

Summary
Although promising, dendritic cell (DC) vaccines still provide limited clinical benefits, mainly due to the immunosuppressive tumor microenvironment (TME) and the lack of tumor-associated antigens (TAAs). Oncolytic virus therapy is an ideal strategy to overcome immunosuppression and expose TAAs; therefore, they may work synergistically with DC vaccines. In this study, we demonstrate that oncolytic virus M1 (OVM) can enhance the antitumor effects of DC vaccines across diverse syngeneic mouse tumor models by increasing the infiltration of CD8+ effector T cells in the TME. Mechanically, we show that tumor cells counteract DC vaccines through the SIRPα-CD47 immune checkpoint, while OVM can downregulate SIRPα in DCs and CD47 in tumor cells. Since OVM upregulates PD-L1 in DCs, combining PD-L1 blockade with DC vaccines and OVM further enhances antitumor activity. Overall, OVM strengthens the antitumor efficacy of DC vaccines by targeting the SIRPα-CD47 axis, which exerts dominant immunosuppressive effects on DC vaccines.
Graphical abstract
Highlights
-
•
OVM effectively activates DC vaccines and initiates tumor-specific T cell responses
-
•
OVM reactivates tumor-suppressed DCs by downregulating both SIRPα and CD47
-
•
Adding PD-L1 blockade to OVM plus DC vaccine regimen further enhances antitumor efficacy
Dan et al. shows that the oncolytic virus M1 can boost the antitumor effect of DC vaccines by disrupting the SIRPα-CD47 immune checkpoint between tumor cells and DCs. This combination therapy also works better with PD-L1 blockade, offering a new strategy to fight cancer.
Introduction
Immunotherapy has attracted considerable attention in cancer therapy due to its durable control of tumor progression and prolongation of survival.1 Dendritic cells (DCs), known as the professional antigen-presenting cells, have an excellent capacity to take up, process, and present tumor antigens, prime naive T cell activation, and link innate immunity with adaptive immune responses.2 Due to these features, DCs have become a kind of promising clinical tool for cancer therapy.3,4 In 2010, the first DC-based autologous cancer vaccine, sipuleucel-T (Provenge), was approved by the US Food and Drug Administration (FDA) for treatment of prostate cancer.5 In 2020, the phase II clinical trial of treatment with another DC vaccine, ilixadencel combined with the tyrosine kinase inhibitor sunitinib for patients with renal cell carcinoma (RRC), achieved exciting results, showing an objective response rate and overall survival rate of 42.2% and 54%, respectively.6 To date, over 400 clinical studies based on DC vaccines have been carried out worldwide, aiming to treat various tumor types, including glioblastoma, acute myeloid leukemia, metastatic colorectal cancer, pancreatic cancer, high-risk melanoma, etc.7 Most clinical studies employ monocyte-derived DCs (Mo-DCs), which are induced from monocytes by granulocyte/monocyte colony-stimulating factor (GM-CSF) and interleukin-4 (IL-4), in the generation of DC vaccines because of the high purity of induction and relative ease at obtaining enough cells from peripheral blood.
While DC vaccines can induce tumor-specific T cells with minimal side effects, their efficacy remains limited, mainly owing to the immunosuppressive tumor microenvironment (TME) and the insufficient expression of tumor antigens.3,8,9 On the one hand, tumor cells reduce the expression of DC chemoattractant or cytokines to limit the survival, migration, and infiltration of DCs and produce inhibitory factors to suppress the differentiation and maturation of DCs. Moreover, immune checkpoints (such as programmed cell death receptor 1 [PD-1], programmed cell death-ligand 1 [PD-L1], and T cell immunoglobulin mucin-3 [TIM-3]) expressed on the surface of DCs also cripple their function.10,11 On the other hand, the low immunogenicity of tumors and the low exposure of tumor-associated antigens (TAAs) make it harder to identify ideal tumor-specific antigens, which are critical in the preparation of DC vaccines. These effects result in poor T cell priming function of endogenous DCs in the TME and may restrain the antitumor efficacy of DC vaccines. Hence, overcoming the inhibitory effects of the TME and promoting the release of TAAs are potential strategies for improving the therapeutic effect of DC vaccines.
Oncolytic virotherapy, a strategy that utilizes naturally existing or genetically modified viruses to selectively kill tumor cells without toxicity to normal cells, has shown promising effects in clinical and preclinical studies.12 Oncolytic virotherapy has also been reported to reverse tumor immunosuppression, promote antigen release and presentation, generate antitumor immunity, and achieve notable efficacy in combination with other cancer immunotherapies.13 Therefore, oncolytic viruses may have the capacity to alleviate the inhibitory effects of the TME and strengthen the efficacy of DC vaccines. Furthermore, oncolytic viruses can induce the antiviral type I interferon (IFN) pathway and promote IFN-α/β secretion, which are essential for the function of DCs and the transition to adaptive immune responses.14,15 Alphavirus M1 (OVM) is a novel natural oncolytic virus targeting matrix remodeling-associated 8 (MXRA8)-overexpressing and zinc-finger antiviral protein (ZAP)-deficient tumor cells16,17 and is nonpathogenic to nonhuman primates.18 In addition, a series of studies also reported that OVM can significantly increase T cell infiltration in the TME and further improve the antitumor effects of immune checkpoint blocking therapy in various tumor-bearing mouse models.19,20 However, whether OVM possesses the potential to enhance the antitumor efficacy of DC vaccines has not been studied.
In this study, we found that OVM strongly improves the antitumor effect of a DC vaccine against various syngeneic mouse tumor models. Furthermore, we identified the signal regulatory protein α (SIRPα)/CD47 axis as the key immunosuppressive mechanism for how tumor cells inhibit the maturation of DCs and the effects of DC vaccines. Treatment with OVM downregulates both SIRPα on the surface of DCs and CD47 on the surface of tumor cells, thus alleviating the inhibition of DCs and restoring their function to prime the T cell response. What’s more, OVM can also induce an increase in the expression of PD-L1 in DCs, and blocking PD-L1 further enhances the efficacy of combination therapy with OVM and DC vaccine. These findings highlight the crucial role of OVM as a dual inhibitor of the SIRPα/CD47 immune checkpoint axis and suggest the potential of an OVM plus DC vaccine to be developed as the arsenal for cancer treatment in the future.
Results
OVM enhances the efficacy of DC vaccine
Our previous findings that OVM is capable of activating DCs and triggering a systemic immune response in syngeneic mouse tumor models prompted us to investigate the possibility that OVM may enhance the antitumor activity of a DC vaccine derived from GM-CSF-stimulated bone marrow-derived DCs (BMDCs). As pulsing DCs with appropriate TAAs is the key step in preparing DC vaccines,4 we first tested the combination of DC vaccine and OVM in a model with a well-known antigen, ovalbumin (OVA). Immunocompetent C57BL/6 mice with subcutaneous B16-F10-OVA melanoma received intravenous injection of OVM once a day for 5 consecutive days, a single intratumoral injection of the OVA-peptide-loaded and lipopolysaccharide (LPS)-activated DC vaccine, or a combination of both (Figure S1A). Compared with either monotherapy, the combination treatment showed a significant delay in tumor growth and an increased survival time, implying that OVM was able to enhance the antitumor efficacy of DC vaccine (Figures 1A and S1B).
Figure 1.

OVM enhances the therapeutic efficacy of a DC vaccine
(A) C57BL/6J mice were implanted subcutaneously in the right flank with B16-F10-OVA cells and treated with vehicle (n = 9), OVM (n = 10), DC vaccine (stimulated by OVA and LPS) (n = 10), or OVM plus DC vaccine (n = 10) (tumor volumes were approximately 50 mm3). OVM was administered via tail vein injection for 5 consecutive days. And DC vaccine was administered through intratumoral injection once. Left: tumor growth curves. Center: the relative tumor proliferation rate (T/C %). Right: Kaplan-Meier survival curves.
(B) The mean fluorescence intensity (MFI) values of the activation markers CD69 and CD44 on the OT-I CD8+ T cells stimulated by differently treated DCs. DCs were subjected to one of the following treatments for 24 h: control (Ctrl), OVA (1 μg/mL, as positive control), OVM (1 MOI), or B16-F10-OVA oncolysate (1 mL). After treatment, the DCs were washed with PBS and cocultured with spleen-derived CD8+ T cells from OT-I mice at a 1:2 ratio for 48 h; n = 3.
(C) The MFI of CD69 and CD44 on the OT-I CD8+ T cells was assessed after stimulation by DCs at a 1:2 ratio for 48 h. These DCs were treated with UV-inactivated B16-F10-OVA oncolysate (1 mL) or UV-inactivated B16-F10 oncolysate (1 mL) for 24 h; n = 3.
(D–G) C57BL/6J or BALB/c mice were implanted subcutaneously in the right flank with B16-F10 (n = 9), CT-26 (n = 7), RM-1 (n = 8), or Pan02 (n = 7) cells and treated with vehicle, OVM, oncolysate-activated DC vaccine, or OVM combined with DC vaccine. OVM was administered via tail vein injection for 5 consecutive days. And DC vaccine was administered through intratumoral injection twice. Tumor growth curves (left), T/C % (center), and Kaplan-Meier survival curves (right) of B16-F10 (D), CT-26 (E), RM-1 (F), and Pan02 (G) tumor-bearing mouse models.
(H) C57BL/6J mice were implanted subcutaneously in the right flank with B16-F10 cells and treated with vehicle (n = 6) or OVM combined with B16-F10 oncolysate-activated DC vaccine (n = 6). OVM was administered via tail vein injection for 5 consecutive days. And DC vaccine was administered twice using different routes, including tail vein injection, intratumoral injection, or subcutaneous injection near the tumor site. Tumor growth curves (left), T/C % (center), and Kaplan-Meier survival curves (right) are shown. The p values were determined by one-way ANOVA or by the log rank test. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Despite the rapid growth of tumor sequencing data, identifying TAAs for preparation of a DC vaccine is still challenging. Oncolytic viruses may serve as an ideal source of TAAs to prepare a DC vaccine in that they can infect and lyse tumor cells, releasing TAAs into the oncolysate. To prove this, DCs were pretreated with negative control (NC), OVA (positive control), OVM, or oncolysate from OVM-infected B16-F10-OVA cells first, and then cocultured with OT-I CD8+ T cells (Figure S1C). Only the OVA- or oncolysate-treated DCs, and not the OVM-treated DCs, appreciably induced upregulation of CD69 and CD44 expression on OT-I CD8+ T cells (Figures 1B and S1D), indicating that OVA antigens were released by OVM-treated B16-F10-OVA cells, and these antigens were successfully presented by DCs to stimulate T cells. To further exclude the possibility that DCs were stimulated by the live OVM rather than TAAs in the oncolysates, DCs were incubated with UV-inactivated oncolysates derived from OVM-treated B16-F10-WT or B16-F10-OVA cells and then cocultured with OT-I CD8+ T cells (Figure S1E). Only DCs loaded with oncolysate from B16-F10-OVA cells, and not B16-F10-WT cells, successfully activated OT-I CD8+ T cells, as evidenced by the elevated expression of CD69 and CD44 (Figures 1C and S1F). Moreover, we evaluated the effects of LPS and oncolysate on the activation of DCs and found no significant difference between them (Figure S1G). These data indicate that OVM infection facilitated the release of TAAs, and the oncolysate-stimulated DCs can prime and activate T cells to initiate antitumor immunity.
The successful preparation of functional DC vaccines with oncolysate activation prompted us to further explore the combination effect of oncolysate-stimulated DC vaccine and OVM on refractory cancers, such as larger B16-F10 (tumor volume ∼100 mm3) melanoma, CT-26 colon cancer, RM-1 prostatic cancer, and Pan02 pancreatic cancer (Figure S1H). A similar enhancement of the DC vaccine by OVM was observed in all the tested animal models (Figures 1D–1G and S1J–S1M). In addition, as reported in our previous study, OVM can be intravenously administered and will accumulate in tumor sites,16,21 whereas the delivery methods for DC vaccines, including subcutaneous, intravenous, intralymphatic, and intratumoral injection, have been described, but the optimal route of administration remains undetermined.3 Therefore, we investigated whether the administration route of the DC vaccine affects the therapeutic efficacy of combination with OVM. In the B16-F10 tumor-bearing mouse model, OVM was injected via the tail vein, while an equivalent dose of the oncolysate-stimulated DC vaccine was delivered through intravenous, intratumoral, and subcutaneous injections (Figure S1I). By monitoring tumor growth curves and the T:C ratio, we found that intratumoral administration of the DC vaccine yielded a slightly better result, although no significant differences were observed in terms of survival rates (Figures 1H and S1N). These data reveal that the combined use of OVM and DC vaccine markedly improves the antitumor efficacy.
