

Abstract
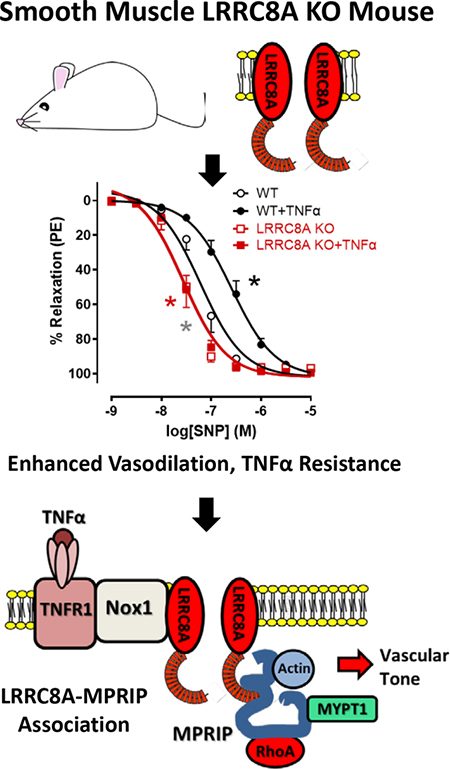
Leucine Rich Repeat Containing 8A (LRRC8A) volume regulated anion channels (VRACs) are activated by inflammatory and pro-contractile stimuli including tumor necrosis factor alpha (TNFα), angiotensin II and stretch. LRRC8A associates with NADPH oxidase 1 (Nox1) and supports extracellular superoxide production. We tested the hypothesis that VRACs modulate TNFα signaling and vasomotor function in mice lacking LRRC8A exclusively in vascular smooth muscle cells (VSMCs, Sm22α-Cre, Knockout). Knockout (KO) mesenteric vessels contracted normally but relaxation to acetylcholine (ACh) and sodium nitroprusside (SNP) was enhanced compared to wild type (WT). 48 hours of ex vivo exposure to TNFα (10ng/ml) enhanced contraction to norepinephrine (NE) and markedly impaired dilation to ACh and SNP in WT but not KO vessels. VRAC blockade (carbenoxolone, CBX, 100 μM, 20 min) enhanced dilation of control rings and restored impaired dilation following TNFα exposure. Myogenic tone was absent in KO rings. LRRC8A immunoprecipitation followed by mass spectroscopy identified 33 proteins that interacted with LRRC8A. Among them, the Myosin Phosphatase Rho-Interacting protein (MPRIP) links RhoA, MYPT1 and actin. LRRC8A-MPRIP co-localization was confirmed by confocal imaging of tagged proteins, Proximity Ligation Assays, and IP/western blots. siLRRC8A or CBX treatment decreased RhoA activity in VSMCs, and MYPT1 phosphorylation was reduced in KO mesenteries suggesting that reduced ROCK activity contributes to enhanced relaxation. MPRIP was a target of redox modification, becoming oxidized (sulfenylated) after TNFα exposure. Interaction of LRRC8A with MPRIP may allow redox regulation of the cytoskeleton by linking Nox1 activation to impaired vasodilation. This identifies VRACs as potential targets for treatment or prevention of vascular disease.
Keywords: Vasodilation, Inflammation, LRRC8A anion channel, Myosin Phosphatase Rho-Interacting protein, Vascular smooth muscle cell
Graphical Abstract
Introduction
Chronic vascular inflammatory condition induces cardiovascular diseases. Serum levels of tumor necrosis factor alpha (TNFα)(1), interleukin 1-beta (IL-1β)(2) and the pro-inflammatory vasoconstrictor angiotensin II (AngII)(3) are chronically elevated and inflammation correlates with severity of disease(4). In vascular smooth muscle cells (VSMCs) these signaling pathways all depend upon activation of the Nox1 NADPH oxidase(5, 6). As blood pressure rises, increased wall tension promotes further inflammation(7) in a reactive oxygen species (ROS)-dependent manner(8). In resistance vessels, stretch activates the autoregulatory “myogenic” contractile response of VSMCs that helps to normalize local blood flow. This response is also Nox1(9) and ROS-dependent(10). While acutely adaptive, myogenic tone can drive further increases in vascular resistance and support a positive feedback loop of inflammation and hypertension that drives disease progression(11). Thus, ROS that originate as Nox1-derived superoxide (O2−•) are critical mediators of both inflammatory and bio-mechanical signaling in VSMCs.
O2−• can increase vascular tone by inactivation of nitric oxide (NO•). It has also been linked to activation of the RhoA small GTPase, the primary regulator of Rho-associated kinase (ROCK) which is hyperactivated in multiple forms of experimental hypertension(12). ROCK regulates the calcium sensitivity of VSMC contraction by phosphorylating and inhibiting Myosin Phosphatase Target Subunit 1 (MYPT1), the regulatory component of the myosin light chain phosphatase (MLCP) complex. This increases MLC phosphorylation and promotes crossbridge cycling with actin.
Volume-regulated anion channels (VRACs) are composed of Leucine Rich Repeat Containing 8 (LRRC8) family proteins and are essential elements of a multi-protein signaling complex that also includes the type 1 TNFα receptor (TNFR1)(13), Nox1 and the redox-sensitive MAPKKK5, also known as Apoptosis Signaling-related Kinase 1 (ASK1)(14). Loss of either LRRC8A, the LRRC8 isoform that is required for all VRACs(15) or its LRRC8C partner(16) impaired Nox1 activation by TNFα. This disrupted multiple endpoints of downstream signaling in VSMCs including TNFα receptor endocytosis and Mitogen-Activated Protein Kinase (MAPK) and NF-κB activation. The basis for functional dependence of Nox1 on anion current is unknown. Extracellular scavenging of O2−• by superoxide dismutase (SOD) interfered with TNFα signaling, while catalase was without effect(15). Thus, O2−• from Nox1 clearly plays a role beyond serving as a substrate for conversion to H2O2.
Since loss or block of LRRC8A channels so effectively protected cultured VSMCs from TNFα-induced inflammation(15) we wanted to study this effect in intact blood vessels and created VSMC-specific LRRC8A knockout mice. KO mesenteric blood vessels had normal contractile responses but displayed remarkably enhanced vasodilation via multiple mechanisms, and completely lacked myogenic tone. TNFα signaling was completely abrogated. Immunoprecipitation of LRRC8A and mass spectroscopy identified Myosin Phosphatase Rho-Interacting Protein (MPRIP, a.k.a. M-RIP or p116Rip) as a binding partner for LRRC8A. MPRIP is a scaffolding protein that also binds to RhoA, MYPT1 and actin and is essential for regulation of the MLCP complex. Reduction of RhoA activity and ROCK-dependent MYPT1 phosphorylation in VSMCs with reduced LRRC8A protein levels or VRAC inhibition suggests that LRRC8A colocalizes TNFR1/Nox1-dependent O2−• production with the regulatory machinery controlling MLCP activity. This supports the novel concept that inflammatory signaling impacts VSMC contractility via redox signaling that depends upon LRRC8A anion channels.
Methods
Animals
Floxed LRRC8A mice (C57B6/129 mixed background) were provided by the Dr. Rajan Sah lab (Washington University, St. Louis, MO). VSMC-specific LRRC8A knockout (KO) mice were created by crossing these animals with SM22α-Cre mice (Jackson Laboratory, Bar Harbor, ME). Wild Type (WT) and KO littermates underwent experimentation at 12–17 weeks of age. For studies involving pharmacologic inhibition of VRAC, 12–15 week-old male C57BL/6 mice were obtained from Jackson Labs. All procedures were performed in accordance with the Guiding Principles in the Care and Use of Animals, approved by the Vanderbilt University Institutional Animal Care and Use Committee. The animals were housed on a 12-hour light/dark cycle and fed a standard chow diet with water ad libitum. Prior to blood vessel harvesting, mice were euthanized by placement in a sealed chamber with 100% CO2 provided at a flow rate of 2 L/min for 3 min. At that time, mice were monitored till they had > 1 min of respiratory cessation after which they underwent cervical dislocation. PCR analysis of tail-tip DNA was used to identify WT and LRRC8A KO mice.
Functional evaluation of mesenteric arteries
First- or second-order branches of the superior mesenteric arteries (~100 μM inner diameter) were excised, cleaned of fat and connective tissue and cut into 1 to 2 mm length-rings in an ice-cold physiological salt solution (PSS) consisting of 130 mmol/L NaCl, 4.7 mmol/L KCl, 1.18 mmol/L KH2PO4, 1.18 mmol/L MgSO4·7H2O, 1.56 mmol/L CaCl2·2H2O, 14.9 mmol/L NaHCO3, 5.6 mmol/L glucose, and 0.03 mmol/L EDTA. Mesenteric rings were either immediately mounted in wire myographs (Danish Myo Technology A/S, Aarhus, Denmark) or incubated in Dulbecco’s Modified Eagle Medium (DMEM; Life Technologies, Grand Island, NY) containing 0.5 % fetal bovine serum (FBS; Corning) with or without TNFα (10 ng/L) for 2 days. Incubated rings were subsequently mounted in wire myographs containing warmed (37°C), oxygenated (95% O2/5% CO2) PSS and allowed to equilibrate for 45 min under a passive force of 2 – 2.5 mN. Arterial viability was assessed by stimulation with 120 mmol/L KCl. After washing, rings were stimulated with phenylephrine (PE, 10−6 mol/L), followed by exposure to acetylcholine (ACh, 10−5 mol/L). A more than 80% relaxation response to ACh was taken as evidence of an intact endothelial layer. Contractile responses were then assessed by cumulative exposure to KCl (20–120 mM), PE (10−9 to 10−5 mol/L) or NE (10−9 to 10−5 mol/L). Endothelium-dependent relaxation was assessed on PE-contracted (10−6 mol/L) rings by cumulative addition of ACh (10−8 to 3×10−5 mol/L), while endothelium-independent relaxation was tested using sodium nitroprusside (SNP, 10−9 to 10−5 mol/L). Other vasodilators evaluated included BAY60–2770 (10−9 to 10−6 mol/L), Forskolin (10−9 to 10−6 mol/L), and Y-27632 (10−8 to 10−5 mol/L). Contractions were recorded as changes in tension (mN) from baseline, expressed as a percentage of the response to 120mM KCl. Relaxation was expressed as a percentage of the stable contraction produced by PE in each ring immediately prior to the first dose of vasodilator. Return to the tension recorded before addition of PE was considered to be 100% relaxation.
