

Summary
Standard methods of mixture analysis involve subjecting a dried crime scene sample to a “bulk” DNA extraction method such that the resulting isolate compromises a homogenized DNA mixture from the individual donors. If, however, instead of bulk DNA extraction, a sufficient number of individual cells from the mixed stain are subsampled prior to genetic analysis then it should be possible to recover highly probative single source, non-mixed scDNA profiles from each of the donors. This approach can detect low DNA level minor donors to a mixture that otherwise would not be identified using standard methods and can also resolve rare mixtures comprising first degree relatives and thereby also prevent the false inclusion of non-donor relatives. This literature landscape review and associated commentary reports on the history and increasing interest in current and potential future applications of scDNA in forensic genomics, and critically evaluates opportunities and impediments to further progress.
Subject areas: Biological sciences, Genomics, Bioinformatics
Graphical abstract
Biological sciences, Genomics, Bioinformatics
Introduction
Single cell genomics is a powerful tool in modern biology and medicine that is transforming our understanding of cellular biology and disease. Many biological processes, including development, disease progression, and the immune response, are influenced by the diversity of individual cells within tissues.1 Single cell genomic analysis (scDNA for short) allows researchers to analyze the genetic and epigenetic differences between individual cells and gain insights into the cellular heterogeneity underlying these processes, both in normal and disease states. Some cell types, including incipient tumor or other disease state cells, are present in very small numbers within tissues, making them difficult to identify and study using traditional methods.2 In contrast scDNA enables the detection and characterization of rare cell types, which can be crucial for not only understanding disease progression but informing potential therapeutic intervention.3 Forensic genetic analysis relies on the use of technologies such as DNA profiling to facilitate human identification (HID) in criminal and other medical legal matters, including humanitarian efforts with unidentified human remains from victims of accident, natural disasters, or other mass fatality events. In a forensic setting, scDNA has the potential to aid in investigations where limited DNA samples are available from persons of interest as well as deconvoluting the DNA profiles of multiple contributors (e.g., victims and perpetrators) to crime scene stains. The latter deconvoluted single source DNA profiles, can be used for direct human identification or inferring the biogeographical ancestry, DNA phenotypes and genetic genealogy of individual donors. This literature landscape review and associated commentary reports on the history and potential future applications of scDNA in forensic genomics by critically evaluating opportunities and impediments for further progress.
Current forensic genetic analysis methodology
The principal genetic analysis methodology currently used by forensic laboratories involves targeting ∼20–30 specifically chosen polymorphic short tandem repeat biomarker loci spread across the autosomal chromosomes (aSTRs).4 The STRs used in forensic science typically have tandem repeat sizes of 3–5 bp and individuals differ in the number of repeats that they have (i.e., alleles) within and between loci.5 The analysis begins by isolating DNA from potential evidentiary biological material from crime scenes, individuals, or weapons. Biological evidence can comprise anything of a biological nature from which DNA can be extracted, such as blood, semen, saliva, touch deposits ("touch DNA"), hair, teeth, and bones. After multiplex PCR amplification of the STR targeted alleles using fluorescently labeled primers, the resulting amplimers are separated according to allele size and detected via capillary electrophoresis (CE). Interpretation of the results typically proceeds by comparing the resulting DNA profiles with known reference profiles from persons of interest (POI) or searching against a DNA database. If a crime scene DNA profile matches a POI’s reference profile (i.e., could have had a common origin), the evidentiary weight of the match is estimated using a likelihood ratio (LR) approach.6 For example, the LR comprises the ratio of the probability of obtaining the crime scene DNA profile if it originates from the POI versus the probability of the DNA profile if, instead, it originated from a random unrelated individual. LRs with good quality single source matching DNA profiles (i.e., unmixed with any other individual) are typically > 1015 (i.e., log10 (LR) > 15). Apart from aSTRs, other biomarkers are currently employed in certain specialized cases. STRs located on the male sex chromosome (Y-STRS) can be useful in sexual assault cases.7,8 Mitochondrial DNA (mtDNA) sequence variation in the control region is useful for bones and teeth in missing persons identification and has traditionally been interrogated by Sanger sequencing.9,10 The implementation of massively parallel sequencing (MPS), due to its high suitability to multiplex any number of targeted markers, enables the implementation of combined marker types (e.g., Y-STRs, aSTRs, and SNPs).11,12,13 The ability of MPS to multiplex a large number of SNP biomarkers further facilitates the implementation of nuclear and mitochondrial genome SNP analysis.14 SNPs are especially useful for degraded DNA, unidentified human remains identification, and are de rigueur in the new field of forensic investigative genetic genealogy.15,16
Even during the early stages of forensic DNA, single cell analysis was being posited as a potential method to analyze smudged fingerprints, skin flakes, individual sperm, and low template samples potentially deposited on weapons.17,18,19 Findlay et al. demonstrated in 1997 the potential use of single cells in forensic science obtaining full DNA profiles in 50% of buccal cells tested using a 6 locus aSTR assay while some degree of recovery was obtained in 91% of the cells tested.17 The authors noted the presence of a significant degree of confounding now-well-characterized low DNA template stochastic effects including preferential amplification and allele drop out.17 In 1988 Li et al. were able to genotype SNPs from two genetic loci in single sperm cells and in diploid cell lines using high cycle PCR number (50 cycles) and dot probe hybridization detection.18 Also, in 1988 Jeffreys et al. demonstrated the potential for human identification with the amplification and analysis of hypervariable minisatellites ("DNA fingerprints") in micromanipulated single buccal and epithelial cells.19 Though technologies have drastically improved since these early papers, the low template nature of single cells is still responsible for many of the challenges associated with scDNA. These challenges are particularly exacerbated when it comes to “touch” DNA or shed skin cells largely composed of anucleated corneocytes. However, even in such challenging samples, probative information can still be obtained. In 2011 Schneider et al. demonstrated the utility of such an approach examining tape lifts from true crime investigations and subsequently analyzed hundreds of individual shed skin cells/cell agglomerates.20 In each of the two cases described, a full suspect DNA profile was obtained. Additional studies examining mock casework scenarios have demonstrated the ability to retrieve probative genotype information from skin/skin and skin/fabric contact incidences21,22,23 as well as tape lifts collected from firearms.24
The problem of DNA mixtures
One of the biggest problems in forensic DNA evidence analysis is the presence of mixed DNA from two or more individuals, with each additional donor adding to the complexity and difficulty in resolving individual donor genotypes.25 DNA mixtures often comprise a significant number of cases in a forensic laboratory due to an increased level of sensitivity in the latest commercial STR systems compared to their predecessors. Due to overlapping alleles present from two or more individuals, the presence of DNA donors in different proportions, and PCR artifacts accompanying low DNA template donors, insufficient (or no) probative STR genotype information from some or all of the POIs may be obtained, thus weakening the value of the evidence. Although the advent of continuous model probabilistic genotyping (PG) approaches has facilitated recovery of donor genotypes from mixtures, this can still fail to recover probative genotypes from all donors in complex mixtures.26,27 It is important to understand that any loss of probative STR genotype information (as exhibited by low LR values) due to mixture complexity not only could prevent the detection of a perpetrator’s DNA signature when actually present, but could fail to definitively exclude a falsely implicated suspect whose DNA is not actually present in the mixture.
PG software helps to account for various confounding DNA mixture attributes such as allele drop-out and drop-in.28 However, as previously mentioned, probative information cannot always be obtained for every donor, whether that is due to a large number of contributors (NOC), the extreme minor presence of a donor, or a mixture being composed of related individuals. Previous reports,29,30,31 have demonstrated how complex mixtures can routinely appear to be composed of fewer donors. This can result in an underestimation of the NOC. It is well reported, that underestimating the NOC of a mixture often results in the false exclusion of known minor donors.32,33,34,35,36,37 However, even when the NOC is accurately determined, probative results are still not always obtained.30,44,101 To illustrate the loss of probative genetic information expected from one or more donors to mixtures, a PG software system (STRmix v2.8) validated for standard DNA mixture analysis in the authors’ laboratory was used to determine the limits of the system in extracting probative information from standard mixtures (Figure 1). For a 5-person mixture, donor 1 is typically the most major contributor and donor 5 is the most minor contributor. Thus, with a 5-person mixture, the data from this study indicate the two most minor donors routinely fall below an LR of 106 (i.e., the threshold for extremely strong support38,39) with even the 3rd donor’s LR hovering around that threshold. Even with a more simple 3-person mixture the expectation is that, with some mixtures, the 3rd donor LR will be less than the 106 threshold.
Figure 1.

Average log (LR) of contributors to 2–5 person mixtures obtained by standard PG mixture analysis
Average log (LR) values (+/− 1 SD) for standard mixtures (n=50 selected from in-house prepared mixtures and the PROVEDIt database40,41); blue circle (2-person), orange circle (3-person), gray circle (4-person), yellow circle (5-person); dashed line (internationally recognized consensus threshold that needs to be met to provide “extremely strong” or “very strong” support for the inclusionary hypothesis pertaining to the POI).38,39 X axis: donor number.