Combination of OVM and DC vaccine increases the infiltration and lymphocytotoxicity of T cells in the TME
Considering that one of the most important functions of DCs is to initiate an adaptive immune T cell response, we wondered whether combining the DC vaccine with OVM virotherapy can change the proportions of T cells in the TME. Advanced B16-F10-OVA tumor-bearing mice were randomized to receive OVM, DC vaccine, or both, and tumors were harvested the day after the last dose to analyze the infiltration and composition of T cells (Figure S2A). As expected, mice treated with the combination therapy had the smallest tumor volume (Figure S2B). Accordingly, the proportion of CD3+ T cells among viable cells in the combination group was the highest among the four tested groups (Figures 2A and 2C), with CD8+ T cells being the predominantly increased subset of T cells (Figures 2B and 2D–2G). The expression of activation marker CD44 on the surface of CD4+ T and CD8+ T cells was significantly increased compared with DC vaccine alone (Figures 2H and 2I). Further phenotypic analysis of tumor-infiltrating T cells also revealed a significantly increased amount of both central memory T (TCM) cells and effector memory T cells (TEM), including CD4+ and CD8+ T cells, in the combination group, suggesting the strongest tumor killing activity and antitumor memory effect (Figures 2J–2M and S2C). The exhausted PD-1+ CD4+ and CD8+ T cells (Tex) and regulatory CD4+ T cells (Tregs), in contrast, were markedly downregulated following treatment with OVM and DC vaccine (Figures 2N–2P), reflecting the activated antitumor immune microenvironment as well. In summary, these data demonstrate that the combination of OVM and DC vaccine strongly increases the infiltration of T cells, especially CD8+ T cells, in the TME and promotes the activation and memory phenotypes, whereas it decreases the exhausted phenotype of T cells.
Figure 2.

Combination of OVM and DC vaccine increases the infiltration and lymphocytotoxicity of T cells in the TME
(A–P) C57BL/6J mice were implanted subcutaneously in the right flank with B16-F10-OVA cells on day 0 and were treated with vehicle, OVM, OVA and LPS-activated DC vaccine, or OVM plus DC vaccine. Mice were sacrificed on the second day after the last dose administration, and immune cells that had infiltrated the TME were analyzed by flow cytometry. (A) Representative plots of the proportion of total T cells (CD45+ CD3+) among live cells in the TME. (B) Representative plots of the proportions of CD4+ T cells and CD8+ T cells among CD45+ CD3+ T cells in the TME. (C–E) Statistical data for the proportion of total T cells (CD45+ CD3+, C), CD4+ T cells (CD45+ CD3+ CD4+, D), and CD8+ T cells (CD45+ CD3+ CD8+, E) among live cells in the TME. (F and G) Proportion of CD4+ T cells (F) and CD8+ T cells (G) among CD3+ T cells. (H and I) MFI of CD44 on CD4+ T cells (H) and CD8+ T cells (I) in the TME. (J–M) Proportions of CD4+ CD44+ CD62L+ TCM cells (J), CD8+ CD44+ CD62L+ TCM cells (K), CD4+ CD44+ CD62L− TEM cells (L), and CD8+ CD44+ CD62L− TEM cells (M) among live cells in the TME. TCM, central memory T cells; TEM, effector memory T cells. (N and O) Proportion of PD-1+ cells among CD4+ T cells (N) and CD8+ T cells (O). Tex, exhausted T cells. (P) Proportion of CD25+ FOXP3+ cells among CD4+ T cells.
(Q and R) (Q) Lymphocytes isolated from tumor tissue samples of mice in the indicated groups were cocultured with B16-F10-OVA cells in an E:T = 1:3 ratio for 48 h to evaluate lymphocytotoxicity. (R) IFN-γ secretion was analyzed in the supernatants of lymphocytes from TILs cocultured with B16-F10-OVA cells. E, effector cells (lymphocytes); T, target cells (B16-F10-OVA cells). The bars show the mean ± SD values and the p values were determined by one-way ANOVA in (C)–(R).
(S) Tumor growth curves of B16-F10 tumor-bearing C57BL/6J mice that were subjected to the following treatments: isotype control, OVM combined with B16-F10 oncolysate-activated DC vaccine, OVM plus DC vaccine and anti-CD4 antibody, or OVM plus DC vaccine and anti-CD8 antibody; n = 6 in every group.
(T and U) Batf3-KO mice and their WT littermates were implanted subcutaneously in the right flank with B16-F10 cells on day 0 and treated with vehicle (n = 5), OVM (n = 6), B16-F10 oncolysate-stimulated DC vaccine (n = 5), or OVM combined with DC vaccine (n = 6). (T) Tumor growth curves of Batf3-KO mice. (U) The comparison of combined treatment groups between Batf3-KO mice and WT mice (n = 7). The p values in graphs (S)–(U) were determined by one-way ANOVA at the final time point as indicated in the graphs. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
To deeply investigate whether T cells activated by the OVM and DC vaccine combo have enhanced antitumor activity, the lymphocytotoxicity of tumor-infiltrating lymphocytes (TILs) was determined by measuring the viability of adhered B16-F10 tumor cells in a coculture system. As expected, TILs from the combination group had the strongest killing effect on tumor cells (Figure 2Q). We also measured IFN-γ secretion in the coculture supernatant, and likewise, the IFN-γ secretion capability of TILs in the combination group was higher than that in either monotherapy group (Figure 2R). These data strongly suggest that OVM primes the immune system to trigger a more potent antitumor T cell response in the TME when administered in combination with DC vaccine.
Considering the increased infiltration of T cells in the TME, we aimed to elucidate the role of T cells in the combination therapy. We employed anti-CD4 or anti-CD8 antibodies to deplete CD4+ or CD8+ T cells in B16-F10 tumor-bearing mice and then administered the OVM plus DC vaccine treatment (Figure S2D). The results demonstrated that depleting CD4+ T cells did not affect the therapeutic efficacy of OVM + DC vaccine, while the complete abrogation of the antitumor effect occurred upon the depletion of CD8+ T cells (Figures 2S and S2E). This finding indicates that the therapeutic effect of OVM and DC vaccine is dependent on CD8+ T cells rather than CD4+ T cells. Due to the crucial role of endogenous type 1 conventional DCs (cDC1s) in the activation and migration of CD8+ T cells,22,23 we sought to investigate their contribution to the antitumor effect of combination therapy. To further explore this, we utilized a Batf3-knockout (Batf3-KO) mouse model (Figure S2F), which fails to develop cDC1s selectively but keeps the other antigen-presenting cells normal in vivo. Interestingly, we observed that the combination of DC vaccine and OVM can delay the tumor growth in both wild-type (WT) and Batf3-KO mice similarly, whereas the antitumor effects of monotherapy with either DC vaccine or OVM were significantly offset in Batf3-KO mice (Figures 2T–2U and S2G–S2J). These results indicate that CD8+ T cells are activated in a cDC1s-independent manner and play a critical role in the therapeutic effect of OVM combined with the DC vaccine.
The above findings demonstrate that OVM boosts the DC vaccine to recruit and activate antitumor T cells, especially CD8+ T cells in the TME. In addition, endogenous cDC1s are not essential for the efficacy of the OVM plus DC vaccine combo.
OVM reactivates the tumor-suppressed DC vaccines
We next sought to figure out the mechanism of how OVM improves the antitumor activity of DC vaccines. It is considered that DCs are usually inhibited in the TME and cannot function normally. Indeed, by coculturing BMDCs with multiple tumor cells (co-DCs) to mimic the TME, we found that the levels of DC maturation markers, including the costimulatory molecules CD86 and CD83 and the major histocompatibility complex (MHC) II molecule, were markedly decreased (Figures 3A–3F and S3A–S3F), suggesting that the maturation of DCs was impaired in the TME. Meanwhile, the function of co-DCs to prime T cell responses was also severely damaged, as evidenced by the activation levels of both CD4+ T cells and CD8+ T cells cocultured with co-DCs being significantly lower than those with induced DCs (iDCs), showing decreased expression of the T cell activation markers CD69 and CD44 and reduced secretion of IFN-γ, perforin, and granzyme B (Figures 3G and 3H). We further evaluated whether the ability of co-DCs to elicit antigen-specific T cell responses was also weakened. OVA was added to DCs cocultured with or without B16-F10 tumor cells, and a lower expression level of MHC I molecules specifically presenting OVA257–264 peptide (H2Kb-OVA257–264) in co-DCs than in iDCs was observed (Figure 3I). Moreover, by coculturing the isolated co-DCs pulsed with OVA with OT-I CD8+ T cells (Figure 3J), which specifically recognize OVA257–264, the ability of co-DCs to prime OT-I CD8+ T cells was much lower than that of the iDCs (Figure 3K). Consistently, transcriptome analysis showed the impaired activation, differentiation, and function (including antigen presentation and inflammatory cytokine secretion) of co-DCs compared with iDCs (Figure S3J).
Figure 3.

OVM reactivates tumor-suppressed DC vaccines
(A–C) The expression of maturation markers (CD86, CD83, and MHC II) in mouse CD11c+ DCs after being cocultured at a 3:1 ratio with B16-F10 cells (A) or CT-26 cells (B) for 48 h or with Pan02 cells for 72 h (C); n = 3.
(D–F) The expression of CD86 and MHC II in human CD11c+ DCs after being cocultured at a 3:1 ratio with HCT-8 cells (D), HCT-116 cells (E), or SW-620 cells (F) for 24 h; n = 3.
(G and H) DCs were untreated or cocultured with B16-F10 cells for 48 h, separated with magnetic beads, and cocultured with spleen-derived T cells at a 1:5 ratio for 96 h; n = 3 in per group. (G) The MFI values of CD69 and CD44 on CD4+ (left) or CD8+ (right) T cells. (H) The concentrations of IFN-γ, perforin, and granzyme B in the coculture system.
(I) DCs were untreated or cocultured with B16-F10 cells and treated with 1 μg/mL OVA for 24 h. The MFI of H-2Kb OVA257-264 on CD11c+ DCs was determined by flow cytometry; n = 4.
(J and K) DCs were untreated or cocultured with B16-F10 cells for 24 h and stimulated with 1 μg/mL OVA for another 24 h. DCs were separated with magnetic beads and cocultured with spleen-derived CD8+ T cells from OT-I mice at a 1:2 ratio for 48 h; n = 3 per group. (J) Schematic diagram. (K) Representative histograms (left) and MFI values (right) of CD69 and CD44 on OT-I CD8+ T cells are shown. The data in (A)–(G) are presented as the means ± SD, and p values were determined by unpaired Student’s t test (A)–(I) or one-way ANOVA (K).
(L–N) (L) C57BL/6J mice were implanted subcutaneously in the right flank with B16-F10-OVA cells on day 0 and administered one dose of vehicle, OVA-loaded DC vaccine (activated by OVA and LPS), or co-DC vaccine by intratumoral injection on day 6 (tumor volumes were approximately 50 mm3). OVA-loaded DCs were cocultured with B16-F10 cells for 48 h and sorted by magnetic beads to prepare the co-DC vaccine. (M) Tumor growth curves and (N) Kaplan-Meier survival curves are shown; n = 9. The p values were determined by one-way ANOVA at the final time point as indicated in the graphs or by the log rank test, as appropriate.
(O) The changes in CD86, CD83, and MHC II expression on mouse CD11c+ DCs cocultured with B16-F10 cells for 24 h and stimulated with control (Ctrl), LPS (1 μg/mL, as a positive control), or OVM (1 MOI) for another 24 h.
(P) The changes in CD86 and MHC II expression on human DCs cocultured with HCT-8 cells for 24 h and stimulated with control (Ctrl) or OVM (1 MOI) for another 24 h.
(Q) The concentrations of TNF-α, IL-12p70, and IFN-β in the coculture supernatant of graph (O).
(R and S) CD11c+ DCs were cocultured with B16-F10 cells for 24 h and stimulated with OVM (1 MOI) for another 24 h, separated with magnetic beads, and cocultured with spleen-derived T cells at a 1:5 ratio for 96 h. (R) The MFI of CD69 and CD44 on CD4+ (left) or CD8+ (right) T cells. (S) The concentrations of IFN-γ, perforin, and granzyme B in the co-DC or OVM-treated coculture system; n = 3. One-way ANOVA was used to determine the significance of differences between groups in the graphs of (O)–(S). All data are presented as the means ± SD. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Next, we verified whether the inhibitory effects of tumor cells on DCs sabotaged the antitumor efficacy of DC vaccines in vivo. Subcutaneous B16-F10-OVA tumor-bearing C57BL/6 mice were randomized to receive intratumoral injections of OVA-loaded DC vaccines that were cocultured with or without B16-F10 cells (Figure 3L). The DC vaccines significantly inhibited the tumor growth and improved the survival of B16-F10-OVA tumor-bearing mice, whereas co-DC based vaccines completely lost the antitumor effect (Figures 3M and 3N), indicating that tumor cells can greatly weaken the efficacy of DC vaccines in vivo.
