory recall (N = 5, Retrieval: two-tailed paired t-test (no tone versus tone) p = 0.017, t = 3.92 df = 4. BL stimulation: two-tailed paired t-test (no BL versus BL) p = 0.005, t = 5.53 df = 4.).
(C) Blue light stimulation of the mPFC (BL+, 1mW) allows the recruitment of the same ensemble of cells activated during conditioning. It produces significant freezing (vGATE-ONmPFC), comparable to the fear-conditioned response observed in the cued test (vGATE-OFFmPFC) for memory recall (N = 5, Retrieval: two-tailed paired t-test (no tone versus tone) p = 0.0014, t = 7.95 df = 4; BL stimulation: two-tailed paired t-test (no BL versus BL) p = 0.025, t = 3.47 df = 4). ∗p < 0.05, ∗∗p < 0.01 by two-tailed paired t-test. Data are presented as mean ± s.e.m. Scale bars, 100 mm.
Discussion
Here, by genetic and combinatorial blocking of synaptic output, we demonstrate that cued-fear memory engram is sequentially printed from the BLA to the mPFC and, subsequently, to the other brain regions. We further demonstrate that presynaptic mPFC and postsynaptic BLA NMDARs are necessary for the sequential printing of memory engrams for flexible memory retrieval.
Direct evidence for “sequential” printing of fear memory engrams across brain regions
The amygdala and the medial prefrontal cortex are crucial in organizing fear memories. It is, however not known if cued fear memory representations at the circuit level follow a specific neuroanatomical pathway. When we precisely targeted BLA neurons using an advanced genetic method for virus-delivered inducible silencing of synaptic transmission (vINSIST-2) (Figure 1A), we found that consistent with previous findings,74 genetic silencing (with dsTeTxLC) of BLA synaptic output before fear conditioning, but not afterward, impaired fear memory expression (Figures 1B and 1C), suggesting that fear memory engrams are formed in the BLA, and subsequently could be printed from BLA to a different brain region(s). Given the strong reciprocal connectivity67 between BLA and mPFC, we selectively blocked synaptic output from the mPFC. Surprisingly, this manipulation did not interfere with fear memory expression (Figures 1D and 1E). We thus hypothesized that fear memory engrams could be distributed between different brain regions; BLA, mPFC and other region(s) could be used as alternative pathways for memory retrieval. For decisive testing of this hypothesis, we constitutively blocked mPFC synaptic output to prevent this pathway for fear memory retrieval and inducibly blocked BLA output after fear conditioning. We discovered that under such a condition fear memory expression was impaired (Figure 2A). However, the simultaneous blockade of BLA and mPFC synaptic outputs after fear conditioning did not interfere with fear expression (Figure 2B). These results support the hypothesis that cued-fear memory engram is first formed in the BLA and then printed to the mPFC and subsequently to other brain region(s).
N-methyl-D-aspartate receptor-dependent systems plasticity is required for cued-fear memory formation
Previous pharmacological studies using NMDAR-specific antagonists have revealed the essential role of NMDAR-dependent plasticity in the BLA in fear memory acquisition and expression.47,48 However, in those studies, not only postsynaptic BLA NMDARs were blocked, but also the presynaptic NMDARs that provide input to the BLA from other brain regions, such as the mPFC.
With advanced genetic tools for inducible gene deletion (Figure 3A), we targeted the BLA (Figures 3B, 3C, and S4) and validated the specific NMDAR impairment in acute brain slices (Figure S5).
We found that BLA-specific removal of NMDAR by Grin1 gene deletion either before (Figure 3D) or after fear conditioning did not interfere with fear expression (Figure S6A). Similarly, Grin1-specific deletion in the mPFC did not affect fear expression. However, when NMDARs were removed simultaneously from the BLA and the mPFC before fear conditioning, fear expression was impaired (Figure 3F), but not if the deletion was induced after fear conditioning (Figure S6B). These results demonstrate that BLA-mPFC NMDAR pre-post synaptic communication is needed for cued-fear memory formation but not expression. Importantly, the pharmacological block of NMDARs locally in the amygdala47,48 and double Grin1 gene deletion in BLA and mPFC have provided comparable results, suggesting that the presynaptic mPFC and postsynaptic BLA NMDARs are the key determinants of systems plasticity in fear memory formation (Figure 3G). In support, we found that in vivo long-term potentiation of the presynaptic mPFC to postsynaptic BLA synapse was impaired in the double Grin1 gene knockout mice (Figure 3H), supporting the roles of postsynaptic and presynaptic NMDARs in synaptic plasticity as described in previous studies.44,45,51,52,53,54,55,75,76,77,78,79
Moreover, our BLA/mPFC double Grin1 knockout mice displayed changes in network activity and frequency-dependent functional crosstalk between the amygdala and the mPFC during behavior (Figure 4). Similar to our findings, previous studies have shown that blocking NMDARs disrupt network synchrony and impair cognitive processes with elevated baseline gamma frequency bands, likely perturbed by loss of NMDAR in parvalbumin neurons. Cortical NMDAR is also needed for the development of theta and other oscillations.78,79,80,81,82 It is thought that an intricate balance of excitation and inhibition generates network oscillations and phase coherence between interacting brain regions, which might serve as a substrate for binding sensory perceptions and experiences in generating memories.70
Optogenetic activation of engram cells for fear memory retrieval
To investigate whether fear memory engrams were effectively formed and stored in the BLA and mPFC, we used a method for virus-delivered genetic activity-induced tagging of cell ensembles (vGATE) (Figure 5A) that has been functionally validated and applied to discover contextual fear memory engram in the hypothalamus.72 Here, we used the vGATE method to express hChR2 in BLA and mPFC-tagged engram neurons for optogenetic recall of fear memory. Optogenetic activation of memory engram-tagged cells either in the BLA or in the mPFC was sufficient for fear memory recall (Figures 5B and 5C). Thus, our results convincingly demonstrate that fear memory engram is sequentially printed from the BLA to the mPFC and preserved in both regions. Moreover, our work is consistent with multi-trace memory theory for the formation of distributed memory engrams across brain regions and the finding that memory engrams are temporally printed across the different brain regions for retrieval83 and are quite stable.84
A proposal for the “multi-trace systems consolidation” mechanism
Our finding that mPFC presynaptic NMDAR and BLA postsynaptic NMDAR are needed for fear memory formation is a significant leap forward that adds a circuit mechanism for cued-fear memory and perhaps the other memory forms. It is well established that synaptic and homeostatic plasticity mechanisms44,85,86 play crucial roles in modulating synaptic connections by LTP and long-term depression (LTD). The NMDAR has emerged as a critical molecular plasticity switch in these processes.44,75,76 Recent evidence suggests that presynaptic NMDARs are highly relevant in evoked neurotransmitter release and long-term presynaptic plasticity. Cortical presynaptic NMDAR activates calcium-dependent BDNF release, which in turn binds to its cognate presynaptic TrkB receptor in eliciting LTP.55,57,58
We proposed that the BLA postsynaptic and mPFC presynaptic NMDARs likely play a pivotal role in the printing of BLA engram neurons.39 This proposed mechanism operating in the BLA-mPFC synapse for the sequential printing of synaptic memory engram across brain regions needs to be investigated with greater detail in the future.
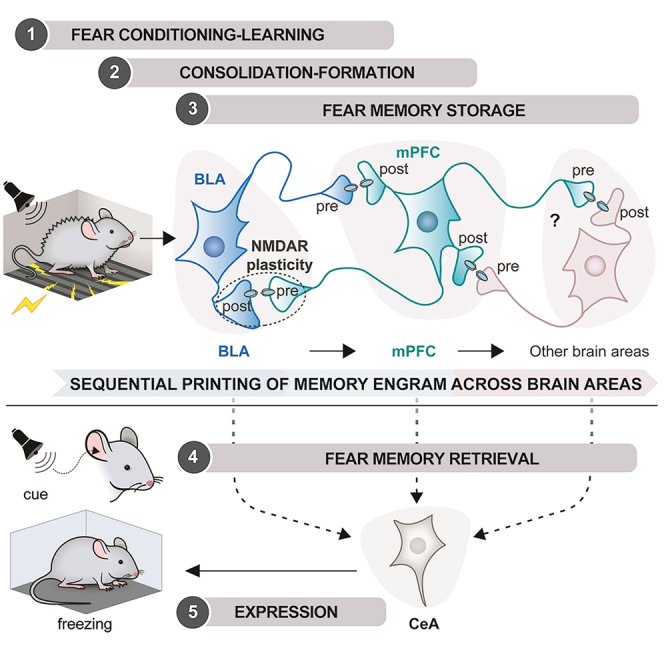
Based on our findings and published work, we propose a “multi-trace systems consolidation” mechanism involving NMDAR-dependent plasticity for the sequential printing of memory engram from the BLA to the mPFC and across the different brain region(s) (see Graphical Abstract): (1) during cued fear conditioning, sensory signals from cortical structures are integrated into the BLA to form a fear memory engram, (2) the BLA engram synaptic input onto mPFC neurons actively recruits them, (3) the excited presynaptic mPFC terminals make connections with the potentiated postsynaptic BLA neurons, (4) BLA postsynaptic NMDAR and presynaptic NMDAR are both needed for synaptic plasticity, and memory formation, and (5) memory engram is printed from the BLA to the mPFC and then to another brain region(s) for memory storage, providing alternative pathways for memory retrieval.