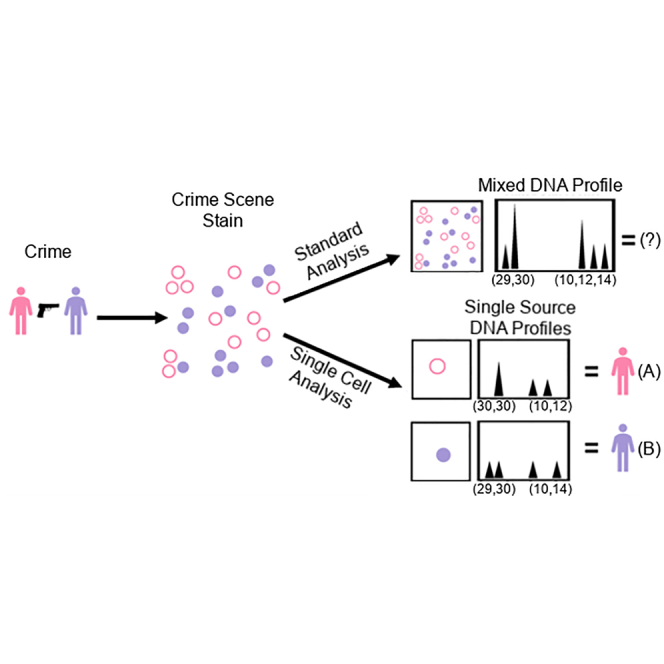
Single cell analysis and mixture deconvolution
In theory, the inevitable loss of potentially critical genotype information from donors in some complex DNA mixtures using standard methods could be minimized by the use of single cell genetic analysis, the subject of this review. Standard methods of mixture analysis involve subjecting a dried crime scene mixed DNA sample to a “bulk” DNA extraction method such that the resulting DNA isolate compromises a homogenized DNA mixture from the individual donors. The DNA for genetic analysis will thus comprise a single bulk isolate that has a defined multi-donor composition characterized by the relative weight ratio of the amount of DNA contributed by each donor to the original mixture. If, however, instead of bulk DNA extraction, a sufficient number of individual cells from the mixed stain are subsampled prior to genetic analysis then it should be possible to recover highly probative single source, non-mixed scDNA profiles from each of the donors.42 This potential ability to fully deconvolute complex mixtures into their constituent donors is the main impetus for the current interest in the potential of single cell forensic genomics. scDNA analysis can also permit the detection of a POI in a mixture that otherwise could not be detected at all by standard bulk mixture analysis. This is the case where there is at least one very minor donor that constitutes <2% of the mixture, which does arise in criminal investigations.43,44 The inability to detect the minor donor in this situation is due to the PCR amplification process in that the preponderant major donor(s) kinetically out compete(s) the minor donor(s) for critical PCR reactants (e.g., DNA polymerase, dNTPs, etc.) to such an extent that insufficient, if any, minor donor amplicon products are produced.44
Cell enrichment and genetic analysis workflows
In single cell analysis workflows, a distinction should be made between cell sub-population enrichment and the actual genetic analysis at the single cell level (i.e., scDNA) (Figure 2). For example, a cell enrichment method is used routinely in forensic genetics today to reduce DNA mixture complexity in sexual assault cases by separating sperm cells (from one or more donors) from a female victim’s vaginal epithelial cells via a differential extraction (DE) process45 before a subsequent bulk genetic analysis of the separated cell populations (Figure 2A) (reviewed by Clark et al.46). Genetic analysis at the single cell level requires a single cell capture method (e.g., micromanipulation) for subsampling the mixture for genetic analysis (Figure 2C). However, for some case type scenarios, the mixed cell populations targeted for single cell capture and scDNA analysis can be first reduced in complexity from the bulk cell population via an enrichment process (e.g., DE) (Figure 2B). In this review we only briefly describe the enrichment methodology47 for forensic genomics use as it pertains to be able to subsequently perform genetic analysis of single cells. Cell capture and subsequent scDNA analysis by STR-CE forms the main focus due to it being the principal genetic analysis method carried out to date, although other reported methodological applications such as mtDNA and MPS-based SNPs are discussed.
Figure 2.

Single cell enrichment and genetic analysis workflows in forensic genomics
(A) The standard STR-CE bulk genetic analysis of a complex four-person mixture (diploid cells from each of the individual donors are indicated by different circle colors) is shown in which the bulk sample undergoes a co-extraction of DNA from all four contributors. The genetic analysis results obtained from two (of the twenty or more) STR loci tested are shown in the electropherogram (epg). The peaks on the x axis represent the alleles (designated by the number of tandem repeats detected), and their signal heights on the y axis (measured in relative fluorescence units, RFU) indicate their relative proportions in the mixture. Two alleles (of maternal and paternal origin) are contributed by each donor at each STR locus, who will either be heterozygous (two different alleles) or homozygous (two indistinguishable identical-by-state alleles). In this four-person example there are a total of eight alleles per locus contributed by the contributors. However due to overlapping alleles from some of the donors and homozygosity in one of the donors at each locus in this particular mixture, four (locus 1) or five (locus 2) CE-distinguishable alleles are obtained, and fully deconvoluting the mixture into four individual donor genotypes is challenging.
(B) The use of a bulk enrichment process (using MACS, FACS, or DE) prior to the genetic analysis reduces the complexity of the original bulk mixture (into two subpopulations in this instance), but mixture deconvolution, albeit simpler, is still required.
(C) Direct single cell subsampling from the bulk mixture (using the DEPArray digital cell sorting system, laser capture microscopy [LCM] or micromanipulation [MM]) and subsequent genetic analysis of single cells can result in complete deconvolution of the mixture into single source genotype information from each of the donors. It is possible to combine bulk enrichment and single cell genetic analysis (thin arrow) (e.g., sperm and vaginal epithelial cells) but not all cell types and mixtures can be processed in this manner at this time.
Cell population enrichment studies
Differential extraction (DE)
As previously mentioned, the differential extraction is a staple method in forensic laboratories to analyze samples from sexual assault scenarios. DE was first reported in 1985 by Gill et al.45 During the DE process, sperm and epithelial cells are preferentially lysed and separated due to their biochemical differences, which is believed to include the presence in sperm cell membranes of disulfide-rich crosslinked proteins that, unlike vaginal epithelial cells, require a strong reducing agent for lysis. A variation of DE referred to as acoustic differential extraction (ADE) has been reported using a microfluidic device to separate sperm and epithelial cells significantly reducing the time of analysis.48,49
Fluorescence-activated cell sorting (FACS)
Various methods have been employed utilizing a similar cell separation concept to DE including sampling methods to separate sub populations of cells based on tissue specific, antibody specific, or chromosome specific features. One such method is fluorescence-activated cell sorting (FACS). FACS enables separation of fluorescent antibody-labeled cells often relying on cell differences based on size, surface phenotype, cytoplasm, and DNA content.47 FACS enables bulk cell enrichment typically separating two distinct cell population types. Reports have demonstrated FACS use in separating epithelial cells,50 sperm,51 blood,52 and hormone specific cell fractions.53 Though FACS is a useful bulk cell enrichment process, imperfect separation can occur due to fluorescent crosstalk between populations. Furthermore, if a mixed sample is composed of donors from the same cell population (e.g., tissue, antibody, and chromosome) then separation into single source cell populations is not possible.
Magnetic activated cell sorting (MACS)
Magnetic activated cell sorting (MACS) can be utilized to separate cell populations based on cell specific antigens utilizing immune-magnetic beads usually coupled with antibodies.47 MACS has been utilized to separate epithelial and sperm cell populations54,55 as well as ABO blood groups.56 In 2016, MACS was used in combination with FACS to separate individual sperm based on ABO blood groups.57
Cell capture
Laser capture
In 2003, Elliot et al. used laser capture micro dissection (LCM) to capture spermatozoa from post coital microscope slides outperforming DE.58 LCM (sometimes referred to as LCD) utilizes a light microscope and UV laser to cut out the specific cell or cells of interest. Subsequent studies have demonstrated further success with LCM59,60,61,62 even utilizing LCM in conjunction with chromosome specific probes.63,64 PG approaches have also been applied to laser captured cells resulting in cell population subsamples (10x 20-cell subsamples) with various weight ratios resulting in the recovery of more genotype information as opposed to standard DNA extractions.65 In 2020, England et al. demonstrated the ability to use the ForenSeq DNA Signature Prep Kit with laser micro-dissected cells to obtain aSTR and Y-STR profiles. However, at least 25 cells were required.66
Digital cell sorting (DEPArrayTM)
Much of the scDNA work being conducted today uses the DEPArrayTM digital cell sorter that permits the recovery of single cells.67 DEPArrayTM is a semiautomated digital cell sorting based approach which relies on fluorescent labeling and dielectrophoretic (DEP) cages with programmed changes in AC current to collect individual or small cell pools. Using the DEPArrayTM system, Anslinger et al. have demonstrated the ability to recover and genetically analyze white blood cells from cold cases up to 27 years old,68 deconvolute blood/blood mixtures,69 and chimeric mixtures due to blood transplantation.70 Fontana et al., separated epithelial, blood, and sperm by creating small cell pools ranging from 5 to 50 cells.67 Meloni et al. have also separated white blood cells (1–10 cells)71 while Williamson et al. have utilized the DEPArrayTM system to analyze post-coital sperm containing samples.72
Micromanipulation
The most simplistic, and thus more affordable, cell separation technique is micromanipulation, though some degree of manual dexterity skill is required depending on the method used. Various micromanipulation-based approaches have been utilized including manual20 approaches with micro/picopipets,18,19,73 tungsten needles,22,23,74,75 micromanipulators,75,76 and optical tweezers.77
Forensic genetic analysis of single cells
The DNA profiling of single cells offers a number of challenges due to the limited amount of gDNA available for analysis, which is estimated to be ∼6.6 pg for diploid cells (e.g., white blood cells, vaginal and buccal epithelial cells) and ∼3.3 pg for haploid cells (spermatozoa).78 In the forensic context scDNA analysis is therefore classified as low template DNA (LTDNA) which is normally regarded as involving the analysis of <100 pg of gDNA.79,80 Thus, scDNA analysis requires the development of enhanced DNA typing procedures that can overcome, or adequately deal with statistically, the stochastic PCR artifacts characteristic of LTDNA. These include for aSTR genotyping, allele or locus drop out, allele drop in, unbalanced heterozygote allelic signals and increased stutter.79,81 Stutter is the presence of an additional weaker (∼15%) artifactual allelic signal that originates from the accompanying bona fide progenitor allele but is predominantly one repeat shorter. This is caused by slipped strand mispairing during the PCR process and needs to be distinguished from a true allele from a second donor in a mixture. Different labs use different enhanced DNA methods for scDNA analysis. What follows are some of the technical considerations usually needed to be addressed to develop a scDNA analysis workflow, illustrated by some of the authors’ own experience in this area.