For quantitation of myogenic tone, dissected mesenteric artery rings were placed in ice-cold physiologic salt solution (PSS) consisting of 109 mmol/L NaCl, 34 mmol/L NaHCO3, 4.7 mmol/L KCl, 0.9 mmol/L MgSO4, 1.0 mmol/L KH2PO4, 11.1 mmol/L dextrose, and 2.5 mmol/L CaCl2 and bubbled with 95% O2/5% CO2. The vessels were then transferred to custom microvessel perfusion chambers (Instrumentation and Model Facility, The University of Vermont, Burlington, VT) filled with 4 ml of chilled, preoxygenated PSS. The excised vessel preparation was positioned and secured on ~120 μm pipette tips. The perfusion chamber was placed on an inverted microscope equipped with a video camera and an image-capture system (IonOptix, Milton, MA) to continuously record intraluminal diameter. Optical markers used to detect the lumen diameter were positioned at the narrowest point of the mounted vessel. Bath temperature was held constant at 36.5–37.5 °C as monitored by a thermal microprobe tip placed adjacent to the mounted vessel. Chambers were perfused with warmed oxygenated PSS at 6 ml/minute. Vessels were allowed to equilibrate at ~37°C for 50 min, then exposed to 50 mM KCl (×1) to verify vessel reactivity, washed, and then pressurized in a stepwise manner from 20 to 140 mm Hg in 20mm increments and diameter recorded. Vessels were then returned to baseline pressure (20mm) in three steps and switched to calcium-free PSS containing the vasodilator papaverine (10−4 M, Sigma-Aldrich) for 60 min. Pressure ramps from 20–140 mmHg were then repeated under calcium-free conditions. Vessels were held at each pressure until a stable baseline diameter was reached (typically 10–15 min).
Cell Culture
Primary mesenteric or aortic VSMCs were isolated from mouse mesenteric arteries or aortas by the explant method. Briefly, first- and second-order mesenteric artery branches or thoracic aortas were excised and cleaned of adherent fat. The endothelial layer was removed by passing a wire or a pin through the lumen and the arteries were cut into 2–4 mm segments or square sections in ice-cold PSS. The segments were then placed in a culture dish and maintained in DMEM containing 30% FBS and 1% penicillin/streptomycin in a humidified incubator at 37°C, 5% CO2 atmosphere. After a week, the segments were removed and cells that had the migrated onto the dish were maintained in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 1X minimum essential medium non-essential amino acids, 1X vitamins, and 20 mM HEPES. Cells were confirmed to be VSMCs by immunostaining for smooth muscle α-actin.
HEK293T cells were obtained from the American Tissue Culture Collection and maintained at 37° C in 5% CO2 in DMEM, supplemented with 10% fetal bovine serum (FBS, Hyclone) and 0.1% Pen/Strep (25U/ml, Life Technologies).
Mass Spectroscopy
Protein from cultured aortic VSMCs from WT and LRRC8A KO mice was immunoprecipitated with LRRC8A antibody (#A304–175A, Bethyl Laboratories) cross linked to Dynabeads™ Protein G (#10003D, ThermoFisher Scientific). Anti-LRRC8A antibody was immobilized on protein G beads in phosphate buffer (pH 8.0) for 30 min at 4°C on a nutator, cross-linked with 25mM dimethyl pimelimidate (#150945, MP Biomedicals) in 200mM triethanolamine (Alfa Aesar), pH 8.2 for 30min, followed by blocking with 100mM ethanolamine (Acros Organics), pH 8.2, both at room temperature. The beads were washed with phosphate buffered saline (PBS, pH 7.4) then resuspended in PBS + 0.1% Tween 20, 5mM NaN3. LRRC8A WT and KO VSMCs were lysed in PPLB, nutated at 4°C for 30min, and centrifuged for 30 min at 20,000 × g. Supernatants were incubated with antibody-conjugated beads for 1.5 hrs at 4°C on a nutator. Beads were then washed with PPLB and proteins eluted into 1X Laemmli buffer with 5% SDS. Mass Spectroscopy was performed on two independent sets of protein isolates by the Vanderbilt Mass Spectroscopy Core Lab. Candidate proteins were sorted for those that were identified 4 or more times in WT but never appeared in samples from LRRC8A KO cells. Proteins were also considered to be of interest if the frequency of reads in WT cells was greater than 4x higher than that seen in KO cells. Functional connections between the 35 proteins identified were assessed using string-db.org.
Western blot analysis
Intact mesenteric artery and venous beds including omentum and fat were dissected free from the bowel and incubated in 5ml chambers containing PSS (95% O2/5% CO2, 37°C) for 30–40 min. Tissues were then exposed to PE (2 × 10−6M) for 3 min followed by SNP (3 × 10−8 M) for 3 min before being flash frozen in liquid nitrogen and stored at −80 °C. They were subsequently ground in a liquid nitrogen-chilled mortar and pestle, and proteins extracted in phosphoprotecting lysis buffer PPLB (50mM Tris base, 150mM NaCl, 1mM EDTA, 10% glycerol, 1mM DTT, 1% Triton X-100, 0.1% Na-DOC, 0.1% SDS, 10mM β-glycerophosphate, 20mM para-nitrophenyl phosphate, 2mM sodium pyrophosphate, 1mM Na3VO4, 5mM NaF, 10 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride (PMSF) at pH 7.4), centrifuged for 20 min at 20,000g. Protein extracts (60 – 80 μg) were separated by electrophoresis on a polyacrylamide gel and transferred to nitrocellulose membranes. Nonspecific binding was inhibited by blocking buffer (LI-COR) for 1 hour at room-temperature. Membranes were then incubated with primary antibodies in Tris-buffered saline solution with Tween 20 (0.1%) overnight at 4 °C. Antibodies were as follows: LRRC8A (#A304–175A, Bethyl Laboratories, Montgomery, TX), p-MYPT1 (#4563), MYPT1 (#2634) from Cell Signaling Technology (Danvers, MA), alpha smooth muscle (sm)-actin (#ab8211, Abcam, Waltham, MA). After incubation with fluorescent secondary antibodies for 2 hours in room-temperature, signals were developed using the Odyssey Imaging System and Image Studio software (LI-COR Biosciences, Lincoln, NE).
Immunoprecipitation mapping of MPRIP-LRRC8A interaction
A C-terminal tagged clone of human MPRIP (DDK (Flag), Genscript, Cat # OHu00028D) was truncated by insertion of an additional Flag tag followed by a premature stop codon using the QuickChange site-directed mutagenesis kit (Agilent). Internal regions of the protein (aa213–545, aa389–545) were PCR amplified using primers flanked by HindIII and DDK-Stop-EcoR1 sites and inserted into pcDNA3.1+ by restriction digestion and ligation. These clones were co-expressed with full length LRRC8A (Myc-DDK, Origene, Cat # RC508632 or an untagged version in which the Flag tag was removed by insertion of a premature stop) in HEK293 cells following transfection using Lipofectamine 2000 (Life Technologies). After 24 hours cells were lysed in PPLB, nutated at 4°C for 30min, and centrifuged for 30 min at 20,000 × g. Supernatants were incubated with antibody (2 μg) for 1.5 hrs at 4°C then incubated with protein-G sepharose on a nutator for 1 hr. Beads were washed with PPLB, resuspended in SDS sample buffer and boiled. The associated proteins were analyzed by western blotting. Antibodies for IP were; anti-Myc (Clone 9e10, Vanderbilt Antibody Core) or anti-LRRC8A (#A304–175A, Bethyl Laboratories). Western blotting was performed using anti-MPRIP (#14396, Cell Signaling Technology) or anti-Flag (F1804, Sigma).
Proximity ligation assay (PLA)
PLA were performed using a commercially available kit according to the manufacturer’s instructions (Duolink PLA, MilliporeSigma, St. Louis, MO). 96-well glass bottom plates were coated with poly-L-lysine (2x) for 20 min then washed with Dulbecco’s phosphate-buffered saline (D-PBS). The plates were then treated with glutaraldehyde 50% (1000x) for 15 min, washed with D-PBS, and lastly coated with gelatin (40x), then washed again with D-PBS. Afterwards, 70% ethanol was applied for 30 min followed by a 30 min wash in medium. Cells were seeded in the plate and allowed to grow over 24 hrs. The cells were exposed to 4% formaldehyde in PBS for 15 min, permeabilized with 0.1% TritonX-100 in PBS for 15 min, and then incubated with 40 μL of Duolink blocking solution (MilliporeSigma) at 37 °C for 1 h. Primary antibodies were applied in Duolink antibody diluent at a 1:100 dilution: LRRC8A mouse mAb (clone #8H9, Abnova, Taipei City, Taiwan), MPRIP rabbit mAb (#14396, Cell Signaling Technology). Plates were then sealed and incubated overnight at 4 °C prior to development per the Duolink manufacturer’s directions. Signal was imaged for both the Duolink Orange fluorescent probe (ex 554/em 576) and BioTracker 488 (ex 500/em 515, MilliporeSigma) in Live Cell Imaging Solution (Thermo Fischer Scientific) using an inverted confocal microscope (Zeiss LSM 880, Carl Zeiss AG, Oberkochen, Germany). Fluorescent reactions and cell number were counted by averaging two random fields per well using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Tagged Protein Imaging
HEK293T cells were grown on glass coverslips and transfected with C-terminal meGFP-tagged LRRC8A and C-terminal mCherry-tagged MPRIP using Lipofectamine 2000 (Life Technologies). After 24 hours live images were captured using a Zeiss LSM880 Airyscan confocal microscope.
siRNA transfection and RhoA activation
VSMCs were grown on 6-well plates and transfected with either scrambled siRNA or siLRRC8A (Dharmacon, Lafayette, CO, 100 nM) incubated with Lipofectamine 2000 (Life Technologies) in serum-free medium for 15 min. The resultant complex of siRNA-Lipofectamine 2000 was added to cells in DMEM containing 5% FBS and then maintained for 3 days. Cells were stimulated by PE (2 × 10−6 M) for 3 min followed by SNP (3 × 10−8 M) for 3 min before the assay. RhoA activity was assessed using the G-LISA RhoA activation assay kit (#BK124, Cytoskeleton, Inc., Denver, CO) according to the manufacturer’s specifications.
MPRIP Sulfenylation
A cell permeable, biotin-labeled Cys-OH trapping probe (3-(2,4-dioxocyclohexyl) propyl, DCP-Bio1; MilliporeSigma) was used to quantify MPRIP sulfenylation. Live LRRC8A WT and KO VSMCs were incubated with 125 μM DCP-Bio1 for 1 hour in DMEM containing 0.5% FBS before exposure to TNFα (10ng/ml) for 3 min. Cells were then lysed (50 mM Tris, pH 7.0, 50 mM NaCl, 2 mM EDTA, 1 mM NaF, 1mM Na3VO4, 2.5 mM Sodium pyrophosphate, 1% Triton-X, 100 μM NEM, 100 μM IAA, protease inhibitor, 100 μM PMSF). DCP-biotin labelled proteins were pull-downed with streptavidin beads overnight, washed, and analyzed by western blot with anti-MPRIP.
