

Abstract
Advances in genetic tools and sequencing technology in the past few years have vastly expanded our understanding of the genetics of neurodevelopmental disorders. Recent high-throughput sequencing analyses of structural brain malformations, cognitive and neuropsychiatric disorders, and localized cortical dysplasias have uncovered a diverse genetic landscape beyond classic Mendelian patterns of inheritance. The underlying genetic causes of neurodevelopmental disorders implicate numerous cell biological pathways critical for normal brain development.
Keywords: neurodevelopment, brain malformations, somatic mutations, autism
INTRODUCTION
The complexity of the human brain is astounding. During development, distinct cell types must proliferate, differentiate into various fates, migrate to their proper locations, and integrate into a cohesive circuitry. If all goes well, at the end of development, 85 billion neurons (8) will form the highly specified and specialized human brain, capable of complex language, cognition, and emotion. These elaborate and uniquely human processes have intrigued scientists and philosophers alike. To understand how these processes occur, physicians and scientists have often focused on disorders of brain function. By studying how brain development and function go awry, we can better understand the critical components of normal development and function.
Human genetics is a powerful tool to dissect brain development because it is increasingly clear that humans as a population show mutations in every gene—if not every codon—of the genome (21, 134, 135), providing a rich opportunity to identify disease-causing genes essential for development. Historically, geneticists have relied heavily on Mendelian principles of inheritance, based on the premise that each gene possesses a specific function that, when perturbed, causes a specific set of disease symptoms (26). However, the era of next-generation, high-throughput deep sequencing has changed the way we study human genetics (79). The ability to sequence massive amounts of DNA rapidly, cost effectively, and at greater sequence read depth has led to an explosion of computationally based gene identification, in which the tens of thousands of genetic variants in an individual’s exome are filtered by multiple layers of criteria (including inheritance model, type of mutation, absence in control populations, allelic frequency, and predicted pathogenicity) to rapidly arrive at a small subset of candidate disease genes (88, 89). Compared with the years required to identify disease-causing mutations two decades ago, we can now identify variants throughout an individual’s exome in a matter of weeks. These techniques have led to a surge in the identification of novel disease genes and new alleles in known disease genes.
In particular, high-throughput sequencing has allowed us to more readily capture mutations causing subtle or atypical phenotypes, including hypomorphic and less-penetrant alleles. As a result, novel mutations in known disease-causing genes are being linked to phenotypes separate from their typical Mendelian phenotype. Recent studies in neurodevelopment, reviewed here, indicate that the genetic and phenotypic diversity of known disease genes is even broader than previously anticipated and shed light on the structural and connective complexity of the brain.
MENDELIAN DISORDERS OF NEURODEVELOPMENT HIGHLIGHT PATHWAYS CRITICAL FOR CORTICOGENESIS
The normal human cortex lies at the surface of the brain and is composed of six distinct histological layers. Development begins from neuroepithelial progenitors lining the lateral ventricles that divide to expand the progenitor pool and then give rise to intermediate progenitors, which subsequently divide and give rise to neurons (see sidebar Overview of Neuronal Development along with Figure 1). The neurons migrate from the proliferative ventricular zones toward the pial surface of the brain to form the layered cortex, where the connections between neurons form and mature (78). The convolutions of the brain surface, known as gyri and sulci, are thought to be due to the dramatic expansion of neurons in number and surface area, relative to the volume of the brain (109, 145).
OVERVIEW OF NEURONAL DEVELOPMENT.
The neocortex comprises two major cell types: neurons and glia. Neurons are the signaling cells of the nervous system, and glia perform myriad functions to support the function of neurons. Neurons can be further subdivided into excitatory projection neurons and inhibitory interneurons. The development of excitatory neurons differs from that of inhibitory neurons.
Projection neurons are born from polarized neuroepithelial cells lining the lateral ventricle, which divide and differentiate into radial glial progenitors, with cell bodies along the lateral ventricle and a long basolateral fiber extending to the pial surface (78) (see Figure 1). Radial glia subsequently divide to give rise to intermediate progenitors and outer radial glial progenitors, forming the proliferative ventricular zones of the brain. These progenitors divide and give rise to excitatory projection neurons, which migrate along the radial processes of the radial glia toward the pial surface and form the cortical plate. Early-born neurons form the deepest layers of the developing cortex, and later-born neurons migrate to the more superficial surface to form the upper layers of the cortex, eventually forming a highly organized, laminar structure. After neurogenesis is complete, progenitors give rise to glia (48). In contrast, inhibitory interneurons are born in the ventral regions of the developing brain, in the ganglionic eminences, and first migrate tangentially into the dorsal telencephalon. Upon reaching the dorsal cortex, they turn and migrate radially into the cortical plate and integrate with the excitatory projection neurons (138).
Mutations in various genes have been shown to interfere with the proliferation and migration of neurons. Microcephaly genes, including WDR62 and NDE1, discussed in this review, are implicated in ventricular zone progenitor proliferation, and lissencephaly genes, including LIS1, DCX, and ARX, affect neuronal migration from the ventricular zones to the cortical plate. In addition, polyalanine tract expansions in ARX affect interneuron migration, resulting in imbalances between excitation and inhibition, which are thought to be the cause of epilepsy. Cognitive and neuropsychiatric disorders are also believed to be due to perturbations in synapse function.
Figure 1.
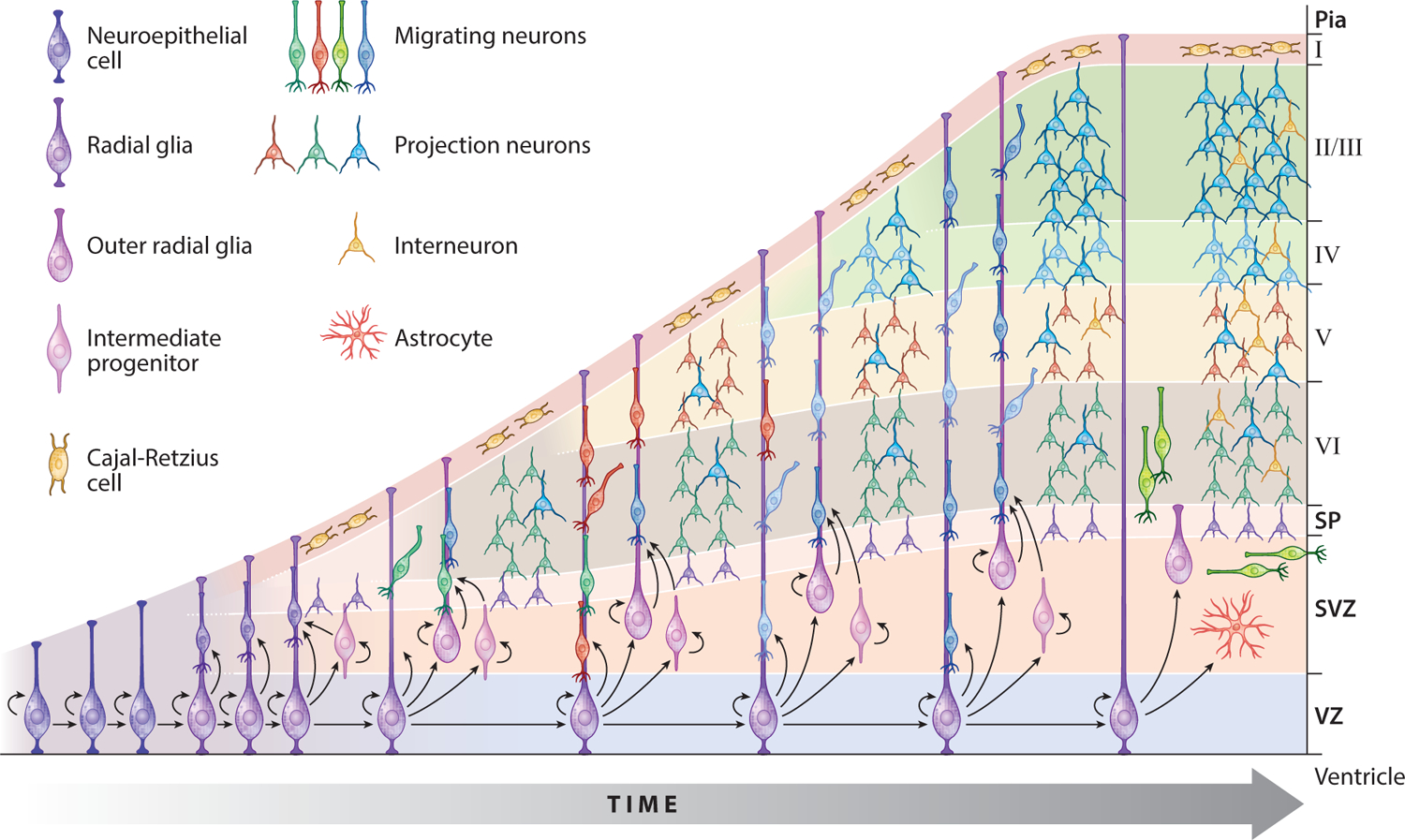
Schematic of neocortical development. The development of the cortex begins at the ventricular zone (VZ) for excitatory projection neurons and at the ganglionic eminences for inhibitory interneurons. Both progenitor populations undergo multiple rounds of proliferation before differentiating into neurons and migrating into the cortical plate, where they integrate into functional circuitry. (For additional details, see sidebar Overview of Neuronal Development.) Abbreviations: SP, subplate; SVZ, subventricular zone. Adapted from Greig et al. (48) with permission.
