

Summary
Neurovascular dysfunction is a preclinical manifestation of diabetic complications, including diabetic retinopathy (DR). Herein, we report that a transfer RNA-derived RNA fragment, tRF-3001a, is significantly upregulated under diabetic conditions. tRF-3001a downregulation inhibits Müller cell activation, suppresses endothelial angiogenic effects, and protects against high-glucose-induced retinal ganglion cell injury in vitro. Furthermore, tRF-3001a downregulation alleviates retinal vascular dysfunction, inhibits retinal reactive gliosis, facilitates retinal ganglion cell survival, and preserves visual function and visually guided behaviors in STZ-induced diabetic mice and db/db diabetic mice. Mechanistically, tRF-3001a regulates neurovascular dysfunction in a microRNA-like mechanism by targeting GSK3B. Clinically, tRF-3001a is upregulated in aqueous humor (AH) samples of DR patients. tRF-3001a downregulation inhibits DR-induced human retinal vascular endothelial cell and Müller cell dysfunction in vitro and DR-induced retinal neurovascular dysfunction in C57BL/6J mice. Thus, targeting tRF-3001a-mediated signaling is a promising strategy for the concurrent treatment of vasculopathy and neuropathy in diabetes mellitus.
Keywords: diabetic retinopathy, neurovascular dysfunction, transfer RNA-derived RNA fragment, vasculopathy, neuropathy
Graphical abstract
Highlights
-
•
tRF-3001a expression is significantly upregulated under diabetic conditions
-
•
tRF-3001a inhibitor decreases Müller activation and exerts anti-angiogenic effects
-
•
Injection of tRF-3001a antagomir alleviates retinal neurovascular dysfunction
-
•
tRF-3001a regulates retinal neurovascular dysfunction in a microRNA-like mechanism
Zhu et al. report that a transfer RNA-derived RNA fragment, tRF-3001a, is involved in retinal neurovascular dysfunction in diabetic mice. tRF-3001a downregulation alleviates diabetes-induced retinal neurodegeneration and vascular dysfunction and preserves visual function and visually guided behaviors. Fine-tuning of tRF-3001a level is promising for concurrent treatment of retinal vasculopathy and neuropathy.
Introduction
Diabetes mellitus is a metabolic disorder characterized by hyperglycemia and glucose intolerance. Globally, diabetes mellitus has a high prevalence, and more than 400 million people suffer from this disorder.1 Long-term hyperglycemia can lead to serious injuries to a series of body systems, especially blood vessels and nerves.2 Diabetic vasculopathy has been regarded as the major causes of end-stage renal failure, blindness, and atherosclerosis, accounting for the disabilities and mortality in diabetic patients.3 Moreover, about 50% of diabetic patients have obvious neuropathy, showing serious injuries to sensory neurons, motor neurons, and autonomic nervous system.4 Thus, the pathogenesis of diabetes mellitus is not only associated with vascular dysfunction but also associated with neurodegeneration. Current treatment modalities for diabetes and its complications involve diet control, physical activity, tobacco use cessation, and complication medications.5,6 However, these modalities cannot completely reverse or halt the progression of vasculopathy and neurodegeneration due to metabolic memory. Thus, further understanding of the mechanism leading to vasculopathy and neuropathy contributes to the development of novel preventive and therapeutic targets in diabetes mellitus.
Epigenetic mechanisms can regulate gene expression without altering DNA sequence. They can shape and store the molecular responses of the cells for gene-environment interactions.7 The pathogenesis of diabetes mellitus is highly influenced by environmental factors, such as nutritional status, growth factors, and oxidant stress, which can cause the dysregulation of epigenetic mechanism.8,9 Small non-coding RNAs are a class of important epigenetic regulators involved in gene regulatory network, including microRNAs (miRNAs), piwi-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), and transfer RNA-derived RNA fragments (tRFs).10 Among them, tRFs are a class of 13- to 48-nucleotide (nt) products of the precursor or mature transfer RNAs generated by ribonuclease cleavage under the pathological condition.11 tRFs have been implicated in several biological processes, such as gene expression, protein synthesis, and RNA processing.12,13 Dysregulation of tRFs has been implicated in the pathogenesis of cancers, metabolic disorders, and neurological disorders.14,15,16,17 Thus, tRFs are shown as promising diagnostic markers and therapeutic targets for human disorders. However, few studies have investigated the involvement of tRFs in the process of vasculopathy and neuropathy in diabetes mellitus.
The retina is the extension of the central nervous system and the most prevalent site of hyperglycemic injury. Diabetic retinopathy (DR) is a leading cause of vision disability in working age.18 DR is generally recognized as a microvascular complication of diabetes, and its diagnosis is highly dependent on the detection of vascular lesions.19 However, a considerable proportion of DR patients still fail to achieve obvious visual improvement. In fact, the pathogenesis of DR is also accompanied by retinal neurodegeneration. Previous study has reported that retinal neurodegeneration sometimes occurs before retinal vasculopathy.20 Thus, DR has been gradually recognized as a neurovascular disease that affects both retinal vascular cells and neuronal cells. The neurovascular unit (NVU) is composed of vascular cells, glial cells, and neurons.21 All component cells in the NVU are intimately connected and control the integrity of ocular architecture and function following metabolic stress. NVU impairment has become a crucial event in DR.22,23 However, the mechanism underlying NVU impairment has not been fully elucidated.
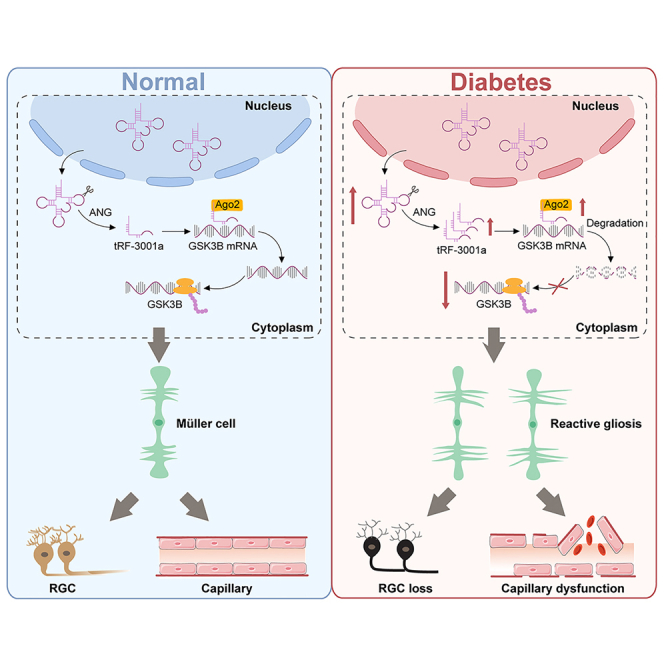
In this study, we investigated the role of a tRF, tRF-3001a, in diabetes-induced retinal neurovascular dysfunction. tRF-3001a is a 3′-tRF-LeuAAG/TAG fragment, and its sequence is 5′- ATCCCACCGCTGCCACCA-3′. The results show that the levels of tRF-3001a expression are significantly upregulated following diabetic stress. Reduction of tRF-3001a expression can alleviate retinal vascular dysfunction and suppress retinal neurodegeneration. Mechanistically, tRF-3001a regulates retinal neurovascular dysfunction in a miRNA-like mechanism. Clinically, the levels of tRF-3001a expression are upregulated in aqueous humor (AH) samples of DR patients. Thus, tRF-3001a is a promising regulator of retinal neurovascular dysfunction and a promising target for concurrent treatment of vasculopathy and neuropathy in diabetes mellitus.
Results
Expression level of tRF-3001a is upregulated following diabetic stress
To evaluate the potential role of tRFs in DR, we focused on tRF-3001a and investigated its expression pattern following diabetic stress in vivo and in vitro. C57BL/6J mice received repeated intraperitoneal injections of streptozotocin (STZ) to establish a diabetic model. qRT-PCR assays demonstrated that the levels of tRF-3001a expression were upregulated in diabetic retinas compared with the non-diabetic controls (Figure 1A). We next determined the expression pattern of tRF-3001 following diabetic stress in vitro. Primary Müller cells, retinal endothelial cells (ECs), retinal ganglion cells (RGCs), pericytes, and retinal pigment epithelium (RPE) cells were exposed to high glucose (25 mM) or oxidative stress (H2O2, 100 μM) to mimic diabetes-related stress. The levels of tRF-3001a expression were markedly upregulated in Müller cells, RGCs, and ECs following diabetic stress (Figures 1B and 1C). We also examined tRF-3001a expression in clinical samples, including fibrovascular membranes from DR patients and idiopathic epiretinal membranes from non-diabetic patients. tRF-3001a expression obviously increased in fibrovascular membranes from DR patients (Figure 1D). Together, the above-mentioned results indicate that tRF-3001a is a potential regulator of the pathogenesis of DR.
Figure 1.

Expression level of tRF-3001a is upregulated following diabetic stress
(A) Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assays were conducted to compare tRF-3001a expression between non-diabetic retinas and diabetic retinas following 2, 4, or 6 months of STZ injection (n = 6, ∗p < 0.05, Student’s t test).
(B and C) Primary Müller cells, primary retinal ganglion cells (RGCs), retinal endothelial cells (ECs), pericytes, and RPEs were exposed to high glucose (HG, 25 mM) or oxidative stress (H2O2, 100 μM) or were left untreated (Ctrl) for 48 h. The levels of tRF-3001a expression were examined by qRT-PCR assays (n = 4, ∗p < 0.05, Student’s t test).
(D) tRF-3001a expression in fibrovascular membranes of diabetic patients and idiopathic epiretinal membranes of non-diabetic patients was examined by qRT-PCR assays (n = 10, ∗p < 0.05, Student’s t test).
(E) Western blots and quantitative analysis were conducted to assess the levels of ANG and Dicer expression in non-diabetic retinas and diabetic retinas following 2-, 4-, and 6-month diabetes induction (n = 6, ∗p < 0.05, one-way ANOVA followed by post hoc Bonferroni test).
(F and G) Müller cells were transfected with ANG siRNA1-3, Dicer siRNA1-3, or negative control (NC) siRNA for 48 h. qRT-PCR assays were conducted to detect the levels of ANG and Dicer expression (n = 4, ∗p < 0.05, one-way ANOVA followed by post hoc Bonferroni test).
