

Summary
HBV is a small, enveloped DNA virus that replicates by reverse transcription via RNA intermediate. Current anti-HBV treatment regiments that include interferon α and nucleos(t)ide analogs have insufficient efficiency, are of long duration and can be accompanied by systemic side-effects. The HBV RNaseH is essential for viral replication, however is unexploited as a drug target against HBV. RNaseH inhibitors that actively block viral replication would represent an important addition to the potential new drugs for treating HBV infection. Here we describe two methods to measure the activity of RNaseH inhibitors. DNA oligonucleotide-directed RNA cleavage assay allows low-throughput screening of compounds for potential anti-HBV RNaseH activity in vitro. Analysis of preferential inhibition of plus-polarity DNA strand synthesis by HBV RNaseH inhibitors in cell culture model of HBV replication can be used to validate the efficiency of these compounds to block viral replication.
Keywords: Hepatitis B virus, Ribonuclease H, Inhibitors, DNA-oligonucleotide directed RNA cleavage assay, HBV core particle DNA, quantitative PCR
1. Introduction
The hepatitis B virus (HBV) is small, enveloped DNA virus that replicates by reverse transcription via RNA intermediate (1–3). The encapsidated viral genome consists of a 3.2 kb partially double-stranded relaxed circular DNA (rcDNA). HBV is hepatotropic virus and upon hepatocyte entry the DNA-containing core particle is translocated into the nucleous where the rcDNA is converted into a covalently closed circular DNA (cccDNA). The cccDNA is the template for viral mRNA and pre-genomic RNA synthesis (4) and is responsible for HBV persistence (5). It is believed that persistence of cccDNA in infected cell is the major mechanism of HBV chronicity (6, 7).
HBV infection remains a major public health problem despite the availability of a prophylactic vaccine (8–10). More than 250 million people worldwide are chronically infected with HBV (11) and are at an increased risk for developing end-stage liver disease and hepatocellular carcinoma (12).
Treatment of HBV infection include the use of IFN-α and nucleos(t)ide analog chain terminators such as lamivudine, adefovir, tenofovir disoproxil fumarate and entecavir (13, 14). However, while these drugs can slow progression of HBV-induced disease, they are only partially effective and only very rarely lead to a full elimination of viral infection (15–17). For example, the first direct-acting anti-HBV drug, lamivudine, is very effective and well tolerated (18). However after 5 years of therapy 75% of the treated patients develop resistant HBV strains (19). Moreover, due to the overlapping polymerase and HBsAg open reading frames some of the resistance mutations also led to mutations to the HBsAg that can cause reduced binding to anti-HBs antibody (20). Clearly, new drugs with different targets and mechanisms for anti-HBV therapy are urgently needed.
Promising therapeutic approaches that directly target the virus-infected cells, as well as immunotherapeutic strategies that activate the HBV-specific adaptive immune response or innate intrahepatic immunity are being developed (9, 21–27). The HBV polymerase, the only protein with enzymatic activity encoded by the virus, is a multifunctional enzyme. The reverse transcriptase (RT) domain possesses nucleotidyl transfer activity and synthetizes viral DNA during reverse transcription of single-stranded pre-genomic RNA to rcDNA. The RNaseH domain of the HBV polymerase hydrolyzes the RNA strand of RNA/DNA hybrids that are generated during reverse transcription to enable synthesis of double-stranded DNA. Both activities are necessary for viral replication, however, the currently clinically available direct-acting anti-HBV drugs – the nucleos(t)ide analogs – target HBV RT, whereas RNaseH inhibitors are yet to be developed. Therefore, RNaseH is an attractive target for new anti-HBV drugs that might provide new approach to treat patients resistant to currently available anti-HBV therapies as well as may be used in combination with current drugs to increase effectiveness of treatment and prevent the development of resistance (28). Recently we developed a low throughput screening strategy using recombinant HBV RNaseH and identified several promising compounds that effectively inhibit both HBV RNaseH activity and viral replication (29–33).
