

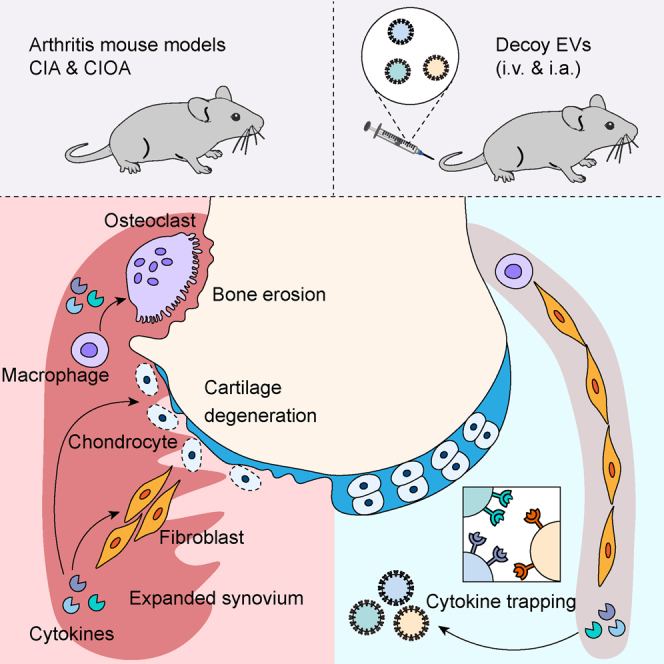
Summary
The interleukin 6 (IL6) signaling pathway plays pleiotropic roles in regulating the inflammatory milieu that contributes to arthritis development. Here, we show that activation of IL6 trans-signaling induces phenotypic transitions in tissue-resident cells toward an inflammatory state. The establishment of arthritis increases the serum number of extracellular vesicles (EVs), while these EVs express more IL6 signal transducer (IL6ST, also known as gp130) on their surface. Transferring these EVs can block IL6 trans-signaling in vitro by acting as decoys that trap hyper IL6 and prevent inflammatory amplification in recipient arthritic mice. By genetically fusing EV-sorting domains with extracellular domains of receptors, we engineered EVs that harbor a higher quantity of signaling-incompetent decoy receptors. These exogenous decoy EVs exhibit significant potential in eliciting efficient anti-inflammatory effects in vivo. Our findings suggest an inherent resistance of decoy EVs against inflammation, highlighting the therapeutic potential of efficient decoy EVs in treating inflammatory diseases.
Keywords: decoy extracellular vesicles, inflammatory-driven arthritis, IL6 trans-signaling, bone and joint homeostasis, EV therapy
Graphical abstract
Highlights
-
•
IL6 trans-signaling drives robust cytokine production and promotes osteoclastogenesis
-
•
The emergence of IL6ST-bearing EVs was along with arthritis onset
-
•
Transferred IL6ST-bearing EVs act as decoys to prevent inflammatory amplification
-
•
Engineered decoy EVs hold strong potential to elicit efficient anti-inflammatory effects
Liang et al. reveal an inherent resistance of EVs against inflammatory progression and propose a potential therapeutic strategy for treating inflammatory arthritis by the engineering of decoy EVs capable of efficiently sequestering cytokines.
Introduction
Inflammatory arthritis constitutes a group of articular conditions characterized by tissue inflammation, cartilage destruction, and systemic manifestations. Cytokines such as interleukin 6 (IL6), tumor necrosis factor alpha (TNFα), and IL1β can initiate a cascade of responses characterized by the induction of a broad spectrum of pro-inflammatory cytokines and aberrant functions of tissue-resident cells,1 instigating the joint microenvironment out of physiological homeostasis. Current therapies for inflammatory arthritis are insufficient in preventing progressive joint erosion and curing the disease completely. First-line drug agents, such as methotrexate, have a wide range of side effects experienced by a portion of rheumatoid arthritis (RA) patients during treatment.2,3 Capable of efficiently suppressing cytokines and their cascade reaction, anti-cytokine therapy has emerged as an attractive therapeutic strategy for inflammatory arthritis.1
IL6 signaling plays a key role in the inflammatory cascade in arthritis. The pathway activated by IL6 and membrane-bound IL6R (mIL6R) is considered cis-signaling, while the activity elicited by IL6 and soluble IL6R (sIL6R) is referred to as trans-signaling.4 Formation of both complexes results in trans-phosphorylation and activation of JAK, which subsequently phosphorylates STAT3, leading its dimerization and translocation to the nucleus.4 Aberrant activation of IL6 signaling is implicated in the pathogenesis and progression of both RA and osteoarthritis (OA).5 So far, several phase III clinical trials found that blockade of IL6 with tocilizumab or sarilumab improved RA prognosis and clinical score, leading to their approval as a first-line treatment.6 The recent identification of distinct homeostatic or pathogenic IL6-induced signaling pathways has introduced the concept of selectively inhibiting the deleterious effects of IL6 trans-signaling while preserving the homeostatic bioactivities of IL6 cis-signaling for regeneration of tissue microenvironment.4,7 However, the impact of selectively inhibiting IL6 trans-signaling on inflammatory arthritis remains uncertain.
Extracellular vesicles (EVs) are nanosized particles released by virtually all types of cells into the extracellular space or body fluids.8 Apart from the generally recognized roles in intercellular or interorgan communications, a growing number of studies are illuminating the potential of these vesicles to functionally load and deliver biomolecules. We and others have uncovered that cargos carried by EVs can modulate signal transduction in both physiological bone remodeling and pathological bone metastasis,9,10,11 highlighting the potential therapeutic application of EVs in bone-related diseases. Besides, genetic manipulation and chemical modification have been employed to engineer the surface receptors or luminal cargos of EVs, aiming to overcome their suboptimal in vivo distribution and to enhance their functional efficacy.8 However, the underlying mechanisms of EV cargo sorting are independent of parental cells,8,12,13 implicating the challenge of poor controllability of cargo for EV therapy. This is particularly important in EV engineering, especially how to assemble target proteins efficiently and stably on the surface of EVs, to precisely improve the therapeutic effect while reducing the interference of other unnecessary EV cargos.14,15
In this study, we investigate the biological effects of IL6 trans-signaling on several tissue-resident cells of articular joint, including chondrocytes, osteoclasts, and synovial fibroblasts, all of which are keystone mediators of the disturbed processes implicated in arthritis. We confirm that the emergence of IL6ST-bearing decoy EVs was along with arthritis onset, while adoptive transfer of these EVs can prevent inflammatory amplification in recipient arthritic mice. Furthermore, we develop several EVs that harbor respective decoy receptors through genetic engineering technology. The antagonistic capacity of these decoy EVs against cytokines and their therapeutic effects on inflammatory arthritis were further confirmed both in vitro and in vivo.
Results
Response of IL6 trans-signaling by tissue-resident cells of articular joint
Most cells respond to both classical and trans-signaling of IL6, but their activation levels are largely dominated by the ratio between mIL6R and IL6ST on cell membrane.4,7 We first detected surface levels of both mIL6 and IL6ST by flow cytometry. We found that chondrocytes express abundant IL6ST even higher than oncostatin M receptor (OSMR) or IL1R, while mIL6R was barely detectable (Figures 1A and 1B). Comparable consequences were also observed in bone marrow macrophages (BMMs), a major origin of bone resorbing osteoclasts (Figures S1A and S1B). Gene set enrichment analysis of a previously published RNA expression dataset16 showed that treatment of IL6/sIL6R complex upregulated nine gene sets associated with cytokine synthesis and substance metabolism, while it downregulated four gene sets related to skeletal development and endochondral osteogenesis (Figure S2A). Key involvement of IL6 trans-signaling on production of multiple cytokines was further independently confirmed by stimulating mouse cartilage explants with hyper IL6 (Figures 1C and 1D), a fusion protein of IL6 and sIL6R able to selectively activate IL6 trans-signaling in IL6ST-expressing cells.17 Substantial evidence was also observed when sgp130Fc and hyper IL6 were applied together, for which the massive cytokine production was completely blocked (Figures 1C and 1D). Upon stimulation of hyper IL6, protein expression of MMP inhibitor family (TIMP) 1 and 3 in explant lysates increased in parallel with ADAMTS4/5 and MMP1, even though the hypertrophy marker RUNX2 was not altered (Figure S1C). Compared with wild-type mice, il6stfl/fl Col2a1-cre mice developed a phenotype with attenuated cartilage degeneration and reduced proteoglycan loss after injection of collagenase into the knee joint (Figures S1D–S1G). Of note, IL6ST knockout did not affect membranous IL6R (mIL6R) expression, but it reduced NITEGE staining, suggesting a strong dependence of aggrecanases activation on IL6 trans-signaling (Figure S1H).
Figure 1.

Response of IL6 trans-signaling by tissue-resident cells of articular joint
(A and B) Flow cytometric histogram (A) and quantitative gMFI (B) showing the expression of respective receptors in primary mouse chondrocytes. n = 3 biological replicates. gMFI, geometric mean fluorescence intensity.
(C and D) Experimental design (C) and heatmap (D) showing the secretion of interleukins, chemokines, and matrix enzymes from cartilage explants (n = 3) treated with hyper IL6, sgp130Fc, or their combination at the indicated doses. Each cell of heatmap represents the average expression value of cytokines from three technical replicates. Cytokines were measured using Luminex assay.
(E) Representative TRAP stain images of BMMs (isolated from wild-type, il6st−/−, and tnsrsf11a−/− mice) treated with 50 ng/mL hyper IL6 or 100 ng/mL RANKL for 5 days. Scale bars, 500 μm. These micrographs are representative of three biological replicates for each group.
(F) Surface area of TRAP-positive BMMs in (E) was measured using ImageJ software. Each dot represents the pixel size of an identified osteoclast.
(G) Representative TRAP stain images of hyper IL6-induced BMMs treated with DMSO (<0.1%), 20 μg/mL AG490, or 5 μmol/L Stattic. Scale bars, 500 μm. These micrographs are representative of three biological replicates for each group.
(H) Surface area of TRAP-positive BMMs in (G) was measured using ImageJ software. Each dot represents the pixel size of an identified osteoclast.
The data are representative of two (A–D) or three (E–H) independent experiments with biologically independent samples. All data are presented as mean ± SEM. In (F) and (H), statistical significance was calculated using Kruskal-Wallis test with Dunn’s multiple comparisons test.
IL6 trans-signaling regulates osteoclastogenesis through two major ways, by amplifying the response of osteoclasts to RANKL stimulation18 or by inducing osteoblasts to secrete soluble RANKL.19 We found that 50 ng/mL hyper IL6 was sufficient to induce the formation of TRAP-positive multinucleated cells in the absence of RANKL, where sgp130Fc effectively blocked hyper IL6 but not RANKL-induced osteoclast fusion (Figures 1E and 1F). The expression of pSTAT3 in BMMs was controlled by hyper IL6 in a dose-dependent manner (Figure S1I), suggesting that IL6 trans-signaling controls a RANKL-independent osteoclastogenic pathway. Indeed, cellular responsiveness to hyper IL6 was attributed to membranous IL6ST, evidence that osteoclastogenesis was suppressed upon knockout of IL6ST but not RANK (Figures 1E and 1F). Corroborative evidence for this was observed when wild-type BMMs were treated with tyrphostin AG490 (a JAK2 inhibitor) and Stattic (a STAT3 inhibitor), in which both reagents decreased multinucleated TRAP-positive cell number (Figures 1G and 1H). Given that the significant inflammatory response in synovium is a typical feature of RA, we also tested the ability of synovial tissue in response to IL6 signaling using a collagen-induced arthritis (CIA) mouse model. The fluorescence intensities of both IL6ST and mIL6R in synovial fibroblasts were markedly higher in the late phase of the disease than those in the pre-symptomatic phase (Figures S1J–S1L), implying an increased sensitivity of synovium in response to IL6 trans-signaling. Together, the above data implied the different biological effects of IL6 trans-signaling on multiple cell types, indicating trans-signaling as a potential therapeutic target for arthritis pathology.