Diseases have been identified that affect each stage of neurodevelopment (Figure 2). For example, microcephaly (“small brain”) is caused by defects in progenitor proliferation, resulting in a decreased number of neurons and smaller brain size. The identification of autosomal recessive primary microcephaly (MCPH) genes reflects the changing dynamics of human genetics as a result of high-throughput sequencing. Initial linkage analyses more than a decade ago identified six loci for MCPH (107), and subsequent positional cloning revealed causative mutations in genes encoding centrosomal and pericentriolar proteins (including MCPH1, CDK5RAP2, ASPM, and CENPJ) (15–17, 49, 61, 68, 90, 119, 122, 143). However, several loci, notably MCPH2 and MCPH4, remained elusive (63, 108), as the linked intervals simply contained too many genes. With the advent and power of high-throughput sequencing, three groups nearly simultaneously discovered WDR62 as the causative disease gene at the MCPH2 locus (15, 90, 143). More than a dozen new null and hypomorphic WDR62 alleles were identified and connected WDR62 to a wide array of structural brain defects beyond primary microcephaly. The causative disease gene at the MCPH4 locus also required more than a decade to identify, with studies using high-throughput techniques eventually finding two separate genes at this locus, CASC5 and CEP152, that cause microcephaly (42, 49). In addition to the six classic MCPH loci, more novel MCPH genes have been identified in the past few years, including STIL, CEP135, and CEP63 (60, 68, 122).
Figure 2.
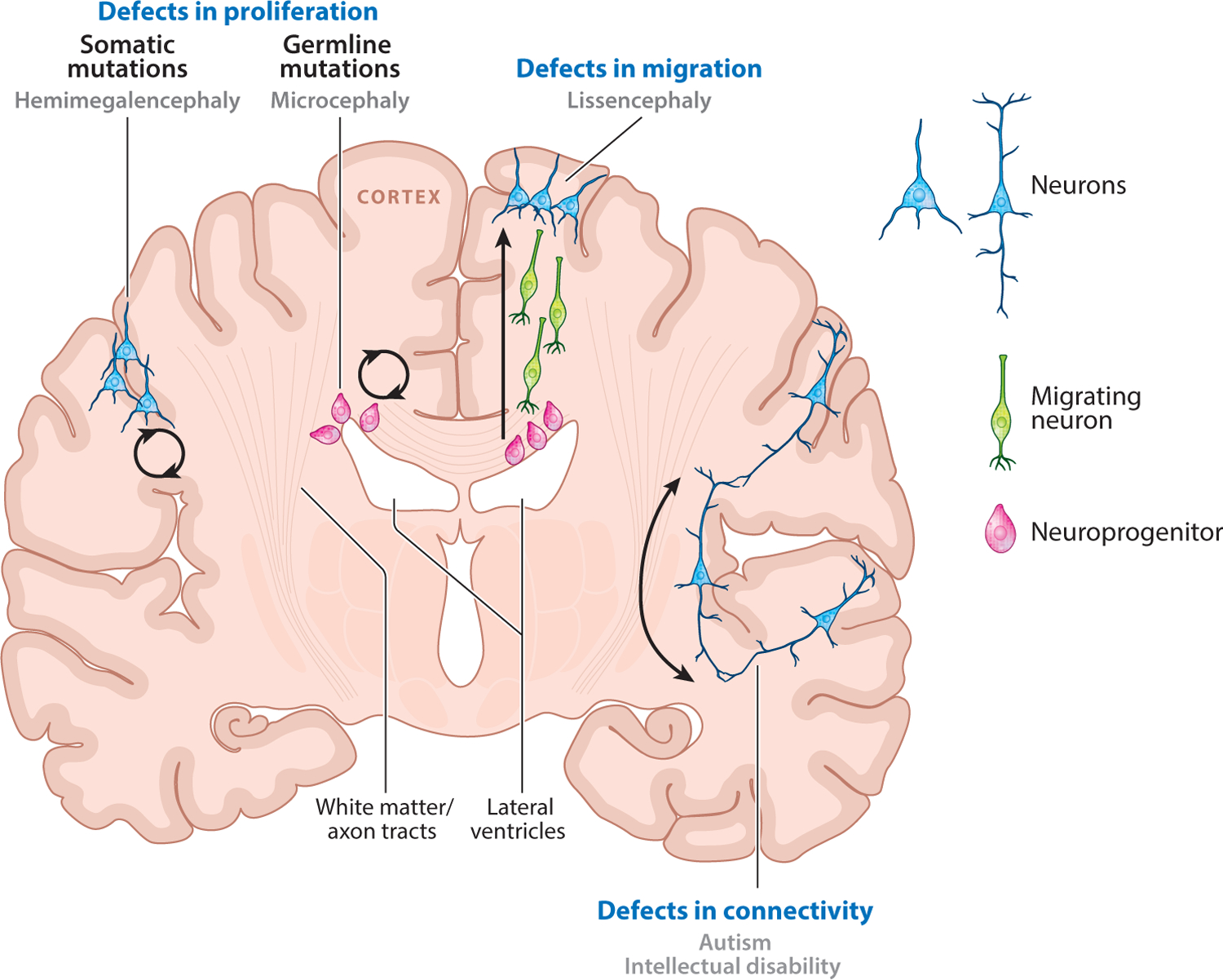
Schematic showing how neurodevelopmental disorder mutations affect genes that function at different stages of neurodevelopment. Defects in the proliferation of progenitors in the ventricular zone or the cortex can lead to microcephaly or hemimegalencephaly, respectively. Defects affecting the migration of neurons from the ventricular zone to the cortex give rise to lissencephaly. Connectivity defects are thought to underlie autism, intellectual disability, and neuropsychiatric disorders.
A common theme arising from genetic studies of microcephaly is the role of the centrosome in neuronal proliferation. The centrosome is a key microtubule-organizing center that helps maintain the cellular cytoskeleton (18, 92). During meiosis and mitosis, centrosomes nucleate the microtubule spindles to coordinate the segregation of duplicated chromosomes. Nearly all of the identified MCPH genes encode centrosomal proteins, or proteins required for proper chromosomal segregation. Given the colocalization of microcephaly proteins to the interphase centrosome and mitotic spindle poles, and the similarity of their mutant phenotypes, it is tempting to posit that they may function together in a complex or as part of a common pathway to regulate neuronal progenitor proliferation.
Genetic advances resulting from new sequencing techniques have also expanded our understanding of neuronal migration disorders in recent years. Lissencephaly is a striking brain malformation defined as a loss in gyral patterning on the surface of the brain, with concomitant thickening of the cortex. This thickening is caused by defective neuronal migration resulting in disorganization within the cortex, as neurons no longer form the normal laminated structure (139). The first disease gene, LIS1 (also known as PAFAH1B1), was identified 20 years ago (76, 105), and the second, DCX, encoding the doublecortin protein, was discovered 5 years later (29, 43). The majority of lissencephaly cases are attributable to mutations in these two genes (99).