With early tagging of neural circuits,85,87 the generation of BLA-mPFC memory engrams31,35 and other brain regions,83 even in an evolutionarily older brain structure such as the hypothalamus,72 provide multiple pathways for flexible memory retrieval.27,28,35,88 These findings prompted us to propose that memory engrams are sequentially printed across the different brain regions following the “multi-trace systems consolidation” mechanism.14,15,42
Conclusion
We have demonstrated that (1) cued-fear memory engram is sequentially printed from BLA to mPFC and subsequently to other brain regions (s), (2) presynaptic mPFC NMDAR and postsynaptic BLA NMDAR coordinate the formation of cued fear memory, (3) NMDAR-dependent BLA-mPFC circuits regulate changes in network activity and coherence between amygdala and mPFC during behavior, and (4) optogenetic stimulation of engram cells in either BLA or mPFC is sufficient to induce memory recall.
Our work has fulfilled the critical criteria of the synaptic plasticity memory hypothesis,89 namely NMDAR-dependent systems synaptic plasticity as “necessity,” network activity changes during behavior as “detectability,” and optogenetic recall of memory as “sufficiency." We conclude that cued fear memory engrams are sequentially printed across the different brain regions by the “multi-trace systems consolidation” mechanism.
Our findings of altered circuit dynamics in double Grin1 gene knockout in the BLA and mPFC would provide crucial insight into cognitive dysfunction, psychosis, and other psychiatric disorders such as autism spectrum disorder,90 posttraumatic disorder (PTSD),91,92,93,94 and schizophrenia.95 The genetic toolbox applied in the current study is adequately applicable for understanding the operating principles of brain circuits for the various neurobiological processes and pathological conditions and has the potential to facilitate the design of circuit-targeted therapeutics to treat brain diseases.
Limitations of the study
In this work, we performed circuit-based studies and revealed that the cued-fear memory is sequentially printed from BLA to mPFC and, subsequently, to other brain regions. Moreover, we demonstrate that presynaptic mPFC NMDAR and postsynaptic BLA NMDAR are essential for memory formation, likely facilitated by NMDAR-dependent LTP between the mPFC to BLA synapse. However, other points need to be further investigated in the future: (1) Developing novel genetic tools to inactivate the NMDAR receptor selectively in the presynaptic mPFC or postsynaptic BLA compartments would be necessary. Despite this, based on the previous studies with the local injection of NMDARs antagonist in the BLA, we can safely conclude that the presynaptic mPFC NMDAR and postsynaptic BLA NMDARs are the critical determinants for cued-fear conditioning. (2) Although we have unequivocally demonstrated by virus-delivered genetic intervention studies that memory engram is formed in the BLA and then printed to the mPFC and subsequently to other brain regions (s), it would be experimentally challenging to identify the third brain region(s), where memory engram is printed from mPFC. (3) Our vGATE-assisted tagging is based on c-fos. However, we recognize that other types of immediate-early genes are likely participating in the organization of memory engram. In addition, it is still unclear what the minimum number of temporally selected engram neurons would be sufficient to induce memory recall when artificially reactivated. Moreover, vGATE-assisted tagging of cells was performed during fear conditioning and not later after weeks. Regardless, if the tagged neurons during the early phase of cued-fear conditioning were not needed during the later time, the memory engram trace could be lost. This was not the case. (4) It would be necessary to anatomically trace the synaptic printing of memory engram from BLA to mPFC. This would require monosynaptic trans-synaptic tracing with the development of new sets of genetic tools. (5) It would also be interesting to perform simultaneous activity imaging with cellular resolution in the BLA and mPFC using mini-scopes to establish neuronal activity changes during cued-fear conditioning and later memory retrieval over time.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-synaptobrevin-2 | Abcam | Cat# ab3347, RRID:AB_2212462 |
| Mouse monoclonal anti-β-tubulin | Sigma-Aldrich | Cat# T8328, RRID:AB_1844090 |
| Horseradish peroxidase-linked anti-rabbit secondary ab | Vector Laboratories | Cat# PI-1000-1, RRID:AB_2916034 |
| Mouse monoclonal anti-NeuN | Millipore | MAB337, RRID:AB_2313673 |
| Rabbit polyclonal anti-Cre recombinase | Covance | PRB-106C |
| Anti-rabbit, Cy3-conjugated secondary ab | Jackson Immuno Research Labs | RRID:AB_2338000 |
| Anti-mouse, FITC-conjugated secondary ab | Jackson Immuno Research Labs | RRID:AB_2338594 |
| Rabbit monoclonal anti-GluN1 | Millipore | AB9864R, RRID:AB_10807557 |
| Bacterial and virus strains | ||
| rAAV-PhSYN-rtTA | Dogbevia et al.61 | N/A |
| rAAV-Ptetbi-iCre/tdTOM | Dogbevia et al.,61 | N/A |
| rAAV-Ptetbi-dsTeTxLC | This paper | N/A |
| pAAV-(tetO)7-Pfos-rtTA | Hasan et al.,72 | N/A |
| rAAV-Ptetbi-Cre/YC3.60 | Hasan et al.,72 | N/A |
| rAAV-PCAG-FLEX-hChR2-mCherry | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| doxycycline | Sigma-Aldrich | D3447-500MG |
| Critical commercial assays | ||
| ECL™ Prime Western Blotting Detection Reagent | GE Healthcare | GERPN2236 |
| Experimental models: Cell lines | ||
| HEK293 cells | ATCC | https://www.atcc.org/products/crl-1573/ |
| Primary hippocampal neurons | Stoppini et al.96 | N/A |
| Experimental models: Organisms/strains | ||
| C57Bl/6N mice | Charles River |
Mus musculus Strain Code 027 |
| Grin12lox mice | Niewoehner et al., 2007, available at EMMA>:< B6.129-Grin1tm1Rsp/Kctt EM:09220 |
Mus musculus Strain: B6.129 |
| Oligonucleotides | ||
|
Grin1 oligo 1 for in situ: 5’-CCAG TGTGCTCCGAGGGATCTCCTTC TTGACCA GAATGGTC-3’; |
Hasan et al.45 | N/A |
|
Grin1 oligo 2 for in situ: 5’TCGCT GTTCACCTTAAATCGGCCAAAG GGACTGAAGCGGTCCAGCAG-3’ |
This paper | N/A |
|
Grin1 oligo 3 for in situ: 5’-GATAC GAGCAGAGAAACTCCGGGGGG CACCTTCCCCAATGCCAGAGTT-3’ |
This paper | N/A |
|
Gria1 oligo for in situ: 5’-GTCACT GGTTGTCTGATCTCGTCCTTCTT CAAACTCTTCACTGTG-3’ |
This paper | N/A |
| Recombinant DNA | ||
| PhSYN-rtTA | Dogbevia et al.61 | N/A |
| Ptetbi-iCre/tdTOM | Dogbevia et al.61 | N/A |
| Ptetbi-dsTeTxLC | This paper | N/A |
| (tetO)7-Pfos-rtTA | Hasan et al.,72 | N/A |
| Ptetbi-Cre/YC3.60 | Hasan et al.,72 | N/A |
| PCAG-FLEX-hChR2-mCherry | This paper | N/A |
| Software and algorithms | ||
| Video capture system (Sony HDR-SR12E) | For LFP | https://www.sony.co.uk/electronics/support/hard-drive-camcorders-hdr-sr-series/hdr-sr12e/manuals/ |
| Multi-Clamp 700B | Patch clamp | https://www.autom8.com/shop/mdcaxon-instruments/microelectrode-amplifiers/molecular-devices-multiclamp-700b/ |
| Clampex 10.0 | Molecular Devices, Palo Alto, CA | https://info.moleculardevices.com/ |
| CED 1401 Plus (or maybe it’s not a software?) | Cambridge Electronics Design | |
| Spike2 software | Cambridge Electronics Design | https://ced.co.uk/products/spkovin/ |
| Chronux toolbox | National Institute of Mental Health | http://chronux.org/ |
| Sigmaplot 11 | Systat Software GmbH | https://systatsoftware.com/sigmaplot/ |
| PRISM- GraphPad | GraphPad Software, La Jolla, CA, USA | https://www.graphpad.com/ |
Resource availability
Lead contact
Further information and requests for resources, reagents and genetic tools should be directed to and will be fulfilled by the lead contact: mazahir.t.hasan@gmail.com.
Materials availability
-
•
Mouse lines used in this study are available for purchase at the EMMA European Mutant Mice Archive.
-
•
This study did not generate new unique reagents.
-
•
Information and requests regarding genetic tools should be made to the lead contact: mazahir.t.hasan@gmail.com.
Experimental model and subject details
C57Bl/6N (Charles River, Sulzfeld, Germany) and gene-targeted Grin12lox mice35 were housed under standard conditions (21 ± 2°C of temperature, 50 ± 10% humidity) in a 12 h light/dark cycle with food and water ab libitum. Mice were housed in standard laboratory cages with sawdust as bedding material and paper towels and cardboard houses for environmental enrichment in groups of 4–6 per cage. Mice used for the behavioral experiments were a mixture of both males and females with an age ranging from 8 to 24 weeks.
After surgery, mice were housed individually and were allowed to recover before the commencement of behavioral experiments. All surgical procedures and experiments were conducted during the day. Efforts were made to minimize animal suffering and to reduce the number of animals.
