

Abstract
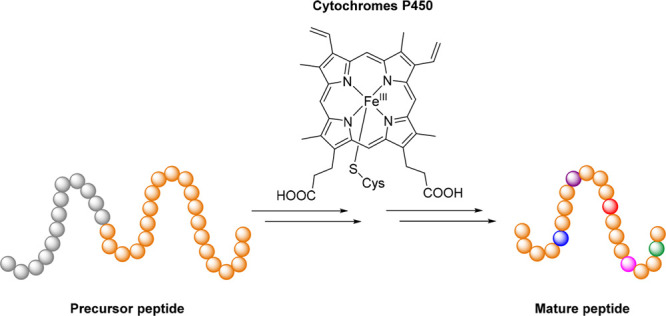
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a class of exponentially increased natural products with characteristic chemical structures, topologies, and biosynthetic mechanisms as well as exceptional bioactivities including antibacteria, antitumors, and antiviruses. The biosynthesis of RiPP proceeds via a ribosomally assembled precursor peptide that undergoes varied post-translational modifications to generate a mature peptide. Cytochrome P450 (CYP or P450) monooxygenases are a superfamily of heme-containing enzymes that span a wide range of secondary metabolite biosynthetic pathways due to their broad substrate scopes and excellent catalytic versatility. In contrast to the enormous quantities of RiPPs and P450s, the P450 associated RiPP biosynthesis is comparatively limited, with most of their functions and timings remaining mysterious. Herein, this Review aims to provide an overview on the striking roles of P450s in RiPP biosyntheses uncovered to date and to illustrate their remarkable functions, mechanisms, as well as remaining challenges. This will shed light on novel P450 discovery and characterizations in RiPP biosyntheses.
Keywords: Cytochrome P450, RiPP, post-translational modification, peptide, natural product, secondary metabolite, oxygenation, cross-link
1. Introduction
Natural products occupy a prominent place in the pharmaceutical industry since they have acted as the most crucial source of new drugs over the past four decades and are still the best choice for novel bioactive structure discovery.1 Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a universally acknowledged class with unique chemical structures/topologies and intriguing physiological activities such as antibacteria, antitumors, antiviruses, and so on.2,3 In addition, advances in bioinformatics, molecular biology, and chemical analytics have led to an explosive growth of structurally diverse RiPPs, which include more than 40 subgroups according to their structural features and post-translational modifications (PTMs).4
Distinct biosynthetic logic is employed for the generation of RiPPs. The biosynthesis originates from a ribosomally synthesized precursor peptide, which is bipartite and typically consists of an N-terminal leader peptide for the recruitment of different PTM enzymes and a C-terminal core peptide that constitutes the RiPP structural backbone,4,5 albeit unconventional arrangements as the widely spread multicore-containing precursor peptide and N-terminal core peptide for structure constitution with C-terminal follower peptide for recognition of PTM enzymes (e.g., bottromycins and triculamin/alboverticillin) are also found.6−8 Leader peptide dependent PTMs act at the forefront on the full length precursor peptide, followed by leader peptide cleavage by a dedicated protease/peptidase to release the modified core peptide, which further undergoes leader peptide independent PTMs and excretes to extra cells. In many cases, the leader peptide independent PTMs are nonessential and the modified core peptide is exported directly as a mature peptide (Figure 1). Copious PTMs have been unraveled to date, correlated with varied chemical structures and excellent bioactivities of RiPPs.4,5
Figure 1.
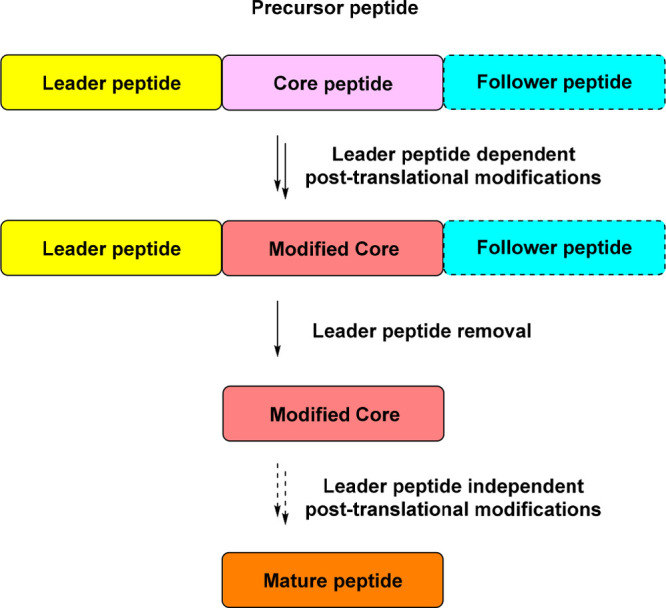
Typical biosynthetic logic of RiPPs. In many cases, the leader peptide independent PTMs are nonessential for RiPP maturation.
Cytochromes P450 (CYPs or P450s) represent a large superfamily of heme-thiolate monooxygenases that carry out various catalytic roles. P450 takes the name from the diagnostic red shift of the spectroscopic absorption maximum at 420 nm to 450 nm when exposed to carbon monoxide in ferrous form.9−12 The catalytic center iron is chelated with four nitrogen atoms of protoporphyrin IX, an axial thiolate from the strictly conserved cysteine residue of P450 and a resting water (or hydroxide ion) on the distal side, which is displaced by substrate binding after P450 catalytic cycle initiation.9−12 Redox proteins like eukaryotic NADPH-cytochrome P450 oxidoreductase (CPR) and bacterial/mitochondrial NAD(P)H-ferredoxin reductase (FdR)/ferredoxin (Fdx) partners are required for most P450s, termed multicomponent P450s to shuttle reducing equivalents.13,14 The heme receives the first electron from redox proteins to afford ferrous species, followed by dioxygen binding and the second electron acceptance, after which the distal oxygen of the nascent ferric peroxo species is protonated to produce the ferric hydroperoxy intermediate known as compound 0. Heterolytic cleavage of the O–O bond in compound 0 results in the highly reactive intermediate compound I. This species proceeds with hydrogen atom abstraction of the substrate to produce iron-(IV) hydroxyl intermediate termed compound II. The nascent substrate radical then rebinds the hydroxyl group connected with iron to generate the oxygenated product and reproduce the P450 enzyme for the next catalytic cycle (Figure 2).9−12
Figure 2.

Widely accepted catalytic cycle of P450s. The oxygen atoms, substrates, and redox partners are highlighted in red, blue, and purple, respectively. The shunt pathways are shown in light gray.
While P450s impress by their tremendously functional versatility that is involved in the biosynthesis of abundant secondary metabolites including polyketides (PKs), nonribosomal peptide (NRPs), cyclodipeptides (CDPs), and terpenes (Figure 3),10 the involvement of P450s in the biosynthesis of RiPPs is comparatively limited and few reviews have been summarized for this field to my knowledge (Table 1). While this Review was under preparation, Kunakom et al. published an excellent review about P450s involved in bacterial RiPP biosynthesis, with emphases on the distribution of P450s in bacterial genomes and the biosyntheses of atropitides, biarylitides, cittlins, as well as nocathiacin that harbors rich P450s.15 Herein, this Review provides a more comprehensive description of all the P450s with functionalities in the PTMs of RiPPs to date according to different classifications (Table 1), including P450s involved in some eukaryotic RiPP biosyntheses. It is hoped that this Review could draw much attention to the discovery and characterizations of novel P450s in RiPP biosyntheses, a field that has long been neglected.
Figure 3.

P450-mediated reactions (highlighted in blue) involved in some secondary metabolites including PKs, NRPs, CDPs, and terpenes.
Table 1. Summary of Cytochromes P450 Associated with the Biosyntheses of RiPPs.
| P450 | associated RiPP (RiPP subclass) | plausible catalytic function | leader/follower peptide-(in)dependent | ref |
|---|---|---|---|---|
| TpdJ1 | thiomuracins (1–7) (monocyclic thiopeptides) | hydroxylation or stochastic oxidations | unknown | (17, 21) |
| TpdJ2 | thiomuracins (1–7) (monocyclic thiopeptides) | hydroxylation or stochastic oxidations | unknown | (17, 21) |
| PbtO | GE2270s (8–16) (monocyclic thiopeptides) | hydroxylation | unknown | (20) |
| GetJ | GE37468 (19) (monocyclic thiopeptide) | consecutive oxidations | unknown | (21, 22) |
| BerH | berninamycins (22–29) (monocyclic thiopeptides) | hydroxylation | unknown | (27) |
| GenH | geninthiocins (30–33) (monocyclic thiopeptides) | hydroxylation | unknown | (30) |
| SulN | sulfomycins (34–36) (monocyclic thiopeptides) | unknown | unknown | (34) |
| Lit1 | litoralimycins (37, 38) (monocyclic thiopeptides) | unknown | unknown | (35) |
| SchY | Sch40832 (46) (bicyclic thiopeptide) | epoxidation and rearrangement | independent | (36) |
| TsrPa | thiostrepton (50) (bicyclic thiopeptide) | epoxidation | dependent | (39) |
| TsrRa | thiostrepton (50) (bicyclic thiopeptide) | dihydroxylation | dependent | (39) |
| NotF | nocardithiocin (55) (monocyclic thiopeptide) | hydroxylation(s) | unknown | (43) |
| NotH | nocardithiocin (55) (monocyclic thiopeptide) | hydroxylation(s) | unknown | (43) |
| NosBb | nosiheptide (61) (bicyclic thiopeptide) | hydroxylation | independent | (44) |
| NosCb | nosiheptide (61) (bicyclic thiopeptide) | hydroxylation | independent | (44) |
| NocVc | nocathiacin (62) (bicyclic thiopeptide) | hydroxylation and ether formation | unknown | (48) |
| NocUc | nocathiacin (62) (bicyclic thiopeptide) | hydroxylation | unknown | (49) |
| NocTc | nocathiacin (62) (bicyclic thiopeptide) | oxygenation | unknown | |
| PerX | persiathiacin (63) (bicyclic thiopeptide) | hydroxylation | independent | (50) |
| BytO | YYH (71) and YFH (72) (biarylitides) | C–C biaryl cross-link (A–B cross-link) | dependent | (51) |
| BytO | myxarylin (76) (biarylitide) | C–N biaryl cross-link (A–N–B cross-link) | dependent | (52) |
| P450Blt | 79 (biarylitide) | C–N biaryl cross-link (A–N–B cross-link) | dependent | (53) |
| P450Blt | 82 (biarylitide) | C–O biaryl cross-link (A–O–B cross-link) | dependent | (53) |
| CitB | cittilins (85, 86) (cittilins) | C–C biaryl cross-link and C–O biaryl cross-link | dependent | (55) |
| TrpB | tryptorubins (89, 90) (atropitides/atropopeptides) | C–C cross-link and C–N cross-links | dependent | (58) |
| AmyB | amyxirubins (91, 92) (atropitides/atropopeptides) | C–C cross-link and C–N cross-links | dependent | (58) |
| CihB | cihunamides (94–97) (bitryptides) | C–N biaryl cross-link | dependent | (59) |
| ScnB | 99 (bitryptides) | C–N biaryl cross-link | dependent | (60) |
| MciB | 101 | C–C biaryl cross-link | dependent | (60) |
| MibO | microbisporicins (102–105) (lanthipeptides) | dihydroxylation | unknown | (63) |
| NtaM | nocathioamides (106–108) (lanthipeptides) | consecutive oxygenations and imide formation | unknown | (64) |
| BotCYP | bottromycin (115) (bottromycins) | oxidative decarboxylation | independent | (73) |
| McmId | mechercharmycins (119, 120) (polyazole cyclopeptides) | hydroxylation | dependent | (80) |
| Pgm7 | pheganomycins (126–129) | hydroxylation | unknown | (81) |
| GmP450-29 | α-amanitin (130) | hydroxylation | unknown | (83) |
| OphB1/OphM1, OphB2/OphM2 | omphalotins (132–140) | hydroxylations and C–N cross-link | unknown | (84, 85) |
| UstC | ustiloxin B (143) | C–S cross-link | unknown | (87) |
Homologous proteins of TsrP and TsrR are also encoded in the BGCs of siomycin (51) (ref (41)), thiopeptin (52) (refs (37, 38)), Sch18640 (53) (ref (42)), and saalfelduracin (54) (ref (42)).