Inflammatory stimulation promotes the release of IL6ST-bearing decoy EVs
To determine whether elevated membranous IL6ST induced by inflammation involves to the vesicular IL6ST level, we tested the level of IL6ST decoy EVs in two RA mouse models—K/BxN serum transfer-induced arthritis (STA) and CIA—and an experimental autoimmune myositis (EAM) mouse model. In all three disease models, we observed a moderate increase of EV numbers in both serum and liver compared to controls (Figure 2A). We further found an increase in the molecular weight band of IL6ST in EV fractions isolated from the serum of CIA mice compared with unimmunized dilute brown non-agouti (DBA)/1J mice (Figures 2B and 2C), even if these EVs were morphologically comparable and shared similar particle size distribution (Figures 2D and S2B). We further quantified the IL6ST number at the single-EV level using a previously reported flow cytometry-based method.20 IL6ST numbers in blood from K/BxN STA (237.6 ± 24.46), CIA (148.4 ± 13.46), and EAM (56.32 ± 8.66) mouse models were higher than that in control DBA/1J mice (29.94 ± 5.65) (Figures 2E and S2C). We then tested whether these released EVs could serve as a decoy to bind and inhibit hyper IL6. We found that EVs isolated from blood of K/BxN STA and CIA mice, but not from control DBA/1J mice, were able to inhibit the hyper IL6-induced trans-signaling in a human secreted embryonic alkaline phosphatase (SEAP) reporter cell model (Figures 2F and S2D). Importantly, we demonstrated that EVs from blood of K/BxN STA mice alleviated arthritis score in CIA mouse model, whereas preincubation of EVs with IL6ST monoclonal antibodies (mAbs) mitigated the protective effects of EVs (Figures 2G–2I). These results showed that EVs function as decoy to sequestrating IL6/sIL6R heterodimeric complexes, suggesting a natural protection role of EVs against inflammation in vivo.
Figure 2.

Inflammatory stimulation promotes the release of IL6ST-bearing decoy EVs
(A) EV quantification in blood and liver samples from DBA/1J (n = 3), K/BxN STA (n = 6), CIA (n = 6), or EAM mice (n = 3) using nanoparticle tracking analysis (NTA).
(B and C) Protein lysates were collected from serum EVs from DBA/1J, K/BxN STA, CIA, and EAM mice. Representative western blot (B) and quantification analysis (C) of IL6ST are shown. β-tubulin was used as a loading control. n = 3 mice each group.
(D) Representative transmission electron micrographs of serum EVs from indicated mouse models. Scale bars, 100 nm.
(E) Total estimated number of IL6ST receptors per EV from serum of DBA/1J (n = 2), K/BxN STA (n = 6), CIA (n = 6), or EAM mice (n = 3) estimated based on obtained MFI values after fluorescence calibration of with Quantibrite polyethylene (PE) beads by flow cytometry.
(F) Relative intensities of SEAP signal in HEK-Blue IL6 reporter cells induced by 10 ng/mL hyper IL6 and treated with 5 × 108 EV isolated from DBA/1J, K/BxN STA, CIA, or EAM mice. Data were normalized to HEK-Blue IL6 reporter cells treated with mock. Treatment with 200 ng/mL sgp130Fc was used as a positive control. n = 3 biological triplicates. Each dot represents the average value of three technical replicates.
(G) Experimental design for EV transfer from K/BxN STA model mice to recipient CIA mice.
(H and I) Clinical arthritis score over time (H) and at the endpoint (day 52) (I) in CIA mice treated with saline (n = 3), K/BxN STA mouse EVs (n = 5), or K/BxN STA mouse EVs preincubated with IL6ST mAb (n = 3) or IgG control (n = 3) recorded at the indicated time points. Each mouse was intravenously injected with 5 × 1010 EVs. mAb, monoclonal antibody.
The data are representative of two independent experiments with biologically independent samples (A–I). Data are presented as mean ± SEM.
(A, C, E, F, and I) Statistical significance was calculated using one-way ANOVA with Tukey’s post hoc test for multiple comparisons.
Construction of engineered IL6ST decoy EVs by surface display of chimeric proteins
The presence of IL6ST-bearing decoy EVs implies a promising targeting strategy, wherein the decoy efficiency of EVs can be improved through genetic engineering of IL6ST decoy receptors using a surface display technique. Two EV-sorting proteins, CD63 (a four-transmembrane EV hallmark) and syntenin (a cytosolic adaptor of the single transmembrane domain protein syndecan), were selected as display scaffolds based on their (1) high expression in EVs and localization at EV membrane, as determined by a number of previous reports8; (2) short amino acid chains and small molecular weight to facilitate engineering and expression; (3) low cytotoxicity to parental cells; and (4) reported tolerance to fusion with other proteins.21 Genetic constructs were established by fusing the fluorescein-tagged extracellular domain of IL6ST with the transmembrane helix of PDGFRβ and to the N terminus of CD63 or syntenin, respectively (Figure 3A). Donor HEK293T cells were respectively expressed with the crystal structure of two chimeric proteins, CD63-eGFP-IL6ST and IL6ST-mCherry-syntenin, containing N-terminal domains and C-terminal domains, extracellular, transmembrane, and intracellular domains of each protein (Figure 3B). In this conceptual design, expression of chimeric proteins displayed signaling-incompetent IL6ST receptors on the EV membrane, while eGFP and mCherry were kept outside the EV surface functioning as fluorescent reporters and distribution trackers for chimeric proteins (Figure 3C). HEK293T cells were transiently transfected with plasmids expressing different constructs, and engineered EVs were isolated by differential ultracentrifugation in combination with sucrose cushion ultracentrifugation. Common EV morphology with lipid bilayer vesicles of engineered EVs was identified using transmission electron microscopy, as well as a size distribution in line with NTA (Figures S2E and S2F). In addition, EV number per cell increased by 35% after expressing IL6ST-syntenin construct (Figure S2G), indicating that fusion with IL6ST did not suppress the regulation of syntenin on endosomal sorting complex required for transport (ESCRT)-dependent EV biogenesis pathway.13 We found that trans-signaling, instead of classical signaling, was efficiently blocked by engineered EVs, highlighting the specificity of trans-signaling inhibition by engineered EVs (Figure 3D). Western blot probing for the respective fused fluorescent proteins or IL6ST corroborated the expression of fusion proteins and the functionality of cytokine decoy EVs (Figure 3E). Fused fluorescent modules efficiently tracked cellular internalization of decoy EVs, as evidenced by an increase in intracellular fluorescence intensities over time (Figures 3F and 3G).
Figure 3.

Construction of engineered IL6ST decoy EVs by surface display of chimeric proteins
(A) Schematic of DNA constructs expressing IL6ST-eGFP-CD63 and IL6ST-mCherry-syntenin.
(B) Predicted accurate model building for the intracellular, extracellular, and transmembrane domains of IL6ST-eGFP-CD63 (pLDDT = 84.25), or IL6ST-syntenin (pLDDT = 81.22). A, Ig-like C2-type; B, fibronectin type-Ⅲ 1; C, fibronectin type-Ⅲ 2; D, fibronectin type-Ⅲ 3; E, fibronectin type-Ⅲ 4; F, fibronectin type-Ⅲ 5; pLDDT greater than 70 indicates that the predicted structure has a high degree of confidence. pLDDT, predicted local-distance difference test.
(C) Schematic diagram showing two types of IL6ST decoy EVs obtained by fusing IL6ST to EV-sorting proteins in tandem with fluorescent proteins.
(D) Relative intensities of SEAP signal in HEK-Blue IL6 reporter cells induced by 10 ng/mL of either hyper IL-6 (left) or IL6 (right) and treated with wild-type EVs, IL6ST-CD63 EVs, or IL6ST-syntenin EVs at the indicated doses. n = 3 biological replicates. Data were normalized to cells treated with wild-type EVs.
(E) Protein lysates was collected from IL6ST decoy EVs or their donor cells. Western blot analysis was performed examining IL6ST, mCherry and eGFP. β-tubulin was used as a loading control. Specific bands corresponding to the predicted molecular weight of chimeric proteins are indicated by the black boxes.
(F) Representative fluorescence images showing IL6ST decoy EVs labeled with eGFP (green) or mCherry (red) are internalized by HEK293 cells. Scale bars, 10 μm.
(G) The number of eGFP or mCherry particles per cell in (F). The dot plot represents the number of eGFP or mCherry particles from individual cells. n = 20 cells each group.
The data are representative of three independent experiments with biologically independent samples (D–G). Data are presented as mean ± SEM.
(D) Statistical significance was calculated using two-way ANOVA with Dunnett’s multiple comparisons test.
(G) Statistical significance was calculated using Kruskal-Wallis test with Dunn’s multiple comparisons test.
Co-expression of chimeric proteins optimizes the display of decoy receptors
Given the limited expression of any kind of sorting proteins in individual EVs, the utilization of single sorting domains may impede efficient use of EV surface space for displaying decoy receptors. To address this issue, we transfected both chimeric constructs into HEK293 with the aim of enhancing the display efficiency of surface receptors (Figure 4A). The survival rate of donor cells transfected with respective chimeric construct or co-transfection was comparable to that of controls, with a cell viability greater than 92% (Figure S3A), indicative of the low predicted cytotoxicity of these constructs and expressed proteins, reducing the rate of apoptotic EV secretion to the conditioned media. A high degree of colocalization of the two chimeric proteins, i.e., the overlap between the red and green signals at the pixel level, was observed almost throughout the cytoplasm but not in the nucleus (Figures S3B and S3C). We also observed enrichment of IL6ST labeling dots outside the bilayer membrane of engineered EVs (Figure 4B). Quantification of IL6ST epitopes of EVs from cells co-transfected with chimeric constructs was significantly higher than that of respective transfection (Figure 4C), suggesting a high display efficiency by co-transfection. We further quantified the IL6ST receptor number at the single EV level. The number of IL6ST of IL6ST-syntenin EVs (568.3 ± 46.13) or IL6ST-CD63 EVs (603.6 ± 21.78) was higher than that of wild-type EVs (23.4 ± 7.19), while this was increased further after co-transfection of chimeric constructs (947 ± 38.52) (Figures 4D and S3D). Engineering the decoy receptors did not affect the general content of EVs, as the pan-EV markers expression of IL6ST-CD63/syntenin EVs was comparable to controls (Figure S3E). In the presence of IL6ST-CD63/syntenin EVs, the hyper IL6-induced cytokine secretion by cartilage explants was almost completely blocked (Figure 4E). Phosphorylation of STAT3 induced by hyper IL6 was decreased by approximately 75% upon a high dose of IL6ST-CD63/syntenin EVs (5 × 109 EV per mL) (Figure S3F). Importantly, both secretion of matrix-degrading enzymes and IL6-induced osteoclastogenesis were significantly attenuated by IL6ST-CD63/syntenin EVs, compared with either IL6ST-syntenin EVs or IL6ST-CD63 EVs (Figures 4F–4H). A relative high dose (3 × 1010 EV per mL) of engineered EVs was well tolerated whether by cell lines or primary BMMs/chondrocytes (Figures S3G–S3I). In another separate in vivo experiment for toxicity evaluation, mice treated with a high dose of IL6ST-CD63/syntenin EVs (3 × 1011) displayed similar levels in body weight, hepatic function, and hematological parameters at the endpoint, compared to either wild-type EVs or saline treatment (Figure S3J–S3R). Together, these data clearly support the notion that co-expression of chimeric constructs optimized the display of decoy receptors and increased biological efficacy of engineered decoy EVs.