All experiments were performed in accordance with European Union (2010/276:33–79/EU) guidelines and with the animal welfare guidelines of the Max Planck Society and Spanish (BOE 34:11370-421, 2013) regulations for the care of laboratory animals. The project received approval from the local authorities (Regierungspräsidium Karlsruhe), under project licenses 35–9185.81/G71-10, 35–9185.81/G171-10, and by the local ethics committee of the Pablo de Olavide University (Seville, Spain).
Method details
Inducible gene deletion method
For inducible gene deletion in the brain of Grin1 floxed (Grin12lox) mice,45 we used two rAAVs: the first virus (rAAV-PhSYN-rtTA2-nM2) is equipped with the human synapsin promoter (PhSYN) to drive the expression of the doxycycline (Dox)-controlled reverse tetracycline transactivator (rtTA2-nM2). The second virus (rAAV-Ptetbi-iCre/tdTOM) has a bidirectional tet promoter (Ptetbi) to simultaneously express two responder genes, encoding for the Cre recombinase and a red fluorescent protein, tdTomato (tdTOM). Two-three weeks after the virus injection, mice were treated with Dox by a single intraperitoneal injection and also kept on Dox in drinking water (2% Dox, 5% sucrose). Dox treatment activates the expression of tdTOM and Cre recombinase for the inducible, region-specific knock-out.
The vINSIST-2 method
We developed an advanced method for Dox-controlled inducible silencing of synaptic transmission between connected circuits. We engineered the tetanus toxin light chain protein (TeTxLC)60 with a half-life time of several minutes. The destabilized TeTxLC (dsTeTxLC) cleaves the synaptic vesicle protein, synaptobrevin-2, which is essential for evoked neurotransmitter release. This is the next-generation technology for virus-deliver inducible silencing of synaptic transmission (Version-2) or vINSIST-2 (Dogbevia et al., manuscript in preparation). In brief, the Ptetbi is used to express dsTeTxLC (rAAV-Ptetbi-dsTeTxLC). Using a second rAAV equipped with the rtTA under the human synapsin promoter (rAAV-PhSYN-rtTA), dsTeTxLC expression is switched-ON with Dox and switched-OFF in the absence of Dox, respectively, for silencing and un-silencing of synaptic transmission.
The vGATE method
We developed an advanced genetic method for virus-delivered Activity-induced Tagging of cell Ensembles or vGATE.72 We engineered a synthetic c-fos promoter with upstream heptameric tetracycline (tet) operators, (tetO)7 in AAV (virus 1; pAAV-(tetO)7-Pfos-rtTA). Next, an AAV equipped with the Ptetbi expressing the Cre-recombinase was linked to a genetically-encoded calcium indicator (YC3.60) (virus-2; rAAV-Ptetbi-Cre/YC3.60) for Cre-dependent expression hChR2-mCherry (virus-3; rAAV-PCAG-FLEX-hChR2-mCherry). In the vGATE method, the c-fos promoter drives rtTA expression only when neurons are activated. The rtTA generated by transient c-fos promoter activity binds to the upstream (tetO)7 only in the presence of Dox. This way, the rtTA drives its own expression, thus establishing an autoregulatory loop, even when the induced c-fos promoter activity declines to baseline levels as neuronal activity subsides. Therefore, only in the presence of Dox can the rtTA activate the expression of the Ptetbi to express any gene of choice, for example, the Cre recombinase, for permanent tagging of activated cells via a Cre-dependent FLEX cassette.
AAV production and purification
Transfection of HEK293 cells for virus production
Virus preparation was done as described previously.97 In brief, HEK293 cells were grown in DMEM supplemented with 10 % FCS and 50 mg/l penicillin/streptomycin in 5 % CO2 at 37°C. Transfection was performed by calcium phosphate method with 50 μg total DNA per 15 cm plate. Plasmids corresponding to rAAV constructs were co-transfected with pDp1 and pDp2 (ratio 2:1:1) helper plasmids in HEK293 cells.85 Forty-eight hours after transfection, cells were harvested by scraping and pulled into 50 ml falcon tubes. The cells were resuspended in 45 ml of 20 mM Tris 150 mM NaCl buffer (pH 8.0), frozen immediately in liquid nitrogen, and stored overnight at -70 C. Cells were thawed at room temperature (RT) and incubated at 37°C with 40 U/ml of Benzonase (for the degradation of unpacked DNA) and 0.5 % NaDOC for 60 min with frequent mixing. Lysed cells were spun at 3900 rpm for 15 min, and the supernatant was collected into a new falcon tube and frozen at -70°C overnight. The next day, the supernatant was thawed and spun for 15 min at 3900 rpm. The supernatant was run through a pre-equilibrated 1 ml heparin column (from Amersham, Freiburg, Germany). The column was serially washed with 20 ml of 100 mM NaCl 20 mM Tris (pH 8), 1 ml of 200 mM NaCl 20 mM Tris (pH 8), and 1 ml of 300 mM NaCl 20 mM Tris (pH 8). The virus was eluted serially with 1.5 ml of 400 mM NaCl 20 mM Tris.HCl (pH 8), 3 ml of 450 mM NaCl 20 mM Tris (pH 8), and 1.5 ml of 500 mM NaCl 20 mM Tris.HCl (pH 8). The eluted virus solution was collected into a 15 ml Amicon Ultra concentrator. The virus was washed at least two times with PBS by filling the tubes and spinning at 3200 rpm for 2 min. The virus was concentrated further into a final 250 μl and filtered through a 0.2 μm Acrodisc column. 10 μl of the purified virus was loaded on 10 % SDS gel. Purified viruses were stored at -80°C in 10 μl volumes for long-term storage. Dissociated neurons were infected with serially diluted viruses to determine the functional virus titer, and fluorescently labeled cells were counted.
Coomassie blue staining of proteins
10 μl of the purified virus was loaded onto a 10 % SDS PAGE with 2.5 μl of 5x Lämli buffer. The gel was run at 130 V and 400 mA for about an hour. The gel was stained for 45 min with Coomassie blue and de-stained for another 45 min. Successful virus purification resulted in the observation of 3 bands on the gel corresponding to the viral proteins (87 kDa VP1, 73 kDa VP2, and 62 kDa VP3).
Determination of infectious virus titer
Primary hippocampal neurons were prepared from E18 rat embryos and plated at 5 x 104 in cell culture multi-well dishes (24 wells). Four days after preparation, the primary cultures were infected with different volumes of rAAVs. Two weeks after infection, the highest dilution at which fluorescent cells were present was used for titer determination by counting. Six 10x magnification fields were photographed, and the number of fluorescent neurons was counted. This number was multiplied by the ratio of the total well surface area to the 10x field area and divided by the volume of virus applied. This yielded the number of neurons infected per microliter of the virus.
Analysis of AAV-mediated gene expression in rat organotypic brain slices and dissociated neurons
Organotypic brain slices were prepared from rat embryos according to an existing protocol.96 Slices were infected with different rAAVs by directly dropping 0.5-1 μl of the viral cocktail onto the tissue. Tissue media were changed 5 days after infection, and subsequent media changes were done every 2-3 days. After two weeks of virus infection, tissues were either harvested for Western blotting, quantitative luciferase assay, or fluorescent microscopy. On the other hand, dissociated neurons were infected by adding 1 μl of the virus cocktail into the media. Two weeks after virus addition, cells were lysed for quantitative luciferase assay or used for viral titer determination.
Immunoblot analysis
Two weeks after the virus infection, rat organotypic slices were harvested in cold lysis buffer and homogenized by sonication. Protein concentrations were determined by Bradford assay. Samples of 15 μg of the protein lysates from both rAAV infected and uninfected tissues and lysates were separated by SDS-PAGE (15% separating and 6% stacking gels). Transfer onto nitrocellulose membranes was done overnight at 30V and 90mA followed by blocking of the membrane in 5-10% fat-free milk in 1x PBS for 1 hour. Western blots were probed with the following primary antibodies either overnight at 4°C or for 2 hours at room temperature (RT): synaptobrevin-2 (1:1000, rabbit polyclonal Abcam) and β-tubulin (1:1000, mouse monoclonal, Sigma-Aldrich). As a secondary antibody, we used the horseradish peroxidase-linked anti-rabbit (1:15000, Vector Laboratories, Peterborough, UK). Western blots were detected by ECLplus (GE Healthcare).
Stereotaxic injections
Mice were anesthetized with isoflurane and placed in a stereotaxic device. The mice received inhaled isoflurane during all surgeries. Respiratory rate and absence of the tail pinch response were monitored regularly, and a heating pad was used to prevent hypothermia. Stereotaxic viral injections were performed as described previously.45,61,62 Mice received bilateral viral infusion with the following coordinates: for the mPFC, AP 1.75 mm; ML, 0.3 mm; DV, 1.3/1.8 mm; for the BLA, AP, -1.8 mm; ML, 3.55 mm; DV, 3.7 mm. All coordinates were measured from bregma and the tip of the brain.
Doxycycline treatment
The stock solution was prepared to dissolve doxycycline (Sigma-Aldrich) 5 mg/ml in 0.9% NaCl. A single dose of Dox was administered intraperitoneally (10 μl/g body weight), and where mentioned, Dox was also provided in the drinking water (2 mg/ml supplemented with 5% sucrose).
Behavioral analysis
Except for the burrowing test, mice were transported to a dimly illuminated testing room adjacent to the animal’s room and left undisturbed for at least 30 min before testing. At the end of each trial, each apparatus was accurately cleaned up with ethanol 2% and water.
