

Abstract
GLS4, a potent antiviral drug candidate, has been widely studied and entered into phase II clinical trials. Nevertheless, the therapeutic application of GLS4 is limited due to poor water solubility, short half-life, and low bioavailability. In order to improve the hydrophilicity and pharmacokinetic (PK) properties of GLS4, herein, we retained the dominant fragments, and used a scaffold hopping strategy to replace the easily metabolized morpholine ring of GLS4 with diverse sizes of spiro rings consisting of hydrogen bond donor and acceptor substituents. Potent in vitro anti-HBV activity and low cytotoxicity were observed for compound 4r (EC50 = 0.20 ± 0.00 μM, CC50 > 87.03 μM), which was more potent than the positive control lamivudine (EC50 = 0.37 ± 0.04 μM, CC50 > 100.00 μM) in this assay and was about a quarter as effective as GLS4 (EC50 = 0.045 ± 0.01 μM, CC50 > 99.20 μM). Preliminary structure-activity relationship (SAR) analysis and molecular docking studies were carried out to explore potential interactions and binding mode between compounds and target protein. In terms of the physicochemical properties, 4r was predicted to be consistent with the rule-of-five, which means 4r may have favourable absorption and permeation. Finally, ADMET and PK characteristics of 4r and GLS4 were predicted to be comparable in most aspects, implying that the two compounds may have similar profiles in vivo.
Keywords: HBV, Capsid protein inhibitors, Heteroaryldihydropyrimidine, Scaffold hopping, GLS4
1. Introduction
Hepatitis B is a severe infectious disease caused by the hepatitis B virus (HBV). HBV can be transferred through blood, mother to child, sexual contact, sharing of dirty needles, and skin mucosa impairment. Hepatitis B infection can cause acute or chronic viral hepatitis, where chronic infection leads to cirrhosis, liver failure, liver cancer or hepatocellular carcinoma[1]. The World Health Organization statistics revealed that, approximately 296 million people suffer from chronic HBV infection globally, accounting for about 4% of the world's population[2]. Every year, about 820,000 deaths occur due to chronic viral hepatitis associated liver diseases.
At present, HBV treatment mainly relies on interferon and nucleot(s)ide analogues. The six NRTIs (nucleoside reverse transcriptase inhibitors) approved by the FDA for HBV treatment are shown in Figure 1, but neither of these two types of therapies can completely resolve HBV infections. Tolerance of interferon is poor and treatment rarely clears the virus from patients. Additionally, when nucleot(s)ide analogues are withdrawn, viral tiers often rebound to pre-treatment levels. Therefore, research on promising HBV inhibitors is a top priority.
Figure 1.

The chemical structures of U.S. FDA approved anti-HBV NRTIs.
HBV inhibitors currently in clinical and pre-clinical research target key steps in the HBV replicative cycle. Notably, capsid (core) protein allosteric modulators (CpAM) have attracted significant attention due to the versatility of its targets. Cp (core protein) plays key roles in each stage of the HBV replicative cycle, including subcellular transport and release of the HBV genome[3], pre-genomic RNA encapsidation and viral DNA synthesis[4], capsid assembly and transport[5]. Moreover, recent findings suggest that Cp can regulate cccDNA and host gene expression[6]. Thus, Cp has become an attractive target for mechanism-based antiviral therapy.
Several types of HBV CpAMs exist with various chemical moieties such as heteroaryl-dihydropyrimidines (HAP), sulfamoylbenzamide (SBA), phenylacrylamides, etc. The chemical structures of representative HBV CpAM candidate drugs (GLS4, NVR-010-001-E2, NVR3-778 and AT-130) are in Figure 2.
Figure 2.

Chemical structures of representative HBV CpAM candidates.
Heteroaryldihydropyrimidines (HAPs), such as GLS4 and NVR-010-001-E2, can bind at the dimer-dimer interface of Cp and inhibit HBV replication by inducing the assembly of aberrant capsids and decreasing capsid stability. In addition, the crystal structure of HBV Cp and the HAP analogue NVR-010-001-E2 has been solved, (Figure 3). The crystal structure provides understanding of the mechanism of action of HAPs and facilitates structure-based drug design. Many studies have established a structure-activity relationship for HAP analogues[7-10], which is consistent with the compound binding mode shown by the crystal structure.
Figure 3.

Crystal structure of ligand NVR-010-001-E2 and Cp (PDB code: 5E0I). The viral Cp is displayed as cartoon, while NVR-010-001-E2 and adjacent residues are shown as stick format. Hydrogen bonds are displayed as yellow dotted lines. Key protein residues are labelled.
The pyrimidine core of a HAP locates in the binding pocket and anchors to the protein by the hydrogen bond of one N atom and side chain of Trp102. Therefore, presence of ─NH group at C1 position of the dihydropyrimidine parent ring is essential for potent activity, and the substitution of ─NH resulted in reduction or complete loss of activity. Other substitutions of the core form additional interactions to the reciprocal sub-pocket. A thiazole moiety at the C2 position is favourable in a hydrophobic environment composed of aromatic residues Trp102, Phe23, Phe122, and Tyr118. The 2-fluoro-4-bromophenyl moiety sits at the large hydrophobic pocket provided by Pro25 and the hydrophobic face of Asp29, Leu30, Thr33, Trp102, Ile105, and Ser106. The ester group fits into a small cavity formed by Thr33 and Leu37 on one side and Thr109 and Phe110 on the other side. The C4 and C5 positions are suitable of halogen-substituted phenyl and ester groups respectively. The C6 position locates in an open area of the solvent interface, where various substituents bearing five- and six-membered fused rings can be accommodated.
The HAP molecule GLS4 is currently in clinical phase II studies. The potency of GLS4, with a 50% effective concentration (EC50) value of 0.012 μM, is appreciably better than lamivudine (EC50 = 0.325 μM), and it is also active against adefovir-resistant HBV mutant strains[11]. A double-blind, randomized, parallel, entecavir-controlled study assessed GLS4’s antiviral activity and tolerability. After 28 days of treatment with GLS4, HBV DNA, HBsAg and pregenomic RNA showed a strong downward trend in patients[12]. However, GLS4 has poor water solubility (KS = 14 μM, kinetic solubility), short half-life (t1/2 = 1.5 h), and low bioavailability (F = 14%)[13]. Also, the morpholine ring of GLS4 is easily metabolized[14]. Therefore, GLS4 is mainly used in combination with ritonavir to extend its t1/2 by blocking GLS4’s degradation by CYP450[15]. However, this may increase the risk of drug-drug interactions and adverse reactions.
The spiro ring compound represents a twisted structure of two or more rings linked together by one common atom. The spiro structure occupies more space in the three-dimensional structure than a simple monocyclic or aromatic ring moiety. Due to its relative rigidity, the three-dimensional structure can change the lipophilicity and hydrophilicity, improve activity, increase half-life, and decrease the toxicity of drug molecules to a certain extent. Therefore, investigation of spiro ring containing compounds has increased within drug discovery[16, 17]. Examples include: i) Spiro ring-containing fragments can improve activity. The non-structural protein NS5A plays an important role in the HCV replication cycle, and participates in the replication, assembly, and secretion of HCV, thus attracting a lot of attention. Gilead has discovered an HCV NS5A protein inhibitor effective against both genotypes 1a and 1b (GT1a EC50 = 56 pM; GT1b EC50 = 4 pM). Further structural optimization of HCV NS5A inhibitors lead to the discovery of Ledipasvir bearing azaspiro[2.4]heptane ring with improved activity, (GT1a EC50 = 31 pM; GT1b EC50 = 4 pM) [18]. ii). The spiro ring also can increase the half-life of a compound. Aprepitant, the first neurokinin 1 (NK1) receptor antagonist, developed by Merck, has a good control effect on nausea and vomiting caused by chemotherapy. However, it is a moderate CYP3A4 inhibitor and CYP2C9 inducer with a half-life of 9-13h[19, 20]. Modifying Aprepitant led to discovery of Rolapitant (Company: Schering-Plough) bearing azaspiro[4.5]decane, which is a high-affinity selective NK1 functional antagonist (Figure 4). Rolapitant has a relatively long half-life (7.5 days) and no inhibitory effect on CYP3A4, reducing the concerns of use with CYP3A4 substrates[21].
Figure 4.

The structure and activity of spiro ring fragment application.
To address these current limitations of GLS4, we introduced spiro ring fragments at the solvent exposed C6 position of GLS4. A bridged ring structure (Figure 5) with excellent activity was previously reported in a patent (EC50 = 0.001 μM)[22]; the spiro ring structure was therefore designed to contain a N atom, consistent with the morpholine ring of GLS4. The morpholine ring was replaced with different spiro ring fragments bearing hydrogen bond donor or acceptor substitutions, with or without a "tail" structure. This was anticipated to improve the activity, half-life, and pharmacokinetic properties of newly designed molecules. Figure 5 illustrates the design pipeline of HAP-spiro series compounds.
Figure 5.

The design pipeline of the novel HAP-spiro series analogues.
2. Results
2.1. Chemistry
Scheme 1 describes the stepwise synthesis of the desired HAP-spiro derivatives. Initially, intermediate 2 was achieved by performing the "Biginelli" cyclization reaction between 2-thiazolecarboxamidine hydrochloride, 2-bromo-4-fluorobenzaldehyde and ethyl acetoacetate in the presence of sodium acetate and ethanol under reflux conditions. Further bromination of intermediate 2 was carried out by using N-bromosuccinimide in dichloromethane and obtained the essential intermediate 3. Then the N-spiro ring fragment was mixed with NaH in tetrahydrofuran, and then intermediate 3 was added to generate 4(a-c). At the same time, 4(d-r) were obtained from intermediate 3 using K2CO3 as the base and KI as the catalyst to react with the N-spiro ring fragment in acetonitrile. Finally, removal of a tert-butoxycarbonyl group of 4 (a-k) in the presence of trifluoroacetic acid in dichloromethane at room temperature led to the formation of 5 (a-k).
Scheme 1.

Reagents and reaction conditions: (i) 2-bromo-4-fluorobenzaldehyde, ethyl acetoacetate, CH3COONa, EtOH, 80°C; (ii) N-bromosuccinimide, DCM, 40 °C ; (iii) NH-Spiro ring fragment, NaH, THF, r.t.; or K2CO3, KI, CH3CN, 75 °C, 1 h; (iv) CF3COOH, DCM, 10h, r.t..
2.2. Anti-HBV activity evaluation
HepDES19 cells are a HepG2 (human hepatoblastoma) cell line derivative stably transfected with a HBV genotype D genome under the control of tetracycline-repressible promoter[23]. HepDES19 cells were induced to replicate HBV by withdrawing tetracycline. Compounds were added and the cells were incubated for three days. The cells were lysed and harvested as described previously[12]. Strand preferential quantitative polymerase chain reaction (qPCR) was used to measure the compound's effectiveness in inhibiting HBV replication. This assay measures effects of HBV inhibitors on each DNA strand separately and can resolve DNA elongation inhibitors from compounds that work by other mechanisms because the viral plus-polarity DNA strand is preferentially suppressed by replication inhibitors, but the two strands are suppressed in concert by other classes of inhibitors, such as the CpAMs. Cytotoxicity was assessed using MTS assays that measures mitochondrial function. The lead compound GLS4 and the approved drug lamivudine (3TC) were selected as positive controls.
A total of 29 HAP-spiro derivatives were synthesized and screened for their in vitro anti-HBV DNA replication activity in cell culture. Thirteen compounds were found to have a significant inhibitory effect on the expression of HBV DNA (<70%) at 20 and 10 μM, as represented in Table 1. These compounds were further evaluated for their in vitro anti-HBV activity. Each compound was tested in three independent 9-point EC50 and CC50 assays. The average EC50, CC50, standard deviation (SD) and selective index (SI) were calculated, respectively, in Table 2.
Table 1.
Preliminary evaluation of target compounds in inhibiting HBV DNA replication.
| Compd. | R1 or R2 | Plus DNAa(%) (20μM)b |
Minus DNAa(%) (20μM)b |
Plus DNAa(%) (5μM)b |
Minus DNAa(%) (5μM)b |
|---|---|---|---|---|---|
| 4a |

|
3.5 | 1.8 | 2.4 | 2.5 |
| 4b |

|
0.8 | 0.8 | 3.3 | 2.9 |
| 4c |

|
1.8 | 1.3 | 2.7 | 2.9 |
| 4n |

|
3.0 | 2.5 | 2.8 | 2.3 |
| 4o |

|
2.4 | 2.7 | 3.4 | 4.0 |
| 4p |

|
2.4 | 2.5 | 2.7 | 3.4 |
| 4q |

|
2.0 | 2.2 | 3.0 | 2.8 |
| 4r |

|
3.4 | 3.4 | 3.8 | 3.1 |
| 5a |

|
108.4 | 85.3 | - | - |
| 5b |

|
109.2 | 81.4 | - | - |
| 5c |

|
119.8 | 68.9 | - | - |
| 3TC | 4.0 | 11.8 | - | - | |
| DMSO | 100.0 | 100.0 | 100.0 | 100.0 | |
| Compd. | R1 or R2 | Plus DNA (%) (10μM)b |
Minus DNA (%) (10μM)b |
Plus DNA (%) (3μM)b |
Minus DNA (%) (3μM)b |
| 4d |