STR amplification kit
Forensic STR analysis requires a robust system compatible with direct PCR which includes sufficient markers to allow for highly probative genotyping results even with partial profiles. Much of the illustrative work described in this review was conducted in the authors’ own laboratory using the GlobalFilerTM Express (GFE) system which contains 21 aSTR markers and was designed specifically for direct PCR. Nevertheless, we have additionally had success utilizing the VeriFilerTM Plus and PowerPlex® Fusion 6C systems (PPF6C) (each containing 23 aSTR markers). The PPF6C system has associated direct PCR protocols easily modified for single cell use.82,83 Various reports on single cell analysis have been conducted using PPF6C,72,84,85 as well as fewer STR marker kits PowerPlex® ESXfast (16 aSTR loci),68,69,70 PowerPlex® ESX 1771,74 and IndentifilerTM Plus (15 aSTR loci).21,86,87 Y-STRs have additionally been analyzed via direct PCR using YFilerTM (17 Y-STR markers),88 YFilerTM Plus (25 Y-STR markers),24 and PowerPlex® Y23 (23 Y-STR loci).74
PCR cycles
scDNA analysis often employs low copy number (LCN) techniques. Perhaps the most common of these techniques is increasing PCR cycle number to improve the limit of detection.79 However, increased PCR cycle numbers are known to increase the risk of contamination from extraneous environmental DNA and, as a result, some laboratories are wary of implementing them.89 Due to single cell analysis being optimized for very low DNA input concentrations, any minute contamination can result in significant amplification of the contaminating DNA. However, comparison of the individual subsample profiles to a staff database could easily detect contaminated subsamples. Single cell genotyping results have been obtained with as few as 28 cycles (i.e., using standard PCR cycle numbers) though most single cell studies use 30–34 cycles. As expected, higher cycle numbers often correlate with decreased allelic drop-out rates.84
Reaction volume
A second factor often modified for scDNA analysis is PCR reaction volume. A reduced reaction volume can not only improve sensitivity but can further reduce the cost associated with scDNA by reducing the reagent amount utilized per cell sample.90 Typically, scDNA reaction volumes between 12.5 μL and 5 μL are utilized which often equates to a half or more volume reduction compared to most standard forensic STR DNA workflows that utilize 25 μL reaction volumes.91,92,93
Direct lysis/PCR additives
Due to the low template nature of single cells, direct PCR is typically conducted in which captured cells are recovered into a lysis solution (e.g., DEPArrayTM Lyse Prep Kit [Menarini, Silicon Biosystems], forensicGEM® lysis solution [ZyGEM forensicGEM® Saliva kit, VWR, Suwanne, GA, USA], Prep-n-GoTM Lysis buffer [ThermoFisher Scientific, Carlsbad, CA, USA], PunchSolutionTM [Promega, Madison, WI, USA], or Casework Direct [Promega, Madison, WI, USA]) and then, in the same tube, PCR reactants are added and STR amplification proceeds. Direct PCR is not only quick but also prevents loss of gDNA through the multi-step extraction and quantitation procedures.94 The quantitation of DNA prior to genetic analysis, as required by National DNA Standards in the US (the FBI’s Quality Assurance Standards, QAS),95 is determined by the number of nucleated cells used in the amplification reaction.
Laboratories should determine the optimal lysis method for scDNA analysis, since the STR genotyping efficacy can vary with different commercial products and sample types. As an illustration of the efficacy of different lysis methods, Figure 3 provides an example of a comparison in the authors' laboratory of the extent of the allele recovery from single buccal epithelial cells using the GFE (GlobalFilerTM Express, ThermoFisher Scientific, Carlsbad, CA) STR set, when Prep-n-GoTM Lysis buffer, PunchSolutionTM, and Casework Direct lysis solution is utilized. Lysis with PunchSolutionTM and Casework Direct provided comparable results, with Prep-n-GoTM not performing as well for this application. PunchSolutionTM, however, requires amplification tubes to be opened during lysis to allow for evaporation and potentially increases the risk of DNA contamination. Thus, Casework Direct was found to be the most optimal of the lysis methods tested for our laboratory’s scDNA analysis workflow.
Figure 3.

Comparison of lysis solutions for single cell allele recovery with the GlobalFiler Express (GFE) STR system
Prep-n-Go Buffer (GFE PnG) (green), PunchSolution™, (GFE Punch) (blue), and Casework Direct (burgundy) with single cells. n = number of single cells tested.
The use of PCR additives may further aid in single cell analysis. Commercial amplification kits such as GFE require the addition of a master mix additive to aid in inhibition with direct PCR. Although, GFE contains a proprietary additive added prior to amplification, co-incorporation of the AmpSolution (Promega, Madison, WI, USA) additive was found to further improve allele recovery (data not shown), although it should be noted, that PunchSolution and Casework Direct still provided superior recovery. The basis of allele recovery improvement due to the presence of additives is not always known although there may be some indication as to their purpose. For example, the AmpSolution improvement seen in the aforementioned instance is presumably due to it containing a surfactant which prevents small DNA molecules from adhering to the PCR tube.
Buffers
Depending upon the cell separation method, various buffer solutions may be specified in the protocol. For example, the digital cell sorting method DEPArray requires cells to be collected via PBS buffer to prevent cytolysis.96 However, reports have indicated that using TE−4 to suspend cells and create cell suspensions of known weight ratios for validation purposes actually improved allele recovery as opposed to PBS.97 Sheth et al., further demonstrated reducing PBS concentration during DEPArray’s volume reduction and extraction processes improved single cell EPG results.85
Clustering cell genotypes/consensus profiling
As stated previously single cell analysis is an extreme example of low template DNA (LTDNA), with each diploid cell containing ∼6.6 pg gDNA. LTDNA, with all of the aforementioned PCR artifacts, therefore, should benefit from multiple amplifications of the same DNA extract. Caragine et al.98 and Benschop et al.99 have each demonstrated the ability of replicate amplifications to aid in the reconstitution of an LTDNA profile using a consensus-based approach. Similarly, many forensic scDNA publications have demonstrated the benefits of replicate/consensus reporting for single cells.24,68,69,72,74,87,100,101,102,103,104 As demonstrated in Figure 4, a consensus approach typically allows for an allele to be “called” if it is present in at least 2 replicates thus helping to account for the stochastic effects of drop-out and drop-in. Standard DNA analysis often employs a stochastic threshold or a threshold at which a sister allele of a heterozygous pair, has the potential to drop-out as well as >60–70% peak height balance for a heterozygous pair. Such a threshold is not realistic with single cell analysis especially since it is not uncommon for one allele of a heterozygous pair to be present at a high RFUs while its sister allele has dropped out entirely.97 Drop-in, on the other hand, is often present at higher RFUs with single cell analysis and can be similar in peak height to true alleles in the sample.71 By combining multiple individually typed cells (or cell subsets) much, if not all, of individual donors’ genotypes can be fully reconstituted. In order to apply a consensus/replicate based approach to mixtures, a method must first be employed to identify related partial genotypes that have a common origin whether by a standard visual inspection of the resulting genotypes,24,69,74,87 by using clustering algorithms (e.g., EM, K-means, or MBC/EESCIt)105,106 or by a PG related approach.97,101,102,107,108,109
Figure 4.

Replicate analysis and consensus DNA profiling
In this illustration, four LTDNA replicates were genotyped and the results at 4 STR loci are shown. The consensus genotype is shown at the bottom, which is based upon the appearance of the same allele in at least two replicates.
PG considers various facets of DNA profile complexity such as allele height, height balance, drop-in, drop-out, degradation, and stutter artifacts to provide a probability for the profile data given a specific POI genotype.110,111,112 Although designed for standard mixture analysis, PG has been applied to single cell analysis.97 The use of PG to analyze individual cells allows for an initial screening of the subsamples (i.e., either excluding or including each suspected POI according to LR). Subsamples with inclusionary LRs (i.e., log (LR)s ≥ 1) for a specific donor can then be combined into a single analysis using the PGS’ replicate analysis function (somewhat similar, but not identical, in concept to that seen in Figure 4). For instances in which a POI reference is not available, clustering of samples could be conducted using the mixture to mixture and common donor applications of software such as DBLR™.107,108,109 Similarly, model-based clustering (MBC) has been developed by Duffy et al. in which subsample LRs are averaged across clusters generally resulting in log(LR)s > 5.106 Ge et al. has further demonstrated clusters of ≥5 diploid cells have ≥97.6% accuracy to the donor aSTR profile.105
A consensus-based aSTR approach becomes more difficult when dealing with haploid sperm cells as only half the original donor’s aSTR DNA profile can be obtained within an individual cell therefore typically requiring a larger number of sperm compared to diploid cells to obtain a full aSTR profile. Though, only half of the individually recovered sperm will contain a Y chromosome, full Y-STR profiles have been obtained with multiple single sperm subsamplings in multi-sperm mixtures.103,104 This is particularly beneficial for samples that would otherwise result in Y-STR mixtures as there is currently no commercially available PG system to deconvolute Y-STRs, although, at least one (“STRmixY”) is in development.113,114 The reconstitution of full aSTR profiles from haploid cells may become much simpler if single cell analysis were performed using a whole genome amplification (WGA) or NGS approach like the ForenSeq Signature Prep kit which provides both aSTR, X-STR, and Y-STR results for a single sample. Theunissen et al. used WGA and then clustered sperm based on donor X- and Y-STR profiles which were then utilized to create consensus aSTR profiles. They demonstrated that a full aSTR profile could be reconstituted using 11 sperm while 7 sperm resulted in 96% of the donor aSTR profile.74,115,116 There remains a degree of skepticism within the forensic community about the efficacy of WGA for the types of low level, and possibly degraded or otherwise damaged, samples encountered in forensic investigations. However, the renewed interest in the method generated by these recent publications may result in further useful forensic applications.117 In a recent non-empirical, theoretical study, Ge et al. using simulated mixtures demonstrate clusters of ≥7 haploid cells have ≥97.6% accuracy to the donor aSTR profile.105
In 2011, Pereira et al., demonstrated the ability to type mtDNA from individual sperm in post coital samples.118 Similar to single cell Y-STR analysis, such an approach may be beneficial in multi-individual sperm containing mixtures. Single sperm MPS whole mtDNA analysis could especially be useful for cases in which male perpetrators belong to the same paternal line but different maternal lines. In such instances perpetrators cannot be differentiated via Y-STR analysis since they would possess the same Y-STR profile.