Burrowing test
A burrowing test was performed to detect deterioration in the ability to perform “Activities of daily living” (ADL) as described previously.64 Briefly, a 200mm long, gray plastic, 68mm diameter grey tube filled with 200g of food pellet was placed against the wall in the mouse home cage, and the amount of food burrowed out was measured after 2 h and overnight as an indication of the ability to perform daily tasks that would come naturally to rodents. A wild-type C57Bl/6N mouse should burrow around 60g in the first 2h of testing and around 150g overnight.
Open-field test
Mice were placed in the center of a 50 cm x 50 cm square gray opaque plastic arena illuminated with 1000 lux and allowed to explore for 5–15 min. A video tracking system monitoring the arena from the top was used to measure the distance traveled by the mice in 1 min bins, as well as the percentage of time spent exploring the central 25 cm x 25 cm area of the arena.
Elevated plus maze test
The apparatus consisted of a plus-shaped platform elevated 1 m from the ground with 38 cm long arms and a .5 cm x 3.5 cm intersection. Two opposed arms were flanked by 3 cm high walls that protected the mice from falling, while the other two arms were open. The mice were placed on the central intersection of the maze, and the amount of time spent in the closed arms and the number of entries into the closed arms were measured over 10 min. The central area of the cross was considered part of the closed arms.
Light-dark box test
Mice were kept under dark conditions for 30 min before the start of the experiment. The apparatus consisted of a plastic box (40 cm long, 30 cm wide, 36 cm high) consisting of two symmetrical compartments communicated by a small opening (3.5 cm wide, 3 cm high): one of them with white walls and brightly lit with 1000 lux; the other with black walls and a black lid to create a dark interior. Mice were first placed in the light compartment, and the amount of time spent exploring each of the two chambers was measured for 5 min, as well as the number of entries into the dark compartment.
Passive avoidance test
The passive avoidance apparatus consisted of a dark chamber with a metallic grid floor placed on top of a tower. The chamber has an entrance that leads to a small platform, which is elevated 1 m above the ground and illuminated with 1000 lux from the top. A habituation session was conducted, in which the mice were placed on the platform and allowed to enter the dark chamber and explore for 3 min. In the acquisition phase, mice were placed on the platform and allowed to explore for 1 min. If they did not enter within this time, mice were returned to the home cage, and the procedure was repeated after 5 min. The latency to enter was measured, and once the mouse was inside, the entrance was blocked, and a 2 s 0.7 mA foot shock was administered, after which mice remained an additional 1 min inside before returning to their home cage. In the test session, mice were again placed on the open platform, and the latency to enter the dark chamber was measured.
Visual-association swimming task
The apparatus consisted of a small trapezoidal pool (36 cm and 24 cm sides, 137 cm long, 50 m high) and a large trapezoidal pool (70 cm and 24 cm sides, 137 cm long) filled with water at a depth of 15 cm, in which a 14 cm high platform could be completely submerged. The water temperature was kept at 21°C.
Fear conditioning and evaluation
A three-shock acquisition protocol was performed into the fear-conditioning box (TSE; 25 cm wide, 25 cm long, 35 cm high) with metallic grid floor, transparent plexiglass wall, 70% ethanol smell, and 800 lux illumination (2 bulbs) (context A). Mice were allowed to explore for 3 min, after which a 30 s, 80 dB, 7.5 kHz tone (CS) was presented. The last 2 s of the tone overlapped with a 0.4 mA foot shock (US). The CS-US pairing was repeated twice more with 2 min intervals between each presentation and following the last one. Therefore, the fear conditioning acquisition test lasted lasting total 10.5 min. The cued fear test was performed in the fear conditioning box equipped with a different floor, a plastic opaque grey one, black plastic walls, an acetic acid smell, and 400 lux illumination (only one bulb) (context B). Mice were allowed to explore for 3 min, after which the CS was presented continuously for another 3 min (total duration 6 min). Freezing was assessed every 5 s by a blind observer and defined as the absence of visible movement, except for respiration. The percentage of freezing was calculated over the total time of the cued test before and during the CS presentation (180 +180 s).
Electrophysiology
Standard procedures98 were used to prepare 300 μm-thick coronal slices from 9- to 13-month-old male ControlBLA or Grin1ΔBLA mice following a protocol approved by the Veterinary Department of the Canton of Basel-Stadt. Briefly, the brain was dissected in ice-cold artificial CSF (ACSF), mounted on an agar block, and sliced with a vibratome (Leica VT 1000; Leica, Wetzlar, Germany) at 4°C. Slices were maintained for 45 min at 37°C in an interface chamber containing ACSF equilibrated with 95% O2/5% CO2 and containing the following (in mM): 124 NaCl, 2.7 KCl, 2 CaCl2, 1.3 MgCl2, 26 NaHCO3, 0.4 NaH2PO4, 18 glucose, 4 ascorbate, and then for at least 45 min at room temperature before being transferred to a super-fusing recording chamber. Whole-cell recordings from BLA projection neurons were performed at 35°C. Neurons were visually identified with infrared video microscopy using an upright microscope equipped with a 40x objective (Olympus, Tokyo, Japan). Patch electrodes (3–5 MΩ) were pulled from borosilicate glass tubing and normally filled with a solution containing the following (in mM): 133 K-gluconate, 7 KCl, 10 HEPES, 10 phosphocreatine-Na2, 4 Mg-ATP, and 0.3 Na-GTP (pH adjusted to 7.2 with KOH or CsOH, respectively, 280-290 mOsm).
Electrode implantation for in vivo electrophysiology
Mice received bilateral viral infusion as described previously45 with the following coordinates: AP, 1.75; ML, 0.3; DV, 1.3/1.8 mm to target the mPFC and AP, -1.8; ML, 3.55; DV, 3.7 mm to target the BLA. All coordinates were measured from bregma and the tip of the brain. Two weeks later, animals (n = 10 KO and n = 10 WT) were re-anesthetized and implanted bilaterally with recording electrodes in the mPFC (1.75 mm anterior to bregma, 0.3 mm lateral, and 1.5–2.2 mm from brain surface) and in the amygdala (1.5 mm posterior to bregma, 2.5 mm lateral, and 4 mm from brain surface).96 Electrodes were made from 50 μm, Teflon coated, tungsten wire (Advent Research). A bare silver wire was affixed to the bone as ground. All the implanted wires were soldered to two four-pin sockets (RS Amidata) that were fixed to the skull with small bone screws and dental cement. LTP was evoked at the mPFC-amygdala synapse by high-frequency stimulation (HFS) of the ipsilateral mPFC in alert-behaving mice, followed by pair of pulses presented with an interval of 40 ms.
Fear conditioning and local field potential recordings
Experiments were started 3 weeks after the first surgical step. For fear conditioning, we followed the three-shock acquisition protocol. Experiments were carried out in a Skinner box (MED Associates) adapted for fear conditioning. The box (12.5 × 13.5 × 18.5 cm) was provided with a metallic grid floor, a front transparent Plexiglas wall, 70% ethanol smell, and 800 lux illumination (context A). Mice were subjected to the same above-described delay fear conditioning protocol. LFPs were recorded at the implanted sites during the whole duration of the tests. LFPs were recorded with Grass P511 differential amplifiers with a bandwidth of 0.1-10 KHz (Grass-Telefactor). All training sessions were recorded with a video capture system (Sony HDR-SR12E) synchronized to LFP recordings.
Optogenetic
Optical fiber cannula implantation
Mice received bilateral viral infusion (vGATE) and fiber optic cannula implantation (0.39 NA, 200 μm diameter; ThorLabs) in either the mPFC or the BLA. For mPFC, a fiber optic cannula (length: 2 mm) was implanted slantwise to reach the top of the infected area: coordinates: AP: 1.75 mm; ML, 1.25 with an angle of 16 degrees; whereas for BLA, 4mm long fiber optic cannula was implanted bilaterally using the same coordinates used for infection. In this way, the fibers could illuminate the infected area because the length of the cannula also comprises the thickness of the skull. Improperly targeted mice were excluded from the analysis; they did not show BL stimulation-induced changes in behavior (Table S1). Following surgical recovery, the animals were intensively handled by the operator and habituated to the optical fibers in a home-cage environment for 10 min per day for 2 days.
Optical stimulation
On the day of the experiment, the fiber optic cannulas implanted on animals were connected to the optical fibers (200 μm core, 0.39 numerical aperture, 1 m long, Thorlabs), through a sleeve. The animals were allowed to recover from this handling for 5 min in their cage before behavioral testing. 24 hours before the acquisition (context A), mice received an intraperitoneal injection of Dox (Day 1) to activate the vGATE system (see above). On day 3, mice were exposed to context B for retrieval cued test. During these two tests, no blue light stimulation was given. After ten days, when the animal body was completely cleared from Dox, mice were again exposed to context B, and blue-light (BL) stimulation was given instead of the tone during the session. BL illumination of cells expressing ChR2 was performed using PlexBright Optogenetic Stimulation System (Plexon, USA) for 180s (which represents the entire duration of the tone in the cued retrieval test) at 30Hz with 10ms pulses. BL intensity was calibrated at the fiber-tops to 3 mW power to bilaterally activate BLA cells that expressed c-fos during learning and, therefore, ChR2 at the time of the test (n = 5 mice), and to 1 mW for mPFC cells (n = 5 mice). The other 2 groups of mice were excluded from the analysis because of incorrect or undetectable targeting (n= 4,4). The excluded mice underwent the same BL stimulations during the fear retrieval test and were useful as controls to evaluate the effect of BL stimulation on behavior.