Given the above results demonstrating that tumor cells have the ability to inhibit the maturation and activation of DCs and eliminate the antitumor effect of DC vaccine, whether OVM can relieve this inhibition, reactivate tumor-suppressed DCs, and restore their ability to initiate T cell response has become an open question. An upregulated expression of maturation markers in B16-F10 or HCT-8 cell-suppressed co-DCs was observed after OVM infection in vitro, and LPS was used as the positive control to stimulate the maturation of DC (Figures 3O, 3P, S3G, and S3H). Moreover, OVM led to increased secretion of inflammatory cytokines, including TNF-α, IL-12p70, and IFN-β, which are important for innate immune activation, antiviral response, and DC survival (Figure 3Q). We further tested whether OVM can restore the function of co-DCs in T cell priming. DCs were cocultured with B16-F10 cells and then infected with OVM. After magnetic cell sorting, the stimulated co-DCs were cocultured with spleen-derived naive T cells. Both the flow cytometry and the ELISA results showed that treatment with OVM resulted in significant upregulation of CD69 and CD44 on CD4+ T cells and CD8+ T cells (Figure 3R), as well as a substantial increase in the secretion of the cytokines, including IFN-γ, perforin, and granzyme B, into the coculture supernatants (Figure 3S). Transcriptomic analysis by RNA sequencing (RNA-seq) revealed that the gene sets related to the inflammatory cytokine secretion, immune activation, and DC function pathways were markedly upregulated in OVM-treated co-DCs as well (Figure S3K). These data collectively suggest that OVM can reactivate co-DCs that were inhibited by tumor cells.
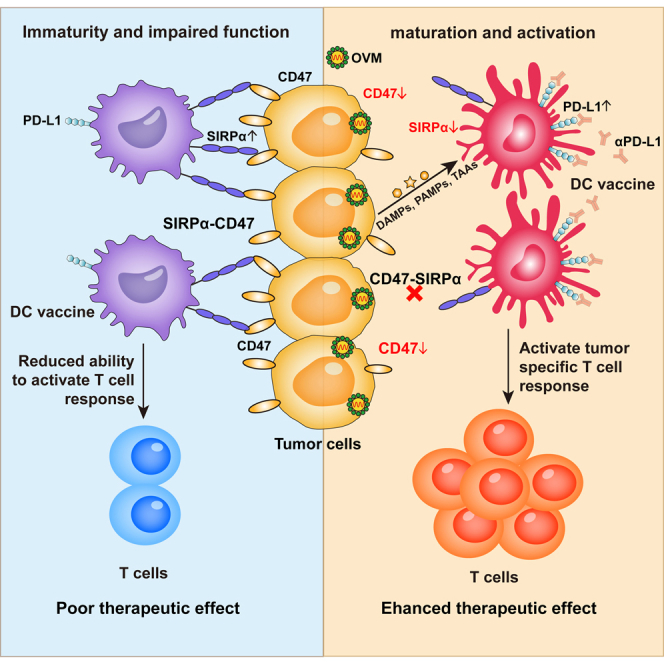
In summary, the above results indicate that tumor cells can inhibit the maturation and activation of DCs and eliminate the anticancer effect of DC vaccine, while OVM can reactivate the tumor-suppressed DCs and restore their functions, thereby enhancing the therapeutic effect of DC vaccines.
Tumor cells inhibit DCs through the SIRPα-CD47 pathway
The reactivation of tumor-suppressed DCs by OVM prompted us to delineate the underlying molecular mechanisms. With the tumor-derived conditioned medium (CM) and a Transwell coculture system (Figure 4A), we found that the inhibition of DCs by tumor cells was mediated through direct cell-cell contact rather than transfer of secreted molecules. Specifically, only co-DCs that were directly contacted by tumor cells showed significantly reduced expression of costimulatory and MHC II molecules, whereas DCs cultured in tumor-derived CM or Transwell chambers (T-DCs) showed no such inhibition (Figures 4B–4F). In addition, the levels of secreted inflammatory cytokines, including TNF-α and IL-12p70, were decreased in the supernatant of co-DCs but not T-DCs (Figure 4G). To further explore the possibility that coculturing with tumor cells may trigger the secretion of inhibitory factors by DCs, we seeded DCs in the upper well of a Transwell chamber and seeded both DCs and B16-F10 tumor cells in the lower well, thus concurrently obtaining co-DCs and T-DCs (Figure 4A). Only co-DCs were inhibited, as their expression of CD86, CD83, and MHC II was suppressed, while T-DCs remained unaffected (Figure 4H). Taken together, these results indicate that tumor cells inhibit DCs in a contact-dependent manner. Regarding the contact-dependent inhibition, we assumed that cell membrane proteins such as receptor-ligand pairs may be involved in the inhibitory effect of tumor cells against DCs. This hypothesis is supported by the reduced inhibitory effect of DCs cocultured with trypsin-digested B16-F10 cells (Figures 4I and 4J).
Figure 4.

Tumor cells inhibit DCs in a contact-dependent manner
(A) Diagrammatic sketch of the coculture experiment described in (B)–(H).
(B and C) The expression of CD86, CD83, and MHC II on DCs after being cultured with B16-F10-conditioned medium (CM) (B) or CT-26-CM (C) for 48 h.
(D–G) DCs were cocultured with B16-F10, CT-26, or Pan02 cells in direct contact (co-DCs) or in Transwell inserts (T-DCs). The MFI and proportions of CD86, CD83, and MHC II are shown in (D)–(F), and the levels of TNF-α and IL-12p70 in the coculture supernatant determined by ELISA are shown in (G).
(H) The DC activation/maturation phenotypes of DCs cocultured with B16-F10 cells under both contact-dependent (C + T, co-DC) and contact-independent (C + T, T-DC) conditions for 48 h.
(I and J) The maturation of DCs after being cocultured with B16-F10 cells (Ctrl) or trypsin-digested B16-F10 cells (Trypsin) for 6 or 24 h. These data are presented as the means ± SD and were analyzed by unpaired Student’s t test (B and C) or one-way ANOVA (D–H and J); n = 3 per group. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
To identify the potential receptors/ligands, we first compared the transcriptomes of co-DCs (cocultured with B16-F10 cells) and iDCs. Gene set enrichment analysis (GSEA) showed that genes related to the immunoreceptor tyrosine-based activation motif (ITAM) pathway were significantly downregulated in co-DCs, while those related to the protein phosphatase were upregulated (Figure 5A). Almost all immune cells contain an ITAM and an immunoreceptor tyrosine-based inhibitory motif (ITIM) to mediate the transduction of activation or inhibition signals.24 Quantitative polymerase chain reaction (qPCR) analysis verified that the mRNA levels of genes downstream of the ITAM pathway were reduced in co-DCs (Figure 5B). In accordance with the mRNA expression, the phosphorylation levels of kinases downstream of the ITAM pathway (including spleen tyrosine kinase [SYK], phospholipase C-γ1 [PLCγ1] and PLCγ2) were strikingly decreased in co-DCs (Figure 5C), suggesting the activation of phosphatases in the ITIM pathway.
Figure 5.

Tumor cells inhibit DCs through SIRPα-CD47
(A) Protein kinase/phosphatase-related GSEA pathways significantly enriched in B16-F10-cocultured DCs (co-DCs) compared with untreated DCs (iDCs).
(B) qPCR analysis comparing iDCs and co-DCs cocultured with B16-F10 cells for 48 h. These genes participate in the ITIM/ITAM downstream pathway; n = 3.
(C) Representative western blot pictures (left) and quantitative statistics (right) of the phosphorylation of SIRPα-related downstream kinases; n = 3.
(D) Heatmap of top 10 changes in ITIM-containing receptors between the co-DC and the iDC group.
(E) The immune checkpoint expression changes between co-DCs and the untreated iDC group; n = 6 in SIRPα, PD-1, and CTLA-4; n = 5 in TIM-3; n = 3 in PIR-B and CD33.
(F) SIRPα-KO mouse- or wild-type (WT) mouse-derived DCs were cocultured with B16-F10 cells, and the maturation of CD11c+ DCs was determined by flow cytometry; n = 3.
(G and H) WT mouse-derived DCs were cocultured with B16-F10-WT or B16-F10-CD47 KO cells at a 3:1 ratio for 48 h. The proportion of cells with high expression levels of CD80, CD86, and CD83 (G) and secretion levels of TNF-α (H) was determined to assess the DC activation/maturation phenotype; n = 3 in per group.
(I–L) C57BL/6J mice were implanted subcutaneously in the right flank with B16-F10 cells on day 0 and administered one dose of vehicle, oncolysate-stimulated DC vaccine, or oncolysate-stimulated SIRPα-KO DC vaccine by intratumoral injection on day 6 (tumor volumes were approximately 50 mm3). (I) The levels of TNF-α and IL-12 in the tumor interstitial fluid. On the fifth day after treatment, fresh tumor tissues were collected, weighed, and incubated at 37°C for 2 h in 1 mL of PBS per gram of tumor tissue to obtain the tumor interstitial fluid for ELISA; n = 5. (K) Tumor growth curves and (L) Kaplan-Meier survival curves are shown; n = 6.
(M–O) C57BL/6J mice were implanted subcutaneously in the right flank with B16-F10-WT or B16-F10-CD47 KO cells on day 0 and administered one dose of vehicle or B16-F10 oncolysate-stimulated DC vaccine by intratumoral injection on day 6. (N) Tumor growth curves (n = 6) and (O) Kaplan-Meier survival curves (n = 7) are shown. The p values were determined by unpaired Student’s t test (B, C, E), one-way ANOVA (F–I), one-way ANOVA at the final time point (K, N), or log rank test (L, O). n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
To identify the particular ITIM-containing receptors that mediated this inhibition of DCs by tumor cells, we compared the mRNA expression of common receptors with ITIM motifs between co-DCs and iDCs25,26,27 and found that SIRPα, which is known as a myeloid immune checkpoint on macrophages,28 was strongly upregulated in co-DCs (Figure 5D). Consistently, flow cytometry analysis demonstrated that the protein expression of SIRPα on co-DCs was notably increased compared with that on iDCs, while the expression of other well-known immune checkpoints (including PIR-B, CD33, PD-1, TIM-3, and CTLA-4) showed no difference between co-DCs and iDCs (Figures 5E and S3I). The above findings suggest that SIRPα may be the inhibitory receptor via which tumor cells suppress DCs and impair the efficacy of DC vaccines.
To determine whether tumor cells inhibit DCs by binding SIRPα with CD47, we first prepared DCs from SIRPα-KO mice or WT littermates (SIRPα-KO DCs or WT DCs, respectively) (Figure S4A). After coculturing with B16-F10, costimulatory molecules CD86, CD83, and MHC II on the surface of WT DCs were significantly reduced, while those on the SIRPα-KO DCs remained unchanged or even upregulated (Figures 5F and S4D). In addition, we also generated a CD47-KO B16-F10 cell line (B16-F10-CD47-KO), which grew at a rate similar to that of B16-F10-WT cells in vitro (Figures S4B and S4C). DCs were cocultured with B16-F10-WT or B16-F10-CD47-KO cells, and the expression of CD86 and CD83 on DCs cocultured with B16-F10-CD47-KO cells was not decreased compared with those on B16-F10-WT cells nor was the secretion of TNF-α (Figures 5G, 5H, and S4E). These results indicate that the inhibition of DCs by tumor cells is mainly dependent on the SIRPα-CD47 axis.
Given that SIRPα-KO DCs are resistant to the immunosuppression induced by B16-F10, we further explored whether the DC vaccine based on SIRPα-KO DCs had a better therapeutic effect than the WT DC vaccine in the B16-F10 subcutaneous model (Figure 5J). Compared with the animals receiving the DC vaccine based on WT DCs, the animals that received the DC vaccine based on SIRPα-KO DCs exhibited higher levels of IL-12 in the TME and displayed slower tumor growth and longer survival times (Figures 5I–5L and S4F). Moreover, we also established B16-F10-CD47-KO tumor-bearing mouse models to cancel the SIRPα-CD47-mediated inhibition on the DC vaccine (Figure 5M). As expected, B16-F10-CD47-KO cell-derived tumors displayed slower growth and higher sensitivity to the DC vaccine than the B16-F10-WT tumors (Figures 5N–5O and S4G). These data suggest that alleviating the inhibitory effect of tumor cells on DCs through targeting the SIRPα-CD47 pathway effectively improved the potency of the DC vaccine.
Collectively, these results indicate that tumor cells inhibit DCs through the contact-dependent SIRPα-CD47 pathway, which impairs the efficacy of the DC vaccine. Blocking the SIRPα-CD47 pathway may abolish this suppressive effect, reactivate DCs, and enhance the potency of the DC vaccine.
OVM reactivates the tumor-inhibited DCs by downregulating the expression of both SIRPα and CD47
As tumor cells inhibit DC function and abolish the antitumor effect of the DC vaccine through the SIRPα-CD47 pathway, we wondered whether OVM could activate tumor-suppressed DCs by inhibiting the SIRPα-CD47 pathway. Both the mRNA expression and protein levels of CD47 in tumor cells were markedly reduced by OVM infection (Figures 6A and 6B). Meanwhile, OVM can also significantly downregulate the mRNA and protein expression of SIRPα, which was induced in co-DCs (Figures 6C and 6D). Similarly, by using the B16-F10 mouse melanoma model (Figure 6E), we confirmed that treatment with OVM significantly reduced SIRPα expression in SIRPα+ DCs and CD47 expression in live cells in the TME, simultaneously, without downregulation of SIRPα in spleen-derived DCs in vivo (Figure 6F). What’s more, SIRPα expression was also distinctly decreased to a lower level in the DC vaccine plus OVM combination group than in the DC vaccine group (Figures 6E and 6G). These findings reveal the capacity of OVM to reduce the expression of SIRPα-CD47 checkpoint molecules.