Probabilistic genotyping of single cells
Validation of the PG software systems STRmix and EuroForMix for single cell STR analysis (scPG) has been reported by Huffman et al.97,101 As previously mentioned, subsamples originating from the same donor can be combined into a single analysis using a PG replicate analysis function or similar replicate extension.119 This often results in donor LRs comparable to the reciprocal of the random match probability (RMP),97,101,102 which indicates full recovery of the tested STR genotype information from the sample. While previous reports97,101 have demonstrated improved donor LRs for complex equimolar 2- to 6-person mixtures by single cell subsampling (compared to standard analysis), further significant improvement is now obtainable due to adjustments made to the lysis solution utilized (as previously mentioned). This greatly reduces the number of single cell subsamples needed to obtain highly probative DNA profiles. To illustrate the gain in donor LR information typically obtained when using an optimized scPG method, Figure 5 provides the results obtained in our laboratory from an equimolar 6-person mixture when analyzed using STRmix by standard methods (gray bars) and single cell replicates (blue bars). The maximum information achievable is demonstrated by each individual donor’s RMP (black bars) as determined from a single source reference profile. For the specific 6-person mixture provided, only 30x 1-cell subsamples were collected (a decrease from 30x 1-cell and 2-cell subsamples previously needed). Samples were lysed with PunchSolution, and amplified using the GlobalFiler Express amplification kit. Donor log(LR)s ranged from 1 to 8 with standard analysis and 14 to 27 with single cell replicates.
Figure 5.

Significantly increased contributor log(LR)s recovery from single cells collected from a 6-person mixtures by direct single cell subsampling, compared to standard bulk mixture analysis.
The scPG approach can further be applied to small cell number subsamples which enables a larger survey of an overall crime scene stain as opposed to single cells alone. This was demonstrated by the direct single cell subsampling (DSCS) approach in which 5-cell subsamples were collected from a bulk mixture with an original weight ratio of 1:50 (i.e., containing an extreme minor donor undetectable by standard methods). These 5-cell subsamples either resulted in single source DNA profiles from the major contributor or new “mini-mixtures” with reduced weight ratios (compared to the original bulk mixture) that could subsequently be analyzed by PG.44 This approach provided probative information about an extreme minor donor with an LR ∼1011 which surpasses the evidentiary threshold for “very strong support.”38,39 The further application of common donor analysis (via DBLR) increased discrimination power to ∼1023 by taking into account the shared major component of the mini-mixture profiles (as determined from both the original bulk analysis and the single-source profiles obtained).109
Another application of scPG is with familial mixtures. Some complex familial mixtures comprising several first-degree relative donors can yield false inclusions and exclusions using standard bulk STR mixture analysis.34,120 However, scDNA analysis conducted in some of these complex familial mixtures can prevent the false inclusion of non-donor relatives.102
MPS applications to forensic scDNA analysis
A few reports have started appearing in the literature on using MPS-based approaches for single cell forensic analysis. Although each of them describes a number of technical issues, expected when a transformative methodology is beginning to be applied to scDNA forensic analysis, that will need to be addressed moving forward.
Dipenbroek et al. demonstrated the ability to use single cell semiconductor ion sequencing on white blood cells recovered from autopsy blood using digital cell sorting for ancestry (using bootstrapping admixture analysis across seven root populations) and DNA phenotyping (hair, skin and eye color) with both cell pools and single cells.121 Single cell analysis with sufficient genotyping information for accurate ancestry and phenotyping inference was only achieved after library amplification prior to sequencing and the use of consensus profiling akin to that described earlier for STRs. In 2023, Kulhankova et al. in a visionary study demonstrated the ability of single cell transcriptome sequencing (scRNA-seq) to fully deconvolute complex 2–9 person blood-blood mixtures using forensically useful SNPs in RNA transcripts instead of in gDNA, thereby making use of scRNA-seq technology. They were able to recover and genotype identity SNPs (for identification of the mixture donors) as well as recover investigative intelligence information about the donors including sexing (for males, Y chromosome: number of reads aligning to Y chromosome; for females, the inactive X chromosome: presence of the XIST non-coding RNA) and the maternal/paternal/biparental biogeographical ancestry of the donors (using mtDNA and Y chromosomal haplotypes and genome-wide SNPs).122 However, as the authors admit the current iteration of the method will have limited forensic utility, since the scRNA-seq platform used (10X genomics) requires live cells for their genetic separation, and RNA may not be universally suitable for all cell types and environmental conditions due to restricted RNA expression of many of the required SNP biomarkers and increased susceptibility to environmental hydrolysis compared to gDNA. Live cells are not recovered from crime scene evidence which comprises deposited dead ex vivo cells in the dehydrated state. Alternative single cell platforms not reliant on live cells or RNA, and increased coverage and deeper sequencing of SNPs are suggestions for further development of this more extensive genomics approach than heretofore.
Future directions
Not only can single cell genetic analysis be used to deconvolute complex mixtures including those mixtures involving first degree related individuals, but it may also help to inform an analyst on how to analyze the original bulk mixture.102 This can occur by either providing an additional metric to help determine the number of contributors to the mixture or by providing a full single-source donor DNA profile from one or more donors.102 The latter could thus permit the recovery of more probative genotype information from the mixture using standard PG bulk mixture analysis (due to being able to condition the mixture on the presence of now known individual(s) genotype(s) obtained from scPG).
Although in recent years substantial progress has been made in forensic single cell genetic analysis, further developments in several areas are necessary, particularly in the field of automation, before this methodology can be incorporated into the forensic scientist’s routine casework armamentarium. As can be appreciated from the foregoing sections, single cell analysis remains labor intensive, with even the current semi-automated approaches such as digital cell sorting being quite a lengthy process as well as expensive. Thus, the development of a microfluidics droplet based forensic DNA marker genotyping system that uses single molecule nanoliter reactions (thus dramatically reducing the cost of genotyping per cell) would significantly help drive the field toward casework implementation.123,124,125,126,127,128 At least one system that can do this (scDNA-seq), for clinical samples, has now become commercially available.129,130 Successful DNA profiling of touch deposits via single cells or agglomerates of groups of cells originating from a single individual in expected binary mixtures (e.g., victim and POI) has been reported.13,21,24 However, there is a paucity of studies in which scDNA analysis (akin to the blood, buccal or sperm cells described previously) was used to fully deconvolute touch deposits into their constituent genotypes. Touch deposits in the form of complex mixtures are encountered frequently in forensic casework, with the different donors contributing varying amounts of DNA, including some, or all, with low template DNA and in different states of degradation, depending upon when the deposit took place and the environmental conditions impacting it over time.131 The DNA in touch deposits occur either packaged in cells or as cell free DNA (cfDNA) with not all of the cells present containing DNA that would be amenable to scDNA analysis. Moreover, there appears to be some difficulty in recovering single epithelial cells from touch deposits via the commonly used digital cell sorting DEPArray platform, perhaps due to their size and/or shape. There may be some concern that using scDNA analysis as a mixture deconvolution tool would fail to find a mixture donor whose DNA was only present on the cfDNA fraction. It is unclear whether touch deposits contain a cfDNA fraction that is not reflected in the cellular components, although at least one report indicates there appears to be an equivalency in DNA profiling results between cfDNA and the cellular components.132 In order to further investigate the potential of scDNA analysis to resolve touch DNA mixtures, the DNA content of each of the cellular and cfDNA components present on a variety of touch deposits (of different mixture ratios, number of contributors, time since deposition and environmental influences) needs to be measured directly as well as determining the deconvolution success rates in bona fide touch deposits and whether cfDNA from a donor can be found in the absence of cells from the same donor.
The question sometimes arises as to whether the DNA typing results obtained from single cells would have limited utility to be entered into uploadable, searchable crime scene and criminal databases such as CODIS. However, when multiple cells are combined using a replicate analysis function of STRmix or the common donor analysis of DBLR, high quality profiles with component interpretation weights of >99% are often achieved.109 This use would have to be accompanied by extensive community validation of the low template methods employed which admittedly would seem to place a burden on the part of the caseworking laboratory. In reality, however, many laboratories are de facto using low template methods currently due to the use of high sensitivity kits and the fact that PG deals routinely with the stochastic effects common to low template donors to mixtures. The path to CODIS or other regulated criminal database acceptability may be long and narrow for single cell approaches employing high sensitivity DNA typing methods, but it is not impossible, since there are published guidelines on how to validate them.133
As mentioned briefly earlier, there is an ongoing effort in forensic genomics, due to the advent of MPS technology, to extend the capabilities and utility of genetic analysis beyond that of the genetic markers currently employed for human identification (principally CE separated STRs and, to a much lesser extent, mtDNA control region SNPs) to include SNPs for human identification, biogeographical ancestry, DNA phenotyping (i.e., inferring external visible traits such as eye, skin and hair color, etc), whole mtDNA sequencing and epigenetic analysis of differentially methylated sites (DMS) for biological age prediction.121,122,134 The principal advantage of these MPS approaches is that more actionable donor information can be recovered from the biological evidence, including from degraded samples that are intractable to standard analysis. The transition toward SNP based genotyping systems in forensic genomics would face a significant challenge when applied to criminal casework, namely due to the frequent occurrence of complex mixtures and the limited number of alleles at each locus, (i.e., two). While simple two person mixtures may be amenable to complete donor SNP genotype deconvolution via standard bulk mixture DNA analysis135,136,137 it’s going to be extremely challenging, if not impossible, to fully deconvolute more complex mixtures, although some progress in partial deconvolution of complex SNP mixtures has been made.134,135,138 Thus, a direct single cell subsampling method (with or without an enrichment process) prior to SNP analysis may be required to accomplish this. A few reports have started appearing in the literature on using MPS-based approaches for single cell forensic analysis. Each of them describes a number of technical issues expected when a transformative methodology is beginning to be applied to scDNA forensic analysis that will need to be addressed moving forward. This could herald a new era of scDNA analysis and applications for SNP genotyping in DNA mixtures, including in the developing field of forensic investigative genetic genealogy (FIGG).135
Acknowledgments
The authors would like to thank the State of Florida for seed funding for portions of the work described that was carried out in the authors' laboratory at the University of Central Florida, Orlando, Florida, USA.