Histology
At the end of the experiments, mice were deeply anesthetized by isoflurane (Baxter) inhalation and transcardially perfused with ice-cold PBS (pH 7.5) and ice-cold 4% paraformaldehyde (PFA, Merck) in PBS. Brains were removed and post-fixed overnight in 4% PFA. Fixed brains were either agarose-embedded for sectioning with a vibratome (VT1000S, Leica, Wetzlar, Germany) or cryoprotected in 30% sucrose in PBS (0.1 M, pH 7.4) and then frozen for cryostat (Leica Instruments) sectioning. Brain coronal sections (50 μm thick) of prefrontal and amygdalar regions were used for different types of analysis.
For in situ hybridization, mice were euthanized by cervical dislocation, and brains rapidly froze on dried ice. Cryostat coronal 14-μm-thick slices were cut from unfixed frozen brains, slide-mounted, and then fixed with 4% paraformaldehyde and stored in ethanol at 4°C until required.
in situ hybridization
Sections were processed according to the protocol reported.99 Roughly 40-/45-mer oligonucleotides complementary to NMDA receptor GluN1 subunit (Grin1 gene) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor AMPA receptor (AMPAR) subunit, GluA1 (Gria1 gene) mRNAs were radio-labelled with [α33P] dATP by Hartmann Analytic GmbH. To increase the signal for GluN1, a mixture of three different oligonucleotide probes was used as probes (see key resources table). Each oligonucleotide recognizes a different sequence of the mRNA. Radio-labeled probes were diluted in a hybridization buffer, applied to the brain sections, and hybridized overnight at 42°C. The next day excess probes were washed off, and after dehydration, the sections were exposed to X-ray film (Amersham Hyperfilm ECL). Autoradiograms were then scanned to obtain global images of hybridized brain slices.
Double fluorescence immunostaining for NeuN and Cre antigens
Free-floating 50-μm-thick coronal sections were blocked for 1 h at room temperature (RT) in blocking buffer (3% normal goat serum and 1% TritonX-100 in PBS) and then incubated for 24h at 4°C with primary mouse monoclonal antibody against NeuN (Millipore) and rabbit polyclonal antibody against Cre recombinase (Covance) both diluted 1:1000 in 1% NGS, 0.3% TritonX-100 in PBS. The day after sections were incubated for 2 h RT with the appropriate species-specific secondary antibody (Cy3-conjugated or FITC-conjugated, Jackson Immuno Research) diluted 1:800. Finally, DAPI (Sigma, 1:5000) was added for 15 min before the final washing steps with PBS. Slices were mounted, dehydrated, and cover-slipped with aqua polymount (Polyscience). Pictures were collected using a fluorescence Leica microscope.
Fluorescence immunostaining for GluN1
This immunostaining requires a first step for antigen retrieval with sodium citrate buffer (10mM, pH6) at 95°C for 40 min. After cooling down to room temperature (approximately 20 min), sections were blocked 1 hour with 1% BSA and 0.5% TritonX-100 in PBS. Monoclonal rabbit anti-GluN1 antibody (AB9864R, Millipore) diluted 1:500 in 0.2% TritonX-100 in PBS was applied overnight at 4°C. The day after, the sections were washed in PBS and then incubated for 2 h with a Cy3-conjugated anti-rabbit secondary antibody (1:800, Jackson Immuno Research). DAPI (Sigma, 1:5000) was added for 15 min before the final washing steps with PBS. Slices were mounted on glass slides using 80% glycerol in PBS, and cover slips were fixed with nail polish. Section pictures were collected using a fluorescence Leica microscope.
DAB immunohistochemistry for GluN1
The antigen retrieval step (see above) was followed by endogenous Peroxidase blocking by 15 min incubation with hydrogen peroxide 0.5% in PBS. Afterward, sections were blocked with a solution made of 0.3% Triton-X 100, 1% BSA, and 2% NGS in PBS for 1 h and incubated overnight at 4°C with primary rabbit anti-GluN1 antibody (AB9864R, Millipore) at 1:1000 dilution. The day-after sections were incubated for 2h with a peroxidase-conjugated anti-rabbit secondary antibody (Vector Laboratories, 1:600). Peroxidase was reacted with 0.04% diaminobenzidine (DAB) and 0.003% hydrogen peroxide in the dark. After washing, brain slices were mounted on slides and air-dried overnight. DAB-developed slices were cover-slipped with aqua mounting medium and analyzed with a Leica brightfield microscope.
Nissl staining
To determine the proper location of recording electrodes, slices were mounted on gelatinized glass slides using the Nissl technique with 0.1% toluidine blue as described previously.100 In brief, fixed brain sections were washed in 0.1 M PBS, dehydrated in ethanol solutions with increasing concentrations (70, 80, 90, 96, 100%, three times, 30 min each), cleared in xylol (three times, 15 min and overnight), incubated (twice at 60°C, overnight and 2 h), and finally embedded in paraffin. Paraffin was removed by incubation of the slices with RotI-histol (2 × 10 min, Carl Roth GMBH) and subsequent treatment with ethanol 100, 96, and 70% (3 min, each) and rinsing in H2O. Staining was performed in 0.1% cresyl violet solution for 5–10 min followed by short rinsing in H2O and two brief washes (in 96% ethanol). After dehydration in 100% ethanol (twice, 3 min each) slices were cleared in RotI-histol (twice, 3 min each) and mounted in a permanent mounting medium (Eukit, Sigma-Aldrich).
Quantification and statistical analysis
In vitro slice patch-clamp electrophysiology data analysis
Data were recorded with a Multi-Clamp 700B, filtered at 2 kHz, and digitized at 10 kHz. In all experiments, series resistance was monitored before and after the experiment by applying a depolarizing current pulse of 10 pA throw the seal test provided by Clampex 10.0. Only access resistances below 15 MΩ were accepted; if they changed by 15%, the data were not included in the analysis. Data were acquired with Clampex 10.0 and analyzed with Clampfit 10.0 (Molecular Devices, Palo Alto, CA). Evoked EPSCs were elicited by stimulation of cortical afferent fibers with a bipolar twisted platinum/10% iridium wire (25 μm diameter) as previously described,98 recording in voltage-clamp mode at -70 mV for the AMPA and at +40 mV for the NMDA component. All experiments were performed in the presence of the non-competitive GABA-A receptor blocker picrotoxin (100 μM). The AMPA component was calculated by subtracting the peak of the fast eEPSC with the basal value of the trace right before the stimulus artifact (this value is shown with a gray dashed line in the figure) at -70 mV; the NMDA component was calculated at 50 ms after the peak of the AMPA current at 40 mV. NMDA/AMPA ratio was calculated by dividing the NMDA by the peak of the AMPA component.
In vivo local field potential data collection and analysis
Recordings of LFPs and moving images were stored digitally on a computer through an analog-to-digital converter (CED 1401 Plus, Cambridge Electronics Design). Collected data were sampled at 5 kHz and low-pass filtered at 0-100 Hz. Data were analyzed offline with the Spike2 software (Cambridge Electronics Design) for quantification of animal performance in the modified Skinner box. Power spectra and time-frequency spectrograms and coherence of selected LFP recordings were computed by using the multi-taper methods (5 tapers (K) and time-bandwidth (TW) of 3) from Chronux toolbox (http://chronux.org) for MATLAB. These computed results were processed for statistical analysis using Sigmaplot 11 (Systat Software). Unless otherwise indicated, data are always represented as the mean ± s.e.m. Only animals completing all the experimental steps (good LFP recordings, identifiable freezing behavior in the video recordings, and proper location of recording electrodes) were included in the analysis. For figure representation and analysis, we selected the following frequency bands: delta, 1.5-4 Hz; theta, 4–12 Hz; beta, 10–30 Hz; and gamma, 30–100 Hz. Acquired data were analyzed using one-way or two-way repeated-measures ANOVA. All of them were followed by Holm–Sidak or Tukey's post hoc testing depending on the previous statistical test.
In vivo LTP between mPFC and BLA: data analysis
Field EPSPs evoked at mPFC-BLA synapses were recorded across a high-impedance probe (2 × 1012Ω, 10 pF) with Grass P511 differential amplifiers (Grass-Telefactor, West Warwick, RI, USA), at a bandwidth of 0.1 Hz-10 kHz. Electrical stimulus applied to the mPFC area consisted of 100 μs, square, biphasic (positive-negative) pulses presented paired with 40 ms of inter-pulse interval. Stimulus intensities were ≤ 0.4 mA. Prior to LTP induction, we have 15 min of baseline recordings (3 per min). The stimulus intensity was set < 40% of peak fEPSP values. Then, each animal was presented with a high-frequency stimulation (HFS) protocol consisting of five trains (200 Hz, 100 ms) of pulses at a rate of 1/s. This protocol was presented 6 times in total, at intervals of 1 min. Evolution of fEPSPs after the HFS protocol was followed for 60 min at the same stimulation rate (3 per min). Additional post-HFS sessions (30 min) were carried out for three additional days.