Figure 4.

Co-expression of chimeric proteins optimizes the display of decoy receptors
(A) Schematic diagram of the arrangement at the EV membrane of IL6ST fused to the sorting proteins in tandem with fluorescent proteins.
(B) Representative transmission electron micrographs of decoy EVs with nanogold-labeled antibodies staining of IL6ST. Scale bars, 100 nm. Nanogold-labeled IgG antibodies was used as a negative control. These micrographs are representative of over 10 such images for each group.
(C) Quantification of nanogold-labeled IL6ST epitopes at the EV membrane. The dot plot represents the number of nanogold particles from individual micrographs. n = 10 images each group.
(D) Total estimated number of IL6ST receptors per EV estimated based on obtained MFI values after fluorescence calibration of with Quantibrite PE beads by flow cytometry. n = 3 biological replicates.
(E) Heatmap showing the expression of interleukins, chemokines, and matrix enzymes in the supernatant of mouse cartilage explants treated with 50 ng/mL hyper IL6 and increasing doses of decoy EVs. Each cell of heatmap represents the average expression value of cytokines from three biological replicates. Cytokines were measured using Luminex assay.
(F) Quantitative analysis showing the expression of six matrix enzymes in the supernatant of mouse cartilage explants treated with 50 ng/mL hyper IL6 and 3 × 109/mL decoy EVs. Each dot represents the average expression value of cytokines from three biological replicates. Cytokines were measured using Luminex assay.
(G) Representative TRAP stain images of BMMs treated with 50 ng/mL hyper IL6 and 5 × 109 decoy EVs for 5 days. Scale bars, 500 μm. These micrographs are representative of three biological replicates for each group.
(H) Mature osteoclasts with three or more nuclei per well (n = 3) were counted.
The data are representative of two (B–F) or three (G) independent experiments with biologically independent samples. All data are presented as mean ± SEM.
(C and D) Statistical significance was calculated using one-way ANOVA with Tukey’s post hoc test for multiple comparisons.
(F) Statistical significance was calculated using one-way ANOVA with Dunnett’s multiple comparisons test.
(H) Statistical significance was calculated using two-way ANOVA with Tukey’s post hoc test for multiple comparisons.
Efficient sorting of decoy tumor necrosis factor receptor-1 (TNFR1) and IL1RⅡ into engineered EVs through chimeric protein construction
The pathogenesis of inflammatory arthritis involves the intricate interplay of multiple cytokines, thereby posing a challenge for therapeutic interventions targeting individual cytokines.22 This drives us to sequentially engineer decoy EVs that antagonize other cytokines. We constructed chimeric proteins targeting IL1β and TNFα signaling to efficiently display their signaling-incompetent receptors outside the EV membrane (Figures 5A, S4A, and S4B). TNFR1 was selected for restraint of TNFα signaling based on previous research experience. IL1RⅡ was selected because it does not bind with interleukin 1 receptor antagonist (IL1Ra), a natural competitive inhibitor of IL1β, which hinders the binding of IL1β to IL1RI and impairs the neutralization of free IL1β by decoy receptors.23 In all cases, the engineered EVs were 50–200 nm in diameter and enriched in the classical EV markers (Figure S4C). Expressing the chimeric constructs increased the respective geometric mean fluorescence intensity (gMFI) of IL1RⅡ and TNFR1 on the cell surface by 1.9-fold and 1.7-fold compared with controls, while this increase was smaller than that of directly transfecting cells with IL1RⅡ and TNFR1 expression vectors (2.63-fold and 2.96-fold, respectively) (Figures 5B–5E). Importantly, however, EVs from cells expressing chimeric constructs showed higher numbers of decoy receptors than cells transfected with expression vectors (Figures 5F and 5G), suggesting that EV-sorting proteins as molecular scaffolds are essential for the effective display of decoy receptors. This proposal was also corroborated by immunogold electron microscopy probing the respective epitopes, showing an evident accumulation of decoy receptors around the engineered EV surface (Figures 5H and 5I). We confirmed efficient tracking of decoy EVs by fused fluorescent modules by investigating EV internalization by HEK293T cells (Figure S4D). We further confirmed the dose-dependent inhibition of engineered decoy EVs on TNFα and IL1β signaling transduction using well-established reporter cells of TNFα and IL1β (Figures 5J, 5K, and S4E). Besides, we confirmed that decoy IL1RⅡ EVs specifically inhibited the activation of osteoclastogenic Tregs driven by IL1β (Figures 5L, 5M, and S4F).24 Neither of these two engineered EVs had negative effects on cell viability at a dose of 5 × 109 EV per mL (Figure S4G).
Figure 5.

Effective sorting of decoy TNFR1 and IL1RⅡ into engineered EVs by chimeric protein construction
(A) Schematic diagram of the arrangement at the EV membrane of TNFR1 or IL1RⅡ fused to the sorting proteins in tandem with fluorescent proteins.
(B and C) Flow cytometric histogram (B) and TNFR1 gMFI (C) in HEK293T cells transfected TNFR1 expression vector or TNFR1 chimeric constructs. n = 3 biological triplicates. Data were normalized to transfection of empty vector.
(D and E) Flow cytometric histogram (D) and IL1RⅡ gMFI (E) in HEK293T cells transfected with IL1RⅡ expression vector or IL1RⅡ chimeric constructs. n = 3 biological triplicates. Data were normalized to transfection of empty vector.
(F and G) Total estimated number of TNFR1 (F) and IL1RⅡ (G) per EV estimated based on obtained MFI values after fluorescence calibration of with Quantibrite PE beads by flow cytometry. n = 7 technical replicates.
(H) Representative transmission electron micrographs of decoy EVs with nanogold-labeled antibodies staining of TNFR1 and IL1RⅡ. Scale bars, 100 nm. These micrographs are representative of over eight such images for each group.
(I) Quantification of nanogold-labeled TNFR1 or IL1RⅡ epitopes at the EV membrane. The dot plot represents the number of nanogold particles from individual micrographs. n = 8 images each group.
(J) Schematic of reporter assay for testing the antagonistic ability of TNFR1 decoy EVs or their donor cells to TNFα.
(K) Relative intensities of SEAP signal in 5 ng/mL TNFα-induced HEK-Blue TNFα reporter cells co-cultured with EV donor cells at the indicated densities or treated with TNFR1 decoy EVs at the indicated doses. n = 3 biological replicates. Data were normalized to cells treated with wild-type HEK293T cells or wild-type EVs.
(L) Sorted Treg cells induced by 10 ng/mL IL1β were co-cultured with BMMs and treated with 5 × 109/mL IL1RⅡ decoy EVs. TRAP staining was performed after 7 days of co-culture. Scale bars, 500 μm. These micrographs are representative of three biological replicates for each group.
(M) Surface area of TRAP-positive BMMs in (L) was measured using ImageJ software. Each dot represents the pixel size of an identified osteoclast.
The data are representative of two (B–I) or three (J–M) independent experiments with biologically independent samples. All data are presented as mean ± SEM. In (C), (E), (F), and (G), statistical significance was calculated using one-way ANOVA with Tukey’s post hoc test for multiple comparisons. In (I), statistical significance was calculated using Student’s two-sided t test. In (K), statistical significance was calculated using two-way ANOVA with Sidak’s multiple comparisons test. In (M), statistical significance was calculated using Kruskal-Wallis test with Tukey’s post hoc test for multiple comparisons.
Decoy EVs reduced knee joint inflammation in collagenase-induced osteoarthritis mice
Inflammation-driven OA is a common subtype of clinical OA, characterized by degradation of extracellular matrix and dysregulated anabolism of chondrocytes induced by a high level of multiple cytokines. We therefore sought to detect whether decoy EVs have therapeutic potential in experimental OA mice. To generate stable cells that enable production scale-up and to reduce variability emanating from transient transfection of cells, we stably engineered HEK293T producer cells to produce IL6ST, TNFR1, and IL1RⅡ decoy EVs using lentiviral transduction. Previous studies have indicated that fluorescent markers such as eGFP are immunogenic across several models, which may potentially confound the interpretation of in vivo experimental data when we use immunocompetent animals.25,26 To avoid the eGFP and mCherry fluorophores from altering our arthritis model, we removed fluorophores from the chimeric constructs and reacquired decoy EVs for the subsequent animal experiments (Table S2). The antagonistic ability of decoy EVs to respective cytokines was validated using reporter cell lines (Figures S5A–S5D). We did not observe obvious therapeutic effects upon intraarticular (i.a.) injection of decoy EVs in mice with anterior cruciate ligament transection (ACLT) surgery, as evidenced by comparable OA score and serum levels of inflammatory cytokines between treatment and control groups (Figures S5E and S5F). However, i.a. injection of collagenase-induced osteoarthritis (CIOA) mice with either IL6ST decoy EVs or combinatorial decoy EVs reduced destruction of articular cartilage, with a significant decrease in the number of interarticular osteophytes and improved bone morphological parameters of subchondral bone (Figures 6A–6D and S5G–S5I). Decreased levels of IL1β, IL6, and TNFα parallelly corroborated the antagonism of these decoy EVs against cytokines (Figure S5J). Notably, the therapeutic disparities of decoy EVs in these two models may be attributed to a more profound and prolonged inflammation induced by CIOA, while ACLT is likely to elicit a chronic and low-grade inflammation (Figure S5K), as previously proposed.27 Additionally, treatment with decoy EVs resulted in a reduction of phosphorylated STAT3 activation both in synovium and cartilage of CIOA mice, as well as a decrease in MMP-13 expression by chondrocytes (Figure S5L).
Figure 6.

Decoy EV treatment relieves disease phenotypes of several arthritis mouse models
(A) Experimental design for assessing effects of decoy EV treatment on knee OA progression using a CIOA mouse model.
(B) Clinical Osteoarthritis Research Society International (OARSI) score of disease progression in CIOA mice intraarticularly injected with saline (n = 3), 3 × 1011 wild-type EVs (n = 3), 3 × 1011 IL6ST decoy EVs (n = 6), or 3 × 1011 combinatorial decoy EVs (n = 6) at the endpoint (day 42). The combinatorial EVs were composed of IL6ST, TNFR1, and IL1RⅡ decoy EVs in equal proportions (1011 of each type).