This chapter describes two protocols for measuring activity of HBV RNaseH inhibitors. The first is used for initial screening of compounds for anti-HBV RNaseH activity in vitro in a DNA oligonucleotide-directed RNA cleavage assay (29, 33). In this assay, the activity of recombinant HBV RNaseH is determined by its ability to cleave a 32P-labeled RNA substrate in the form of an RNA:DNA heteroduplex. The RNA fragments products of the reaction are then resolved by urea polyacrylamide gel electrophoresis and detected by autoradiography (Figure 1A). Inhibition of RNaseH activity is assessed by quantifying the intensity of cleaved RNA bands after compound addition to the reaction mixture and comparing to the activity of vehicle-treated enzyme (Figure 1B and 1C). The second protocol is used to measure inhibition of HBV replication in cell culture by RNaseH inhibitors (33). This method includes isolation of HBV core particles DNA and measurement of preferential inhibition of plus-polarity DNA strand synthesis by quantitative PCR (Figure 2). This is because during HBV replication inside of viral capsid particles the plus-polarity DNA can only be made when the RNaseH removes the pgRNA from newly synthetized minus-polarity DNA strand and generates the short RNA primer for synthesis of plus-polarity DNA strand (34–37). Thus, the amount of plus-polarity DNA is strictly dependent on HBV RNaseH activity. In contrast, synthesis of the minus-polarity DNA strand is largely unaffected by RNaseH inhibitors (29–32, 38). Antiviral activity of RNaseH inhibitors is tested by treating cells replicating HBV (e.g. HepDES19 cells, which contain a tetracycline-repressible expression cassette for a replication-competent HBV genotype D genome (39)) with the compounds, and determining the amount of HBV minus- and plus-polarity DNA strands in core particles (31, 32).
Figure 1. Oligonucleotide-directed RNA cleavage assay.
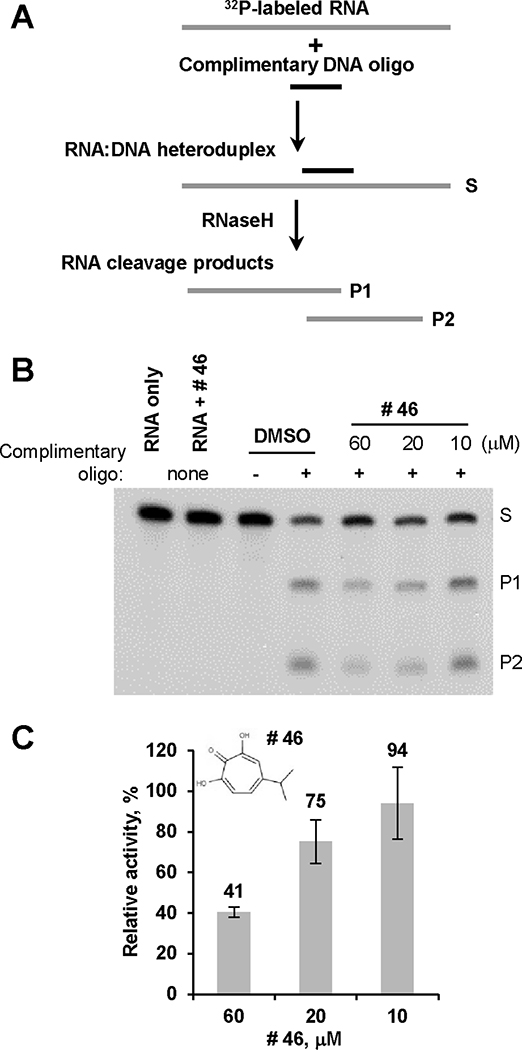
A. Schematic representation of oligonucleotide-directed RNA cleavage assay. Internally radiolabeled RNA is bound to a complementary DNA oligonucleotide to form the RNA:DNA heteroduplex substrate for RNaseH. The RNaseH cleaves the RNA within the RNA:DNA heteroduplexes. RNA, grey line; DNA, black line; S, substrate; P1 and P2, RNaseH cleavage products of RNA. B. Example of primary compound screening assay for HBV RNaseH inhibition by an oligonucleotide-directed RNA cleavage assay. Activity of HBV RNaseH is assessed in the presence of different concentration of compound of interest (compound # 46, β-thujaplicinol is shown) or in the presence of DMSO as a vehicle control. Reaction products are resolved by denaturing polyacrylamide electrophoresis and detected by autoradiography. S, substrate, 32P-labeled 264 nt RNA derived from the Duck Hepatitis B Virus genome (DRF+ RNA); P1, cleavage product 1; P2, cleavage product 2; “-”, a non-complementary DNA oligonucleotide as a specificity control; “+ ”, complementary DNA oligonucleotide; RNA only, includes RNA but no enzyme as a control for RNA quality; RNA + #46, control for compound solution purity and the lack of compound-induced non-specific RNA cleavage, no enzyme present. C. Example of quantitation of compound anti-HBV RNaseH activity. The intensity of P2 band was quantified using ImageJ. Non-specific background values were determined from the incorrect oligonucleotide negative control lane and subtracted from all experimental values. Relative activity of HBV RNaseH is presented as the percentage of intensity of P2 band with compound present in the reaction relative to the intensity of P2 band in the control vehicle-containing reaction. Data are mean values ± one standard deviation from three independent experiments. Insert, structure of compound #46, β-thujaplicinol.
Figure 2. Replication inhibition assay.