Figure 6.

OVM enhances the therapeutic efficacy of DC vaccine by downregulating the expression of SIRPα and CD47
(A and B) The (A) mRNA levels and (B) MFI values of CD47 in tumor cells that were infected with OVM for 24 h.
(C) Sirpa expression changes in B16-F10-cocultured co-DCs and OVM-treated co-DCs; n = 3 in (A)–(C).
(D) MFI values of SIRPα on DCs that were co-cultured with B16-F10, CT-26, or HCT-8 cells for 48 h or cocultured with tumor cells for 24 h and then treated with vehicle (Ctrl) or OVM (1 MOI) for another 24 h.
(E and F) C57BL/6J mice were implanted subcutaneously in the right flank with B16-F10 cells on day 0 and administered accordingly with vehicle, OVM, B16-F10 oncolysate-stimulated DC vaccine, or OVM plus DC vaccine. (E) Schematic diagram of tumor inoculation and treatment. (F) The MFI of CD47 on live cells and the MFI of SIRPα on SIRPα+ DCs (gate: CD45+ CD11c+ MHC II+ CD103- CD11b+ SIRPα+) in the TME and spleen (gate: CD45+ CD11c+ MHC II+ SIRPα+) of B16-F10 tumor-bearing mice treated with vehicle (n = 5 or 6) or OVM (n = 6).
(G) MFI of SIRPα on the surface of SIRPα+ DCs (gate: CD45+ CD11c+ MHC II+ CD103- CD11b+ SIRPα+) in the TME from B16-F10 tumor bearing C57BL/6J mice treated with vehicle (n = 6), OVM (n = 7), B16-F10 oncolysate-stimulated DC vaccine (n = 9), or OVM plus DC vaccine (n = 8). These data are presented as the means ± SD and were analyzed by one-way ANOVA (A, B, and G) and unpaired Student’s t test (C, D, and F).
(H and I) B16-F10-WT tumor-bearing C57BL/6J mice were treated with vehicle, OVM, OVM plus B16-F10 oncolysate-stimulated DC vaccine, or OVM combined with B16-F10 oncolysate-activated SIRPα-KO DC vaccine. B16-F10-CD47-KO tumor-bearing C57BL/6J mice were treated with vehicle, OVM, or OVM plus DC vaccine; n = 7. (H) Schematic diagram of the treatment regimen of B16-F10-WT and B16-F10-CD47-KO tumor-bearing C57BL/6J mice. (I) Tumor growth curves. The p values were determined by one-way ANOVA at the final time point.
(J) The T:C ratios of B16-F10 and B16-F10-CD47-KO tumor-bearing mice from graph (I) and Figure S10E after receiving the corresponding treatments. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Owing to the decreased mRNA levels of SIRPα and CD47 after OVM treatment, it is reasonable to suspect that OVM may regulate SIRPα and CD47 expression at the transcription level, possibly through transcription factors (TFs). GSEA of transcriptome data from co-DCs and OVM-treated co-DCs showed that genes regulated by MYC were significantly downregulated after OVM treatment (Figure S5A), suggesting that the transcription activity of MYC was suppressed. Consistently, a decreased mRNA expression of MYC was observed in co-DCs treated with OVM (Figure S5B). We then looked into Encyclopedia of DNA Elements (ENCODE) TF target datasets and found that MYC is one of the potential TFs of SIRPα. Moreover, through analysis of The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) databases, we found that the expression of MYC was positively correlated with the expression of SIRPα in a variety of tumor tissues (Figure S5C). These findings suggested that MYC may be involved in the downregulation of SIRPα in OVM-treated co-DCs. Meanwhile, MYC has been reported to mediate CD47 transcription and trigger high levels of CD47 protein.29,30 Similarly, the expression between MYC and CD47 has shown significantly positive correlations in various tumors (Figure S5D), and we also observed a significant downregulation of MYC expression in OVM virus-infected tumor cells (Figure S5E), indicating that OVM may reduce the transcription levels of CD47 by reducing MYC expression as well. Overall, these data provide hints that OVM can decrease the expression of MYC in both co-DCs and tumor cells, thereby reducing the levels of both SIRPα and CD47.
Next, we sought to figure out whether the enhanced effect of OVM on the DC vaccine depends on the inhibition of the SIRPα-CD47 pathway. Although combination of OVM and DC vaccine exhibited significant antitumor potency in B16-F10 melanoma, the depletion of SIRPα on DC vaccines, the KO of CD47 on B16-F10 cells, or even the simultaneous KO of both SIRPα and CD47 failed to further potentiate this effect (Figures 6I, 6J, and S6B–S6F), strongly suggesting that OVM can fully overcome CD47-SIRPα inhibition by downregulating both molecules.
PD-L1 blockade potentiates the antitumor efficacy of the combination regimen consisting of OVM and DC vaccine
While OVM can downregulate the expression SIRPα and CD47 to enhance the antitumor effect of the DC vaccine, further transcriptional analysis of common immune checkpoints showed that PD-L1 expression in DCs was dramatically upregulated by OVM (Figure 7A). Flow cytometry data also confirmed this result that, after OVM treatment, PD-L1 expression was markedly increased, whereas SIRPα expression was decreased (Figure 7B). Moreover, we examined the expression of PD-L1 in the TME of tumor-bearing mice treated with OVM, DC vaccine, or their combination. We observed that the combination therapy increased PD-L1 expression on CD11c+ DCs more than any single therapy, while OVM alone and the combination therapy also elevated PD-L1 expression on CD45− cells (Figure 7C). PD-L1 not only interacts with PD-1 on activated T cells to induce T cell exhaustion but also binds in a cis conformation with CD80 on DCs and prevents DCs from priming the T cell response.10,31 These observations suggested that PD-L1 blockade may contribute to additional antitumor effects in the context of combined therapy with DC vaccine and OVM. Thus, we added an anti-PD-L1 antibody (αPD-L1) to the DC vaccine plus OVM regimen to treat B16-F10 or CT-26 tumor-bearing mice (Figure S7A). Treatment with αPD-L1 significantly potentiated the antitumor activity of the DC vaccine plus OVM combination regimen. Among all the tested treatments, the triple therapy of OVM, DC vaccine, and αPD-L1 antibody achieved the most potent tumor suppression and survival benefit (Figures 7D, 7E, and S7B–S7E). To further evaluate the therapeutic potential of our strategy in more advanced cancer stages, we used a larger tumor model (∼250 mm3) and applied the same treatment regimen (Figure S7A). Remarkably, the triple therapy still effectively inhibited tumor growth and significantly prolonged the survival of the mice (Figures 7F and S7F). This evidence powerfully suggests that the effect of combination treatment with the DC vaccine and OVM can further be augmented by targeting PD-L1.
Figure 7.

PD-L1 blockade potentiates the antitumor efficacy of the combination regimen consisting of DC vaccine and OVM
(A) Heatmap of common immune checkpoints between B16-F10-cocultured co-DCs and OVM-treated co-DCs from RNA-seq.
(B) MFI of PD-L1 and SIRPα (as a control) on iDCs, co-DCs (Ctrl), and OVM-treated co-DCs (OVM); n = 3.
(C) The MFI of PD-L1 on the CD45− cells and CD45+ CD11c+ DCs in the TME from B16-F10 tumor-bearing mice that received corresponding treatments. C57BL/6J mice were implanted subcutaneously in the right flank with B16-F10 cells on day 0 and administered accordingly when the tumor volumes reached approximately 50 mm3 with vehicle, OVM, B16-F10 oncolysate-stimulated DC vaccine, or OVM plus DC vaccine. On the seventh day after the last dose administration of OVM, the tumor tissues were harvested to detect the PD-L1 expression; n = 4 in the vehicle group and n = 5 in the other groups.
(D–F) C57BL/6J or BALB/c mice were implanted subcutaneously in the right flank with (D and F) B16-F10 or (E) CT-26 cells on day 0 and treated with the indicated drugs when the tumors reached the appropriate volume (B16-F10 and CT-26, ∼100 mm3; advanced B16-F10, ∼250 mm3). Tumor growth curves (left), T/C % curves (center), and Kaplan-Meier survival curves (right) are shown. The p values were determined by one-way ANOVA at the final time point or by the log rank test.
(G–L) C57BL/6J mice were implanted subcutaneously in the right flank with B16-F10 and treated with the indicated drugs when the tumors reached the appropriate volume. Mice were sacrificed on the third day after the last dose administration of αPD-L1, and infiltrated immune cells in the TME were analyzed by flow cytometry; n = 5. (G–J) Proportions of (G) CD4+ CD44+ CD62L+ TCM cells, (H) CD8+ CD44+ CD62L+ TCM cells, (I) CD4+ CD44+ CD62L− TEM cells, and (J) CD8+ CD44+ CD62L− TEM cells among live cells in the TME. TCM, central memory T cells; TEM, effector memory T cells. (K and L) Proportions of PD-1+ cells among (K) CD4+ T cells and (L) CD8+ T cells. Tex, exhausted T cells. The p values were determined by one-way ANOVA. n.s., not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
To explore how the TME changes after administration of PD-L1 blockade, we analyzed the phenotypic changes of T cells in TME after αPD-L1 therapy. On the third day after the last administration of αPD-L1 treatment (the seventh day post-OVM plus DC vaccine treatment) (Figure S7G), B16-F10 tumor-bearing mice were sacrificed, and tumor tissues were collected for flow cytometry analysis. The results revealed a significant increase in the proportion of CD45+ cells in the TME of the treated groups. Notably, in the triple combination group (OVM + DC + αPD-L1), T cells accounted for approximately 20% of CD45+ cells, whereas in the OVM + DC group, T cells represented only 5% of CD45+ cells, despite an overall increase in the proportion of T cells in the live cell population (Figures S7H and S7I). Furthermore, the OVM + DC group exhibited a significant increase in the proportion of TCM in the early stage (Figures 2J and 2K), but this proportion was reduced in the later stage (Figures 7G and 7H). In contrast, the triple combination group showed a substantial increase in both TCM and TEM, with TEM being the predominant subset (Figures 7G–7J and S7I). In addition, we also investigated the proportion of Tex cells. Similarly, in the early stage of OVM + DC treatment, exhausted CD8+ T cells significantly decreased compared with the vehicle or single-agent group (Figure 2O), while in the later stage, the proportion of CD8+ Tex in CD8+ T cells was not significantly different from that in the vehicle group, while in the triple combination group, the proportion of Tex cells decreased within the CD4+ and CD8+ T cell population (Figures 7K, 7L, and S7J). These results collectively suggest that although the combination treatment of OVM plus DC vaccine significantly inhibits tumor growth in the early stage, the upregulation of PD-L1 exerts inhibitory effects on T cells. The addition of αPD-L1 therapy can further alleviate T exhaustion, leading to a more robust and long-lasting antitumor effect.
Discussion
In 2011, Ralph Steinman was awarded the Nobel Prize for Medicine or Physiology for his discovery of DCs and their importance. In recent decades, DCs have been described as sentinels that mediate innate and adaptive immune responses. DC-based vaccination is also expected to achieve great clinical efficacy.3 To date, a search for the keywords “dendritic cell vaccine” and “tumor” in the clinical research database ClinicalTrials.gov (https://clinicaltrials.gov/) has uncovered a total of 422 clinical trial registrations, including 12 phase III and phase IV clinical trial registrations, with indications for melanoma, non-Hodgkin’s lymphoma, malignant glioma, metastatic colorectal cancer, prostate cancer, ovarian cancer, etc. Due to the limited number of naturally occurring cDC1s in the body, and the lower purity of cDC1s obtained through in vitro induction, despite their crucial role in activating CD8+ T cells, the majority of DCs used in clinical trials are derived from CD14+ monocytes or CD34+ hematopoietic progenitor cells stimulated with GM-CSF, IL-4, and other cytokines.32 However, although their safety has been fully confirmed in the clinical trials conducted to date, DC vaccines have not yet provided robust clinical benefits in large patient populations, with objective response rates rarely exceeding 15%.3,4 A crucial reason for this deficiency is immune suppression in the TME. Therefore, how to break the inhibition of the TME on DC vaccines and improve their effectiveness has become an important problem to be solved in the research on DC vaccines. Our research shows that tumor cells inhibit the maturation and function of DCs in a SIRPα-CD47-dependent manner and thus abolish the efficacy of DC vaccines. Alphavirus OVM can alleviate this inhibition by bidirectionally downregulating SIRPα and CD47, thereby greatly improving the therapeutic effect of DC vaccines in multiple tumor models.