|
91.1 | 109.6 | 93.0 | 74.5 |
| 4e |

|
92.5 | 76.1 | 85.3 | 85.0 |
| 4f |

|
60.9 | 60.2 | 87.7 | 83.7 |
| 4g |

|
111.2 | 116.6 | 87.8 | 97.2 |
| 4h |
|
101.6 | 87.0 | 92.0 | 80.1 |
| 4i |

|
16.8 | 28.8 | 79.9 | 67.1 |
| 4j |

|
54.7 | 56.5 | 115.8 | 85.3 |
| 4k |

|
105.0 | 96.5 | 93.3 | 85.7 |
| 4l |

|
18.3 | 37.8 | 58.4 | 44.1 |
| 4m |

|
22.4 | 34.8 | 18.9 | 21.1 |
| 5d |

|
105.6 | 97.7 | 79.2 | 67.1 |
| 5e |

|
105.2 | 99.6 | 85.2 | 72.5 |
| 5f |

|
112.8 | 93.4 | 82.9 | 70.8 |
| 5g |

|
114.3 | 104.7 | 73.8 | 69.9 |
| 5h |

|
88.4 | 86.4 | 68.7 | 69.9 |
| 5i |

|
105.7 | 88.7 | 81.6 | 76.0 |
| 5j |

|
82.2 | 88.8 | 88.2 | 84.5 |
| 5k |
|
87.4 | 94.8 | 88.8 | 85.9 |
| GLS4 | 17.7 | 22.6 | 21.1 | 18.1 | |
| 3TC | 16.2 | 54.6 | 23.9 | 79.1 | |
| DMSO | 100.0 | 100.0 | 100.0 | 100.0 |
The replication inhibition assay employed independently detects the HBV plus-polarity and minus-polarity DNA strands. Capsid inhibitors suppress both strands in concert, so the two values can be considered to be technical repetitions of the quantitative PCR assay on the same sample using different but functionally equivalent primer sets.
In this qualitative primary screening two concentrations per compound were used, either 20 μM and 5 μM or 10 μM and 3 μM. The first screening assay employed 10μM and 3μM but these concentrations did not differentiate between inhibitors and non-inhibitors adequately. While under the concentration of 20 μM, DNA levels detected in the presence of the inhibitors was less than 10% and that of the inactive compound was around 100%. This provided a better prioritization for advancing the compounds into the more laborious EC50 assays.
Table 2.
Anti-HBV activity results of the newly synthesized target compounds inhibiting HBV DNA replication and cytotoxicity. All EC50 and CC50 values are reported in μM.
| Compd. | Average EC50 (μM) | Average CC50 (μM) | SI |
|---|---|---|---|
| 4a | 13.27 ± 0.70 | 21.13 ± 7.47 | 1.59 |
| 4b | 5.17 ± 0.78 | > 83.37 | > 16.13 |
| 4c | 10.00 ± 0.71 | 22.27 ± 6.42 | 2.23 |
| 4f | 10.37 ± 2.64 | > 100.00 | > 9.65 |
| 4i | 4.83 ± 0.86 | > 100.00 | > 20.69 |
| 4j | 8.83 ± 2.15 | > 100.00 | > 11.32 |
| 4l | 3.43 ± 0.66 | 58.36 ± 5.60 | 17.00 |
| 4m | 0.68 ± 0.21 | 48.75 ± 9.15 | 71.34 |
| 4n | 2.60 ± 0.73 | 69.30 ± 25.42 | 26.65 |
| 4o | 3.80 ± 0.78 | > 83.40 | > 21.94 |
| 4p | 1.17 ± 0.05 | > 78.83 | > 67.37 |
| 4q | 2.10 ± 0.08 | > 88.43 | > 42.11 |
| 4r | 0.20 ± 0.00 | > 87.03 | > 435.15 |
| 3TC | 0.37 ± 0.04 | > 100.00 | > 267.86 |
| GLS4 | 0.045 ± 0.01 | > 99.20 | > 2220.90 |
Most of the newly synthesized HAP-spirocyclic derivatives exhibited significant anti-HBV activity. After preliminary activity screening, 13 compounds (4a, 4b, 4c, 4f, 4i, 4j, 4l, 4m, 4n, 4o, 4p, 4q, 4r) inhibited HBV with EC50s of 0.20 - 13.27 μM. Compound 4r was the most potent molecule with an EC50 value of 0.20 ± 0.00 μM, which was better than the positive control lamivudine (EC50 = 0.37 ± 0.04 μM) and only moderately weaker than GLS4 (EC50 = 0.045 ± 0.01 μM). Moreover, 4r had low cytotoxicity (CC50 > 87.03 μM). Taken together, 4r merits further investigation as a potential lead compound.
Based on the antiviral activity results, the preliminary structure-activity relationships (SAR) of the HAP-spiro series are as follows: a) The size of the spiro ring plays a significant role in the antiviral activity. When the sipro ring is too large or too small, the observed reduction in activity is very prominent. For instance, compounds 4d and 5d with [5.5]undecane, 4k and 5k with [3.3]heptane; b) Incorporation of substitution at the spiro ringtail resulted in potent activity compared to those without substitution at the spiro ringtail. For instance, 4i displayed potent activity (EC50 = 4.83 ± 0.86 μM) due to the substitution at the spiro ringtail. This is in contrast to 5i, which is inactive or less potent without substitution at the spiro ringtail; c) The introduction of oxygen atoms or carbonyl substitutions on the spiro ring promotes the activities. For example, 4r with two oxygen atoms on the spiro ring (EC50 = 0.20 ± 0.00 μM) and 4m with two carbonyl substitutions (EC50 = 0.68 ± 0.21 μM) are more potent than others; d) Increasing the number of carbonyl substitutions on the sipro ring could improve antiviral activities. For example, 5e with no carbonyl groups (inactive at initial screening) < 4l with one carbonyl group (EC50 = 3.43 ± 0.66 μM) < 4m with two carbonyl groups (EC50 = 0.68 ± 0.21 μM); e). The position of carbonyl substitution on the ring also played a key role in the activity. Placing the carbonyl group near the parent dihydropyrimidine ring resulted in less active and more toxic compounds, whereas placing it far away from the parent ring was more beneficial as it increased activity and decreased toxicity. For instance, 4n bearing carbonyl group far from the parent ring showed good activity (EC50 = 2.60 ± 0.73 μM, CC50 = 69.30 ± 25.42 μM), while 5b and 5c (carbonyl group close to the parent ring) are inactive, and 4a and 4c demonstrated relative higher cytotoxicity (CC50 = 21.13 ± 7.47 μM and 22.27 ± 6.42 μM, respectively).
2.3. Molecular docking studies
HAPs can bind to the HBV capsid protein dimer interface to misdirect capsid assembly, thereby blocking the replication cycle of HBV. To understand the binding mode of the target compounds and HBV capsid protein, 4r and capsid crystal structure (PDB code: 5E0I) were docked using the molecular docking software SYBYL-X 2.0 and displayed by PyMOL software as represented in Figure 6.
Figure 6.