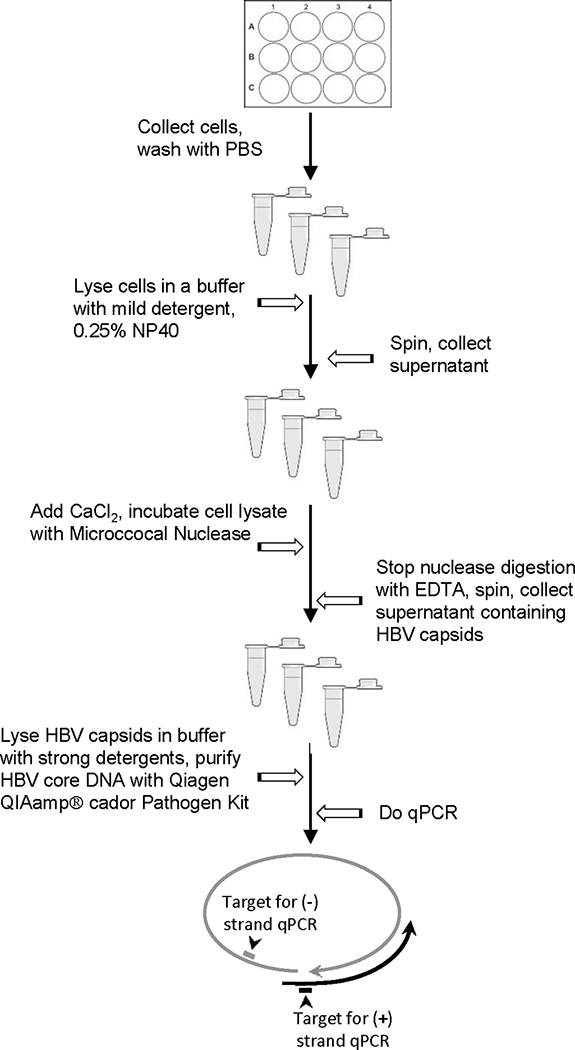
Plus-polarity DNA is measured by amplifying HBV DNA across the gap in the minus-polarity DNA strand. Minus-polarity DNA strands are measured by amplifying sequences downstream of the 3’ end of the vast majority of plus-polarity DNA strands in viral capsids.
2.1. In Vitro Enzymatic Assay for Screening HBV RNaseH Inhibitors
2.2. Materials
10% dimethyl sulfoxide (DMSO).
Diethylpyrocarbonate (DEPC) H2O (see Note 1).
10x RH buffer: 500 mM Tris-HCl, pH 7.5, 50 mM MgCl2, 1000 mM NaCl, 20 mM DTT (see Note 2).
0.5% octylphenoxy poly(ethyleneoxy)ethanol (NP-40).
RNAseOut™ Recombinant Ribonuclease Inhibitor (Life Technologies, Cat. # 10777-019).
5x TBE buffer: 1.1 M Tris-HCl, 900 mM Boric Acid, 25 mM EDTA, pH 8.3.
-
in vitro-transcribed using MEGAscript® Kit (Ambion) 32P labelled RNA (DRF+) (40), sequence:
5’GGGAACAAAAGCUUGCAUGCCUGCAGGUCGACUCUAGAGGAUCCCCACUUUGUCCCGAGCAAAUAUAAUCCUGCUGACGGCCCAUCCAGGCACAGACCGCCUGAUUGGACGGCUUUUCCAUACACCCCUCUCUCGAAAGCAAUAUAUAUUCCACAUAGGCUAUGUGGAACUUAAGAAUUACACCCCUCUCCUUCGGAGCUGCUUGCCAAGGUAUCUUUACGUCUACAUUGCUGUUGUCGUGUGUGACUGUGGGUACCGAGCUCG’ (see Note 3).
Complimentary DNA oligo, D2507: 5’GTTCCACATAGCCTATGTGG3’; non-complimentary control DNA oligo, D2526: 5’CCACATAGGCTATGTGGAAC3’; 1 μg/μL (see Note 4).
6% Sequencing Acrilamide solution, for 100 mL: 5x TBE, 20 mL,5.7 g acrylamide, 0.3 g N,N’-methylenebisacrilamide, 48 g urea, H2O to 100 mL (see Note 5).
Ammonium persulfate (APS): 10% solution in water (see Note 6).
N,N,N′,N′-Tetramethylethylenediamine (TEMED).
Sequencing Loading Buffer: 98% Formamide, 10 mM EDTA, 0.025% xylen cyanol, 0.025% bromphenol blue (see Note 6).
2.3. Methods
2.3.1. Oligonucleotide-directed RNA cleavage assay
Thaw frozen compound solutions. Vortex, then briefly centrifuge. Dilute test compound to 10x desired concentration in 10% DMSO in DEPC H2O.
-
Prepare Master Mix. Calculate the total volume required for each component: volume for 1 reaction × the total number of reactions.