Behavioral data analysis and statistics
All experiments and analyses were conducted experimenter-blind. Unless otherwise indicated, data are always represented as the mean ± s.e.m. A P-value of < 0.05 was considered statistically significant. Unpaired two-tailed Student’s t-test was used to compare two independent groups for the Open field test. Two-way repeated-measures ANOVA followed by the Bonferroni post-test for each trial was used to analyze the Visual Association test. For fear conditioning analysis, we used repeated-measures ANOVA to compare the degree of fear conditioning to the auditory cue (tone fear difference = CS-novel context freezing), with cue and genotype as factors (PRISM, GraphPad Software, La Jolla, CA, USA). If a significant difference was detected, groups were further compared with Bonferroni post hoc tests. The graphs present freezing bars before and during the tone (novel context freezing and CS).
Quantitative analysis of vGATE-tagged cells
Fifty mm thick brain sections were counterstained with 1:10000 4′,6-Diamidine-2′-phenylindole dihydrochloride DAPI (Aldrich, Milano, Italy) for 20 min, then rinsed in PBS, mounted and cover-slipped on a slide. Pictures were taken with a NIKON ECLIPSE (Nikon Instruments, Firenze, Italy) confocal microscope at 40X magnification, with each focal plane 1 mm thick. Twelve microns thick stacks were acquired for each picture and analyzed with ImageJ using the cell counter plugin and selecting cells within the stack (http://rsbweb.nih.gov/ij/index.html). The number of ChR2-tdTOM positive cells in each picture was divided by the number of DAPI-stained nuclei, and the percentage of recruited cells was calculated. This analysis was restricted to the infected area that was defined by the most external hChR2-positive cells in each acquired field. A total of 6 fields belonging to three different sections were averaged for each animal.
Acknowledgments
We thank Simone Hundemer for molecular biology and virus production, Judith Müller, Annette Herold and Gorka Kortabarria Perez for technical assistance, M. Sanchez-Enciso, J.M. González-Martín and J.A. Santos-Naharro for their help in animal handling and care. A special thanks to Andreas Lüthi and Benedetto Sacchetti for support with electrophysiology experiments, fear conditioning/optogenetic experiments, respectively, and to both in critically reading the article and providing input. We thank SciStyle for schematics. This work was funded by the Max Planck Society (I.B., G.K.D., M.T., R.S. & M.T.H.), Schloessman Foundation (M.T.H.), Fritz Thyssen Stiftung (M.T.H.), the Ingeborg Ständer Fouindation, ERA-NET NEURON (TopDown PTSD) (M.T.H), Grant PCIN-2017-120 MCIN/AEI/10.13039/501100011033/ERDF/NextGenerationEU/PRTR” (M.T.H.), Grant PID2021-124013OB-I00 MCIN/AEI/10.13039/501100011033 (M.T.H.), RTI2018-101624-B-I00 (M.T.H.) and National Institute of Health (M.T.H.), Spanish Ministry of Science and Innovation (BFU2017-82375-R), the Junta de Andalucía (Spain, BIO-122), the Spanish Tatiana Pérez de Guzmán el Bueno Foundation to A.G. and J.M.D.-G, and I.B. received funding from the SFB636 (R.S.).
Author contributions
Conceptualization, M.T.H.; Methodology, M.T.H.; Validation, G.K.D, M.T. and M.T.H.; Investigation, I.B, F.R.A., M.T.R.-B, A.C.G., M.C., J.M.D.G., P.B., A.O. and P.M.; Data curation, I.B., M.C., M.T., P.B., A.C.G. and J.M.D.G; Writing – Original Draft, M.T.H. and I.B.; Writing–Review & Editing, M.T.H., I.B., M.C., M.E.L. and R.S; Funding Acquisition, R.S., J.M.D.G and M.T.H; Resources, R.S. and J.M.D.G; Supervision, M.T.H.
Declaration of interests
The authors declare no competing interests.
Published: September 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108050.
Contributor Information
Ilaria Bertocchi, Email: ilaria.bertocchi@unito.it.
José Maria Delgado-García, Email: jmdelgar@upo.es.
Mazahir T. Hasan, Email: mazahir.t.hasan@gmail.com.
Supplemental information
Data and code availability
-
•
Data: data reported in this paper is available from the lead contact upon request.
-
•
Code: this paper does not report original code.
-
•
All other items: all additional information required about data and results reported in this paper is available from the lead contact upon request.
References
- 1.Gale G.D., Anagnostaras S.G., Godsil B.P., Mitchell S., Nozawa T., Sage J.R., Wiltgen B., Fanselow M.S. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J. Neurosci. 2004;24:3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Do Monte F.H., Quirk G.J., Li B., Penzo M.A. Retrieving fear memories, as time goes by. Mol. Psychiatr. 2016;21:1027–1036. doi: 10.1038/mp.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kandel E.R., Dudai Y., Mayford M.R. The molecular and systems biology of memory. Cell. 2014;157:163–186. doi: 10.1016/j.cell.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Squire L.R., Bayley P.J. The neuroscience of remote memory. Curr. Opin. Neurobiol. 2007;17:185–196. doi: 10.1016/j.conb.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tulving E. Multiple memory systems and consciousness. Hum. Neurobiol. 1987;6:67–80. [PubMed] [Google Scholar]
- 6.Tonegawa S., Morrissey M.D., Kitamura T. The role of engram cells in the systems consolidation of memory. Nat. Rev. Neurosci. 2018;19:485–498. doi: 10.1038/s41583-018-0031-2. [DOI] [PubMed] [Google Scholar]
- 7.Vetere G., Kenney J.W., Tran L.M., Xia F., Steadman P.E., Parkinson J., Josselyn S.A., Frankland P.W. Chemogenetic Interrogation of a Brain-wide Fear Memory Network in Mice. Neuron. 2017;94:363–374.e4. doi: 10.1016/j.neuron.2017.03.037. [DOI] [PubMed] [Google Scholar]
- 8.Harris A.Z., Gordon J.A. Long-range neural synchrony in behavior. Annu. Rev. Neurosci. 2015;38:171–194. doi: 10.1146/annurev-neuro-071714-034111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva A.J., Zhou Y., Rogerson T., Shobe J., Balaji J. Molecular and cellular approaches to memory allocation in neural circuits. Science. 2009;326:391–395. doi: 10.1126/science.1174519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCullough K.M., Morrison F.G., Ressler K.J. Bridging the Gap: Towards a cell-type specific understanding of neural circuits underlying fear behaviors. Neurobiol. Learn. Mem. 2016;135:27–39. doi: 10.1016/j.nlm.2016.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGaugh J.L. Memory--a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- 12.Asok A., Leroy F., Rayman J.B., Kandel E.R. Molecular Mechanisms of the Memory Trace. Trends Neurosci. 2019;42:14–22. doi: 10.1016/j.tins.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortega-de San Luis C., Ryan T.J. Understanding the physical basis of memory: Molecular mechanisms of the engram. J. Biol. Chem. 2022;298 doi: 10.1016/j.jbc.2022.101866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frankland P.W., Bontempi B. The organization of recent and remote memories. Nat. Rev. Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- 15.Nadel L., Moscovitch M. Memory consolidation, retrograde amnesia and the hippocampal complex. Curr. Opin. Neurobiol. 1997;7:217–227. doi: 10.1016/s0959-4388(97)80010-4. [DOI] [PubMed] [Google Scholar]
- 16.Fanselow M.S., Poulos A.M. The neuroscience of mammalian associative learning. Annu. Rev. Psychol. 2005;56:207–234. doi: 10.1146/annurev.psych.56.091103.070213. [DOI] [PubMed] [Google Scholar]
- 17.Krabbe S., Gründemann J., Lüthi A. Amygdala Inhibitory Circuits Regulate Associative Fear Conditioning. Biol. Psychiatr. 2018;83:800–809. doi: 10.1016/j.biopsych.2017.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Janak P.H., Tye K.M. From circuits to behaviour in the amygdala. Nature. 2015;517:284–292. doi: 10.1038/nature14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ressler R.L., Maren S. Synaptic encoding of fear memories in the amygdala. Curr. Opin. Neurobiol. 2019;54:54–59. doi: 10.1016/j.conb.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sears R.M., Schiff H.C., LeDoux J.E. Molecular mechanisms of threat learning in the lateral nucleus of the amygdala. Prog. Mol. Biol. Transl. Sci. 2014;122:263–304. doi: 10.1016/B978-0-12-420170-5.00010-6. [DOI] [PubMed] [Google Scholar]
- 21.Tovote P., Fadok J.P., Lüthi A. Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci. 2015;16:317–331. doi: 10.1038/nrn3945. [DOI] [PubMed] [Google Scholar]
- 22.Mátyás F., Lee J., Shin H.S., Acsády L. The fear circuit of the mouse forebrain: connections between the mediodorsal thalamus, frontal cortices and basolateral amygdala. Eur. J. Neurosci. 2014;39:1810–1823. doi: 10.1111/ejn.12610. [DOI] [PubMed] [Google Scholar]