(H and I) Müller cells were transfected with negative control (NC) siRNA, ANG siRNA, or Dicer siRNA, and exposed to high glucose (HG, 25 mM) for 48 h. qRT-PCR assays were conducted to detect the levels of tRF-3001 expression (n = 4, ∗p < 0.05, one-way ANOVA followed by post hoc Bonferroni test). See also Table S1.
tRF-3001a is usually generated by the cleavage of the mature tRNA by angiogenin (ANG) or Dicer.11 To further explore the mechanism for the upregulation of tRF-3001a following diabetic stress, we detected the levels of ANG and Dicer expression in retinal tissues from non-diabetic and diabetic mice. Western blot analysis revealed that compared with non-diabetic retinas, the protein levels of ANG expression significantly increased in diabetic retinas. By contrast, Dicer expression was not altered following diabetes induction (Figure 1E). To determine whether ANG or Dicer is required for the production of tRF-3001a, Müller cells were transfected with ANG siRNA or Dicer siRNA to reduce the levels of ANG and Dicer expression (Figures 1F and 1G). Under high-glucose stress, transfection with ANG siRNA but not Dicer siRNA led to reduced tRF-3001a expression in Müller cells (Figures 1H and 1I). These results indicate that induction of tRF-3001a expression following high-glucose stress is mainly mediated through the increased cleavage of mature tRNA by ANG.
tRF-3001a regulates retinal vascular cell and neuronal cell function in vitro
The aforementioned results have revealed that tRF-3001a expression was dysregulated in Müller cells, ECs, and RGCs following diabetic stress. Notably, the greatest upregulation of tRF-3001a expression was detected in Müller cells. Thus, we first examined the role of tRF-3001a in Müller cells. Müller cells were primarily isolated, and the purity was determined by glial fibrillary acidic protein (GFAP) and glutamine synthetase (GS) staining. The results showed that the percentage of GS-positive or GFAP-positive Müller cells was greater than 90% (Figure S1). Subsequently, tRF-3001a inhibitors or tRF-3001a mimics were transfected into Müller cells to regulate tRF-3001a levels. The efficiency of overexpression and knockdown of tRF-3001a is shown in Figure 2A.
Figure 2.

tRF-3001a regulates Müller cell function following diabetic stress in vitro
(A) Primary Müller cells were transfected with the negative control (NC) mimics, tRF-3001a mimics, NC inhibitors, or tRF-3001a inhibitors or were left untreated (Ctrl) for 24 h. The levels of tRF-3001a were determined by qRT-PCRs (n = 4, ∗p < 0.05 versus Ctrl, one-way ANOVA followed by Bonferroni test).
(B–F) Primary Müller cells were transfected with NC mimics, tRF-3001a mimics, NC inhibitors, or tRF-3001a inhibitors or were left untreated (WT) for 6 h and then exposed to high glucose (25 mM) for 48 h. The group without high-glucose exposure was taken as the control (Ctrl) group. Cell viability was examined by CCK-8 assays (B, n = 4). Cell proliferation ability was examined by EdU staining and quantitated. EdU, green; DAPI, blue. Scale bar, 20 μm (C, n = 4). TUNEL assays were used to detect the apoptosis of Müller cells. TUNEL, green; DAPI, blue. Scale bar, 50 μm (D, n = 4). The dead or dying cells were detected by Calcein-AM/PI staining. Calcein-AM, green; PI, red. Scale bar, 20 μm (E, n = 4). Rhodamine 123 staining was used to detect the change of mitochondrial membrane potentials (ΔΨm) in Müller cells. Rhodamine 123, green; DAPI, blue. Scale bar, 20 μm (F, n = 4). ∗p < 0.05 versus Ctrl; #p < 0.05 between the marked groups. The significant difference was evaluated by one-way ANOVA followed by post hoc Bonferroni test. See also Figures S1, S2, and S3.
In response to high-glucose stress, Cell Counting Kit-8 (CCK-8) assays showed that transfection of tRF-3001a mimics enhanced the viability of Müller cells, while transfection of tRF-3001a inhibitors reduced the viability of Müller cells (Figure 2B). 5-Ehynyl-2′-deoxyuridine (EdU) assays demonstrated that transfection of tRF-3001a mimics boosted the proliferation ability of Müller cells, while downregulation of tRF-3001a level by tRF-3001a inhibitors significantly reduced the proliferation ability of Müller cells (Figure 2C). TUNEL assays, calcein-AM/propidium iodide (PI) double staining, and rhodamine 123 staining assays were conducted to investigate the role of tRF-3001a in cell apoptosis following high-glucose stress. Transfection of tRF-3001a mimics obviously retarded the development of high-glucose-induced apoptosis in Müller cells as shown by decreased number of TUNEL-positive cells (Figure 2D), decreased number of PI-positive cells (Figure 2E), and increased mitochondrial depolarization (Figure 2F). By contrast, downregulation of tRF-3001a by tRF-3001a inhibitors accelerated the development of Müller apoptosis (Figures 2D–2F).
Müller cells can wrap around retinal vessels and neurons and control retinal homeostasis by regulating vessel cell and neuronal cell function.24 We employed the co-culture assays to investigate Müller cell-EC crosstalk or Müller cell-RGC crosstalk upon high-glucose stress in vitro. Co-culture with Müller cells boosted the proliferation, migration, tube formation, and sprouting ability of ECs. However, downregulation of tRF-3001a in Müller cells partially eliminated the pro-angiogenic effects of Müller cells on ECs as shown by decreased proliferation, migration, tube formation ability, and reduced endothelial sprouting length (Figure S2).
Primary RGCs were isolated, and the crosstalk between RGCs and Müller cells was also investigated. As shown in Figure S3, the percentage of TUJ1-positive RGCs was about 90%. TUNEL staining and CCK-8 assays showed that co-culture with Müller cells could aggravate high-glucose-induced RGC injury following 24 h exposure to high glucose, suggesting that Müller cells exerted the detrimental effects on RGC function. Transfection of tRF-3001a mimics led to abnormal activation of Müller cells, which could further aggravate high-glucose-induced RGC injury as shown by increased TUNEL-positive RGC numbers and reduced RGC viability (Figure S3). Collectively, the above-mentioned results suggest that tRF-3001a is a potential regulator of retinal vascular cell and neuronal cell function in vitro.
tRF-3001a regulates diabetes-induced retinal vascular dysfunction in vivo
DR is a microvascular complication of diabetes.25 We next investigated whether tRF-3001a regulated diabetes-induced retinal vascular dysfunction in vivo. STZ-induced diabetic mice or db/db diabetic mice received an intravitreal injection of negative control (NC) agomir, tRF-3001a agomir, NC antagomir, or tRF-3001a antagomir. The efficacy of tRF-3001a overexpression and knockdown was examined by qRT-PCR assays, as shown in Figures 3A and S4A. Evans blue assays were used to detect the degree of retinal vascular permeability. Compared with STZ-induced diabetic mice, intravitreal injection of tRF-3001a agomir led to a marked increase in retinal vascular permeability following 2-, 4-, and 6-month induction of diabetes. By contrast, intravitreal injection of tRF-3001a antagomir had an opposite effect on retinal vascular permeability, showing reduced retinal vascular leakage (Figure 3B). Hyperglycemia can destroy retinal vasculature and cause pericyte loss and acellular capillary formation. Trypsin digest and periodic acid Schiff (PAS) assays revealed that intravitreal injection of tRF-3001a agomir significantly increased the number of acellular capillaries. By contrast, intravitreal injection of tRF-3001a antagomir obviously alleviated retinal vascular leakage and reduced the number of acellular capillaries at 2, 4, and 6 months after diabetes induction (Figure 3C). A similar event was observed in db/db diabetic mice. Compared with the NC antagomir group, intravitreal injection of tRF-3001a antagomir led to a marked reduction of retinal vascular permeability and acellular capillary number, exhibiting similar effects as aflibercept on retinal vascular dysfunction (Figures S4B and S4C).
Figure 3.

tRF-3001a regulates diabetes-induced retinal vascular dysfunction in vivo
(A) STZ-induced diabetic C57BL/6J mice received intravitreal injections of negative control (NC) agomir, tRF-3001a agomir, NC antagomir, or tRF-3001a antagomir or were left untreated (DR) once a month for 6 months. The non-diabetic C57BL/6J mice were taken as the control (Ctrl) group. qRT-PCRs were conducted to determine the levels of tRF-3001a expression (n = 6).
(B) The mice were infused with Evans blue dye for 2 h. The fluorescence signal of flat-mounted retina was observed under a 4× lens. Evans blue leakage was quantified following 2-, 4-, and 6-month diabetes induction. The representative images were shown at 6 months after diabetes induction (n = 6). Scale bar, 500 μm.
(C) Retinal trypsin digestion was used to detect retinal acellular capillaries. Red arrow indicates acellular capillaries. Quantification analysis was averaged from 15 randomly selected fields per retina. The representative images are shown (n = 6). Scale bar, 10 μm.
(D) qRT-PCR assays were conducted to detect the expression of VEGF, IL-6, IL-1β, ICAM-1, and TNF-α mRNA (n = 6).
(E) ELISA assays were conducted to examine the expression of VEGF, IL-6, IL-1β, ICAM-1, and TNF-α protein in retinal lysates (n = 4). ∗p < 0.05 versus Ctrl; #p < 0.05 between the marked groups. The significant difference was evaluated by one-way ANOVA followed by post hoc Bonferroni test. See also Figures S4 and S5.
An oxygen-induced retinopathy (OIR) model was also employed to assess the role of tRF-3001a in retinal neovascularization in vivo. 7-day-old C57BL/6J mice with their mothers were exposed to 75% oxygen for 5 days following the injections of NC antagomir, tRF-3001a antagomir, NC agomir, or tRF-3001a agomir or left untreated (WT) and were then exposed to room air condition until post-natal day 17 (P17). Compared with the WT group, injection of tRF-3001a antagomir obviously reduced the angiogenic area and avascular area in OIR retinas, while injection of tRF-3001a agomir aggravated pathological retinal neovascularization at P17 in OIR retinas (Figure S5).