(C and D) Representative micro-CT images of sagittal views of subchondral bone medial compartment (C) and quantification of aberrant calcified tissue volume (Cal Tis. V) (D) in CIOA mice treated with saline (n = 3), 3 × 1011 wild-type EVs (n = 3), 3 × 1011 IL6ST decoy EVs (n = 6), or 3 × 1011 combinatorial decoy EVs (n = 6) at the endpoint (day 42). Black arrows indicate osteophytes on the edge of the tibial plateau.
(E) Experimental design for assessing the biodistribution of decoy EVs in diseased tissues of a CIA mouse model. 5 × 1010 EVs were injected intravenously into mice in a single dose at day 10 or day 26, respectively. The distribution of decoy EVs in synovium and bone marrow was detected using NTA 6 h after EV injection.
(F) EV numbers in synovium and bone marrow from sham mice (n = 3) or CIA mice (n = 6) were measured using NTA.
(G) Experimental design for assessing the protective effects of decoy EVs on arthritis onset in a CIA mouse model. Mice were intravenously injected with saline, 2 mg/kg methotrexate, 3 × 1011 wild-type EVs, or 3 × 1011 combinatorial decoy EVs from day 3 to day 21 after the first immunization.
(H) The incidence of arthritis in (H) was observed and recorded from day 21 to the endpoint (day 52).
(I) Clinical arthritis score showing disease progression in CIA mice treated with saline (n = 3), 2 mg/kg methotrexate (n = 5), 3 × 1011 wild-type EVs (n = 5), or 3 × 1011 combinatorial decoy EVs (n = 12) over time.
(J) Experimental design for assessing the therapeutic potential of decoy EVs on arthritis symptoms in a CIA mouse model.
(K) Clinical arthritis score of disease progression in CIA mice treated with saline (n = 3), 3 × 1010 combinatorial decoy EVs (n = 10), or 3 × 1011 combinatorial decoy EVs (n = 10) over time. The combinatorial EVs were composed of IL6ST, TNFR1, and IL1RⅡ decoy EVs in equal proportions.
(L) Heatmap and quantification of serum IL1β, TNFα, and IL6 in CIA mice treated with saline (n = 3), 3 × 1010 decoy EVs (n = 10), or 3 × 1011 decoy EVs (n = 10). Saline treatment was used as a negative control. Cytokines were measured using Luminex assay.
(M) Schematic diagram showing proposed mechanism of action for therapeutic potential of decoy EVs in inflammatory arthritis.
All data are presented as mean ± SEM. In (B) and (D), statistical significance was calculated using one-way ANOVA with Tukey’s post hoc test for multiple comparisons. In (F) and (L), statistical significance was calculated using Student’s two-sided t test. In (I) and (K), statistical significance was calculated using two-way ANOVA with Dunnett’s multiple comparisons test.
Decoy EVs safely and efficiently alleviate systemic inflammatory arthritis
Systemic administration of exogenous EVs can send them to several specific organs or tissues, even solid tumors.28,29 Here we established a CIA mouse model to mimic systemic RA features and confirmed the different tissue biodistribution of intravenously injected EVs between sham and CIA mice. We respectively assessed EV accumulation in CIA mice 10 days after the first immunization and 5 days after the second immunization, where EV number was increased in synovium and bone marrow of CIA mice at both points of time compared to sham mice (Figures 6E and 6F). Nevertheless, this is still not sufficient to reason the increased EV uptake efficiency by CIA tissue, as the EV yield of cells in a disease state could be increased compared to that of healthy condition.30 We therefore detected the fluorescent trackers of decoy EVs 26 days after the first immunization and found a markedly increased eGFP+/mCherry+ EV population in the CIA group compared to control (Figure S5M). These data suggest an increased uptake quantity of EVs by CIA tissue and enable us to further study the biological functions of decoy EVs in vivo.
CIA mice were treated with decoy EVs before and after the onset of arthritis symptoms, so that the protective and therapeutic effects of EVs could be discriminated. In the protective treatment experiment, 3 × 1011 combinatorial decoy EVs showed comparable efficacy with 2 mg/kg methotrexate, with decreased RA incidence rate and reduced clinical arthritis score at the endpoint compared to saline treatment (Figures 6G–6I). Compared with methotrexate treatment, a decrease in serum alanine transferase (ALT) activity was evident after decoy EV treatment along with reduced steatosis and inflammatory infiltration of hepatic sections (Figures S6A and S6B), suggesting a favorable protection of liver function by decoy EVs. In another separate experiment for therapeutic evaluation, we treated CIA mice with two different doses of decoy EVs after symptom onset (Figure 6J). We observed a significant reduction of clinical arthritis score by 3 × 1011 decoy EVs at the endpoint (Figure 6K). Significant reductions of hallmark cytokines corroborated the effective antagonism of decoy EVs against inflammation (Figure 6L). Further bone morphological analysis revealed extensive erosion coupled with re-mineralization around the ankle joints of mice treated with saline, observed as dents and knobs on the bone surface, whereas bone surfaces of mice treated with high dose of decoy EVs (3 × 1011 each mouse) were smooth in appearance (Figure S6C), indicating a decrease in erosions and remodeling events. Corroborative evidence for this also came from (1) a decreased number of erosion pores and improved bone morphological parameters of the calcaneus and (2) reduced expression of osteoclast markers in paw lysate (Figures S6D–S6F). Taken together, these data suggest that combinatorial decoy EVs effectively target multiple cytokine signals to regulate molecular and cellular events, making it a promising strategy in the management of inflammatory arthritis (Figure 6M).
Discussion
Tissue-resident cells can respond to inflammatory stimuli in various manners, which primarily depends on their membrane receptor distribution. In this study, we identified that IL6 trans-signaling can drive robust cytokine production from chondrocytes and promote osteoclastogenesis from BMMs. Both chondrocytes and BMMs have significantly higher membrane IL6ST while almost absent mIL6R, reflecting their stronger ability in response to trans-signaling than classic signaling. The above, along with the observation that synovial fibroblasts express increased IL6ST with arthritis progression, prompted us to ascertain the host response to inflammatory milieu in vivo. We found that the number of serum EVs in either K/BxN STA or CIA mice was much higher than that in healthy mice. These findings are partially in line with recent studies,31,32 which proposed that inflammation may also promote EV release. We further demonstrated that IL6ST was enriched on EVs derived from inflammatory milieu; these IL6ST-bearing EVs can prevent inflammatory amplification in recipient arthritic mice by acting as cytokine decoys. In view of the special transduction mode of IL6 trans-signaling, these EVs are likely to bind and inhibit the in vivo heterodimeric complex formed by IL6 and sIL6R. Therefore, on the one hand, cytokines induce target cells to enter an aberrant state that contributes to cascade reaction and tissue damage; on the other hand, the host can mount a protective response against inflammation by releasing EVs carried with IL6ST decoy receptors. Consistent with our findings, a recent study has reported that Schwann cells can selectively sequestered TNFR1 into EVs upon stimulation by TNFα, while these EVs can buffer against the toxic effects of TNFα.33 These suggest that decoy receptors on EV membrane can be diverse, and their types might vary according to distinct inflammatory milieus. In addition, a recent study proposed a critical role of EVs in innate immune response to bacterial infection by acting as decoys that trap membrane-acting virulence factors, such as pore-forming toxins, to prevent damage of target tissues.34 Therefore, it is reasonable to hypothesize that tissues might release EVs loaded with neutralizing molecules in response to external stimuli. One of the potential mechanisms that mediates the decoy EV secretion might be secretory autophagy, which is recently described as a mechanistically coordinated process for extracellular cargo secretion by EVs.30,34 Studies have also demonstrated a strong association between secretory autophagy and both bacterial infection and inflammation.34,35 Although this spontaneous process may not reverse disease progression, EVs marked with neutralizing factors still have great potential as biomarkers for disease surveillance and assessment.
Discovery of IL6ST-bearing decoy EVs provides the possibility to treat disease using engineered EVs with enhanced neutralizing efficacy. For this purpose, we designed engineering strategies for surface display of biologics on EVs. Generally, EV surface engineering is performed by genetically overexpressing the target proteins in parental cells to increase the protein abundance of the EV membrane. However, it should be pointed that gene expression and EV cargo sorting are not synchronized, since EV cargo sorting is independently and tightly controlled by a series of mechanisms headed by ESCRT machinery.12,13 Our data also demonstrated that expression vectors of both TNFR1 and IL1RⅡ show an efficient expression of membrane receptor in parental cells, whereas this high efficiency cannot be reproduced at the vesicular level. Using the concept of chimeric proteins, we fused the cytokine-binding domains to the EV-sorting domains for efficient sequestration of decoy receptors into EV membrane. Syntenin interacts with apoptosis-linked gene-2-interacting protein X (ALIX) to directly prompt the intraluminal budding and abscission of endosomal membranes,12,13 while CD63 is a tetraspanin steadily accumulated in multivesicular bodies (MVBs).8,12 Hence, syntenin and CD63 are initially localized on the surface of intraluminal vesicles (ILVs) and are secreted along with ILVs upon fusion of MVBs with the plasma membrane.8,12 Our data confirmed that syntenin and CD63 as cargo sorting domains in the fusion protein can efficiently carry decoy receptors into EVs.
We examined display efficiency of decoy receptors in donor cells transfected with chimeric constructs and expression vectors of respective receptors. Compared with expression vectors of IL1RⅡ and TNFR1, chimeric constructs displayed fewer plasma membrane receptors, but robustly increased the number of EV membrane receptors. One step further, by co-expressing distinctive chimeric proteins, we increased the number of EV membrane receptors and improved cytokine trap efficiency of decoy EVs. The effective display of decoy receptors also contributes to reducing EV dose for treatment, minimizing the interference of other unnecessary EV cargos. Another engineering strategy is allowing EVs from a single origin to simultaneously inhibit more than two cytokines.20 However, the irregular elevation of distinct cytokines should be noted, which is determined by various factors such as disease types, disease stages, and individual differences among patients or among animals. Therefore, decoy EVs expressing a single type of receptor have a strong antagonistic ability to a specific cytokine, while combinations of certain types of decoy EVs could tailor precise, individualized strategies for therapy, allowing better therapeutic outcomes to be achieved with minimal doses of EVs.
In this study, we fused fluorescent modules into the extracellular region of chimeric constructs to track the subcellular expression of these chimeric proteins in parental cells. Generally, fusion with other proteins does not affect the luminescent function of eGFP and mCherry,21,36 both of which have relative short amino acid chains with small molecular weight. Based on this, incorporation of fluorescent modules allows us to visually assess whether the target chimeric sequences were efficiently expressed, especially since recent studies have proposed that chimeric proteins with large molecular weight may have low or virtually no expression.37,38 Our data confirmed that chimeric proteins were fully expressed in parental cells, while colocalization of eGFP and mCherry also suggested that CD63 and syntenin in chimeric proteins still have biological function. On the other hand, fluorescent proteins could also efficiently track EVs in vitro (Figure 3F). Unlike the transient fluorescent dyes commonly used for EV labeling such as PKH67 and DiD, conformational stabilities of eGFP and mCherry enable them to remain stable for long periods both in vitro and in vivo. Expression of certain fluorescent proteins such as dsRed may be significantly cytotoxic.39 Our data showed that, in all cases, expression of chimeric proteins induced only minimal cytotoxicity.