PyMOL displays the binding mode of compound 4r and HBV capsid crystal structure. Compound 4r is shown in yellow, and the cavity composed of different residues are shown in different colours such as green, blue, and purple. The residues are labelled around the sticks. The HBV capsid protein is shown in the grey cartoon.
According to the virtual docking results, there are additional predicted interactions between the substituents on the HAP parent ring of 4r and the protein to improve the affinity between the compound and the protein. The 2'-fluoro-4'-bromine phenyl group at C4 position is located in the large hydrophobic pocket formed by the residues of Pro25, Asp29, Leu30, Thr33 and Trp102, and the fluorine atom interacts with the Val124 of the adjacent subunit. The C2 thiazole ring extends into the hydrophobic cavity composed of aromatic residues of Phe23, Trp102, Tyr118 and Phe122. The thiazolyl group can form a π-π stacking interaction with the phenyl group of Phe23. The C5 ester group is predicted to be located at the top of the cavity formed by Thr109, Phe110, Thr33 and Leu37. The ethyl group is suitable in the hydrophobic sub-pocket, large or polar moiety may decrease the activity or even cause inactivity. The substituent at the C6 is located in the wide-open area of the solvent interface. The C6 spiro ring can be used as a rigid linker to deliver the hydrophobic tail to the solvent. In this open solvent region, hydrophilic substituent groups or atoms on the spiro ring can improve water solubility.
2.4. In silico prediction of physicochemical properties
The rule of 5 (Ro5) is an empirical rule that helps to predict compounds with good drug-like properties. The Ro5 provides guidance on molecular weight (MW), hydrogen bond donors and acceptors, calculated partition coefficient, and number of rotatable bonds for potential small molecule therapeutics[24]. To further investigate the drug-like properties of 4r compared to GLS4, in silico predictions of physicochemical properties were conducted via online software, shown in Table 3 (http://www.swissadme.com/)[25].
Table 3.
In silico prediction of physicochemical properties of 4r and GLS4.
| Parameter itemsa | Acceptable | 4r | GLS4 |
|---|---|---|---|
| natoms | - | 35 | 31 |
| MW (g/mol) | < 500 | 565.46 | 509.39 |
| nON | < 10 | 8 | 7 |
| nOHNH | < 5 | 1 | 1 |
| nrotb | <10 | 7 | 7 |
| cLogPb | CLogP < 5 (MLogP < 4.15) | iLOGP = 4.60 | iLOGP = 4.00 |
| XLOGP3 = 2.86 | XLOGP3 = 2.54 | ||
| WLOGP = 2.75 | WLOGP = 2.24 | ||
| MLOGP = 2.42 | MLOGP = 2.18 | ||
| SILICOS-IT LOGP= 5.53 | SILICOS-IT LOGP = 5.26 | ||
| Consensus LOGP = 3.63 | Consensus LOGP = 3.24 | ||
| nViol | <2 | 1 | 1 |
| TPSA (Å2)c | <140 | 113.52 | 104.29 |
| Log Sd | - | Log S (ESOL) = −4.92 | Log S (ESOL) = −4.40 |
| Moderately soluble | Moderately soluble | ||
| Log S (Ali) = −4.90 | Log S (Ali) = −4.38 | ||
| Moderately soluble | Moderately soluble | ||
| Log S (SILICOS-IT) = −7.25 | Log S (SILICOS-IT) = −6.76 | ||
| Poorly soluble | Poorly soluble |
natoms = number of non-hydrogen atoms; MW = molecular weight; nON = number of hydrogen bond acceptors; nOHNH = number of hydrogen bond donors; cLog P = calculated Log P; nrotb = number of rotatable bonds; TPSA = topological polar surface area; nViol = number of violations.
iLOGP is obtained by an in-house physics-based method implemented from [26]; XLOGP3 is obtained by an atomistic and knowledge-based method calculated by XLOGP program[27], version 3.2.2, courtesy of CCBG, Shanghai institute of organic chemistry; WLOGP is obtained by an atomistic method implemented from [28]; MLOGP is obtained by a topological method implemented from [29-31]; SILICOS-IT LOGP is obtained by a hybrid fragmental / topological method calculated by FILTER-IT program, version 1.0.2, courtesy of SILICOS-IT, http://www.silicos-it.com[32]; Consensus LOGP is average of all five predictions of Log P.
ESOL is a topological method implemented from [33]; Ali is a topological method implemented from [34]; SILICOS-IT is a fragmental method calculated by FILTER-IT program, version 1.0.2, courtesy of SILICOS-IT, http://www.silicos-it.com[32]. Soluble class (Log S): insoluble < −10 < poorly < −6 < moderately < −4 < soluble < −2 < very < 0 < highly.
In general, compounds that violate two or more of the Ro5 are likely to possess poor drug like properties[35]. Both 4r and GLS4 are consistent with Ro5; however, both exceed the MW the limit of 500 Da. Though, it is commonly accepted that topological polar surface area (TPSA), cLog P, and numbers of hydrogen bond donors is more important than MW[35]. TPSA, which indicates the membrane permeation and absorption of compounds, was predicted to be 113.52 Å2 for 4r and 104.29 Å2 for GLS4. The predicted values of cLog P were obtained in five different methods, four of which were within the acceptable range for both compounds tested. Taken together 4r has similar predicted physiochemical properties to GLS4 and is anticipated to share both permeability and adsorption characteristics.
Solubility is a critical factor for the development of anti-HBV agents to ensure drug candidate’s oral pharmacokinetic (PK) properties. Three prediction methods were used in order to appraise the solubility of both GLS4 and 4r. Both compounds were predicted to be moderately soluble based on topologic methods, but were predicted to be poorly soluble base on fragmental methods. 4r and GLS4 received similar predicted solubility, however GLS4 received a slightly more favourable Log S than 4r. Suggesting that the addition of the spiro ring did not increase the solubility of the GLS4 derivative.
2.5. Prediction of inhibition on CYP450
Cytochrome P450 (CYP450) is a superfamily of drug-metabolizing enzymes in the liver that may be inhibited or induced by therapeutics resulting in metabolism-mediated drug-drug interactions (DDI) and/or impacting plasma concentrations of other therapeutics. GLS4 is readily metabolized by CYP450 and has poor PK properties[36]. The first-in-human trial of GLS4 reported that the steady-state concentration of GLS4 was lower than the effective concentration 90% (EC90 = 55.7 ng/ml) [37]. However, addition of ritonavir, a strong inhibitor of CYP3A4, significantly increased the plasma concentration of GLS4 from 2.40 to 49.8 ng/ml. While, the PK properties of GLS4 can be improved clinically with ritonavir, the potential use of combination therapy may increase the risk of DDI and adverse reactions. Therefore, it is important to investigate the metabolic behavior of GLS4 analogues.
We utilized 3 different tools [25, 38, 39] to evaluate GLS4 and 4r as potential inhibitors or substrates of major CYP450 enzymes, as shown in Table 4. While, there were discrepancies in the predicted results of different methods, the consensus results of GLS4 were consistent with those previously reported (1A2 IC50 > 50 μM, 2C9 IC50 =16 μM, 2C19 IC50 = 4 μM, 2D6 IC50 = 52 μM, 3A4 IC50 = 2 μM)[13].
Table 4.
Prediction of inhibition on CYP450 of compounds.
| CYP isozyme |
Inhibitor / substrate |
4r | GLS4 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Method 1a |
Method 2b |
Method 3c |
Consensus | Method 1 a |
Method 2b |
Method 3c |
Consensus | ||
| 1A2 | Inhibitor | − | − | − | − | − | − | + | − |
| Substrate | − | ND | ND | − | − | ND | ND | − | |
| 2C9 | Inhibitor | + | − | + | + | + | + | + | + |
| Substrate | − | ND | + | ND | − | ND | + | ||
| 2C19 | Inhibitor | + | − | + | + | + | + | + | + |
| Substrate | − | ND | ND | − | − | ND | ND | − | |
| 2D6 | Inhibitor | + | + | − | + | − | + | + | + |
| Substrate | + | ND | + | + | + | ND | + | + | |
| 3A4 | Inhibitor | + | + | + | + | + | + | + | + |
| Substrate | + | ND | + | + | + | ND | + | + | |
−: Non-inhibitor/substrate, the predicted probability is less than or equal to 0.5 (minimum value is 0); +: inhibitor/substrate, the predicted probability is greater than 0.5 (maximum value is 1).
ND: not determined.
method 1 is utilizing the online software Molinspiration (https://www.molinspiration.com/)
method 2 is utilizing the online software SwissADME (http://www.swissadme.ch/index.php#)
method 3 is utilizing the online software admetSAR (http://lmmd.ecust.edu.cn/admetsar2/).
Compounds 4r and GLS4 had similar predicted metabolic profiles for the five subtypes of CYP450 enzymes. 4r and GLS4 were both likely to be inhibitors of 2C9, 2C19, 2D6 and 3A4. Additionally, 4r and GLS4 were predicted to be sensitive metabolic substrates of 2D6 and 3A4. Taken together these predictions suggest that 4r may act similarly to GLS4 in future metabolic studies.
2.6. Prediction of pharmacokinetic properties
GLS4 exhibited poor PK properties both in animals (T1/2 = 1.5 ± 0.1 h, F = 14% in female Balb/c mice[13]; T1/2 = 17.9 h, F = 23.8% in Beagle dogs[7]) and humans (T1/2 = 1.09 – 15.8 hours[12]). Two online tools, http://admet.scbdd.com/calcpre/index/[38] and http://lmmd.ecust.edu.cn/admetsar2/ [39] were utilized to predict the PK properties of GLS4 and 4r, as shown in Table 5. The prediction of GLS4 is comparable to the reference reported (T1/2 = 1.5 ± 0.1 h, F = 14%)[13]. The predicted half-life, plasma protein binding, and acute oral toxicity for 4r was similar to those of GLS4. These predictions suggest that both GLS4 and 4r possess similar PK properties.
Table 5.
Prediction of pharmacokinetic properties of GLS4 and 4r.
| 4r | GLS4 | |
|---|---|---|
| T1/2 (h)a | 1.594 | 1.498 |
| F (%)a | < 20 | < 20 |
| Plasma protein binding (%)b | 1.155 | 1.24 |
| Acute oral toxicity (kg/mol)b | 2.575 | 2.521 |
Half-life (T1/2) and bioavailability (F) prediction were obtained using http://admet.scbdd.com/calcpre/index/
plasma protein binding and acute oral toxicity were obtained via http://lmmd.ecust.edu.cn/admetsar2/.
3. Conclusion