Retinal vascular injury can lead to vessel inflammation and accelerate the progression of DR.26 We further employed qRT-PCRs and ELISA assays to detect the expression changes of inflammatory factors. The results showed that intravitreal injection of tRF-3001a agomir aggravated diabetes-induced retinal inflammation at 6 months after STZ injection, showing enhanced expression levels of vascular endothelial growth factor (VEGF), interleukin-6 (IL-6), IL-1β, intercellular cell adhesion molecule 1 (ICAM-1), and TNF-α (tumor necrosis factor α). Conversely, injection of tRF-3001a antagomir obviously alleviated retinal vascular inflammation (Figures 3D and 3E). Likewise, intravitreal injection of tRF-3001a antagomir also reduced diabetes-induced retinal inflammation in db/db diabetic mice, showing similar effects as aflibercept on retinal inflammation (Figure S4D). Taken together, these results demonstrate that tRF-3001a has emerged as a critical regulator of retinal vascular function under diabetic condition.
tRF-3001a regulates diabetes-induced retinal neuronal dysfunction in vivo
Retinal neurodegeneration is another pathological feature in DR. Long-term diabetes can trigger retinal neurodegeneration by destroying the structure and function of retinal NVU.27 To reveal the biological relevance of tRF-3001a in retinal neurodegeneration, retinal electroretinography assays were conducted to examine retinal neuronal function in STZ-induced diabetic mice. The results showed that diabetes led to reduced amplitude and increased latency of b waves. Injection of tRF-3001a agomir further aggravated retinal neuronal dysfunction as shown by lower b-wave amplitude and greater b-wave latency compared with the mice following 2-, 4-, and 6-month induction of diabetes. By contrast, intravitreal injection of tRF-3001a antagomir could ameliorate visual function and partially reverse retinal neuronal dysfunction, as shown by greater b-wave amplitude and lower b-wave latency compared with DR group (Figure 4A).
Figure 4.

tRF-3001a regulates diabetes-induced retinal neuronal dysfunction in vivo
(A) Electrophysiology was performed to detect retinal neuronal function in non-diabetic mice (Ctrl), STZ-induced diabetic mice injected with negative control (NC) agomir, tRF-3001a agomir, NC antagomir, and tRF-3001a antagomir following 2-, 4-, or 6-month diabetes induction. The representative images were shown at 4 months after diabetes induction (n = 6). The amplitudes and latency of b waves were statistically calculated (n = 6).
(B and C) Immunofluorescence and quantitative analysis of GFAP staining (B) or GS staining (C) were conducted to detect retinal reactive gliosis along with the representative images (n = 6). Scale bar, 50 μm.
(D and E) Immunofluorescence and quantitative analysis of NeuN staining (D) or TUJ1 staining (E) were conducted to detect RGC survival. The representative images are shown (n = 6). Scale bar, 50 μm.
(F) Retinal whole mounts following TUJ1 staining were observed from the peripheral area. RGC survival rate was calculated by dividing the average number of TUJ1-positive cells in one field in the injured retina by that in the uninjured (Ctrl) retina (n = 6). Scale bar, 20 μm ∗p < 0.05 versus Ctrl; #p < 0.05 between the marked groups. The significant difference was evaluated by one-way ANOVA followed by post hoc Bonferroni test. See also Figures S6, S7, and S8.
Reactive gliosis and neuronal apoptosis are the most important histological features of DR.28 Immunostaining assays revealed that compared with the NC agomir group, intravitreal injection of tRF-3001a agomir could aggravate retinal reactive gliosis, as shown by increased GFAP and GS staining (Figures 4B and 4C), and could decrease RGC survival, as shown by the reduced number of NeuN- and TUJ1-positive RGCs (Figures 4D and 4E). By contrast, injection of tRF-3001a antagomir had the opposite effects as shown by reduced reactive gliosis and increased RGC survival compared with the corresponding group (Figures 4B–4E). Moreover, we conducted whole-mount immunofluorescence to detect RGC survival. The results also demonstrated that intravitreal injection of tRF-3001a antagomir contributed to RGC survival (Figure 4F). A similar event was observed in db/db diabetic mice (Figure S6).
In addition, retinal slices were stained with retinal marker proteins to label other retinal cells, including calbindin (ganglion cells, amacrine, and horizontal cells), calretinin (ganglion cells and amacrine cells), rhodopsin (rod and cone photoreceptor), and protein kinase Cα (PKCα; bipolar cells), in STZ-induced diabetic mice and db/db diabetic mice. Compared with diabetic retinas, intravitreal injection of tRF-3001a agomir or antagomir altered the number of calbindin-labeled cells or calretinin-labeled cells in the ganglion cell layer (GCL). However, the number of calbindin-labeled cells in the inner nuclear layer (INL), horizontal cells, amacrine cells, or calretinin-labeled cells in the INL layer were not affected (Figures S7 and S8). Rhodopsin and PKCα immunostaining revealed that intravitreal injection of tRF-3001a agomir or antagomir had no effects on the signaling of photoreceptors and bipolar cells (Figures S7 and S8). Collectively, the above-mentioned results show that tRF-3001a regulates diabetes-induced retinal neuronal function through affecting retinal reactive gliosis and RGC survival.
tRF-3001a regulates diabetes-induced visual impairment in vivo
Retinal neurovascular dysfunction can lead to visual impairment. Given the difficulty in direct measurement of visual acuity in mice, the vision-related behavioral tests, including visual cliff test, dark-light preference test, and Morris water maze test, were used to evaluate the degree of visual impairment in mice.29 6 months after STZ injection, the visual cliff test was conducted to determine the effect of tRF-3001a expression change on the ability for discriminating cliff side and safe side (Figure 5A). The result showed that there was no significant difference among DR group, DR + NC agomir, and DR + NC antagomir group in discriminating cliff side and safe side. tRF-3001a agomir group had a greater propensity to choose cliff side than DR group. By contrast, intravitreal injection of tRF-3001a antagomir greatly improved the discriminating ability compared with the diabetic group, showing a similar discriminating ability for cliff side and safe side as the non-diabetic group (Figure 5B). Given the innate tendency for the avoidance of bright environment, a dark-light preference test was used to estimate light perception of mice (Figure 5C). Compared with DR group, intravitreal injection of tRF-3001a antagomir significantly increased the preference for dark environments in diabetic mice as shown by increased duration time. By contrast, intravitreal injection of tRF-3001a agomir led to decreased duration time in the dark environment (Figure 5D). Morris water maze test was used to examine the visual discrimination acuity (Figure 5E). Intravitreal injection of tRF-3001a antagomir in DR mice could obviously shorten the average time for reaching at the safety platform compared with diabetic mice. However, intravitreal injection of tRF-3001a agomir greatly increased the average time for reaching at the safety platform in DR mice (Figure 5F). A similar event was observed in db/db diabetic mice (Figure S9). Collectively, the aforementioned results indicate that tRF-3001a is shown as a therapeutic target for visual impairment.
Figure 5.

tRF-3001a regulates diabetes-induced visual impairment in vivo
Vision-related behavioral tests were used to evaluate the degree of visual impairment in non-diabetic mice (Ctrl), STZ-induced diabetic mice (WT), or STZ-induced diabetic mice injected with negative control (NC) agomir, tRF-3001a agomir, NC antagomir, or tRF-3001a antagomir at 6 months after diabetes induction. The schematic diagram for visual cliff test is shown (A). The statistical results display the number (left y axis) and percentage (right y axis) of cliff/safe side choosers (B, n = 6). The schematic diagram of dark-light preference test is shown (C). The statistical result shows the percentage of time in the dark chamber within 5 min (D, n = 6). The schematic diagram of Morris water maze test is shown (E). The statistical result shows the time required until the mice reached the visible platform for 8 days (F, n = 6). ∗p < 0.05 between the marked groups. The significant difference was evaluated by one-way ANOVA followed by post hoc Bonferroni test. See also Figure S9.
tRF-3001a-GSK3B signaling axis regulates retinal neurovascular dysfunction in vitro and in vivo
We next determined the mechanism of tRF-3001a-mediated retinal neurovascular dysfunction. qRT-PCR assays revealed that tRF-3001a was mainly expressed in the cytoplasm of Müller cells (Figure S10A). RNA immunoprecipitation (RIP) assays demonstrated that tRF-3001a could be immunoprecipitated by Ago2 but not IgG (Figure S10B). Ago2 is a crucial protein involved in miRNA-mediated biology.30 Due to the direct binding between Ago2 and tRF-3001a, we speculated that tRF-3001a played its role at the post-transcriptional level in a miRNA-like mechanism. The potential target genes of tRF-3001a were predicted by the miRDB database (https://mirdb.org/). Pathway analysis of the potential target genes were conducted using the Reactome pathway database (https://reactome.org/) (Figure S10C). GSK3B, NSF, and TP53RK were predicted as the potential targets of tRF-3001a due to their roles in vascular dysfunction or neurodegeneration. Transfection of tRF-3001a mimics led to a marked reduction of GSK3B expression, whereas transfection of tRF-3001a inhibitors significantly increased the level of GSK3B expression. By contrast, transfection of tRF-3001a mimics or inhibitors had no effects on the expression levels of NSF and TP53RK (Figure S10D). We further conducted luciferase assays to verify the direct interaction between tRF-3001a and GSK3B. Transfection of tRF-3001a mimics significantly decreased the luciferase activity of wild-type 3′-UTR of GSK3B, while the luciferase activity of mutant 3′-UTR of GSK3B was not altered following the transfection of tRF-3001a mimics (Figure S10E). RNA pull-down assays demonstrated that GSK3B was greatly enriched in the tRF-3001a-captured fraction compared with the tRF-1001 (NC)-captured fraction (Figure S10F).
Since the maximal increase of tRF-3001a expression was detected in Müller cells following diabetic stress, we thus investigated the role of tRF-3001a-GSK3B signaling axis in glial activities. CCK-8 and EdU assays revealed that transfection of tRF-3001a mimics led to enhanced viability of Müller cells and accelerated the proliferation of Müller cells. Silencing of GSK3B could mimic the effects of tRF-3001a mimics, while overexpression of GSK3B could reverse the effects of tRF-3001a mimics on Müller cell function (Figures 6A and 6B). We also investigated the role of tRF-3001a-GSK3B signaling axis in endothelial angiogenic activities. Transfection of tRF-3001a mimics played a pro-angiogenic role in endothelial cell function as shown by increased cell viability and proliferation. Silencing of GSK3B could mimic the pro-angiogenic effects of tRF-3001a mimics. Overexpression of GSK3B could eliminate the pro-angiogenic effects of tRF-3001a mimics on endothelial cell function (Figure S11).