Homologous proteins of NosB and NosC are also encoded in the BGCs of nocathiacin (62) (refs (46, 47)) and persiathiacin (63) (ref (50)).
Homologous proteins of NocV, NocU and NocT are also encoded in the BGC of persiathiacin (63) (ref (50)).
2. P450s in Thiopeptide Biosynthesis
Nearly half of the P450s uncovered in RiPP PTMs are related to thiopeptide biosynthesis. As a well-known group of highly modified polythiazolyl RiPPs, thiopeptides feature an enzymatic [4 + 2] cycloadditional pyridine ring central to multiple azol(in)es and dehydroamino acids.16 According to the number of macrocyclic scaffolds, thiopeptides are divided into monocyclic members such as berninamycin-like thiopeptides, and side-ring appending bicyclic members including thiostrepton-like and nosiheptide-like thiopeptides.
2.1. Monocyclic Thiopeptides
In monocyclic thiopeptides, the P450 enzyme usually introduces a hydroxyl (or epoxide) group at a single specific location within the macrocycle scaffold. Due to the limited research of P450s in monocyclic thiopeptides, little is known about the timings of these P450s and whether the PTMs are leader peptide dependent.
Thiomuracins (1–7) are Nonomuraea derived 29-membered monocyclic thiopeptides that target elongation factor thermo unstable (EF-Tu) to hinder it from chaperone aminoacylated tRNAs to the ribosome and result in inhibition against Gram-positive bacteria like Enterococcus faecalis and Staphylococcus aureus. Apart from the class-defining characteristics of thiopeptides, additional PTMs, for instance, β-hydroxylation in Phe5 and varying oxidative degrees in Ile8 are included in thiomuracins (Figure 4). Two P450s termed TpdJ1 (or TbtJ1) and TpdJ2 (or TbtJ2) are deduced for the oxidative PTMs, albeit lacking any experimental evidence.17,18 Additionally, TpdJ2 T247A and TpdJ1 T234A/C340S could be recruited to selectively cyclopropanate dehydroalanines (Dhas) in several linear and cyclic thiopeptide analogues with the addition of ethyl diazoacetate (EDA) as the source of the carbon atom and convert them to l-amino-2-cyclopropane carboxylic acids (ACCAs).19
Figure 4.

Chemical structures of monocyclic thiopeptides. The moieties formed by P450s are highlighted in blue. Consecutive oxidations are catalyzed by P450s in thiomuracins (1–7) and GE37468 (19), while P450s catalyze monohydroxylations in GE2270s (8–16), berninamycins (22–29), and geninthiocins (30–33). The functions of P450s in sulfomycin (34–36) and litoralimycin (37, 38) biosyntheses remain elusive. The timings of all the P450s are still mysterious.
Similar β-hydroxylation in Phe and oxygenation in Ile are unveiled in other 29-membered thiopeptides GE2270s (8–16) and GE37468 (19), respectively. The function of the exclusive P450 gene pbtO (or tpdQ) in the GE2270s biosynthetic gene cluster (BGC) was investigated in vivo. Two congeners 17 and 18 in a 1:1 ratio instead of GE2270s are produced in pbtO mutated strain. Compound 17 carries an unmodified Phe8 residue, while 18 is its O-desmethyl derivative (Figure 4). These results underpin the notion that PbtO catalyzes Phe8 β-hydroxylation, without which the O-methylation of thiazole D would be relatively diminished. Besides, it is highly efficient for the PbtO-mediated hydroxylation, as no deoxy-Phe congener has been detected in the wild type producing strain.20
Unlike the stochastic oxidation in thiomuracins, Ile8 residue in GE37468 (19) is oxygenated to cyclic hemiaminal β-methyl-δ-hydroxy-proline (mhP) as its exclusive structure, a moiety also found in thiomuracin I (7). The precursor sequences of both GE37468 and thiomuracins imply that the unique mhP originates from Ile rather than the structurally similar Pro residue. This is further confirmed by isotopic feeding with 13C6-l-Ile. Tandem mass spectrometry analysis localizes the labeled carbons to the mhP8 moiety. A subsequent getJ knockout experiment generates an mhP8Ile variant of GE37468, intimating that GetJ, the only P450 encoded in the BGC, is responsible for the consecutive oxygenations of Ile8 (Figure 4). The structural constraint introduced by mhP8 is a requirement for the biological activities of GE37468, since mhP8Ile and mhP8Ala variants dramatically reduce the antibiotic activities against Methicillin-resistant S. auereus (MRSA) MW2 and Bacillus subtilis 168.21 Oxygenation of the Ile8Leu variant also occurs to yield δ-OHLeu (20) as the major product along with a minor product γ-methyl-δ-hydroxyproline (γ-mhP, 21), both of which show a slight decrease of bioactivities against MRSA MW2, MRSA COL, and S. subtilis PY79.22
Berninamycins (22–29) represent the 35-membered monocyclic thiopeptides featuring a unique oxazolyl-thiazolyl-pyridine core. In total 11 berninamycins have been isolated from a number of Streptomyces, 8 of which encompass a β-OHVal7 residue (berninamycins A, C, D, E, G, H, I, K, Figure 4).23−26 Moreover, a congener bearing two hydroxylations at Val7 was determined by MS/MS fragmentation studies, albeit in a trace amount of yield.27 Analysis of the berninamycin BGC identifies a P450 gene berH that is presumed for the hydroxylation at Val7 after scaffold macrocyclization.27 A comparable β-OHVal (β–OHVal6) is also observed in thiocillins. While no P450 gene is identified in the BGC of thiocillins, the nonheme iron-dependent dioxygenase TclD is likely to be the candidate for Val6 hydroxylation.28
Geninthiocins (30–33) share almost the same scaffold macrocyclization including the β-OHVal7 residue with berninamycin A (22), which differs on the first oxazol ring by lacking the additional methyl group.29−33 This difference results from the fifth amino acid in the core peptide, which is Ser in geninthiocins instead of Thr in berninamycins (Figure 4). Coinciding with the structural similarities between geninthiocins and berninamycins, their BGCs resemble with each other closely. GenH, the P450 enzyme in geninthiocin BGC, is homologous to BerH with an identity of 83.00% and is most probably involved in Val7 hydroxylation.30 Besides, the BGCs of sulfomycins (34–36) and litoralimycins (37, 38) (Figure 4), another two 35-membered monocyclic thiopeptides, also encode P450 enzymes SulN and Lit1, of which the functions remain elusive.34,35 Since the lit cluster also encodes sulfomycin precursor (Lit11), chances are that the P450 enzyme Lit1 participates in the biosyntheses of sulfomycins rather than litoralimycins.
2.2. Bicyclic Thiopeptides
Compared with monocyclic thiopeptides, the P450 modifications in bicyclic thiopeptides have drawn much attention. Sch40832 (46) contains an extraordinary imidazopiperidine moiety, the most heavily functionalized central domain characteristic of thiopeptides. The establishment of this central heterocycle ring had remained enigmatic until recently. Sequence alignment of the Sch40832 BGC (sch cluster) with other BGCs including thiopeptin (52) (tpp cluster) and thiostrepton (50) (tsr cluster) indicates schW, schX, and schY that involve in the PTMs for imidazopiperidine biosynthesis.36 Follow-up characterization reveals that the F420-dependent oxidoreductase SchZ reduces the dehydropiperidine (39) to piperidine ring (40), a phenomenon also found in the biosynthesis of thiopeptin,37,38 after which the P450 SchY functions as a leader peptide independent monooxygenase to epoxidize the thiazole moiety adjacent to the nascent piperidine. The nitrogen atom in piperidine nucleophilically attacks the C4′ to open the epoxide and cleave the C–S bond to afford imidazopiperidine equipped with a mercapto group as well as an aldehyde group (42). This highly reactive intermediate is followed with aldehyde reduction and mercapto methylation to generate a stable imidazopiperidine heterocycle (45) (Figure 5A).36
Figure 5.

(A) SchY catalyzes an epoxidation to generate intermediate 41, which is further rearranged to form the imidazopiperidine moiety in the biosynthesis of Sch40832 (46). (B) TsrR and TsrP are involved in the biosynthesis of thiostrepton (50) for dihydroxylation at Ile10 and epoxidation of quinaldic acid moiety, respectively. (C) Chemical structures of other dihydroxylation containing thiopeptides (51–55). The moieties formed by P450s are highlighted in blue.
In addition to SchY, another two P450s, SchP and SchR, are also involved in the biosynthesis of Sch40832 (46). They show moderate sequence identities to TsrP and TsrR (52.62% and 50.00%, respectively) that are involved in thiostrepton biosynthesis. In-frame deletions of tsrP and tsrR imply that TsrP mediates the epoxidation of the thiostrepton quinaldic acid moiety and TsrR catalyzes the dihydroxylation at Ile10. The dihydroxylation is a checkpoint in thiostrepton maturation prior to macro-ring closure, whereas TsrP recognizes the leader peptide-containing substrate 47 after quinaldic acid loading and macro-ring closure.39 Following with this epoxidation, a dual function α/β-hydrolase TsrI cleaves the leader peptide of 48 to release the primary amine in N-terminal core peptide that further attacks the epoxide in quinaldic acid and closes the side ring of thiostrepton (Figure 5B).40 Undoubtedly, both TsrR and TsrP are leader peptide dependent oxygenases, despite the exact timing of TsrR being controversial.