In this study, 29 spiro ring-containing heteroaryldihydropyrimidine (HAP) analogues were designed, synthesized, and evaluated as HBV capsid protein inhibitors. The structures of all compounds were experimentally verified. The experimental HBV CpAM GLS4 was selected as both the lead and positive compound, while the approved nucleoside analogue lamivudine served as positive control. In vitro activity assays revealed antiviral potency and low cytotoxicity of 4r (EC50 = 0.20 ± 0.00 μM, CC50 > 87.03 μM), which is better than lamivudine (EC50 = 0.37 ± 0.04 μM , CC50 > 100.00 μM), and about one quarter the activity of GLS4 (EC50 = 0.045 ± 0.01 μM, CC50 > 99.20 μM). Moreover, a preliminary SAR analysis of spiro ring-containing HAPs revealed that the size and substitution of spiro ring, position and number of carbonyl groups on the spiro ring, and number of oxygen atoms had a significant influence on the activity. According to physicochemical properties forecasting, 4r was predicted to be consistent with Ro5, therefore likely to possess favourable absorption and permeation. ADMET and PK characteristics predictions were also performed, including interactions with CYP450 enzymes, half-life, plasma protein binding and acute toxicity, which revealed 4r’s possibly comparable in vivo behaviors to GLS4 rather than significant differences. In conclusion, compound 4r is the best inhibitor of this spiro ring containing HAPs, and further modification could lead to get more potent anti-HBV inhibitors consisting of HAPs.
4. Experimental section
4.1. Chemistry
4.1.1. Synthetic procedures and analytical data
The solvents and key reagents were purchased from commercial suppliers. TLC was performed on Silica Gel GF254 and visualized with UV light (254 nm and 365 nm). Flash column chromatography was performed on columns packed with silica gel 60 (200-300 mesh). All melting points (M.p.) were determined on a micro melting point apparatus. Mass spectra were performed on an LC Autosampler Device: Standard G1313A instrument by electrospray ionization. 1H NMR and 13C NMR spectrum were obtained on a Bruker AV-400 spectrometer (Bruker BioSpin, F€allanden, Switzerland) in the solvent DMSO-d6 and CDCl3. Chemical shifts were expressed in ppm, using tetramethylsilane (TMS) as an internal standard, and J values were reported in hertz (Hz).
4.1.2. General procedure for the synthesis of Ethyl4-(2-bromo-4-fluorophenyl)-6-methyl-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (2)
2-Thiazolecarboxamidine hydrochloride (1.0 g, 6.11 mmol), 2-bromo-4-fluorobenzaldehyde (1.86 g, 9.16 mmol) and sodium acetate (1.0 g, 1.22 mmol) were dissolved in absolute ethanol (100 mL) and to this mixture, added ethyl acetoacetate (1.2 mL, 9.20 mmol) with stirring at room temperature, and then refluxed at 80°C for 8 h. After reaction completion, salts were filtered, and the resulted liquid was cooled to room temperature, afforded precipitated yellow crystals (2). The solvent of the remaining mother liquor was removed under reduced pressure. Water (60 mL) and ethyl acetate (25 mL × 3) were added and extracted. The organic phases were collected, combined, and extracted with saturated sodium chloride solution (25 mL). The organic phases were dried with anhydrous magnesium sulfate. After filtering, it was separated by flash column chromatography with developing solvent PE: EA = 10:1. The obtained solid was recrystallized by using a dichloromethane-n-hexane system to achieve the yellow coloured product 2. Yield: 82.0 %; mp: 158-160°C; 1H NMR (400 MHz, DMSO-d6): δ 9.92 (s, 1H), 7.97 (d, J = 2.8 Hz, 1H), 7.89 (s, 1H), 7.59 – 7.50 (m, 1H), 7.42 – 7.31 (m, 1H), 7.23 (t, J = 8.3 Hz, 1H), 5.98 (s, 1H), 3.94 (q, J = 6.9 Hz, 2H), 2.48 (s, 3H), 1.03 (t, J = 7.0 Hz, 3H); EI-MS: 424.3 [M+H]+; C17H15BrFN3O2S [423.01].
4.1.3. General procedure for the synthesis of Ethyl4-(2-bromo-4-fluorophenyl)-6-(bromomethyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (3)
Intermediate 2 (1.86 g, 4.39 mmol) was dissolved in dichloromethane (50 mL), and NBS (1.95 g, 1.10 mmol) was added slowly by stirring at room temperature. Then the reaction mixture was refluxed for 1.5 h. After the reaction completion, the solvent was removed under reduced pressure. Water (60 mL) and ethyl acetate (25 mL × 3) were added to the crude and extracted. The organic phases were collected, combined, and extracted with saturated sodium chloride solution (25 mL). The organic phases were dried with anhydrous magnesium sulfate. After filtering, it was separated by flash column chromatography with a solvent system of PE: EA = 10:1. The obtained solid was recrystallized by using a dichloromethane-n-hexane system to achieve 3. Yield: 65.0 %; mp: 125-127 °C; 1H NMR (400 MHz, CDCl3): δ 7.83 (d, J = 2.4 Hz, 1H), 7.64 – 7.34 (m, 3H), 7.32 (d, J = 7.5 Hz, 1H), 7.01 (d, J = 7.0 Hz, 1H), 6.12 (d, J = 38.9 Hz, 1H), 4.93 (d, J = 8.1 Hz, 1H), 4.59 (d, J = 8.1 Hz, 1H), 4.12 (d, J = 6.8 Hz, 2H), 1.16 (t, J = 7.0 Hz, 3H); EI-MS: 499.90 [M-H]−; C17H14Br2FN3O2S [500.92]
4.1.4. General procedure for the synthesis of Ethyl4-(2-bromo-4-fluorophenyl)-6-(morpholinomethyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (GLS4)
Intermediate 3 (1.0 g, 2.00 mmol) was dissolved in DMF (50 mL), then morpholine (346 μL, 4.00 mmol) was added, and the mixture was stirred overnight at room temperature. After completion of the reaction, the solvent was removed under reduced pressure. Water (50 mL) and ethyl acetate (25 mL × 3) were added to the crude and extracted. The organic phases were collected, combined, and extracted with saturated sodium chloride solution (25 mL). The organic phases were dried with anhydrous magnesium sulfate. After filtering, it was separated by flash column chromatography with the solvent system of PE: EA = 4:1. The obtained solid was recrystallized by using a dichloromethane-n-hexane system to achieve GLS4. Yield: 79.0 %; mp: 124-127 °C; 1H NMR (400 MHz, DMSO-d6): δ 9.69 (s, 1H), 8.04 (d, J = 3.1 Hz, 1H), 7.95 (d, J = 2.5 Hz, 1H), 7.57 (dd, J = 8.5, 1.9 Hz, 1H), 7.40 (dd, J = 8.6, 6.2 Hz, 1H), 7.22 (td, J = 8.5, 2.4 Hz, 1H), 6.04 (s, 1H), 4.03 – 3.85 (m, 4H), 3.68 (t, J = 4.2 Hz, 4H), 2.55 (t, J = 7.2 Hz, 4H), 1.06 (t, J = 7.1 Hz, 3H); EI-MS: 509.15 [M+H]+; C21H22BrFN4O3S [508.06].
4.1.5. General procedure for the synthesis of 4(a-c)
To a stirring mixture of NaH (60%, 1.33 mmol) and different N-Boc substituted spiro rings (0.442 mmol) in 20 mL of THF was added 3 (220 mg, 0.440 mmol) in THF, and the reaction mixture was stirred at room temperature for 0.5h. After completion of the reaction, the solvent was removed under reduced pressure. Water (50 mL) and ethyl acetate (25 mL × 3) were added to the crude and extracted. The organic phases were collected and combined and extracted once more with saturated sodium chloride solution (25 mL). The organic phases were dried with anhydrous magnesium sulfate. After filtering, it was separated by flash column chromatography with developing solvent PE: EA = 4:1. The obtained solid was recrystallized by using a dichloromethane-n-hexane system to achieve the desired products.
tert-butyl 6-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-7-oxo-2,6-diazaspiro[3.4]octane-2-carboxylate (4a)
Yield: 65.6 %; mp: 92-93°C; 1H NMR (400 MHz, DMSO-d6): δ 9.36 (d, J = 241.0 Hz, 1H, dihydropyrimidine-H), 8.08 – 7.83 (m, 2H, thiazole-H), 7.57 (d, J = 8.6 Hz, 1H, Ph-H), 7.43 (ddd, J = 11.4, 8.7, 6.2 Hz, 1H, Ph-H), 7.30 – 7.19 (m, 1H, Ph-H), 6.12 – 5.85 (m, 1H, dihydropyrimidine-CH), 4.68 – 4.53 (d, J = 2.1 Hz, 2H, dihydropyrimidine-CH2), 4.00 (q, J = 7.1 Hz, 2H, CH2CH3), 3.94 – 3.77 (m, 4H, CCH2N), 3.76 – 3.64 (m, 2H, NCH2), 2.67 (d, J = 9.5 Hz, 2H, COCH2), 1.37 (d, J = 1.3 Hz, 9H, Boc), 1.08 (dt, J = 9.0, 7.1 Hz, 3H, CH3); 13C NMR (100 MHz, DMSO-d6): δ 174.82, 173.12, 165.58, 161.65 (d, J = 252.5 Hz), 155.94, 150.48, 144.81, 143.84, 140.67 (d, J = 3.4 Hz), 131.96 (d, J = 8.3 Hz), 126.48, 123.02 (d, J = 9.6 Hz), 120.08 (d, J = 29.5 Hz), 116.30 (d, J = 21.2 Hz), 105.11, 99.60, 79.09, 60.21, 58.94, 58.34, 58.00, 52.07, 44.87, 34.26, 28.49, 14.52; EI-MS: 648.18 [M+H]+; C28H31BrFN5O5S [647.12].
tert-butyl 7-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-6-oxo-2,7-diazaspiro[4.4]nonane-2-carboxylate (4b)
Yield: 11.2 %; mp: 85-87°C; 1H NMR (400 MHz, CDCl3) δ 8.92 (s, 1H, dihydropyrimidine-H),7.84 (d, J = 3.1 Hz, 1H, thiazole-H), 7.51 (s, 1H, thiazole-H), 7.44 – 7.28 (m, 2H, Ph-H), 7.04 (s, 1H, Ph-H), 6.12 (d, J = 41.0 Hz, 1H, dihydropyrimidine-CH), 4.91 (d, J = 26.0 Hz, 2H dihydropyrimidine-CH2), 4.07 (m, 2H, CH2CH3), 3.64 (s, 2H, CCH2NCO), 3.50 (s, 2H, CONCH2CH2C), 3.40 (s, 2H, NCH2CH2C), 2.31 (m, 2H, NCH2CH2C), 2.10 (s, 2H, CCH2CH2NCO), 1.47 (s, 9H, Boc), 1.15 (t, J = 9.0, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 175.69, 165.55, 161.61 (d, J = 252.5 Hz), 155.85, 153.89, 144.79, 144.36, 143.91,140.39 (d, J = 3.0 Hz), 131.99 (d, J = 8.1 Hz), 125.28, 123.07 (d, J = 10.2 Hz), 120.06 (d, J = 24.2 Hz), 116.25 (d, J = 21.6 Hz), 99.78, 78.81, 60.35, 60.24, 56.50, 50.03, 49.11, 45.63, 44.97, 34.91, 31.43, 28.63, 14.52; C29H33BrFN5O5S [661.14]; EI-MS: 662.16 [M+H]+, 684.17 [M+Na]+.
tert-butyl 7-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-8-oxo-2,7-diazaspiro[4.4]nonane-2-carboxylate (4c)
Yield: 53.4 %; mp: 119-120°C; 1H NMR (400 MHz, DMSO-d6): δ 9.63 (s, 1H, dihydropyrimidine-H), 7.97 (d, J = 31.4 Hz, 2H, thiazole-H), 7.57 (d, J = 8.5 Hz, 1H, Ph-H), 7.41 (d, J = 6.0 Hz, 1H, Ph-H), 7.25 (m, 1H, Ph-H), 6.02 – 5.92 (m, 1H, dihydropyrimidine-CH), 4.71 (s, 2H, dihydropyrimidine-CH2), 4.06 – 3.92 (m, 2H, CH2CH3), 3.40 (m, 4H, CH2NBocCH2), 3.27 (s, 2H, NCH2C), 2.41 (d, J = 13.0 Hz, 2H, COCH2C), 1.90 (m, 2H, CCH2CH2N), 1.45 – 1.26 (m, 9H, Boc), 1.07 (dt, J = 14.2, 7.1 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 173.29, 165.59, 161.31 (d, J = 252.5 Hz), 154.05, 150.48, 144.78, 144.30, 143.96, 140.57 (d, J = 3.0 Hz), 131.38 (d, J = 9.0 Hz), 125.24, 123.01 (d, J = 10.1 Hz), 120.13 (d, J = 24.4 Hz), 116.28 (d, J = 21.4 Hz), 99.82, 78.85, 60.31, 60.22, 58.34, 56.49, 52.11, 45.08, 44.96, 41.60, 35.57, 28.60, 14.52; EI-MS: 662.16 [M+H]+, 684.12 [M+Na]+, C29H33BrFN5O5S [661.14].