Reaction mixture for one reaction:
3.5 μL DEPC H2O
2 μL 0.5% NP-40
2 μL 10x RH buffer
0.5 μL RNAseOut
3 μL DNA oligo, 1 μg/μL
2 μL Test compound or 10% DMSO
6 μL HBV RNaseH
1 μL RNA, 0.02 μg/μL.
Assemble Master Mix in the following order: H2O, NP-40, RH buffer, RNAseOut. Add all components to 1.5 mL microcentrifuge tube, vortex briefly to mix, then centrifuge the tubes briefly to spin down the contents and eliminate air bubbles.
Assemble reactions on ice. Aliquot 8 μL Master Mix to each reaction tube. Add 3 μL of the appropriate DNA oligo to each reaction tube. Add 2 μL test compound or DMSO to each reaction tube. Add 6 μL HBV RNaseH protein to each reaction tube (see Note 7). Vortex tubes to mix, then briefly centrifuge the tubes. Initiate the reaction by adding 1 μL of 32P-radiolaballed RNA diluted 10x with DEPC H2O immediately before the experiment to each reaction tube, so that the final concentration of RNA in the reaction is 0.001 μg/μL. Vortex briefly to mix, then centrifuge the tubes briefly to spin down the contents and eliminate air bubbles (see Note 8).
Incubate reactions at 42°C for 90 min.
Stop the 20 μL RNAseH reactions with 80 μL 1x formamide loading buffer per reaction.
2.3.2. Denaturing Polyacrylamide Gel Electrophoresis
Warm the 6% Sequencing Acrylamide solution (6% acrylamide/bis-acrylamide [19:1]/6M urea in 1x TBE) to near room temperature and check to be sure the urea has not crystallized before pouring a gel.
Set up a vertical mini-gel. Prepare 6% Sequencing Acrylamide mix – for each 10 cm × 11 cm mini-gel mix 20 mL 6% Sequencing Acrylamide solution, 150 μL 10% APS and 20 μL TEMED. Pour a single phase gel and insert the comb. Let the gel solidify at room temperature, the gel will be ready to use in approximately 40 minutes.
Remove casting clamps, rinse the plates carefully with dH2O and remove the comb. Mount the gel in a mini-gel rig, using 1x TBE as the running buffer. Immediately rinse the residual unpolymerized acrylamide and urea from the wells using a syringe and needle.
Pre-run the gel for 5 min. at 40 mA (220–230V).
Heat samples to >90°C for 3–5 min. and chill them immediately on ice. Rinse the wells in the gel again with a syringe and needle to remove urea that has diffused into the wells. Promptly load 50 μL of sample per well.
Electrophorese at 40 mA (220–230V) until bromophenol blue is near the bottom of the gel. Cut the bromophenol blue band from the bottom of the gel and discard it in the radioactive waste.
Soak gel in dH2O with shaking for 30 min. to remove the urea. Change the water 2 or 3 times during the soaking phase.
Dry the gel at 80°C under vacuum for 1 hour.
Detect radiolabeled RNA by standard autoradiography. Analyze film images and quantitate using ImageJ software (http://rsb.info.nih.gov/ij/).
Calculate inhibition activity of compounds as the percentage of intensity of P2 band (Figure 1B) with compound present in the reaction relative to the intensity of P2 band in the control vehicle containing reaction as follows: % Inhibition = 100% − [(relative intensity of P2 band in the presence of inhibitor/relative intensity of P2 band in control vehicle treated sample)*100%] (see Note 9).
3.1. Cell culture based assay for measuring antiviral activity of HBV RNaseH inhibitors
3.2. Materials
1 M Tris-HCl, pH 7.5.
0.5 M ethylenediamine tetraacetic acid (EDTA), pH 8.0.
5 M NaCl.
1 M CaCl2.
Core Preparation Lysis Buffer: 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 0.25% octylphenoxy poly(ethyleneoxy)ethanol (NP-40), 50 mM NaCl, and 8% sucrose.
Micrococcal Nuclease (MN) (2000 U/μL, NEB #M0247S), MN buffer (NEB #B0247S).
QIAamp® cador® Pathogen Mini Kit (Qiagen, Cat. No 54104).
TaqMan® Fast Advanced Master Mix (Applied Biosystems, Cat. No 4444556).
Primers and probe (IDT Inc.) for the plus-polarity HBV DNA strand: 5’CATGAACAAGAGATGATTAGGCAGAG3’; 5’GGAGGCTGTAGGCATAAATTGG3’; 5’/56-FAM/CTGCGCACC/ZEN/AGCACCATGCA/3IABkFQ.
Primers and probe for the minus-polarity HBV DNA strand: 5’GCAGATGAGAAGGCACAGA3’; 5’CTTCTCCGTCTGCCGTT3’; 5’/56-FAM/AGTCCGCGT/ZEN/AAAGAGAGGTGCG/3IABkFQ.