Other thiostrepton-like thiopeptides such as siomycin (51),41 thiopeptin (52),37,38 Sch18640 (53), and saalfelduracin (54)42 also contain similar quinaldic acid and dihydroxylation at isoleucine (Figure 5C), similar to the previously mentioned thiostrepton (50). In accordance with their structural similarities, homologous proteins of TsrP and TsrR are encoded in their BGCs, ranging from 52% to 74% for TsrP and from 50% to 79% for TsrR in sequence identities. It is reasonable to speculate that these homologous proteins play analogical roles in thiostrepton-like thiopeptide biosyntheses. Besides, isoleucine dihydroxylation is also found in nocardithiocin (55), a monocyclic thiopeptide from Nocardia pseudobrasiliensis IFM 0761. The BGC of nocardithiocin encodes two P450s (NotF and NotH) with unknown functions due to their low identities with TsrP and TsrR.43 At least one of the P450s is deduced for this dihydroxylation of Ile8 and the other one for the hydroxylation at dehydroalanine (Dha4) (Figure 5C).
Nosiheptide (61) is a bioactive thiopeptide against Gram-positive bacteria and is widely used as a feed additive for animals. Its BGC includes two P450 genes nosB and nosC, reminiscent of the hydroxylations in the central pyridine ring and Glu6. Inactivation of nosB and nosC completely abolishes the production of nosiheptide and results in the generation of 60 and 57, respectively (Figure 6A). Compared with nosiheptide, 60 lacks the hydroxy group in Glu6, while 57 misses the hydroxy group in pyridine ring as well as the NosA-catalyzed enamine dealkylation in the C-terminus, indicating that the enamine dealkylation is dependent on pyridine hydroxylation but is not affected by nosB deletion. In vitro characterization further proved their leader peptide independent oxygenation activities as NosB installs the Glu6 hydroxylation at the γ-position on 60, the intermediate isolated from the ΔnosAB mutant (59) and the intermediate isolated from the ΔnosBC mutant (56) in a stereoselective manner, while NosC hydroxylates the pyridine ring on 56 and 57.44 In addition to the previously reported substrate (58),45 NosA also tolerates 59 as a substrate. Therefore, the entire biosynthetic process of nosiheptide results from different routes in a cooperative or synergetic way owing to the broad substrate specificities of NosABC (Figure 6A).44
Figure 6.

(A) Proposed oxidative routes for the maturation of nosiheptide (61). NosB and NosC are responsible for the hydroxylations of Glu6 and the central pyridine ring in leader peptide independent manners, respectively. (B) Chemical structures of nocathiacin I (62) and persiathiacin A (63). The functions of NocB and PerX are validated in vitro and are leader peptide independent. While the functions of NocU and NocV are validated in vivo and the timings of these two P450s are obscure for the moment. (C) Chemical transformation catalyzed by NocV. This P450 oxygenates β-hydroxylation on Glu6 (65) and then oxidatively couples the nascent β-hydroxyl with the methyl group in MIA to form the third cyclic ring via an ether linkage (66). The moieties formed by P450s are highlighted in blue.
The BGC of nocathiacin (62), a more functionalized analogue of nosiheptide (61), shows significant resemblances in the gene sequence and arrangement with nosiheptide, suggesting great similarities in their biosyntheses. Three addtional P450 oxygenases, i.e., NocT, NocU, and NocV, are assumed for multiple oxidations toward the maturation of nocathiacin apart from NocB and NocC that show moderate sequence identities with NosB and NosC (47.10% and 67.45%, respectively) (Figure 6B).46 NocB was confirmed to leader peptide independently hydroxylate Glu6 of bicyclic intermediate 60 at the γ-position with a reduced activity compared to NosB, probably for the reason that NocB favors the tricyclic intermediate with a rigid core structure as its native substrate.47 This molecular rigidity is attributed to NocV which stereoselectively oxygenates β-hydroxylation on Glu6 (65), after which the nascent β-hydroxyl oxidatively couples with the methyl group in the 3-methyl-2-indolic acid (MIA) moiety to form the third cyclic ring via an ether linkage (66) (Figure 6C). Moreover, the enhancement in molecular rigidity has a marked improvement on its antimicrobial activity.48 The electron-rich indole ring is prone to electrophilic aromatic substitution since the lone pair electrons in the indole nitrogen take part in the aromatic system to increase its electron density. This results in the unique N-hydroxylation of the indolic moiety catalyzed by NocU since the more reactive 3-position and 5-position of the indolic moiety are blocked by methylations (Figure 6B).49 It is noteworthy that the timings of NocV and NocU are obscure for the moment, with the possibilities that they act in the early stage of PTMs in leader peptide dependent manners.48,49 Due to the homology of NocC with NosC, NocC is proposed for hydroxylation at the central pyridine ring, and the remaining P450 NocT is presumed for the oxygenation at the dehydrated Thr4 residue, albeit these hypotheses need further certifications (Figure 6B).
Persiathiacins (63) from Actinokineospora sp. UTMC 2475 and Actinokineospora sp. UTMC 2448 are polyglycosylated nosiheptide analogues bearing additional functionalities. Interestingly, the putative persiathiacin BGC encodes six P450s, of which PerB, PerC, PerT, PerU, and PerV show sequence similarities with NocB, NocC, NocT, NocU, and NocV, respectively. Benefiting from the high resemblance of nocathiacin (62) and persiathiacin (63) in structures and biosynthetic machinery, these five P450s are assumed to perform analogous functions as their homologues in nocathiacin biosynthesis. The remnant P450 PerX catalyzes thiazole hydroxylation of nosiheptide both in vivo and in vitro (Figure 6B), although the structure of the product has not been fully characterized.50 Persiathiacins (63) by far may represent the highest number of P450s involved in RiPP biosynthesis. The PerX catalyzed hydroxylation is prone to be leader peptide independent, but whether nosiheptide is the physiological substrate is unclear.
3. P450s in Biarylitide Biosynthesis
Biarylitides are a family of ribosomally synthesized and N-terminally blocked tripeptides with one cross-link between the aromatic side chains in the first and third core peptide amino acids. The cross-link endows biarylitides with rigidity and stability, along with potentially restricted rotation and planar chirality. Their BGCs are compact, commonly with an 18-bp gene (bytA) encoding a pentapeptide precursor, the smallest precursor ever reported, and an adjacent P450 gene (bytO) for post-translational cross-linking ahead of trimming the additional two amino acids at the N-terminus by a nonspecific protease.51 The founding members of biarylitides are YYH (71) and YFH (72) isolated from Planomonospora with a C–C biaryl cross-link (A–B cross-link) (Figure 7). The N-termini of these tripeptides are further acetylated by a yet-unknown acetyltransferase to generate maturated biarylitides.51 Nevertheless, the first myxobacterial biarylitide termed myxarylin (MeYLH, 76) is found to be an N-methylated tripeptide that displays a C–N biaryl cross-link (A–N–B cross-link) other than the C–C biaryl cross-link (Figure 7). An S-adenosyl-methionine (SAM) dependent methyltransferase BytZ is encoded in the BGC of myxarylin for the methylation at the N-terminal free amine as the final tailoring step.52
Figure 7.

Biosyntheses of biarylitides via distinct cross-links. All the cross-links are leader peptide dependent and occur between two aryl groups. Precursor 80 is a non-natural substrate for P450Blt. The P450 catalyzed cross-links are highlighted in blue.
The detailed biosynthetic mechanism comes from the investigation of the biarylitide 79 produced by Micromonospora sp. MW-13. In vitro characterization of the CYP1251 C3 (hereafter designated as P450Blt) toward MRYLH precursor peptide (77) was performed using the electron transport system from Rhodopseudomonas palustris CGA009 (PuR/PuxB). This characterization demonstrates an A–N–B cross-link between tyrosine and histidine, akin to myxarylin (76) (Figure 7). The two amino acids leader peptide is essential for the pentapeptide precursor catalyzation, since the removal of Met dramatically diminishes the activity, and removal of Met-Arg completely abolishes the activity of P450Blt. Modification of Met(−2) via formylation or omission the sulfur atom by a Met(−2)norleucine (Nle) replacement is well tolerated. While sulfoxidation or truncation of Met(−2) side chain to Ala significantly decreases the efficiency. These variants imply that the interactions between P450Blt and Met(−2) are likely hydrophobic and the pentapeptide cyclization is much rapider than sulfoxidation. The Arg(−1) residue also shows some effect on the peptide binding to P450Blt, as Ser and Asn replacements noticeably affect the cyclization of precursor peptide. The substitutions of Leu2 to amino acids with limited side chain size like Val, Ile, Nle, Ser, Cys, and even the non-natural amino acids acetamidomethyl protected Cys (AcmCys) and 2-amino-4-pentynoic acid are considerable tolerance, while substitutions to Asp, Trp, and diaminopropanoic acid (Dap) considerably hinder this process.53
P450Blt seems to be strictly specific for the residues involved in the cross-link (i.e., Tyr3 and His5), since none of the d-Tyr, d-His, His/Tyr reverse, Tyr3Hpg (4-hydroxyphenylglycine), His5Hpg and replacement of His5 imidazole side chain with a thiazole moiety or replacement of the C-terminal carboxylate of His5 with an amide could be accepted. However, the substitution of His5 to Trp (MRYLW, 80) is cyclized quite effectively, albeit the alteration of a cross-linking residue and the size increase of Trp. Most importantly, this exchange surprisingly alters the type of cross-link with the Tyr phenol oxygen connected to the Trp indole ring via an A–O–B cross-link instead of the former A–B or A–N–B cross-link (Figure 7). Other pentapeptides with different amino acids in position 2 (e.g., MRYGW, MRYAW, MRY-Nle-W) exhibit an identical ether linkage. These findings support the potential plasticity in P450 for the generation of alternate ring linkages in biarylitides.53
4. P450s in Cittilin and Atropitide (Atropopeptide) Biosyntheses
In addition to biarylitides, P450s also act as class-defining modification enzymes in leader peptide dependent manners in other classes such as cittilins, atropitide (atropopeptide), and related RiPPs. Cittilins (85, 86) are the second myxobacterial RiPPs that have been biosynthetically characterized. Cittilin A (86) potently inhibits the carbon storage regulator protein A (CsrA)-RNA interaction of bacteria and dysregulates multiple virulence-relevant processes as a potential anti-infective drug without killing the bacterial pathogens.54 The genome of Myxococcus xanthus points toward a ribosomal synthesis of cittilins from a precursor peptide (CitA, 83) with a C-terminal amino acid sequence YIYY. Downstream of citA are citB and citC encoding a P450 enzyme responsible for the cyclization of tetrapeptide and a SAM-dependent methyltransferase responsible for hydroxyl methylation, respectively. Their functions are validated by a vanillate-inducible promoter system inserted upstream of citA and gene disruption experiments of citB and citC. The production of cittilins is influenced by the supplementation of vanillate in the citA mutant. And cittilins are wholly abolished in the genetic disruption of citB, whereas disruption of citC results in the exclusive production of cittilin B (85) with an enhanced yield. Just like biarylitides, the cittilin operon also lacks a gene encoding protease or peptidase that commonly excises the leader peptide and releases the modified core peptide. This process is achieved by a prolyl endopeptidase (MX PEP) located outside of cittilin BGC. Subsequent in vitro investigation supports that CitB catalyzes the biaryl and aryl-oxy-aryl linkages as the first PTM in a leader peptide dependent manner, followed by proteolytic excision of leader peptide by MX PEP to form cittilin B (85) and methylation of cittilin B by CitC as the final step to give the major derivative cittilin A (86) (Figure 8A). In silico analysis uncovers analogous BGCs from M. hansupus mixupus and Streptomyces sp. Ncost-T10-10d with alternative core peptide sequences YHYY and YSYY, respectively.55
Figure 8.