4.1.6. General procedure for the synthesis of 4(d-r)
Intermediate 3 (0.608 g, 1.21 mmol), different spiro ring fragments (1.21 mmol), potassium carbonate (0.25 g, 1.82 mmol) and potassium iodide (0.30 g, 1.82 mmol) were dissolved in 25mL acetonitrile and refluxed at 75°C for 1 hour. After completion of the reaction, the solvent was removed under reduced pressure. Water (50 mL) and ethyl acetate (25 mL × 3) were added to the crude and extracted. The organic phases were collected and combined and extracted once more with saturated sodium chloride solution (25 mL). The organic phases were dried with anhydrous magnesium sulfate. After filtering, it was purified by flash column chromatography with the solvent system of PE: EA = 4:1. The obtained solid was recrystallized using a dichloromethane-n-hexane system to achieve the desired products 4(d-r).
tert-butyl 9-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-3,9-diazaspiro[5.5]undecane-3-carboxylate (4d)
Yield: 39.0 %; mp: 77-80°C; 1H NMR (400 MHz, DMSO-d6): δ 9.69 (s, 1H, dihydropyrimidine-H), 8.00 (s, 1H, thiazole-H), 7.93 (s, 1H, thiazole-H), 7.56 (d, J = 8.1 Hz, 1H, Ph-H), 7.37 (m, 1H, Ph-H), 7.22 (s, 1H, Ph-H), 6.02 (s, 1H, dihydropyrimidine-CH), 4.08 – 3.79 (m, 4H, dihydropyrimidine-CH2, CH2CH3), 3.31(s, 4H, 2×NCH2), 2.50(s, 4H, 2×BocNCH2), 1.53 (s, 4H), 1.39 (s, 9H, Boc), 1.23 (s, 2H, CCH2), 1.05 (t, J = 6.9 Hz, 3H, CH2CH3), 0.85(s, 2H, CCH2); 13C NMR (100 MHz, DMSO-d6): δ 165.65, 162.54, 161.25 (d, J = 248.3 Hz), 154.45, 147.45, 144.44, 144.07, 140.69 (d, J = 3.3 Hz), 131.32 (d, J = 8.8 Hz), 125.14, 123.05 (d, J = 9.9 Hz), 120.05 (d, J = 24.2 Hz), 115.93 (d, J = 20.9 Hz), 97.23, 78.86, 59.80, 58.68, 56.42, 49.26, 38.71, 35.72, 29.48, 28.58, 14.46; EI-MS: 676.97 [M+H]+, C31H39BrFN5O4S [675.19].
tert-butyl 8-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-2,8-diazaspiro[4.5]decane-2-carboxylate (4e)
Yield: 71.0 %; mp: 167-170°C; 1H NMR (400 MHz, DMSO-d6): δ 9.69 (s, 1H, dihydropyrimidine-H), 8.03 (s, 1H, thiazole-H), 7.95 (s, 1H, thiazole-H), 7.57 (dd, J = 8.6, 2.5 Hz, 1H, Ph-H), 7.39 (dd, J = 8.8, 6.0 Hz, 1H, Ph-H), 7.23 (dd, J = 9.8, 7.5 Hz, 1H, Ph-H), 6.03 (s, 1H, dihydropyrimidine-CH), 3.97 (t, J = 6.9 Hz, 2H, CH2CH3), 3.33 (s, 2H,), 3.30 (s, 2H, CCH2NBoc), 3.12 (s, 2H, CH2CH2NBoc), 2.46 (s, 4H, 2×NCH2), 1.73 (q, J = 7.5 Hz, 2H, CCH2CH2NBoc), 1.58 (t, J = 5.4 Hz, 4H, NCH2CH2CCH2CH2N), 1.41 (s, 9H, Boc), 1.06 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.65, 162.53, 161.25 (d, J = 248.2 Hz), 156.12, 147.25, 144.37, 144.11, 140.67 (d, J = 3.4 Hz), 131.34 (d, J = 9.0 Hz), 125.16, 123.05 (d, J = 9.8 Hz), 120.06 (d, J = 24.3 Hz), 115.95 (d, J = 21.3 Hz), 97.31, 78.90, 59.84, 58.66, 56.24, 50.68, 35.61, 33.04, 28.56, 14.45; EI-MS: 661.97 [M+H]+, C30H37BrFN5O4S [661.17].
tert-butyl 7-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-2,7-diazaspiro[3.5]nonane-2-carboxylate (4f)
Yield: 38.0 %; mp: 103-106°C; 1H NMR (400 MHz, DMSO-d6): δ 9.66 (s, 1H, dihydropyrimidine-H), 8.02 (s, 1H, thiazole-H), 7.95 (s, 1H, thiazole-H), 7.57 (d, J = 8.5, 2.2 Hz, 1H, Ph-H), 7.38 (dd, J = 9.0, 6.1 Hz, 1H, Ph-H), 7.23 (dd, J = 9.5, 7.2 Hz, 1H, Ph-H), 6.03 (s, 1H, dihydropyrimidine-CH), 3.95 (q, J = 8.6, 7.8 Hz, 2H, CH2CH3), 3.58 (s, 4H, CH2NBocCH2), 2.45 (s, 4H, CH2NCH2), 1.76 (t, J = 5.4 Hz, 4H, CH2CCH2), 1.39 (d, J = 1.7 Hz, 9H, Boc), 1.05 (td, J = 7.0, 1.7 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.65, 162.53, 161.25 (d, J = 247.1 Hz), 156.12, 147.25, 144.37, 144.11, 140.67 (d, J = 3.1 Hz), 131.34 (d, J = 8.8 Hz), 125.16, 123.04 (d, J = 9.7 Hz), 120.06 (d, J = 23.9 Hz), 115.95 (d, J = 20.9 Hz), 97.31, 78.90, 59.84, 58.66, 56.24, 50.68, 35.61, 33.04, 28.56, 14.45; EI-MS: 648.04 [M+H]+; C29H35BrFN5O4S [647.16]
Ethyl 4-(2-bromo-4-fluorophenyl)-6-((2-((tert-butoxycarbonyl) amino)-7-azaspiro [3.5]nonan-7-yl)methyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (4g)
Yield: 35.9 %; mp: 153-156°C; 1H NMR (400 MHz, DMSO-d6): δ 9.69 (s, 1H, dihydropyrimidine-H), 8.03 (s, 1H, thiazole-H), 7.94 (s, 1H, thiazole-H), 7.43 – 7.34 (m, 1H, Ph-H), 7.23 (dd, J = 9.7, 7.1 Hz, 1H, Ph-H), 7.10 (d, J = 8.0 Hz, 1H, Ph-H), 6.02 (s, 1H, dihydropyrimidine-CH), 3.97 – 3.82 (m, 5H, CH2CH3, dihydropyrimidine-CH2, NHBoc), 2.46 (s, 2H, CH2NCH2), 2.38 (s, 2H, CH2NCH2), 2.11 (t, J = 9.3 Hz, 2H, CH2CH2CCH2CH2), 1.72 – 1.48 (m, 6H, CH2CH2CCH2CH2, CCH2CHCH2C), 1.38 (d, J = 1.9 Hz, 9H, Boc), 1.05 (td, J = 7.1, 1.9 Hz, 3H, CH2CH3) ; EI-MS: 662.20 [M+H]+; 13C NMR (100 MHz, DMSO-d6): δ 165.65, 162.55, 161.24 (d, J = 248.3 Hz), 155.01, 147.44, 144.37, 144.13, 140.71 (d, J = 3.3 Hz), 131.35 (d, J = 8.7 Hz), 125.12, 123.04 (d, J = 9.6 Hz), 120.05 (d, J = 24.3 Hz), 115.95 (d, J = 20.9 Hz), 97.13, 77.94, 59.79, 58.66, 56.48, 51.05, 50.79, 36.34, 31.16, 28.73, 14.47; EI-MS: 662.20 [M+H]+; C30H37BrFN5O4S [661.17].
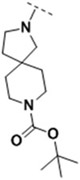
tert-butyl 2-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-2,8-diazaspiro[4.5]decane-8-carboxylate (4h)
Yield: 68.7 %; mp: 91-94°C; 1H NMR (400 MHz, DMSO-d6): δ 9.65 (s, 1H, dihydropyrimidine-H), 7.97 (d, J = 12.0, 2H, thiazole-H), 7.57 (dd, J = 8.5, 2.5 Hz, 1H, Ph-H), 7.39 (dd, J = 8.7, 6.1 Hz, 1H, Ph-H), 7.24 (dd, J = 9.5, 7.1 Hz, 1H, Ph-H), 6.03 (s, 1H, dihydropyrimidine-CH), 4.05 – 3.95 (m, 4H, CH2CH3, dihydropyrimidine-CH2), 3.25 (s, 4H, CH2NBocCH2), 2.84 (d, J = 7.8 Hz, 1H, NCH2CH2C), 2.71 (d, J = 7.9 Hz, 1H, NCH2CH2C), 2.60 (d, J = 9.2 Hz, 1H, NCH2C), 2.40 (d, J = 9.1 Hz, 1H, NCH2C), 1.68 (t, J = 7.0 Hz, 2H, NCH2CH2C), 1.54 (td, J = 13.8, 11.6, 5.1 Hz, 4H, CH2CH2CCH2CH2), 1.39 (s, 9H, Boc), 1.06 (t, J = 7.2 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.64, 162.42, 161.22 (d, J = 240.9 Hz), 154.38, 147.75, 144.44, 143.92, 140.63 (d, J = 3.4 Hz) , 131.34 (d, J = 8.5 Hz), 125.24, 123.05 (d, J = 9.6 Hz), 120.07 (d, J = 24.3 Hz), 115.98 (d, J = 21.1 Hz), 96.75, 78.92, 65.02, 59.83, 58.61, 53.60, 53.50, 40.94, 36.60, 28.56, 14.48; EI-MS: 662.65 [M+H]+; C30H37BrFN5O4S [661.17].
tert-butyl 7-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-2,7-diazaspiro[4.4]nonane-2-carboxylate (4i)
Yield: 89.4 %; mp: 118-121°C; 1H NMR (400 MHz, DMSO-d6): δ 9.65 (d, J = 16.9 Hz, 1H, dihydropyrimidine-H), 7.98 (d, J = 17.4 Hz, 2H, thiazole-H), 7.57 (dd, J = 8.4, 2.4 Hz, 1H, Ph-H), 7.39 (t, J = 7.7 Hz, 1H, Ph-H), 7.27 – 7.18 (m, 1H, Ph-H), 6.03 (s, 1H, dihydropyrimidine-CH), 4.07 – 3.92 (m, 4H, dihydropyrimidine-CH2, CH2CH3), 3.23 (d, J = 10.2 Hz, 4H, CH2NBocCH2), 2.94 – 2.56 (m, 4H, CH2NCH2), 1.97 – 1.78 (m, 4H, NCH2CH2CCH2), 1.36 (d, J = 21.2 Hz, 9H, Boc), 1.06 (t, J = 7.2 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.64, 162.48, 161.22 (d, J = 240.9 Hz), 154.05, 144.48, 143.81, 140.65 (d, J = 3.4 Hz), 131.32 (d, J = 8.5 Hz), 125.26, 123.06 (d, J = 7.6 Hz), 120.08 (d, J = 26.0 Hz), 115.97 (d, J = 26.7 Hz), 96.87, 78.67, 63.81, 59.86, 58.60, 53.74, 53.52, 48.69, 45.58, 45.29, 40.18, 36.52, 28.65, 14.46; EI-MS: 648.67 [M+H]+; C29H35BrFN5O4S [647.16].
tert-butyl 6-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-2,6-diazaspiro[3.4]octane-2-carboxylate (4j)
Yield: 51.0 %; mp: 158-161°C; 1H NMR (400 MHz, DMSO-d6): δ 9.57 (s, 1H, dihydropyrimidine-H), 7.95 (p, J = 2.9 Hz, 2H, thiazole-H), 7.56 (dt, J = 8.6, 2.3 Hz, 1H, Ph-H), 7.42 – 7.36 (m, 1H, Ph-H), 7.23 (td, J = 8.5, 4.3 Hz, 1H, Ph-H), 6.02 (d, J = 1.6 Hz, 1H, dihydropyrimidine-CH), 4.01 (s, 2H, dihydropyrimidine-CH2), 3.95 (dt, J = 7 .9, 6.2 Hz, 2H, CH2CH3), 3.84 (d, J = 18.2 Hz, 4H, CH2NBocCH2), 2.77 (tt, J = 27.2, 9.1 Hz, 4H, CH2NCH2), 2.07 (t, J = 7.4 Hz, 2H, NCH2CH2C), 1.37 (d, J = 1.7 Hz, 9H, Boc), 1.05 (td, J = 7.1, 1.7 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.63, 162.49, 161.25 (d, J = 247.9 Hz), 155.92, 147.54, 144.36, 143.75, 140.69 (d, J = 3.3 Hz), 131.37 (d, J = 8.8 Hz), 125.33, 123.04 (d, J = 9.7 Hz), 120.05 (d, J = 24.3 Hz), 115.98 (d, J = 21.0 Hz), 96.83, 78.90, 64.72, 59.84, 58.58, 53.61, 53.40, 36.46, 28.53, 14.47; EI-MS: 634.37 [M+H]+; C28H33BrFN5O4S [633.14].
tert-butyl 6-((6-(2-bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3,6-dihydropyrimidin-4-yl)methyl)-2,6-diazaspiro[3.3]heptane-2-carboxylate (4k)
Yield: 41.0 %; mp: 166-169°C; 1H NMR (400 MHz, DMSO-d6): δ 9.43 (s, 1H, dihydropyrimidine-H), 7.99 (dd, J = 30.9, 2.7 Hz, 2H, thiazole-H), 7.57 (dd, J = 8.6, 2.6 Hz, 1H, Ph-H), 7.35 (dd, J = 8.7, 5.9 Hz, 1H, Ph-H), 7.23 (t, J = 8.5 Hz, 1H, Ph-H), 6.00 (d, J = 1.9 Hz, 1H, dihydropyrimidine-CH), 3.97 (t, J = 6.9 Hz, 8H, dihydropyrimidine-CH2, CH2CH3, CH2NBocCH2), 3.49 (s, 4H, CH2NCH2), 1.42 – 1.35 (m, 9H, Boc), 1.06 (ddd, J = 8.3, 6.5, 1.7 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.56, 162.43, 161.23 (d, J = 242.9 Hz), 155.79, 147.09, 144.31, 144.06, 140.58 (d, J = 3.5 Hz), 131.23 (d, J = 8.4 Hz), 125.23, 123.02 (d, J = 9.8 Hz), 120.10 (d, J = 24.8 Hz), 115.95 (d, J = 21.4 Hz), 97.21, 79.01, 64.56, 59.87, 58.40, 56.79, 33.50, 28.52, 14.46; EI-MS: 620.10 [M+H]+; C27H31BrFN5O4S [619.13].
ethyl 4-(2-bromo-4-fluorophenyl)-6-((1-oxo-2,8-diazaspiro[4.5]decan-8-yl)methyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (4l)
Yield: 48.0 %; mp: > 200°C; 1H NMR (400 MHz, DMSO-d6): δ 9.62 (s, 1H, dihydropyrimidine-H), 8.05 (s, 1H, thiazole-H), 7.95 (s, 1H, thiazole-H), 7.56 (s, 1H, Ph-H), 7.42 – 7.37 (m, 1H, Ph-H), 7.23 (t, J = 8.4 Hz, 1H, Ph-H), 6.04 (s, 1H, dihydropyrimidine-CH), 4.05 – 4.02 (m, 2H, dihydropyrimidine-CH2), 3.96 (d, J = 6.9 Hz, 2H, CH2CH3), 3.18 (t, J = 6.8 Hz, 2H, CONHCH2), 2.90 (d, J = 11.5 Hz, 1H, CONHCH2CH2), 2.77 (d, J = 11.6 Hz, 1H, CONHCH2CH2), 2.29 (d, J = 31.2 Hz, 2H, CH2NCH2), 1.97 (s, 2H, CH2NCH2), 1.80 (d, J = 11.8 Hz, 2H, CH2CCH2), 1.45 – 1.36 (m, 2H, CH2CCH2), 1.05 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 180.66, 165.66, 162.59, 161.24 (d, J = 248.3 Hz), 147.29, 144.32, 144.13, 140.75 (d, J = 3.2 Hz), 131.39 (d, J = 8.8 Hz), 125.17, 123.03 (d, J = 9.7 Hz), 120.05 (d, J = 24.2 Hz), 115.96 (d, J = 20.9 Hz), 97.34, 65.49, 59.82, 58.64, 50.53, 50.35, 41.63, 38.71, 38.37, 32.36, 32.25, 14.46; EI-MS: 576.53 [M+H]+; C25H27BrFN5O3S [575.10].