In a separate experiment, we examined the remission of RA phenotype by treatment with either decoy EVs or methotrexate. We showed that therapeutic potential of decoy EVs is comparable with methotrexate, and both therapies can effectively slow down RA onset in the protective treatment model. However, benefits of methotrexate in RA therapy are achieved along with histological changes including steatosis and hepatic fibrosis (Figure S6B), reflecting a wide range of side effects suffered by a portion of RA patients during methotrexate treatment.2,3 In contrast, mice that received decoy EV treatment retained their physiological hepatic structure, suggesting minimal hepatotoxicity and high biocompatibility of EVs. Our EV treatment is based on IL6ST decoy EVs or a combination of three types of decoy EVs against respective inflammatory pathways, showing that decoy EVs against multiple cytokines are generally more effective in remission of arthritis phenotype than decoy EVs against the single IL6 trans-signaling. These results, along with recent studies,22,40 strongly suggest the importance of broadening the anti-cytokine profile, especially in several clinical trials that have proposed that mAbs against a single cytokine such as IL1β and TNFα could not significantly improve synovitis, bone and cartilage destruction, and arthritic pain in arthritis patients.41,42 Notably, decoy EVs failed to reproduce their therapeutic effects on ACLT mice, highlighting that different arthritis models have their specific advantages in exploring arthritis pathogenesis. That is, ACLT mimics human post-traumatic OA with slowly progressing and lower cytokine levels, while CIOA induces rapid and severe joint degeneration with intense inflammation.27 Our data suggest that therapeutic outcomes of decoy EVs depend on the degree of inflammatory infiltration. The irregularity of cytokine elevation in different diseases or different models also implies the need to combine decoy EVs with precise and customized strategies in hope of achieving better outcomes. Additionally, treatment strategies should consider the blockade of other cytokines that play crucial roles in driving arthritis, such as IL17, IL18, and TGFβ.43,44
In summary, our findings show an inherent resistance of EVs against inflammatory progression and propose a potential therapeutic strategy for treating inflammatory arthritis by the engineering of decoy EVs capable of efficiently sequestering cytokines. In addition, the observed therapeutic differences among disease models suggest the importance of customized combinations and doses of decoy EVs in anti-cytokine therapy for arthritis treatment.
Limitations of the study
First, the present study did not investigate the underlying mechanisms by which inflammatory stimulation induces the release of IL6ST-bearing EVs. Although these EVs were detected in mouse serum, their tissue origin and parental cells remain undefined. Further investigation is required to clarify the specific molecular mechanisms involved in this process. Second, it should be noted that the decoy EVs employed in this study lacked specific targeting capabilities toward bone, joint, or muscle tissues. So far, there is limited research dedicated to enhancing the bone-targeting efficacy of EVs. Although it is reported that intravenous administration of EVs derived from specific cell types can naturally accumulate in bone tissue, there is currently insufficient evidence to substantiate the claim that these EVs still have acceptable bone-targeting capacity even after engineering.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-GAPDH | Abcam | Cat# ab9485; RRID: AB_307275 |
| Rabbit anti-IL6ST | Abcam | Cat# ab283685 |
| Rabbit anti-eGFP | Bioss | Cat# bs-2194R; RRID: AB_10881247 |
| Rabbit anti-mCherry | Bioss | Cat# bs-41161R |
| Rabbit anti-TIMP1 | Bioss | Cat# bs-0415R; RRID: AB_10856775 |
| Rabbit anti-TIMP3 | Abcam | Cat# ab277794 |
| Rabbit anti-ADAMTS4 | Bioss | Cat# bs-4191R; RRID: AB_11073215 |
| Rabbit anti-ADAMTS5 | Bioss | Cat# bs-3573R; RRID: AB_10856276 |
| Rabbit anti-RUNX2 | Bioss | Cat# bs-1134R; RRID: AB_10856062 |
| Rabbit anti-MMP1 | Bioss | Cat# bs-0424R; RRID: AB_10858058 |
| Rabbit anti-STAT3 | Bioss | Cat# bsm-52235R |
| Rabbit anti-pSTAT3 | Bioss | Cat# bs-1658R;RRID: AB_10855117 |
| Goat anti-rabbit IgG | Bioss | Cat# bs-0295G; RRID: AB_10856483 |
| Rabbit anti-mIL6R | Bioss | Cat# bs-23660R |
| Rabbit anti-MMP13 | Bioss | Cat# bs-10250R |
| Rabbit anti-IL6ST | BioLegend | Cat# 149401; RRID: AB_2565294 |
| Rabbit anti-mIL6R | BioLegend | Cat# 115803; RRID: AB_313674 |
| Rabbit anti-IL1RⅡ | Abcam | Cat# ab212208 |
| Mouse anti-TNFR1 | Abcam | Cat# ab194814; RRID: AB_2889242 |
| Goat anti-rabbit IgG H&L (Alexa Fluor® 488) | Abcam | Cat# ab150077; RRID: AB_2630356 |
| Goat anti-mouse IgG H&L (Alexa Fluor® 488) | Abcam | Cat# ab150113; RRID: AB_2576208 |
| Rabbit anti-CD81 | Abcam | Cat# ab109201; RRID: AB_10866464 |
| Rabbit anti-CD9 | Abcam | Cat# ab307085 |
| Rabbit anti-TSG101 | Abcam | Cat# ab125011; RRID: AB_10974262 |
| Rabbit anti-HSP27 | Abcam | Cat# ab12351; RRID: AB_299035 |
| Rabbit anti-β Tubulin | Abcam | Cat# ab6046; RRID: AB_2210370 |
| Rabbit anti-TNFR1 | Abcam | Cat# ab223352 |
| Rabbit anti-IL1RⅡ | Abcam | Cat# ab273025 |
| Biological samples | ||
| Primary mouse chondrocytes | C57BL/6 mice in this study | N/A |
| Mouse bone marrow macrophages | C57BL/6 mice in this study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase D | Roche | Cat# roche.11088858001 |
| Fetal bovine serum | Hyclone | Cat# SH30406.02 |
| Recombinant mouse M-CSF | R&D Systems | Cat# 416-ML-050/CF |
| Recombinant mouse RANKL | R&D Systems | Cat# 462-TR-010/CF |
| Recombinant human TNFα | R&D Systems | Cat# 210-TA-020 |
| Recombinant human IL1β | Sigma-Aldrichs | Cat# SRP3083 |
| Recombinant human gp130Fc | R&D Systems | Cat# 671-GP-100 |
| Recombinant mouse gp130Fc | R&D Systems | Cat# 468-MG-100 |
| Recombinant mouse hyper IL6 | R&D Systems | Cat# 9038-SR-025 |
| Penicillin-streptomycin | Hyclone | Cat# SV30010 |
| Complete Freund’s Adjuvant | Chondrex | Cat# 7001 |
| Incomplete Freund’s Adjuvant | Chondrex | Cat# 7002 |
| Chicken collagen-II | Chondrex | Cat# 20011 |
| RIPA buffer | Thermo-Scientific | Cat# 89901 |
| Paraformaldehyde | Servicebio | G1101 |
| EDTA | Servicebio | G1105 |
| Safranin O-Fast Green staining solution | Servicebio | G1053 |
| EV-depleted FBS | System Biosciences | Cat# EXO-FBS-250A-1 |
| Critical commercial assays | ||
| QUANTI-Blue™ Solution | InvivoGen | rep-qbs |
| TRAP stain kit | Sigma-Aldrich | Cat# SCR004 |
| BCA assay kit | Thermo Fisher | Cat# 23225 |
| Mouse Luminex Discovery Assay kit | R&D Systems | Cat# LXSAMSM |
| Exostep™ kit | Immunostep | Cat# ExoS-25-C9 |
| BD Quantibrite ™ Beads PE Fluorescence Quantitation Kit | BD | Cat# 340495 |
| Deposited data | ||
| Mouse primary chondrocytes RNA expression dataset | Liu et al.16 | Table S2 |
| Experimental models: Cell lines | ||
| HEK293T cells | ATCC | CRL-3216; RRID: CVCL_0063 |
| HEK-Blue™ IL6 Reporter cells | InvivoGen | hkb-hil6; RRID: CVCL_UF60 |
| HEK-Blue™ TNFα Reporter cells | InvivoGen | hkb-tnfdmyd; RRID: CVCL_UF25 |
| HEK-Blue™ IL1β Reporter cells | InvivoGen | hkb-il1bv2; RRID: CVCL_A8CL |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Laboratory Animal Center of TMMU | this manuscript |
| BALB/c mice | Laboratory Animal Center of TMMU | this manuscript |
| DBA/1J mice | Laboratory Animal Center of TMMU | this manuscript |
| Col2a1-cre mice | Jackson Laboratories | RRID:IMSR_JAX:003554 |
| il6stfl/fl mice | Cyagen Biosciences | S-CKO-03111 |
| Oligonucleotides | ||
| Primer used, see Table S3 | N/A | N/A |
| Recombinant DNA | ||
| IL6ST-eGFP-CD63 | This Paper | N/A |
| IL6ST-mCherry-syntenin | This Paper | N/A |
| TNFR1-eGFP-CD63 | This Paper | N/A |
| TNFR1-mCherry-syntenin | This Paper | N/A |
| IL1RⅡ-eGFP-CD63 | This Paper | N/A |
| IL1RⅡ-mCherry-syntenin | This Paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 9.4.0 | GraphPad | http://www.graphpad-prism.cn/; RRID: SCR_002798 |
| FlowJo v.10 | Tree Star, Inc. | https://www.flowjo.com/; RRID: SCR_008520 |
| AlphaFold2 | DeepMind | https://github.com/deepmind/alphafold |
| Pymol v2.5 | Schrödinger, Inc. | ; RRID: SCR_000305 |
| CT Analyser | Bruker | http://bruker-micro-ct-software/ |
| ImageJ v1.54f | National Institutes of Health | https://imagej.net/; RRID: SCR_003070 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Qinyu Ma (maqinyu@tmmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
3–7 days old C57BL/6 mice, 8–10 weeks old C57BL/6 mice, DBA/1J mice and BALB/c mice were purchased from Laboratory Animal Centre of Third Military Medical University (Chongqing, CHINA). Col2a1-cre and il6stfl/fl mice were obtained from Jackson Laboratories and Cyagen Biosciences. The Col2a1-cre transgenic mice were backcrossed with il6stfl/fl mice to generate chondrocyte-specific conditional knockout mice (il6stfl/flCol2a1-cre). Mice were given food and water ad libitum and maintained under 12 h light, 12 h dark cycles. Mice were euthanized according to the AVMA Guidelines for the Euthanasia of Animals. All animal breeding and experiments were approved by the Institutional Animal Care and Use Committee of Third Military Medical University.
Cell culture and reagents
Primary chondrocytes were isolated from newborn C57BL/6 mice (3–7 days) as described previously.45 Briefly, after mice were euthanized, articular cartilage from femoral heads, femoral condyles and tibial plateau was carefully dissected and subjected to overnight digestion with 0.5 mg/mL collagenase D. Chondrocytes were collected the next day and seeded on a culture dish (5 × 103 cells per cm2). Mouse bone marrow cells were isolated from hind limbs (femurs and tibias) of C57BL/6 mice and incubated with M-CSF (50 ng/mL) for 96 h to obtain BMMs, as described previously.10,46 HEK293T cells were purchased from the ATCC cell bank. All cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, and maintained at 37°C in 5% CO2. All cells were confirmed as free from mycoplasma contamination.