Biosyntheses of cittilins (A), atropopeptides (atropitides) (B) and the structures of amyxirubins (C), cihunamides, compounds 99 and 101 (D). All the P450s above catalyze leader peptide dependent multicoupling or monocoupling reactions. The core peptides in precursors are highlighted in brown. The nascent chemical bonds are highlighted in blue.
Tryptorubins A and B (89, 90) that differ by an N-terminal alanine residue are rigid polycyclic globular peptides isolated from a Fungus-derived Streptomycete.56 Total synthesis of tryptorubin A strikingly unveiled that the identical chemical connectivity of tryptorubin matches two distinct noninterconvertible configurations. These isomers are termed noncanonical atropisomers, which distinguishs them from other common stereochemical definitions such as point chirality, E/Z isomerism, and canonical (single axially chiral) atropisomerism. Just one of the atropisomeric configurations (tryptorubin A) is isolated in nature, and the other atropisomer (atrop-tryptorubin A) is only obtained via chemical synthesis.57 The tryptorubin-like noncanonical atropisomers are designated atropitides or the alternative name atropopeptides.4,58 Tryptorubins were initially postulated to be NRPs but were subsequently revised to be RiPPs.56,57 Gene cluster comparison reveals that the BGCs of the aforementioned biarylitides and cittilins highly resemble the BGC of tryptorubins, which encompasses only a precursor peptide and a P450 as the sole modifying enzyme for the atropospecific cross-links of one carbon–carbon and two carbon–nitrogen bonds. Besides, the specific peptidase for leader peptide cleavage is also absent in the direct vicinity.57,58 After the P450 (TrpB) modifications on precursor peptide TrpA (87), a two-step maturation process is proposed for the generation of tryptorubin B (90), which involves a genome encoded protease to remove the leader peptide to form tryptorubin A and another peptidase to further hydrolyze the N-terminal alanine residue for the formation of tryptorubin B (Figure 8B).58
Publicly available genome sequences were investigated to find genes resembling the BGC of tryptorubins. Fourty-seven putative atropitide precursor peptides have been identified in total.57,58 Among the six core peptide residues, Ala1, Trp2, Tyr3, and Trp5 are highly conserved, while the positions 4 and 6 are somewhat flexible.58 Furthermore, two additional atropitides termed amyxirubin A and B (91, 92) with the core peptide sequence (A)WYIWW have been isolated and structurally characterized very recently (Figure 8C). These atropitides, together with tryptorubins, display proliferative and promigratory properties in endothelial cells.58 Moreover, re-examining the genomes accessible in the GenBank database further showed another 33 putative atropitide precursor peptide genes which are neighboring trpB homologous P450 genes in this review (Figure S1). The trpB gene in Xanthomonas cucurbitae ATCC 23378 is truncated by a single base substitution (TGG to TAG) at codon 105, giving rise to a stop codon (TAG) and the impossibility to produce atropitide. Multiple sequence alignment of the putative atropitide precursor peptides highlights the KSLK motif in leader peptides, though the KS residues are not strictly conserved. In addition, most of the TrpA homologues contain Tyr3 and Trp5, two aromatic residues that are atropospecifically cross-linked with the exclusively conserved Trp2 residue in core peptides. Nonetheless, Trp5 in some of the homologues is replaced by Tyr, along with a non-Tyr residue in position 3. Besides, it is surprising that the precursor peptides in Xanthomonas albilineans strains exhibit an eight-residue core peptide (AWALYISY). The cross-links in this octapeptide are mysterious and fascinating.
During the evaluation of this Review, cihunamides A–D (94–97) and the peptide fragment 99 with atypical Trp-Trp linkages along with the peptide fragment 101 containing a Tyr-Trp linkage were reported.59,60In silico analysis of the cihunamides producing strain reveals two adjacent genes encoding a precursor peptide (CihA, 93) with the C-terminal sequence “TWNIWYS” and a P450 (CihB), which is very closely related to the atropitide family members in sequences. Nonetheless, cihunamides exhibit a distinct C–N linkage between two tryptophans and lack the class-defining feature of atropitides, (i.e., noncanonical atropisomerism resulted from multiple covalent linkages) (Figure 8D). Coexpression of cihA and cihB in Escherichia coli suggests that the CihB enzyme is sufficient for the core macrocyclization of CihA in the absence of additional redox partners.59
Another survey of 21,911 actinobacteria genomes generated 1957 potential BGCs in total, which are composed of a P450 gene and a precursor peptide gene with at least two aromatic residues (Trp, Tyr, or His) encoded in the C-terminus. The BGCs (scn and mci) from Streptomyces sp. CNQ-509 and Micromonospora citrea DSM 43903 were heterologously expressed in E. coli, and two peptide fragments (99 and 101) were identified and characterized after trypsin digestions. Fragment 99 exhibits an almost identical Trp-Trp cross-link with cihunamides, while a Tyr-Trp linkage is identified in fragment 101. Furthermore, both P450 enzymes ScnB and MciB display broad substrate selectivities against key ring-forming residues. But it should be noted that neither 99 nor 101 is the corresponding natural products of the two BGCs. The corresponding natural products are not detected from the wildtype strains.60
The newfound cihunamides and 99, 101 reveal canonical atropisomerism with a single biaryl linkage and are beyond the scope of atropitide (atropopeptide). Very recently, the name “bitryptides” is suggested to define the RiPPs with biaryl linkages between two tryptophan residues.59 The bitryptide group includes tryptorubins, amyxirubins as well as cihunamides and 99, but 101 and some tryptorubin congeners that lack the second tryptophan residue (as shown in Figure S1) are excluded according to this definition. A more proper name is suggested to summarize this expanded RiPP class.
5. P450s in Lanthipeptide Biosynthesis
Despite representing one of the largest classes of RiPPs,61,62 to my knowledge, only two lanthipeptides under the name of microbisporicins (NAI-107) (102–105) and nocathioamides (106–108) require P450 modifications thus far.63,64 Microbisporicins (102–105) contain chlorinated tryptophan and (di)hydroxyproline residues (Figure 9), and the latter is attributed to the P450 protein MibO, albeit the timing remains enigmatic.63 The hydroxylation state at Pro14 shows little effect on its antibacterial activity.65 Besides, oxidation in the first thioether bridge 3–7 is found in microbisporicin congeners (microbisporicin B1 and B2 (104, 105), Figure 9), as observed with other lanthipeptides like actagardine, NAI-802, and the following mentioned nocathioamides.64−67 Whether this sulfoxidation is catalyzed by MibO is uncertain, as a similar sulfoxide could be generated in an acidic aqueous solution during extraction other than genuine enzymatic reaction.68 Besides, the hydroxyproline residue has been identified in another lantibiotic daspyromycin B. Nonetheless, this hydroxylation seems to be a detoxification step generated by the heterologous host.69
Figure 9.
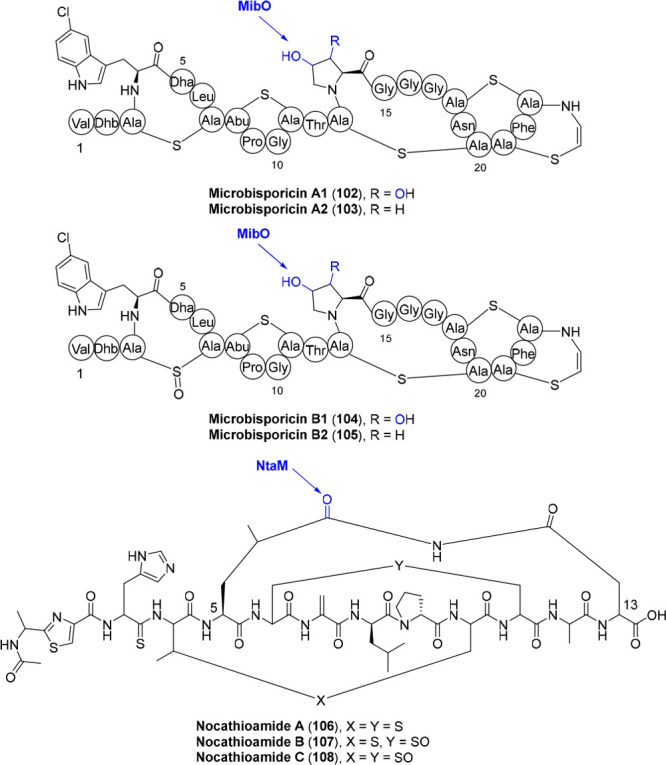
Structures of microbisporicins (102–105) and nocathioamides (106–108). MibO is proposed to oxygenate the Pro14 residue to form (di)hydroxylation, and NtaM is predicted for the multioxygenations of one of the δ-carbon atoms in Leu5 to deliver a carboxyl group. The exact functions and timings of these P450s are elusive. The proposed oxygenations catalyzed by P450s are highlighted in blue.
Nocathioamides (106–108) define a novel class of chimeric lanthipeptides combined with a thioamide that is exceptionally unveiled in thioamitides as well as some thiopeptides, and a thazole ring that is also perceived in linear azol(in)e-containing peptides (LAPs), cyanobactins, and pyritides (Figure 9). Nonetheless, the most intriguing characteristic of these tribrid RiPPs is the imide linkage between the fifth (Leu5) and thirteenth (Asn13) residues of the core peptide. The P450 enzyme NtaM is predicted for the multioxygenations of one of the δ-carbon atoms in Leu5 to deliver a carboxyl group, which further interlinks with the amide in Asn13 to form imide.64 The exact function and timing of NtaM remain largely enigmatic and are worthy of further investigation.
6. P450s in Other RiPP Biosyntheses
Other RiPPs related P450s are difficult to be classified and are described in this section. Bottromycins (115) are a unique class of RiPPs with promising antibacterial activities against several problematic multidrug-resistant pathogens. The biosynthesis of bottromycin possesses a list of fantastic features, including the atypical constitution of precursor peptide (N-terminal core peptide connected with a follower peptide at the C-terminus), the reversible macroamidine formation, along with the follower peptide excision catalyzed by a stand-alone YcaO protein BotCD and the amidohydrolase BotAH, respectively. Of note, BotAH acts as a YcaO accessory protein of BotCD to stop the reverse hydrolyzation of macroamidine (111) (Figure 10A).7,70,71 In particular, the P450 protein BotCYP performs an oxidative decarboxylation of 113 to convert the C-terminal thiazoline to the aromatic thiazole ring (114), a process frequently ascribed to flavin-dependent dehydrogenase. BotCYP shows a stereopreference for the d-Asp7 substrate (113) which is stemmed from the reversible epimerization catalyzed by an atypical α/β hydrolase BotH (Figure 10A). This P450′s activity provides stereochemical resolution for the epimers and pulls the epimerization equilibrium to the side of d-Asp7 product (113).71−73
Figure 10.