Figure 6.

tRF-3001a-GSK3B signaling axis regulates retinal neurovascular dysfunction in vitro and in vivo
(A and B) Müller cells were transfected with negative control (NC) mimics (Ctrl), tRF-3001a mimics, NC siRNA, GSK3B siRNA, tRF-3001a plus GSK3B overexpression vector, or tRF-3001a plus null vector for 24 h. Cell viability was examined by CCK-8 assays (A, n = 4). Cell proliferation was examined by EdU staining. EdU, green; DAPI, blue. Scale bar, 20 μm (B, n = 4).
(C–G) STZ-induced diabetic mice received intravitreal injections of NC agomir, tRF-3001a agomir, NC shRNA, GSK3B shRNA, tRF-3001a plus GSK3B overexpression vector, or tRF-3001a plus null vector for 2 months. Retinal reactive gliosis (C; scale bar, 50 μm), RGC degeneration (D; scale bar, 50 μm), retinal vasopermeability (E and F; scale bar, 500 μm), and retinal acellular capillaries (G) were detected to evaluate the role of tRF-3001a-GSK3B signaling axis in retinal neurovascular dysfunction in vivo (n = 5). ∗p < 0.05 versus Ctrl group, #p < 0.05 between the marked groups; one-way ANOVA followed by post hoc Bonferroni test. See also Figures S10, S11, and S12.
Immunostaining assays were conducted to determine the role of tRF-3001a-GSK3B signaling axis in glial reactivity and RGC survival in STZ-induced diabetic mice and db/db diabetic mice. Compared with NC agomir group, intravitreal injection of tRF-3001a agomir could aggravate retinal reactive gliosis as shown by increased GFAP signaling (Figures 6C and S12A) and accelerated the degeneration of RGCs (Figures 6D and S12B) as shown by reduced number of TUJ1-positive cells. Silencing of GSK3B could mimic the effects of tRF-3001a agomir on retinal neurodegeneration, while overexpression of GSK3B could interrupt the detrimental effects of tRF-3001a agomir on retinal neuronal function as shown by decreased retinal glial reactivity and reduced RGC degeneration (Figures 6C, 6D, S12A, and S12B).
Evans blue assays and PAS assays revealed that intravitreal injection of tRF-3001a agomir aggravated retinal vascular dysfunction as shown by increased vascular leakage and increased number of acellular capillaries in STZ-induced diabetic mice and db/db diabetic mice. GSK3B silencing could mimic the effects of tRF-3001a agomir on retinal vascular dysfunction. By contrast, overexpression of GSK3B could interrupt the detrimental effects of tRF-3001a agomir on retinal vascular function (Figures 6E–6G and S12C–S12E). Together, these results indicate that tRF-3001a-GSK3B signaling axis is involved in retinal neurovascular dysfunction.
Clinical relevance of tRF-3001a-mediated signaling in neurovascular disease
To reveal the clinical relevance of tRF-3001a-mediated signaling, we compared the levels of tRF-3001a expression in the patients with DR and cataract. qRT-PCR assays revealed that tRF-3001a expression was obviously upregulated in AH samples of DR patients (Figure 7A). By contrast, GSK3B had an opposite expression pattern compared with tRF-3001a, which was downregulated in AH samples of DR patients (Figure 7B). Another cohort of patients, anti-VEGF treatment cohort, were selected to investigate the effects of anti-VEGF treatment on tRF-3001a expression. Anti-VEGF treatment led to reduced levels of tRF-3001a expression in AH samples of DR patients (Figure 7C). Human retinal vascular endothelial cells (HRVECs) or Müller cells were cultured with AH samples from DR patients. EdU assays revealed that pretreatment with AH samples led to increased proliferative ability. By contrast, tRF-3001a inhibitors significantly reduced the proliferation ability of HRVECs and Müller cells (Figures 7D and 7E).
Figure 7.

Clinical implication of tRF-3001a-mediated signaling in neurovascular disease
(A and B) Aqueous humor (AH) samples were collected from the patients with DR (n = 30 eyes) and the patients with cataract (Ctrl, n = 30 eyes). qRT-PCRs and ELISA assays were conducted to detect the levels of tRF-3001a and GSK3B expression. ∗p < 0.05 versus Ctrl group, Mann-Whitney U test.
(C) AH samples were collected from DR patients before (Ctrl group) or after anti-VEGF treatment. qRT-PCRs were conducted to detect the levels of tRF-3001a expression (n = 20 eyes). ∗p < 0.05 versus Ctrl group; Mann-Whitney U test.
(D and E) HRVECs or Müller cells were cultured with the AH samples from DR patients, AH samples plus negative control (NC) inhibitor, or AH samples plus tRF-3001a inhibitors or were left untreated (Ctrl) for 48 h. EdU assays were conducted to determine the role of tRF-3001a in cell proliferation (n = 4; ∗p < 0.05 versus Ctrl group, #p < 0.05 versus AH group; one-way ANOVA followed by post hoc Bonferroni test).
(F–H) C57BL/6J mice received intravitreal injections of AH samples from DR patients, AH samples plus NC antagomir, or AH samples plus tRF-3001a antagomir or were left untreated (Ctrl) for 14 days. Retinal vasopermeability (F; scale bar, 500 μm), reactive gliosis (G; scale bar, 50 μm), and RGC degeneration (H; scale bar, 50 μm) were examined to evaluate the role of tRF-3001a in retinal neurovascular dysfunction (n = 5, ∗p < 0.05 versus Ctrl group, #p < 0.05 versus AH group, one-way ANOVA followed by post hoc Bonferroni test). To visualize whole retinal vasculature in (F), the tiles-canning technique was used whereby multiple overlapping images were acquired using a 4X objective with similar gain settings. The composite images were constructed by arraying the individual images in Photoshop software. The representative composite images and statistical results are shown. See also Table S2.
The mice received intravitreal injections of PBS or AH samples from DR patients without or with tRF-3001a antagomir to induce retinal vascular dysfunction. Injection of AH samples from DR patients led to increased retinal vasopermeability, while injection of tRF-3001a antagomir significantly reduced AH sample-induced retinal vasopermeability (Figure 7F). Injection of AH samples from DR patients led to abnormal activation of glial cells and increased RGC degeneration in the murine retinas, whereas injection of tRF-3001a antagomir could reduce AH sample-induced retinal reactive gliosis and protect RGCs against AH sample-induced RGC degeneration (Figures 7G and 7H).
Discussion
Neurovascular dysfunction has emerged as a key clinical manifestation of DR, which is characterized by retinal vascular dysfunction and neurodegeneration.31 However, the mechanism of neurovascular dysfunction has not fully understood. Herein, we report that a tRNA-derived fragment, tRF-3001a, is obviously induced following diabetic stress. Inactivation of tRF-3001a-mediated signaling retards retinal vascular dysfunction, suppresses retinal reactive gliosis, protects RGCs against from diabetic injuries, and preserves visual function and visually guided behaviors. Overall, this study reveals that tRF-3001a is a promising target for neurovascular protection and vision preservation.
The retina is the extension of brain and has similar pathological mechanisms as central nervous system diseases. It can be observed noninvasively. In addition, due to the anatomical and functional features, retinal changes may precede those in the central nervous system, which makes the retina an appealing platform for studying neurodegeneration.32,33 The retina consists of a series of cell types, including neurons, vascular cells, microglia, and glial cells. In fact, these cells constitute the NVU and often face similar pathological stimuli under diabetic condition.34,35 We observe that long-term diabetes causes the dysfunction of retinal NVU, including retinal vascular dysfunction and neurodegeneration. As a sensor of hyperglycemia, tRF-3001a is obviously induced under diabetic condition in vitro and in vivo. tRF-3001a expression is also upregulated in the clinical samples of DR patients. Induction of tRF-3001a acts as an inducer of retinal glial activation and endothelial angiogenic effects, thereby causing retinal neurodegeneration, vascular leakage, and acellular capillary generation. Thus, reduction of tRF-3001a level is critical for controlling the progression of DR.
In the NVU, neuronal cells and vascular cells are tightly integrated and functionally coupled to meet energy demand.36 Intact NVU can ensure a homeostatic environment for neuronal function and protect them against different insults. NVU dysfunction is recognized as a major cause of vision impairment in DR.29 Induction of tRF-3001a is shown as a stress response upon diabetic stress. Increased tRF-3001a contributes to abnormal activation of ECs, showing excessive endothelial growth and increased vascular permeability. Vascular dysfunction can affect the clearance of harmful substances and contribute to retinal neuronal injury.37 Reduced tRF-3001a level will reconstitute the microenvironment of the NVU, alleviate retinal vascular dysfunction, and retard diabetes-induced complications.