Statistical analysis
Values are mean ± standard error of the mean (SEM), and ‘n’ represent the number of animals used in the experiments, or of independently performed experiments in cultured cells. Concentration-response curves were fitted using a nonlinear interactive fitting program (Graph Pad Prism 9; GraphPad Software, San Diego, CA), and two pharmacological parameters were obtained: the maximal effect generated by the agonist (or Emax) and EC50 (molar concentration of agonist producing 50% of the maximum response). Statistical differences were calculated by t-test or one-way ANOVA. Post hoc testing was performed using Newman-Keuls analysis to compare all groups. Two-way ANOVA was used to compare pressure-response curves under normal versus calcium-free conditions and Bonferroni post-hoc analysis was performed when significant differences were found. A P value less than 0.05 was considered statistically significant.
Results
Vasomotor function in LRRC8A null vessels.
Freshly isolated mesenteric vessel segments from WT and VSMC-specific LRRC8A KO male mice were studied by wire myography. Compared to WT, KO vessels displayed indistinguishable contractile responses to depolarization by KCl or adrenergic receptor activation by phenylephrine (PE, Fig. 1A and B). Sustained contractile responses to KCl and PE are almost completely dependent on influx of extracellular calcium(17). Following contraction with PE (10−6 M) KO vessels were more responsive to relaxation induced by acetylcholine (ACh, LogEC50, WT −6.2 ± 0.11 vs. KO −6.7 ± 0.07, p < 0.05) which elicits an endothelium-dependent relaxation response that is multi-factorial (Fig. 1C). An important component of the response is production of nitric oxide (NO•) by endothelial cells. To selectively evaluate responsiveness to NO•, relaxation in response to sodium nitroprusside (SNP) was evaluated (Fig. 1D). Relaxation to SNP was also enhanced in KO vessels (LogEC50, WT −8.1 ± 0.11 vs. KO −8.6 ± 0.13, p < 0.05).
Figure 1. LRRC8A KO augments vasodilation in mesenteric arteries.
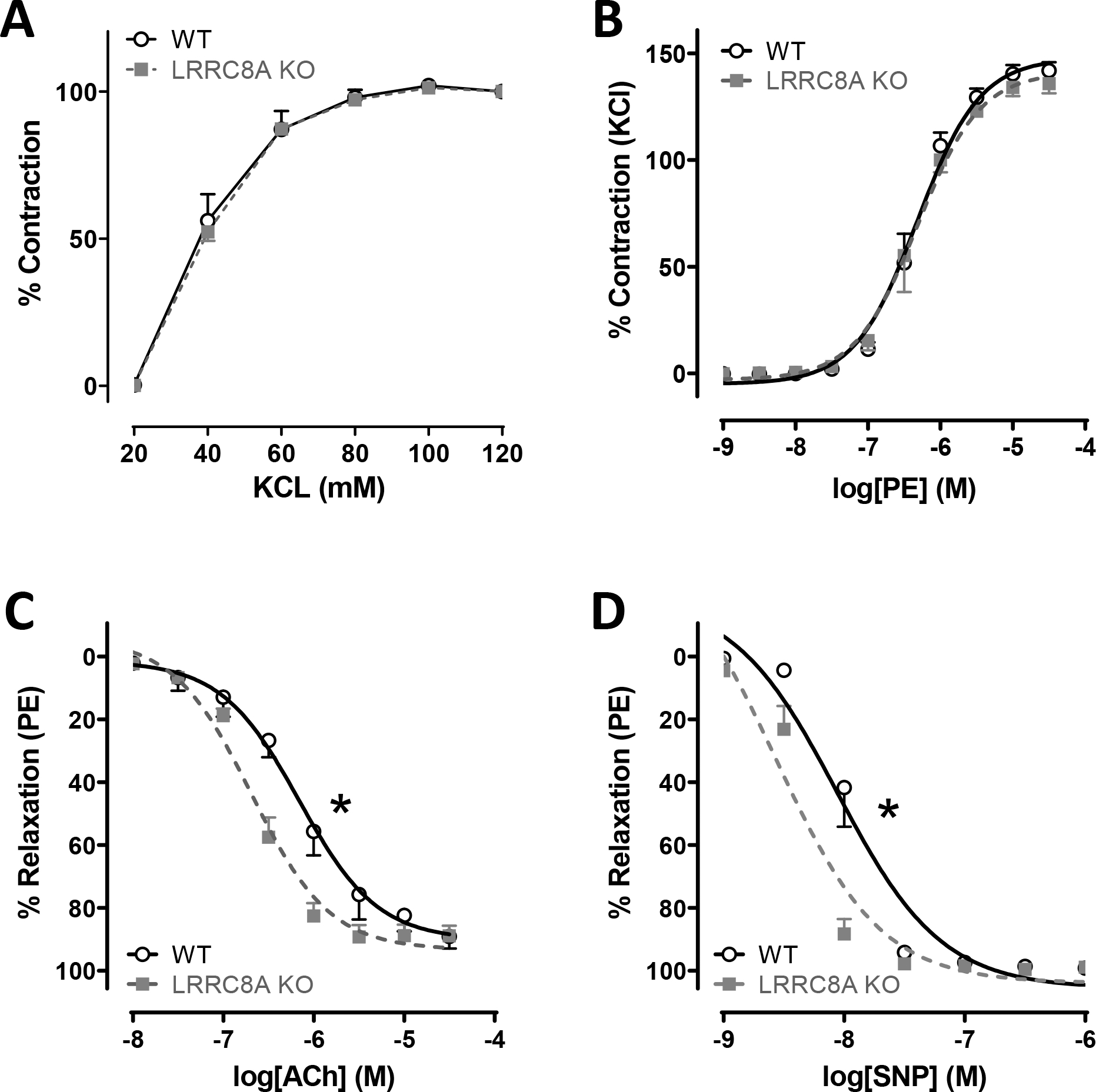
In freshly isolated mesenteric arteries, contractile responses to A) KCl (n = 3) and B) phenylephrine (PE, n = 4) were unaltered. Relaxation in response to C) acetylcholine (ACh, WT n = 6, KO n = 8) and D) sodium nitroprusside (SNP, WT n = 6, KO n = 5) was significantly augmented in KO compared to WT vessels. * Indicates p < 0.05 for EC50.
To determine if potentiation of vasodilation in LRRC8A KO vessels was related to loss of ion channel function, these experiments were repeated in WT vessel segments exposed to the VRAC inhibitor, carbenoxolone (CBX, 10−4 M) or vehicle (water) for 15 minutes prior to assessment of vasodilator function. As with KO vessels, CBX enhanced dilation to ACh (Fig. 2A and Suppl. Table S1). Because the response to ACh in mesenteric vessels reflects a summation of signaling via multiple dilator mechanisms, pharmacological tools were used to isolate those components (Fig. 2B and C and Suppl. Table S1). In control vessels (Fig. 2B) blocking prostaglandin production with indomethacin (5 μM) did not alter relaxation to ACh. Endothelium-dependent VSMC hyperpolarization can be inhibited by blockade of potassium channels with a mixture of apamin (50 nM) and charybdotoxin (ChTX, 50 nM). This combination decreased ACh sensitivity in control rings, making them approximately as sensitive as CBX-treated rings. NO• production was inhibited using L-NAME (100 μM) and this profoundly impaired relaxation in control vessels where the maximal relaxation was significantly reduced. The response of CBX-treated rings to ACh (Fig. 2C) was also insensitive to indomethacin while apamin + ChTX caused a small rightward shift in the D/R curve but preserved a higher sensitivity compared to control. The most remarkable effect of CBX was on relaxation responses in L-NAME-treated rings. NO synthase inhibition still caused a small decrease in ACh sensitivity, but the ability of ACh to cause a full relaxation response was preserved. In summary, CBX-treated vessels relaxed significantly better than control under all conditions tested.
Figure 2. carbenoxolone (CBX) increases vasodilation in male mesenteric arteries.
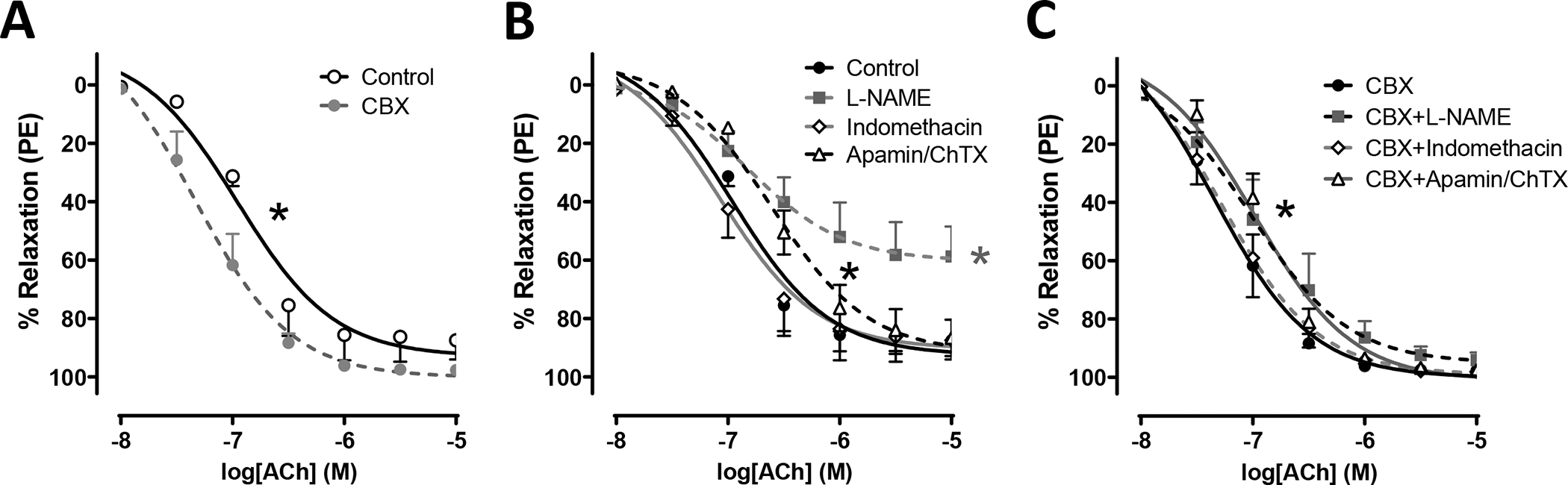
A) 15 min incubation in CBX (100 μM) significantly enhanced relaxation responses to ACh. * Indicates p < 0.05 for EC50. B) Effect of inhibiting; NO synthase with L-NAME (100 μM, 30 min), prostaglandin synthesis with indomethacin (5 μM, 20 min), or potassium channels with Apamin (50 nM, 20 min) + Charybdotoxin (ChTX; 50 nM, 20 min), on ACh-induced relaxation under control conditions in WT vessels. C) Effect of the same drugs on the ACh response in WT vessels treated with CBX for 15 minutes. CBX-treatment enhances relaxation compared to untreated controls under all conditions tested. Black* indicates p < 0.05 Apamin/ChTX vs. Control (B) or CBX (C) for EC50. Gray* indicates p < 0.05 L-NAME vs. Control for Emax (n = 4 to 5).