Method details
Reporter cell lines
HEK-Blue IL6 Reporter cells, HEK-Blue TNFα Reporter cells and HEK-Blue IL1β Reporter cells were used to detect the activation of their respective cytokine-mediated signaling pathways. To detect the suppression of IL6ST decoy EVs to IL6 trans-signaling, 1 × 104 HEK-Blue IL6 cells were planted in 96-well plate. After 24 h, cells were treated with IL6ST decoy EVs in the presence of 10 ng/mL hyper IL6 and incubated at 37°C in 5% CO2 for 6 h. QUANTI-Blue Solution (InvivoGen) was prepared according to the instructions. 20 μL cell supernatant and 180 μL QUANTI-Blue Solution were mixed in 96-well plates to form a 200 μL reaction system. After incubation at 37°C for 2 h in the dark, SEAP levels in the supernatant were quantified with optical density at 630 nm by a Microplate reader (Bio-Rad iMark). To detect the suppression of decoy EVs to TNFα or IL1β signaling transduction, HEK-Blue IL1β cells or HEK-Blue TNFα cells were administered with 5 ng/mL TNFα or 10 ng/mL IL1β, in the presence of respective decoy EVs. After 6 h, SEAP levels were detected with optical density at 630 nm using QUANTI-Blue Solution.
Mouse models
For CIA mouse model, DBA/1J mice were intradermally injected with 100 μL Complete Freund’s Adjuvant (CFA) and chicken collagen-Ⅱ (CⅡ) emulsion at the base of the tail. The booster injection was administrated on day 21 with CⅡ and IFA. For EAM mouse model, BALB/c mice were immunized by intradermal injection of 200 μg myosin-binding protein C emulsified with CFA containing Mycobacterium tuberculosis on the back of mice on days 0, 7, and 14. The mice were given 500 ng pertussis toxin intraperitoneally immediately after the first immunization. For K/BxN STA mouse model, DBA1/J mice were injected with 150 μL of serum from K/BxN mice on days 0 and 2 as described previously.47 For experimental OA mouse models, mice were either induced a chronic and low-grade inflammation with ACLT surgery, or an acute and profound inflammation with CIOA as described previously.48,49 To harvest cartilage explants, the femoral head of 8–10 weeks old C57BL/6 mice were collected and cultured with FBS-free DMEM for 24 h. Subsequently, the cartilage explants were cultured with hyper IL6 in the presence or absence of sgp130Fc. After 72 h, the supernatant was collected for Luminex assay. In the protective treatment experiment, CIA mice were intravenously administrated with saline, 2 mg/kg methotrexate, 3 × 1011 wild-type EVs and 3 × 1011 combinatorial decoy EVs every three days from day 3 to day 21 after the first immunization. In the therapeutic treatment experiment, CIA mice were intravenously administrated with saline, 3 × 1010 decoy EVs and 3 × 1011 decoy EVs every three days from day 22 to day 40. The clinical RA score was recorded as described previously.50
In vivo toxicity detection
To detect EV toxicity in vivo, male C57BL/6 mice were randomly administrated intravenous or intraperitoneal injections of saline, wild-type EVs, and IL6ST decoy EVs. Mice were administrated injections at five-day intervals. The body weight of mice was measured on day 1 and 16 post the initial EV injection, while whole blood samples were collected from the orbital cavity for routine and biochemical analyses. Subsequently, mice were euthanized, liver samples were collected, fixed in 4% paraformaldehyde, and embedded in paraffin for subsequent histological experiments.
Osteoclast differentiation and TRAP staining assay
3 × 103 BMMs were seeded in 96-well plates and cultured with DMEM at 37°C for 24 h. After adhesion, BMMs were stimulated with 50 ng/mL hyper IL6 or 100 ng/mL RANKL, respectively, and incubated at 37°C in 5% CO2 for 5 days. TRAP staining assay was performed to evaluate osteoclast formation using a TRAP stain kit. Briefly, cells were fixed with 4% paraformaldehyde for 20 min and washed with PBS for three times. The TRAP staining solution was prepared according to the manufacturer’s instructions. Cells were incubated with staining solution in the dark for 1 h, followed by PBS washing and microscopic observation.
Plasmid design and construction
All protein sequences were obtained from UniProt (entry IDs: IL6ST: Q00560; TNFR1: P25118; IL1RⅡ: P27931; CD63: P08962; syntenin: O00560; eGFP: C5MKY7; mCherry: X5DSL3; PDGFRβ: P09619) to generate chimeric proteins (protein sequences available on Table S1; S2). Sequences were linked together by a “GGGGS” flexible linker. The molecular 3D structure of chimeric proteins was predicted using AlphaFold2 and visualized using PyMOL software. To construct the expression vectors of chimeric constructs, protein sequences were reversely translated into DNA sequences using codon optimization. Then DNA inserts were synthesized (Gen 9) and cloned into a pCMV6-AC-Myc DNA vector at BamHI and XhoI sites using PCR amplification, enabling recombinant protein expression under the control of a CMV promoter. All cloned sequences were identified by DNA sequencing. All constructs were sourced through GenScript. For Transient plasmid transfection, 5 × 105 HEK293T cells were seeded on 6-well plates and transfected with 3 μg CD63-eGFP-IL6ST or IL6ST-mCherry-syntenin DNA plasmid using 1 mg/mL Polyethylenimine Linear (PEI) MW40000 with a 3:1 ratio of PEI to DNA (w/w), respectively. After 48 h of cell transfection, cell culture supernatants were collected for EV isolation.
Lentivirus transfection and cell transfection
HEK293T cells were co-transfected with transfer plasmids containing sorting proteins fused to fluorescent proteins, packaging plasmids and envelope vectors. Cell supernatant was collected after 72 h of co-transfection and ultracentrifuged at 50,000 g for 2 h at 4°C for lentivirus purification. The pelleted lentiviral particles were then resuspended in PBS and stored at −80°C. To obtain stable cell lines, 5 × 105 HEK293T cells were transfected with 5 μL CD63-eGFP-IL6ST or IL6ST-mCherry-syntenin virus respectively. For stably production of IL6ST-CD63/syntenin EVs, 5 × 105 HEK293T cells were engineered by co-transfection with 2.5 μL CD63-eGFP-IL6ST and 2.5 μL IL6ST-mCherry-syntenin virus. The stable expression cells were selected with puromycin (CD63-eGFP-IL6ST) or neomycin (IL6ST-mCherry-syntenin), starting at 48 h after infection, for at least 10 days prior to other experiments.
Luminex assay
For cytokine detection in vitro, the supernatant of mouse cartilage explants was collected and centrifuged at 16,000g for 15 min. Samples were then treated with Mouse Luminex Discovery Assay kit according to the manufacturer’s instructions, and the expression of multiple cytokines was tested using a Bio-Rad Bio-Plex analyzers. For cytokine detection in mouse serum, blood samples were collected and clotted at room temperature for 1 h, followed by centrifugation at 2,000 g for 20 min. Serum samples were subsequently collected and subjected to analysis using a Mouse Luminex Discovery Assay kit and a Bio-Rad analyzer. All standards and samples were set up in triplicate, averaged, and then the mean fluorescence intensity of the blank samples was subtracted. A standard curve was created to calculate the final concentration.
Western blotting
Cell and EV samples were collected, washed with PBS, and subsequently suspended in RIPA buffer supplemented with 10 × protease inhibitor. Tissue homogenate was pelleted twice at 10,000 g for 10 min at 4°C. Protein concentration was determined using a BCA assay kit, followed by dilution of the total protein with 5× loading buffer and incubation at 100°C for 10 min. For SDS-PAGE electrophoresis, 30 μg of proteins were loaded onto SDS-PAGE gel and run at 120 V for 1 h. Proteins were then transferred to an ImmunoBlot PVDF membrane using the Bio-Rad transfer system at 100 V for 120 min. Then the PVDF membrane was soaked in 5% BSA blocking solution and blocked for 2 h at room temperature. Membranes were incubated overnight at 4°C with primary antibodies: rabbit anti-GAPDH at 1:10,000, rabbit anti-IL6ST at 1:2000, rabbit anti-eGFP at 1:1,000, rabbit anti-mCherry at 1:1,000, rabbit anti-TIMP1 at 1:1,000, rabbit anti-TIMP3 at 1:1,000, rabbit anti-ADAMTS4 at 1:500, rabbit anti-ADAMTS5 at 1:500, rabbit anti-RUNX2 at 1:1,000, rabbit anti-MMP1 at 1:1,000, rabbit anti-STAT3 at 1:1,000, rabbit anti-pSTAT3 at 1:1,000. Membranes were washed three times with TBST and probed with secondary antibody Goat Anti-Rabbit IgG for 60 min at RT. Following another washing with TBST, membranes were exposed using ChemiDoc XRS+ imaging system (Bio-Rad).
Cell analysis using flow cytometry
Cells were planted into 35 mm Petri dishes. After 24 h, cells were digested with 0.02% EDTA and washed with PBS for three times and collected by centrifugation at 1,000g for 10 min at 4°C. 1 × 105 cells were resuspended in 50 μL flow cytometry staining buffer and stained with antibodies against IL6ST (1:200), mIL6R (1:200), OSMR (1:200), TNFR1(1:50) and IL1R (1:200) at 4°C for 30 min. Cells were then washed with flow cytometry staining buffer for three times and incubated with secondary antibody (Alexa Fluor 488, Goat anti-IgG (H + L)) for 30 min at 4°C in the dark. Finally, cells were washed with PBS for three times and resuspended for flow cytometry analysis using a CytoFLEX (Beckman Coulter).
EV analysis using flow cytometry
A Exostep kit was used for flow cytometry analysis of EVs. Briefly, 1 × 108 EV resuspended in 50 μL PBS were mixed with 50 μL superparamagnetic capture beads overnight at room temperature. Then, the bead-bound EVs tube was incubated with 5 μL primary antibodies for 1 h at 4°C. The samples were then washed with 1× assay buffer and magnetic beads were collected and incubated with 5 μL secondary antibody for 30 min at 4°C. After incubation, the samples were washed with 1× assay buffer. Magnetic beads were collected, resuspended in 300 μL 1× assay buffer and placed on ice to for further flow cytometry analysis. To quantify the number of decoy receptors on the EV membrane, BD Quantibrite Beads PE Fluorescence Quantitation Kit was used. Isolated EVs were incubated with PE-labelled antibodies against respective receptors for 1 h at room temperature. BD Quantibrite PE tube was run at the same instrument settings and the MFI values were converted into the number of PE molecules per bead. By using known ratios of MFI to PE molecules, the number of decoy receptor on EVs was estimated.
Micro-CT
Mice were euthanized at the respective endpoints and their knee and ankle joints were harvested. After fixation with 4% paraformaldehyde for 24 h, the joints were scanned using a Bruker MicroCT Skyscan 1272 system (Kontich, Belgium). After dataset reconstruction and localization, analysis of the calcaneus surface erosion was performed using a customized script in ImageJ. Osteophyte development and all other bone morphological parameters were assessed using the CT Analyser (Kontich, Belgium) analysis software.