Müller cells are the important components of the NVU in the retina. They are highly vulnerable and are easily activated following diabetic stress. During the pathogenesis of DR, retinal reactive gliosis is mainly mediated by Müller cells.22 A greater increase in tRF-3001a expression was detected in Müller cells than other retinal cells, suggesting that Müller cells are the most sensitive cells upon diabetic stress. Co-culture with Müller cells increases the proliferation, migration, and tube formation ability of ECs, showing a pro-angiogenic role of Müller cells in endothelial function. Meanwhile, co-culture with Müller cells aggravates high-glucose-induced RGC injuries, displaying a detrimental role of Müller cells in RGC function. tRF-3001a induction can lead to the abnormal activation of Müller cells. Transfection of tRF-3001a mimics in Müller cells plays a greater pro-angiogenic role in endothelial function and exerts a greater harmful role in RGC function in vitro. Moreover, intravitreal injection of tRF-3001a agomir aggravates retinal reactive gliosis and reduces the survival of RGCs. Thus, tRF-3001a-mediated retinal glial activation is a critical event in DR.
tRFs have been identified as a novel class of small non-coding RNAs. They have similar expression abundance as miRNAs and are highly conserved across different organisms.38 Previous studies have revealed that tRFs regulate gene expression at the post-transcriptional level in a miRNA-like mechanism or RNAi-like mechanism, causing the degradation or translational repression of target mRNAs.39,40 In this study, RIP assays, luciferase activity assays, and RNA pull-down assays have confirmed the direct interactions between tRF-3001a and GSK3B. GSK3B is a serine-threonine kinase and a negative regulator of glucose homeostasis. This gene has been reported to be involved in energy metabolism, inflammation, endoplasmic reticulum stress, mitochondrial dysfunction, and apoptotic pathways.41,42 We show that tRF-3001a-GSK3B axis can regulate retinal neurovascular dysfunction in vitro and in vivo. GSK3B overexpression reverses the effects of tRF-3001a mimics on Müller activation and reverses the effects of tRF-3001a mimics on endothelial angiogenic effects. Moreover, GSK3B overexpression partially interrupts tRF-3001a-mediated effects on glial cell reactivity and RGC survival and alleviates tRF-3001a-mediated retinal vascular leakage and acellular capillary generation. Thus, it is not surprising that tRF-3001a-GSK3B axis is involved in retinal neurovascular dysfunction.
Intraocular injection of anti-VEGF drugs, such as bevacizumab, ranibizumab, or aflibercept has revolutionized the therapy for DR. However, anti-VEGF drugs often have injection risks and unexpected adverse events. Furthermore, long-term injections can increase the chance of treatment complications, such as endophthalmitis, retinal detachment, high intraocular pressure, and hemorrhage.43,44 The discovery of gene silencing via RNA interference (RNAi) has revolutionized the way in which gene expression is regulated. Gene silencing via small RNAs has demonstrated great potential as the therapeutic strategy for eliminating disease gene expression.45 Herein, we report a novel regulatory mechanism underlying retinal neurovascular dysfunction, which is regulated by tRF-3001a-mediated signaling axis. Based on the signaling axis, we can design an inhibitor of tRF-3001a, which is a promising small molecular drug. Generally, the small molecular drug is cheaper than the great molecular antibody drugs. Moreover, the small molecular drug easily penetrates into intracellular targets and extracellular targets. Currently, several hurdles remain to be overcome before clinical adoption, including off-target effects, toxicity due to endogenous saturation and carrier, limited duration of silencing, and effective targeted delivery.46 In the future, we will improve small RNA delivery by designing novel carriers or conducting chemical modification of small RNA molecules to reduce the potential off-target effects.
Taken together, this study not only provides the insights into post-transcriptional regulatory mechanism of DR but also offers the clue for the unanswered questions such as which molecules act as the sensor for diabetic stress and which molecules can coordinately regulate retinal vascular dysfunction and neurodegeneration. Furthermore, a great improvement in visual function and visually guided behavior in tRF-3001a-deficient retinas suggests that targeting tRF-3001a is a promising strategy for treating retinal vasculopathy and neuropathy. Future efforts might be directed at designing drugs for tRF-3001a-mediated signaling and determining the roles for treating ocular neurovascular disorders.
Limitations of the study
Although the result that tRF-3001a regulates retinal neurovascular dysfunction is encouraging, there are still some limitations in this study. tRF-3001a is expressed mainly in Müller cells, and the most obvious upregulation of tRF-3001a is also observed in Müller cells following diabetic stress. However, nonspecific knockdown or overexpression not only affects tRF-3001a expression in Müller cells but also in other retinal cells. The possible off-target effects are required to be examined to avoid the overestimation of tRF-3001a function resulting from nonspecific intervention or incomplete inhibition. In addition, we report the potential role of tRF-3001a for the concurrent treatment of vasculopathy and neuropathy in diabetes mellitus. However, we should not ignore off-target effects caused by the unintended interactions between small RNAs and cell components, potential immune responses, or saturation effects of endogenous RNA interference. Designing a novel RNA delivery system and improving the modifications of small RNAs are still required to reduce the potential off-target effects.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-GFAP | Abcam | Cat# ab68428; RRID:AB_1209224 |
| Rabbit anti-GS | Abcam | Cat# ab228590 |
| Rabbit anti-NeuN | Abcam | Cat# ab177487; RRID:AB_2532109 |
| Rabbit anti-TUJ1 | Abcam | Cat# ab18207; RRID:AB_444319 |
| Rabbit anti-PKCα | Abcam | Cat# ab32376; RRID:AB_777294 |
| Mouse anti-Rhodopsin | Abcam | Cat# ab5417; RRID:AB_304874 |
| Mouse anti-Calbindin | Santa Cruz | Cat# sc-365360; RRID:AB_10841576 |
| Mouse anti-Calretinin | Santa Cruz | Cat# sc-365956; RRID:AB_10846469 |
| Rabbit anti-Angiogenin | proteintech | Cat# 18302-1-AP; RRID:AB_2274028 |
| Rabbit anti-Dicer | abcam | Cat# ab227518 |
| Rabbit anti-β-Αctin | ZEN BIO | Cat# 380624; RRID:AB_2893488 |
| Alexa Fluor 488 goat anti-mouse IgG | Thermo Fisher Scientific | Cat# A32723; RRID:AB_2633275 |
| Alexa Fluor 594 goat anti-mouse IgG | Thermo Fisher Scientific | Cat# A-11005; RRID:AB_2534073 |
| Alexa Fluor 488 goat anti-rabbit IgG | Thermo Fisher Scientific | Cat# A-11008; RRID:AB_143165 |
| Alexa Fluor 594 goat anti-rabbit IgG | Thermo Fisher Scientific | Cat# A-11012; RRID:AB_2534079 |
| Biological samples | ||
| Fibrovascular membranes tissue samples from DR patients | the Affiliated Eye Hospital of Nanjing Medical University | N/A |
| Epiretinal membranes tissue samples from patients with idiopathic macular holes | the Affiliated Eye Hospital of Nanjing Medical University | N/A |
| Aqueous humor samples from DR patients | the Affiliated Eye Hospital of Nanjing Medical University | N/A |
| Aqueous humor samples from cataract patients | the Affiliated Eye Hospital of Nanjing Medical University | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 3000 | Life Technologies | Cat# 13778150 |
| STZ | Biofroxx | Cat# 2196GR001 |
| Evans blue dye | Sigma Aldrich | Cat# E2129 |
| Trypsin | BioFroxx | Cat# 1004GR025 |
| Glycogen Periodic Acid Schiff (PAS/Hematoxylin) Stain Kit | Solarbio | Cat# G1281 |
| OCT medium | Thermo Scientific | Cat# 6502 |
| Papain Dissociation System | Worthington | Cat# K003150 |
| DMEM | Gibco | Cat# 21013024 |
| FBS | Gibco | Cat# 10099141 |
| 0.025% trypsin | Sigma | Cat# T9201 |
| Insulin | Sigma-Aldrich | Cat# I6634 |
| Progesterone | Sigma-Aldrich | Cat# P0130 |
| Selenite | Sigma-Aldrich | Cat# S5261 |
| Transferrin | Sigma-Aldrich | Cat# T8158 |
| Forskolin | Sigma-Aldrich | Cat# F6886 |
| CNTF | PeproTech | Cat# 450-13 |
| TRIzol | Life Technologies | Cat# 15596026 |
| PowerUp™ SYBR™ Green Master Mix | Thermo Fisher Scientific | Cat# A25742 |
| CCK-8 | Beyotime | Cat# C0038 |
| Calcein-AM | AAT Bioquest | Cat# 22002 |
| PI | Biofroxx | Cat# 1246MG100 |
| Rhodamine123 | Solarbio | Cat# R8030 |
| Methyl cellulose solution | Sigma-Aldrich | Cat# M0512-100G |
| Basement membrane matrix | Coring | Cat# 356234 |
| Critical commercial assays | ||
| rtStar™ tRF and tiRNA pretreatment kit | Arraystar | Cat# AS-FS-005 |
| rtStar™ First-strand cDNA Synthesis kit | Arraystar | Cat# AS-FS-003 |
| SuperScript IV First-Strand Synthesis System | Thermo Fisher Scientific | Cat# 18091050 |
| BeyoClick™EdU detection kit | Beyotime | Cat# C0071S |
| In situ Cell Death Detection Kit | Roche | Cat# 12156792910 |
| Experimental models: Cell lines | ||
| HRVEC | CSC | N/A |
| human retinal pericytes | CSC | N/A |
| ARPE-19 | ATCC | RRID:CVCL_0145 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice | Animal Core Facility of Nanjing Medical University | N/A |
| BKS-Leprem2Cd479/Gpt (db/db) mice | GemPharmatech | strain NO.T002407 |
| Oligonucleotides | ||
| Primers for qRT-PCR, see Table S4 | Generay | N/A |
| Sequence information for the relevant RNAs, see Table S3 | RiboBio | N/A |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/cn/ |
| ImageJ | National Institutes of Health (NIH) | https://imagej.net/ij/index.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Biao Yan (biao.yan@fdeent.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Clinical sample collection
The clinical samples were obtained from 2020 to 2022 at the Affiliated Eye Hospital of Nanjing Medical University. The clinical studies conformed to the Declaration of Helsinki. The usage of patient samples was approved by the Ethical Committee of the Affiliated Eye Hospital, Nanjing Medical University. All involved patients were given the written informed consents prior to clinical sample collection. The fibrovascular membranes were collected from the patients with proliferative DR. The epiretinal membranes were collected from the patients with idiopathic macular holes (as control) at the time of pars plana vitrectomy. The relevant information about the patients is shown in Table S1.
Aqueous humor (AH) samples were obtained from DR patients (n = 30). and cataract patients (n = 30). About 100 μL of AH samples were collected under a surgical microscope with a sterile syringe to avoid the injurires to iris and anterior lens capsule to prevent protein contamination. The samples were cooled in ice, centrifuged for 15 min at 4°C to remove cells, and stored in liquid nitrogen. The inclusion criteria: (1) the patients diagnosed as DR and required vitrectomy; (2) without intravitreal anti-VEGF or steroid injections within six months. The exclusion criteria: (1) retinal diseases besides DR, e.g., retinal vein occlusion and neovascular AMD; (2) other ocular diseases such as glaucoma; (3) infection and intraocular inflammation; and (4) intraocular surgery during the previous 6 months. The relevant information about the patients is shown in Table S2.