Author contributions
Investigation, K.H. and J.B.; Resources, J.B.; Writing – Original Draft Preparation, K.H. and J.B.; Writing – Review and Editing, K.H. and J.B.
Declaration of interests
No competing interests to disclose.
References
- 1.Macaulay I.C., Voet T. Single Cell Genomics: Advances and Future Perspectives. PLoS Genet. 2014;10:e1004126. doi: 10.1371/journal.pgen.1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heitzer E., Auer M., Gasch C., Pichler M., Ulz P., Hoffmann E.M., Lax S., Waldispuehl-Geigl J., Mauermann O., Lackner C., et al. Complex Tumor Genomes Inferred from Single Circulating Tumor Cells by Array-CGH and Next-Generation Sequencing. Cancer Res. 2013;73:2965–2975. doi: 10.1158/0008-5472.CAN-12-4140. [DOI] [PubMed] [Google Scholar]
- 3.Philippron A., Depypere L., Oeyen S., De Laere B., Vandeputte C., Nafteux P., De Preter K., Pattyn P. Evaluation of a marker independent isolation method for circulating tumor cells in esophageal adenocarcinoma. PLoS One. 2021;16:e0251052. doi: 10.1371/journal.pone.0251052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan J.Y.Y., Tan Y.P., Ng S., Tay A.S., Phua Y.H., Tan W.J., Ong T.Y.R., Chua L.M., Syn C.K.C. A preliminary evaluation study of new generation multiplex STR kits comprising of the CODIS core loci and the European Standard Set loci. J. Forensic Leg. Med. 2017;52:16–23. doi: 10.1016/j.jflm.2017.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Butler J.M. 1st ed. Academic Press; 2009. Fundamentals of Forensic DNA Typing. [Google Scholar]
- 6.Evett I.W., Weir B.S. Sinauer Associates; 1998. Interpreting DNA Evidence: Statistical Genetics for Forensic Scientists. [Google Scholar]
- 7.Roewer L., Andersen M.M., Ballantyne J., Butler J.M., Caliebe A., Corach D., D’Amato M.E., Gusmão L., Hou Y., de Knijff P., et al. DNA commission of the International Society of Forensic Genetics (ISFG): Recommendations on the interpretation of Y-STR results in forensic analysis. Forensic Sci. Int. Genet. 2020;48:102308. doi: 10.1016/j.fsigen.2020.102308. [DOI] [PubMed] [Google Scholar]
- 8.Roewer L. Y-chromosome short tandem repeats in forensics—Sexing, profiling, and matching male DNA. WIREs Forensic Sci. 2019;1 doi: 10.1002/wfs2.1336. [DOI] [Google Scholar]
- 9.Melton T., Holland C., Holland M. 2012. Forensic Mitochondrial DNA Analysis: Current Practice and Future Potential. [PubMed] [Google Scholar]
- 10.Tagliabracci A., Turchi C. The Human Mitochondrial Genome. Elsevier; 2020. mtDNA exploitation in forensics; pp. 145–169. [DOI] [Google Scholar]
- 11.Ballard D., Winkler-Galicki J., Wesoły J. Massive parallel sequencing in forensics: advantages, issues, technicalities, and prospects. Int. J. Legal Med. 2020;134:1291–1303. doi: 10.1007/s00414-020-02294-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollard C., Ausset L., Chantrel Y., Jullien S., Clot M., Faivre M., Suzanne É., Pène L., Laurent F.X. Automation and developmental validation of the ForenSeq ™ DNA Signature Preparation kit for high-throughput analysis in forensic laboratories. Forensic Sci. Int. Genet. 2019;40:37–45. doi: 10.1016/j.fsigen.2019.01.010. [DOI] [PubMed] [Google Scholar]
- 13.Montano E.A., Bush J.M., Garver A.M., Larijani M.M., Wiechman S.M., Baker C.H., Wilson M.R., Guerrieri R.A., Benzinger E.A., Gehres D.N., Dickens M.L. Optimization of the Promega PowerSeq™ Auto/Y system for efficient integration within a forensic DNA laboratory. Forensic Sci. Int. Genet. 2018;32:26–32. doi: 10.1016/j.fsigen.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Syndercombe Court D. Mitochondrial dna in forensic use. Emerg. Top. Life Sci. 2021;5:415–426. doi: 10.1042/ETLS20210204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge J., Budowle B. Forensic investigation approaches of searching relatives in DNA databases. J. Forensic Sci. 2021;66:430–443. doi: 10.1111/1556-4029.14615. [DOI] [PubMed] [Google Scholar]
- 16.Tillmar A., Sturk-Andreaggi K., Daniels-Higginbotham J., Thomas J.T., Marshall C. The FORCE Panel: An All-in-One SNP Marker Set for Confirming Investigative Genetic Genealogy Leads and for General Forensic Applications. Genes. 2021;12:1968. doi: 10.3390/genes12121968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Findlay I., Taylor A., Quirke P., Frazier R., Urquhart A. DNA fingerprinting from single cells. Nature. 1997;389:555–556. doi: 10.1038/39225. [DOI] [PubMed] [Google Scholar]
- 18.Li H.H., Gyllensten U.B., Cui X.F., Saiki R.K., Erlich H.A., Arnheim N. Amplification and analysis of DNA sequences in single human sperm and diploid cells. Nature. 1988;335:414–417. doi: 10.1038/335414a0. [DOI] [PubMed] [Google Scholar]
- 19.Jeffreys A.J., Wilson V., Neumann R., Keyte J. Amplification of human minisatellites by the polymerase chain reaction: towards DNA fingerprinting of single cells. Nucleic Acids Res. 1988;16:10953–10971. doi: 10.1093/nar/16.23.10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider H., Sommerer T., Rand S., Wiegand P. Hot flakes in cold cases. Int. J. Legal Med. 2011;125:543–548. doi: 10.1007/s00414-011-0548-7. [DOI] [PubMed] [Google Scholar]
- 21.Farash K., Hanson E.K., Ballantyne J. Single source DNA profile recovery from single cells isolated from skin and fabric from touch DNA mixtures in mock physical assaults. Sci. Justice. 2018;58:191–199. doi: 10.1016/j.scijus.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 22.Farash K., Hanson E.K., Ballantyne J. Enhanced Genetic Analysis of Single Human Bioparticles Recovered by Simplified Micromanipulation from Forensic & #8216;Touch DNA& #8217; Evidence. J. Vis. Exp. 2015:52612. doi: 10.3791/52612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanson E.K., Ballantyne J. Nucleic Acid Detection. 2013. “Getting Blood from a Stone”: Ultrasensitive Forensic DNA Profiling of Microscopic Bio-Particles Recovered from “Touch DNA” Evidence; pp. 3–17. [DOI] [PubMed] [Google Scholar]
- 24.Huffman K., Hanson E., Ballantyne J. Y-STR mixture deconvolution by single-cell analysis. J. Forensic Sci. 2023;68:275–288. doi: 10.1111/1556-4029.15150. [DOI] [PubMed] [Google Scholar]
- 25.Butler J.M., Iyer H., Press R., Taylor M.K., Vallone P.M., Willis S. DNA Mixture Interpretation: A NIST Scientific Foundation Review. NISTIR 8351- DRAFT. 2021. https://nvlpubs.nist.gov/nistpubs/ir/2021/NIST.IR.8351 1.