The identification of LIS1 and DCX mutations utilized the traditional genetic tools of karyotyping, linkage analysis, and positional cloning, based on clues from large cohorts of patients and families. As is often the case with Mendelian diseases of severe phenotypes, mutations in LIS1 and DCX represent loss-of-function alleles. Subsequent identification of additional LIS1 alleles revealed genic deletions, missense mutations, and nonsense mutations (76). To date, all null germline mutations in LIS1 cause classic lissencephaly, following the Mendelian “one gene, one phenotype” phenomenon. Germline mutations in DCX, which is X-linked, also result in stereotyped phenotypes—lissencephaly in males and double cortex in females. Loss-of-function nonsense mutations, frameshifts, genic deletions, and splicing alleles are distributed across the entire length of the protein (10, 135), whereas missense mutations cluster predominantly around two functional microtubule-binding domains and disrupt tubulin binding, also rendering the protein functionally null (112, 126). Thus, loss-of-function missense alleles can identify specific residues critical for protein structure and function.
Since the discovery of LIS1 and DCX, an amazing variety of additional genetic causes of lissencephaly have been discovered at increasing rates via high-throughput sequencing, including de novo mutations in TUBA1A, encoding a neuronal alpha-tubulin (65). Most recently, de novo mutations in DYNC1H1 (encoding a dynein heavy-chain isoform), KIF2A (encoding a kinesin heavy chain), and TUBG1 (encoding gamma-tubulin) have also been found in association with microcephaly and pachygyria (“thick gyri”), a milder disease on the lissencephaly spectrum (102).
These discoveries add to a growing body of literature highlighting the role of cytoskeletal proteins in lissencephaly and deepening our understanding of the underlying cell biology (see sidebar The Role of the Cytoskeleton in Neurodevelopment). The complex of cytoplasmic dynein with Lis1 (36) and homologs Nde1 and Ndel1, both of which are centrosomal proteins (37, 91, 114), has long been known to be essential for neuronal migration (120, 125, 130, 141), and Nde1-null mice display reduced brain size and defects in cortical neuron migration (38). Recent genetic studies, nearly a decade after the initial characterization of the mouse phenotype, have identified mutations in NDE1 in patients with microlissencephaly, confirming a link between the underlying cell biological pathways of microcephaly and lissencephaly (2, 12). Doublecortin, a microtubule-associated protein that stabilizes microtubules against depolymerization (39, 44), is also required for proper neuronal migration (11), and recent biochemical and cell biological studies have focused on doublecortin as an adaptor that directly binds to tubulin molecules, facilitating the interaction of kinesins (and potentially also dyneins) at the microtubule interface (75).
THE ROLE OF THE CYTOSKELETON IN NEURODEVELOPMENT.
Cytoskeletal rearrangements are crucial for every aspect of neurodevelopment, from the regulation of cell division and migration, to the growth of extensive dendritic arbors and axonal branches, to the transport of cargo along those fibers. Several lines of evidence support a role for the centriolar organization of microtubules in regulating neuronal progenitor mitosis. Centrioles are microtubule-based structures that comprise the cores of centrosomes. During the G0/G1 phase of ciliated cells, centrioles form the basal bodies required for the proper generation of cilia and flagella. Signaling pathways transduced via primary cilia, such as the sonic hedgehog and Wnt pathways, are known to serve important functions in neuronal fate determination and regulation of cortical size. Prior to entry into the cell cycle, cilia and flagella are resorbed, and the centrioles are released from the basal body to nucleate the mitotic spindle and coordinate the segregation of chromosomes during mitosis (92).
Additionally, the genetic evidence that mutations in genes encoding microtubule-associated proteins (including LIS1 and DCX as well as genes encoding dyneins, kinesins, and tubulin isoforms) cause disease points to a role for the microtubule structure itself. In particular, Dcx regulates microtubule stability, and perturbations cause defective migration, as rapid cytoskeletal rearrangements are necessary during migration. Additionally, motor proteins are required both for the trafficking of specific cargo and for the generation of force during structural rearrangements. For example, coupling of the nucleus to the centrosome during cell migration is necessary and depends on the Lis1-dynein-Nde1/Ndel1 complex.
Finally, recent studies have identified mutations in different genes encoding tubulin subunits and kinesin proteins associated with cortical brain malformations and neurological disorders beyond microcephaly and lissencephaly, ranging from polymicrogyria to nodular heterotopia to eye movement abnormalities. These include mutations in TUBB2B (encoding a beta-tubulin isoform) (62), TUBB3 and TUBB5 (encoding neuronal beta-tubulin isoforms) (22, 128), and KIF5C (encoding a neuron-specific kinesin) (102). Notably, unlike the almost exclusively loss-of-function mutations in LIS1 and DCX, the identified tubulin, dynein, and kinesin alleles are nearly all heterozygous de novo missense changes. To date, null homozygous or heterozygous alleles—which would suggest loss of function or haploinsufficiency as the cause of disease—have not been identified, raising the possibility that these mutations harbor a specific dominant-negative effect (22, 102) and that loss of function or haploinsufficiency may be incompatible with life. Given the rare, sporadic nature of these mutations, high-throughput sequencing was instrumental in their identification.
ALLELIC DIVERSITY BROADENS THE SPECTRUM OF NEURODEVELOPMENTAL PHENOTYPES
Since the initial identification of the classic loss-of-function LIS1 and DCX mutations, missense mutations as well as somatic and germline mosaic mutations have been found to cause variable brain phenotypes. These include somatic LIS1 mutations and somatic DCX mutations in males, causing double cortex (45, 121), and missense DCX mutations in females, causing intellectual disability (ID) and cryptogenic epilepsy (50). Although these examples were by far the minority, they hinted that subtle perturbations in protein function or in a limited subset of cells may manifest differently from the complete loss-of-function phenotype, and they foreshadowed the broadening of genotype–phenotype correlations revealed in recent years.
An early example of phenotypic diversity occurs in the gene ARX, encoding Aristaless-related homeobox protein. Null alleles of ARX classically cause a syndrome of X-linked lissencephaly with agenesis of the corpus callosum and ambiguous genitalia with early lethality (OMIM 300215) (64, 66, 132), whereas missense changes and polyalanine tract expansions cause X-linked ID with various forms of epilepsy (115, 124) and seizures, infantile spasms, and mild to moderate ID with or without dystonia, ataxia, or autism (1, 14, 33, 123, 131). There is a general correlation between genotype and disease: Truncating and null mutations cause the most severe malformations, whereas missense and polyalanine tract repeat alleles cause ID and seizures, often without any identifiable structural involvement, although the severity of ID can vary widely (40, 64, 80).
Another outstanding example of the power of human genetics to define biochemically related proteins while showing a range of phenotypic diversity is the dystroglycanopathies, a group of disorders causing muscular dystrophy with variable central nervous system (CNS) involvement. The primary defect is in O-glycosylation of alpha-dystroglycan, which interferes with its ability to bind extracellular matrix ligands (84). Mutations of multiple genes in the glycosylation pathway have been identified as causative mutations for a wide spectrum of dystroglycanopathies (47), and recently, two missense mutations in the gene encoding alpha-dystroglycan were shown to cause disease (41, 52), punctuating the genetic links to a biochemical pathway.
The severity of dystroglycanopathies varies widely, from congenital onset with profound brain and eye malformations and early infantile death to adult-onset muscular dystrophy with no brain or eye involvement (47). Furthermore, mutations in the same gene can cause widely different diseases (25, 81). For example, FKRP, encoding Fukutin-related protein, was first identified as causing severe congenital skeletal muscle defects without brain or eye abnormalities (23). However, mutations in FKRP were soon thereafter identified as causing milder muscular dystrophies with an age of presentation as late as adulthood (24); Walker–Warburg syndrome (OMIM 613153) and muscle-eye-brain disease (OMIM 613153), both of which have severe CNS manifestations; and other dystroglycanopathies with CNS abnormalities (13, 77, 82, 129). In fact, this has prompted a reclassification of dystroglycanopathies based on the genetic underpinnings rather than the constellation of phenotypes (46, 47).
The broad phenotypic spectrum caused by mutations in the same gene is a constant theme in recent genetic studies of neurodevelopmental disorders. WDR62 patients exhibit variable gyral simplification, cortical lamination, and abnormalities of the corpus callosum (15, 90, 143). Mutations in DYNC1H1 can also cause Charcot–Marie–Tooth neuropathy (OMIM 614228) (136) and peripheral neuropathy in a form of spinal muscular atrophy (OMIM 158600) (53), and DYNC1H1 patients with cortical malformations have variable neuropathy (102). Finally, TUBB3 mutations, which cause eye movement disorders affecting the oculomotor nerves, show variable white-matter tract abnormalities (128).