Cell isolation and culture
Human retinal Müller cells were obtained from 4 to 16 h postmortem donor eyes. None of the donors had a known history of ocular diseases. Primary mouse Müller cells were isolated from the postnatal day 11–12 mice. Briefly, the enucleated eyes were held in DMEM medium on ice. The retinas were detached from the posterior eyeballs. The dissected retinas were minced with a micro-scissor. Retinal tissues were dissociated using the Worthington Papain Dissociation System. Subsequently, retinal cells were passed through a 30-μm cell strainer and cultured at 37°C. The medium was replaced every 3 days. Cell purity was identified by cell morphology and immunostaining with glial acidic fibrillary protein (GFAP) and glutamine synthetase (GS). Primary Müller cells were cultured in the medium containing DMEM, 1% GlutaMax, 1% Penicillin/Streptomycin, and 10% FBS.
Primary RGCs were isolated from C57BL/6 mouse pups at postnatal days 0–3. The collected retinas were dissociated with papain solution (15 unit/ml) and collagenase (70 unit/ml) for 15 min. The suspensions of retinal cells were incubated with anti-macrophage serum (1:100) for 1 h at 37°C to eliminate macrophages and microglial cells. The non-adherent cells were transferred to 100-mm petri dishes pre-conjugated with anti-Thy1.2 antibody to purify RGCs for 1 h at 37°C. The dishes were then rinsed with PBS to remove the non-adherent cells. After a rinse in the panning buffer (Dulbecco’s-PBS, 0.02% bovine serum albumin, 5 μg/mL insulin), the adherent RGCs were released from the plate by incubation with 0.025% trypsin for 5 min at 37°C and seeded at a density of 2.0×105 cells/well. The purity of RGCs was determined by staining with TUJ1 antibody. Primary RGCs were cultured in DMEM medium supplemented with insulin (2.0 mM), progesterone (40 nM), selenite (60 nM), transferrin (100 nM), CNTF (40 ng/mL), and forskolin (6 μM).
Human retinal vascular endothelial cells (HRVECs) and human retinal pericytes were obtained from Cell System Crop (CSC, Kirkland, WA, USA). Human retinal pigment epithelial cell line (ARPE-19 cells) were obtained from ATCC (Lot Number 63478793; Manassas, VA). HRVECs were cultured in endothelial growth medium (EGM2-MV) supplemented with 5% fetal bovine serum (FBS). Pericytes were cultured in DMEM medium supplemented with 10% FBS. ARPE-19 cells were cultured in DMEM/F-12 medium supplemented with 10% FBS.
Mouse models
The animal experiments were approved by the Animal Ethics and Experimentation Committee of Nanjing Medical University. The animals were breed according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Great efforts were made to minimize the pain, suffering, and discomforts to the animals. Eight-week-old male C57BL/6J mice (weighing 20–25 g) were obtained from the Animal Core Facility of Nanjing Medical University (Nanjing, Jiangsu, China). They were fasted for 6 h and received an intraperitoneal injection of streptozotocin (STZ, 50 mg/kg) over 5 days47 The weight and age-matched controls were injected with the equivalent amount of buffer. The level of blood glucose was measured with the One Touch Ultra Glucometer. Diabetes induction was confirmed when blood glucose levels were ≥16.7 mmol/L. Three-month-old male BKS-Leprem2Cd479/Gpt (db/db) mice were obtained from GemPharmatech (Nanjing, China). The fasting blood glucose levels of db/db mice were ≥16 mmol/L. Age-matched heterozygotes mice without diabetes were used as the controls (db/m). They were housed in a controlled environment: 12-h light/dark cycle, room temperature of 25 ± 2°C, and humidity of 50 ± 10%.
Method details
Evans blue dye assay
Retinal vascular permeability was determined by measuring the leakage of Evans blue dye from retinal vessels.48 Briefly, Evans blue (45 mg/kg) was injected via femoral vein and circulated for 2 h. An 18-gauge cannula was inserted through the ventricle into the ascending aorta to flush intravascular dye. The cornea, lens, and vitreous humor were removed. Then, the retinas were dissociated. One eye from each mouse was used for the detection of Evans blue fluorescence and the other eye was used for the extraction of Evans blue dye. The retinas were fixed in 4% PFA in PBS for 30 min at room temperature, and then dried for 5 h. Evans blue dye was extracted by incubating the retina in formamide (0.2 mL per retina) for 12 h at 78°C. Then, the resulting suspensions were centrifuged at 12,000 g for 45 min. The absorbance at 620 and 740 nm (background) was measured by a spectrophotometer. Each sample was measured 3 times and averaged. The amount of Evans blue dye was calculated from the standard curve and normalized using the following formula.
| Retinal Evans blue concentration (mg/mL)/retinal weight (mg) |
| Blood Evans blue concentration (mg/mL) × circulation time (h) |
Retinal trypsin digestion
The eyes were enucleated, fixed in 10% neutral formaldehyde for 24 h, and washed in the distilled water for 30 min. The retinas were digested with 3% trypsin (1:250) at 37°C for 1 h and shaken gently until the translucent vascular network was observed. The samples were air-dried overnight and stained with periodic acid-Schiff and hematoxylin. Acellular capillaries were counted in 15 randomly selected fields using a fluorescence microscope (Olympus IX 73 DP80, Japan).49
Immunofluorescent staining
The eyes were cryoprotected in 30% sucrose in 0.1 M phosphate-buffered saline solution (PBS) for 48 h. Then, the eyecups were embedded with the Optimal Cutting Temperature (OCT) medium and 10-μm tissue sections were collected with a cryostat (Thermo Scientific). After blocking with 5% BSA for 1 h, the sections were incubated with the primary antibody overnight at 4°C. After incubation, the sections were washed 3 times in PBS for 5 min. Then, the sections were incubated with the fluorophore-conjugated secondary antibody for 2 h at room temperature. Finally, the sections were counterstained with DAPI and observed under a fluorescence microscope (Olympus IX 73 DP80, Japan).
Whole flat-mount staining
The eyes were enucleated and fixed in 4% paraformaldehyde (PFA) for 1 h. After washing with PBS, the anterior segment and neuroretina were removed. The retinas were incubated in 4% PFA for additional 30 min and dissected into the petal shape as whole flat-mount. The intact retinas were blocked with 5% BSA for 1 h and incubated with TUJ1 antibody overnight at 4°C. After incubation, the retinas were washed 3 times in PBS for 5 min each time and incubated with the Alexa Fluor 488 goat anti-mouse IgG (1:500) for 2 h at room temperature. The images were captured from the peripheral areas. TUJ1-positive cells were counted using ImageJ software and RGC survival rate was calculated by dividing average number of TUJ1 positive cells in one field in the injured retina by that in the uninjured retina. Only cells in ganglion cell layer were counted.
Intravitreal injection
The mice were randomly divided into six groups: (1) normal control group; (2) DR group; (3) DR + NC agomir group, diabetic eyes with 2 μg of NC agomir injection; (4) DR+tRF-3001a agomir group, diabetic eyes with 2 μg of tRF-3001a agomir injection; (5) DR + NC antagomir group, diabetic eyes with 2 μg of NC antagomir injection; (6) DR+tRF-3001a antagomir group, diabetic eyes with 2 μg of tRF-3001a antagomir injection. The mice were anesthetized with the mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg). Then, topical 1% tropicamide (Akorn Pharmaceuticals, Illinois, USA) and 0.3% Genteal (Novartis, Texas, USA) was applied to the cornea. The injections were conducted once a month using a 33-gauge needle (Hamilton Bonaduz AG, Bonaduz, Switzerland). The needle was inserted from the limbus with a 45° injection angle into the vitreous, avoiding the lens and directing needle tip to vitreous cavity around optic nerve head. To prevent infection, ofloxacin ointment (Santen, Osaka, Japan) was applied.
Cell transfection
tRF-3001a mimics, tRF-3001a inhibitors, and their corresponding negative control were purchased from RiboBio Co., Ltd. (Guangzhou, China). When the confluence reached about 80% confluence, these cells were transfected with the mimics/inhibitors or the corresponding negative controls using Lipofectamine 3000 (Life Technologies, Waltham, MA) according to the manufacturer’s protocol. The sequences are shown in Table S3.
Cell co-culture assay
Cell co-culture system was used to detect the crosstalk between Müller cells and HRVECs or the crosstalk between Müller cells and RGCs. Briefly, HRVECs or RGCs at the bottom chamber and Müller cells on the top chamber were separated in a transwell culture system (0.4-μm pore size; Millipore, PTHT24H48). HRVECs (2×105 cells/well) or RGCs (2.5×105 cells/well) were seeded in 24-well plates with 500 μL of specific cell medium. After these cells adhered for 6–8 h, the medium was changed into 1 mL of DMEM/F12 with 10% FBS. Then, Müller cells (1×105 cells/well) were cultured on the top chamber and co-cultured with HRVECs or RGCs at 37°C in 95% humidified atmosphere containing 5% CO2 for an additional 24 h.
RNA extraction and quantitative reverse transcription-PCR (qRT-PCR)
Total RNAs were isolated from the retinas, cells, or clinical samples using TRIzol reagent. RNA integrity was determined using the NanoDrop ND-1000 Spectrophotometer (Agilent, USA) and the concentration of RNAs was determined by a spectrophotometer at 260 nm. To detect tRF-3001a expression, the rtStar tRF and tiRNA pretreatment kit was used to remove RNA modification. Then, rtStar First-strand cDNA Synthesis kit was used to reversely transcribe RNAs into cDNAs. For other genes, RNA samples were reversely transcribed into cDNAs with the SuperScript IV First-Strand Synthesis System. qPCR assays were conducted using the PowerUp SYBR Green Master Mix: 95°C for 1 min followed by 42 cycles of 95°C for 15 s and 60°C for 30 s tRF-3001a expression was normalized by U6 and other genes were normalized by β-actin. 2−ΔΔCt relative expression method was used to detect relative gene expression. The primer sequences were shown in Table S4.