To assess the impact of VSMC LRRC8A KO on TNFα signaling, male mice mesenteric vessel segments were incubated in tissue culture for 48hrs in the presence of TNFα (10ng/ml) or vehicle. In contrast to PE responsiveness in freshly isolated tissue, KO rings incubated in tissue culture for 48hrs displayed a slightly but significantly decreased maximal contractile response to norepinephrine (NE) compared to WT rings, Fig. 3A). Vehicle-exposed cultured vessels from KO mice were also more sensitive to both ACh (Fig. 3B) and SNP (Fig. 3C). TNFα markedly enhanced contractile responses to NE but this effect was less pronounced in KO vessels. TNFα significantly impaired relaxation of WT tissues in response to both ACh and SNP but had no effect on the responses of KO vessels. Impaired relaxation of WT rings following TNFα exposure could result from impaired endothelial dilator production and/or reduced VSMC responsiveness. Vessels from both groups would be expected to have intact endothelial cell responsiveness to TNFα and therefore should display a similar degree of endothelial dysfunction. The degree to which endothelial dysfunction contributes to this in vitro model of TNFα-induced inflammation is not discernable by these experiments. However, the clear lack of detrimental impact of TNFα on the response of KO vessels to SNP likely reflects a combination of impaired VSMC TNFα signaling and enhanced baseline vasodilator function in KO VSMCs.
Figure 3. Disruption of LRRC8A function protects against TNFα-induced vascular dysfunction.
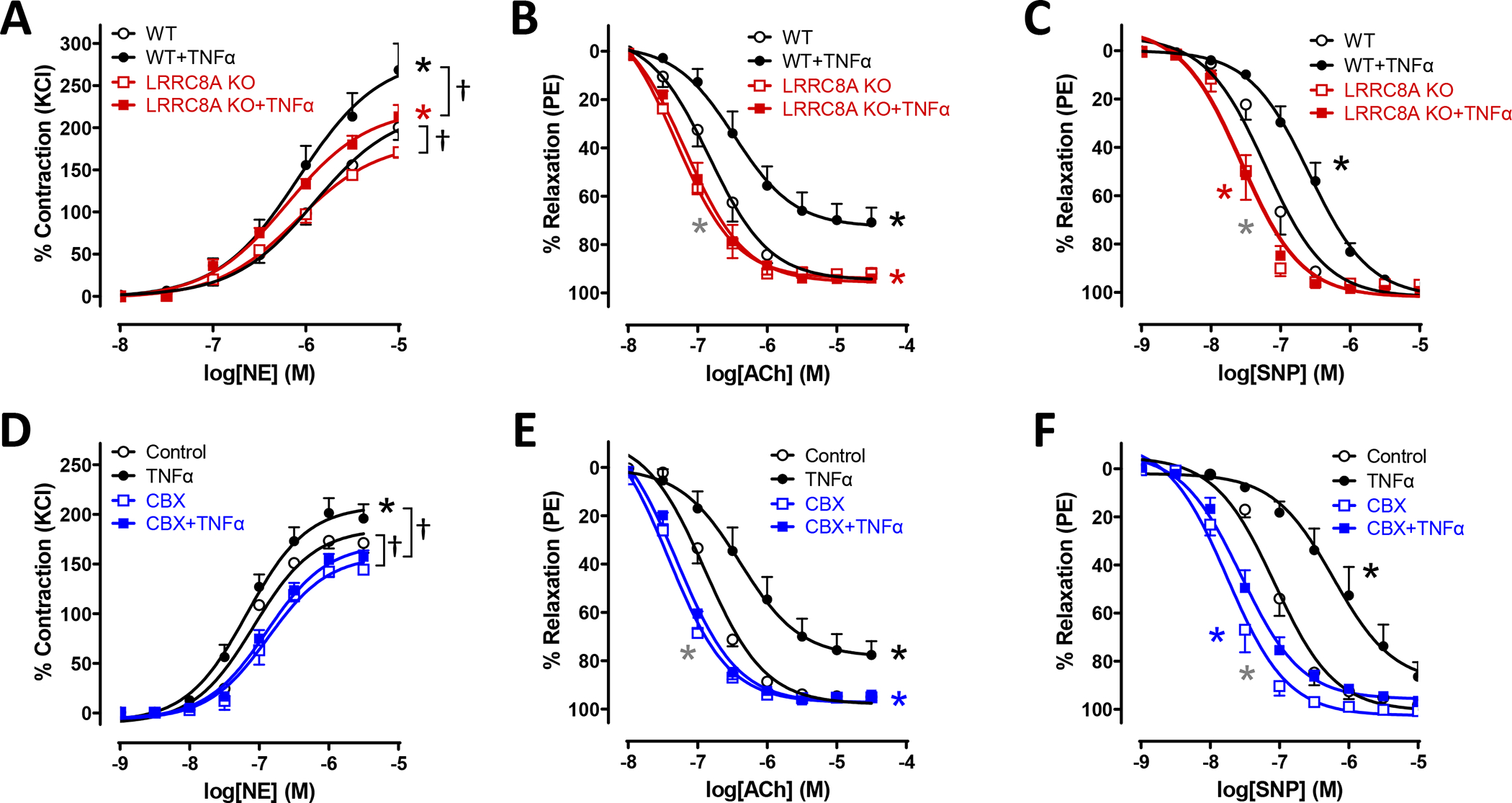
WT and LRRC8A KO male mesenteric vessels were incubated in tissue culture for 48 hrs in media ± TNFα (10ng/ml). Contractile responses to A) NE and relaxation to B) ACh and C) SNP were assessed. A) The maximal response to NE was significantly greater in control WT rings than in KO. TNFα increased the maximum response to NE in rings from both groups but accentuated the difference between WT and KO. C and D) KO vessels relaxed significantly better than WT and were completely protected from impairment following TNFα exposure as seen in WT vessels (n = 6 – 8). D) Contractile responses to NE in WT vessels were increased by TNFα exposure and this difference was abrogated by CBX which modestly but significantly reduced contraction of all vessels. E) and F) Vessel segments from WT mice were exposed to TNFα for 48hrs and then following mounting for tension recording to CBX (100μM, 20min) or vehicle. CBX effectively reversed impaired dilation after TNFα exposure (n = 5). A) * Indicates p < 0.05 for WT vs. WT+TNFα (black*) or KO vs. KO+TNFα (red*) for Emax. † Indicates p < 0.05 for Emax. B) * Indicates p < 0.05 for WT vs. WT + TNFα (black*) for EC50 and Emax, WT + TNFα vs. KO + TNFα (red*) for EC50 and Emax, or WT vs. KO (gray*) for EC50. C) * Indicates p < 0.05 for WT vs. WT + TNFα (black*) for EC50, WT + TNFα vs. KO + TNFα (red*) for EC50, or WT vs. KO (gray*) for EC50. D) * Indicates p < 0.05 for Control vs. TNFα (black*) for Emax. † Indicates p < 0.05 for Emax. E) * Indicates p < 0.05 for Control vs. TNFα (black*) for EC50 and Emax, TNFα vs. CBX+TNFα (blue*) for EC50 and Emax, or Control vs. CBX (gray*) for EC50. F) * Indicates p < 0.05 for Control vs. TNFα (black*) for EC50, TNFα vs. CBX+TNFα (blue*) for EC50, or Control vs. CBX (gray*) for EC50.
TNFα-induced VSMC dysfunction after 48hrs could be related to VSMC “damage”, such as protein or lipid oxidation, or to changes in gene expression. Alternatively, TNFα could drive ongoing inactivation of NO• by O2−• or persistently stimulate O2−•-dependent signaling. The latter mechanisms might be rapidly reversible. We addressed this question by applying CBX treatment for only 20 min in the muscle bath following 48 hours of TNFα exposure. This brief period of VRAC inhibition by CBX provided protection from the adverse effects of cytokine exposure (Fig. 3D, E and F) that was very similar to that provided by LRRC8A KO. CBX-mediated rescue of enhanced vasoconstriction to NE and impaired vasodilation in response to both ACh and SNP suggests that VRAC inhibition disrupts an ongoing signal that interferes with normal VSMC function.
Relaxation of LRRC8A KO vessels might be enhanced by protection from NO• inactivation. This would be consistent with our previous demonstration of reduced O2−• production(15). We reasoned that if this was the case, relaxation in response to vasodilator mechanisms that are downstream of, or unrelated to NO•, would be unaltered. We therefore assessed vasodilator responsiveness to agents that alter the function of three different kinases that phosphorylate MYPT1 and regulate MLCP activity. Following contraction with PE, WT and KO vessels were exposed to; 1) BAY60–2770, which directly activates soluble guanylyl cyclase, increases intracellular cyclic GMP, and activates protein kinase G (PKG), 2) Forskolin, which activates adenylyl cyclase, increases cyclic AMP levels and activates protein kinase A (PKA), or 3) Y-27632, which inhibits Rho-associated coiled-coil kinase (ROCK, Fig. 4). All three agents were fully effective vasodilators (~100% relaxation), and responsiveness was unaffected by TNFα exposure in either WT or KO tissues. This suggests that the mechanism(s) by which TNFα impairs vasodilation are upstream of MLCP and the contractile proteins. In contrast, the ED50 for all three drugs was significantly lower in KO compared to WT vessels. Thus, the mechanism of enhanced relaxation in LRRC8A KO vessels is downstream of PKG, PKA and ROCK. This strongly implicated a fundamental change in MYPT1 regulation of MLCP.
Figure 4. Loss of LRRC8A enhances relaxation by multiple mechanisms.