3.3. Methods
3.3.1. Cell Culture and Compound Treatment
Day 0. Plate 0.3 × 106 HepDES19 (tetracycline-inducible, tet-off) cells (39) per well in 12-well plates in 2 mL of DMEM/F12 supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 U/mL streptomycin plus 1 μg/mL tissue-culture grade tetracycline.
Day 1. 24 hours later replace medium with 2 mL fresh medium without tetracycline to induce HBV replication.
Day 3. 48 hours later replace medium with 1 mL medium without tetracycline containing the desired concentration of the test compounds. Balance the dimethyl sulfoxide (DMSO) concentration for all samples and limit DMSO to ≤ 1% v/v. Also use DMSO as vehicle control, final concentration ≤ 1 %.
Day 4. Repeat compound treatment. Change the medium 24 hours later with medium containing the test compounds without tetracycline (see Note 10).
Day 5. Repeat compound treatment. Change the medium 24 hours later with medium containing the test compounds without tetracycline.
Day 6. 24 hours later harvest the cells. Aspirate the medium from the wells. Wash cells two times with phosphate-buffered saline (PBS), add 0.5 mL of trypsin (0.05%), incubate 5 min at 37°C, and collect cells into 1.5 mL tubes. Spin cells at room temperature, 2 min at 2000 × g. Carefully aspirate supernatant, wash cell pellets with 1 mL PBS. Again, spin cells at room temperature, 2 min at 2000 × g. Carefully aspirate supernatant, and loosen the pellet by vortexing.
3.3.2. Extraction of HBV core particles DNA
Add 0.2 mL Core Preparation Lysis Buffer to the cell suspensions to lyse the cells, vortex thoroughly, and incubate at 37° C for 10 min (see Note 11).
Add 2 μL of 1M CaCl2 to each tube (see Note 12).
Centrifuge at 21,000 × g for 5 min at room temperature. Transfer supernatants into new tubes.
Prepare 80 U/μL solution of Micrococcal Nuclease in MN buffer. Add 5 μL of Micrococcal Nuclease solution to each tube. Incubate at 37° C for 60 min (see Note 13).
Centrifuge at 21,000 × g for 5 min at room temperature. Transfer supernatants into new tubes.
Terminate nuclease digestion by adding 20 μL of 0.5 M EDTA per tube. Mix carefully (see Note 14).
-
Extract nucleic acid from core particles using the Qiagen QIAamp® cador® Pathogen Mini Kit according to the manufacturer’s instructions with some modifications described below in steps 7.1 – 7.3.
7.1 Add 20 μL proteinase K into each tube with core particles, vortex, and then briefly centrifuge.
7.2 Add 100 μL Buffer VXL with Carrier RNA in it (1 μg Carrier RNA per 100 μL Buffer VXL) into each tube, vortex.
7.3 Incubate overnight (16 hours) at 37° C (see Note 15).
7.4 Proceed according to the protocol from QIAamp® cador® Pathogen Mini Handbook, p. 21 (https://www.qiagen.com/us/resources/resourcedetail?id=dd291a2f-f2ff-4768-ad97-8e2462fe58c0&lang=en), starting at step 6 of the manufacturer’s protocol.
Elute core-associated nucleic acids with 50 μL buffer AVE. Store at −75° C.
3.3.3. Real-Time qPCR
Thaw frozen HBV core DNA samples on ice. Vortex, then briefly centrifuge.
Thoroughly mix the TaqMan® Fast Advanced Master Mix.
-
Calculate the total volume required for each component: volume for 1 reaction × the total number of reactions.
Reaction mixture for one reaction:
10 μL TaqMan Universal PCR Master Mix
0.2 μL Forward Primer, 20 μM
0.2 μL Reverse Primer, 20 μM
0.4 μL TaqMan probe, 2 μM
5.2 μL H2O
4 μL sample DNA.
Prepare two PCR Master Mix solutions (one for measuring (+) polarity strand of HBV core particles DNA and the second one for measuring (–) polarity DNA). Add all components, except for sample DNA, to 1.5 mL microcentrifuge tubes, vortex briefly to mix, then centrifuge the tubes briefly to spin down the contents and eliminate air bubbles.
Add the appropriate volume of PCR Master Mix (16 μL) to each well of an optical reaction plate (MicroAmp Fast Optical 96-well reaction plate, Applied Biosystems, Cat. No 4346906).
Add 4 μL of HBV core DNA samples or standard HBV DNA (see Note 16) to each well, cover the reaction plate with Optical Adhesive Covers (Applied Biosystems, Cat. No 4360954), then centrifuge the plate briefly to spin down the contents and eliminate air bubbles.