(A) Biosynthetic pathway of bottromycin A2 (115). The thiazol(in)e ring involved in BotCYP-catalyzed decarboxylation is highlighted in blue. (B) Proposed biosynthetic pathway of mechercharmycins (119, 120) and chemical structures of other polyazole cyclopeptides (121–124). McmI is assumed for the leader and follower peptides dependent β-hydroxylation at Phe. (C) Chemical structures of pheganomycins (126–129). Pgm7 is deduced for the hydroxylation of the methyl group. For (B) and (C), the proposed hydroxylations catalyzed by P450s are highlighted in blue.
Polyazole cyclopeptides such as mechercharmycins (119, 120), YM-216391 (121), aurantizolicin (122), curacozole (123), and telomestatin (124) are characterized by a continuum of thiazoles and oxazoles, and usually exhibit potent cytotoxicities against cancer cells (Figure 10B).74−79 Conserved P450 genes are uncovered in the BGCs of polyazole cyclopeptides containing distinctive 5-phenyloxazole, i.e., YM-216391 (121), aurantizolicin (122), curacozole (123), and mechercharmycins (119, 120), but are absent in the BGC of telomestatin (124) that lacks the 5-phenyloxazole moiety.80 Thus, the P450 in mechercharmycin biosynthesis (McmI) is expected for the leader and follower peptide dependent β-hydroxylation at Phe after the regioselective dehydration and consecutive heterocyclizations. The β-hydroxylated Phe1 is eventually converted to the unique 5-phenyloxazole via heterocyclization and dehydrogenation, a process which is worthy of further investigation (Figure 10B).80
Pheganomycins (126–129) are characteristic RiPPs that show the N-terminal of the precursor peptide connected with a nonribosomally derived and nonproteinogenic amino acid, (S)-2-(3,5-dihydroxy-4-hydroxymethyl)phenyl-2-guanidinoacetic acid (125).81 Apparently, the two phenolic hydroxyl groups in 125 are formed by Pgm8–11 that are homologous to DpgA–D involved in vancomycin and teicoplanin biosynthesis.81,82 Whereas the P450 protein Pgm7 is anticipated for the generation of the remaining hydroxyl in the methyl group (Figure 10C), this modification and its timing remain to be verified.81
While an array of P450s take part in the biosyntheses of bacterial RiPPs, the number of fungal RiPPs related P450s is really limited. To date, only three fungal RiPPs, i.e., α-amanitin (130), omphalotins (132–140), and ustiloxin B (143) involve P450 modifications. In the flanking region of α-amanitin precursor gene locates four P450 genes, one of which termed GmP450−29 was successfully disrupted. This mutant yields an amatoxin analogue (θ-amanitin) (131) lacking hydroxyl groups at the C-4 position of proline as well as the C-5 position of isoleucine, alluding that GmP450−29 is responsible for one or both of the oxygenations (Figure 11A).83 The functions of the other three P450s are yet to be elucidated.
Figure 11.

(A) Chemical structures of α-amanitin (127) and θ-amanitin (128). GmP450-29 is responsible for one or both oxygenations on proline and isoleucine. (B) Chemical structures of omphalotins A–I (129–137). The two P450s encoded in the omphalotin BGC are presumed for the C–N cross-link of Trp1 and consecutive oxygenations in Trp1, Val7, Ile8, and Gly9. (C) UstC-catalyzed C–S cross-link and chemical structure of ustiloxin B (140). The PTMs catalyzed by P450s are highlighted in blue.
Omphalotins (132–140) are the founding members of a novel family of RiPPs, of which the precursor peptide is fused to the C-terminal of its N-methyltransferase for autocatalytic backbone N-methylations. Only the methyltransferase attached precursor peptide OphMA and the peptidase OphP are needed for the reconstitution of omphalotin A (132). The additional two P450s (OphB1/OphM1 and OphB2/OphM2) and an O-acyltransferase (OphD/OphC) are speculated for the PTMs in omphalotins B–I (133–140) (Figure 11B).84,85 The C–N cross-link of Trp1 and consecutive oxygenations in Trp1, Val7, Ile8, and Gly9 are presumed by the two P450 enzymes. Ustiloxin B (143) potently inhibits mitosis and microtubule assembly. Little is known about the exact function of the P450 enzyme UstC encoded in its BGC. It is hypothesized that UstC catalyzes a C–S cross-link of the mercapto group in cysteine and the aromatic ring in tyrosine residue (141) (Figure 11C).86,87 The functions and timings of these fungal RiPPs related P450s need to be further investigated in the future.
7. Conclusions and Prospects
At present, only a handful of P450s have been uncovered in RiPP biosynthesis, and almost all of them are associated with oxygenations or intramolecular cross-linking/coupling reactions. Despite a wide range of P450 enzymes involve in the biosynthesis of monocyclic and bicyclic thiopeptides, only NosB, NosC involved in nosiheptide (61) biosynthesis, NocB (the homologue of NosB) involved in nocathiacin (62) biosynthesis and SchY involved in Sch40832 (46) biosynthesis are fully characterized to date.36,44,47 They perform hydroxylation or epoxidation reactions without the assistance of leader peptides. Due to the dramatically reshaped structures and complicated PTMs, most of the P450s involved in thiopeptide biosynthesis lack of sufficient studies, and more work will be required to unequivocally establish the functions, especially the timings of P450s. Apart from thiopeptides, oxygenations are also uncovered in the biosyntheses of microbisporicins (102–105) and nocathioamides (106–108), the only two lanthipeptides including P450 catalysis, and the 5-phenyloxazole containing polyazole cyclopeptides (119–123), pheganomycins (126–129), α-amanitin (130), and omphalotins (133–140). These oxygenations warrant further studies to elucidate the functions, mechanisms, and timings of these intriguing P450 enzymes.
Apart from tailoring reactions, P450s also act as class-defining modification enzymes in some novel classes of RiPPs including biarylitide, cittilin, atropitide (atropopeptide) and several related peptides. All the P450s involved in class-defining modifications are leader peptide dependent and are responsible for varied coupling reactions. It is hard to imagine that different functional groups in atropitides (atropopeptides) could be cross-linked via an exclusive P450 in chemoselective, regioselective, stereoselective, and atroposelective manners, indicative of the flexibility of P450s. Besides, P450s also catalyze non-class-defining cross-links in the biosyntheses of omphalotins (133–140) and ustiloxin B (143), but their functions and timings are ill-understood as well.
It is worth mentioning that the vast majority of intramolecular coupling reactions in RiPP PTMs are catalyzed by a totally different class of enzymes–radical SAM (rSAM) enzymes.88−92 Both P450s and rSAM enzymes readily abstract a hydrogen radical from the substrates during catalytic processes and generate highly reactive radical species, which further undergo cross-links with other intramolecular positions to forge new chemical bonds in the product scaffolds. Their resemblance makes it hard to distinguish the cross-links from each other by RiPP chemical structures. As is shown in Figure 2, the iron-porphyrin complex in P450 enzymes reacts with oxygen in the presence of external electrons to form the high-valent compound I that abstracts the hydrogen radical. Nevertheless, the hydrogen radical abstraction by rSAM enzymes is quite different. These enzymes rely on the [4Fe-4S] cluster to transfer an external electron to SAM, which is homolytically cleaved to release the byproduct methionine and yield the reactive 5′-deoxyadenosyl radical intermediate (5′dA·) for the hydrogen radical abstraction from the inactivated substrate.93,94 In contrast to the oxygen requirement for P450 catalysis, most of the rSAM enzymes only function effectively under anaerobic conditions due to the oxygen sensitivity of the [4Fe-4S] cluster.95 Moreover, all the unveiled cross-links by P450s are between aromatic residues (i.e., Tyr, His, Trp) (Figures 7 and 8), indicating that it is difficult for P450s to abstract a hydrogen radical from inactivated carbons. However, given that the bond dissociation energy for the 5′-dAdo· C5′-H bond is higher than the inactivated sp3 C–H bonds,94 it is unsurprising that rSAM enzymes could install new chemical bonds in unconventional positions and this may interpret the fact that nature chooses rSAM enzymes instead of P450s for most of the RiPP cross-linking reactions.
The P450 BotCYP involved in bottromycin (115) biosynthesis is more likely to be an individual case. It has an assigned biosynthetic function for the leader peptide independent aromatization of thiazoline via an oxidative decarboxylation.73 In addition to bacterial RiPPs, several eukaryotic RiPPs also include P450 modifications.96 Probably owing to the membrane-anchored nature of eukaryotic P450s as well as their redox partners,11,12 their functions and mechanisms are largely unexplored.
As a result of the exceeding catalytic ability of P450s, it could be very difficult to discriminate the leader peptide dependent P450s from the leader peptide independent P450s, or to distinguish the RiPP-associated P450s from the typical non-RiPP-associated P450s.60 Besides, the low accuracy in precursor peptide annotation, the lack of a universal feature across distinct RiPP classes, and the ubiquitous non-RiPP-related P450 enzymes hinder the discovery of novel RiPPs modified by P450s.60,97 Nevertheless, in silico analysis has shown promise in the exploration of unknown RiPP-associated P450s. Adam et al. selected all the members of PF00067 (cytochrome P450) and analyzed their corresponding BGCs to exclude the non-RiPP-related P450 enzymes. This analysis revealed that in bacteria approximately 1800 P450s (∼1.5%) are predicted to lie in or near to BGCs for different RiPPs, such as lantipeptides, thiopeptides, bacteriocins, lasso peptides, linaridins, cynaobactins, and so on.73 As mentioned above, He et al. identified 1957 potential BGCs composed of a P450 gene and a precursor peptide gene with at least two aromatic residues (Trp, Tyr, or His) encoded in the C-terminus. AlphaFlod2-multimer was then implemented to measure the precursor-enzyme binding properties to filter out the non-RiPP BGCs. This resulted in the identification of two novel RiPP BGCs (scn and mci) encoding P450s.60
To identify additional members of the RiPP-associated P450s, a sequence similarity network (SSN) and genome neighborhood network (GNN) were generated using the Enzyme Function Initiative Enzyme Similarity Tool (EFI-EST).98,99 The PF00067 family was used in the initial SSN analysis with UniRef50 cluster ID sequences instead of UniProt IDs, an E-value of 10–50, and a sequence alignment score threshold of 40. The SSN result was subsequently analyzed by GNN with a neighborhood size of 10, and the Pfam family hub-nodes were utilized for further analysis. A number of P450s were revealed adjacent to typical RiPP PTM enzymes, such as lantipeptide dehydratase, lanthionine synthetase, YcaO cyclodehydratase, consistent with the biosyntheses of thiopeptides and lantipeptides (Table S1). Besides, some P450s neighboring PqqD family proteins were also suspected for RiPP PTMs. In particular, the P450s adjacent to the emerging family of PTM enzymes termed domain of unknown function 692 (DUF692) or multinuclear nonheme iron dependent oxidative enzymes (MNIOs) are intriguing too. Members of MNIOs usually catalyze unprecedented chemical transformations in RiPP biosynthesis, and only three members have been functionally characterized.100 It should be noted that, apart from the P450s shown in Table S1, other P450s in this GNN may also be involved in RiPP PTMs. Since many PTM-installing enzymes are homologous to other cellular machinery, it is difficult to determine whether these P450s are RiPP PTM enzymes. Further investigations will be required for the discovery of new P450 modified RiPPs and the functional assignments of novel P450s.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.3c00026.