In fact, the same mutation in the same gene can, less commonly, cause different diseases. Previously, it was generally believed that the range of phenotypes was due to allelic differences. However, the same alanine repeat expansion in ARX causing X-linked ID without structural brain abnormalities was recently reported to cause agenesis of the corpus callosum with periventricular heterotopia, frontal polymicrogyria, and interhemispheric cyst (97). Identical alleles of POMGNT1 (140), another member of the glycosyl transfer pathway, cause different dystroglycanopathy severities (30), even within the same family (127), and a familial DYNC1H1 allele affects children more severely than a mother who harbors the same mutation (102).
The growing diversity of disease-associated phenotypes identified in recent years highlights the complexity of neurodevelopmental disorders (Figure 3). This may be partially attributable to the fact that new sequencing methods capture more rare, hypomorphic, and less-penetrant alleles, whereas prior linkage studies were necessarily biased toward functionally null and highly penetrant mutations. Taken together, these data clearly indicate that other genetic and nongenetic factors that modify disease phenotypes are much more prevalent than previously thought.
Figure 3.
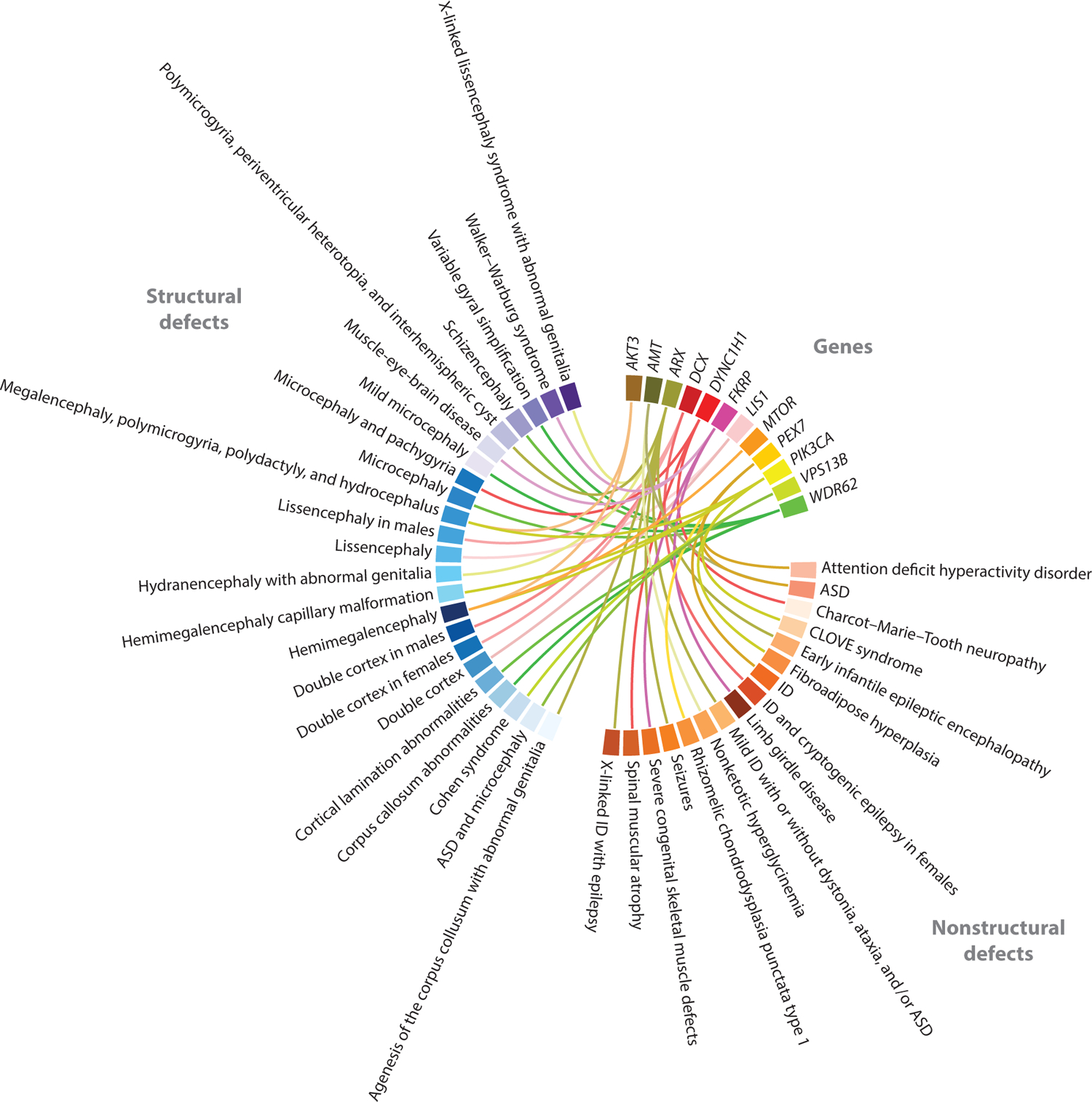
Circle plot illustrating the allelic and phenotypic diversity of a subset of neurodevelopmental disease genes. Mutations in the same gene cause diverse neurodevelopmental phenotypes. Advances in high-throughput sequencing techniques have expedited the identification of hypomorphic and somatic mutations, increasing the spectrum of phenotypes associated with any single gene. Phenotypes are grouped into structural brain defects and abnormalities without any structural brain defects. Abbreviations: ASD, autism spectrum disorder; CLOVE, congenital lipomatous overgrowth, vascular malformations, and epidermal nevi; ID, intellectual disability.
A RANGE OF ALLELES IN MENDELIAN DISEASE GENES CAUSE COGNITIVE AND NEUROPSYCHIATRIC DISORDERS
The plummeting cost of sequencing (137) and concomitant introduction of high-throughput sequencing technologies paved the way for studies of neurodevelopmental diseases that do not have structural abnormalities and are of unclear inheritance. Several large-scale studies using whole-exome sequencing have identified a high rate of de novo mutations in nonsyndromic cognitive disorders, including ID. Unlike inherited mutations, de novo mutations are present in a child but not in that child’s parents, and likely arose in the germline of the parent. De novo mutations in ID consist of large copy-number variants that delete one allele of many genes (27, 55, 58) and more recently were found to also include isolated point mutations (28, 104).
Another example where modern sequencing techniques have elucidated the genetic underpinnings of nonsyndromic cognitive disorders is autism spectrum disorders (ASDs). ASDs are a complex and genetically heterogeneous group of diseases characterized by impaired social interaction, communication deficits, limited interests, and stereotyped behaviors (59). Multiple genetic and nongenetic factors contribute toward the phenotype, increasing the challenge of studying ASDs. Furthermore, ASDs commonly occur in sporadic individuals without significant family history and would have been impossible to study using traditional methods of linkage analysis and positional cloning. Only with the advent of sequencing techniques in which we can survey broad regions of the genomes of many individuals have we been able to probe the genetic landscape of ASDs. Several recent studies have shown the importance of de novo copy-number variants (72, 100, 110, 117) and de novo point mutations in ASDs (7, 83, 87, 94–96, 111).
A second theme that has emerged from whole-exome sequencing studies is the contribution of recessive hypomorphic mutations to ASDs. These mutations partially inactivate genes that are known to cause more severe phenotypes when completely inactivated. Unlike de novo point mutations, which inactivate only one of two copies of a typical autosomal gene, leaving one copy of the gene still functional, both alleles are mutated in recessive, hypomorphic mutations causing ASDs. For example, ASD mutations have been identified in AMT, encoding aminomethyltransferase, which is widely expressed in human tissues (69). Mutations in AMT cause nonketotic hyperglycinemia (NKH; OMIM 605899) (5), a recessive syndrome resulting from impaired glycine cleavage. Depending on the severity of AMT mutations, NKH symptoms can vary from progressive lethargy, hypotonia, severe seizures, and death within the first year of life to delayed age of onset, developmental delay, and behavioral problems (51). More recently, hypomorphic mutations in AMT have been identified in patients with a range of neurodevelopmental symptoms, including ASDs, language and motor delays, ID, and severe epilepsy (142). The biochemical abnormalities that are typically detected in classic NKH often escape clinical detection methods in milder atypical NKH (4, 31). Functional assays to characterize the ASD AMT mutations demonstrated that the enzymatic activity of the mutant AMT was partially reduced, confirming that the affected children had a mild form of undiagnosed NKH presenting as an ASD with seizures (142).