ethyl 4-(2-bromo-4-fluorophenyl)-6-((1,3-dioxo-2,8-diazaspiro[4.5]decan-8-yl)methyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (4m)
Yield: 56.0 %; mp: > 200°C; 1H NMR (400 MHz, DMSO-d6): δ 11.17 (s, 1H, CONHCO), 9.61 (s, 1H, dihydropyrimidine-H), 8.05 (t, J = 2.3 Hz, 1H, thiazole-H), 7.96 (t, J = 2.5 Hz, 1H, thiazole-H), 7.57 (dd, J = 8.6, 2.5 Hz, 1H, Ph-H), 7.40 (dd, J = 9.1, 5.8 Hz, 1H, Ph-H), 7.23 (dd, J = 9.7, 7.2 Hz, 1H, Ph-H), 6.03 (d, J = 1.7 Hz, 1H, dihydropyrimidine-CH), 3.97 (t, J = 7.0 Hz, 2H, dihydropyrimidine-CH2), 3.94 – 3.84 (m, 2H, CH2CH3), 2.93 (d, J = 11.7 Hz, 1H, CH2NCH2), 2.78 (d, J = 11.7 Hz, 1H, CH2NCH2), 2.61 (s, 2H, COCH2), 2.31 (t, J = 11.6 Hz, 1H, CH2NCH2), 2.22 (t, J = 11.8 Hz, 1H, CH2NCH2), 1.90 (q, J = 11.9, 11.5 Hz, 2H, CH2CCH2), 1.70 – 1.56 (m, 2H, CH2CCH2), 1.06 (td, J = 7.1, 1.8 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 184.20, 177.67, 165.69, 161.29 (d, J = 246.4 Hz), 155.82, 147.14, 144.38, 144.14, 140.73 (d, J = 3.2 Hz), 131.41 (d, J = 8.2 Hz), 125.25, 120.09 (d, J = 24.3 Hz), 116.01 (d, J = 21.0 Hz), 97.51, 60.26, 59.90, 58.68, 56.33, 50.18, 49.98, 43.99, 33.11, 14.49; EI-MS: 590.42 [M+H]+ ; C25H25BrFN5O4S [589.08].
ethyl 4-(2-bromo-4-fluorophenyl)-6-((8-oxo-2,7-diazaspiro[4.4]nonan-2-yl)methyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (4n)
Yield: 58.9 %; mp: 150-152°C; 1H NMR (400 MHz, DMSO-d6): δ 9.62 (d, J = 3.4 Hz, 1H, dihydropyrimidine-H), 8.00 – 7.93 (m, 2H, thiazole-H), 7.56 (dd, J = 8.4, 2.6 Hz, 1H, Ph-H), 7.46 – 7.31 (m, 1H, Ph-H), 7.26 – 7.19 (m, 1H, Ph-H), 6.02 (s, 1H, dihydropyrimidine-CH), 4.04 (s, 2H, dihydropyrimidine-CH2), 3.95 (q, J = 7.0 Hz, 2H, CH2CH3), 2.89 – 2.56 (m, 6H, CH2NCH2, NHCH2), 2.32 – 2.22 (m, 2H, COCH2), 1.95 – 1.87 (m, 2H, NCH2CH2C), 1.05 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 176.29, 165.63, 162.45, 161.24 (d, J = 243.7 Hz), 147.70, 144.19, 143.98, 140.64 (d, J = 2.9 Hz), 131.30 (d, J = 8.5 Hz), 125.24, 123.05 (d, J = 9.9 Hz), 120.08 (d, J = 24.3 Hz), 115.97 (d, J = 21.1 Hz), 96.80, 65.46, 59.84, 58.59, 53.93, 53.58, 53.42, 45.78, 43.58, 37.04, 14.47; EI-MS: 562.36 [M+H]+; C24H25BrFN5O3S [561.08].
ethyl 4-(2-bromo-4-fluorophenyl)-6-((3-oxo-2,8-diazaspiro[4.5]decan-8-yl)methyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (4o)
Yield: 75.5 %; mp: 140-142°C; 1H NMR (400 MHz, DMSO-d6): δ 10.16 (s, 1H, CONH), 9.66 (s, 1H, dihydropyrimidine-H), 7.98 (d, J = 29.5 Hz, 2H, thiazole-H), 7.54 (d, J = 5.8 Hz, 1H, Ph-H), 7.40 (m, 1H, Ph-H), 7.31 (m, 1H, Ph-H), 6.02 (s, 1H, dihydropyrimidine-CH), 4.55 (s, 1H, dihydropyrimidine-CH2), 3.95 (d, J = 6.5 Hz, 3H, dihydropyrimidine-CH2, CH2CH3), 3.54 (s, 1H, NHCH2), 3.24 (s, 1H, NHCH2), 3.08 (s, 2H, CH2NCH2), 2.08 (s, 2H, CH2NCH2), 1.91 (s, 2H, CH2CCH2), 1.64 (s, 2H, CH2CCH2), 1.14 – 0.99 (m, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 175.95, 165.62, 162.51, 161.17 (d, J = 235.4 Hz), 158.06, 144.41, 144.14, 140.70 (d, J = 5.1 Hz), 131.42 (d, J = 9.0 Hz), 125.24, 122.43 (d, J = 17.1 Hz), 120.13 (d, J = 25.9 Hz), 116.25 (d, J = 23.1 Hz), 97.28, , 60.67, 59.87, 58.62, 56.50, 52.27, 50.70, 37.07, 36.43, 14.45; EI-MS: 576.43 [M+H]+; C25H27BrFN5O3S [575.10].
ethyl 4-(2-bromo-4-fluorophenyl)-6-((1-oxo-2,7-diazaspiro[3.5]nonan-7-yl)methyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (4p)
Yield: 32.8 %; mp: 135-137°C; 1H NMR (400 MHz, CDCl3): δ 9.69 (s, 1H, dihydropyrimidine-H), 7.86 (s, 1H, thiazole-H), 7.47 (s, 1H, thiazole-H), 7.31 (s, 2H, Ph-H), 7.00 (s, 1H, Ph-H), 6.16 (s, 1H, dihydropyrimidine-CH), 5.69 (s, 1H, CONH), 4.03 (s, 2H, CH2CH3), 3.22 (s, 2H, NHCH2), 2.17- 2.04 (m, 4H, CH2NCH2) 1.43 - 1.10 (m, 7H, CH2CCH2, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 170.81, 165.37, 162.52, 161.30 (d, J = 245.8 Hz), 158.38, 144.39, 144.20, 140.64 (d, J = 3.6 Hz), 131.52 (d, J = 15.5 Hz), 125.29, 122.94 (d, J = 14.9 Hz), 120.12 (d, J = 25.7 Hz), 116.04 (d, J = 11.5 Hz), 97.33, 60.23, 58.62, 56.49, 56.28, 51.16, 47.63, 31.43, 14.56; EI-MS: 562.40 [M+H]+; C24H25BrFN5O3S [561.08].
ethyl 6-((7-benzyl-6,8-dioxo-2,7-diazaspiro[4.4]nonan-2-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (4q)
Yield: 55.7 %; mp: 74-78°C; 1H NMR (400 MHz, CDCl3): δ 9.48 (d, J = 9.7 Hz, 1H, dihydropyrimidine-H), 7.86 (dd, J = 10.1, 3.1 Hz, 1H, thiazole-H), 7.43 (s, 1H, thiazole-H), 7.30 (t, J = 7.2 Hz, 6H, Ph-H), 7.26 – 7.21 (m, 1H, Ph-H), 7.00 – 6.89 (m, 1H, Ph-H), 6.17 (d, J = 4.2 Hz, 1H, dihydropyrimidine-CH), 4.66 (d, J = 10.0 Hz, 2H, dihydropyrimidine-CH2), 4.17 (d, J = 2.6 Hz, 2H, Ph-CH2), 4.07 – 3.99 (m, 2H, CH2CH3), 3.25 – 2.87 (m, 6H, CH2NCH2, CCH2CO), 2.55 (m, 1H, NCH2CH2C), 1.96 (dtd, J = 12.4, 7.5, 4.4 Hz, 1H, NCH2CH2C), 1.17 – 1.08 (m, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 181.41, 176.32, 165.63, 162.52, 161.25 (d, J = 243.4 Hz), 144.45, 144.27, 140.87 (d, J = 3.4 Hz), 131.51 (d, J = 8.7 Hz), 128.99, 127.86, 127.84, 127.78, 127.75, 125.23, 123.03 (d, J = 9.8 Hz), 120.01 (d, J = 24.6 Hz), 115.94 (d, J = 20.7 Hz), 96.98, 59.86, 58.59, 54.10, 53.35, 49.74, 43.84, 42.04, 37.11, 14.45; EI-MS: 666.49 [M+H]+; C31H29BrFN5O4S [665.11].
ethyl 6-((1,4-dioxa-8-azaspiro[4.5]decan-8-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (4r)
Yield: 24.1 %; mp: 142-150°C; 1H NMR (400 MHz, DMSO-d6): δ 9.74 (s, 1H, dihydropyrimidine-H), 7.99 (dd, J = 34.5, 3.1 Hz, 2H, thiazole-H), 7.57 (dd, J = 8.6, 2.6 Hz, 1H, Ph-H), 7.39 (dd, J = 8.7, 6.2 Hz, 1H, Ph-H), 7.22 (td, J = 8.5, 2.6 Hz, 1H, Ph-H), 6.03 (s, 1H, dihydropyrimidine-CH), 4.00 – 3.84 (m, 8H, CH2CH3, dihydropyrimidine-CH2, OCH2CH2O), 2.61 (s, 4H, CH2NCH2), 1.73 (d, J = 5.2 Hz, 4H CH2CCH2), 1.05 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.65, 162.54, 161.25 (d, J = 248.2 Hz), 147.29, 144.41, 144.12, 140.65 (d, J = 3.3 Hz), 131.37 (d, J = 8.7 Hz), 125.21, 123.05 (d, J = 9.7 Hz), 120.06 (d, J = 24.4 Hz), 115.96 (d, J = 20.9 Hz), 106.33, 97.18, 64.17, 59.83, 58.65, 56.50, 55.79, 51.70, 35.27, 14.46; EI-MS: 565.58 [M+H]+; C24H26BrFN4O4S [564.08].
4.1.7. General procedure for the synthesis of 5(a-k)
Compound 4 (a-k) was dissolved in 25 mL DCM, and then 2.5 mL TFA was added and stirred at room temperature overnight. After the reaction, the excess solvent was removed under reduced pressure. Water (50 mL) and DCM (25 mL × 3) were added and extracted. The organic phases were collected, combined, and extracted with saturated sodium chloride solution (25 mL) and dried with anhydrous magnesium sulfate. It was filtered and concentrated under reduced pressure and purified by preparative TLC with the solvent system of DCM: MeOH = 10:1. Compound 5 (a-k) was obtained by recrystallization using a dichloromethane-n-hexane system.
ethyl 4-(2-bromo-4-fluorophenyl)-6-((7-oxo-2,6-diazaspiro[3.4]octan-6-yl)methyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5a)
Yield: 92.0 %; mp: 160-178°C; 1H NMR (400 MHz, DMSO-d6): δ 8.82 (s, 1H, dihydropyrimidine-H), 8.01 (s, 2H, thiazole-H), 7.57 (s, 1H, Ph-H), 7.43 (s, 1H, Ph-H), 7.27 (s, 1H, Ph-H), 5.98 (d, J = 38.6 Hz, 1H, dihydropyrimidine-CH), 4.68 – 4.55 (m, 2H, dihydropyrimidine- CH2), 4.00 (d, J = 6.9 Hz, 2H, CH2CH3), 3.95 – 3.57 (m, 6H, CH2NCH2, NCH2C), 2.72 (s, 2H, COCH2), 1.08 (d, J = 6.9 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 172.62, 165.42, 162.89, 161.34 (d, J = 247.0 Hz), 155.53, 144.82, 143.98, 140.45 (d, J = 3.6 Hz), 132.02 (d, J = 11.4 Hz), 125.25, 123.00 (d, J = 5.9 Hz), 120.09 (d, J = 24.0 Hz), 116.33 (d, J = 24.8 Hz), 105.24, 60.28, 58.52, 57.83, 56.49, 56.20, 52.08, 44.76, 37.80, 14.50; EI-MS: 548.23 [M+H]+; C23H23BrFN5O3S [547.07].
ethyl 4-(2-bromo-4-fluorophenyl)-6-((1-oxo-2,7-diazaspiro[4.4]nonan-2-yl)methyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5b)
Yield: 45.8 %; mp: 168-170°C; 1H NMR (400 MHz, CDCl3): δ 8.95 (d, J = 42.5 Hz, 1H, dihydropyrimidine-H), 7.82 (s, 1H, thiazole-H), 7.50 (s, 1H, thiazole-H), 7.30 (m, 2H, Ph-H), 7.09 (s, 1H, Ph-H), 6.12 (d, J = 36.3 Hz, 1H, dihydropyrimidine-CH), 4.84 (m, 2H, dihydropyrimidine- CH2), 4.08 (m, 2H, CH2CH3), 3.90 (m, 2H, NCH2CH2C), 3.25 (m, 2H, NCH2CH2C), 2.84 (m, 2H, CCH2CH2NH), 2.42 (s, 4H, CH2NCH2), 1.13 (s, 3H, CH2CH3); EI-MS: 562.44 [M+H]+; C24H25BrFN5O3S [561.08].
ethyl 4-(2-bromo-4-fluorophenyl)-6-((3-oxo-2,7-diazaspiro[4.4]nonan-2-yl)methyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5c)
Yield: 94.0 %; mp: 135-140°C; 1H NMR (400 MHz, CDCl3): δ 8.97 (s, 1H, dihydropyrimidine-H), 7.84 (dd, J = 7.9, 3.2 Hz, 1H, thiazole-H), 7.52 (d, J = 3.4 Hz, 1H, thiazole-H), 7.46 – 7.27 (m, 2H, Ph-H), 7.12 – 6.95 (m, 1H, Ph-H), 6.12 (d, J = 39.3 Hz, 1H, dihydropyrimidine-CH), 4.92 – 4.66 (m, 2H, dihydropyrimidine-CH2), 4.11 – 3.99 (m, 2H, CH2CH3), 3.66 – 3.47 (m, 2H, NCH2C), 3.33 (m, 2H, CCH2CH2NH), 3.25 – 3.12 (m, 2H, CCH2NH), 3.12 – 3.04 (m, 1H, NH), 2.76 – 2.49 (m, 2H, COCH2C), 1.96 (t, J = 7.5 Hz, 2H, CCH2CH2NH), 1.13 (td, J = 7.0, 4.6 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6) δ 173.32, 165.57, 162.90, 161.53 (d, J = 231.2 Hz), 150.52, 144.84, 143.99, 140.57 (d, J = 3.0 Hz), 132.03 (d, J = 7.7 Hz), 125.28, 123.68 (d, J = 8.5 Hz), 120.09 (d, J = 24.8 Hz), 116.06 (d, J = 25.5 Hz), 105.26, 60.27, 56.49, 55.58, 52.10, 47.31, 44.68, 36.94, 29.49, 14.45; EI-MS: 562.45 [M+H]+; C24H25BrFN5O3S [561.08].
ethyl 6-((3,9-diazaspiro[5.5]undecan-3-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5d)
Yield: 54.0 %; mp: 185-187°C; NMR (400 MHz, DMSO-d6): δ 9.67 (s, 1H, dihydropyrimidine-H), 9.11 (s, 1H, NH), 8.01 (d, J = 3.2 Hz, 1H, thiazole-H), 7.95 (d, J = 3.2 Hz, 1H, thiazole-H), 7.57 (dd, J = 8.5, 2.7 Hz, 1H, Ph-H), 7.42 – 7.31 (m, 1H, Ph-H), 7.23 (t, J = 8.6 Hz, 1H, Ph-H), 6.03 (s, 1H, dihydropyrimidine-CH), 4.01 – 3.82 (m, 4H, dihydropyrimidine- CH2, CH2CH3), 3.22 (s, 4H, CH2NHCH2), 3.01 (t, J = 5.6 Hz, 4H, CH2NCH2), 1.66 (t, J = 5.8 Hz, 4H, CH2CCH2), 1.57 (d, J = 5.5 Hz, 4H, CH2CCH2), 1.05 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6) δ 165.64, 162.49, 161.26 (d, J = 247.6 Hz), 144.42, 144.06, 140.62 (d, J = 2.9 Hz), 131.31 (d, J = 12.1 Hz), 125.94, 123.04 (d, J = 9.5 Hz), 120.07 (d, J = 26.3 Hz), 116.00 (d, J = 20.0 Hz), 100.79, 59.85, 58.62, 49.08, 31.75, 30.86, 29.55, 29.45, 29.16, 28.73, 27.02, 22.57, 14.47; EI-MS: 576.91 [M+H]+; C26H31BrFN5O2S [575.14].