- 23.Silva B.A., Burns A.M., Gräff J. A cFos activation map of remote fear memory attenuation. Psychopharmacology (Berl) 2019;236:369–381. doi: 10.1007/s00213-018-5000-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silva B.A., Gross C.T., Gräff J. The neural circuits of innate fear: detection, integration, action, and memorization. Learn. Mem. 2016;23:544–555. doi: 10.1101/lm.042812.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herry C., Ciocchi S., Senn V., Demmou L., Müller C., Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- 26.Cambiaghi M., Grosso A., Likhtik E., Mazziotti R., Concina G., Renna A., Sacco T., Gordon J.A., Sacchetti B. Higher-Order Sensory Cortex Drives Basolateral Amygdala Activity during the Recall of Remote, but Not Recently Learned Fearful Memories. J. Neurosci. 2016;36:1647–1659. doi: 10.1523/JNEUROSCI.2351-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Do-Monte F.H., Quiñones-Laracuente K., Quirk G.J. A temporal shift in the circuits mediating retrieval of fear memory. Nature. 2015;519:460–463. doi: 10.1038/nature14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goshen I., Brodsky M., Prakash R., Wallace J., Gradinaru V., Ramakrishnan C., Deisseroth K. Dynamics of retrieval strategies for remote memories. Cell. 2011;147:678–689. doi: 10.1016/j.cell.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 29.Kitamura T., Ogawa S.K., Roy D.S., Okuyama T., Morrissey M.D., Smith L.M., Redondo R.L., Tonegawa S. Engrams and circuits crucial for systems consolidation of a memory. Science. 2017;356:73–78. doi: 10.1126/science.aam6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herry C., Johansen J.P. Encoding of fear learning and memory in distributed neuronal circuits. Nat. Neurosci. 2014;17:1644–1654. doi: 10.1038/nn.3869. [DOI] [PubMed] [Google Scholar]
- 31.McDonald A.J. Cortical pathways to the mammalian amygdala. Prog. Neurobiol. 1998;55:257–332. doi: 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 32.Kwon J.T., Jhang J., Kim H.S., Lee S., Han J.H. Brain region-specific activity patterns after recent or remote memory retrieval of auditory conditioned fear. Learn. Mem. 2012;19:487–494. doi: 10.1101/lm.025502.112. [DOI] [PubMed] [Google Scholar]
- 33.Sparks F.T., Spanswick S.C., Lehmann H., Sutherland R.J. Neither time nor number of context-shock pairings affect long-term dependence of memory on hippocampus. Neurobiol. Learn. Mem. 2013;106:309–315. doi: 10.1016/j.nlm.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 34.Squire L.R., Stark C.E.L., Clark R.E. The medial temporal lobe. Annu. Rev. Neurosci. 2004;27:279–306. doi: 10.1146/annurev.neuro.27.070203.144130. [DOI] [PubMed] [Google Scholar]
- 35.Arruda-Carvalho M., Clem R.L. Pathway-selective adjustment of prefrontal-amygdala transmission during fear encoding. J. Neurosci. 2014;34:15601–15609. doi: 10.1523/JNEUROSCI.2664-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milad M.R., Quirk G.J. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 37.Rozeske R.R., Valerio S., Chaudun F., Herry C. Prefrontal neuronal circuits of contextual fear conditioning. Gene Brain Behav. 2015;14:22–36. doi: 10.1111/gbb.12181. [DOI] [PubMed] [Google Scholar]
- 38.Marek R., Sun Y., Sah P. Neural circuits for a top-down control of fear and extinction. Psychopharmacology (Berl) 2019;236:313–320. doi: 10.1007/s00213-018-5033-2. [DOI] [PubMed] [Google Scholar]
- 39.Lee J.H., Kim W.B., Park E.H., Cho J.H. Neocortical synaptic engrams for remote contextual memories. Nat. Neurosci. 2023;26:259–273. doi: 10.1038/s41593-022-01223-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nonaka A., Toyoda T., Miura Y., Hitora-Imamura N., Naka M., Eguchi M., Yamaguchi S., Ikegaya Y., Matsuki N., Nomura H. Synaptic plasticity associated with a memory engram in the basolateral amygdala. J. Neurosci. 2014;34:9305–9309. doi: 10.1523/JNEUROSCI.4233-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGaugh J.L. Memory consolidation and the amygdala: a systems perspective. Trends Neurosci. 2002;25:456. doi: 10.1016/s0166-2236(02)02211-7. [DOI] [PubMed] [Google Scholar]
- 42.Pedraza L.K., Sierra R.O., Crestani A.P., Quillfeldt J.A., de Oliveira Alvares L. Sequential learning during contextual fear conditioning guides the rate of systems consolidation: Implications for consolidation of multiple memory traces. Hippocampus. 2017;27:518–528. doi: 10.1002/hipo.22708. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X., Ren Q., Guo A. Parallel pathways for cross-modal memory retrieval in Drosophila. J. Neurosci. 2013;33:8784–8793. doi: 10.1523/JNEUROSCI.4631-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Citri A., Malenka R.C. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- 45.Hasan M.T., Hernández-González S., Dogbevia G., Treviño M., Bertocchi I., Gruart A., Delgado-García J.M. Role of motor cortex NMDA receptors in learning-dependent synaptic plasticity of behaving mice. Nat. Commun. 2013;4:2258. doi: 10.1038/ncomms3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunt M.J., Kasicki S. A systematic review of the effects of NMDA receptor antagonists on oscillatory activity recorded in vivo. J. Psychopharmacol. 2013;27:972–986. doi: 10.1177/0269881113495117. [DOI] [PubMed] [Google Scholar]
- 47.Kim J.J., DeCola J.P., Landeira-Fernandez J., Fanselow M.S. N-methyl-D-aspartate receptor antagonist APV blocks acquisition but not expression of fear conditioning. Behav. Neurosci. 1991;105:126–133. doi: 10.1037//0735-7044.105.1.126. [DOI] [PubMed] [Google Scholar]
- 48.Rodrigues S.M., Schafe G.E., LeDoux J.E. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J. Neurosci. 2001;21:6889–6896. doi: 10.1523/JNEUROSCI.21-17-06889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maren S., Aharonov G., Stote D.L., Fanselow M.S. N-methyl-D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav. Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- 50.Walker D.L., Davis M. Amygdala infusions of an NR2B-selective or an NR2A-preferring NMDA receptor antagonist differentially influence fear conditioning and expression in the fear-potentiated startle test. Learn. Mem. 2008;15:67–74. doi: 10.1101/lm.798908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banerjee A., Larsen R.S., Philpot B.D., Paulsen O. Roles of Presynaptic NMDA Receptors in Neurotransmission and Plasticity. Trends Neurosci. 2016;39:26–39. doi: 10.1016/j.tins.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buchanan K.A., Blackman A.V., Moreau A.W., Elgar D., Costa R.P., Lalanne T., Tudor Jones A.A., Oyrer J., Sjöström P.J. Target-specific expression of presynaptic NMDA receptors in neocortical microcircuits. Neuron. 2012;75:451–466. doi: 10.1016/j.neuron.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farb C.R., Ledoux J.E. Afferents from rat temporal cortex synapse on lateral amygdala neurons that express NMDA and AMPA receptors. Synapse. 1999;33:218–229. doi: 10.1002/(SICI)1098-2396(19990901)33:3<218::AID-SYN6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 54.Larsen R.S., Smith I.T., Miriyala J., Han J.E., Corlew R.J., Smith S.L., Philpot B.D. Synapse-specific control of experience-dependent plasticity by presynaptic NMDA receptors. Neuron. 2014;83:879–893. doi: 10.1016/j.neuron.2014.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Urban-Ciecko J., Wen J.A., Parekh P.K., Barth A.L. Experience-dependent regulation of presynaptic NMDARs enhances neurotransmitter release at neocortical synapses. Learn. Mem. 2014;22:47–55. doi: 10.1101/lm.035741.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamada K., Nabeshima T. Interaction of BDNF/TrkB signaling with NMDA receptor in learning and memory. Drug News Perspect. 2004;17:435–438. doi: 10.1358/dnp.2004.17.7.863702. [DOI] [PubMed] [Google Scholar]
- 57.Park H., Popescu A., Poo M.M. Essential role of presynaptic NMDA receptors in activity-dependent BDNF secretion and corticostriatal LTP. Neuron. 2014;84:1009–1022. doi: 10.1016/j.neuron.2014.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu B., Gottschalk W., Chow A., Wilson R.I., Schnell E., Zang K., Wang D., Nicoll R.A., Lu B., Reichardt L.F. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J. Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duguid I.C., Smart T.G. In: Biology of the NMDA Receptor. Van Dongen A.M., editor. 2009. Presynaptic NMDA Receptors. [Google Scholar]
- 60.Reus-García M.M., Sánchez-Campusano R., Ledderose J., Dogbevia G.K., Treviño M., Hasan M.T., Gruart A., Delgado-García J.M. The Claustrum is Involved in Cognitive Processes Related to the Classical Conditioning of Eyelid Responses in Behaving Rabbits. Cerebr. Cortex. 2021;31:281–300. doi: 10.1093/cercor/bhaa225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dogbevia G.K., Marticorena-Alvarez R., Bausen M., Sprengel R., Hasan M.T. Inducible and combinatorial gene manipulation in mouse brain. Front. Cell. Neurosci. 2015;9:142. doi: 10.3389/fncel.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dogbevia G.K., Roβmanith M., Sprengel R., Hasan M.T. Flexible, AAV-equipped Genetic Modules for Inducible Control of Gene Expression in Mammalian Brain. Mol. Ther. Nucleic Acids. 2016;5:e309. doi: 10.1038/mtna.2016.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mingat J., Micoud M. [Passage of doxycycline across the blood-brain barrier] Nouv. Presse Med. 1980;9:136–137. [PubMed] [Google Scholar]
- 64.Deacon R. Assessing burrowing, nest construction, and hoarding in mice. J. Vis. Exp. 2012:e2607. doi: 10.3791/2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gewirtz J.C., Falls W.A., Davis M. Normal conditioned inhibition and extinction of freezing and fear-potentiated startle following electrolytic lesions of medical prefrontal cortex in rats. Behav. Neurosci. 1997;111:712–726. doi: 10.1037//0735-7044.111.4.712. [DOI] [PubMed] [Google Scholar]