Another example of a metabolic disease gene where hypomorphic mutations result in ASDs is PEX7, encoding a peroxisomal receptor, peroxisome biogenesis factor 7, that is required for protein import into the peroxisome. PEX7 is ubiquitously expressed, and mutations in the gene classically result in rhizomelic chondrodysplasia punctata (RCDP) type 1 (OMIM 215100), a metabolic syndrome of severe ID, growth retardation, skeletal dysplasia, and cataracts, with most cases not surviving beyond two years of age (19, 20, 86). Patients with partial loss-of-function mutations in PEX7 often lack the skeletal abnormalities typically associated with RCDP and can have ASD, severe attention deficit hyperactivity disorder, or ID (19). Mutations in another neurometabolic disease gene, PAH, encoding phenylalanine hydroxylase, the cause of phenylketonuria (OMIM 261600), were one of the earliest-described causes of ASD (144).
Cohen syndrome (OMIM 216550) is a disorder of ID, microcephaly, motor abnormalities, hypotonia and joint laxity, facial dysmorphisms, retinal dystrophy, and intermittent neutropenia (56). Cohen syndrome is caused by recessive mutations in VPS13B, encoding vacuolar protein sorting 13 homolog B (yeast), which is widely expressed in human tissues (67). There is significant variability in the spectrum of Cohen syndrome clinical features (85, 118), but many patients with biallelic null mutations in VPS13B can present with typical ASD features. In addition, milder forms of Cohen syndrome, often resulting from missense mutations suggestive of partial loss of function, have been identified that are associated with ASD, microcephaly, mild dysmorphic features, and joint laxity (32).
With advances in sequencing technology and the introduction of clinical sequencing, it is anticipated that additional mild hypomorphic mutations in syndromic disease genes will be identified in ASDs and other neuropsychiatric disorders. Notably, syndromic disease genes with hypomorphic ASD mutations are ubiquitously expressed (20, 67, 69) and cause multiorgan system disease with complete loss of function. This is in contrast to more traditional Mendelian disorders of neurodevelopment, such as lissencephaly caused by mutations in DCX and ARX, where gene expression is limited mostly to the nervous system (29, 43, 66, 98, 124). The restricted expression patterns of DCX and ARX explain the tissue-specific phenotypes caused by loss-of-function alleles. For hypomorphic alleles of ubiquitously expressed genes causing ASDs, an explanation for the prevalence of neurological phenotypes remains elusive, although one explanation could be the selective sensitivity of the brain to mild perturbations in the levels of these proteins compared with that of other organs.
The significant variability in the phenotypes that can be associated with developmental syndromes makes genetic diagnosis of the milder atypical forms presenting as ASDs and other nonmal-formation neurodevelopmental diseases challenging. Clinical sequencing should help overcome this challenge in at least a subset of cases, increasingly driving genetic rather than phenotypic classification. However, functional validation of candidate hypomorphic mutations will remain critical.
SOMATIC MUTATIONS CAUSE LOCALIZED BRAIN MALFORMATIONS
Some of the most remarkable findings about complex patterns of mutation have come from the realization that some de novo mutations occur after fertilization and might not be present in all tissues, but nonetheless affect the brain. Early confirmation came with the identification of somatic mutations in LIS1 and DCX (45, 121). However, these mutations were found in relatives of known DCX patients. It would take another decade to develop techniques capable of identifying somatic mutations in an unbiased manner. Key breakthroughs allowing the study of somatic mutations have included deep sequencing (to capture alleles present at low frequencies in bulk tissue) and single-cell sorting and genomic amplification techniques (to isolate and allow sequencing of individual cells).
Recently, somatic mutations in the PI3K-AKT-mTOR pathway were identified as causing hemimegalencephaly (71, 101, 106). Hemimegalencephaly is an overgrowth disorder of one hemisphere of the brain, often causing severe epilepsy that requires resection of the entire hemisphere (3, 6, 54, 113, 116). The sporadic nature of the disease and localized brain involvement is consistent with a noninherited pattern and highly suggestive of somatic mutation (9).
Studies of the resected diseased tissue identified mutations in the PI3K-AKT-mTOR pathway present in a subset of cells from the brain that were either absent from circulating lymphoblasts (101) or present at lower levels (106). Based on the sequencing of single cells, the proportion of affected alleles was estimated at 39% of neurons and 27% of nonneuronal cells (35). Surprisingly, a mutation in only a small proportion of affected cells is capable of causing disease of the entire tissue. Germline activating mutations in the PI3K-AKT-mTOR pathway have not been identified and are likely incompatible with life. In support of this hypothesis, the mutations identified to date in genes of this pathway that are associated with numerous other overgrowth syndromes, including Proteus syndrome (asymmetric overgrowth of the hands and feet, nevi, adipose tissue, and vascular malformations; OMIM 176920) and CLOVE syndrome (congenital lipomatous overgrowth, vascular malformations, and epidermal nevi; OMIM 612918), are also mosaic in nature (70, 73, 74).
The identification of somatic mutations in hemimegalencephaly raises the question of whether somatic mutations also cause focal neurological diseases, including focal cortical dysplasias. Tuberous sclerosis complex (TSC) is a neurocutaneous syndrome causing growth of nonmalignant hamartomas in multiple organ systems, including cortical tubers in the brain that are similar to focal cortical dysplasias. TSC is caused by mutations in TSC1 and TSC2, which are components of the mTOR pathway (34, 133), and somatic second-hit mutations overlying a germline mutation are common in non-nervous-system hamartomas (57, 93) and can cause cortical tubers, albeit at low frequencies (103).
Whether somatic mutations also cause diseases of connectivity without structural abnormalities, including neuropsychiatric diseases and ASDs, where de novo mutations are common, remains unanswered. For focal malformations, tissue resection is often necessary to control epilepsy (3, 6, 54, 113, 116) and provides a means for postoperative diagnostic confirmation. In hemimegalencephaly patients with other nonbrain malformations, low levels of mosaicism were detected in blood and saliva samples (106), suggesting that in cases where somatic mutations occur early during embryogenesis and affect multiple organ systems, noninvasive diagnosis is possible. Additionally, given the recent evidence from recessive autism mutations in which germline hypomorphic mutations preferentially affect cognitive function, it is possible that systemic mosaicism may also preferentially affect the brain, even in the absence of gross malformations. Studies of brain-specific mosaicism, however, will require access to brain tissue and may limit our ability to detect late, organ-specific somatic mutations in disease.
CONCLUSIONS
The examples reviewed here illuminate how the landscape of human genetics is expanding, from classic Mendelian inherited diseases and mutations of high penetrance, to more complex genes causing variable phenotypes, to de novo somatic mutations detectable only in subsets of cells. The genotype–phenotype boundaries are clearly breaking down. These discoveries would not have been possible without great advances in sequencing technology that led to an exponential drop in costs and expanded sequencing coverage of the genome, as well as the ability to capture high read depth to detect low-level mutations. Many of the more complex genes discussed here were originally identified as Mendelian disorder genes, with new technology aiding in the identification of additional alleles. With whole-exome sequencing becoming the new standard for genetic studies, we may find that the identification of hypomorphic alleles will soon precede the identification of classic null alleles.
The broad spectrum of genes that cause neurodevelopmental disorders when mutated illustrates the high sensitivity of the developing CNS compared with other organ systems. The fact that hypomorphic mutations in ubiquitously expressed Mendelian disease genes cause neurodevelopmental disorders without the typical multiorgan syndromes supports this hypothesis. Initial studies of lissencephaly syndromes revealed the structural complexity of the brain, and later reports of epilepsy and autism genetics revealed the complexity of connectivity in the brain. Given the absence of structural malformations in most cases of epilepsy, autism, and ID, it seems that connectivity is the most susceptible aspect of neurodevelopment, with diseases of connectivity manifesting before defective neuronal proliferation, differentiation, or migration can cause structural abnormalities.
In the examples presented here, mutations in many different genes often give rise to similar phenotypes. Such data suggest an overlap in cellular mechanisms and pathways. Indeed, lissencephaly genes not only are required for the same cellular process of neuronal migration, but also encode proteins that bind microtubules. Similarly, microcephaly proteins localize to the centrosome and are hypothesized to regulate mitosis, and autism genes overlap significantly with synaptic and metabolic genes. Although the precise pathways of these diseases have not yet been elucidated, recent work on hemimegalencephaly and the perturbation of the PI3K-AKT-mTOR pathway suggests that this will be true for other diseases as well.