Histological analysis
Mouse joints were fixed with 4% paraformaldehyde after the skin and muscle were removed. The joints were decalcified with EDTA for 4 weeks and embedded in paraffin. Continuous coronal sections (5 μm thick) were made of the entire knee or ankle joint. Each of five sections from both groups was stained with Safranin O-Fast Green to observe cartilage destruction. The OARSI scoring system (grade 0–6) and the Mankin scoring system were used to assess the cartilage degeneration of knee joint. Cartilage damage was determined by measuring 3–5 evenly distributed thicknesses in the medial tibial plateau. For immunohistochemical staining, sections were dried, deparaffinized in xylene, rehydrated with ethanol gradients. Sections were incubated with 0.3% H2O2 methanol for 5 min to block endogenous peroxidase activity. After blocking with 10% FBS, sections were incubated with rabbit anti-NITEGE (1:200), rabbit anti-mIL6R (1:200) and rabbit anti-MMP13 (1:200) for 1 h at room temperature, followed by incubated with biotinylated secondary antibody for 30 min at 37°C. DBA substrate solution was added on the slides to reveal the color of antibody staining. German semi-quantitative scoring system was used to evaluate the IHC scores, as described in previously.9
Purification of EVs
EV isolation was performed using a series of differential ultracentrifugation in combination with sucrose cushion ultracentrifugation steps as described previously.10,51 For EV purification from cell culture supernatant, cells were first cultured with medium containing 10% EV-depleted FBS (System Biosciences). After 24 h, the supernatant was collected and centrifuged at 1,000 g for 15 min and 3,000 g for 15 min to remove cell debris and apoptotic bodies (Beckman Coulter, Allegra X-15R). Subsequently, the supernatant was centrifuged at 18,000 g for 30 min to further remove any large particles. The supernatant was ultracentrifuged at 110,000 g for 70 min at 4°C (Type 70 Ti rotor) to pellet EVs. The pelleted EVs were washed in 1× particle-free PBS, loaded over 30% sucrose solution (diluted with D2O) slowly without mixing the two layers, and centrifuged at 110,000 g, 4°C for 2 h (SW 32 Ti rotor). For tissue samples, EVs were collected as previously described.52 Briefly, the tissues were gently dissociated into small pieces and incubated with collagenase D and DNase I for 30 min at 37°C. After filtration through a 70 μm pore size filter, EVs were collected through differential ultracentrifugation in combination with sucrose cushion ultracentrifugation. For serum EVs, blood samples were collected from mice, left for 1 h at room temperature, and centrifuged at 1,000 g for 20 min at 4°C. Ultracentrifugation operations were conducted using an Optima XE-90 (Beckman Coulter). After elution with PBS, EV samples were concentrated using an Amicon Ultra-4 10 kDa centrifugal filter (Millipore) and stored at −80°C for further experiments.
Characterization of the purified EVs
We characterized and quantified the separated EVs through several approaches including western blot analysis of well-established pan-EV markers (CD9, CD81, TSG101 and HSP27), transmission electron microscopy and NTA according to Minimal Information 2018 for Studies of Extracellular Vesicles (MISEV2018) guidelines.53 EV samples were resuspended with 10× RIPA buffer and lysed by sonication. Protein concentration was determined using BCA assay. After SDS-PAGE and blotting, the membrane was incubated overnight at 4°C with primary antibodies against CD81 (1: 1,000), CD9 (1: 1,000), TSG101 (1: 2,000), HSP27 (1: 1,000) and β-tubulin (1: 500). Membranes were washed and incubated with secondary antibody. After washing with TBST, membranes were exposed using ChemiDoc XRS+ imaging system (Bio-Rad). NTA was performed to evaluate the distribution size and concentration of EV. EV samples were diluted with PBS to a final volume of 1 mL. NTA measurement was operated using Nanosight NS300 according to the manufacturer’s software manual as previously described.10
Transmission electron microscopy
EV samples were resuspended in PBS to the optimal concentration and mixed with 4% paraformaldehyde at 1:1 ratio. A drop of 10 μL resuspended EVs was added to Formvar-carbon coated grids and fixed for 20 min at room temperature. The grids were placed on 100 μL PBS droplet and washed twice for 3 min, then transferred to 100 μL 50 mM glycine droplet for 3 min and repeated three times. For Immunogold staining, the grids were blocked in 100 μL 5% BSA blocking buffer for 10 min and washed six times with PBS. The grids were incubated with 20 μL primary antibodies against IL6ST, TNFR1, IL1RⅡ and IgG for 30 min. Then, the grids were incubated with gold-labeled secondary antibody for 30 min, transferred to 1% glutaraldehyde for 2 min, and stained with uranyl acetate for 15 s. Grids were dried naturally and viewed with FEI Tecnai 20 at an accelerating voltage of 80 kV, digital images were obtained.
RT-qPCR
Mouse paws were segmented into small pieces and immersed in 2 mL TRIzol reagent. Tissue homogenization was performed using a low-temperature tissue grinder machine. The supernatant was collected by centrifugation at 12,000 g for 10 min at 4°C. Subsequently, total RNA was extracted according to the manufacturer’s instructions of TRIZOL reagent. RNA concentration was measured using NanoDrop2000. The cDNA was synthesized by reverse transcription using PrimeScript RT Reagent Kit. SYBR Premix Ex Tag Ⅱ was used for PCR reaction. The primer sequences were listed in Table S3.
Quantification and statistical analysis
Statistical analyses and graphing in this study were performed using GraphPad Prism software (Version Prism 9.4.0). Values are expressed as mean ± SEM. Number of animals or independent replicates are denoted in the figure legend. A Student’s two-sided t test or two-tailed Mann-Whitney U test was performed for two-group comparisons. Multiple comparisons were performed using an ANOVA or Kruskal-Wallis test, followed by Sidak’s, Dunn’s, Dunnett’s or Tukey’s post hoc tests for intergroup comparisons. p < 0.05 were considered statistically significant.
Acknowledgments
This work was supported by a grant from the National Natural Science Foundation of China (82202710 to Q.M.) and a grant from the Chongqing Postdoctoral Program for Innovative Talents (CQBX202202 to Q.M.).
Author contributions
Conceptualization, Q.M.; methodology, M.L., K.W., X.W., and Q.M.; software, M.L., K.W., and Q.M.; formal analysis, M.L., K.W., X.W., X.G., H.T., H.X., P.Y., L.Z., C.D., S.D., and Q.M.; investigation, M.L., K.W., X.W., X.G., H.T., J.W., Z.M., J.X., F.L., and Q.M.; resources, M.L., X.W., H.X., and J.W.; writing – original draft, M.L. and Q.M.; writing – review & editing, M.L., F.L., and Q.M.; visualization, M.L., K.W., H.X., P.Y., L.Z., C.D., S.D., and Q.M.; supervision, F.L. and Q.M.; project administration, F.L. and Q.M.; funding acquisition, Q.M.
Declaration of interests
The authors report no conflict of interest.
Published: October 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101228.
Contributor Information
Fei Luo, Email: luofly1009@hotmail.com.
Qinyu Ma, Email: maqinyu@tmmu.edu.cn.
Supplemental information
Data and code availability
-
•
Previous published datasets that were reanalyzed are available under https://doi.org/10.1016/j.joca.2015.05.011.16
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Chen Z., Bozec A., Ramming A., Schett G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat. Rev. Rheumatol. 2019;15:9–17. doi: 10.1038/s41584-018-0109-2. [DOI] [PubMed] [Google Scholar]
- 2.Cronstein B.N., Aune T.M. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat. Rev. Rheumatol. 2020;16:145–154. doi: 10.1038/s41584-020-0373-9. [DOI] [PubMed] [Google Scholar]
- 3.Wang W., Zhou H., Liu L. Side effects of methotrexate therapy for rheumatoid arthritis: A systematic review. Eur. J. Med. Chem. 2018;158:502–516. doi: 10.1016/j.ejmech.2018.09.027. [DOI] [PubMed] [Google Scholar]
- 4.Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012;8:1237–1247. doi: 10.7150/ijbs.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Usón J., Balsa A., Pascual-Salcedo D., Cabezas J.A., Gonzalez-Tarrio J.M., Martín-Mola E., Fontan G. Soluble interleukin 6 (IL-6) receptor and IL-6 levels in serum and synovial fluid of patients with different arthropathies. J. Rheumatol. 1997;24:2069–2075. [PubMed] [Google Scholar]
- 6.Aletaha D., Smolen J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA. 2018;320:1360–1372. doi: 10.1001/jama.2018.13103. [DOI] [PubMed] [Google Scholar]
- 7.Hunter C.A., Jones S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015;16:448–457. doi: 10.1038/ni.3153. [DOI] [PubMed] [Google Scholar]
- 8.Kalluri R., LeBleu V.S. The biology, function, and biomedical applications of exosomes. Science. 2020;367 doi: 10.1126/science.aau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Q., Liang M., Wu Y., Luo F., Ma Z., Dong S., Xu J., Dou C. Osteoclast-derived apoptotic bodies couple bone resorption and formation in bone remodeling. Bone Res. 2021;9:5. doi: 10.1038/s41413-020-00121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Q., Liang M., Wu Y., Dou C., Xu J., Dong S., Luo F. Small extracellular vesicles deliver osteolytic effectors and mediate cancer-induced osteolysis in bone metastatic niche. J. Extracell. Vesicles. 2021;10 doi: 10.1002/jev2.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadu L., Krishnan R.H., Akshaya R.L., Das U.R., Satishkumar S., Selvamurugan N. Exosomes in bone remodeling and breast cancer bone metastasis. Prog. Biophys. Mol. Biol. 2022;175:120–130. doi: 10.1016/j.pbiomolbio.2022.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Mathieu M., Martin-Jaular L., Lavieu G., Théry C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019;21:9–17. doi: 10.1038/s41556-018-0250-9. [DOI] [PubMed] [Google Scholar]
- 13.Baietti M.F., Zhang Z., Mortier E., Melchior A., Degeest G., Geeraerts A., Ivarsson Y., Depoortere F., Coomans C., Vermeiren E., et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012;14:677–685. doi: 10.1038/ncb2502. [DOI] [PubMed] [Google Scholar]
- 14.Herrmann I.K., Wood M.J.A., Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021;16:748–759. doi: 10.1038/s41565-021-00931-2. [DOI] [PubMed] [Google Scholar]
- 15.Wiklander O.P.B., Brennan M.Á., Lötvall J., Breakefield X.O., El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X., Liu R., Croker B.A., Lawlor K.E., Smyth G.K., Wicks I.P. Distinctive pro-inflammatory gene signatures induced in articular chondrocytes by oncostatin M and IL-6 are regulated by Suppressor of Cytokine Signaling-3. Osteoarthritis Cartilage. 2015;23:1743–1754. doi: 10.1016/j.joca.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 17.Fischer M., Goldschmitt J., Peschel C., Brakenhoff J.P., Kallen K.J., Wollmer A., Grötzinger J., Rose-John S. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 1997;15:142–145. doi: 10.1038/nbt0297-142. [DOI] [PubMed] [Google Scholar]