- 66.Sacchetti B., Baldi E., Lorenzini C.A., Bucherelli C. Differential contribution of some cortical sites to the formation of memory traces supporting fear conditioning. Exp. Brain Res. 2002;146:223–232. doi: 10.1007/s00221-002-1165-y. [DOI] [PubMed] [Google Scholar]
- 67.Vertes R.P. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- 68.Karalis N., Dejean C., Chaudun F., Khoder S., Rozeske R.R., Wurtz H., Bagur S., Benchenane K., Sirota A., Courtin J., Herry C. 4-Hz oscillations synchronize prefrontal-amygdala circuits during fear behavior. Nat. Neurosci. 2016;19:605–612. doi: 10.1038/nn.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Concina G., Cambiaghi M., Renna A., Sacchetti B. Coherent Activity between the Prelimbic and Auditory Cortex in the Slow-Gamma Band Underlies Fear Discrimination. J. Neurosci. 2018;38:8313–8328. doi: 10.1523/JNEUROSCI.0540-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keil J., Senkowski D. Neural Oscillations Orchestrate Multisensory Processing. Neuroscientist. 2018;24:609–626. doi: 10.1177/1073858418755352. [DOI] [PubMed] [Google Scholar]
- 71.Reijmers L.G., Perkins B.L., Matsuo N., Mayford M. Localization of a stable neural correlate of associative memory. Science. 2007;317:1230–1233. doi: 10.1126/science.1143839. [DOI] [PubMed] [Google Scholar]
- 72.Hasan M.T., Althammer F., Silva da Gouveia M., Goyon S., Eliava M., Lefevre A., Kerspern D., Schimmer J., Raftogianni A., Wahis J., et al. A Fear Memory Engram and Its Plasticity in the Hypothalamic Oxytocin System. Neuron. 2019;103:133–146.e8. doi: 10.1016/j.neuron.2019.04.029. [DOI] [PubMed] [Google Scholar]
- 73.Liu X., Ramirez S., Pang P.T., Puryear C.B., Govindarajan A., Deisseroth K., Tonegawa S. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature. 2012;484:381–385. doi: 10.1038/nature11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilensky A.E., Schafe G.E., LeDoux J.E. Functional inactivation of the amygdala before but not after auditory fear conditioning prevents memory formation. J. Neurosci. 1999;19:RC48. doi: 10.1523/JNEUROSCI.19-24-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bouvier G., Larsen R.S., Rodríguez-Moreno A., Paulsen O., Sjöström P.J. Towards resolving the presynaptic NMDA receptor debate. Curr. Opin. Neurobiol. 2018;51:1–7. doi: 10.1016/j.conb.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 76.Costa R.P., Mizusaki B.E.P., Sjöström P.J., van Rossum M.C.W. Functional consequences of pre- and postsynaptic expression of synaptic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017;372 doi: 10.1098/rstb.2016.0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Monday H.R., Castillo P.E. Closing the gap: long-term presynaptic plasticity in brain function and disease. Curr. Opin. Neurobiol. 2017;45:106–112. doi: 10.1016/j.conb.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carlén M., Meletis K., Siegle J.H., Cardin J.A., Futai K., Vierling-Claassen D., Rühlmann C., Jones S.R., Deisseroth K., Sheng M., et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol. Psychiatr. 2012;17:537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goonawardena A.V., Heiss J., Glavis-Bloom C., Trube G., Borroni E., Alberati D., Wallace T.L. Alterations in High-Frequency Neuronal Oscillations in a Cynomolgus Macaque Test of Sustained Attention Following NMDA Receptor Antagonism. Neuropsychopharmacology. 2016;41:1319–1328. doi: 10.1038/npp.2015.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee J., Hudson M.R., O'Brien T.J., Nithianantharajah J., Jones N.C. Local NMDA receptor hypofunction evokes generalized effects on gamma and high-frequency oscillations and behavior. Neuroscience. 2017;358:124–136. doi: 10.1016/j.neuroscience.2017.06.039. [DOI] [PubMed] [Google Scholar]
- 81.Pinault D. N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol. Psychiatr. 2008;63:730–735. doi: 10.1016/j.biopsych.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 82.Gu Z., Alexander G.M., Dudek S.M., Yakel J.L. Hippocampus and Entorhinal Cortex Recruit Cholinergic and NMDA Receptors Separately to Generate Hippocampal Theta Oscillations. Cell Rep. 2017;21:3585–3595. doi: 10.1016/j.celrep.2017.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roy D.S., Park Y.G., Kim M.E., Zhang Y., Ogawa S.K., DiNapoli N., Gu X., Cho J.H., Choi H., Kamentsky L., et al. Brain-wide mapping reveals that engrams for a single memory are distributed across multiple brain regions. Nat. Commun. 2022;13:1799. doi: 10.1038/s41467-022-29384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ryan T.J., Roy D.S., Pignatelli M., Arons A., Tonegawa S. Memory. Engram cells retain memory under retrograde amnesia. Science. 2015;348:1007–1013. doi: 10.1126/science.aaa5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lesburguères E., Gobbo O.L., Alaux-Cantin S., Hambucken A., Trifilieff P., Bontempi B. Early tagging of cortical networks is required for the formation of enduring associative memory. Science. 2011;331:924–928. doi: 10.1126/science.1196164. [DOI] [PubMed] [Google Scholar]
- 86.Zenke F., Gerstner W. Hebbian plasticity requires compensatory processes on multiple timescales. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017;372 doi: 10.1098/rstb.2016.0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Luboeinski J., Tetzlaff C. Memory consolidation and improvement by synaptic tagging and capture in recurrent neural networks. Commun. Biol. 2021;4:275. doi: 10.1038/s42003-021-01778-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Penzo M.A., Robert V., Tucciarone J., De Bundel D., Wang M., Van Aelst L., Darvas M., Parada L.F., Palmiter R.D., He M., et al. The paraventricular thalamus controls a central amygdala fear circuit. Nature. 2015;519:455–459. doi: 10.1038/nature13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martin S.J., Grimwood P.D., Morris R.G. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu. Rev. Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 90.Fortier A.V., Meisner O.C., Nair A.R., Chang S.W.C. Prefrontal circuits guiding social preference: Implications in autism spectrum disorder. Neurosci. Biobehav. Rev. 2022;141 doi: 10.1016/j.neubiorev.2022.104803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bailey C.R., Cordell E., Sobin S.M., Neumeister A. Recent progress in understanding the pathophysiology of post-traumatic stress disorder: implications for targeted pharmacological treatment. CNS Drugs. 2013;27:221–232. doi: 10.1007/s40263-013-0051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chambers R.A., Bremner J.D., Moghaddam B., Southwick S.M., Charney D.S., Krystal J.H. Glutamate and post-traumatic stress disorder: toward a psychobiology of dissociation. Semin. Clin. Neuropsychiatry. 1999;4:274–281. doi: 10.10153/SCNP00400274. [DOI] [PubMed] [Google Scholar]
- 93.Fenster R.J., Lebois L.A.M., Ressler K.J., Suh J. Brain circuit dysfunction in post-traumatic stress disorder: from mouse to man. Nat. Rev. Neurosci. 2018;19:535–551. doi: 10.1038/s41583-018-0039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Colucci P., Marchetta E., Mancini G.F., Alva P., Chiarotti F., Hasan M.T., Campolongo P. Predicting susceptibility and resilience in an animal model of post-traumatic stress disorder (PTSD) Transl. Psychiatry. 2020;10:243. doi: 10.1038/s41398-020-00929-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zick J.L., Blackman R.K., Crowe D.A., Amirikian B., DeNicola A.L., Netoff T.I., Chafee M.V. Blocking NMDAR Disrupts Spike Timing and Decouples Monkey Prefrontal Circuits: Implications for Activity-Dependent Disconnection in Schizophrenia. Neuron. 2018;98:1243–1255.e5. doi: 10.1016/j.neuron.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stoppini L., Buchs P.A., Muller D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 97.Heindorf M., Hasan M.T. Fluorescent Calcium Indicator Protein Expression in the Mouse Brain Using Recombinant Adeno-Associated Viruses. Cold Spring Harb. Protoc. 2015;2015:697–709. doi: 10.1101/pdb.prot087635. [DOI] [PubMed] [Google Scholar]
- 98.Humeau Y., Shaban H., Bissière S., Lüthi A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature. 2003;426:841–845. doi: 10.1038/nature02194. [DOI] [PubMed] [Google Scholar]
- 99.Wisden W., Morris B.J. In situ hybridization with oligonucleotide probes. Int. Rev. Neurobiol. 2002;47:3–59. doi: 10.1016/s0074-7742(02)47051-1. [DOI] [PubMed] [Google Scholar]
- 100.Bertocchi I., Eltokhi A., Rozov A., Chi V.N., Jensen V., Bus T., Pawlak V., Serafino M., Sonntag H., Yang B., et al. Voltage-independent GluN2A-type NMDA receptor Ca(2+) signaling promotes audiogenic seizures, attentional and cognitive deficits in mice. Commun. Biol. 2021;4:59. doi: 10.1038/s42003-020-01538-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: data reported in this paper is available from the lead contact upon request.
-
•
Code: this paper does not report original code.
-
•
All other items: all additional information required about data and results reported in this paper is available from the lead contact upon request.