The future of genetics is high-throughput sequencing, and clinical centers and geneticists are rapidly embracing whole-exome sequencing as a diagnostic tool. Thus, over the next few years, the rate of gene discovery will continue to accelerate, with an exponential explosion of novel genetic findings.
ACKNOWLEDGMENTS
W.F.H. is supported in part by National Institutes of Health (NIH) Medical Science Training Program grant T32GM007753 and by National Institute of Neurological Disorders and Stroke grant F31NS083111. M.H.C. is supported in part by a Nancy Lurie Marks Postdoctoral Fellowship award. C.A.W. is supported by grants from the National Institute of Mental Health (R01MH083565 and 1RC2MH089952), the National Institutes of Neurological Disorders and Stroke (R01NS35129, R01NS032457, and R01NS079277), the Simons Foundation, the Paul G. Allen Family Foundation, and the Manton Center for Orphan Disease Research. C.A.W. is an investigator of the Howard Hughes Medical Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Absoud M, Parr JR, Halliday D, Pretorius P, Zaiwalla Z, Jayawant S. 2010. A novel ARX phenotype: rapid neurodegeneration with Ohtahara syndrome and a dyskinetic movement disorder. Dev. Med. Child Neurol 52:305–7 [DOI] [PubMed] [Google Scholar]
- 2.Alkuraya FS, Cai X, Emery C, Mochida GH, Al-Dosari MS, et al. 2011. Human mutations in NDE1 cause extreme microcephaly with lissencephaly. Am. J. Hum. Genet 88:536–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez RM, García-Díaz L, Márquez J, Fajardo M, Rivas E, et al. 2011. Hemimegalencephaly: prenatal diagnosis and outcome. Fetal Diagn. Ther 30:234–38 [DOI] [PubMed] [Google Scholar]
- 4.Applegarth DA, Toone JR. 2001. Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Mol. Genet. Metab 74:139–46 [DOI] [PubMed] [Google Scholar]
- 5.Applegarth DA, Toone JR. 2004. Glycine encephalopathy (nonketotic hyperglycinaemia): review and update. J. Inherit. Metab. Dis 27:417–22 [DOI] [PubMed] [Google Scholar]
- 6.Aronica E, Becker AJ, Spreafico R. 2012. Malformations of cortical development. Brain Pathol 22:380–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, et al. 2010. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am. J. Hum. Genet 87:316–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, et al. 2009. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol 513:532–41 [DOI] [PubMed] [Google Scholar]
- 9.Baek ST, Gibbs EM, Gleeson JG, Mathern GW. 2013. Hemimegalencephaly, a paradigm for somatic postzygotic neurodevelopmental disorders. Curr. Opin. Neurol 26:122–27 [DOI] [PubMed] [Google Scholar]
- 10.Bahi-Buisson N, Souville I, Fourniol FJ, Toussaint A, Moores CA, et al. 2013. New insights into genotype-phenotype correlations for the doublecortin-related lissencephaly spectrum. Brain 136:223–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. 2003. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci 6:1277–83 [DOI] [PubMed] [Google Scholar]
- 12.Bakircioğlu M, Carvalho OP, Khurshid M, Cox JJ, Tüysüz B, et al. 2011. The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am. J. Hum. Genet 88:523–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beltrán-Valero de Bernabé D, Voit T, Longman C, Steinbrecher A, Straub V, et al. 2004. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet 41:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, et al. 2002. ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum. Mol. Genet 11:981–91 [DOI] [PubMed] [Google Scholar]
- 15.Bilgüvar K, Öztürk AK, Louvi A, Kwan KY, Choi M, et al. 2010. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467:207–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, et al. 2002. ASPM is a major determinant of cerebral cortical size. Nat. Genet 32:316–20 [DOI] [PubMed] [Google Scholar]
- 17.Bond J, Roberts E, Springell K, Lizarraga SB, Lizarraga S, et al. 2005. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet 37:353–55 [DOI] [PubMed] [Google Scholar]
- 18.Bornens M. 2012. The centrosome in cells and organisms. Science 335:422–26 [DOI] [PubMed] [Google Scholar]
- 19.Braverman N, Chen L, Lin P, Obie C, Steel G, et al. 2002. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum. Mutat 20:284–97 [DOI] [PubMed] [Google Scholar]
- 20.Braverman N, Steel G, Obie C, Moser A, Moser H, et al. 1997. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat. Genet 15:369. [DOI] [PubMed] [Google Scholar]
- 21.Brenner S. 2003. Nature’s gift to science (Nobel lecture). ChemBioChem 4:683–87 [DOI] [PubMed] [Google Scholar]
- 22.Breuss M, Heng JI-T, Poirier K, Tian G, Jaglin XH, et al. 2012. Mutations in the β-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep 2:1554–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, et al. 2001. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am. J. Hum. Genet 69:1198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, et al. 2001. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet 10:2851–59 [DOI] [PubMed] [Google Scholar]
- 25.Clement E, Mercuri E, Godfrey C, Smith J, Robb S, et al. 2008. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann. Neurol 64:573–82 [DOI] [PubMed] [Google Scholar]
- 26.Collins FS. 1995. Positional cloning moves from perditional to traditional. Nat. Genet 9:347–50 [DOI] [PubMed] [Google Scholar]
- 27.Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, et al. 2011. A copy number variation morbidity map of developmental delay. Nat. Genet 43:838–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, et al. 2012. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med 367:1921–29 [DOI] [PubMed] [Google Scholar]
- 29.des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, et al. 1998. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92:51–61 [DOI] [PubMed] [Google Scholar]
- 30.Diesen C, Saarinen A, Pihko H, Rosenlew C, Cormand B, et al. 2004. POMGnT1 mutation and phenotypic spectrum in muscle-eye-brain disease. J. Med. Genet 41:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dinopoulos A, Matsubara Y, Kure S. 2005. Atypical variants of nonketotic hyperglycinemia. Mol. Genet. Metab 86:61–69 [DOI] [PubMed] [Google Scholar]
- 32.Douzgou S, Petersen MB. 2011. Clinical variability of genetic isolates of Cohen syndrome. Clin. Genet 79:501–6 [DOI] [PubMed] [Google Scholar]
- 33.Ekşioğlu YZ, Pong AW, Takeoka M. 2011. A novel mutation in the aristaless domain of the ARX gene leads to Ohtahara syndrome, global developmental delay, and ambiguous genitalia in males and neuropsychiatric disorders in females. Epilepsia 52:984–92 [DOI] [PubMed] [Google Scholar]
- 34.Eur. Chromosome 16 Tuberous Scler. Consort. 1993. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75:1305–15 [DOI] [PubMed] [Google Scholar]
- 35.Evrony GD, Cai X, Lee E, Hills LB, Elhosary PC, et al. 2012. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151:483–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faulkner NE, Dujardin DL, Tai CY, Vaughan KT, O’Connell CB, et al. 2000. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat. Cell Biol 2:784–91 [DOI] [PubMed] [Google Scholar]
- 37.Feng Y, Olson EC, Stukenberg PT, Flanagan LA, Kirschner MW, Walsh CA. 2000. LIS1 regulates CNS lamination by interacting with mNudE, a central component of the centrosome. Neuron 28:665–79 [DOI] [PubMed] [Google Scholar]
- 38.Feng Y, Walsh CA. 2004. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 44:279–93 [DOI] [PubMed] [Google Scholar]
- 39.Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, et al. 1999. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23:247–56 [DOI] [PubMed] [Google Scholar]
- 40.Friocourt G, Poirier K, Rakić S, Parnavelas JG, Chelly J. 2006. The role of ARX in cortical development. Eur. J. Neurosci 23:869–76 [DOI] [PubMed] [Google Scholar]
- 41.Geis T, Marquard K, Rödl T, Reihle C, Schirmer S, et al. 2013. Homozygous dystroglycan mutation associated with a novel muscle-eye-brain disease-like phenotype with multicystic leucodystrophy. Neurogenetics 14:205–13 [DOI] [PubMed] [Google Scholar]
- 42.Genin A, Désir J, Lambert N, Biervliet M, Van Der Aa N, et al. 2012. Kinetochore KMN network gene CASC5 mutated in primary microcephaly. Hum. Mol. Genet 21:5306–17 [DOI] [PubMed] [Google Scholar]
- 43.Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, et al. 1998. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92:63–72 [DOI] [PubMed] [Google Scholar]
- 44.Gleeson JG, Lin PT, Flanagan LA, Walsh CA. 1999. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23:257–71 [DOI] [PubMed] [Google Scholar]
- 45.Gleeson JG, Minnerath S, Kuzniecky RI, Dobyns WB, Young ID, et al. 2000. Somatic and germline mosaic mutations in the doublecortin gene are associated with variable phenotypes. Am. J. Hum. Genet 67:574–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Godfrey C, Clement E, Mein R, Brockington M, Smith J, et al. 2007. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 130:2725–35 [DOI] [PubMed] [Google Scholar]