Cell Counting Kit-8 (CCK-8) assay
Cell viability was determined by CCK-8 assay. Briefly, the cells were seeded into 96-well plates (1×105 cells/well). After the required treatment, 10 μL of 10% CCK-8 reagent was added to each well and incubated at 37°C for 1.5 h. The absorbance was measured in triplicates at the wavelength of 450 nm by a microplate reader (Molecular Devices, FilterMax F5).
5-Ethynyl-2′-deoxyuridine (EdU) assay
To detect cell proliferation, EdU assays were conducted using the BeyoClick EdU detection kit. About 2.0×105 cells were seeded onto the 24-well plates and incubated with 50 mM of EdU medium for 2 h at 37°C. Then, EdU assays were conducted according to the manufacturer’s instruction. Finally, cell nuclei were stained with DAPI (0.5 μg/mL). All images were captured under a fluorescent microscope (Olympus IX-73). Cell proliferation was evaluated by the percentage of EdU-positive cells using ImageJ.
Calcein-AM and propidium iodide (PI) double staining
Calcein-AM is membrane permeable and can stain the living cells, whereas PI is membrane-impermeable and can stain the dead cells. After the required treatment, the cells were incubated with 10 μM of Calcein-AM and PI solution at 37°C for 20 min in the dark. After washing with PBS buffer, the living cells (green) were observed at 490 nm excitation filter, while the dead cells (red) were observed at 545 nm excitation filter. The images were captured under an Olympus IX-73 microscope.
TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay
Cell apoptosis was detected by an In situ Cell Death Detection Kit. Briefly, the cells were seeded on the 24-well plate at a density of 1 × 105 cells/well. After high glucose treatment, they were stained with TUNEL (5 μM) according to the manufacturer’s instruction. Next, DAPI was used for nuclei staining. TUNEL-positive cells were observed by an Olympus IX-73 fluorescence microscopy.
Rhodamine 123 staining
The mitochondrial membrane potential (ΔΨm) was determined by Rhodamine 123 staining. Briefly, the cells were seeded on the 24-well plates (2×105 cells/well), incubated with Rhodamine123 (10 μM) at 37°C for 30 min in the dark, and washed with PBS twice. The fluorescent images were captured by an Olympus IX-73 microscope.
Cell migration assay
HRVECs (1×105 cells) were plated in the transwell insert (8-μm pore size; Millipore, PTEP24H48) in a 24-well plate. The upper chamber was loaded in 100 μL of serum-free culture medium and the bottom of the lower chamber was filled with 600 μL of DMEM plus 10% FBS. After 14 h incubation, the transwell chambers were fixed with 10% methanol for 15 min and stained with 0.1% crystal violet for 20 min at room temperature. Then, these cells on the upper side of the membranes were gently removed using a cotton swab. All images were observed under a light microscope.
Endothelial cell sprouting assay
HRVECs (8×104 cells) were resuspended in 5 mL of methyl cellulose solution (2.5 mg/mL) in EGM2-MV supplemented with 2% FBS. 25 μL of cell suspensions were pipetted onto a 15 cm culture dish (JET BIOFIL, TCD010150). To form the spheroids, the culture dishes were turned upside-down and incubated at 37°C and 5% CO2 for 24 h. The spheroids were embedded in a collagen matrix containing VEGF-A (25 ng/mL) and FGF2 (25 ng/mL) and cultured at 37°C and 5% CO2 for 24 h. The length of all spouts per spheroid was calculated by software analysis (FIJI/ImageJ).
Tube formation assay
HRVECs (2.5×105/well) were seeded onto 24-well plate pre-coated with the basement membrane matrix. They were cultured at 37°C in a humidified incubator supplied with 5% CO2 for 12 h. Tube formation was observed under an Olympus IX-73 microscope and analyzed by the Angiogenesis Analyzer plug-in on ImageJ software.
Electroretinogram (ERG) assay
After dark adaptation for 12 h, the mice were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg). Then, their pupils were dilated with 1% tropicamide. A recording electrode was placed on the central cornea. The reference needle electrode was placed cutaneously around the eye and a ground electrode was attached to the tail. To avoid drying and optimize the electrical recording, a topical drop (1% carboxymethylcellulose) was applied to the cornea. After recording a scotopic intensity series, the photopic flash responses were measured with the increased light stimuli. The amplitude of b-wave was measured from the trough of a-wave to the peak of b-wave and b-wave latency were also obtained. ERG recordings were conducted using the Roland Consult Color Ganzfeld Q450C recording machine.
Visual cliff test
Visual cliff test was conducted to evaluate the discrimination ability of visual depth. Two clear plexiglass boxes (40 cm × 20 cm × 50 cm) were placed side by side. A glass plate was placed on the plexiglass box as the platform. A visual cliff was formed using the blank-and-white checkered paper (checker size: 2.5 cm × 2.5 cm). In addition, a center platform (40 cm × 4 cm × 4 cm) was taped in the middle to form a cliff side or a safe side. The mice were placed on the center platform and the choices to cliff side or safe side were recorded. Each mouse was given 10 tests for 10-min trial. The boxes and central platform were cleaned after each trial. Finally, the number of times that the mice chose shallow safe side or deep cliff side was counted and converted into the selection percentage.
Morris water maze experiment
Morris water maze experiment was carried out to detect visual discrimination acuity and spatial cognitive function. A circular pool (120 cm in diameter) was filled with water. The depth of water was 45 cm and water temperature was maintained between 22°C and 25°C. The visible escape platform (8 cm × 8 cm) was located at 1–2 cm higher than water surface with a red flag to increase the visibility. The test was performed for 8 consecutive days: 7-day training and 1-day test. On the training day, the mouse was placed into the water for 5 min in advance for water adaptation. Then, the mouse was required to search for the platform within 1 min. The time required until the mouse found the platform was recorded. If the mouse failed to find the platform within 1 min, it was guided manually to reach at the platform and the escape time was regarded as 1 min. After resting on the platform for 15 s, the mouse was trained again. Five times were conducted on the training day. On day 8, only one test was conducted and the escape time was recorded.
Dark light preference test
The apparatus consisted of a light, open topped, opaque, plexiglas box (30 × 40 × 30 cm), and a smaller black, closed topped, opaque, plexiglas dark box (30 × 20 × 30 cm) containing a small opening (5 cm × 5 cm) for mouse to freely pass. The light chamber was brightly illuminated by a 100 W desk lamp, whereas the black chamber had no illumination. Before the experiment, each mouse was placed in the light/dark box in the dark for 12 h and allowed to explore freely for 10 min in the dark box for environment adaption. Next, the mouse was placed in the light/dark box with its head facing the dark chamber. The times of entering dark chamber within 5 min was recorded. After each test, the odor was eliminated by alcohol spraying.
Quantification and statistical analysis
Statistical analysis was conducted using the GraphPad Prism 8 software (GraphPad Software, San Diego, CA). All data were expressed as means ± standard deviation (SD). For the normally distributed data, Student’s t test was conducted for pairwise comparison and one-way analysis of variance (ANOVA) followed by post hoc Bonferroni test was conducted for multiple group comparisons. For the non-normally distributed data, Mann-Wallis U test was conducted for pairwise comparison and Kruskal-Wallis test followed by post hoc Bonferroni test was conducted for multiple group comparisons. p < 0.05 was considered statistically significant.
Acknowledgments
This study was funded by grants from the National Natural Science Foundation of China (nos. 82225013 and 81970809 to B.Y. and nos. 81570859 and 82070983 to Q.J.).
Author contributions
B.Y. and Q.J. designed this study. J.-Y.Z., W.Y., X.-S.N., M.-D.Y., W.B., T.-J.Y., and Z.-R.Z. performed the experiments, prepared figures, and drafted the manuscript; J.-Y.Z., W.Y., and X.-S.N. developed the methods and data analysis; J.-Y.Z., W.Y., and M.-D.Y. acquired and interpreted the data; J.-Y.Z. and B.Y. wrote and revised the paper. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: September 26, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101209.
Contributor Information
Qin Jiang, Email: jiangqin710@126.com.