Currently, the therapeutic strategy of combining oncolytic viruses with other immunotherapies is a hot research topic.33 A total of 71 clinical trials involving the combination of oncolytic virotherapy have been conducted or are about to be conducted, including with DC vaccines.34 However, the phase III clinical trial of T-VEC plus ipilimumab versus placebo plus ipilimumab did not demonstrate improved efficacy in patients with advanced melanoma. Therefore, there is still an unmet need for more effective oncolytic virotherapy combinations.35 Oncolytic virotherapy is believed to induce the release of damage-associated molecular patterns (DAMPs), trigger a proinflammatory cytokine cascade, and stimulate the activation of the innate immune system. Furthermore, oncolytic virus-induced oncolysis leads to tumor-associated neoantigen release, resulting in a robust tumor-specific adaptive immune response.12,36 In our research, in addition to alleviating the inhibitory activity of tumor cells against DC vaccines and improving the therapeutic effect of DC vaccines in tumor-bearing mice, OVM is used to prepare DC vaccines because of its ability to release TAAs. The choice of antigen for DC vaccine preparation is critical, and therapeutic vaccines usually either use one verified tumor antigen or a small group of validated antigens as peptide antigens or use whole tumor cells or lysates containing a wide spectrum of antigens.3,37 A large number of clinical trials have been conducted using one or more synthetic TAAs to pulse DC vaccines, but this strategy has shown limited efficacy,37 possibly due in part to loss of antigen expression for rapid progression of tumor variants or human leukocyte antigen (HLA) allotype limitation. Furthermore, RNA-seq followed by antigen synthesis seems to be an effective method for preparing DC vaccines, but identifying useful neoantigens is time consuming. In contrast, the preparation of antigens from whole-tumor lysates is relatively simple, and the limitations of this approach are circumvented by providing a broad library of tumor antigens. Moreover, autologous whole-tumor lysate antigens are not restricted to specific HLA haplotypes. However, whole-tumor lysates also contain self-antigens as well as other factors that inhibit DC maturation.3,4,38 In our study, oncolysates from tumor cells infected with OVM strongly activate DCs,19 and the inclusion of TAAs elicits the tumor-specific T cell response (Figures 1B and 1C). Furthermore, DC vaccines prepared with OVM-infected tumor oncolysates can also play a role in treating tumors and have a combinatory effect with OVM (Figures 1D–1G). These data prove the feasibility of using OVM-infected tumor oncolysates for activation of DC vaccines, a strategy that can not only provide TAAs but also promote DC maturation.
Tumor cells affect the activity of DCs by secreting cytokines; only a few reports have proposed that DC inhibition by tumor cells is mediated by receptor-ligand interactions. Here, we report that the activation of DCs cocultured with tumor cells is markedly inhibited in a contact-dependent manner rather than by cytokine secretion (Figure 4). More importantly, this inhibitory effect is mediated by the SIRPα-CD47 checkpoint, and either knocking out SIRPα on DCs or knocking out CD47 in B16-F10 tumor cells protected DCs from suppression by tumor cells, exhibiting a further improved efficacy of the DC vaccine in tumor-bearing mice (Figures 5J–5O). SIRPα-CD47, the “don’t eat me” signal, serves mainly to regulate macrophage phagocytosis. Although early work proved that CD47-Fc treatment suppresses the phenotypic and functional maturation of DCs, little is known about whether tumor cells inhibit DCs through the SIRPα-CD47 axis.39,40,41 In some subsequent studies, CD47 blockade was found to induce antitumor responses in a manner dependent on SIRPα+ CD11c+ DCs rather than macrophages.42,43 Collectively, these results emphasize the importance of SIRPα-CD47 blockade on DCs and provide strong evidence for the effectiveness of SIRPα-CD47 therapy in targeting DCs in addition to targeting macrophages.
CD47 is not only highly expressed on the surface of various tumor cells, but also widely expressed in normal cells, such as red blood cells and platelets.28,44,45 CD47-targeting drugs not only mediate the killing of tumor cells but also inevitably cause damage to normal cells, leading to severe side effects like red blood cell aggregation. However, reducing the affinity of CD47 antibodies may compromise their ability to activate a significant antitumor response, thus sacrificing the therapeutic efficacy of the drug. In addition, the high expression of SIRPα in the central and peripheral nervous systems also raises the potential for neurologic side effects when targeting SIRPα. Consequently, the development of CD47-targeting drugs has encountered obstacles, with several clinical studies being urgently halted due to safety concerns. Balancing safety and effectiveness is a critical scientific challenge that urgently needs to be addressed in the development of CD47-targeting drugs. Our results demonstrate that OVM can not only kill tumor cells directly, supply TAAs, and activate antitumor immune responses, but also downregulate the expression of both SIRPα on the surface of DCs and CD47 on tumor cells in vitro and in vivo (Figures 6A–6G). When using the SIRPα-KO DC vaccine and in the B16-F10-CD47 KO tumor-bearing mouse model, blocking either SIRPα or CD47 improved the therapeutic effects of the DC vaccine (Figures 5J–5O). However, coadministration of the DC vaccine with OVM in the B16-F10-WT model achieved the same efficacy as blocking either SIRPα or CD47 (Figures 6I, 6J, and S6E). These results strongly suggest the use of OVM as an inhibitor of the SIRPα-CD47 immune checkpoint in DC vaccines. OVM can specifically target tumor cells and selectively downregulate CD47 expression only on tumor cells, thereby avoiding off-target effects and potential toxicity. Interestingly, in our previous study, treatment with OVM was shown to downregulate PD-1 expression on T cells,19 prompting us to consider the potential of OVM as an inhibitor of various immune checkpoints and the selection of immune checkpoint inhibitors that would be the best partners in combination with OVM. Moreover, our findings suggest that the mechanism by which OVM downregulates SIRPα-CD47 may be mediated by the TF MYC (Figure S5), and this hypothesis needs to be elucidated in future work.
In summary, our study shows that OVM enhances the efficacy of a DC vaccine by downregulating SIRPα-CD47 expression, providing a theoretical basis for clinical trials of OVM as a powerful adjuvant of DC vaccines. Finally, a combination of OVM, a DC vaccine, and anti-PD-L1 antibodies can be a promising therapeutic strategy for malignancies.
Limitations of the study
While this study demonstrates the potential antitumor effects of combined treatment with OVM and DC vaccines, these in vivo therapeutic outcomes predominantly rely on syngeneic tumor mouse models, which, although informative, might have some inherent limitations in recapitulating the complex heterogeneity and microenvironment of clinical tumors in human patients. Testing in additional preclinical models, including humanized mouse with patient-derived xenografts and genetically engineered mouse models of human cancer, may strengthen the translational relevance. Moreover, deeper analysis of the tumor immune microenvironment and TCR clonality could provide insights into the elicited adaptive immunity.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Mouse CD11c APC, clone N418 | eBioscience | Catalog#:17-0114-82; RRID: AB_469346 |
| anti-Mouse CD86 eFluor 450, clone GL1 | eBioscience | Catalog#: 48-0862-82; RRID: AB_2574031 |
| anti-Mouse CD83 PE, clone Michel-17 | eBioscience | Catalog#: 12-0831-82; RRID: AB_465758 |
| anti-Mouse MHC II APC eFluor 780, clone M5/114.15.2 | eBioscience | Catalog#: 47-5321-82; RRID: AB_1548783 |
| anti-Mouse OVA257-264 (SIINFEKL) peptide bound to H-2Kb APC, clone eBio25-D1.16 | eBioscience | Catalog#: 17-5743-80; RRID: AB_1311288 |
| anti-Mouse CD45 eFluor 450, clone 30-F11 | eBioscience | Catalog#: 48-0451-82; RRID: AB_1518806 |
| anti-Mouse CD3 PE-Cyanine 5.5, clone 145-2C11 | eBioscience | Catalog#: 35-0031-82; RRID: AB_11219266 |
| anti-Mouse CD8 PE, clone 53-6.7 | eBioscience | Catalog#: 12-0081-82; RRID: AB_465530 |
| anti-Mouse CD4 APC, clone RM4-5 | eBioscience | Catalog#: 17-0042-82; RRID: AB_469323 |
| anti-Mouse CD44 Super Bright 645, clone IM7 | eBioscience | Catalog#: 64-0441-82; RRID: AB_2662590 |
| anti-Mouse CD69 PE Cyanine 7, clone H1.2F3 | eBioscience | Catalog#:25-0691-82; RRID: AB_469637 |
| anti-Mouse CD33 PE, clone 9A11-CD33 | eBioscience | Catalog#:12-0331-82; RRID: AB_2637179 |
| anti-Mouse PIR-A/B APC, clone 10-1-PIR | eBioscience | Catalog#:17-3101-82; RRID: AB_1944406 |
| anti-Mouse PD-1 APC, clone RMP1-30 | eBioscience | Catalog#:17-9981-82; RRID: AB_10852564 |
| anti-Mouse TIM-3 PE, clone RMT3-23 | eBioscience | Catalog#:12-5870-82; RRID: AB_465974 |
| anti-Mouse CD11b FITC, clone M1/70 | eBioscience | Catalog#:11-0112-81; RRID: AB_464934 |
| anti-Mouse CD16/CD32 Monoclonal, Clone 2.4G2, clone | BD | Catalog#: 553141; RRID: AB_394656 |
| anti-Mouse CTLA-4 PE,clone UC10-4F10-11 | BD | Catalog#: 553720; RRID: AB_395005 |
| anti-Mouse CD3 PerCP-Cy™5.5, clone 145-2C11 | BD | Catalog#: 551163; RRID: AB_394082 |
| anti-Mouse CD8 Alexa Fluor 700, clone 53-6.7 | BD | Catalog#: 557959; RRID: AB_396959 |
| anti-Mouse CD4 BV510, clone GK1.5 | BD | Catalog#: 743155; RRID: AB_2741308 |
| anti-Mouse CD62L FITC, clone MEL-14 | BD | Catalog#: 561917; RRID: AB_10893197 |
| anti-Mouse CD44 BV650, clone IM7 | BD | Catalog#: 740455; RRID: AB_2740182 |
| anti-Mouse PD-1 PE, clone J43 | BD | Catalog#: 551892; RRID: AB_394284 |
| anti-Mouse CD274 PE, clone MIH5 | BD | Catalog#: 558091; RRID: AB_397018 |
| anti-Mouse CD25 APC, clone PC61 | BD | Catalog#: 557192; RRID: AB_398623 |
| anti-Mouse Foxp3 PE, clone R16-715 | BD | Catalog#: 563101; RRID: AB_2738006 |
| anti-Human CD11c BV421, clone B-ly6 | BD | Catalog#: 562561; RRID: AB_2737656 |
| anti-Human CD86 APC, clone 2331 (FUN-1) | BD | Catalog#: 555660; RRID: AB_398608 |
| anti-Human MHC II PE, clone G46-6 | BD | Catalog#: 555812; RRID: AB_396146 |
| anti-Human CD47 PE, clone B6H12 | BD | Catalog#: 556046; RRID: AB_396317 |
| anti-Mouse CD11c PE Cyanine7, clone N418 | BioLegend | Catalog#: 117318; RRID: AB_493568 |
| anti-Mouse SIRPα Alexa Fluor 700, clone P84 | BioLegend | Catalog#: 144022; RRID: AB_2650813 |
| anti-Mouse CD103 PE Cyanine7, clone 2E7 | BioLegend | Catalog#: 121425; RRID: AB_2563690 |
| anti-Mouse CD47 PE,clone miap301 | BioLegend | Catalog#: 127508; RRID: AB_1134117 |
| InVivoMAb rat IgG2b isotype control, clone LTF-2 | Bio X cell | Catalog#: BE0090; RRID: AB_1107780 |
| InVivoMAb anti-mouse PD-L1, clone 10F.9G2™ | Bio X cell | Catalog#: BE0101; RRID: AB_10949073 |
| Rat IgG2b isotype control-InVivo, clone LTF-2 | Selleck | Catalog#: A2116; RRID: AB 1107780 |
| Anti-mouse CD4-InVivo, clone GK1.5 | Selleck | Catalog#: A2101; RRID: AB_1107636 |
| Anti-mouse CD8-InVivo, clone GK2.43 | Selleck | Catalog#: A2102; RRID: AB_1125541 |
| DAP12 (D7G1X) Rabbit mAb | CST | Catalog#: 12492 |
| Syk (D3Z1E) XP® Rabbit mAb | CST | Catalog#: 13198 |
| Phospho-Syk (Tyr525/526) (C87C1) Rabbit mAb | CST | Catalog#: 2710 |