- 26.Moretti T.R., Just R.S., Kehl S.C., Willis L.E., Buckleton J.S., Bright J.-A., Taylor D.A., Onorato A.J. Internal validation of STRmix™ for the interpretation of single source and mixed DNA profiles. Forensic Sci. Int. Genet. 2017;29:126–144. doi: 10.1016/j.fsigen.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Bauer D.W., Butt N., Hornyak J.M., Perlin M.W. Validating TrueAllele ® Interpretation of DNA Mixtures Containing up to Ten Unknown Contributors. J. Forensic Sci. 2020;65:380–398. doi: 10.1111/1556-4029.14204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gill P., Gusmão L., Haned H., Mayr W.R., Morling N., Parson W., Prieto L., Prinz M., Schneider H., Schneider P.M., Weir B.S. DNA commission of the International Society of Forensic Genetics: Recommendations on the evaluation of STR typing results that may include drop-out and/or drop-in using probabilistic methods. Forensic Sci. Int. Genet. 2012;6:679–688. doi: 10.1016/j.fsigen.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coble M.D., Bright J.-A., Buckleton J.S., Curran J.M. Uncertainty in the number of contributors in the proposed new CODIS set. Forensic Sci. Int. Genet. 2015;19:207–211. doi: 10.1016/j.fsigen.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 30.Buckleton J.S., Bright J.-A., Gittelson S., Moretti T.R., Onorato A.J., Bieber F.R., Budowle B., Taylor D.A. The Probabilistic Genotyping Software STRmix: Utility and Evidence for its Validity. J. Forensic Sci. 2019;64:393–405. doi: 10.1111/1556-4029.13898. [DOI] [PubMed] [Google Scholar]
- 31.Bright J.-A., Richards R., Kruijver M., Kelly H., McGovern C., Magee A., McWhorter A., Ciecko A., Peck B., Baumgartner C., et al. Internal validation of STRmix™ – A multi laboratory response to PCAST. Forensic Sci. Int. Genet. 2018;34:11–24. doi: 10.1016/j.fsigen.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Francisco D.D.O., Lopez L.F., Gonçalves F.d.T., Fridman C. Casework direct kit as an alternative extraction method to enhance touch DNA samples analysis. Forensic Sci. Int. Genet. 2020;47:102307. doi: 10.1016/j.fsigen.2020.102307. [DOI] [PubMed] [Google Scholar]
- 33.Gill P., Benschop C., Buckleton J., Bleka Ø., Taylor D. A Review of Probabilistic Genotyping Systems: EuroForMix, DNAStatistX and STRmix™. Genes. 2021;12:1559. doi: 10.3390/genes12101559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benschop C.C.G., Nijveld A., Duijs F.E., Sijen T. An assessment of the performance of the probabilistic genotyping software EuroForMix: Trends in likelihood ratios and analysis of Type I & II errors. Forensic Sci. Int. Genet. 2019;42:31–38. doi: 10.1016/j.fsigen.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 35.Bright J.-A., Curran J.M., Buckleton J.S. The effect of the uncertainty in the number of contributors to mixed DNA profiles on profile interpretation. Forensic Sci. Int. Genet. 2014;12:208–214. doi: 10.1016/j.fsigen.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 36.Buckleton J.S., Bright J.-A., Cheng K., Kelly H., Taylor D.A. The effect of varying the number of contributors in the prosecution and alternate propositions. Forensic Sci. Int. Genet. 2019;38:225–231. doi: 10.1016/j.fsigen.2018.11.011. [DOI] [PubMed] [Google Scholar]
- 37.Bille T., Weitz S., Buckleton J.S., Bright J.-A. Interpreting a major component from a mixed DNA profile with an unknown number of minor contributors. Forensic Sci. Int. Genet. 2019;40:150–159. doi: 10.1016/j.fsigen.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 38.European Network of Forensic Science Institutes . 2015. ENFSI guidelines for evaluation and reporting in forensic science. [Google Scholar]
- 39.Scientific Working Group on DNA Analysis Methods Recommendations of the SWGDAM Ad Hoc Working Group on genotyping results reported as likelihood ratios. 2018. https://1ecb9588-ea6f-4feb-971a-73265dbf079c.filesusr.com/ugd/4344b0_dd5221694d1448588dcd0937738c9e46.pdf
- 40.Laboratory for Forensic Technology Development and Integration ProvedIt Database. https://lftdi.camden.rutgers.edu/provedit/files/
- 41.Alfonse L.E., Garrett A.D., Lun D.S., Duffy K.R., Grgicak C.M. A large-scale dataset of single and mixed-source short tandem repeat profiles to inform human identification strategies: PROVEDIt. Forensic Sci. Int. Genet. 2018;32:62–70. doi: 10.1016/j.fsigen.2017.10.006. [DOI] [PubMed] [Google Scholar]
- 42.Garvin A.M., Holzgreve W., Hahn S. Highly accurate analysis of heterozygous loci bysingle cell PCR. Nucleic Acids Res. 1998;26:3468–3472. doi: 10.1093/nar/26.15.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verdon T.J., Mitchell R.J., Chen W., Xiao K., van Oorschot R.A.H. FACS separation of non-compromised forensically relevant biological mixtures. Forensic Sci. Int. Genet. 2015;14:194–200. doi: 10.1016/j.fsigen.2014.10.019. [DOI] [PubMed] [Google Scholar]
- 44.Huffman K., Hanson E., Ballantyne J. Cell Subsampling Recovers Probative DNA Profile Information from Unresolvable/Undetectable Minor Donors in Mixtures. Genes. 2022;13:1117. doi: 10.3390/genes13071117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gill P., Jeffreys A.J., Werrett D.J. Forensic application of DNA ‘fingerprints. Nature. 1985;318:577–579. doi: 10.1038/318577a0. [DOI] [PubMed] [Google Scholar]
- 46.Clark C., Turiello R., Cotton R., Landers J.P. Analytical approaches to differential extraction for sexual assault evidence. Anal. Chim. Acta. 2021;1141:230–245. doi: 10.1016/j.aca.2020.07.059. [DOI] [PubMed] [Google Scholar]
- 47.Suneel P., Poonam R., P J., Maya S N., Priyanka K., Mukul M. Application and utility of alternative methods in isolation of pure cells from forensic biological mixtures in modern-day: a review. J. Forensic Sci. Res. 2021;5:041–047. doi: 10.29328/journal.jfsr.1001026. [DOI] [Google Scholar]
- 48.Clark C.P., Xu K., Scott O., Hickey J., Tsuei A.-C., Jackson K., Landers J.P. Acoustic trapping of sperm cells from mock sexual assault samples. Forensic Sci. Int. Genet. 2019;41:42–49. doi: 10.1016/j.fsigen.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 49.Horsman K.M., Barker S.L.R., Ferrance J.P., Forrest K.A., Koen K.A., Landers J.P. Separation of Sperm and Epithelial Cells in a Microfabricated Device: Potential Application to Forensic Analysis of Sexual Assault Evidence. Anal. Chem. 2005;77:742–749. doi: 10.1021/ac0486239. [DOI] [PubMed] [Google Scholar]
- 50.Verdon T.J., Mitchell R.J., Chen W., Xiao K., van Oorschot R.A.H. FACS separation of non-compromised forensically relevant biological mixtures. Forensic Sci. Int. Genet. 2015;14:194–200. doi: 10.1016/j.fsigen.2014.10.019. [DOI] [PubMed] [Google Scholar]
- 51.Fokias K., Bekaert B. Separation of sperm and epithelial cells based on fluorescence-activated cell sorting. Forensic Sci. Int. Genet. Suppl. Ser. 2022;8:239–241. doi: 10.1016/j.fsigss.2022.10.048. [DOI] [Google Scholar]
- 52.Dean L., Kwon Y.J., Philpott M.K., Stanciu C.E., Seashols-Williams S.J., Dawson Cruz T., Sturgill J., Ehrhardt C.J. Separation of uncompromised whole blood mixtures for single source STR profiling using fluorescently-labeled human leukocyte antigen (HLA) probes and fluorescence activated cell sorting (FACS) Forensic Sci. Int. Genet. 2015;17:8–16. doi: 10.1016/j.fsigen.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Miller J.M., Lee C., Ingram S., Yadavalli V.K., Greenspoon S.A., Ehrhardt C.J. Use of hormone-specific antibody probes for differential labeling of contributor cell populations in trace DNA mixtures. Int. J. Legal Med. 2022;136:1551–1564. doi: 10.1007/s00414-022-02887-x. [DOI] [PubMed] [Google Scholar]
- 54.Li X.-B., Wang Q.-S., Feng Y., Ning S.-H., Miao Y.-Y., Wang Y.-Q., Li H.-W. Magnetic bead-based separation of sperm from buccal epithelial cells using a monoclonal antibody against MOSPD3. Int. J. Legal Med. 2014;128:905–911. doi: 10.1007/s00414-014-0983-3. [DOI] [PubMed] [Google Scholar]
- 55.Alsalafi D., Goodwin W. Capturing spermatozoa for STR analysis of sexual assault cases using anti-sperm antibodies. Forensic Sci. Int. Genet. Suppl. Ser. 2019;7:707–710. doi: 10.1016/j.fsigss.2019.10.146. [DOI] [Google Scholar]
- 56.Yano S., Honda K., Kaminiwa J., Nishi T., Iwabuchi Y., Sugano Y., Kurosu A., Suzuki Y. DNA extraction for short tandem repeat typing from mixed samples using anti-human leukocyte CD45 and ABO blood group antibodies. Forensic Sci. Int. Genet. 2014;10:17–22. doi: 10.1016/j.fsigen.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 57.Xu Y., Xie J., Chen R., Cao Y., Ping Y., Xu Q., Hu W., Wu D., Gu L., Zhou H., et al. Fluorescence- and magnetic-activated cell sorting strategies to separate spermatozoa involving plural contributors from biological mixtures for human identification. Sci. Rep. 2016;6:36515. doi: 10.1038/srep36515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elliott K., Hill D.S., Lambert C., Burroughes T.R., Gill P. Use of laser microdissection greatly improves the recovery of DNA from sperm on microscope slides. Forensic Sci. Int. 2003;137:28–36. doi: 10.1016/S0379-0738(03)00267-6. [DOI] [PubMed] [Google Scholar]
- 59.Sanders C.T., Sanchez N., Ballantyne J., Peterson D.A. Laser Microdissection Separation of Pure Spermatozoa from Epithelial Cells for Short Tandem Repeat Analysis. J. Forensic Sci. 2006;51:748–757. doi: 10.1111/j.1556-4029.2006.00180.x. [DOI] [PubMed] [Google Scholar]
- 60.Vandewoestyne M., Van Hoofstat D., Van Nieuwerburgh F., Deforce D. Automatic detection of spermatozoa for laser capture microdissection. Int. J. Legal Med. 2009;123:169–175. doi: 10.1007/s00414-008-0271-1. [DOI] [PubMed] [Google Scholar]