Run the reaction. For the Applied Biosystems Real-Time PCR 7500 Fast system instrument, use the following parameters: 95°C for 10’, 40 cycles at 95°C for 15”, 60°C for 1’, and detection at 60°C for 1” after each cycle.
Calculate the amount (+) and (–) polarity strands DNA as the percentage relative to the quantity of DNA in DMSO-treated cells to estimate inhibitory activity of compounds (Figure 3).
Figure 3. Inhibition of HBV replication by RNaseH inhibitors.
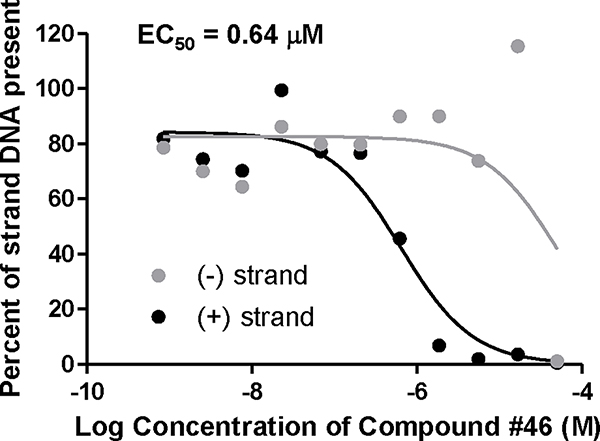
Inhibition of replication by compound #46 was measured against an HBV genotype D isolate in HepDES19 cells. The EC50 value was calculated based on the decline of the plus-polarity DNA strand with GraphPad Prism using three-parameter log(inhibitor) vs. response non-linear curve.
Acknowledgements
Writing of this article was supported in part by R01 AI104494 to J.T. and an ancillary study within the NIDDK-sponsored Hepatitis B Virus Research Network (U01 DK082871 to Adrian Di Bisceglie and J.T.). We thank all previous and past members of Tavis lab for their many contributions to the development and validation of the methods described here.
Notes
DEPC treatment is the most commonly used method for eliminating RNase contamination from water and other solutions. DEPC destroys enzymatic activity by modifying –NH, -SH, and –OH groups in RNases and other proteins. To prepare DEPC water, add 1 mL of DEPC to 1L of H2O. Shake well to disperse the DEPC through the H2O. Incubate at room temperature for at least 12 hours, then autoclave on liquid cycle for 20 min to inactivate the remaining DEPC.
Prepare 10x RH buffer in relatively large batches and freeze aliquots at −20°C.
Use standard protocol provided by manufacturer for in vitro transcription and radiolabel RNA with [32P]UTP. Please refer to the reference (40) for further information. Purify in vitro transcribed radiolabeled RNA using RNeasy MinElute™ Cleanup Kit (Qiagen). Determine concentration of RNA fluorimetrically using QuantiFluor® RNA System (Promega) and dilute to 0.2 μg/μL. Check quality by agarose electrophoresis. Store radiolabeled RNA at −75°C in small aliquots.
You may use any other RNA and complimentary DNA oligonucleotide. Non-complimentary oligonucleotide is included in the assays to determine specificity of RNaseH reaction for heteroduplexes and exclude the potential contribution of contaminating nucleases to the RNA cleavage reaction.
Unpolymerized acrylamide is a neurotoxin. Always wear personal protective equipment when handling acrylamide, avoid skin contact. Filter the acrylamide-urea solution before storage. Store at 4°C protected from light. Urea can precipitate from the solution at 4°C, warm the solution to dissolve urea crystals before preparing the gel. For more details about urea-PAGE see (41).
Store frozen at −20°C in small aliquots.
We expressed the HBV RNaseH in E. coli as a C-terminally hexahistidine-tagged protein (29) or as a maltose-binding protein fusion with a hexahistidine tag at its C-terminus (33). The HBV RNaseH is purified by nickel-affinity chromatography (29), dialyzed in buffer containing 50 mM Hepes, pH 7.3, 0.3 M NaCl, 20% glycerol, and 5mM DTT, and stored under liquid nitrogen in small aliquots.
We recommend including a control reaction without HBV RNaseH into each experiment to monitor the quality of RNA and any unspecific cleavage that might occur due to contamination.
Quantitation of P1 band intensity is unreliable due to a 3’–5’ exonuclease activity of RNaseH.
RNaseH inhibitors can be added only once at day 3 if desired. In this case cells are incubated for two more days without changing the media, and are collected as described at day 6. However, false negative results can arise from instability of a compound at 37°C.
Core Preparation Lysis Buffer is very mild cell lysis buffer that contain 0.25% of NP-40 and will only lyse cellular membrane but not affect HBV nucleocapsids that are resistant to NP-40 detergent (42) or the nucleus. Consequently, low speed centrifugation will separate the lysates from nucleus fraction and unbroken cells.