| PLCγ1 (D9H10) XP® Rabbit mAb | CST | Catalog#: 5690 |
| PLCγ2 (E5U4T) Rabbit mAb | CST | Catalog#: 55512 |
| Phospho-PLCγ2 (Tyr759) (E9E9Y) Rabbit mAb | CST | Catalog#: 50535 |
| GAPDH (14C10) Rabbit mAb | CST | Catalog#: 2118 |
| Phospho-PLCγ1 (Tyr783) (D6M9S) Rabbit mAb | CST | Catalog#: 14008 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Murine GM-CSF | PeproTech | Catalog#: 315-03-250 |
| Recombinant Murine IL-4 | PeproTech | Catalog#: 214-14-100 |
| Ovalbumin | Ovalbumin | Catalog#: A7641 |
| LPS | LPS | Catalog#: tlrl-rslps |
| Critical commercial assays | ||
| Fixable Viability Stain 510 | BD | Catalog#: 564406 |
| Fixable Viability Stain 780 | BD | Catalog#: 565388 |
| Transcription Factor Staining Buffer Set | BD | Catalog#: 562574 |
| EasySepTM mouse CD11c positive selection kit | STEMCELL Technologies | Catalog#: 18780 |
| EasySepTM mouse T cell negative isolation kit | STEMCELL Technologies | Catalog#: 19851 |
| EasySepTM mouse naïve CD8+ T cell negative isolation kit | STEMCELL Technologies | Catalog#: 19858 |
| Mouse tumor lymphocyte infiltration kit | Solarbio | Catalog#: P9000 |
| RBC Lysis Buffer | TIAN GEN | Catalog#: RT122 |
| Mouse TNF-α ELISA Research Reagent Kit | 4A Biotech | Catalog#: CME0004 |
| Mouse IL-6 ELISA Research Reagent Kit | 4A Biotech | Catalog#: CME0006 |
| Mouse IFN-β ELISA Research Reagent Kit | 4A Biotech | Catalog#: CME0116 |
| Mouse TNF-α ELISA Research Reagent Kit | Jingmei Biotech | Catalog#: 02415M1 |
| Mouse IL-12 ELISA Research Reagent Kit | Jingmei Biotech | Catalog#: 11506M1, 11386M1 |
| Mouse granzyme B ELISA Research Reagent Kit | Multi Sciences | Catalog#: EK2173-96 |
| Mouse IFN-γ ELISA Research Reagent Kit | Multi Sciences | Catalog#: EK280/3-96 |
| Mouse perforin ELISA Research Reagent Kit | Elabscience | Catalog#: E-EL-M0890c |
| RevertAid Reverse Transcriptase | Thermo Fisher | Catalog#: EP0441 |
| Super Real Pre Mix SYBR Green | Tiangen | Catalog#: FP204-02 |
| Deposited data | ||
| RNA seq dataset | This paper | GEO: GSE222080 |
| Experimental models: Cell lines | ||
| B16-F10 | Cellcook Biotech | Catalog#: CC9016 |
| CT-26 | Cellcook Biotech | Catalog#: CC9036 |
| RM-1 | Cellcook Biotech | Catalog#: CC9004 |
| Pan02 | Provided by Virotech Co., Ltd. | Catalog#: N/A |
| HCT-8 | ATCC | Catalog#: CCL-244 |
| HCT-116 | National Collection of Authenticated Cell Cultures | Catalog#: TCHu 99 |
| SW-620 | National Collection of Authenticated Cell Cultures | Catalog#: TCHu101 |
| Human-derived BMDCs | ORiCells Biotech | Catalog#: PB-DC001F-C, PB-DC002F-C |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Guangdong Medical Laboratory Animal Center | Catalog#: GDMLAC-019 |
| C57BL/6J | Guang Dong GemPharmatech Co., Ltd. | Catalog#: N000013 |
| Balb/c | Guang Dong GemPharmatech Co., Ltd. | Catalog#: N000020 |
| SIRPα-KO C57 | Provided by Prof. Jun Chen | N/A |
| OT-I C57 | Provided by Virotech Co., Ltd. | N/A |
| Batf3-KO C57 | Provided by Prof. Cliff Yang | N/A |
| Oligonucleotides | ||
| CD47 EasyEdit sgRNA | GenScript | CCCTTGCATCGTCCGTAATG |
| primer | Genewiz | Table S1 for detailed sequence information |
| Software and algorithms | ||
| ImageJ | Image.sc | https://imagej.net/ij/index.html |
| Flowjo | BD | https://www.flowjo.com |
| Grahpad Prism (v9) | Dotmatics | https://www.graphpad.com |
| R | The R Foundation | https://www.r-project.org |
| CytExpert | Beckman Coutler | N/A |
| Adobe Illustrator | Adobe | https://www.adobe.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yuan Lin (liny96@mail.sysu.edu.cn).
Materials availability
All unique/stable reagents generated in this study are available from Guangzhou Virotech Pharmaceutical Technology Co., Ltd.
Experimental model and study participant details
Cell lines
The mouse tumor cell lines B16-F10, CT-26, RM-1, Pan02, and the human tumor cell lines HCT-8, HCT-116, SW-620 were purchased from Guangzhou Cellcook Biotech, National Collection of Authenticated Cell Cultures and American Type Culture Collection. B16-F10-OVA cells were generated from B16-F10 cells by transduction of OVA-loaded lentivirus (GeneCopoeia, China). Human-derived BMDCs (PB-DC001F-C, PB-DC002F-C) were purchased from ORiCells Biotech. B16-F10-CD47 KO cells were generated from B16-F10 cells by transduction of Cas9-loaded lentivirus and EasyEdit sgRNA (sequence: CCCTTGCATCGTCCGTAATG) (GenScript, China). Cells were grown in RPMI-1640 medium (Gibco, 11875093 USA) or Dulbecco's Modified Eagle Medium (Gibco, 11965092, USA) supplemented with 10% (vol/vol) fetal bovine serum (FBS; 10099-141, Gibco, USA) and 1% penicillin/streptomycin (SV30010, HyClone, USA). All cells were cultured at 37 °C in 5% CO2.
Animals
This study primarily used C57BL/6J mice as the main animal model. Six- to eight-week-old female Wild-type C57BL/6J or Balb/c mice were purchased from Guangdong Medical Laboratory Animal Center and Guang Dong GemPharmatech Co., Ltd. SIRPα KO (SIRPα-/-) C57 mice were kindly supplied by the Chen Jun laboratory. OT-I C57 mice were kindly supplied by Guangzhou Virotech Pharmaceutical Technology Co., Ltd. Batf3 KO (Batf3-/-) C57 mice were kindly supplied by the Cliff Yang laboratory. All mice were bred in specific pathogen-free facilities. Studies involving animals were approved by the Animal Ethical and Welfare Committee of Sun Yat-sen University.
Method details
OVM preparations
Alphavirus OVM was provided by Guangzhou Virotech Pharmaceutical Technology Co., Ltd. Preparation of OVM: Briefly, African green monkey kidney-derived Vero cells (ATCC) were seeded in culture bottles, cultured in DMEM medium with 10% FBS and 1% penicillin/streptomycin. When the cell density reached approximately 70%, 100 PFU (Plaque forming unit) of OVM was added to infect Vero cells. After 2 h of infection, the DMEM medium was replaced with VP-SFM (11681020, Gibco, USA) medium supplemented with 10% Non-Essential Amino Acids Solution (11140050, Gibco, USA) and 20% Gibco™ GlutaMAX™ (35050079, Gibco, USA). The OVM-treated Vero cells were incubated for 48∼72 h until a significant amount of cell cytopathic effect was observed. The culture supernatant was collected and cellular debris was removed by centrifugation at 4000 ×g. The supernatant was then filtered through a 0.22 μm filter to obtain OVM stock. The stock was stored at -80 °C and viral titers were determined by a TCID50 (PFU = 0.7×TCID50) assay using baby hamster kidney BHK-21 cells.
Tumor-bearing mouse models
For evaluation of antitumor effects, 1×106 B16-F10-OVA cells, 0.5∼3×106 B16-F10 cells, 1×106 B16-F10-CD47 KO cells, 3×106 CT-26 cells, 1×106 RM-1 cells and 3×106 Pan02 cells were inoculated subcutaneously (s.c.) into the hind flanks of C57BL/6J or Balb/c mice on day 0. After palpable tumors developed (approximately 50-100 mm3), mice were randomized to receive the corresponding treatments. Tumor length and width were measured, and the volume was calculated with the formula length × width2/2. The T/C% is defined as the relative tumor proliferation rate, and T/C% values of less than 40% are considered to indicate an effective response. The T/C% was calculated as follows: T/C% = TRTV /CRTV×100%; Vt, tumor volume after treatment; V0, tumor volume before treatment; RTV, relative tumor volume, RTV = V t /V 0; TRTV, RTV in the treatment group; CRTV, RTV in the control group.
Generation of BMDCs and DC vaccine
Bone marrow cells were collected from the tibias and femurs of C57BL/6J or Balb/c mice and cultured with RPMI-1640 medium containing 10% (vol/vol) FBS, 20 ng/ml mGM-CSF (315-03-250, PeproTech, USA) and 10 ng/ml mIL-4 (214-14-100, PeproTech, USA). Fresh medium supplemented with mGM-CSF and mIL-4 was added on day 3, and half of the culture medium was replaced with fresh medium on day 5. iDCs were harvested on day 7.
To generate OVA-loaded DCs, DCs (106 cells/mL) were pulsed with 1 μg/ml OVA (A7641, Sigma, USA) for 12 h and were then stimulated with 500 ng/ml LPS (tlrl-rslps, InvivoGen, France).To generate oncolysate-loaded DCs, B16-F10, CT-26, Pan02 or RM-1 tumor cells were seeded in 100 mm petri dishes (1∼3×106 cells in 10 ml of RPMI-1640 medium supplemented with mGM-CSF and mIL-4) and infected with OVM (0.1∼1 MOI) for 24∼48 h. After complete killing of tumor cells, the supernatant was collected and filtered through a 0.22 μm filter membrane (Millipore, USA) to remove cell debris, resulting in the tumor-derived oncolysate. The oncolysate was used to induce iDCs at a concentration of 106 cells/mL, and the oncolysate-loaded DC vaccine was harvested after 24 h.
To harvest the DC vaccine, the suspended DCs were collected into centrifuge tubes and centrifuged at 500×g for 5 min, the supernatant is then discarded and the cells are resuspended in PBS, the cells were washed twice by centrifugation at 500×g, and finally, the cells were diluted to a concentration of 107 cells/mL using RPMI-1640 medium without fetal bovine serum or penicillin/streptomycin.
DCs cocultured with B16-F10 cells were purified with a EasySep™ mouse CD11c positive selection kit (18780, STEMCELL Technologies, Canada). SIRPα KO mouse-derived DCs were confirmed with SIRPα-Alexa Fluor 700 (144022, BioLegend, USA).
T cell isolation
T cells were isolated from mouse spleens with a EasySep™ mouse T cell negative isolation kit (19851, STEMCELL Technologies, Canada). CD8+ T cells were isolated from mouse spleens with a EasySep™ mouse naïve CD8+ T cell negative isolation kit (19858, STEMCELL Technologies, USA).
In vivo dosing regimen
OVM (3×106 TCID50 per dose) or vehicle was administered intravenously daily for 5 consecutive days. The DC vaccine (1×106 cells/mouse) was administered via intratumoral injection. The anti-PD-L1 antibody (10 mg/kg/dose, clone 10F.9G2, BE0101, Bio X cell, USA) or the respective isotype control (10 mg/kg/dose, BE0090, Bio X cell, USA) was injected intraperitoneally every two days. For deletion of CD4+ or CD8+ T cells, the anti-CD4 (10 mg/kg/dose, clone GK1.5, A2101, Selleck, USA), anti-CD8 antibody (10 mg/kg/dose, clone GK2.43, A2102, Selleck, USA) or the respective isotype control (10 mg/kg/dose, A2116, Selleck, USA) was injected intraperitoneally every two days, respectively.
Isolation of TILs
B16-F10 tumor-bearing mice were sacrificed on the second day after the completion of OVM administration; tumor tissues were harvested; and single-cell suspensions were prepared by grinding and filtering the tissues through a 40 μm strainer (CSS-010-040, BIOFIL, USA) before lysis of red blood cells (RT122-02, Tiangen Biochemical Technology, China). Single-cell suspensions were washed once with vehicle, and TILs were isolated from tumors using a mouse tumor lymphocyte infiltration kit (P9000, Solarbio, China). Freshly isolated lymphocytes were cocultured with preseeded B16-F10 cells for 2 days. Then, the cells were washed twice with vehicle to remove suspended lymphocytes, and the viability of adherent B16-F10 tumor cells was detected by a MTT assay. In brief, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; 102227, MP Biomedicals, USA) (1 mg/ml final concentration) was added to each well and incubated at 37 °C for 2 h. Then, the MTT-containing medium was removed, dimethyl sulfoxide was added to dissolve the MTT precipitates, and the absorbance at 490 nm was determined using a microplate reader. Lymphocytotoxicity was calculated using the following formula: Lymphocytotoxicity = (Atumor cells-Atumor cells+lymphocytes)/(Atumor cells-Ablank)×100%.