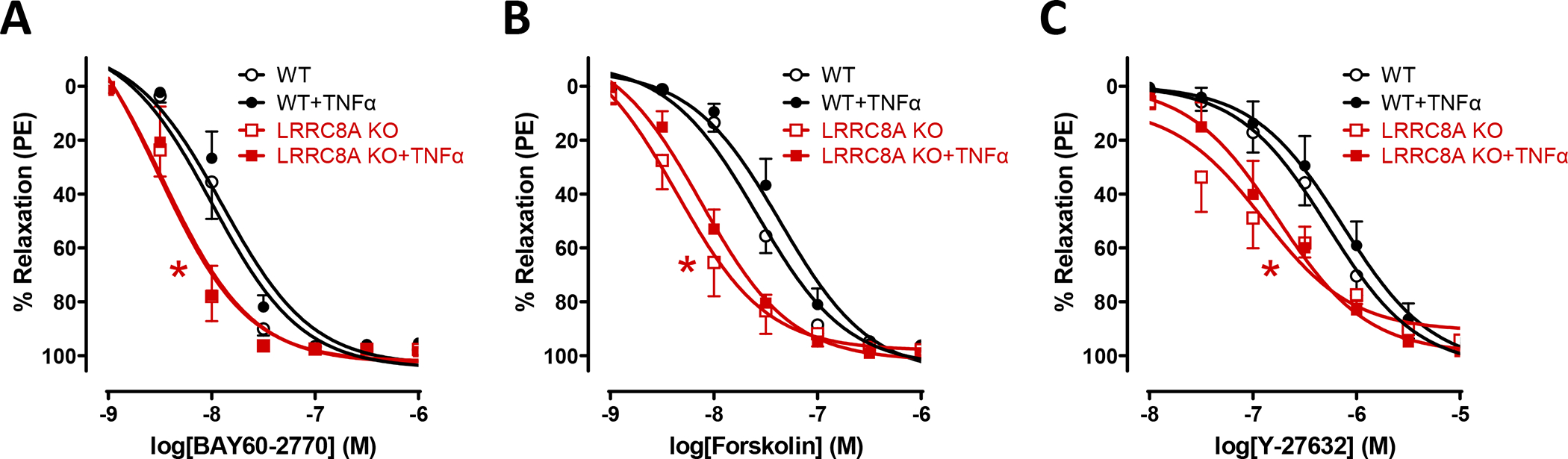
Relaxation responses to activation of A) soluble guanylyl cyclase (BAY60–2770, n = 6 – 7), B) adenylate cyclase (Forskolin, n = 5 – 6) or C) inhibition of Rho kinase (Y-27632, n = 5 – 6). Knockout of LRRC8A in VSMCs significantly enhanced relaxation to all three agents. None of these responses were affected by TNFα exposure in WT or KO vessels. Red* indicates p < 0.05 for WT vs. KO.
Myogenic tone is an innate response of VSMCs to physical stretch. Both Nox1 and ROS have been implicated in its regulation(9, 18). In addition, this response is associated with activation of RhoA/ROCK which enhances calcium sensitivity of the contractile process(19). We reasoned that if loss of LRRC8A impacts vasodilation at the level of MLCP then myogenic tone might also be affected. Pressure myography was used to quantify the response of mesenteric vessel segments to increasing intraluminal pressure (20 – 140 mmHg) in WT (Fig. 5A) and LRRC8A KO (Fig. 5B). A myogenic response was triggered in WT vessels beginning at ~80mmHg as demonstrated by differences in the diameter of vessels tested in normal vs. calcium-free buffer containing a potent vasodilator (papaverine, 10−4 M). The response of WT segments was comparable in magnitude to that previously reported in murine mesenteric vessels(20) but no myogenic tone developed in KO vessels, which displayed exclusively passive dilation in response to increasing luminal pressure.
Figure 5. Myogenic tone is absent in LRRC8A KO mesenteric arteries.
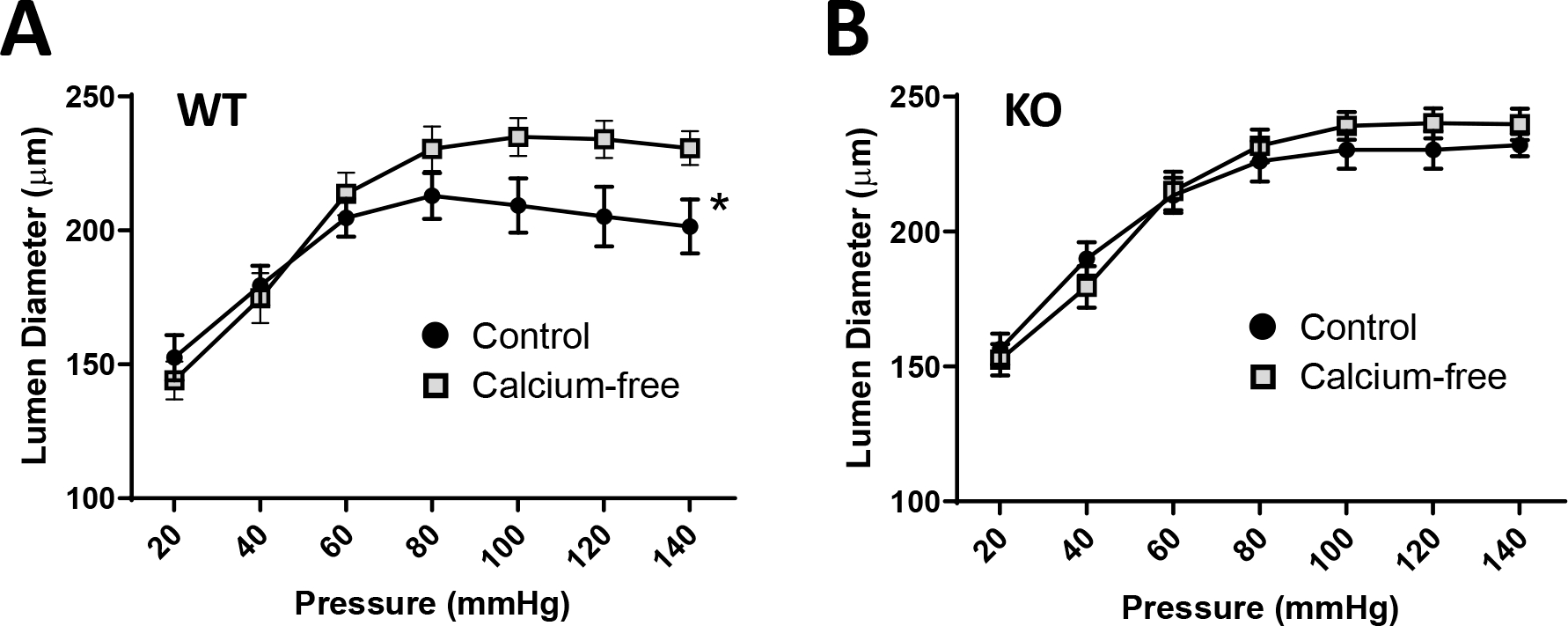
Pressure-induced constriction (the difference in diameter in normal vs. Ca++-free buffer + papaverine) was absent in mesenteric segments from LRRC8A VSMC KO mice. *p < 0.05 by 2-way ANOVA across all pressures (WT n = 11 and KO n = 12).
Identification of LRRC8A-associated proteins.
LRRC8 proteins have cytoplasmic C-terminal LRR domains that are involved in protein-protein interaction(21) and seem likely to confer specific function to VRACs. To explore the mechanism by which LRRC8A impacts vasomotor function we immunoprecipitated proteins from WT and KO VSMCs with an anti-LRRC8A antibody and used mass spectroscopy to identify potential binding partners. Proteins pulled down in KO cells were used to control for non-specific binding. Over two combined experiments (Suppl. Table S2), 33 proteins (Fig 6A, columns 1 and 2) met our initial criteria for LRRC8A-association; at least 5 total reads, ≥ 4 reads in the WT precipitate while either never being identified in the KO, or ≥ 4X as many reads in WT cells. The most frequently identified proteins were LRRC8A and LRRC8C, and LRRC8D was also only found in WT cells, confirming that the pulldown was selective. Analysis of the interrelationship between the proteins was performed using STRING (Fig. 6C). The most associated biological process was “actin filament-based process” (red), the top molecular function was “actin binding” (blue), and the top cellular component was the “actin cytoskeleton” (green, Fig 6B). To further validate the comparison of WT and KO samples we analyzed the frequency of reads for proteins that are highly unlikely to be associated with a plasma membrane protein. This provided a control group of “background” reads that included annexins, collagens, histones, keratins, elongation factors, mitochondrial ATPase subunits, and heat shock, ribonuclear, RNA-binding and ribosomal proteins. Collectively, these reads were present in very similar abundance (WT 584, KO 597, Suppl. Table S2). This suggests a similar degree of non-specific pulldown from the two types of cells. We therefore added to our analysis proteins that had reads in high abundance, such as actin and myosin isoforms, but were identified more than twice as frequently in the WT. These proteins are listed in Fig. 6A, column 3. Including them significantly decreased the estimated false discovery rate for all 3 pathway categories (Fig. 6B, shaded data). The resulting protein-protein interaction network is shown in Fig. 6C and is color coded for pathways of interest.
Figure 6. Identification of LRRC8A-associated proteins.