CaCl2 is needed for subsequent treatment with Micrococcal nuclease which activity is dependent on Ca2+ (43).
Micrococcal nuclease is a relatively non-specific endo-exonuclease that digests double-stranded, single-stranded, circular and linear RNA and DNA substrates. The enzyme is active in the pH range of 7.0 – 10.0 as long as salt concentration is less than 100 mM (43). Micrococcal nuclease treatment is necessary to remove any non-encapsidated RNA and DNA from the lysate that might later interfere with qPCR. HBV core DNA which is protected from Micrococcal nuclease digestion by the viral capsid.
Addition of excess EDTA is necessary to inactivate Micrococcal nuclease.
Buffer VXL contains strong detergents that will lyse HBV core particles and release rcDNA. Proteinase K will degrade proteins in the lysate as it exhibits broad substrate specificity and is active in the presence of detergents, and is routinely used to digest unwanted proteins in DNA preparations (44). We use a long incubation time with Proteinase K to ensure the cleavage of HBV polymerase protein which is covalently attached to the (–) polarity strand HBV DNA. If not removed by digestion with protease, the HBV polymerase that is covalently bound to DNA causes binding to purification column and hence incomplete DNA elution (45).
Double-stranded HBV DNA template for a standard curve is prepared by isolating total DNA from HepDES19 cells using QIAamp DNA Mini Kit (Qiagen, Cat. No 51304) and amplifying 1388 base pairs genomic region of HBV DNA with primers H2900+ (5’-CCGCTTGGGACTCTCTCGTCCC-3’) and HPE180STOP- (5’-TCACCATGGGAAGCTTACTCTTGTTCCCAAGAATATGG-3’).
References
- 1.Summers J, Mason WS (1982) Replication of the genome of a hepatitis B--like virus by reverse transcription of an RNA intermediate. Cell 29, 403–415 [DOI] [PubMed] [Google Scholar]
- 2.Nassal M (2008) Hepatitis B viruses: reverse transcription a different way. Virus Res 134, 235–249 [DOI] [PubMed] [Google Scholar]
- 3.Gerlich WH (2013) Medical virology of hepatitis B: how it began and where we are now. Virol J 10, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seeger C, Mason WS (2015) Molecular biology of hepatitis B virus infection. Virology 10.1016/j.virol.2015.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caruntu FA, Molagic V (2005) CccDNA persistence during natural evolution of chronic VHB infection. Rom J Gastroenterol 14, 373–377 [PubMed] [Google Scholar]
- 6.Werle-Lapostolle B, Bowden S, Locarnini S, et al. (2004) Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 126, 1750–1758 [DOI] [PubMed] [Google Scholar]
- 7.Nassal M (2015) HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 10.1136/gutjnl-2015-309809 [DOI] [PubMed] [Google Scholar]
- 8.Lavanchy D (2005) Worldwide epidemiology of HBV infection, disease burden, and vaccine prevention. J Clin Virol 34 Suppl 1, S1–3 [DOI] [PubMed] [Google Scholar]
- 9.Bertoletti A, Rivino L (2014) Hepatitis B: future curative strategies. Curr Opin Infect Dis 27, 528–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komatsu H (2014) Hepatitis B virus: where do we stand and what is the next step for eradication? World J Gastroenterol 20, 8998–9016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ott JJ, Stevens GA, Groeger J, et al. (2012) Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30, 2212–2219 [DOI] [PubMed] [Google Scholar]
- 12.Ganem D, Prince AM (2004) Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med 350, 1118–1129 [DOI] [PubMed] [Google Scholar]
- 13.Zoulim F, Perrillo R (2008) Hepatitis B: reflections on the current approach to antiviral therapy. J Hepatol 48 Suppl 1, S2–19 [DOI] [PubMed] [Google Scholar]
- 14.Zoulim F, Locarnini S (2013) Optimal management of chronic hepatitis B patients with treatment failure and antiviral drug resistance. Liver Int 33 Suppl 1, 116–124 [DOI] [PubMed] [Google Scholar]
- 15.Zoulim F, Locarnini S (2009) Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 137, 1593–1608 e1591–1592 [DOI] [PubMed] [Google Scholar]
- 16.Scaglione SJ, Lok AS (2012) Effectiveness of hepatitis B treatment in clinical practice. Gastroenterology 142, 1360–1368 e1361 [DOI] [PubMed] [Google Scholar]
- 17.Liaw YF (2013) Impact of therapy on the outcome of chronic hepatitis B. Liver Int 33 Suppl 1, 111–115 [DOI] [PubMed] [Google Scholar]
- 18.Dienstag JL, Perrillo RP, Schiff ER, et al. (1995) A preliminary trial of lamivudine for chronic hepatitis B infection. N Engl J Med 333, 1657–1661 [DOI] [PubMed] [Google Scholar]
- 19.Petersen J, Buti M (2012) Considerations for the long-term treatment of chronic hepatitis B with nucleos(t)ide analogs. Expert Rev Gastroenterol Hepatol 6, 683–693; quiz 694 [DOI] [PubMed] [Google Scholar]