Flow cytometry
For detection of DC maturation after coculture with tumor cells, 1.5×105 B16-F10, CT-26, Pan02, HCT-8, HCT-116 and SW620 tumor cells were inoculated in 6-well plates in advance. After the tumor cells adhered to the walls, 4.5×105 DCs were added and cocultured for 24 h (HCT-8, HCT-116 and SW-620), 48 h (B16-F10 and CT-26) or 72 h (Pan02). The DCs suspended in the supernatant were then collected and washed twice with DPBS. Then, the mouse-derived DCs were stained with 1:100 dilutions of fluorochrome-conjugated anti-Mouse CD11c APC (17-0114-82, eBioscience, USA), anti-Mouse CD86 eFluor 450 (48-0862-82, eBioscience, USA), anti-Mouse CD83 PE (12-0831-82, eBioscience, USA) and anti-Mouse MHC II APC eFluor 780 (47-5321-82, eBioscience, USA) antibodies at 4 °C for 30 min. and the human-derived DCs were stained with anti-Human CD11c BV421 (562561, BD, USA), anti-Human CD86 APC (555660, BD, USA) and anti-Human MHC II PE (555812, BD, USA) antibodies.
To explore the effect of tumor cells on DC antigen presentation ability, 4.5×105 DCs were cocultured with 1.5×105 B16-F10 tumor cells for 24 h and incubated with 1 μg/ml OVA for an additional 24 h. The suspended cells were collected and stained for 2 h at 4 °C with the following antibodies: anti-Mouse CD11c PE Cyanine7 (117318, BioLegend, USA) and anti-Mouse OVA257-264 (SIINFEKL) peptide bound to H-2Kb APC (17-5743-80, eBioscience, USA).
To test the ability of differently treated DCs to activate T cells, DCs and spleen-derived T cells were cocultured at a 1:5 ratio for 96 h. To determine the ability of OVA-loaded DCs to stimulate OI-T cells, DCs were separated with magnetic beads and cocultured with spleen-derived CD8+ T cells from OT-I mice at a 1:2 ratio for 48 h. The suspended cells in the supernatant were collected, washed twice with vehicle and stained with the following fluorescent antibodies: anti-Mouse CD3 PE-Cyanine 5.5 (35-0031-82, eBioscience, USA), anti-Mouse CD8 PE (12-0081-82, eBioscience, USA), anti-Mouse CD4 APC (17-0042-82, eBioscience, USA), anti-Mouse CD44 Super Bright 645 (64-0441-82, eBioscience, USA), and anti-Mouse CD69 PE Cyanine 7 (25-0691-82, eBioscience, USA). Flow cytometric analysis was performed (CytoFLEX, Beckman Coulter, USA).
To detect changes in immune checkpoints on the surface of DCs after coculture with tumor cells, DCs were cocultured with B16-F10 tumor cells for 48 h and collected for staining with the following fluorescent antibodies: anti-Mouse CD11c PE Cyanine7, anti-Mouse SIRPα Alexa Fluor 700 (144022, BioLegend, USA), anti-Mouse CD33 PE (12-0331-82, eBioscience, USA), anti-Mouse PIR-A/B APC(17-3101-82, eBioscience, USA), anti-Mouse PD-1 APC (17-9981-82, eBioscience, USA), anti-Mouse TIM-3 PE (12-5870-82, eBioscience, USA), and anti-Mouse CTLA-4 PE (553720, BD, USA).
To detect the activation of Co-DCs by OVM, 1.5×105 B16-F10 or HCT-8 tumor cells were inoculated in 6-well plates in advance. After the tumor cells adhered to the walls, 4.5×105 DCs were added and cocultured for 24 h. Then treated with vehicle or 1×106 PFU OVM for another 24 h. The DCs suspended in the supernatant were then collected and washed twice with DPBS. After that, DCs were stained with 1:100 dilutions of fluorochrome-conjugated antibodies at 4 °C for 30 min. To detect the SIRPα changes in CTL or OVM treated Co-DCs, DCs were treated according to the above-mentioned method, and were stained with anti-mouse SIRPα Alexa Fluor 700 or anti-Human SIRPα Alexa Fluor 700 (144022, BD, USA).
To detect the infiltration of immune cells and performed phenotypic analysis in the TME, single-cell suspensions of tumor and spleen cells were prepared as described above. The cells were washed with vehicle, stained with Fixable Viability Stain 510 (564406, BD, USA) or Fixable Viability Stain 780 (565388, BD, USA) to exclude cell debris and dead cells and preincubated (15 min, 4 °C) with an anti-Mouse CD16/CD32 Monoclonal (Fc block, Clone 2.4G2, 553141, BD, USA) to block nonspecific binding. The cells were then stained (30 min, 4 °C) with 1:100 dilutions of various combinations of the following fluorochrome-conjugated antibodies: anti-Mouse CD45 eFluor 450 (48-0451-82, eBioscience, USA), anti-Mouse CD3 PerCP-Cy™5.5 (551163, BD, USA), anti-Mouse CD8 Alexa Fluor 700 (557959, BD, USA), anti-Mouse CD4 APC (17-0042-82, eBioscience, USA), anti-Mouse CD4 BV510 (743155, BD, USA), anti-Mouse CD62L FITC (561917, BD, USA), anti-Mouse CD44 BV650 (740455, BD, USA), anti-Mouse PD-1 PE (551892, BD, USA), anti-Mouse CD274 PE (558091, BD, USA), anti-Mouse CD11c APC, anti-Mouse MHC II APC eFluor 780, anti-Mouse CD11b FITC (11-0112-81, eBioscience, USA), anti-Mouse CD103 PE Cyanine7 (121425, BioLegend, USA) and anti-Mouse SIRPα Alexa Fluor 700. To analyze the changes in Treg, cells were stained with the aforementioned method using a viability dye, followed by blocking of Fc receptors, and surface staining with anti-Mouse CD3, anti-Mouse CD4 and anti-Mouse CD25 APC (557192, BD, USA) antibodies. And then, cells were fixed and permeabilized using a Transcription Factor Staining Buffer Set (562574, BD, USA) following the manufacturers' instructions and incubated with anti-Mouse Foxp3 PE (563101, BD, USA) antibody for 1 h at room temperature.
To identify whether OVM downregulates CD47 on the surface of tumor cells after infection in vitro, 2×105 B16-F10, CT-26, Pan02 or HCT-8 tumor cells were inoculated in 6-well plates in advance. After the tumor cells adhered to the walls, they were treated with different titers of OVM for 24 h, and the expression of CD47 was determined by flow cytometry. The cells were fully incubated with trypsin, and the reaction was terminated with complete medium. The cells were then washed twice with vehicle and incubated with a fluorochrome-conjugated anti-Mouse CD47 PE (127508, BioLegend, USA) or anti-Human CD47 PE (556046, BD, USA). For detection of changes in CD47 expression in an in vivo B16-F10 tumor model, tumor tissues were processed into a single-cell suspension similar to that described above and stained with anti-Mouse CD45 eFluor 450 and anti-Mouse CD47 PE antibodies for 30 min at 4 °C.
ELISA
Cell culture supernatants were obtained and centrifuged at 1000 ×g for 10 min at 4 °C to remove cell debris. The concentrations of TNF-α (CME0004, 4A Biotech, China; 02415M1, Jingmei Biotech, China), IL-12 (11506M1, 11386M1, Jingmei Biotech, China), IL-6 (CME0006, 4A Biotech, China), IFN-β (CME0116, 4A Biotech, China), granzyme B (EK2173-96, Multi Sciences, China), perforin (E-EL-M0890c, Elabscience, China) and IFN-γ (EK280/3-96, Multi Sciences, USA) were measured with the corresponding ELISA kits in accordance with the manufacturer’s instructions.
For evaluate the levels of IL-12 in DC vaccine or SIRPα-KO DC vaccine treated tumor-bearing mice, tumor interstitial fluid was collected from freshly resected B16-F10 tumor. Tumor tissue were cut into small pieces, washed once with PBS, and then incubated at 37 °C for 2 h in 1 mL PBS per 1 g of tumor tissue. After incubation, the samples were centrifuged at 4 °C, 1000×g for 15 min, and the supernatant was transferred in new tubes. This process was repeated once more. Afterwards, the supernatant was collected for ELISA analysis.
Quantitative reverse transcription–PCR (qRT–PCR)
Total RNA was extracted with TRIzol reagent (15596018, Life Technologies, USA), and reverse transcription was performed with oligo(dT) primers and RevertAid Reverse Transcriptase (EP0441, Thermo Fisher, USA) according to the manufacturer’s instructions. qPCR was performed with Super Real Pre Mix SYBR Green (FP204-02, Tiangen, China) in an Applied Biosystems 7500 Fast Real-Time PCR System (Life Technologies, USA). Gene expression levels were normalized to those of β-actin. The amplification primers were purchased from Genewiz (China).
Western blot analysis
Cell pellets were lysed using M-PER Mammalian Protein Extraction Reagent (78501, Thermo Scientific, USA). Proteins were separated by SDS/PAGE and analyzed by immunoblotting using primary antibodies from a TREM2 signaling pathway sampler kit (77533T, Cell Signaling Technology, USA), including antibodies specific for DAP12 (D7G1X), Syk (D3Z1E), phospho-Syk (Tyr525/526, C87C1), PLCγ1 (D9H10), phospho-PLCγ1 (Tyr783, D6M9S), PLCγ2 (E5U4T), phospho-PLCγ2 (Tyr759, E9E9Y) and GAPDH (14C10), followed by appropriate HRP-conjugated secondary antibodies. Membranes were visualized on a ChemiDoc XRS+ System (Bio–Rad, USA) using Immobilon Western Chemiluminescent HRP Substrate (WBKLS0500, Millipore, USA).
CCK8 assay
To compare the growth rates between B16-F10-WT and B16-F10-CD47 KO cells, 1×104 B16-F10-WT or B16-F10-CD47 KO cells were inoculated in 48-well plates in advance, and 50 μL per well CCK-8 reagent was added at indicated time points and incubated at 37 °C for 2 h, the absorbance at 450 nm was detected by using a microplate reader.
Source and analysis of TCGA and GTEx datasets
Datasets information:
Pan-cancer datasets from both TCGA and GTEx were retrieved from the UCSC database (https://xenabrowser.net/). The expression data for the genes MYC (ENSG00000136997), CD47 (ENSG00000196776), and SIRPA (ENSG00000198053) in diverse tumor samples were extracted and transformed using the log2(x+0.001) method within the Sangerbox platform (http://sangerbox.com/home.html). Detailed processed gene expression data can be accessed after registering and logging into Sangerbox.
Correlation Analysis:
The MYC expression was matched in the same patients across different cancer types with the expression of either CD47 or SIRPA. The bioinformatics platform (http://www.bioinformatics.com.cn/) was used to perform Pearson correlation analysis on the data, resulting in correlation coefficient scatterplots.
Quantification and statistical analysis
All statistical analyses were performed using GraphPad Prism ver. 6.0. All sample sizes and statistical methods are indicated in the corresponding figure legends. No statistical methods were used to predetermine the sample size. No data generated were excluded. If the data were normally distributed (by the Shapiro–Wilk test) and homoscedastic (by Bartlett’s test), Student’s t test (for two groups) and one-way analysis of variance (ANOVA) (for more than two groups) were used to test the differences in the means between two groups. Most data were analyzed using two-tailed Student’s t test or one-way ANOVA with Dunnett’s test for multiple comparisons. The bars show the mean ± SD values. Tumor volumes were analyzed using Student’s t test or one-way ANOVA at the terminal endpoint as indicated in the generated plots and the bars show the mean ± SEM values. Survival was analyzed by the Kaplan–Meier method, and differences were compared using the log-rank test. Significant differences were indicated as ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Acknowledgments
This work was funded by grants from the National Key R&D Program of China (2021YFA0909800), National Natural Science Foundation of China (82173838, 82172730), and Guangdong Basic and Applied Basic Research Foundation (2020A1515110907, 2021A1515011881, 2022B1515020056) and Fundamental Research Funds for the Central Universities, Sun Yat-sen University (23ykbj001). We acknowledge Prof. Jun Chen and Prof. Cliff Yang of Sun Yat-sen University for their kindly suggestions and generous donations of transgenic mice for the whole work. SIRPα-knockout mice were kindly supplied by the Chen Jun laboratory. OT-I mice were kindly supplied by Guangzhou Virotech Pharmaceutical Technology Co., Ltd. Batf3 knockout mice were kindly supplied by the Cliff Yang laboratory.
Author contributions
Conceptualization, Y. Lin and J.C.; methodology, J.D., Y. Zhong, Y. Zeng, C.W., C.X., J.Z., S.H., and L.H.; investigation, J.D., Y. Zhong, Z.F., and Y. Zeng; visualization, Y. Lin, J.C., W.Z., and J.L.; funding acquisition, J.C., Y. Lin, J.L., G.Y., and Y. Liu; project administration, Y. Lin. and J.C.; writing – original draft, J.D.; writing – review & editing, J.D., W.Z., Y. Lin, J.C., and J.L.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 10, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101229.
Contributor Information
Jiankai Liang, Email: liangjk5@mail.sysu.edu.cn.