- 61.Li C.x., Han J.p., Ren W.y., Ji A.q., Xu X.l., Hu L. DNA Profiling of Spermatozoa by Laser Capture Microdissection and Low Volume-PCR. PLoS One. 2011;6:e22316. doi: 10.1371/journal.pone.0022316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vintiner S.K., Veth J.S., Bright J.-A. A review of DNA profiling success for laser microdissected forensic casework samples. Aust. J. Forensic Sci. 2020;52:282–292. doi: 10.1080/00450618.2018.1510030. [DOI] [Google Scholar]
- 63.Anslinger K., Mack B., Bayer B., Rolf B., Eisenmenger W. Digoxigenin labelling and laser capture microdissection of male cells. Int. J. Legal Med. 2005;119:374–377. doi: 10.1007/s00414-005-0523-2. [DOI] [PubMed] [Google Scholar]
- 64.Feng L., Li C.X., Han J.P., Xu C., Hu L. Isolating cells from female/male blood mixtures using florescence in situ hybridization combined with low volume PCR and its application in forensic science. Int. J. Legal Med. 2015;129:1211–1215. doi: 10.1007/s00414-014-1103-0. [DOI] [PubMed] [Google Scholar]
- 65.Ballantyne J., Hanson E.K., Perlin M.W. DNA mixture genotyping by probabilistic computer interpretation of binomially-sampled laser captured cell populations: Combining quantitative data for greater identification information. Sci. Justice. 2013;53:103–114. doi: 10.1016/j.scijus.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 66.England R., Nancollis G., Stacey J., Sarman A., Min J., Harbison S. Compatibility of the ForenSeq™ DNA Signature Prep Kit with laser microdissected cells: An exploration of issues that arise with samples containing low cell numbers. Forensic Sci. Int. Genet. 2020;47:102278. doi: 10.1016/j.fsigen.2020.102278. [DOI] [PubMed] [Google Scholar]
- 67.Fontana F., Rapone C., Bregola G., Aversa R., de Meo A., Signorini G., Sergio M., Ferrarini A., Lanzellotto R., Medoro G., et al. Isolation and genetic analysis of pure cells from forensic biological mixtures: The precision of a digital approach. Forensic Sci. Int. Genet. 2017;29:225–241. doi: 10.1016/j.fsigen.2017.04.023. [DOI] [PubMed] [Google Scholar]
- 68.Anslinger K., Bayer B. Whose blood is it? Application of DEPArray™ technology for the identification of individual/s who contributed blood to a mixed stain. Int. J. Legal Med. 2019;133:419–426. doi: 10.1007/s00414-018-1912-7. [DOI] [PubMed] [Google Scholar]
- 69.Anslinger K., Graw M., Bayer B. Deconvolution of blood-blood mixtures using DEPArrayTM separated single cell STR profiling. Rechtsmedizin. 2019;29:30–40. doi: 10.1007/s00194-018-0291-1. [DOI] [Google Scholar]
- 70.Anslinger K., Bayer B., von Máriássy D. Application of DEPArrayTM technology for the isolation of white blood cells from cell mixtures in chimerism analysis. Rechtsmedizin. 2018;28:134–137. doi: 10.1007/s00194-017-0221-7. [DOI] [Google Scholar]
- 71.Meloni V., Lombardi L., Aversa R., Barni F., Berti A. Optimization of STR amplification down to single cell after DEPArrayTM isolation. Forensic Sci. Int. Genet. Suppl. Ser. 2019;7:711–713. doi: 10.1016/j.fsigss.2019.10.147. [DOI] [Google Scholar]
- 72.Williamson V.R., Laris T.M., Romano R., Marciano M.A. Enhanced DNA mixture deconvolution of sexual offense samples using the DEPArray™ system. Forensic Sci. Int. Genet. 2018;34:265–276. doi: 10.1016/j.fsigen.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 73.Sheth N., Swaminathan H., Gonzalez A.J., Duffy K.R., Grgicak C.M. Towards developing forensically relevant single-cell pipelines by incorporating direct-to-PCR extraction: compatibility, signal quality, and allele detection. Int. J. Legal Med. 2021;135:727–738. doi: 10.1007/s00414-021-02503-4. [DOI] [PubMed] [Google Scholar]
- 74.Theunissen G.M., Gibb A., Lin P.T., Rolf B., Forat S., Jäger R. DNA profiling of single sperm cells after whole genome amplification. Forensic Sci. Int. Rep. 2021;4:100240. doi: 10.1016/j.fsir.2021.100240. [DOI] [Google Scholar]
- 75.Ostojic L., O’Connor C., Wurmbach E. Micromanipulation of single cells and fingerprints for forensic identification. Forensic Sci. Int. Genet. 2021;51:102430. doi: 10.1016/j.fsigen.2020.102430. [DOI] [PubMed] [Google Scholar]
- 76.Brück S., Evers H., Heidorn F., Müller U., Kilper R., Verhoff M.A. Single Cells for Forensic DNA Analysis-From Evidence Material to Test Tube. J. Forensic Sci. 2011;56:176–180. doi: 10.1111/j.1556-4029.2010.01553.x. [DOI] [PubMed] [Google Scholar]
- 77.Zhang H., Liu K.-K. Optical tweezers for single cells. J. R. Soc. Interface. 2008;5:671–690. doi: 10.1098/rsif.2008.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dolezel J., Bartos J., Voglmayr H., Greilhuber J. Letter to the editor. Cytometry. 2003;51:127–128. doi: 10.1002/cyto.a.10013. author reply 129. [DOI] [PubMed] [Google Scholar]
- 79.Gill P., Whitaker J., Flaxman C., Brown N., Buckleton J. An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci. Int. 2000;112:17–40. doi: 10.1016/S0379-0738(00)00158-4. [DOI] [PubMed] [Google Scholar]
- 80.Gill P. Application of low copy number DNA profiling. Croat. Med. J. 2001;42:229–232. [PubMed] [Google Scholar]
- 81.Steele C.D., Balding D.J. Statistical Evaluation of Forensic DNA Profile Evidence. Annu. Rev. Stat. Appl. 2014;1:361–384. doi: 10.1146/annurev-statistics-022513-115602. [DOI] [Google Scholar]
- 82.Promega Corporation . 2016. PunchSolutionTM Kit Technical Manual. [Google Scholar]
- 83.Promega Corporation . 2020. Casework Direct System Technical Manual. [Google Scholar]
- 84.Watkins D.R.L., Myers D., Xavier H.E., Marciano M.A. Revisiting single cell analysis in forensic science. Sci. Rep. 2021;11:7054. doi: 10.1038/s41598-021-86271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sheth N., Duffy K.R., Grgicak C.M. High-quality data from a forensically relevant single-cell pipeline enabled by low PBS and proteinase K concentrations. J. Forensic Sci. 2022;67:697–706. doi: 10.1111/1556-4029.14956. [DOI] [PubMed] [Google Scholar]
- 86.Farash K., Hanson E.K., Ballantyne J. Enhanced Genetic Analysis of Single Human Bioparticles Recovered by Simplified Micromanipulation from Forensic “Touch DNA” Evidence. J. Vis. Exp. 2015:52612. doi: 10.3791/52612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huffman K., Hanson E., Ballantyne J. Recovery of single source DNA profiles from mixtures by direct single cell subsampling and simplified micromanipulation. Sci. Justice. 2021;61:13–25. doi: 10.1016/j.scijus.2020.10.005. [DOI] [PubMed] [Google Scholar]
- 88.Feng L., Xu C., Zeng X., Zhang H., Yang F., Li W., Tu Z., Li C., Hu L. Y-chromosomal haplotyping of single sperm cells isolated from semen mixtures – a successful identification of three perpetrators in a multi-suspect sexual assault case. Croat. Med. J. 2014;55:537–541. doi: 10.3325/cmj.2014.55.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Budowle B., Eisenberg A.J., Van Daal A. Validity of low copy number typing and applications to forensic science. Croat. Med. J. 2009;50:207–217. doi: 10.3325/cmj.2009.50.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bessekri M.W., Aggoune A., Lazreg S., Bucht R., Fuller V. Forensic Science International : Genetics Supplement Series Comparative study on the effects of reduced PCR reaction volumes and increased cycle number , on the sensitivity and the stochastic threshold of the AmpFlSTR Identifiler 1 Plus kit. Forensic Sci. Int. Genet. Suppl. Ser. 2013;4:e306–e307. doi: 10.1016/j.fsigss.2013.10.156. [DOI] [Google Scholar]
- 91.Schulte J., Marciano M.A., Scheurer E., Schulz I. A systematic approach to improve downstream single-cell analysis for the DEPArray™ technology. J. Forensic Sci. 2023 doi: 10.1111/1556-4029.15344. [DOI] [PubMed] [Google Scholar]
- 92.ThermoFisher Scientific . 2019. GlobalFilerTM and GlobalFilerTM IQC PCR Amplification Kits USER GUIDE. [Google Scholar]
- 93.Promega Corporation . 2019. Use of the PowerPlex® Fusion 6C System to Amplify Extracted DNA. [Google Scholar]
- 94.Ottens R., Templeton J., Paradiso V., Taylor D., Abarno D., Linacre A. Application of direct PCR in forensic casework. Forensic Sci. Int. Genet. Suppl. Ser. 2013;4:e47–e48. doi: 10.1016/j.fsigss.2013.10.024. [DOI] [Google Scholar]
- 95.QAS Guidance Document APPROVED by SWGDAM and Effective the Guidance Document for the FBI Quality Assurance Standards for Forensic DNA Testing and DNA Databasing Laboratories. 2020. [Google Scholar]
- 96.MENARINI silicon biosystems . 2017. DEPArrayTM Forensic Sample Prep Kit: USER MANUAL. [Google Scholar]
- 97.Huffman K., Ballantyne J. Validation of Probabilistic Genotyping Software for Single Cell STR Analysis. Genes. 2023;14:674. doi: 10.3390/genes14030674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Caragine T., Mikulasovich R., Tamariz J., Bajda E., Sebestyen J., Baum H., Prinz M. Validation of Testing and Interpretation Protocols for Low Template DNA Samples Using AmpFℓSTR® Identifiler. Croat. Med. J. 2009;50:250–267. doi: 10.3325/cmj.2009.50.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Benschop C.C.G., van der Beek C.P., Meiland H.C., van Gorp A.G.M., Westen A.A., Sijen T. Low template STR typing: Effect of replicate number and consensus method on genotyping reliability and DNA database search results. Forensic Sci. Int. Genet. 2011;5:316–328. doi: 10.1016/j.fsigen.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 100.Anslinger K., Bayer B. New strategies in the field of mixture deconvolution single cell STR profiling. Forensic Sci. Int. Genet. Suppl. Ser. 2019;7:259–261. doi: 10.1016/j.fsigss.2019.09.099. [DOI] [Google Scholar]