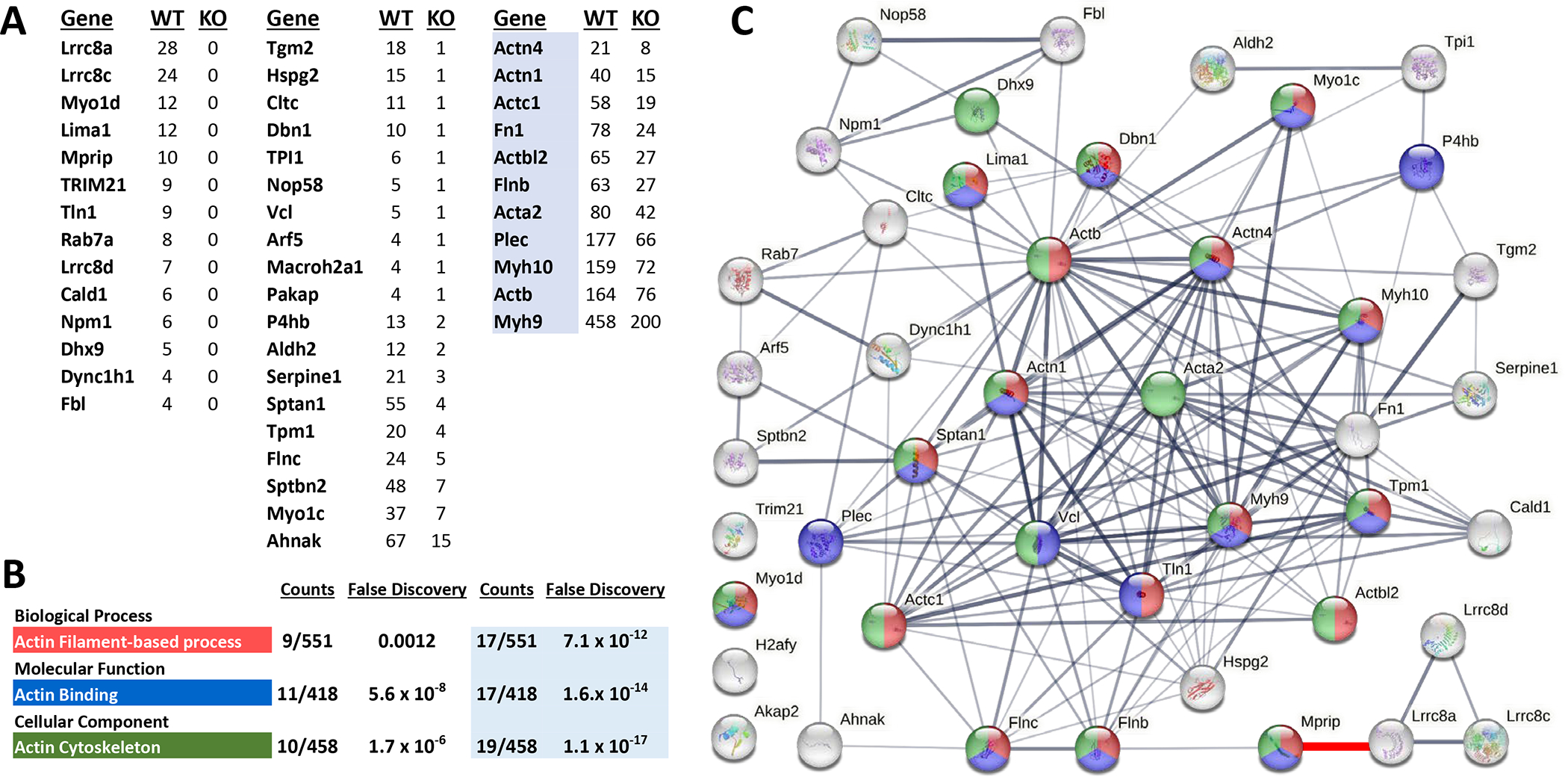
A) Columns 1 and 2 list proteins that met initial criteria for selective immunoprecipitation by anti-LRRC8A as identified by mass spectroscopy (see Suppl. Table S2 for details). Column 3 proteins (blue highlight) met secondary criteria. B) Top result of pathway analysis for three categories of function, associated number of proteins detected for each pathway, and the false discovery rate for each observation. The last 2 columns provide data from a secondary analysis that incorporated the highlighted proteins in column 3. C) Association network generated by string-db.org. Proteins are color coded for their inclusion in the pathways listed in B). The thickness of connecting lines reflects the confidence of the association between proteins. A connection between MPRIP and LRRC8A has been added (red, lower right) to reflect the association demonstrated by the current work.
MPRIP links LRRC8A with the cytoskeleton.
Among the proteins that were uniquely pulled down in WT cells was MPRIP, a scaffolding protein known to associate with stress fibers and colocalize the RhoA small GTPase with the MYPT1 regulatory subunit of MLCP and actin(22–24). Knockdown of MPRIP disrupts RhoA regulation of MLCP(25). We therefore sought to confirm association of LRRC8A with MPRIP and identify the site of LRRC8A binding. Western blotting of the WT and KO LRRC8A immunoprecipitates confirmed the presence of MPRIP in the WT but not the KO pulldown (Fig. 7A). Proximity Ligation Assays (PLA) were employed to further establish colocalization of the two proteins. A clear PLA signal was present in cultured WT but not KO VSMCs. PLA spots were primarily localized to the base of cells where they interacted with the underlying substrate (Fig. 7B and Suppl Fig. 1). Finally, full length MPRIP tagged with C-terminal mCherry was co-expressed with LRRC8A-meGFP in HEK293T cells (Fig. 7C). As previously demonstrated, LRRC8A localizes to the plasma membrane and to prominent intracellular vesicles(26). MPRIP co-localizes with LRRC8A at the plasma membrane in areas where membrane extensions are forming, which appear as linear structures along the cell surface. MPRIP was not associated with LRRC8A that localized to intracellular vesicles. Further demonstration of these relationships is provided by Supplemental videos 1 and 2. Collectively, these data confirm physical interaction between LRRC8A and MPRIP.
Figure 7. Association of LRRC8A and MPRIP.
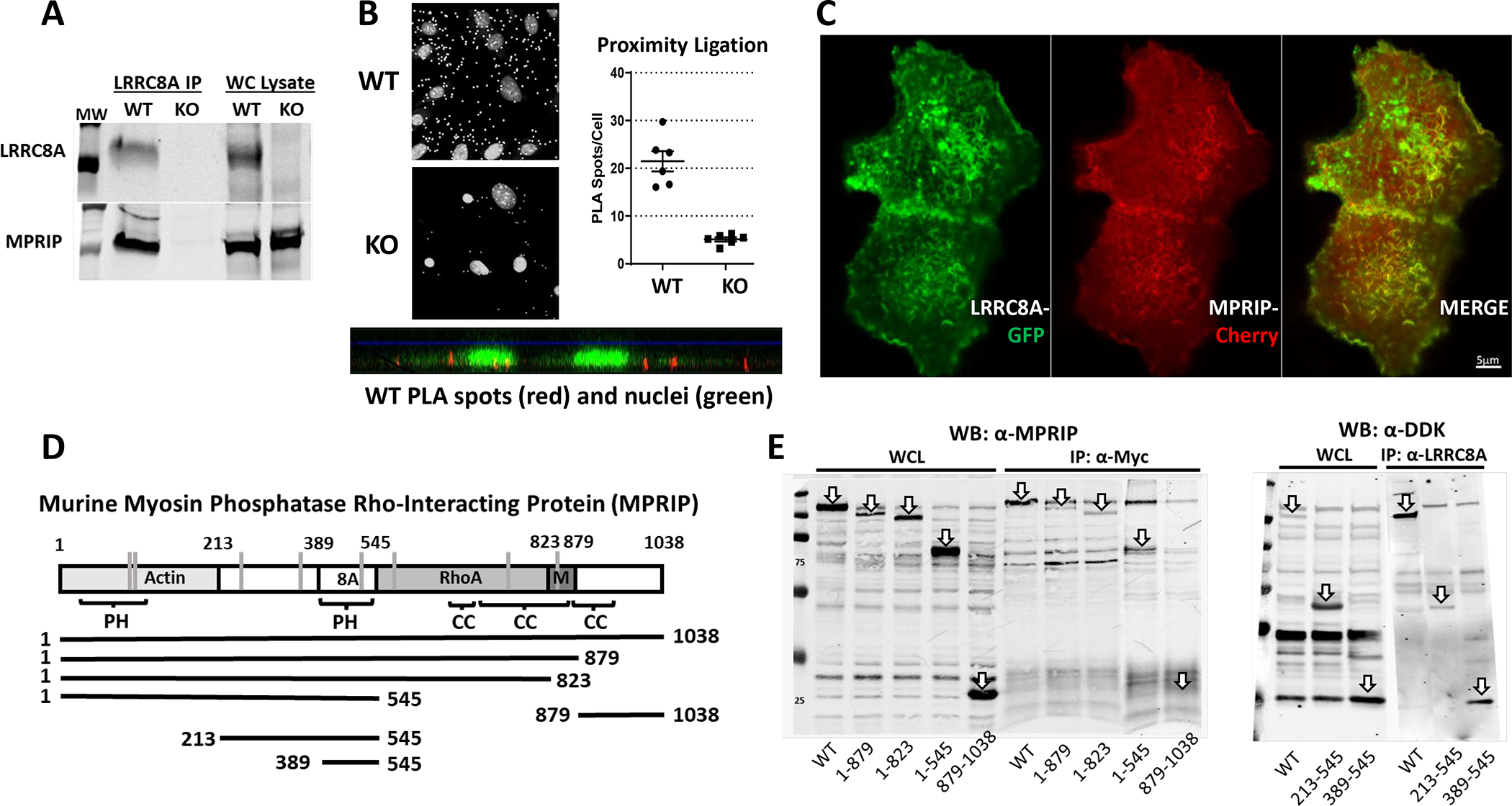
A) Immunoprecipitation (IP) of endogenous LRRC8A from VSMCs confirms the presence of LRRC8A in the whole cell (WC) lysate and precipitated protein from WT but not LRRC8A KO cells. MPRIP is present in the WC lysate from both genotypes but is only pulled down in LRRC8A WT cells. B) Proximity Ligation Assay (PLA) signal from α-MPRIP/α-LRRC8A in WT VSMCs. Small white spots represent PLA signal, the larger spots are nuclei. Counts of spots/nucleus from multiple fields of confluent WT and LRRC8A KO VSMCs (20x). PLA spots were smaller and significantly less numerous in KO cells. The bottom image shows red PLA spots with green nuclei as a side-view of cells. C) Confocal images of HEK293T cells co-expressing LRRC8A-meGFP and MPRIP-mCherry. Note co-localization at the plasma membrane, particularly at ruffled borders but not in LRRC8A-containing intracellular vesicles. D) Sites of protein binding to MPRIP. The first Plextrin Homology (PH) domain (aa 44–152) binds actin. C-terminal coiled-coil domains mediate interaction with RhoA and MYPT1 (M). Below the map are the constructs utilized to determine the site of MPRIP binding to LRRC8A (8A) was localized to aa 389–545. Vertical grey lines represent specific cysteines that can be oxidized based upon the Oxymouse database (Cys103, 120, 235, 361, 506, 571, 723, 830). Susceptible cysteines are present in the binding regions for all 4 protein partners. E) Immunoprecipitation of MPRIP-DDK fragments by full length LRRC8A-Myc in HEK293T cells expressing these constructs. Arrows identify overexpressed proteins, or the site where they would be expected to run (see IP: α-Myc) in whole cell lysates (WCL, left) and in immunoprecipitated protein (IP, right). All C-terminal MPRIP deletion mutants associate with LRRC8A, but the C-terminal MPRIP fragment 879–1038 does not associate with LRRC8A. The 213–545 peptide associated with LRRC8A as did the region between 389 and 545 which contains PH2 (see IP: α-LRRC8A).
The regions of MPRIP that bind its other partners have been previously identified. Actin associates with a Pleckstrin Homology (PH) domain in the N-terminal region (aa 1– 213)(24), RhoA binds to a region between aa 545 and 823(23) and MYPT1 binding occurs adjacent to that (aa 824–879)(27) as outlined in Fig. 7D. We created DDK-tagged C-terminal mutants of MPRIP and co-expressed these with full length myc-tagged LRRC8A in HEK293T cells. Immunoprecipitation with anti-Myc and western blotting with α-MPRIP revealed that LRRC8A associates with the region of MPRIP between the end of the actin binding region and the start of the RhoA binding sequence (aa 213–545). Full length MPRIP also associated with a peptide containing only aa 213–545, as well as a smaller subclone expressing only the region containing the second PH domain (aa 389–545) (Fig. 7E). These experiments define the region of MPRIP interaction with LRRC8A between aa389 and 545 that corresponds to a second PH domain.
LRRC8A modulates RhoA/ROCK/MYPT1 function.