Biao Yan, Email: biao.yan@fdeent.org.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Goedeke L., Perry R.J., Shulman G.I. Emerging pharmacological targets for the treatment of nonalcoholic fatty liver disease, insulin resistance, and type 2 diabetes. Annu. Rev. Pharmacol. 2019;59:65–87. doi: 10.1146/annurev-pharmtox-010716-104727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cole J.B., Florez J.C. Genetics of diabetes mellitus and diabetes complications. Nat. Rev. Nephrol. 2020;16:377–390. doi: 10.1038/s41581-020-0278-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mauricio D., Alonso N., Gratacòs M. Chronic diabetes complications: the need to move beyond classical concepts. Trends Endocrinol. Metabol. 2020;31:287–295. doi: 10.1016/j.tem.2020.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Feldman E.L., Callaghan B.C., Pop-Busui R., Zochodne D.W., Wright D.E., Bennett D.L., Bril V., Russell J.W., Viswanathan V. Diabetic neuropathy. Nat. Rev. Dis. Prim. 2019;5:41. doi: 10.1038/s41572-019-0092-1. [DOI] [PubMed] [Google Scholar]
- 5.Salas-Salvadó J., Becerra-Tomás N., Papandreou C., Bulló M. Dietary patterns emphasizing the consumption of plant foods in the management of type 2 diabetes: a narrative review. Adv. Nutr. 2019;10:S320–S331. doi: 10.1093/advances/nmy102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nauck M.A., Wefers J., Meier J.J. Treatment of type 2 diabetes: challenges, hopes, and anticipated successes. Lancet Diabetes Endocrinol. 2021;9:525–544. doi: 10.1016/S2213-8587(21)00113-3. [DOI] [PubMed] [Google Scholar]
- 7.Cavalli G., Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571:489–499. doi: 10.1038/s41586-019-1411-0. [DOI] [PubMed] [Google Scholar]
- 8.Ling C., Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metabol. 2019;29:1028–1044. doi: 10.1016/j.cmet.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kowluru R.A., Mohammad G. Epigenetic modifications in diabetes. Metabolism. 2022;126 doi: 10.1016/j.metabol.2021.154920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esteller M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 11.Soares A.R., Santos M. Discovery and function of transfer RNA-derived fragments and their role in disease. WIREs. RNA. 2017;8:e1423. doi: 10.1002/wrna.1423. [DOI] [PubMed] [Google Scholar]
- 12.Kim H.K., Fuchs G., Wang S., Wei W., Zhang Y., Park H., Roy-Chaudhuri B., Li P., Xu J., Chu K., et al. A transfer-RNA-derived small RNA regulates ribosome biogenesis. Nature. 2017;552:57–62. doi: 10.1038/nature25005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J., Zhu L., Cheng J., Peng Y. Transfer RNA-derived small RNA: A rising star in oncology. Semin. Cancer Biol. 2021;75:29–37. doi: 10.1016/j.semcancer.2021.05.024. [DOI] [PubMed] [Google Scholar]
- 14.Yang W., Gao K., Qian Y., Huang Y., Xiang Q., Chen C., Chen Q., Wang Y., Fang F., He Q., et al. A novel tRNA-derived fragment AS-tDR-007333 promotes the malignancy of NSCLC via the HSPB1/MED29 and ELK4/MED29 axes. J. Hematol. Oncol. 2022;15:53. doi: 10.1186/s13045-022-01270-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodarzi H., Liu X., Nguyen H.C.B., Zhang S., Fish L., Tavazoie S.F. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell. 2015;161:790–802. doi: 10.1016/j.cell.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puhakka N., Das Gupta S., Vuokila N., Pitkänen A. Transfer RNA-derived fragments and isomiRs are novel components of chronic TBI-induced neuropathology. Biomedicines. 2022;10:136. doi: 10.3390/biomedicines10010136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma U., Conine C.C., Shea J.M., Boskovic A., Derr A.G., Bing X.Y., Belleannee C., Kucukural A., Serra R.W., Sun F., et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–396. doi: 10.1126/science.aad6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benoit S.R., Swenor B., Geiss L.S., Gregg E.W., Saaddine J.B. Eye care utilization among insured people with diabetes in the U.S. Diabetes Care. 2019;42:427–433. doi: 10.2337/dc18-0828. [DOI] [PubMed] [Google Scholar]
- 19.Vujosevic S., Aldington S.J., Silva P., Hernández C., Scanlon P., Peto T., Simó R. Screening for diabetic retinopathy: new perspectives and challenges. Lancet Diabetes Endocrinol. 2020;8:337–347. doi: 10.1016/s2213-8587(19)30411-5. [DOI] [PubMed] [Google Scholar]
- 20.Sohn E.H., van Dijk H.W., Jiao C., Kok P.H.B., Jeong W., Demirkaya N., Garmager A., Wit F., Kucukevcilioglu M., van Velthoven M.E.J., et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. USA. 2016;113:E2655–E2664. doi: 10.1073/pnas.1522014113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segarra M., Aburto M.R., Cop F., Llaó-Cid C., Härtl R., Damm M., Bethani I., Parrilla M., Husainie D., Schänzer A., et al. Endothelial Dab1 signaling orchestrates neuro-glia-vessel communication in the central nervous system. Science. 2018;361 doi: 10.1126/science.aao2861. [DOI] [PubMed] [Google Scholar]
- 22.Simó R., Stitt A.W., Gardner T.W. Neurodegeneration in diabetic retinopathy: does it really matter? Diabetol. 2018;61:1902–1912. doi: 10.1007/s00125-018-4692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammes H.P. Diabetic retinopathy: hyperglycaemia, oxidative stress and beyond. Diabetol. 2018;61:29–38. doi: 10.1007/s00125-017-4435-8. [DOI] [PubMed] [Google Scholar]
- 24.Vecino E., Rodriguez F.D., Ruzafa N., Pereiro X., Sharma S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016;51:1–40. doi: 10.1016/j.preteyeres.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Fowler M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes. 2011;29:116–122. doi: 10.2337/diaclin.29.3.116. [DOI] [Google Scholar]
- 26.Mrugacz M., Bryl A., Zorena K. Retinal vascular endothelial cell dysfunction and neuroretinal degeneration in diabetic patients. J. Clin. Med. 2021;10:458. doi: 10.3390/jcm10030458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spaide R.F. Measurable aspects of the retinal neurovascular unit in diabetes, glaucoma, and controls. Am. J. Ophthalmol. 2019;207:395–409. doi: 10.1016/j.ajo.2019.04.035. [DOI] [PubMed] [Google Scholar]
- 28.Simó R., Hernández C., European Consortium for the Early Treatment of Diabetic Retinopathy EUROCONDOR Neurodegeneration in the diabetic eye: new insights and therapeutic perspectives. Trends Endocrinol. Metabol. 2014;25:23–33. doi: 10.1016/j.tem.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Ji L., Tian H., Webster K.A., Li W. Neurovascular regulation in diabetic retinopathy and emerging therapies. Cell. Mol. Life Sci. 2021;78:5977–5985. doi: 10.1007/s00018-021-03893-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chekulaeva M., Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009;21:452–460. doi: 10.1016/j.ceb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Fu Z., Sun Y., Cakir B., Tomita Y., Huang S., Wang Z., Liu C.-H., S Cho S., Britton W., S Kern T., et al. Targeting neurovascular interaction in retinal disorders. Int. J. Mol. Sci. 2020;21:1503. doi: 10.3390/ijms21041503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stefánsson E., Olafsdottir O.B., Einarsdottir A.B., Eliasdottir T.S., Eysteinsson T., Vehmeijer W., Vandewalle E., Bek T., Hardarson S.H. Retinal oximetry discovers novel biomarkers in retinal and brain diseases. Invest. Ophthalmol. Vis. Sci. 2017;58:BIO227–BIO233. doi: 10.1167/iovs.17-21776. [DOI] [PubMed] [Google Scholar]
- 33.London A., Benhar I., Schwartz M. The retina as a window to the brain—from eye research to CNS disorders. Nat. Rev. Neurol. 2013;9:44–53. doi: 10.1038/nrneurol.2012.227. [DOI] [PubMed] [Google Scholar]
- 34.Metea M.R., Newman E.A. Signaling within the neurovascular unit in the mammalian retina. Exp. Physiol. 2007;92:635–640. doi: 10.1113/expphysiol.2006.036376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng Y., Busch S., Gretz N., Hoffmann S., Hammes H.-P. Crosstalk in the retinal neurovascular unit-lessons for the diabetic retina. Exp. Clin. Endocrinol. Diabetes. 2012;120:199–201. doi: 10.1055/s-0032-1304571. [DOI] [PubMed] [Google Scholar]
- 36.Andreone B.J., Lacoste B., Gu C. Neuronal and vascular interactions. Annu. Rev. Neurosci. 2015;38:25–46. doi: 10.1146/annurev-neuro-071714-033835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feit-Leichman R.A., Kinouchi R., Takeda M., Fan Z., Mohr S., Kern T.S., Chen D.F. Vascular damage in a mouse model of diabetic retinopathy: relation to neuronal and glial changes. Invest. Ophthalmol. Vis. Sci. 2005;46:4281–4287. doi: 10.1167/iovs.04-1361. [DOI] [PubMed] [Google Scholar]
- 38.Sobala A., Hutvagner G. Transfer RNA-derived fragments: origins, processing, and functions. WIREs RNA. 2011;2:853–862. doi: 10.1002/wrna.96. [DOI] [PubMed] [Google Scholar]
- 39.Kim H.K., Yeom J.-H., Kay M.A. Transfer RNA-derived small RNAs: another layer of gene regulation and novel targets for disease therapeutics. Mol. Ther. 2020;28:2340–2357. doi: 10.1016/j.ymthe.2020.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan L., Huang X., Liu Z.-X., Ye Y., Li R., Zhang J., Wu G., Bai R., Zhuang L., Wei L., et al. Inflammatory cytokine-regulated tRNA-derived fragment tRF-21 suppresses pancreatic ductal adenocarcinoma progression. J. Clin. Invest. 2021;131 doi: 10.1172/jci148130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen W., Taylor B., Jin Q., Nguyen-Tran V., Meeusen S., Zhang Y.-Q., Kamireddy A., Swafford A., Powers A.F., Walker J., et al. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat. Commun. 2015;6:8372. doi: 10.1038/ncomms9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryu H.Y., Kim L.E., Jeong H., Yeo B.K., Lee J.-W., Nam H., Ha S., An H.-K., Park H., Jung S., et al. GSK3B induces autophagy by phosphorylating ULK1. Exp. Mol. Med. 2021;53:369–383. doi: 10.1038/s12276-021-00570-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bahrami B., Hong T., Gilles M.C., Chang A. Anti-VEGF therapy for diabetic eye diseases. Asia. Pac. J. Ophthalmol. 2017;6:535–545. doi: 10.22608/apo.2017350. [DOI] [PubMed] [Google Scholar]
- 44.Simó R., Hernández C. Intravitreous anti-VEGF for diabetic retinopathy: hopes and fears for a new therapeutic strategy. Diabetol. 2008;51:1574–1580. doi: 10.1007/s00125-008-0989-9. [DOI] [PubMed] [Google Scholar]
- 45.Jackson A.L., Linsley P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 46.Singh S., Narang A.S., Mahato R.I. Subcellular fate and off-target effects of siRNA, shRNA, and miRNA. Pharm. Res. (N. Y.) 2011;28:2996–3015. doi: 10.1007/s11095-011-0608-1. [DOI] [PubMed] [Google Scholar]
- 47.Li X., Zhu J., Zhong Y., Liu C., Yao M., Sun Y., Yao W., Ni X., Zhou F., Yao J., Jiang Q. Targeting long noncoding RNA-AQP4-AS1 for the treatment of retinal neurovascular dysfunction in diabetes mellitus. EBioMedicine. 2022;77 doi: 10.1016/j.ebiom.2022.103857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shan K., Liu C., Liu B.-H., Chen X., Dong R., Liu X., Zhang Y.-Y., Liu B., Zhang S.-J., Wang J.-J., et al. Circular noncoding RNA HIPK3 mediates retinal vascular dysfunction in diabetes mellitus. Circulation. 2017;136:1629–1642. doi: 10.1161/circulationaha.117.029004. [DOI] [PubMed] [Google Scholar]
- 49.Chou J.C., Rollins S.D., Fawzi A.A. Trypsin digest protocol to analyze the retinal vasculature of a mouse model. J. Vis. Exp. 2013;13 doi: 10.3791/50489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.