- 101.Huffman K., Hanson E., Ballantyne J. Probabilistic genotyping of single cell replicates from complex DNA mixtures recovers higher contributor LRs than standard analysis. Sci. Justice. 2022;62:156–163. doi: 10.1016/j.scijus.2022.01.003. [DOI] [PubMed] [Google Scholar]
- 102.Huffman K., Ballantyne J. Probabilistic Genotyping of Single Cell Replicates from Mixtures Involving First-Degree Relatives Prevents the False Inclusions of Non-Donor Relatives. Genes. 2022;13:1658. doi: 10.3390/genes13091658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Han J.P., Yang F., Xu C., Wei Y.L., Zhao X.C., Hu L., Ye J., Li C.X. A new strategy for sperm isolation and STR typing from multi-donor sperm mixtures. Forensic Sci. Int. Genet. 2014;13:239–246. doi: 10.1016/j.fsigen.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 104.Feng L., Xu C., Zeng X., Zhang H., Yang F., Li W., Tu Z., Li C., Hu L. Y-chromosomal haplotyping of single sperm cells isolated from semen mixtures – a successful identification of three perpetrators in a multi-suspect sexual assault case. Croat. Med. J. 2014;55:537–541. doi: 10.3325/cmj.2014.55.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ge J., King J.L., Smuts A., Budowle B. Precision DNA Mixture Interpretation with Single-Cell Profiling. Genes. 2021;12:1649. doi: 10.3390/genes12111649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Duffy K.R., Lun D.S., Mulcahy M.M., O’Donnell L., Sheth N., Grgicak C.M. Evidentiary evaluation of single cells renders highly informative forensic comparisons across multifarious admixtures. Forensic Sci. Int. Genet. 2023;64:102852. doi: 10.1016/j.fsigen.2023.102852. [DOI] [PubMed] [Google Scholar]
- 107.Bright J.-A., Taylor D., Kerr Z., Buckleton J., Kruijver M. The efficacy of DNA mixture to mixture matching. Forensic Sci. Int. Genet. 2019;41:64–71. doi: 10.1016/j.fsigen.2019.02.020. [DOI] [PubMed] [Google Scholar]
- 108.Taylor D., Kruijver M. Combining evidence across multiple mixed DNA profiles for improved resolution of a donor when a common contributor can be assumed. Forensic Sci. Int. Genet. 2020;49:102375. doi: 10.1016/j.fsigen.2020.102375. [DOI] [PubMed] [Google Scholar]
- 109.Huffman K., Kruijver M., Ballantyne J., Taylor D. Carrying out common DNA donor analysis using DBLR™ on two or five-cell mini-mixture subsamples for improved discrimination power in complex DNA mixtures. Forensic Sci. Int. Genet. 2023;66:102908. doi: 10.1016/j.fsigen.2023.102908. [DOI] [PubMed] [Google Scholar]
- 110.Taylor D., Bright J.-A., Buckleton J. The interpretation of single source and mixed DNA profiles. Forensic Sci. Int. Genet. 2013;7:516–528. doi: 10.1016/j.fsigen.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 111.Bleka Ø., Storvik G., Gill P. EuroForMix: An open source software based on a continuous model to evaluate STR DNA profiles from a mixture of contributors with artefacts. Forensic Sci. Int. Genet. 2016;21:35–44. doi: 10.1016/j.fsigen.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 112.Perlin M.W., Legler M.M., Spencer C.E., Smith J.L., Allan W.P., Belrose J.L., Duceman B.W. Validating TrueAllele ® DNA Mixture Interpretation. J. Forensic Sci. 2011;56:1430–1447. doi: 10.1111/j.1556-4029.2011.01859.x. [DOI] [PubMed] [Google Scholar]
- 113.Taylor D., Curran J., Buckleton J. Likelihood ratio development for mixed Y-STR profiles. Forensic Sci. Int. Genet. 2018;35:82–96. doi: 10.1016/j.fsigen.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 114.Taylor D., Bright J.-A., Buckleton J. Using probabilistic theory to develop interpretation guidelines for Y-STR profiles. Forensic Sci. Int. Genet. 2016;21:22–34. doi: 10.1016/j.fsigen.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 115.Theunissen G.M., Rolf B., Gibb A., Jäger R. DNA profiling of sperm cells by using micromanipulation and whole genome amplification. Forensic Sci. Int. Genet. Suppl. Ser. 2017;6:e497–e499. doi: 10.1016/j.fsigss.2017.09.183. [DOI] [Google Scholar]
- 116.Theunissen G.M.G. 2020. DNA profiling of single sperm cells and single skin flakes in forensics using micromanipulation and whole genome amplification. [Google Scholar]
- 117.Jäger R. New Perspectives for Whole Genome Amplification in Forensic STR Analysis. Int. J. Mol. Sci. 2022;23:7090. doi: 10.3390/ijms23137090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pereira J., Neves R., Forat S., Huckenbeck W., Olek K. MtDNA typing of single-sperm cells isolated by micromanipulation. Forensic Sci. Int. Genet. 2012;6:228–235. doi: 10.1016/j.fsigen.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 119.Bleka Ø., Prieto L., Gill P. EFMrep: An extension of EuroForMix for improved combination of STR DNA mixture profiles. Forensic Sci. Int. Genet. 2022;61:102771. doi: 10.1016/j.fsigen.2022.102771. [DOI] [PubMed] [Google Scholar]
- 120.Kelly H., Coble M., Kruijver M., Wivell R., Bright J.A. Exploring likelihood ratios assigned for siblings of the true mixture contributor as an alternate contributor. J. Forensic Sci. 2022;67:1167–1175. doi: 10.1111/1556-4029.15020. [DOI] [PubMed] [Google Scholar]
- 121.Diepenbroek M., Bayer B., Anslinger K. Pushing the Boundaries: Forensic DNA Phenotyping Challenged by Single-Cell Sequencing. Genes. 2021;12:1362. doi: 10.3390/genes12091362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kulhankova L., Montiel González D., Bindels E., Kling D., Kayser M., Mulugeta E. Single-cell transcriptome sequencing allows genetic separation, characterization and identification of individuals in multi-person biological mixtures. Commun. Biol. 2023;6:201. doi: 10.1038/s42003-023-04557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu Z., Jenkins G., Zhang W., Zhang M., Guan Z., Yang C.J. Single-molecule emulsion PCR in microfluidic droplets. Anal. Bioanal. Chem. 2012;403:2127–2143. doi: 10.1007/s00216-012-5914-x. [DOI] [PubMed] [Google Scholar]
- 124.Geng T., Novak R., Mathies R.A. Single-Cell Forensic Short Tandem Repeat Typing within Microfluidic Droplets. Anal. Chem. 2014;86:703–712. doi: 10.1021/ac403137h. [DOI] [PubMed] [Google Scholar]
- 125.Sánchez Barea J., Lee J., Kang D.-K. Recent Advances in Droplet-based Microfluidic Technologies for Biochemistry and Molecular Biology. Micromachines. 2019;10:412. doi: 10.3390/mi10060412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gao Y., Wu M., Lin Y., Xu J. Acoustic Microfluidic Separation Techniques and Bioapplications: A Review. Micromachines. 2020;11:921. doi: 10.3390/mi11100921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wei C., Yu C., Li S., Meng J., Li T., Cheng J., Li J. A droplet-based multivolume microfluidic device for digital polymerase chain reaction. Sens. Actuators B Chem. 2022;371:132473. doi: 10.1016/j.snb.2022.132473. [DOI] [Google Scholar]
- 128.Geng T., Mathies R.A. Minimizing inhibition of PCR-STR typing using digital agarose droplet microfluidics. Forensic Sci. Int. Genet. 2015;14:203–209. doi: 10.1016/j.fsigen.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 129.Lim J., Chin V., Fairfax K., Moutinho C., Suan D., Ji H., Powell J.E. Transitioning single-cell genomics into the clinic. Nat. Rev. Genet. 2023;24:573–584. doi: 10.1038/s41576-023-00613-w. [DOI] [PubMed] [Google Scholar]
- 130.Nadeu F., Royo R., Massoni-Badosa R., Playa-Albinyana H., Garcia-Torre B., Duran-Ferrer M., Dawson K.J., Kulis M., Diaz-Navarro A., Villamor N., et al. Detection of early seeding of Richter transformation in chronic lymphocytic leukemia. Nat. Med. 2022;28:1662–1671. doi: 10.1038/s41591-022-01927-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Burrill J., Daniel B., Frascione N. A review of trace “Touch DNA” deposits: Variability factors and an exploration of cellular composition. Forensic Sci. Int. Genet. 2019;39:8–18. doi: 10.1016/j.fsigen.2018.11.019. [DOI] [PubMed] [Google Scholar]
- 132.Vandewoestyne M., Van Hoofstat D., Franssen A., Van Nieuwerburgh F., Deforce D. Presence and potential of cell free DNA in different types of forensic samples. Forensic Sci. Int. Genet. 2013;7:316–320. doi: 10.1016/j.fsigen.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 133.Scientific Working Group on DNA Analysis Methods . 2014. Guidelines for STR Enhanced Detection Methods. [Google Scholar]
- 134.Chen M., Zhang J., Zhao J., Chen T., Liu Z., Cheng F., Fan Q., Yan J. Comparison of CE- and MPS-based analyses of forensic markers in a single cell after whole genome amplification. Forensic Sci. Int. Genet. 2020;45:102211. doi: 10.1016/j.fsigen.2019.102211. [DOI] [PubMed] [Google Scholar]
- 135.Mitchell R., Enke S., Eskey K., Ferguson T., Just R. A method to enable forensic genetic genealogy investigations from DNA mixtures. Forensic Sci. Int. Genet. Suppl. Ser. 2022;8:159–161. doi: 10.1016/j.fsigss.2022.10.020. [DOI] [Google Scholar]
- 136.Breslin K., Wills B., Ralf A., Ventayol Garcia M., Kukla-Bartoszek M., Pospiech E., Freire-Aradas A., Xavier C., Ingold S., de La Puente M., et al. HIrisPlex-S system for eye, hair, and skin color prediction from DNA: Massively parallel sequencing solutions for two common forensically used platforms. Forensic Sci. Int. Genet. 2019;43:102152. doi: 10.1016/j.fsigen.2019.102152. [DOI] [PubMed] [Google Scholar]
- 137.Ralf A., Kayser M. Investigative DNA analysis of two-person mixed crime scene trace in a murder case. Forensic Sci. Int. Genet. 2021;54:102557. doi: 10.1016/j.fsigen.2021.102557. [DOI] [PubMed] [Google Scholar]
- 138.Bleka Ø., Eduardoff M., Santos C., Phillips C., Parson W., Gill P. Using EuroForMix to analyse complex SNP mixtures, up to six contributors. Forensic Sci. Int. Genet. Suppl. Ser. 2017;6:e277–e279. doi: 10.1016/j.fsigss.2017.09.084. [DOI] [PubMed] [Google Scholar]