This work was financially supported by the National Natural Science Foundation of China (21907057), the Natural Science Foundation of Jiangsu Province, China (BK20190201), the Future Plan for Young Scholars, and the Fundamental Research Funds (2019GN032) of Shandong University.
The author declares no competing financial interest.
Supplementary Material
References
- Newman D. J.; Cragg G. M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Ongpipattanakul C.; Desormeaux E. K.; DiCaprio A.; van der Donk W. A.; Mitchell D. A.; Nair S. K. Mechanism of action of ribosomally synthesized and post-translationally modified peptides. Chem. Rev. 2022, 122, 14722–14814. 10.1021/acs.chemrev.2c00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G.; Wang Z.-J.; Yan F.; Zhang Y.; Huo L. Recent advances in discovery, bioengineering and bioactivity-evaluation of ribosomally synthesized and post-translationally modified peptides. ACS Bio Med. Chem. Au 2023, 3, 1–31. 10.1021/acsbiomedchemau.2c00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalbán-López M.; Scott T. A.; Ramesh S.; Rahman I. R.; van Heel A. J.; Viel J. H.; Bandarian V.; Dittmann E.; Genilloud O.; Goto Y.; et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep. 2021, 38, 130–239. 10.1039/D0NP00027B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin G. M.; Ding Y. Recent advances in the biosynthesis of RiPPs from multicore-containing precursor peptides. J. Ind. Microbiol. Biot. 2020, 47, 659–674. 10.1007/s10295-020-02289-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz L.; Kazmaier U.; Truman A. W.; Koehnke J. Bottromycins - biosynthesis, synthesis and activity. Nat. Prod. Rep. 2021, 38, 1659–1683. 10.1039/D0NP00097C. [DOI] [PubMed] [Google Scholar]
- Andersen F. D.; Pedersen K. D.; Wilkens Juhl D.; Mygind T.; Chopin P.; B. Svenningsen E.; Poulsen T. B.; Braad Lund M.; Schramm A.; Gotfredsen C. H.; et al. Triculamin: an unusual lasso peptide with potent antimycobacterial activity. J. Nat. Prod. 2022, 85, 1514–1521. 10.1021/acs.jnatprod.2c00065. [DOI] [PubMed] [Google Scholar]
- Dubey K. D.; Shaik S. Cytochrome P450 - the wonderful nanomachine revealed through dynamic simulations of the catalytic cycle. Acc. Chem. Res. 2019, 52, 389–399. 10.1021/acs.accounts.8b00467. [DOI] [PubMed] [Google Scholar]
- Greule A.; Stok J. E.; De Voss J. J.; Cryle M. J. Unrivalled diversity: the many roles and reactions of bacterial cytochromes P450 in secondary metabolism. Nat. Prod. Rep. 2018, 35, 757–791. 10.1039/C7NP00063D. [DOI] [PubMed] [Google Scholar]
- McLean K. J.; Luciakova D.; Belcher J.; Tee K. L.; Munro A. W.. Biological diversity of cytochrome P450 redox partner systems. In monooxygenase, peroxidase and peroxygenase properties and mechanisms of cytochrome P450; Hrycay E. G., Bandiera S. M., Eds.; Springer International Publishing, 2015; pp 299–317. [Google Scholar]
- Li Z.; Jiang Y.; Guengerich F. P.; Ma L.; Li S.; Zhang W. Engineering cytochrome P450 enzyme systems for biomedical and biotechnological applications. J. Biol. Chem. 2020, 295, 833–849. 10.1016/S0021-9258(17)49939-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Du L.; Bernhardt R. Redox partners: function modulators of bacterial P450 enzymes. Trends Microbiol. 2020, 28, 445–454. 10.1016/j.tim.2020.02.012. [DOI] [PubMed] [Google Scholar]
- Chen C.-C.; Min J.; Zhang L.; Yang Y.; Yu X.; Guo R.-T. Advanced understanding of the electron transfer pathway of cytochrome P450s. ChemBioChem. 2021, 22, 1317–1328. 10.1002/cbic.202000705. [DOI] [PubMed] [Google Scholar]
- Kunakom S.; Otani H.; Udwary D. W.; Doering D. T.; Mouncey N. J. Cytochromes P450 involved in bacterial RiPP biosyntheses. J. Ind. Microbiol. Biot. 2023, 50, kuad005. 10.1093/jimb/kuad005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Liu W. Biosynthesis of thiopeptide antibiotics and their pathway engineering. Nat. Prod. Rep. 2013, 30, 218–216. 10.1039/C2NP20107K. [DOI] [PubMed] [Google Scholar]
- Morris R. P.; Leeds J. A.; Naegeli H. U.; Oberer L.; Memmert K.; Weber E.; LaMarche M. J.; Parker C. N.; Burrer N.; Esterow S.; Hein A. E.; Schmitt E. K.; Krastel P. Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J. Am. Chem. Soc. 2009, 131, 5946–5955. 10.1021/ja900488a. [DOI] [PubMed] [Google Scholar]
- Hudson G. A.; Zhang Z.; Tietz J. I.; Mitchell D. A.; van der Donk W. A. In vitro biosynthesis of the core scaffold of the thiopeptide thiomuracin. J. Am. Chem. Soc. 2015, 137, 16012–16015. 10.1021/jacs.5b10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gober J. G.; Ghodge S. V.; Bogart J. W.; Wever W. J.; Watkins R. R.; Brustad E. M.; Bowers A. A. P450-Mediated non-natural cyclopropanation of dehydroalanine-containing thiopeptides. ACS Chem. Biol. 2017, 12, 1726–1731. 10.1021/acschembio.7b00358. [DOI] [PubMed] [Google Scholar]
- Tocchetti A.; Maffioli S.; Iorio M.; Alt S.; Mazzei E.; Brunati C.; Sosio M.; Donadio S. Capturing linear intermediates and C-terminal variants during maturation of the thiopeptide GE2270. Chem. Biol. 2013, 20, 1067–1077. 10.1016/j.chembiol.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Young T. S.; Walsh C. T. Identification of the thiazolyl peptide GE37468 gene cluster from Streptomyces ATCC 55365 and heterologous expression in Streptomyces lividans. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 13053–13058. 10.1073/pnas.1110435108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T. S.; Dorrestein P. C.; Walsh C. T. Codon randomization for rapid exploration of chemical space in thiopeptide antibiotic variants. Chem. Biol. 2012, 19, 1600–1610. 10.1016/j.chembiol.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau R. C. M.; Rinehart K. L. Berninamycins B, C, and D, minor metabolites from Streptomyces bernensis. J. Antibiot. 1994, 47, 1466–1472. 10.7164/antibiotics.47.1466. [DOI] [PubMed] [Google Scholar]
- Kodani S.; Ninomiya A. Isolation of new thiopeptide berninamycin E from Streptomyces atroolivaceus. Asian J. Chem. 2013, 25, 490–492. 10.14233/ajchem.2013.13249. [DOI] [Google Scholar]
- Zhang D.; Wang Y.; Hu X.; Wang X.; Li L.; Gu G.; Zhang B.; Cen S.; You X.; Yu L. Cyclic and linear thiopeptides from soil-derived Streptomyces sp. CPCC 203702 with antiviral and antibacterial activities. Chin. J. Chem. 2021, 39, 3277–3284. 10.1002/cjoc.202100496. [DOI] [Google Scholar]
- De B. C.; Zhang W.; Zhang G.; Liu Z.; Tan B.; Zhang Q.; Zhang L.; Zhang H.; Zhu Y.; Zhang C. Host-dependent heterologous expression of berninamycin gene cluster leads to linear thiopeptide antibiotics. Org. Biomol. Chem. 2021, 19, 8940–8946. 10.1039/D1OB01759D. [DOI] [PubMed] [Google Scholar]
- Malcolmson S. J.; Young T. S.; Ruby J. G.; Skewes-Cox P.; Walsh C. T. The posttranslational modification cascade to the thiopeptide berninamycin generates linear forms and altered macrocyclic scaffolds. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 8483–8488. 10.1073/pnas.1307111110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland Brown L. C.; Acker M. G.; Clardy J.; Walsh C. T.; Fischbach M. A Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 2549–2553. 10.1073/pnas.0900008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun B.-S.; Hidaka T.; Furihata K.; Seto H. Microbial metabolites with tipA promoter inducing activity II. geninthiocin, a novel thiopeptide produced by Streptomyces sp. DD84. J. Antibiot. 1994, 47, 969–975. 10.7164/antibiotics.47.969. [DOI] [PubMed] [Google Scholar]
- Schneider O.; Simic N.; Aachmann F. L.; Rückert C.; Kristiansen K. A.; Kalinowski J.; Jiang Y.; Wang L.; Jiang C.-L.; Lale R.; Zotchev S. B. Genome mining of Streptomyces sp. YIM 130001 isolated from lichen affords new thiopeptide antibiotic. Front. Microbiol. 2018, 9, 3139. 10.3389/fmicb.2018.03139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iniyan A. M.; Sudarman E.; Wink J.; Kannan R. R.; Vincent S. G. P. Ala-geninthiocin, a new broad spectrum thiopeptide antibiotic, produced by a marine Streptomyces sp. ICN19. J. Antibiot. 2019, 72, 99–105. 10.1038/s41429-018-0115-2. [DOI] [PubMed] [Google Scholar]
- Li S.; Hu X.; Li L.; Liu H.; Yu L.; You X.; Jiang B.; Wu L. Geninthiocins C and D from Streptomyces as 35-membered macrocyclic thiopeptides with modified tail moiety. J. Antibiot. 2019, 72, 106–110. 10.1038/s41429-018-0127-y. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Wang J.; Tang Y.; Guo Z.; Bai J.; Wu L.; Su J.; Cen S.; Yu L.; Zhang D. Geninthiocins E and F, two new cyclic thiopeptides with antiviral activities from soil-derived Streptomyces sp. CPCC 200267 using OSMAC strategy. J. Antibiot. 2023, 76, 101–104. 10.1038/s41429-022-00580-0. [DOI] [PubMed] [Google Scholar]
- Du Y.; Qiu Y.; Meng X.; Feng J.; Tao J.; Liu W. A heterotrimeric dehydrogenase complex functions with 2 distinct YcaO proteins to install 5 azole heterocycles into 35-membered sulfomycin thiopeptides. J. Am. Chem. Soc. 2020, 142, 8454–8463. 10.1021/jacs.0c02329. [DOI] [PubMed] [Google Scholar]