ethyl 6-((2,8-diazaspiro[4.5]decan-8-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5e)
Yield: 56.0 %; mp: 121-124°C; 1H NMR (400 MHz, DMSO-d6): δ 9.64 (s, 1H, dihydropyrimidine-H), 9.42 (s, 1H, NH), 8.01 (t, J = 2.4 Hz, 1H, thiazole-H), 7.98 – 7.92 (m, 1H, thiazole-H), 7.57 (dd, J = 8.5, 2.3 Hz, 1H, Ph-H), 7.38 (t, J = 7.5 Hz, 1H, Ph-H), 7.23 (t, J = 8.5 Hz, 1H, Ph-H), 6.02 (s, 1H, dihydropyrimidine-CH), 4.02 – 3.83 (m, 4H, dihydropyrimidine- CH2, CH2CH3), 3.22 (t, J = 7.4 Hz, 2H, CCH2CH2NH), 3.00 (s, 2H, CCH2NH), 1.81 (t, J = 7.4 Hz, 2H, CH2NCH2), 1.66 (s, 4H, CH2NCH2, CH2CCH2), 1.25 (d, J = 12.2 Hz, 4H, CH2CCH2, CCH2CH2NH), 1.05 (t, J = 7.0 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.65, 162.52, 161.27 (d, J = 251.8 Hz), 147.29, 144.39, 144.12, 140.66 (d, J = 3.3 Hz), 131.35 (d, J = 8.4 Hz), 125.24, 123.04 (d, J = 9.6 Hz), 120.06 (d, J = 24.1 Hz), 115.99 (d, J = 20.8 Hz), 97.33, 59.84, 58.63, 56.22, 51.02, 43.41, 34.89, 31.76, 29.50, 29.05, 27.02, 22.57, 14.47; EI-MS: 562.95 [M+H]+; C25H29BrFN5O2S [561.12].
ethyl 6-((2,7-diazaspiro[3.5]nonan-7-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5f)
Yield: 33.0 %; mp: 130-133°C; 1H NMR (400 MHz, DMSO-d6): δ 9.60 (s, 1H, dihydropyrimidine-H), 9.19 (s, 1H, NH), 8.03 – 7.98 (m, 1H, thiazole-H), 7.95 (d, J = 3.4 Hz, 1H, thiazole-H), 7.61 – 7.54 (m, 1H, Ph-H), 7.38 (t, J = 7.4 Hz, 1H, Ph-H), 7.23 (t, J = 8.5 Hz, 1H, Ph-H), 6.03 (s, 1H, dihydropyrimidine-CH), 3.95 (q, J = 7.0 Hz, 2H, CH2CH3), 3.90 – 3.79 (m, 2H, dihydropyrimidine- CH2), 3.71 (s, 2H, CH2NHCH2), 2.46 (s, 2H, CH2NHCH2), 1.86 (d, J = 5.4 Hz, 4H, CH2NCH2), 1.24 (s, 4H, CH2CCH2), 1.05 (t, J = 7.0 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.64, 162.53, 161.28 (d, J = 252.9 Hz), 147.18, 144.33, 144.09, 140.67 (d, J = 3.2 Hz), 131.37 (d, J = 8.8 Hz), 125.25, 123.04 (d, J = 9.7 Hz), 120.11(d, J = 24.2 Hz), 115.99 (d, J = 21.3 Hz), 97.33, 59.84, 58.62, 56.10, 54.54, 50.14, 36.09, 34.86, 14.43; EI-MS: 548.96 [M+H]+; C24H27BrFN5O2S [547.11].
ethyl 6-((2-amino-7-azaspiro[3.5]nonan-7-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5g)
Yield: 65.0 %; mp: 110-113°C; 1H NMR (400 MHz, DMSO-d6): δ 9.68 (s, 1H, dihydropyrimidine-H), 8.05 – 7.98 (m, 1H, thiazole-H), 7.94 (t, J = 2.5 Hz, 1H, thiazole-H), 7.57 (dd, J = 8.6, 2.5 Hz, 1H, Ph-H), 7.37 (dd, J = 8.6, 6.1 Hz, 1H, Ph-H), 7.22 (dd, J = 10.0, 7.5 Hz, 1H, Ph-H), 6.02 (s, 1H, dihydropyrimidine-CH), 3.94 (q, J = 7.0 Hz, 2H, CH2CH3), 3.90 – 3.77 (m, 2H, dihydropyrimidine- CH2), 3.55 (q, J = 7.9 Hz, 2H, CH2NCH2), 2.39 (s, 2H, CH2CHCH2), 2.13 (t, J = 9.8 Hz, 2H, CH2CHCH2), 1.90 (d, J = 1.7 Hz, 1H, CH2CHCH2), 1.79 – 1.67 (m, 2H, CH2CHCH2), 1.63 (t, J = 6.0 Hz, 4H, CH2CCH2), 1.04 (td, J = 7.1, 1.7 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.65, 162.54, 161.25 (d, J = 248.5 Hz), 147.44, 144.37, 144.14, 140.68 (d, J = 3.2 Hz), 131.35 (d, J = 8.7 Hz), 125.22, 123.04 (d, J = 9.3 Hz), 120.06 (d, J = 24.6 Hz), 115.98 (d, J = 21.1 Hz), 97.15, 59.81, 58.63, 56.44, 50.81, 50.58, 41.12, 38.66, 36.21, 31.76, 14.47; EI-MS: 562.87 [M+H]+; C25H29BrFN5O2S [561.12].
ethyl 6-((2,8-diazaspiro[4.5]decan-2-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5h)
Yield: 51.0 %; mp: 85-88°C; 1H NMR (400 MHz, DMSO-d6): δ 9.62 (s, 1H, dihydropyrimidine-H), 7.97 (dd, J = 11.3, 3.0 Hz, 2H, thiazole-H), 7.58 (dd, J = 8.7, 2.5 Hz, 1H, Ph-H), 7.40 (t, J = 7.5 Hz, 1H, Ph-H), 7.24 (t, J = 8.6 Hz, 1H, Ph-H), 6.04 (s, 1H, dihydropyrimidine-CH), 4.02 (s, 2H, dihydropyrimidine-CH2), 3.97 (q, J = 7.1 Hz, 2H, CH2CH3), 3.01 (tq, J = 13.5, 7.9, 7.5 Hz, 4H, CH2NHCH2), 2.90 (d, J = 7.8 Hz, 1H, NCH2C), 2.71 (d, J = 8.9 Hz, 2H, NCH2CH2C), 2.35 (d, J = 9.6 Hz, 1H, NCH2C), 1.92 (d, J = 1.6 Hz, 1H, NCH2CH2C), 1.87 (d, J = 14.0 Hz, 1H, NCH2CH2C), 1.75 (t, J = 7.6 Hz, 4H, CH2CCH2), 1.06 (t, J = 7.0 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.64, 162.44, 161.23 (d, J = 241.8 Hz), 147.65, 144.44, 144.01, 140.59 (d, J = 3.3 Hz), 131.36 (d, J = 7.2 Hz), 125.27, 123.05 (d, J = 10.1 Hz), 120.07 (d, J = 24.3 Hz), 115.99 (d, J = 21.0 Hz), 96.74, 59.84, 58.61, 56.47, 53.39, 41.37, 41.31, 36.63, 33.54, 31.75, 29.55, 29.50, 14.49; EI-MS: 562.91 [M+H]+; C25H29BrFN5O2S [561.12].
ethyl 6-((2,7-diazaspiro[4.4]nonan-2-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5i)
Yield: 66.0 %; mp: 87-88°C; 1H NMR (400 MHz, DMSO-d6): δ 9.60 (s, 1H, dihydropyrimidine-H), 8.00 (d, J = 3.1 Hz, 1H, thiazole-H), 7.96 (d, J = 3.1 Hz, 1H, thiazole-H), 7.57 (d, J = 8.8 Hz, 1H, Ph-H), 7.41 (q, J = 8.1 Hz, 1H, Ph-H), 7.26 (t, J = 8.8 Hz, 1H, Ph-H), 6.03 (s, 1H, dihydropyrimidine-CH), 4.04 (s, 2H, dihydropyrimidine-CH2), 3.97 (q, J = 7.1 Hz, 2H, CH2CH3), 3.26 – 3.07 (m, 4H, CH2NHCH2), 2.92 – 2.61 (m, 4H, CH2NCH2), 2.01 (tq, J = 20.3, 7.1, 6.6 Hz, 3H, NCH2CH2C, NHCH2CH2C), 1.90 – 1.81 (m, 1H, NHCH2CH2C), 1.06 (t, J = 7.1 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.63, 162.47, 161.25 (d, J = 246.3 Hz), 147.50, 144.42, 144.01, 140.60 (d, J = 10.2 Hz), 131.43 (d, J = 23.3 Hz), 125.95, 122.98, 120.07 (d, J = 25.0 Hz), 116.11 (d, J = 25.6 Hz), 96.94, 63.35, 59.88, 58.56, 56.47, 54.50, 48.89, 44.12, 36.21, 34.97, 14.48; EI-MS: 548.37 [M+H]+; C24H27BrFN5O2S [547.11].
ethyl 6-((2,6-diazaspiro[3.4]octan-6-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5j)
Yield: 11.3 %; mp: 127-130°C; 1H NMR (400 MHz, DMSO-d6): δ 9.55 (s, 1H, dihydropyrimidine-H), 8.07 – 7.87 (m, 2H, thiazole-H), 7.59 (dd, J = 14.7, 7.6 Hz, 1H, Ph-H), 7.40 (t, J = 7.4 Hz, 1H, Ph-H), 7.32 (d, J = 6.3 Hz, 1H, Ph-H), 6.03 (s, 1H, dihydropyrimidine-CH), 4.02 (s, 2H, dihydropyrimidine-CH2), 3.96 (q, J = 7.2 Hz, 2H, CH2CH3), 3.82 (t, J = 13.6 Hz, 4H, CH2NHCH2), 2.88 (s, 2H, NHCH2C), 2.74 (dd, J = 16.5, 8.4 Hz, 2H, NCH2CH2C), 2.00 (m, 2H, NCH2CH2C), 1.06 (t, J = 7.0 Hz, 3H, CH2CH3); 13C NMR (100 MHz, DMSO-d6): δ 165.63, 162.51, 161.25 (d, J = 243.2 Hz), 147.33, 144.41, 144.00, 140.60 (d, J = 2.6 Hz), 131.34 (d, J = 8.4 Hz), 125.28, 123.03 (d, J = 11.3 Hz), 120.08 (d, J = 24.2 Hz), 116.08 (d, J = 21.1 Hz), 97.23, 72.81, 70.26, 64.09, 59.89, 58.57, 56.75, 53.29, 43.25, 36.04, 14.46; EI-MS: 534.01 [M+H]+; C23H25BrFN5O2S [533.09].
ethyl 6-((2,6-diazaspiro[3.3]heptan-2-yl)methyl)-4-(2-bromo-4-fluorophenyl)-2-(thiazol-2-yl)-1,4-dihydropyrimidine-5-carboxylate (5k)
Yield: 11.7 %; mp: 147-150°C; 1H NMR (400 MHz, DMSO-d6): δ 9.39 (s, 1H, dihydropyrimidine-H), 9.28 (s, 1H, NH), 8.01 (s, 1H, thiazole-H), 7.96 (s, 1H, thiazole-H), 7.59 (dd, J = 15.5, 7.6 Hz, 1H, Ph-H), 7.37 – 7.29 (m, 2H, Ph-H), 7.25 (d, J = 8.8 Hz, 1H, Ph-H), 6.00 (s, 1H, dihydropyrimidine-CH), 4.23 - 4.08 (m, 4H, dihydropyrimidine- CH2, CH2CH3), 3.96 (d, J = 5.1 Hz, 4H, CH2NHCH2), 3.53 (d, J = 9.9 Hz, 4H, CH2NCH2), 1.06 (t, J = 7.2 Hz, 3H, CH2CH3); EI-MS: 520.08 [M+H]+; C22H23BrFN5O2S [519.07].
4.2. Biological evaluation
4.2.1. Cells employed
HepDES19 cells are a derivative of the HepG2 (human hepatoblastoma) cell line stably transfected with an HBV genotype D genome under the control of a tetracycline-repressible promoter [40]. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 media supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S), and 1 μg/mL tetracycline. Expression of HBV pgRNA was synchronously induced by removing tetracycline from the culture medium.
4.2.2. HBV replication inhibition assay
HBV replication inhibition assays were carried out in the same manner as described previously[41]. HepDES19 cells were seeded in 96-well plates in the absence of tetracycline at 4 x 104 cells per well. Compound dilutions were added 48 hours after induction of HBV replication in 1% DMSO, and cells were incubated for 3 days at 37°C in 5% CO2. Cells were washed with phosphate-buffered saline (PBS) and lysed with lysis buffer (10 mM Tris pH 7.4, 1% Tween20, 150 mM NaCl) and incubated at 20-23°C on an orbital shaker at 350 rpm for 40 minutes. Cellular lysates were centrifuged at 3300 x g for 5 minutes. Supernatants were transferred to a 96-well PCR plate and mixed with 20 units of micrococcal nuclease and 100 μM CaCl2. The lysates were incubated at 37°C for 1 hour, then at 70°C for 10 min to inactivate the nuclease. Qiagen protease (0.005 Anson units) was added to the lysates and incubated overnight before inactivating the protease at 95°C for 10 minutes.
Lysates were used as the template for strand-preferential quantitative polymerase chain reaction (qPCR) analysis as previously described[41]. Breifly, qPCRwas performed with 40 cycles of 95 °C for 15 s and 60 °C for 1 minute employing the Kappa Probe Force universal PCR master mix. The primers and probe for the plus-polarity DNA strand were 5’CATGAACAAGAGATGATTAGGCAGAG3’,5’GGAGGCTGTAGGCATAAATTGG3’, and 5’/56-FAM/CTGCGCACC/ZEN/AGCACCATGCA/3IABkFQ. Primers and probe for the minus-polarity DNA strand were 5’GCAGATGAGAAGGCACAGA3’, 5’CTTCTCCGTCTGCCGTT3’, and 5’/56-FAM/AGTCCGCGT/ZEN/AAAGAGAGGTGCG/3IABkFQ. Fifty percent effective concentrations (EC50) values were calculated for the plus-polarity DNA with GraphPad Prism using a four-parameter log(inhibitor)-versus-response algorithm with the bottom value set to zero.
4.2.3. Cytotoxicity assays
Compound mediated cytotoxicity was measured in HepDES19 cells using the CellTiter 96™ Aqueous Non-Radioactive Cell Proliferation Assay (MTS) as previously described [42]. Cells were seeded at 1 x 104 cells per well in a 96 well plate in the absence of tetracycline. Compounds were applied to the cells two days later in 1% DMSO and incubated for 3 days. Fifty percent cytotoxic concentration (CC50) values were calculated using GraphPad Prism with the four-parameter log(inhibitor)-versus-response algorithm with the bottom value set to zero.
Acknowledgements