- 47.Godfrey C, Foley AR, Clement E, Muntoni F. 2011. Dystroglycanopathies: coming into focus. Curr. Opin. Genet. Dev 21:278–85 [DOI] [PubMed] [Google Scholar]
- 48.Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. 2013. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci 14:755–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guernsey DL, Jiang H, Hussin J, Arnold M, Bouyakdan K, et al. 2010. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am. J. Hum. Genet 87:40–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guerrini R, Moro F, Andermann E, Hughes E, D’Agostino D, et al. 2003. Nonsyndromic mental retardation and cryptogenic epilepsy in women with Doublecortin gene mutations. Ann. Neurol 54:30–37 [DOI] [PubMed] [Google Scholar]
- 51.Hamosh AJM. 2001. Non-ketotic hyperglycinemia. In The Metabolic and Molecular Bases of Inherited Disease, ed. Scriver CR, Sly WS, Childs B, Beaudet AL, Valle D, et al. , pp. 2065–78. New York: McGraw-Hill. 8th ed. [Google Scholar]
- 52.Hara Y, Balci-Hayta B, Yoshida-Moriguchi T, Kanagawa M, Beltrán-Valero de Bernabé D, et al. 2011. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N. Engl. J. Med 364:939–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harms MB, Ori-McKenney KM, Scoto M, Tuck EP, Bell S, et al. 2012. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 78:1714–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hauptman JS, Mathern GW. 2012. Surgical treatment of epilepsy associated with cortical dysplasia: 2012 update. Epilepsia 53(Suppl. 4):98–104 [DOI] [PubMed] [Google Scholar]
- 55.Hehir-Kwa JY, Rodríguez-Santiago B, Vissers LE, de Leeuw N, Pfundt R, et al. 2011. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J. Med. Genet 48:776–78 [DOI] [PubMed] [Google Scholar]
- 56.Hennies HC, Rauch A, Seifert W, Schumi C, Moser E, et al. 2004. Allelic heterogeneity in the COH1 gene explains clinical variability in Cohen syndrome. Am. J. Hum. Genet 75:138–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henske EP, Scheithauer BW, Short MP, Wollmann R, Nahmias J, et al. 1996. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am. J. Hum. Genet 59:400–6 [PMC free article] [PubMed] [Google Scholar]
- 58.Hochstenbach R, van Binsbergen E, Engelen J, Nieuwint A, Polstra A, et al. 2009. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur. J. Med. Genet 52:161–69 [DOI] [PubMed] [Google Scholar]
- 59.Huguet G, Ey E, Bourgeron T. 2013. The genetic landscapes of autism spectrum disorders. Annu. Rev. Genomics Hum. Genet 14:191–213 [DOI] [PubMed] [Google Scholar]
- 60.Hussain MS, Baig SM, Neumann S, Nürnberg G, Farooq M, et al. 2012. A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am. J. Hum. Genet 90:871–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, et al. 2002. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet 71:136–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jaglin XH, Poirier K, Saillour Y, Buhler E, Tian G, et al. 2009. Mutations in the β-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet 41:746–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jamieson CR, Govaerts C, Abramowicz MJ. 1999. Primary autosomal recessive microcephaly: homozygosity mapping of MCPH4 to chromosome 15. Am. J. Hum. Genet 65:1465–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kato M, Das S, Petras K, Kitamura K, Morohashi K-I, et al. 2004. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat 23:147–59 [DOI] [PubMed] [Google Scholar]
- 65.Keays DA, Tian G, Poirier K, Huang G-J, Siebold C, et al. 2007. Mutations in α-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128:45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, et al. 2002. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet 32:359–69 [DOI] [PubMed] [Google Scholar]
- 67.Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, et al. 2003. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am. J. Hum. Genet 72:1359–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumar A, Girimaji SC, Duvvari MR, Blanton SH. 2009. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am. J. Hum. Genet 84:286–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kure S, Kojima K, Kudo T, Kanno K, Aoki Y, et al. 2001. Chromosomal localization, structure, single-nucleotide polymorphisms, and expression of the human H-protein gene of the glycine cleavage system (GCSH), a candidate gene for nonketotic hyperglycinemia. J. Hum. Genet 46:378–84 [DOI] [PubMed] [Google Scholar]
- 70.Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, et al. 2012. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am. J. Hum. Genet 90:1108–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, et al. 2012. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet 44:941–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levy D, Ronemus M, Yamrom B, Lee Y-H, Leotta A, et al. 2011. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70:886–97 [DOI] [PubMed] [Google Scholar]
- 73.Lindhurst MJ, Parker VER, Payne F, Sapp JC, Rudge S, et al. 2012. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat. Genet 44:928–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, et al. 2011. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N. Engl. J. Med 365:611–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu JS, Schubert CR, Fu X, Fourniol FJ, Jaiswal JK, et al. 2012. Molecular basis for specific regulation of neuronal kinesin-3 motors by doublecortin family proteins. Mol. Cell 47:707–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R, Ledbetter DH. 1997. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum. Mol. Genet 6:157–64 [DOI] [PubMed] [Google Scholar]
- 77.Louhichi N, Triki C, Quijano-Roy S, Richard P, Makri S, et al. 2004. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics 5:27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lui JH, Hansen DV, Kriegstein AR. 2011. Development and evolution of the human neocortex. Cell 146:18–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mardis ER. 2011. A decade’s perspective on DNA sequencing technology. Nature 470:198–203 [DOI] [PubMed] [Google Scholar]
- 80.Marsh ED, Golden JA. 2012. Developing models of Aristaless-related homeobox mutations. In Jasper’s Basic Mechanisms of the Epilepsies, ed. Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV. Bethesda, MD: Natl. Cent. Biotechnol. Inf. 4th ed. http://www.ncbi.nlm.nih.gov/books/NBK98176 [PubMed] [Google Scholar]
- 81.Mercuri E, Messina S, Bruno C, Mora M, Pegoraro E, et al. 2009. Congenital muscular dystrophies with defective glycosylation of dystroglycan: a population study. Neurology 72:1802–9 [DOI] [PubMed] [Google Scholar]
- 82.Mercuri E, Topaloglu H, Brockington M, Berardinelli A, Pichiecchio A, et al. 2006. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch. Neurol 63:251–57 [DOI] [PubMed] [Google Scholar]
- 83.Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, et al. 2012. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151:1431–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, et al. 2002. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 418:417–22 [DOI] [PubMed] [Google Scholar]
- 85.Mochida GH, Rajab A, Eyaid W, Lu A, Al-Nouri D, et al. 2004. Broader geographical spectrum of Cohen syndrome due to COH1 mutations. J. Med. Genet 41:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Motley AM, Hettema EH, Hogenhout EM, Brites P, ten Asbroek AL, et al. 1997. Rhizomelic chondrodysplasia punctata is a peroxisomal protein targeting disease caused by a non-functional PTS2 receptor. Nat. Genet 15:377–80 [DOI] [PubMed] [Google Scholar]
- 87.Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, et al. 2012. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485:242–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, et al. 2010. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet 42:790–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, et al. 2010. Exome sequencing identifies the cause of a Mendelian disorder. Nat. Genet 42:30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nicholas AK, Khurshid M, Désir J, Carvalho OP, Cox JJ, et al. 2010. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat. Genet 42:1010–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Niethammer M, Smith DS, Ayala R, Peng J, Ko J, et al. 2000. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28:697–711 [DOI] [PubMed] [Google Scholar]
- 92.Nigg EA, Raff JW. 2009. Centrioles, centrosomes, and cilia in health and disease. Cell 139:663–78 [DOI] [PubMed] [Google Scholar]
- 93.Niida Y, Stemmer-Rachamimov AO, Logrip M, Tapon D, Perez R, et al. 2001. Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am. J. Hum. Genet 69:493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, et al. 2011. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet 43:585–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, et al. 2012. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338:1619–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, et al. 2012. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485:246–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Oegema R, Maat-Kievit A, Lequin MH, Schot R, Nanninga-van den Neste VMH, et al. 2012. Asymmetric polymicrogyria and periventricular nodular heterotopia due to mutation in ARX. Am. J. Med. Genet. A 158A:1472–76 [DOI] [PubMed] [Google Scholar]