The RhoA small GTPase is regulated by Nox1(28) and by O2−• (29). RhoA activates ROCK which phosphorylates MYPT1, inhibits phosphatase activity, and increases the calcium sensitivity the myosin light chain. MYPT1 phosphorylation by ROCK also impairs the ability of protein kinase A (PKA) and protein kinase G (PKG) to phosphorylate MYPT1 at adjacent sites which activates MLCP and triggers vasodilation. We compared RhoA activity in cultured WT VSMCs treated with scrambled or LRRC8A-targeted siRNA or exposed to CBX (100 μM) for 20 min (Fig. 8A). siLRRC8A significantly reduced LRRC8A protein abundance to 71 ± 3.2% of control (n = 6). To mimic conditions in our ring experiments, cells were exposed to PE (2 × 10−6 M) for 3 min followed by SNP (3 × 10−8 M) for an additional 3 min before lysis and assay of RhoA activity. LRRC8A knockdown was associated with significantly lower RhoA activity (siControl: 100 ± 2.0%, siLRRC8A: 89 ± 2.1% of control) as was CBX (Control: 100 ± 3.0%, CBX: 88 ± 4.2% of control). To determine if ROCK activity was affected, we assayed MYPT1 phosphorylation at T853 in acutely isolated, intact mesenteric beds. Mesenteries were exposed to the same conditions utilized for RhoA activity assays (PE followed by SNP), flash frozen and protein isolated for western blotting. Total MYPT1 abundance was similar between WT and KO, but phosphorylation at T853 was significantly reduced in KO mesenteries (Fig. 8B), consistent with diminished activity of RhoA/ROCK. Thus, consistent with the phenotypes of enhanced relaxation and impaired myogenic tone, loss of LRRC8A protein or function impaired Rho/ROCK activity.
Figure 8. RhoA-signaling is LRRC8A-dependent.

A) LRRC8A protein was significantly reduced in cultured mesenteric VSMCs by treatment with siLRRC8A. LRRRC8A knockdown and CBX treatment were associated with similar significant decreases in RhoA activity (n = 6). B) LRRC8A protein abundance is reduced but still detectable in intact mesenteries (fat, vessels and connective tissue) from KO mice (male n = 3, female n = 4). Total MYPT1 protein is unaltered but phosphorylation at T853 (p-MYPT1T853) is significantly reduced in mesenteries from VSMC-specific LRRC8A KO mice. *p < 0.05 compared to siControl or WT.
To further investigate a role for MPRIP in LRRC8A-dependent signaling we utilized siMPRIP to reduce the abundance of the protein in cultured mesenteric and aortic VSMCs (Fig. 9A). MPRIP knockdown was significant in both cell types and was associated with a significant increase in the abundance of LRRC8A protein. This may represent an attempt by the cells to compensate for loss of a downstream signaling partner. In contrast, MPRIP expression was not altered in LRRC8A null VSMCs (Fig. 9B).
Figure 9. MPRIP knockdown increases LRRC8A expression and MPRIP is sulfenylated in response to TNFα.
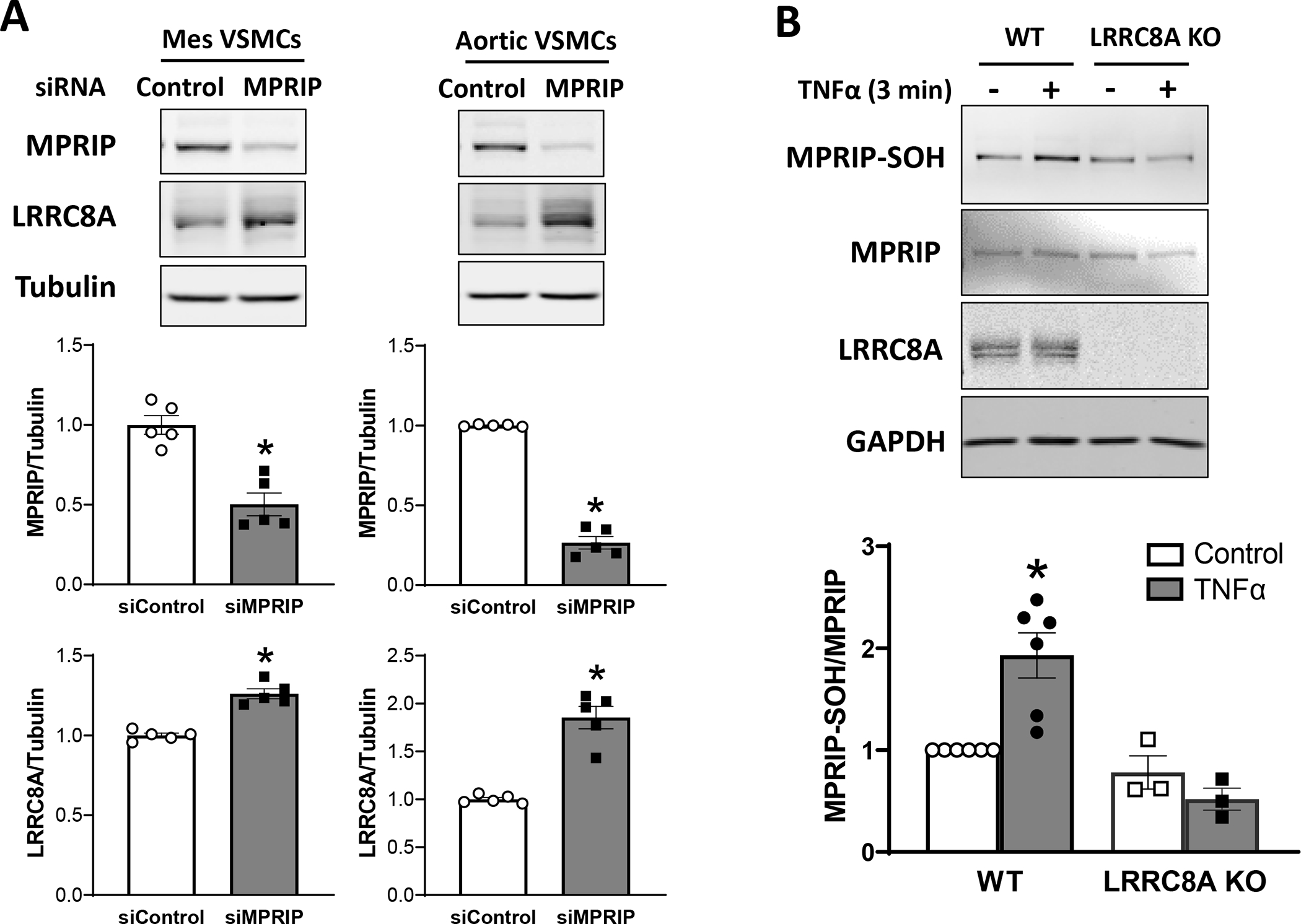
A) siRNA knockdown of MPRIP in cultured mesenteric (Mes) or aortic VSMCs resulted in a significant decrement in the abundance of MPRIP protein. In both cells this was associated with a significant increase in LRRC8A protein. B) A 3 min exposure to TNFα induced sulfenylation of MPRIP (MPRIP-SOH) in WT but not KO VSMCs. The top panels provide representative blots, bar graphs summarize the results of 3 – 6 independent experiments.
The Oxymouse database provides quantitative mapping of the mouse cysteine redox proteome (https://oximouse.hms.harvard.edu). It defines 8 redox-susceptible cysteines in murine MPRIP, including sites within the binding regions for all 4 of its partner proteins (Fig 7D). We hypothesized that association with LRRC8A places of MPRIP in a nanodomain that undergoes significant redox shifts in response to TNFα. We assessed the redox status of MPRIP in live cells by quantification of cysteine sulfenylation with DCP-Bio1 under control conditions and after exposure to TNFα for 3 min. MPRIP was sulfenylated at rest and this increased significantly after TNFα exposure in WT, but not in KO VSMCs (Fig. 9B). Thus, TNFα causes MPRIP oxidation in an LRRC8A-dependent manner. Sulfenylation can alter protein structure and may affect the ability of MPRIP to associate with its binding partners. This may be an important link in the chain of redox-driven events that link TNFα receptor occupation to changes in VSMC contractility.
Discussion
We previously demonstrated that LRRC8A VRACs are required for Nox1 activation by TNFα(15). Here we identify LRRC8A channels as membrane-spanning links in a multi-protein complex that connects Nox1 to the actin cytoskeleton. While VSMC-specific LRRC8A knockout vessels displayed normal contractile responsiveness to depolarization (KCl) or alpha-adrenergic receptor activation (PE), they completely lacked myogenic tone. Vasodilation to ACh and SNP was augmented, and consistent with the requirement of LRRC8A for signaling, KO vessels were protected from TNFα-induced changes in vasomotor function. Identification of cytoplasmic proteins that associate with LRRC8A revealed association with the actin cytoskeleton. Association of LRRC8A with MPRIP at the plasma membrane places it in proximity to RhoA, the MLCP complex and actin, critical regulators of contraction. RhoA activity was reduced by knockdown (siLRRC8A) or inhibition (CBX) of VRACs, as was ROCK-dependent phosphorylation of MYPT1 in KO mesenteric vessels. Collectively, these data suggest that decreased RhoA/ROCK activity contributes to enhanced relaxation and loss of myogenic tone. Importantly, MPRIP becomes oxidized following TNFα exposure, raising the possibility of another level of regulation; dynamic changes in MPRIP association with its partner proteins. While the details of how this multi-protein complex is assembled and regulated remain to be determined, we propose that association of Nox1/LRRC8A with MPRIP colocalizes ROS production with RhoA/ROCK and MYPT1, providing a mechanism by which Nox1-derived superoxide modulates calcium sensitivity.
Nox1 plays a critical role in inflammatory vasculopathy based on the ability of Nox1 knockout to rescue vascular dysfunction in multiple disease states (reviewed in(30)). VRACs support Nox1 activation, as evidenced by the fact that knockdown of LRRC8A or LRRC8C reduced Nox1-dependent extracellular O2−• production and multiple downstream effects of TNFα(15, 16). LRRC8 channels share significant structural similarity with connexins and both channels are inhibited by CBX(31, 32). Connexins provide a pathway for conducted hyperpolarization from endothelial cells to VSMCs(33) and blocking them impairs endothelium dependent VSMC hyperpolarization in response to ACh(31). The concentration of CBX used (100 μM) was shown by others to produce ~75% block of VRAC(30). In endothelium-intact rat hepatic and mesenteric arteries stimulated with 1 μM PE, this concentration of CBX caused a small depolarization of VSMCs but had no effect on ACh-induced hyperpolarization in the hepatic artery and caused only very mild inhibition in the mesenteric(31). This suggests that the majority of voltage-dependent relaxation in these vessels is mediated by activation of K+ channels by endothelial-derived factors(34), rather than by conducted hyperpolarization from the endothelium. This contribution is demonstrated by the partial inhibition of ACh-induced relaxation by apamin + charybdotoxin. However, control and CBX-treated vessels were similarly impacted based on similar degrees of rightward shift in the dose-response relationship (Fig. 2). This suggests that the ability of LRRC8A KO and CBX to enhance relaxation is related to a mechanism that is independent of changes in membrane voltage and calcium influx.