We gratefully acknowledge financial support from the Shandong Provincial Key research and development project (No. 2019JZZY021011), Science Foundation for Outstanding Young Scholars of Shandong Province (ZR2020JQ31), Foreign cultural and educational experts Project (GXL20200015001), Qilu Young Scholars Program of Shandong University and the Taishan Scholar Program at Shandong Province. Biological analyses were supported by NIH grant R01AI122669 to JET.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Worl Health Organization. Fact sheets: Hepatitis B, available from: https://www.who.int/zh/news-room/fact-sheets/detail/hepatitis-b, (accessed on August 2, 2021).
- [2].Worl Health Organization. Global hepatitis report, 2017, available from: https://www.who.int/publications/i/item/global-hepatitis-report-2017, (accessed on August 2, 2021).
- [3].Rabe B, Vlachou A, Panté N, Helenius A, Kann M, Nuclear import of hepatitis B virus capsids and release of the viral genome, Proc. Natl. Acad. Sci. U.S.A 100 (2003) 9849–9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nassal M, The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly, J. virology, 66 (1992) 4107–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Basagoudanavar SH, Perlman DH, Hu J, Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation, J. Virol 81 (2007) 1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Durantel D, Zoulim F, New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus, J. Hepatol 64 (2016) S117–S131. [DOI] [PubMed] [Google Scholar]
- [7].Ren Q, Liu X, Yan G, Nie B, Zou Z, Li J, Chen Y, Wei Y, Huang J, Luo Z, 3-((R)-4-(((R)-6-(2-Bromo-4-fluorophenyl)-5-(ethoxycarbonyl)-2-(thiazol-2-yl)-3, 6-dihydropyrimidin-4-yl) methyl) morpholin-2-yl) propanoic Acid (HEC72702), a Novel Hepatitis B Virus Capsid Inhibitor Based on Clinical Candidate GLS4, J. Med. Chem 61 (2018) 1355–1374. [DOI] [PubMed] [Google Scholar]
- [8].Ren Q, Liu X, Luo Z, Li J, Wang C, Goldmann S, Zhang J, Zhang Y, Discovery of hepatitis B virus capsid assembly inhibitors leading to a heteroaryldihydropyrimidine based clinical candidate (GLS4), Bioorg. Med. Chem 25 (2017) 1042–1056. [DOI] [PubMed] [Google Scholar]
- [9].Qiu Z, Lin X, Zhang W, Zhou M, Guo L, Kocer B, Wu G, Zhang Z, Liu H, Shi H, Discovery and pre-clinical characterization of third-generation 4-H heteroaryldihydropyrimidine (HAP) analogues as hepatitis B virus (HBV) capsid inhibitors, J. Med. Chem 60 (2017) 3352–3371. [DOI] [PubMed] [Google Scholar]
- [10].Qiu Z, Lin X, Zhou M, Liu Y, Zhu W, Chen W, Zhang W, Guo L, Liu H, Wu G, Design and synthesis of orally bioavailable 4-methyl heteroaryldihydropyrimidine based hepatitis B virus (HBV) capsid inhibitors, J. Med. Chem 59 (2016) 7651–7666. [DOI] [PubMed] [Google Scholar]
- [11].Wang XY, Wei ZM, Wu GY, Wang JH, Zhang YJ, Li J, Zhang HH, Xie XW, Wang X, Wang ZH, In vitro inhibition of HBV replication by a novel compound, GLS4, and its efficacy against adefovir-dipivoxil-resistant HBV mutations, Antivir. Ther 17 (2012) 793–803. [DOI] [PubMed] [Google Scholar]
- [12].Zhang H, Wang F, Zhu X, Chen Y, Chen H, Li X, Wu M, Li C, Liu J, Zhang Y, Ding Y, Niu J, Antiviral Activity and Pharmacokinetics of the HBV Capsid Assembly Modulator GLS4 in Patients with Chronic HBV Infection, Clin Infect Dis, 73 (2021) 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li X, Zhou K, He H, Zhou Q, Sun Y, Hou L, Shen L, Wang X, Zhou Y, Gong Z, Design, synthesis, and evaluation of tetrahydropyrrolo [1, 2-c] pyrimidines as capsid assembly inhibitors for HBV treatment, ACS Med. Chem. Lett 8 (2017) 969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhou X, Li L, Deng P, Chen X, Zhong D, Characterization of metabolites of GLS4 in humans using ultrahigh-performance liquid chromatography/quadrupole time-of-flight mass spectrometry, Rapid Commun. Mass Spectrom 27 (2013) 2483–2492. [DOI] [PubMed] [Google Scholar]
- [15].Zhao N, Jia B, Zhao H, Xu J, Sheng X, Luo L, Huang Z, Wang X, Ren Q, Zhang Y, Zhao X, Cui Y, A First-in-Human Trial of GLS4, a Novel Inhibitor of Hepatitis B Virus Capsid Assembly, following Single- and Multiple-Ascending-Oral-Dose Studies with or without Ritonavir in Healthy Adult Volunteers, Antimicrob. Agents. Chemother 64 (2019) e01686–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zheng Y, Tice CM, Singh SB, The use of spirocyclic scaffolds in drug discovery, Bioorg. Med. Chem. Lett 24 (2014) 3673–3682. [DOI] [PubMed] [Google Scholar]
- [17].Zheng Y-J, Tice CM, The utilization of spirocyclic scaffolds in novel drug discovery, Expert. Opin. Drug. Disco 11 (2016) 831–834. [DOI] [PubMed] [Google Scholar]
- [18].Link JO, Taylor JG, Xu L, Mitchell M, Guo H, Liu H, Kato D, Kirschberg T, Sun J, Squires N, Discovery of ledipasvir (GS-5885): a potent, once-daily oral NS5A inhibitor for the treatment of hepatitis C virus infection, J. Med. Chem 57 (2014) 2033–2046. [DOI] [PubMed] [Google Scholar]
- [19].Patel P, Leeder JS, Piquette-Miller M, Dupuis LL, Aprepitant and fosaprepitant drug interactions: a systematic review, Br J Clin Pharmacol, 83 (2017) 2148–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pasricha PJ, Yates KP, Sarosiek I, McCallum RW, Abell TL, Koch KL, Nguyen LAB, Snape WJ, Hasler WL, Clarke JO, Dhalla S, Stein EM, Lee LA, Miriel LA, Van Natta ML, Grover M, Farrugia G, Tonascia J, Hamilton FA, Parkman HP, Aprepitant Has Mixed Effects on Nausea and Reduces Other Symptoms in Patients With Gastroparesis and Related Disorders, Gastroenterology, 154 (2018) 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Duffy RA, Morgan C, Naylor R, Higgins GA, Varty GB, Lachowicz JE, Parker EM, Rolapitant (SCH 619734): a potent, selective and orally active neurokinin NK1 receptor antagonist with centrally-mediated antiemetic effects in ferrets, Pharmacol. Biochem. Behav 102 (2012) 95–100. [DOI] [PubMed] [Google Scholar]
- [22].Lei G, Kou B, Lin X, Shen H, Shi H, Yan S, Zhang W, Zhou M, Zhu W, Novel 6-fused heteroaryldihyfropyrimidines for the treatment and prophylaxis of hepatitis B virus infection, Patent, 2016. [Google Scholar]
- [23].Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo J-T, Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation, J. Virol 81 (2007) 12472–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tinworth CP, Young RJ, Facts, patterns, and principles in drug discovery: Appraising the rule of 5 with measured physicochemical data, J. Med. Chem 63 (2020) 10091–10108. [DOI] [PubMed] [Google Scholar]
- [25].Daina A, Michielin O, Zoete V, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep 7 (2017) 42717, available from: http://www.swissadme.ch/, (accessed on August 2, 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Daina A, Michielin O, Zoete V, iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach, J. Chem. Inf. Model 54 (2014) 3284–3301. [DOI] [PubMed] [Google Scholar]
- [27].Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, Li Y, Wang R, Lai L, Computation of octanol– water partition coefficients by guiding an additive model with knowledge, J. Chem. Inf. Model 47 (2007) 2140–2148. [DOI] [PubMed] [Google Scholar]
- [28].Wildman SA, Crippen GM, Prediction of physicochemical parameters by atomic contributions, J. Chem. Inf. Model. Comput. Sci 39 (1999) 868–873. [Google Scholar]
- [29].Moriguchi I, HIRONO S, LIU Q, NAKAGOME I, MATSUSHITA Y, Simple method of calculating octanol/water partition coefficient, Chemical and pharmaceutical bulletin, 40 (1992) 127–130. [Google Scholar]
- [30].Moriguchi I, Hirono S, Nakagome I, Hirano H, Comparison of reliability of log P values for drugs calculated by several methods, Chem. Pharm. Bull 42 (1994) 976–978. [Google Scholar]
- [31].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug. Deliv. Rev, 23 (1997) 3–25. [DOI] [PubMed] [Google Scholar]
- [32].SILICOS-IT, available from: http://www.silicos-it.com, (accessed on August 2, 2021).
- [33].Delaney JS, ESOL: estimating aqueous solubility directly from molecular structure, J. Chem. Inf. Model. Comput. Sci 44 (2004) 1000–1005. [DOI] [PubMed] [Google Scholar]
- [34].Ali J, Camilleri P, Brown MB, Hutt AJ, Kirton SB, Revisiting the general solubility equation: in silico prediction of aqueous solubility incorporating the effect of topographical polar surface area, Journal of chemical information and modeling, 52 (2012) 420–428. [DOI] [PubMed] [Google Scholar]
- [35].Hou T, Wang J, Zhang W, Xu X, ADME evaluation in drug discovery. 7. Prediction of oral absorption by correlation and classification, J. Chem. Inf. Model 47 (2007) 208–218. [DOI] [PubMed] [Google Scholar]
- [36].Zhou X, Gao Z.-w., Meng J, Chen X.-y., Zhong D.-f., Effects of ketoconazole and rifampicin on the pharmacokinetics of GLS4, a novel anti-hepatitis B virus compound, in dogs, Acta Pharmacol. Sin 34 (2013) 1420–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhao N, Jia B, Zhao H, Xu J, Sheng X, Luo L, Huang Z, Wang X, Ren Q, Zhang Y, A first-in-human trial of GLS4, a novel inhibitor of hepatitis B virus capsid assembly, following single-and multiple-ascending-oral-dose studies with or without ritonavir in healthy adult volunteers, Antimicrob. Agents. Chemother 64 (2020) e01686–01619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].ADMET prediction, available from: http://admet.scbdd.com/calcpre/index/, (accessed on August 2, 2021).
- [39].admetSAR, available from: http://lmmd.ecust.edu.cn/admetsar2/, (accessed on August 2, 2021).
- [40].Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT, Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation, J Virol, 81 (2007) 12472–12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li Q, Edwards TC, Ponzar NL, Tavis JE, A mid-throughput HBV replication inhibition assay capable of detecting ribonuclease H inhibitors, J.Virol. Methods, (2021) 114127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Edwards TC, Lomonosova E, Patel JA, Li Q, Villa JA, Gupta AK, Morrison LA, Bailly F, Cotelle P, Giannakopoulou E, Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles, Antivir. Res 143 (2017) 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]