- Khodamoradi S.; Hahnke R. L.; Mast Y.; Schumann P.; Kämpfer P.; Steinert M.; Rückert C.; Surup F.; Rohde M.; Wink J. Streptomonospora litoralis sp. nov., a halophilic thiopeptides producer isolated from sand collected at Cuxhaven beach. Antonie van Leeuwenhoek 2021, 114, 1483–1496. 10.1007/s10482-021-01609-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B.; Guo H.; Wang H.; Zhao Q.; Liu W. Dissection of the enzymatic process for forming a central imidazopiperidine heterocycle in the biosynthesis of a series c thiopeptide antibiotic. J. Am. Chem. Soc. 2021, 143, 13790–13797. 10.1021/jacs.1c05956. [DOI] [PubMed] [Google Scholar]
- Ichikawa H.; Bashiri G.; Kelly W. L. Biosynthesis of the thiopeptins and identification of an F420H2-dependent dehydropiperidine reductase. J. Am. Chem. Soc. 2018, 140, 10749–10756. 10.1021/jacs.8b04238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Lin Z.; Li Y.; Zheng Q.; Chen D.; Liu W. Insights into the thioamidation of thiopeptins to enhance the understanding of the biosynthetic logic of thioamide-containing thiopeptides. Org. Biomol. Chem. 2019, 17, 3727–3731. 10.1039/C9OB00402E. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Wang S.; Liao R.; Liu W. Precursor-directed mutational biosynthesis facilitates the functional assignment of two cytochromes P450 in thiostrepton biosynthesis. ACS Chem. Biol. 2016, 11, 2673–2678. 10.1021/acschembio.6b00419. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Wang S.; Duan P.; Liao R.; Chen D.; Liu W. An α/β-hydrolase fold protein in the biosynthesis of thiostrepton exhibits a dual activity for endopeptidyl hydrolysis and epoxide ring opening/macrocyclization. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 14318–14323. 10.1073/pnas.1612607113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R.; Duan L.; Lei C.; Pan H.; Ding Y.; Zhang Q.; Chen D.; Shen B.; Yu Y.; Liu W. Thiopeptide biosynthesis featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications. Chem. Biol. 2009, 16, 141–147. 10.1016/j.chembiol.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalen C. J.; Hudson G. A.; Kille B.; Mitchell D. A. Bioinformatic expansion and discovery of thiopeptide antibiotics. J. Am. Chem. Soc. 2018, 140, 9494–9501. 10.1021/jacs.8b03896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K.; Komaki H.; Gonoi T. Identification and functional analysis of the nocardithiocin gene cluster in Nocardia pseudobrasiliensis. PLoS One 2015, 10, e0143264. 10.1371/journal.pone.0143264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Xue Y.; Ma M.; Wang S.; Liu N.; Chen Y. Multiple oxidative routes towards the maturation of nosiheptide. ChemBioChem. 2013, 14, 1544–1547. 10.1002/cbic.201300427. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Guo H.; Zhang Q.; Duan L.; Ding Y.; Liao R.; Lei C.; Shen B.; Liu W. NosA catalyzing carboxyl-terminal amide formation in nosiheptide maturation via an enamine dealkylation on the serine-extended precursor peptide. J. Am. Chem. Soc. 2010, 132, 16324–16326. 10.1021/ja106571g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y.; Yu Y.; Pan H.; Guo H.; Li Y.; Liu W. Moving posttranslational modifications forward to biosynthesize the glycosylated thiopeptide nocathiacin I in Nocardia sp. ATCC202099. Mol. BioSyst. 2010, 6, 1180–1185. 10.1039/c005121g. [DOI] [PubMed] [Google Scholar]
- Wu X.; Huang P.; Xue Y.; Liu W.; Ma M.; Chen Y. The catalytic characteristics of NocB in nocathiacin biosynthesis from Nocardia sp. ATCC 202099. RSC Adv. 2016, 6, 72399–72408. 10.1039/C6RA09571B. [DOI] [Google Scholar]
- Bai X.; Guo H.; Chen D.; Yang Q.; Tao J.; Liu W. Isolation and structure determination of two new nosiheptide-type compounds provide insights into the function of the cytochrome P450 oxygenase NocV in nocathiacin biosynthesis. Org. Chem. Front. 2020, 7, 584–589. 10.1039/C9QO01328H. [DOI] [Google Scholar]
- Guo H.; Bai X.; Yang Q.; Xue Y.; Chen D.; Tao J.; Liu W. NocU is a cytochrome P450 oxygenase catalyzing N-hydroxylation of the indolic moiety during the maturation of the thiopeptide antibiotics nocathiacins. Org. Biomol. Chem. 2021, 19, 8338–8342. 10.1039/D1OB01284C. [DOI] [PubMed] [Google Scholar]
- Dashti Y.; Mohammadipanah F.; Belousoff M.; Vocat A.; Zabala D.; Fage C. D.; Romero-Canelon I.; Bunk B.; Spröer C.; Overmann J.; Cole S. T.; Challis G. L.. Discovery and biosynthesis of persiathiacins: unusual polyglycosylated thiopeptides active against multi-drug resistant tuberculosis. bioRxiv, March 29, 2022, ver. 4. 10.1101/2021.10.24.465558v4 (accessed 2022-09-18). [DOI]
- Zdouc M. M.; Alanjary M. M.; Zarazúa G. S.; Maffioli S. I.; Crüsemann M.; Medema M. H.; Donadio S.; Sosio M. A biaryl-linked tripeptide from Planomonospora reveals a widespread class of minimal RiPP gene clusters. Cell Chem. Biol. 2021, 28, 733–739. 10.1016/j.chembiol.2020.11.009. [DOI] [PubMed] [Google Scholar]
- Hug J. J.; Frank N. A.; Walt C.; Šenica P.; Panter F.; Müller R. Genome-guided discovery of the first myxobacterial biarylitide myxarylin reveals distinct C–N biaryl crosslinking in RiPP biosynthesis. Molecules 2021, 26, 7483. 10.3390/molecules26247483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Marschall E.; Treisman M.; McKay A.; Padva L.; Crüsemann M.; Nelson D. R.; Steer D. L.; Schittenhelm R. B.; Tailhades J.; Cryle M. J. Cytochrome P450Blt enables versatile peptide cyclisation to generate histidine- and tyrosine-containing crosslinked tripeptide building blocks. Angew. Chem., Int. Ed. 2022, 61, e202204957. 10.1002/anie.202204957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer C. K.; Fruth M.; Empting M.; Avrutina O.; Hossmann J.; Nadmid S.; Gorges J.; Herrmann J.; Kazmaier U.; Dersch P.; Müller R.; Hartmann R. W. Discovery of the first small-molecule CsrA-RNA interaction inhibitors using biophysical screening technologies. Future Med. Chem. 2016, 8, 931–947. 10.4155/fmc-2016-0033. [DOI] [PubMed] [Google Scholar]
- Hug J. J.; Dastbaz J.; Adam S.; Revermann O.; Koehnke J.; Krug D.; Müller R. Biosynthesis of cittilins, unusual ribosomally synthesized and post-translationally modified peptides from Myxococcus xanthus. ACS Chem. Biol. 2020, 15, 2221–2231. 10.1021/acschembio.0c00430. [DOI] [PubMed] [Google Scholar]
- Wyche T. P.; Ruzzini A. C.; Schwab L.; Currie C. R.; Clardy J. Tryptorubin A: a polycyclic peptide from a fungus-derived Streptomycete. J. Am. Chem. Soc. 2017, 139, 12899–12902. 10.1021/jacs.7b06176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisberg S. H.; Gao Y.; Walker A. S.; Helfrich E. J. N.; Clardy J.; Baran P. S. Total synthesis reveals atypical atropisomerism in a small-molecule natural product, tryptorubin A. Science 2020, 367, 458–463. 10.1126/science.aay9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanudorn P.; Thiengmag S.; Biermann F.; Erkoc P.; Dirnberger S. D.; Phan T. N.; Fürst R.; Ueoka R.; Helfrich E. J. N. Atropopeptides are a novel family of ribosomally synthesized and posttranslationally modified peptides with a complex molecular shape. Angew. Chem., Int. Ed. 2022, 61, e202208361. 10.1002/anie.202208361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J. S.; Lee H.; Kim H.; Woo S.; Nam H.; Lee J.; Lee J. Y.; Nam S.-J.; Lee S. K.; Oh K.-B.; et al. Discovery and biosynthesis of cihunamides, macrocyclic antibacterial RiPPs with a unique C-N linkage formed by CYP450 catalysis. Angew. Chem., Int. Ed. 2023, 62, e202300998. 10.1002/anie.202300998. [DOI] [PubMed] [Google Scholar]
- He B.-B.; Cheng Z.; Liu J.; Liu R.; Zhong Z.; Gao Y.; Liu H.; Li Y.-X.. Bacterial cytochrome P450-catalyzed post-translational macrocyclization. bioRxiv, May 8, 2023, ver. 1. 10.1101/2023.05.08.539676v1 (accessed 2023-05-28). [DOI] [PubMed]
- Repka L. M.; Chekan J. R.; Nair S. K.; van der Donk W. A. Mechanistic understanding of lanthipeptide biosynthetic enzymes. Chem. Rev. 2017, 117, 5457–5520. 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarksian R.; van der Donk W. A. Divergent evolution of lanthipeptide stereochemistry. ACS Chem. Biol. 2022, 17, 2551–2558. 10.1021/acschembio.2c00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulston L. C.; Bibb M. J. Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13461–13466. 10.1073/pnas.1008285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad H.; Aziz S.; Gehringer M.; Kramer M.; Straetener J.; Berscheid A.; Brötz-Oesterhelt H.; Gross H. Nocathioamides, uncovered by a tunable metabologenomic approach, define a novel class of chimeric lanthipeptides. Angew. Chem., Int. Ed. 2021, 60, 16472–16479. 10.1002/anie.202102571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffioli S. I.; Iorio M.; Sosio M.; Monciardini P.; Gaspari E.; Donadio S. Characterization of the congeners in the lantibiotic NAI-107 complex. J. Nat. Prod. 2014, 77, 79–84. 10.1021/np400702t. [DOI] [PubMed] [Google Scholar]
- Zimmermann N.; Metzger J. W.; Jung G. The tetracyclic lantibiotic actagardine 1H-NMR and 13C-NMR assignments and revised primary structure. Eur. J. Biochem. 1995, 228, 786–797. 10.1111/j.1432-1033.1995.tb20324.x. [DOI] [PubMed] [Google Scholar]