- 18.Feng W., Yang P., Liu H., Zhang F., Li M. IL-6 promotes low concentration of RANKL-induced osteoclastic differentiation by mouse BMMs through trans-signaling pathway. J. Mol. Histol. 2022;53:599–610. doi: 10.1007/s10735-022-10077-7. [DOI] [PubMed] [Google Scholar]
- 19.McGregor N.E., Murat M., Elango J., Poulton I.J., Walker E.C., Crimeen-Irwin B., Ho P.W.M., Gooi J.H., Martin T.J., Sims N.A. IL-6 exhibits both cis- and trans-signaling in osteocytes and osteoblasts, but only trans-signaling promotes bone formation and osteoclastogenesis. J. Biol. Chem. 2019;294:7850–7863. doi: 10.1074/jbc.RA119.008074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta D., Wiklander O.P.B., Görgens A., Conceição M., Corso G., Liang X., Seow Y., Balusu S., Feldin U., Bostancioglu B., et al. Amelioration of systemic inflammation via the display of two different decoy protein receptors on extracellular vesicles. Nat. Biomed. Eng. 2021;5:1084–1098. doi: 10.1038/s41551-021-00792-z. [DOI] [PubMed] [Google Scholar]
- 21.Silva A.M., Lázaro-Ibáñez E., Gunnarsson A., Dhande A., Daaboul G., Peacock B., Osteikoetxea X., Salmond N., Friis K.P., Shatnyeva O., Dekker N. Quantification of protein cargo loading into engineered extracellular vesicles at single-vesicle and single-molecule resolution. J. Extracell. Vesicles. 2021;10 doi: 10.1002/jev2.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridker P.M., MacFadyen J.G., Thuren T., Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur. Heart J. 2020;41:2153–2163. doi: 10.1093/eurheartj/ehz542. [DOI] [PubMed] [Google Scholar]
- 23.Mantovani A., Locati M., Vecchi A., Sozzani S., Allavena P. Decoy receptors: a strategy to regulate inflammatory cytokines and chemokines. Trends Immunol. 2001;22:328–336. doi: 10.1016/s1471-4906(01)01941-x. [DOI] [PubMed] [Google Scholar]
- 24.Levescot A., Chang M.H., Schnell J., Nelson-Maney N., Yan J., Martínez-Bonet M., Grieshaber-Bouyer R., Lee P.Y., Wei K., Blaustein R.B., et al. IL-1beta-driven osteoclastogenic Tregs accelerate bone erosion in arthritis. J. Clin. Invest. 2021;131 doi: 10.1172/JCI141008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ansari A.M., Ahmed A.K., Matsangos A.E., Lay F., Born L.J., Marti G., Harmon J.W., Sun Z. Cellular GFP Toxicity and Immunogenicity: Potential Confounders in in Vivo Cell Tracking Experiments. Stem Cell Rev. Rep. 2016;12:553–559. doi: 10.1007/s12015-016-9670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grzelak C.A., Goddard E.T., Lederer E.E., Rajaram K., Dai J., Shor R.E., Lim A.R., Kim J., Beronja S., Funnell A.P.W., Ghajar C.M. Elimination of fluorescent protein immunogenicity permits modeling of metastasis in immune-competent settings. Cancer Cell. 2022;40:1–2. doi: 10.1016/j.ccell.2021.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang H., Beier F. Mouse models of osteoarthritis: modelling risk factors and assessing outcomes. Nat. Rev. Rheumatol. 2014;10:413–421. doi: 10.1038/nrrheum.2014.46. [DOI] [PubMed] [Google Scholar]
- 28.Cheng L., Hill A.F. Therapeutically harnessing extracellular vesicles. Nat. Rev. Drug Discov. 2022;21:379–399. doi: 10.1038/s41573-022-00410-w. [DOI] [PubMed] [Google Scholar]
- 29.Andaloussi S.E.L., Mäger I., Breakefield X.O., Wood M.J.A. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 30.van der Grein S.G., Defourny K.A.Y., Rabouw H.H., Goerdayal S.S., van Herwijnen M.J.C., Wubbolts R.W., Altelaar M., van Kuppeveld F.J.M., Nolte-'t Hoen E.N.M. The encephalomyocarditis virus Leader promotes the release of virions inside extracellular vesicles via the induction of secretory autophagy. Nat. Commun. 2022;13:3625. doi: 10.1038/s41467-022-31181-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wozniak A.L., Adams A., King K.E., Dunn W., Christenson L.K., Hung W.T., Weinman S.A. The RNA binding protein FMR1 controls selective exosomal miRNA cargo loading during inflammation. J. Cell Biol. 2020;219 doi: 10.1083/jcb.201912074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szul T., Bratcher P.E., Fraser K.B., Kong M., Tirouvanziam R., Ingersoll S., Sztul E., Rangarajan S., Blalock J.E., Xu X., Gaggar A. Toll-Like Receptor 4 Engagement Mediates Prolyl Endopeptidase Release from Airway Epithelia via Exosomes. Am. J. Respir. Cell Mol. Biol. 2016;54:359–369. doi: 10.1165/rcmb.2015-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadri M., Hirosawa N., Le J., Romero H., Martellucci S., Kwon H.J., Pizzo D., Ohtori S., Gonias S.L., Campana W.M. Tumor necrosis factor receptor-1 is selectively sequestered into Schwann cell extracellular vesicles where it functions as a TNFalpha decoy. Glia. 2022;70:256–272. doi: 10.1002/glia.24098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller M.D., Ching K.L., Liang F.X., Dhabaria A., Tam K., Ueberheide B.M., Unutmaz D., Torres V.J., Cadwell K. Decoy exosomes provide protection against bacterial toxins. Nature. 2020;579:260–264. doi: 10.1038/s41586-020-2066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deretic V., Jiang S., Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012;22:397–406. doi: 10.1016/j.tcb.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Courtemanche N., Pollard T.D., Chen Q. Avoiding artefacts when counting polymerized actin in live cells with LifeAct fused to fluorescent proteins. Nat. Cell Biol. 2016;18:676–683. doi: 10.1038/ncb3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia B., Jeon C.O. High-throughput recombinant protein expression in Escherichia coli: current status and future perspectives. Open Biol. 2016;6 doi: 10.1098/rsob.160196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaur J., Kumar A., Kaur J. Strategies for optimization of heterologous protein expression in E. coli: Roadblocks and reinforcements. Int. J. Biol. Macromol. 2018;106:803–822. doi: 10.1016/j.ijbiomac.2017.08.080. [DOI] [PubMed] [Google Scholar]
- 39.Tao W., Evans B.G., Yao J., Cooper S., Cornetta K., Ballas C.B., Hangoc G., Broxmeyer H.E. Enhanced green fluorescent protein is a nearly ideal long-term expression tracer for hematopoietic stem cells, whereas DsRed-express fluorescent protein is not. Stem Cell. 2007;25:670–678. doi: 10.1634/stemcells.2006-0553. [DOI] [PubMed] [Google Scholar]
- 40.Fischer J.A.A., Hueber A.J., Wilson S., Galm M., Baum W., Kitson C., Auer J., Lorenz S.H., Moelleken J., Bader M., et al. Combined inhibition of tumor necrosis factor alpha and interleukin-17 as a therapeutic opportunity in rheumatoid arthritis: development and characterization of a novel bispecific antibody. Arthritis Rheumatol. 2015;67:51–62. doi: 10.1002/art.38896. [DOI] [PubMed] [Google Scholar]
- 41.Kloppenburg M., Peterfy C., Haugen I.K., Kroon F., Chen S., Wang L., Liu W., Levy G., Fleischmann R.M., Berenbaum F., et al. Phase IIa, placebo-controlled, randomised study of lutikizumab, an anti-interleukin-1alpha and anti-interleukin-1beta dual variable domain immunoglobulin, in patients with erosive hand osteoarthritis. Ann. Rheum. Dis. 2019;78:413–420. doi: 10.1136/annrheumdis-2018-213336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fleischmann R.M., Bliddal H., Blanco F.J., Schnitzer T.J., Peterfy C., Chen S., Wang L., Feng S., Conaghan P.G., Berenbaum F., et al. A Phase II Trial of Lutikizumab, an Anti-Interleukin-1alpha/beta Dual Variable Domain Immunoglobulin, in Knee Osteoarthritis Patients With Synovitis. Arthritis Rheumatol. 2019;71:1056–1069. doi: 10.1002/art.40840. [DOI] [PubMed] [Google Scholar]
- 43.Guo Q., Wang Y., Xu D., Nossent J., Pavlos N.J., Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6:15. doi: 10.1038/s41413-018-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Usher K.M., Zhu S., Mavropalias G., Carrino J.A., Zhao J., Xu J. Pathological mechanisms and therapeutic outlooks for arthrofibrosis. Bone Res. 2019;7:9. doi: 10.1038/s41413-019-0047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gosset M., Berenbaum F., Thirion S., Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 2008;3:1253–1260. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]
- 46.Li X., Wang L., Huang B., Gu Y., Luo Y., Zhi X., Hu Y., Zhang H., Gu Z., Cui J., et al. Targeting actin-bundling protein L-plastin as an anabolic therapy for bone loss. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abb7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arandjelovic S., Perry J.S.A., Zhou M., Ceroi A., Smirnov I., Walk S.F., Shankman L.S., Cambré I., Onengut-Gumuscu S., Elewaut D., et al. ELMO1 signaling is a promoter of osteoclast function and bone loss. Nat. Commun. 2021;12:4974. doi: 10.1038/s41467-021-25239-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorenz J., Grässel S. Experimental osteoarthritis models in mice. Methods Mol. Biol. 2014;1194:401–419. doi: 10.1007/978-1-4939-1215-5_23. [DOI] [PubMed] [Google Scholar]
- 49.Zhang H., Lin C., Zeng C., Wang Z., Wang H., Lu J., Liu X., Shao Y., Zhao C., Pan J., et al. Synovial macrophage M1 polarisation exacerbates experimental osteoarthritis partially through R-spondin-2. Ann. Rheum. Dis. 2018;77:1524–1534. doi: 10.1136/annrheumdis-2018-213450. [DOI] [PubMed] [Google Scholar]
- 50.Pongratz G., Anthofer J.M., Melzer M., Anders S., Grässel S., Straub R.H. IL-7 receptor alpha expressing B cells act proinflammatory in collagen-induced arthritis and are inhibited by sympathetic neurotransmitters. Ann. Rheum. Dis. 2014;73:306–312. doi: 10.1136/annrheumdis-2012-202944. [DOI] [PubMed] [Google Scholar]
- 51.Guo J., Wang F., Hu Y., Luo Y., Wei Y., Xu K., Zhang H., Liu H., Bo L., Lv S., et al. Exosome-based bone-targeting drug delivery alleviates impaired osteoblastic bone formation and bone loss in inflammatory bowel diseases. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2022.100881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crescitelli R., Lässer C., Lötvall J. Isolation and characterization of extracellular vesicle subpopulations from tissues. Nat. Protoc. 2021;16:1548–1580. doi: 10.1038/s41596-020-00466-1. [DOI] [PubMed] [Google Scholar]
- 53.Théry C., Witwer K.W., Aikawa E., Alcaraz M.J., Anderson J.D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G.K., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018;7 doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Previous published datasets that were reanalyzed are available under https://doi.org/10.1016/j.joca.2015.05.011.16
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.