Yuan Lin, Email: liny96@mail.sysu.edu.cn.
Supplemental information
Data and code availability
-
•
Data:Gene expression profiles for DCs in this study have been deposited at GEO and are publicly available as of the date of publication (GEO: GSE222080). Tumor gene expression profiles from diverse cancer patients were sourced from TCGA and GTEx datasets, which were obtained from the UCSC (https://xenabrowser.net/). These datasets were extracted and transformed using Sangerbox platform (http://sangerbox.com/home.html), and analyzed using bioinformatics platform (http://www.bioinformatics.com.cn/). The prediction of transcription factors was obtained from the Encyclopedia of DNA Elements (https://www.genome.gov/Funded-Programs-Projects/ENCODE-Project-ENCyclopedia-Of-DNA-Elements). Experimental data reported in our paper is available from the lead contact upon request.
-
•
Code:This paper does not involve any custom code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J. Clin. Invest. 2015;125:3335–3337. doi: 10.1172/JCI83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenbarth S.C. Dendritic cell subsets in T cell programming: location dictates function. Nat. Rev. Immunol. 2019;19:89–103. doi: 10.1038/s41577-018-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabado R.L., Balan S., Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74–95. doi: 10.1038/cr.2016.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harari A., Graciotti M., Bassani-Sternberg M., Kandalaft L.E. Antitumour dendritic cell vaccination in a priming and boosting approach. Nat. Rev. Drug Discov. 2020;19:635–652. doi: 10.1038/s41573-020-0074-8. [DOI] [PubMed] [Google Scholar]
- 5.Cheever M.A., Higano C.S. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011;17:3520–3526. doi: 10.1158/1078-0432.CCR-10-3126. [DOI] [PubMed] [Google Scholar]
- 6.Lindskog M., Laurell A., Kjellman A., Melichar B., Niezabitowski J., Maroto P., Zieliński H., Villacampa F., Bigot P., Bajory Z., et al. A randomized phase II study with ilixadencel, a cell-based immune primer, plus sunitinib versus sunitinib alone in synchronous metastatic renal cell carcinoma. J. Clin. Oncol. 2020;38:11. doi: 10.1016/j.euros.2022.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y., Xiang Y., Xin V.W., Wang X.W., Peng X.C., Liu X.Q., Wang D., Li N., Cheng J.T., Lyv Y.N., et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 2020;13:107. doi: 10.1186/s13045-020-00939-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wculek S.K., Cueto F.J., Mujal A.M., Melero I., Krummel M.F., Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020;20:7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 9.Hosseini R., Asef-Kabiri L., Yousefi H., Sarvnaz H., Salehi M., Akbari M.E., Eskandari N. The roles of tumor-derived exosomes in altered differentiation, maturation and function of dendritic cells. Mol. Cancer. 2021;20:83. doi: 10.1186/s12943-021-01376-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayoux M., Roller A., Pulko V., Sammicheli S., Chen S., Sum E., Jost C., Fransen M.F., Buser R.B., Kowanetz M., et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020;12:eaav7431. doi: 10.1126/scitranslmed.aav7431. [DOI] [PubMed] [Google Scholar]
- 11.Dixon K.O., Tabaka M., Schramm M.A., Xiao S., Tang R., Dionne D., Anderson A.C., Rozenblatt-Rosen O., Regev A., Kuchroo V.K. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature. 2021;595:101–106. doi: 10.1038/s41586-021-03626-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hemminki O., Dos Santos J.M., Hemminki A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020;13:84. doi: 10.1186/s13045-020-00922-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B., Cheng P. Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy. Mol. Cancer. 2020;19:158. doi: 10.1186/s12943-020-01275-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zitvogel L., Galluzzi L., Kepp O., Smyth M.J., Kroemer G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015;15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 15.Brown M.C., Holl E.K., Boczkowski D., Dobrikova E., Mosaheb M., Chandramohan V., Bigner D.D., Gromeier M., Nair S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017;9:eaan4220. doi: 10.1126/scitranslmed.aan4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Y., Zhang H., Liang J., Li K., Zhu W., Fu L., Wang F., Zheng X., Shi H., Wu S., et al. Identification and characterization of alphavirus M1 as a selective oncolytic virus targeting ZAP-defective human cancers. Proc. Natl. Acad. Sci. USA. 2014;111:E4504–E4512. doi: 10.1073/pnas.1408759111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song D., Jia X., Liu X., Hu L., Lin K., Xiao T., Qiao Y., Zhang J., Dan J., Wong C., et al. Identification of the receptor of oncolytic virus M1 as a therapeutic predictor for multiple solid tumors. Signal Transduct. Target. Ther. 2022;7:100. doi: 10.1038/s41392-022-00921-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H., Lin Y., Li K., Liang J., Xiao X., Cai J., Tan Y., Xing F., Mai J., Li Y., et al. Naturally Existing Oncolytic Virus M1 Is Nonpathogenic for the Nonhuman Primates After Multiple Rounds of Repeated Intravenous Injections. Hum. Gene Ther. 2016;27:700–711. doi: 10.1089/hum.2016.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y., Cai J., Liu W., Lin Y., Guo L., Liu X., Qin Z., Xu C., Zhang Y., Su X., et al. Intravenous injection of the oncolytic virus M1 awakens antitumor T cells and overcomes resistance to checkpoint blockade. Cell Death Dis. 2020;11:1062. doi: 10.1038/s41419-020-03285-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W., Liu Y., Hu C., Xu C., Chen J., Chen Y., Cai J., Yan G., Zhu W. Cytotoxic T lymphocyte-associated protein 4 antibody aggrandizes antitumor immune response of oncolytic virus M1 via targeting regulatory T cells. Int. J. Cancer. 2021;149:1369–1384. doi: 10.1002/ijc.33703. [DOI] [PubMed] [Google Scholar]
- 21.Zhu W., Liang J., Tan J., Guo L., Cai J., Hu J., Yan G., Liu Y., Zhang J., Song D., et al. Real-Time Visualization and Quantification of Oncolytic M1 Virus In Vitro and In Vivo. Hum. Gene Ther. 2021;32:158–165. doi: 10.1089/hum.2020.273. [DOI] [PubMed] [Google Scholar]
- 22.Roberts E.W., Broz M.L., Binnewies M., Headley M.B., Nelson A.E., Wolf D.M., Kaisho T., Bogunovic D., Bhardwaj N., Krummel M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell. 2016;30:324–336. doi: 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verneau J., Sautés-Fridman C., Sun C.M. Dendritic cells in the tumor microenvironment: prognostic and theranostic impact. Semin. Immunol. 2020;48:101410. doi: 10.1016/j.smim.2020.101410. [DOI] [PubMed] [Google Scholar]
- 24.Coxon C.H., Geer M.J., Senis Y.A. ITIM receptors: more than just inhibitors of platelet activation. Blood. 2017;129:3407–3418. doi: 10.1182/blood-2016-12-720185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J.B., Ha S.J., Kim H.R. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Front. Pharmacol. 2021;12:681320. doi: 10.3389/fphar.2021.681320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bornhöfft K.F., Goldammer T., Rebl A., Galuska S.P. Siglecs: A journey through the evolution of sialic acid-binding immunoglobulin-type lectins. Dev. Comp. Immunol. 2018;86:219–231. doi: 10.1016/j.dci.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 27.Tang C., Makusheva Y., Sun H., Han W., Iwakura Y. Myeloid C-type lectin receptors in skin/mucoepithelial diseases and tumors. J. Leukoc. Biol. 2019;106:903–917. doi: 10.1002/JLB.2RI0119-031R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Logtenberg M.E.W., Scheeren F.A., Schumacher T.N. The CD47-SIRPα Immune Checkpoint. Immunity. 2020;52:742–752. doi: 10.1016/j.immuni.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia X., Yan B., Tian X., Liu Q., Jin J., Shi J., Hou Y. CD47/SIRPα pathway mediates cancer immune escape and immunotherapy. Int. J. Biol. Sci. 2021;17:3281–3287. doi: 10.7150/ijbs.60782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z., Li B., Li S., Lin W., Wang Z., Wang S., Chen W., Shi W., Chen T., Zhou H., et al. Metabolic control of CD47 expression through LAT2-mediated amino acid uptake promotes tumor immune evasion. Nat. Commun. 2022;13:6308. doi: 10.1038/s41467-022-34064-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun C., Mezzadra R., Schumacher T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson P., Rosendahl N., Radford K.J. Conventional type 1 dendritic cells (cDC1) as cancer therapeutics: challenges and opportunities. Expert Opin. Biol. Ther. 2022;22:465–472. doi: 10.1080/14712598.2022.1994943. [DOI] [PubMed] [Google Scholar]
- 33.Zhu S., Zhang T., Zheng L., Liu H., Song W., Liu D., Li Z., Pan C.X. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021;14:156. doi: 10.1186/s13045-021-01164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwarze J.K., Tijtgat J., Awada G., Cras L., Vasaturo A., Bagnall C., Forsyth R., Dufait I., Tuyaerts S., Van Riet I., Neyns B. Intratumoral administration of CD1c (BDCA-1)(+) and CD141 (BDCA-3)(+) myeloid dendritic cells in combination with talimogene laherparepvec in immune checkpoint blockade refractory advanced melanoma patients: a phase I clinical trial. J. Immunother. Cancer. 2022;10:e005141. doi: 10.1136/jitc-2022-005141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chesney J.A., Ribas A., Long G.V., Kirkwood J.M., Dummer R., Puzanov I., Hoeller C., Gajewski T.F., Gutzmer R., Rutkowski P., et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined With Pembrolizumab for Advanced Melanoma. J. Clin. Oncol. 2023;41:528–540. doi: 10.1200/JCO.22.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li R., Zhang J., Gilbert S.M., Conejo-Garcia J., Mulé J.J. Using oncolytic viruses to ignite the tumour immune microenvironment in bladder cancer. Nat. Rev. Urol. 2021;18:543–555. doi: 10.1038/s41585-021-00483-z. [DOI] [PubMed] [Google Scholar]
- 37.Filin I.Y., Kitaeva K.V., Rutland C.S., Rizvanov A.A., Solovyeva V.V. Recent Advances in Experimental Dendritic Cell Vaccines for Cancer. Front. Oncol. 2021;11:730824. doi: 10.3389/fonc.2021.730824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saxena M., Bhardwaj N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer. 2018;4:119–137. doi: 10.1016/j.trecan.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Q., Wen W., Tang L., Qin C.J., Lin Y., Zhang H.L., Wu H.P., Ashton C., Wu H., Ding J., et al. Inhibition of SIRPα in dendritic cells potentiates potent antitumor immunity. OncoImmunology. 2016;5:e1183850. doi: 10.1080/2162402X.2016.1183850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Latour S., Tanaka H., Demeure C., Mateo V., Rubio M., Brown E.J., Maliszewski C., Lindberg F.P., Oldenborg A., Ullrich A., et al. Bidirectional negative regulation of human T and dendritic cells by CD47 and its cognate receptor signal-regulator protein-alpha: down-regulation of IL-12 responsiveness and inhibition of dendritic cell activation. J. Immunol. 2001;167:2547–2554. doi: 10.4049/jimmunol.167.5.2547. [DOI] [PubMed] [Google Scholar]
- 41.Matozaki T., Murata Y., Okazawa H., Ohnishi H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009;19:72–80. doi: 10.1016/j.tcb.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 42.Xu M.M., Pu Y., Han D., Shi Y., Cao X., Liang H., Chen X., Li X.D., Deng L., Chen Z.J., et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein α Signaling. Immunity. 2017;47:363–373.e5. doi: 10.1016/j.immuni.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y., Zhang M., Wang X., Liu W., Wang H., Yang Y.G. Vaccination with CD47 deficient tumor cells elicits an antitumor immune response in mice. Nat. Commun. 2020;11:581. doi: 10.1038/s41467-019-14102-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Duijn A., Van der Burg S.H., Scheeren F.A. CD47/SIRPα axis: bridging innate and adaptive immunity. J. Immunother. Cancer. 2022;10:e004589. doi: 10.1136/jitc-2022-004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matlung H.L., Szilagyi K., Barclay N.A., van den Berg T.K. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017;276:145–164. doi: 10.1111/imr.12527. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data:Gene expression profiles for DCs in this study have been deposited at GEO and are publicly available as of the date of publication (GEO: GSE222080). Tumor gene expression profiles from diverse cancer patients were sourced from TCGA and GTEx datasets, which were obtained from the UCSC (https://xenabrowser.net/). These datasets were extracted and transformed using Sangerbox platform (http://sangerbox.com/home.html), and analyzed using bioinformatics platform (http://www.bioinformatics.com.cn/). The prediction of transcription factors was obtained from the Encyclopedia of DNA Elements (https://www.genome.gov/Funded-Programs-Projects/ENCODE-Project-ENCyclopedia-Of-DNA-Elements). Experimental data reported in our paper is available from the lead contact upon request.
-
•
Code:This paper does not involve any custom code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.