- Simone M.; Monciardini P.; Gaspari E.; Donadio S.; Maffioli S. I. Isolation and characterization of NAI-802, a new lantibiotic produced by two different Actinoplanes strains. J. Antibiot. 2013, 66, 73–78. 10.1038/ja.2012.92. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liu J.; Tang H.; Qiu Y.; Chen D.; Liu W. Discovery of new thioviridamide-like compounds with antitumor activities. Chin. J. Chem. 2019, 37, 1015–1020. 10.1002/cjoc.201900235. [DOI] [Google Scholar]
- Shi J.; Ma J.-Q.; Wang Y.-C.; Xu Z.-F.; Zhang B.; Jiao R.-H.; Tan R.-X.; Ge H.-M. Discovery of daspyromycins A and B, 2-aminovinyl-cysteine containing lanthipeptides, through a genomics-based approach. Chin. Chem. Lett. 2022, 33, 511–515. 10.1016/j.cclet.2021.06.010. [DOI] [Google Scholar]
- Franz L.; Adam S.; Santos-Aberturas J.; Truman A. W.; Koehnke J. Macroamidine formation in bottromycins is catalyzed by a divergent YcaO enzyme. J. Am. Chem. Soc. 2017, 139, 18158–18161. 10.1021/jacs.7b09898. [DOI] [PubMed] [Google Scholar]
- Sikandar A.; Franz L.; Melse O.; Antes I.; Koehnke J. Thiazoline-specific amidohydrolase PurAH is the gatekeeper of bottromycin biosynthesis. J. Am. Chem. Soc. 2019, 141, 9748–9752. 10.1021/jacs.8b12231. [DOI] [PubMed] [Google Scholar]
- Sikandar A.; Franz L.; Adam S.; Santos-Aberturas J.; Horbal L.; Luzhetskyy A.; Truman A. W.; Kalinina O. V.; Koehnke J. The bottromycin epimerase BotH defines a group of atypical α/β-hydrolase-fold enzymes. Nat. Chem. Biol. 2020, 16, 1013–1018. 10.1038/s41589-020-0569-y. [DOI] [PubMed] [Google Scholar]
- Adam S.; Franz L.; Milhim M.; Bernhardt R.; Kalinina O. V.; Koehnke J. Characterization of the sereoselective P450 enzyme BotCYP enables the in vitro biosynthesis of the bottromycin core scaffold. J. Am. Chem. Soc. 2020, 142, 20560–20565. 10.1021/jacs.0c10361. [DOI] [PubMed] [Google Scholar]
- Sohda K-y.; Nagai K.; Yamori T.; Suzuki K-i.; Tanaka A. YM-216391, a novel cytotoxic cyclic peptide from Streptomyces nobilis. J. Antibiot. 2005, 58, 27–31. 10.1038/ja.2005.2. [DOI] [PubMed] [Google Scholar]
- Skinnider M. A.; Johnston C. W.; Edgar R. E.; Dejong C. A.; Merwin N. J.; Rees P. N.; Magarvey N. A. Genomic charting of ribosomally synthesized natural product chemical space facilitates targeted mining. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E6343–E6351. 10.1073/pnas.1609014113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Z.-F.; Yang M.-J.; Li L.; Jian X.-H.; Yin Y.; Li D.; Pan H.-X.; Lu Y.; Jiang W.; Tang G.-L. Directed production of aurantizolicin and new members based on a YM-216391 biosynthetic system. Org. Biomol. Chem. 2018, 16, 9373–9376. 10.1039/C8OB02665C. [DOI] [PubMed] [Google Scholar]
- Kaweewan I.; Komaki H.; Hemmi H.; Hoshino K.; Hosaka T.; Isokawa G.; Oyoshi T.; Kodani S. Isolation and structure determination of a new cytotoxic peptide, curacozole, from Streptomyces curacoi based on genome mining. J. Antibiot. 2019, 72, 1–7. 10.1038/s41429-018-0105-4. [DOI] [PubMed] [Google Scholar]
- Kanoh K.; Matsuo Y.; Adachi K.; Imagawa H.; Nishizawa M.; Shizuri Y. Mechercharmycins A and B, cytotoxic substances from marinederived Thermoactinomyces sp. YM3–251. J. Antibiot. 2005, 58, 289–292. 10.1038/ja.2005.36. [DOI] [PubMed] [Google Scholar]
- Shin-ya K.; Wierzba K.; Matsuo K.; Ohtani T.; Yamada Y.; Furihata K.; Hayakawa Y.; Seto H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. 10.1021/ja005780q. [DOI] [PubMed] [Google Scholar]
- Pei Z.-F.; Yang M.-J.; Zhang K.; Jian X.-H.; Tang G.-L. Heterologous characterization of mechercharmycin A biosynthesis reveals alternative insights into post-translational modifications for RiPPs. Cell Chem. Biol. 2022, 29, 650–659. 10.1016/j.chembiol.2021.08.005. [DOI] [PubMed] [Google Scholar]
- Noike M.; Matsui T.; Ooya K.; Sasaki I.; Ohtaki S.; Hamano Y.; Maruyama C.; Ishikawa J.; Satoh Y.; Ito H.; Morita H.; Dairi T. A peptide ligase and the ribosome cooperate to synthesize the peptide pheganomycin. Nat. Chem. Biol. 2015, 11, 71–76. 10.1038/nchembio.1697. [DOI] [PubMed] [Google Scholar]
- Chen H.; Tseng C. C.; Hubbard B. K.; Walsh C. T. Glycopeptide antibiotic biosynthesis: enzymatic assembly of the dedicated amino acid monomer (S)-3,5-dihydroxyphenylglycine. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 14901–14906. 10.1073/pnas.221582098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H.; Hallen-Adams H. E.; Lüli Y.; Sgambelluri R. M.; Li X.; Smith M.; Yang Z. L.; Martin F. M. Genes and evolutionary fates of the amanitin biosynthesis pathway in poisonous mushrooms. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2201113119. 10.1073/pnas.2201113119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velden N. S.; Kalin N.; Helf M. J.; Piel J.; Freeman M. F.; Kunzler M. Autocatalytic backbone N-methylation in a family of ribosomal peptide natural products. Nat. Chem. Biol. 2017, 13, 833–835. 10.1038/nchembio.2393. [DOI] [PubMed] [Google Scholar]
- Ramm S.; Krawczyk B.; Mühlenweg A.; Poch A.; Mösker E.; Süssmuth R. D. A self-sacrificing N-methyltransferase is the precursor of the fungal natural product omphalotin. Angew. Chem., Int. Ed. 2017, 56, 9994–9997. 10.1002/anie.201703488. [DOI] [PubMed] [Google Scholar]
- Umemura M.; Nagano N.; Koike H.; Kawano J.; Ishii T.; Miyamura Y.; Kikuchi M.; Tamano K.; Yu J.; Shin-ya K.; Machida M. Characterization of the biosynthetic gene cluster for the ribosomally synthesized cyclic peptide ustiloxin B in Aspergillus flavus. Fungal Genet. Biol. 2014, 68, 23–30. 10.1016/j.fgb.2014.04.011. [DOI] [PubMed] [Google Scholar]
- Ye Y.; Minami A.; Igarashi Y.; Izumikawa M.; Umemura M.; Nagano N.; Machida M.; Kawahara T.; Shin-ya K.; Gomi K.; et al. Unveiling the biosynthetic pathway of the ribosomally synthesized and post-translationally modified peptide ustiloxin B in filamentous fungi. Angew. Chem., Int. Ed. 2016, 55, 8072–8075. 10.1002/anie.201602611. [DOI] [PubMed] [Google Scholar]
- Mahanta N.; Hudson G. A.; Mitchell D. A. Radical S adenosylmethionine enzymes involved in RiPP biosynthesis. Biochemistry 2017, 56, 5229–5244. 10.1021/acs.biochem.7b00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjdia A.; Balty C.; Berteau O. Radical SAM enzymes in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs). Front. Chem. 2017, 5, 87. 10.3389/fchem.2017.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincannon W. M.; Bandarian V.. 5.20 - Radical SAM enzymes involved in modifications of RiPP natural products. In Comprehensive Natural Products III, Liu H.-W., Begley T. P., Eds.; Elsevier, 2020; pp 489–519. [Google Scholar]
- Benjdia A.; Berteau O. Radical SAM enzymes and ribosomally-synthesized and post-translationally modified peptides: a growing importance in the microbiomes. Front. Chem. 2021, 9, 678068. 10.3389/fchem.2021.678068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendauletova A.; Kostenko A.; Lien Y.; Latham J. How a subfamily of radical S-adenosylmethionine enzymes became a mainstay of ribosomally synthesized and post-translationally modified peptide discovery. ACS Bio Med. Chem. Au 2022, 2, 53–59. 10.1021/acsbiomedchemau.1c00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M.-C.; Zou Y.; Watanabe K.; Walsh C. T.; Tang Y. Oxidative cyclization in natural product biosynthesis. Chem. Rev. 2017, 117, 5226–5333. 10.1021/acs.chemrev.6b00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y.; Fu H.; Hyster T. K. Activation modes in biocatalytic radical cyclization reactions. J. Ind. Microbiol. Biot. 2021, 48, kuab021. 10.1093/jimb/kuab021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick J. B.; Duffus B. R.; Duschene K. S.; Shepard E. M. Radical S-adenosylmethionine enzymes. Chem. Rev. 2014, 114, 4229–4317. 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T.; Minami A.; Oikawa H. Recent advances in the biosynthesis of ribosomally synthesized and posttranslationally modified peptides of fungal origin. J. Antibiot. 2023, 76, 3–13. 10.1038/s41429-022-00576-w. [DOI] [PubMed] [Google Scholar]
- Ren H.; Dommaraju S. R.; Huang C.; Cui H.; Pan Y.; Nesic M.; Zhu L.; Sarlah D.; Mitchell D. A.; Zhao H. Genome mining unveils a class of ribosomal peptides with two amino termini. Nat. Commun. 2023, 14, 1624. 10.1038/s41467-023-37287-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R.; Oberg N.; Gerlt J. A. The EFI web resource for genomic enzymology tools: leveraging protein, genome, and metagenome databases to discover novel enzymes and metabolic pathways. Biochemistry 2019, 58, 4169–4182. 10.1021/acs.biochem.9b00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberg N.; Zallot R.; Gerlt J. A. EFI-EST, EFI-GNT, and EFI-CGFP: Enzyme function initiative (EFI) web resource for genomic enzymology tools. J. Mol. Biol. 2023, 435, 168018. 10.1016/j.jmb.2023.168018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayikpoe R. S.; Zhu L.; Chen J. Y.; Ting C. P.; van der Donk W. A. Macrocyclization and backbone rearrangement during RiPP biosynthesis by a SAM-dependent domain-of-unknown-function 692. ACS Cent. Sci. 2023, 9, 1008–1018. 10.1021/acscentsci.3c00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.