- 20.Torresi J, Locarnini S (2000) Antiviral chemotherapy for the treatment of hepatitis B virus infections. Gastroenterology 118, S83–103 [DOI] [PubMed] [Google Scholar]
- 21.Iwamoto M, Watashi K, Tsukuda S, et al. (2014) Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem Biophys Res Commun 443, 808–813 [DOI] [PubMed] [Google Scholar]
- 22.Watashi K, Urban S, Li W, et al. (2014) NTCP and beyond: opening the door to unveil hepatitis B virus entry. Int J Mol Sci 15, 2892–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucifora J, Xia Y, Reisinger F, et al. (2014) Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343, 1221–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang XY, Chen HS (2014) Emerging antivirals for the treatment of hepatitis B. World J Gastroenterol 20, 7707–7717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gebbing M, Bergmann T, Schulz E, et al. (2015) Gene therapeutic approaches to inhibit hepatitis B virus replication. World J Hepatol 7, 150–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seeger C, Sohn JA (2014) Targeting Hepatitis B Virus With CRISPR/Cas9. Mol Ther Nucleic Acids 3, e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kennedy EM, Bassit LC, Mueller H, et al. (2015) Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 476, 196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tavis JE, Gehring AJ, Hu Y (2013) How further suppression of virus replication could improve current HBV treatment. Expert Rev Anti Infect Ther 11, 755–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tavis JE, Cheng X, Hu Y, et al. (2013) The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes. PLoS Pathog 9, e1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Y, Cheng X, Cao F, et al. (2013) beta-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res 99, 221–229 [DOI] [PubMed] [Google Scholar]
- 31.Cai CW, Lomonosova E, Moran EA, et al. (2014) Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antiviral Res 108, 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu G, Lomonosova E, Cheng X, et al. (2015) Hydroxylated tropolones inhibit hepatitis B virus replication by blocking viral ribonuclease H activity. Antimicrob Agents Chemother 59, 1070–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tavis JE, Lomonosova E (2015) The hepatitis B virus ribonuclease H as a drug target. Antiviral Res 10.1016/j.antiviral.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Will H, Reiser W, Weimer T, et al. (1987) Replication strategy of human hepatitis B virus. J Virol 61, 904–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seeger C, Ganem D, Varmus HE (1986) Biochemical and genetic evidence for the hepatitis B virus replication strategy. Science 232, 477–484 [DOI] [PubMed] [Google Scholar]
- 36.Haines KM, Loeb DD (2007) The sequence of the RNA primer and the DNA template influence the initiation of plus-strand DNA synthesis in hepatitis B virus. J Mol Biol 370, 471–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tavis JE, Badtke MP (2009) Hepadnaviral Genomic Replication. In: Cameron CE, Gotte M, and D. RK (Ed.) Viral Genome Replication, Springer, New York, pp. 129–143 [Google Scholar]
- 38.Gerelsaikhan T, Tavis JE, Bruss V (1996) Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J Virol 70, 4269–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo H, Jiang D, Zhou T, et al. (2007) Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81, 12472–12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang C, Yu YT (2013) Synthesis and labeling of RNA in vitro. Curr Protoc Mol Biol Chapter 4, Unit4 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adachi H, Yu YT (2014) Purification of radiolabeled RNA products using denaturing gel electrophoresis. Curr Protoc Mol Biol 105, Unit 4 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi T, Nakagawa S, Hashimoto T, et al. (1976) Large-scale isolation of Dane particles from plasma containing hepatitis B antigen and deomnstration of circular double-stranded DNA molecule extruding directly from their cores. J Immunol 117, 1392–1397 [PubMed] [Google Scholar]
- 43.Cuatrecasas P, Fuchs S, Anfinsen CB (1967) Catalytic properties and specificity of the extracellular nuclease of Staphylococcus aureus. J Biol Chem 242, 1541–1547 [PubMed] [Google Scholar]
- 44.Gross-Bellard M, Oudet P, Chambon P (1973) Isolation of high-molecular-weight DNA from mammalian cells. Eur J Biochem 36, 32–38 [DOI] [PubMed] [Google Scholar]
- 45.Heermann KH, Gerlich WH, Chudy M, et al. (1999) Quantitative detection of hepatitis B virus DNA in two international reference plasma preparations. Eurohep Pathobiology Group. J Clin Microbiol 37, 68–73 [DOI] [PMC free article] [PubMed] [Google Scholar]