- 98.Ohira R, Zhang YH, Guo W, Dipple K, Shih SL, et al. 2002. Human ARX gene: genomic characterization and expression. Mol. Genet. Metab 77:179–88 [DOI] [PubMed] [Google Scholar]
- 99.Pilz DT, Matsumoto N, Minnerath S, Mills P, Gleeson JG, et al. 1998. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum. Mol. Genet 7:2029–37 [DOI] [PubMed] [Google Scholar]
- 100.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, et al. 2010. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466:368–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, et al. 2012. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74:41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, et al. 2013. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet 45:639–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Qin W, Chan JA, Vinters HV, Mathern GW, Franz DN, et al. 2010. Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol 20:1096–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, et al. 2012. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380:1674–82 [DOI] [PubMed] [Google Scholar]
- 105.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, et al. 1993. Isolation of a Miller–Dieker lissencephaly gene containing G protein β-subunit-like repeats. Nature 364:717–21 [DOI] [PubMed] [Google Scholar]
- 106.Rivière J-B, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, et al. 2012. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet 44:934–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roberts E, Hampshire DJ, Pattison L, Springell K, Jafri H, et al. 2002. Autosomal recessive primary microcephaly: an analysis of locus heterogeneity and phenotypic variation. J. Med. Genet 39:718–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Roberts E, Jackson AP, Carradice AC, Deeble VJ, Mannan J, et al. 1999. The second locus for autosomal recessive primary microcephaly (MCPH2) maps to chromosome 19q13.1–13.2. Eur. J. Hum. Genet 7:815–20 [DOI] [PubMed] [Google Scholar]
- 109.Ronan L, Voets N, Rua C, Alexander-Bloch A, Hough M, et al. 2014. Differential tangential expansion as a mechanism for cortical gyrification. Cereb. Cortex In press. doi: 10.1093/cercor/bht082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, et al. 2011. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70:863–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, et al. 2012. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485:237–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sapir T, Horesh D, Caspi M, Atlas R, Burgess HA, et al. 2000. Doublecortin mutations cluster in evolutionarily conserved functional domains. Hum. Mol. Genet 9:703–12 [DOI] [PubMed] [Google Scholar]
- 113.Sasaki M, Hashimoto T, Furushima W, Okada M, Kinoshita S, et al. 2005. Clinical aspects of hemimegalencephaly by means of a nationwide survey. J. Child Neurol 20:337–41 [DOI] [PubMed] [Google Scholar]
- 114.Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, et al. 2000. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron 28:681–96 [DOI] [PubMed] [Google Scholar]
- 115.Scheffer IE, Wallace RH, Phillips FL, Hewson P, Reardon K, et al. 2002. X-linked myoclonic epilepsy with spasticity and intellectual disability: mutation in the homeobox gene ARX. Neurology 59:348–56 [DOI] [PubMed] [Google Scholar]
- 116.Schramm J, Kuczaty S, Sassen R, Elger CE, von Lehe M. 2012. Pediatric functional hemispherectomy: outcome in 92 patients. Acta Neurochir 154:2017–28 [DOI] [PubMed] [Google Scholar]
- 117.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, et al. 2007. Strong association of de novo copy number mutations with autism. Science 316:445–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Seifert W, Holder-Espinasse M, Spranger S, Hoeltzenbein M, Rossier E, et al. 2006. Mutational spectrum of COH1 and clinical heterogeneity in Cohen syndrome. J. Med. Genet 43:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shen J, Eyaid W, Mochida GH, Al-Moayyad F, Bodell A, et al. 2005. ASPM mutations identified in patients with primary microcephaly and seizures. J. Med. Genet 42:725–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shu T, Ayala R, Nguyen M-D, Xie Z, Gleeson JG, Tsai L-H. 2004. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron 44:263–77 [DOI] [PubMed] [Google Scholar]
- 121.Sicca F, Kelemen A, Genton P, Das S, Mei D, et al. 2003. Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology 61:1042–46 [DOI] [PubMed] [Google Scholar]
- 122.Sir J-H, Barr AR, Nicholas AK, Carvalho OP, Khurshid M, et al. 2011. A primary microcephaly protein complex forms a ring around parental centrioles. Nat. Genet 43:1147–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Strømme P, Mangelsdorf ME, Scheffer IE, Gécz J. 2002. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev 24:266–68 [DOI] [PubMed] [Google Scholar]
- 124.Strømme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SME, et al. 2002. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet 30:441–45 [DOI] [PubMed] [Google Scholar]
- 125.Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG. 2004. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol 165:709–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG. 2000. Patient mutations in doublecortin define a repeated tubulin-binding domain. J. Biol. Chem 275:34442–50 [DOI] [PubMed] [Google Scholar]
- 127.Teber S, Sezer T, Kafali M, Manzini MC, Konuk Yüksel B, et al. 2008. Severe muscle-eye-brain disease is associated with a homozygous mutation in the POMGnT1 gene. Eur. J. Paediatr. Neurol 12:133–36 [DOI] [PubMed] [Google Scholar]
- 128.Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, et al. 2010. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140:74–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Topaloglu H, Brockington M, Yuva Y, Talim B, Haliloglu G, et al. 2003. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 60:988–92 [DOI] [PubMed] [Google Scholar]
- 130.Tsai J-W, Chen Y, Kriegstein AR, Vallee RB. 2005. LIS1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages. J. Cell Biol 170:935–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Turner G, Partington M, Kerr B, Mangelsdorf M, Gécz J. 2002. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am. J. Med. Genet 112:405–11 [DOI] [PubMed] [Google Scholar]
- 132.Uyanik G, Aigner L, Martin P, Gross C, Neumann D, et al. 2003. ARX mutations in X-linked lissencephaly with abnormal genitalia. Neurology 61:232–35 [DOI] [PubMed] [Google Scholar]
- 133.van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, et al. 1997. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277:805–8 [DOI] [PubMed] [Google Scholar]
- 134.Walsh CA. 1999. Genetic malformations of the human cerebral cortex. Neuron 23:19–29 [DOI] [PubMed] [Google Scholar]
- 135.Walsh CA, Engle EC. 2010. Allelic diversity in human developmental neurogenetics: insights into biology and disease. Neuron 68:245–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, et al. 2011. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet 89:308–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wetterstrand KA. 2013. DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP) http://www.genome.gov/sequencingcosts
- 138.Wonders CP, Anderson SA. 2006. The origin and specification of cortical interneurons. Nat. Rev. Neurosci 7:687–96 [DOI] [PubMed] [Google Scholar]
- 139.Wynshaw-Boris A, Pramparo T, Youn YH, Hirotsune S. 2010. Lissencephaly: mechanistic insights from animal models and potential therapeutic strategies. Semin. Cell Dev. Biol 21:823–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, et al. 2001. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell 1:717–24 [DOI] [PubMed] [Google Scholar]
- 141.Youn YH, Pramparo T, Hirotsune S, Wynshaw-Boris A. 2009. Distinct dose-dependent cortical neuronal migration and neurite extension defects in Lis1 and Ndel1 mutant mice. J. Neurosci 29:15520–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, et al. 2013. Using whole-exome sequencing to identify inherited causes of autism. Neuron 77:259–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yu TW, Mochida GH, Tischfield DJ, Sgaier SK, Flores-Sarnat L, et al. 2010. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet 42:1015–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zecavati N, Spence SJ. 2009. Neurometabolic disorders and dysfunction in autism spectrum disorders. Curr. Neurol. Neurosci. Rep 9:129–36 [DOI] [PubMed] [Google Scholar]
- 145.Zilles K, Palomero-Gallagher N, Amunts K. 2013. Development of cortical folding during evolution and ontogeny. Trends Neurosci 36:275–84 [DOI] [PubMed] [Google Scholar]