As hypothesized, KO vessels were protected from TNFα-induced injury, but the degree of protection observed was unanticipated as TNFα in known to cause both VSMC and endothelial dysfunction(6). These data suggest that endothelial injury induced by TNFα in this in vitro model was minimal and enhanced contraction and impaired relaxation was almost completely a function of VSMC dysfunction. This interpretation is supported by the fact that responses to ACh and SNP were similarly affected. The ability of short-term exposure to CBX to partially protect hyperresponsiveness to NE and to fully restore both ACh and SNP responsiveness following a 48hr exposure to TNFα was remarkable (Fig. 3). The similarity between the impact of LRRC8A KO and VRAC inhibition suggests that the changes in contractility induced by TNFα are dependent upon VRAC function and caused by a sustained process that does not occur in the KO vessels.
Increased relaxation to ACh and SNP could be related to enhanced NO• responsiveness, and/or to an increase in NO• bioavailability. The latter might be the result of a decrease in O2−• production by Nox1 when LRRC8A is absent or VRACs are blocked(15). However, this mechanism was not supported by the persistence of enhanced vasodilation when proximal steps in NO• signaling were bypassed and PKG was directly activated by BAY60–2770. Enhanced dilation was also not uniquely a property of PKG-dependent signaling as relaxation in response to PKA stimulation with forskolin, or ROCK inhibition by Y-27632 were also potentiated (Fig. 4). These data point to an impact of VRAC on a process that is downstream of PKA, PKG and RhoA/ROCK. These kinases all control MLCP activity via phosphorylation of MYPT1.
NADPH oxidases play critical roles in vascular contraction and mechanosensing(35). Nox-dependent O2−• production is activated in VSMCs by catecholamines(36) and RhoA-dependent calcium sensitization contributes to vasoconstriction in response to both KCl(37) and PE(38). However, in freshly isolated rings (Fig. 1) contractile responses to these agents were unaltered in KO vessels. A change in maximal catecholamine responsiveness was revealed after 2 days in tissue culture and accentuated when this included TNFα exposure (Fig. 3A). Spontaneous tone in rat cremasteric and cerebral is Nox1-dependent(18) and pressure-induced renal afferent arteriolar tone is associated with a burst of oxidant production that is NADPH oxidase-dependent. This response was inhibited by disruption of O2−• (apocynin, diphenylene iodonium, PEG-Superoxide Dismutase, Tempol) but not H2O2 (PEG-catalase) bioavailability(39). Pressurization of mouse mesenteric arteries induced MYPT1 phosphorylation at T853, and myogenic tone was completely blocked by Y-27632(40). Thus, reduced contraction to NE and loss of myogenic tone in LRRC8A KO mesenteric arteries (Fig. 5) is consistent with disruption of a Nox1/LRRC8A/O2−•/RhoA/ROCK signaling axis. In endothelial cells, loss of LRRC8A impaired Akt-dependent eNOS activation by stretch and disrupted the ability of cells to orient to shear stress(41). Importantly, O2−• produced by endothelial Nox2 activates Akt in response to shear stress(42) and Nox2 also physically associates with LRRC8A(43). Future work will be required to sort out the relative contribution of VRACs to endothelial and VSMC dysfunction in pathologic states.
The proteins pulled down by LRRC8A strongly support an association with the actin cytoskeleton. MPRIP was chosen for further investigation as a direct binding partner due to its established connection with proteins that are critical to the regulation of contractility, including actin. The two proteins share a similar pattern of plasma membrane distribution with a particularly strong association in regions of membrane ruffling (Fig. 7C). Importantly, this is where the highest levels of H2O2 were detected by a dynamic, actin-associated sensor(44). Colocalization of proteins with Nox1/LRRC8A may therefore facilitate redox regulation. Actin itself met our second order criteria for pulldown and is known to be redox-regulated, including by Nox-derived ROS(45). Myosin-9 was also detected and has been shown to undergo oxidation during cell adherence and spreading(46). Two unconventional myosins (Myo1c and Myo1d) also associated with LRRC8A. Myo1d binds to and localizes epidermal growth factor receptors (EGFRs) to the plasma membrane(47). EGFR transactivation occurs in response to AngII in an NADPH oxidase and ROS-dependent manner and inhibition of Nox oxidases normalized increased EGFR phosphorylation in spontaneously hypertensive rats(48).
RhoA activation regulates cytosolic calcium concentration by multiple mechanisms including regulation of calcium and potassium channels(10). Reduced RhoA activity induced by siLRRC8A or CBX (Fig. 8A) is consistent with enhanced relaxation. The relatively modest degree of impairment may be due to the fact that RhoA fulfills other roles beyond regulation of ROCK and is also regulated by additional mechanisms. RhoA activity is enhanced by oxidation of specific cysteine residues in its active site which enhances the dissociation rate of GDP(49). In contrast, over-oxidation of these residues can cause formation of an intramolecular disulfide that inhibits RhoA activity(50). In isolated vascular rings, extracellular generation of O2−• using a mixture of xanthine and xanthine oxidase induced vasoconstriction that was blocked by the SOD mimetic tempol, but not by catalase. These responses were associated with an increase in MYPT1 phosphorylation by ROCK(51). This suggests that RhoA can be activated by extracellular O2−• which supports the concept of O2−• influx but does not preclude a role for cytoplasmic H2O2 that is formed after O2−• enters the cell.
MYPT1, the regulatory subunit of MLCP, is the master regulator of calcium sensitivity. It controls activity of the PP1C subunit of the MLCP complex. This phosphatase functions in parallel with the MLC kinase (MLCK), which mediates calcium-calmodulin dependent phosphorylation of the 20kD subunit of MLC. The ratio of kinase to phosphatase activity determines the number of phosphorylated myosin light chains and therefore the contractile state of VSMCs. MYPT1 can thereby modulate the rate of actin-myosin crossbridge cycling at a constant cytoplasmic calcium concentration. MYPT1 is regulated by multiple kinases. ROCK inhibits MYPT1 by phosphorylation at both T696 and T853. Directly adjacent to these sites are serine residues (S695, S852) that are targets for activation by PKA and PKG. Phosphorylation at the ROCK sites is inhibited by the presence of a phosphate at the PKG and PKA sites and vice versa, creating interdependence of MLCP activation and inhibition. Reduced MYPT1 phosphorylation at T853 in LRRC8A KO mesenteries is consistent with a phenotype of enhanced vasodilation, but this may not provide a complete explanation for the functional changes observed when LRRC8A is absent.
MPRIP targets RhoA to stress fibers and facilitates dephosphorylation of myosin by MLCP. Silencing of MPRIP prevented RhoA/ROCK-dependent phosphorylation of MYPT1 but did not interfere with activation of RhoA and ROCK in A7r5 VSMCs(25). Association of MPRIP with MYPT1 enhances phosphatase activity(52) while binding to RhoA is inhibitory(53). These findings point to an important role for MPRIP in complex assembly. Enhanced dilation of KO vessels is likely to be related to reduced RhoA activity but altered association of proteins with MPRIP cannot be ruled out. MPRIP oxidation in response to TNFα (Fig. 9B) adds functional evidence of association with Nox1/LRRC8A and raises that possibility that MPRIP binding to its scaffolded partners may be oxidation-dependent. A contribution of complex assembly could explain why relaxation to SNP was augmented in LRRC8A null mice but was not altered in vessels from Nox1 null mice(54). Future work will be required to determine how redox signals impact MPRIP complex assembly and vasomotor function.
When LRRC8A is missing or VRACs are blocked, extracellular O2−• production by Nox1 in response to TNFα is reduced(15). We have proposed that anion flux through LRRC8A provides charge compensation for electron flow through Nox1 and speculated that VRACs may also serve as a O2−• conductance allowing extracellular O2−• to come back across the plasma membrane(6). While it is not possible to accurately quantify the forces acting on O2−• immediately following its creation, the initial essentially infinite concentration gradient clearly favors O2−• influx, but the concentration of O2−• is very low. However, current flow through NADPH oxidases produces significant membrane depolarization(55). This local event decrements at increasing distance from the oxidase but its potential to impact O2−• influx is maximized by physical association of LRRC8A with Nox1. The concept of a O2−• flux through an anion channel is supported by detection of O2−• efflux from endosomes that is sensitive to anion channel blockers(56). Cytoplasmic dihydroethidium oxidation by extracellular O2−• was measured and inhibited by anion channel blockade or knockdown of Chloride Channel three (ClC-3) expression(57). We recently demonstrated that ClC-3 influences membrane trafficking of LRRC8A, raising the possibility that this effect of ClC-3 was indirect(26).
Summary and significance
Loss of LRRC8A in VSMCs has profound effects on vasomotor function that seem to be dependent on anion channel function, as blockade of LRRC8A channels with CBX recapitulates the effect of removing the protein. We propose that association of LRRC8A with MPRIP provides a critical, membrane-spanning link in redox signaling. A model of Nox1-dependent redox signaling to the cytoskeleton is provided that incorporates MPRIP interaction with all four of its partner proteins (Fig. 10). Extracellular O2−• is created in proximity to LRRC8A VRACs. Local depolarization both activates LRRC8A and drives O2−• influx. Of note, VRACs are also regulated by oxidation(16, 58) which may add an extra layer of regulation to the system. In the cytoplasm, O2−• is deposited into a H+ rich cytoplasmic nanodomain created by Nox1-dependent NADPH metabolism(59). These protons could support local creation of H2O2, as O2−• dismutation is favored by acidic conditions (pKa of H2O2 = 11.75). Association with LRRC8A places MPRIP and RhoA in this oxidized nanodomain. ROCK activity is reduced when LRRC8A is absent or blocked. This reduces MLCP inhibition by ROCK and favors MYPT1 phosphorylation by PKG or PKA which limits contraction and potentiates relaxation. MPRIP is oxidized in response to TNFα which might affect its scaffolding function, thereby impacting MYPT1 and MLCP activity. This suggests that other redox-sensitive proteins within this domain (actin, atypical myosin etc.) may also be subject to Nox1/LRRC8A-dependent oxidation. Localization of LRRC8A-MPRIP is limited to the vicinity of the plasma membrane, particularly in areas of membrane ruffling. It remains to be determined how this influences cell movement. The broad influence of this localized interaction may derive from the fact that this is where receptor-dependent signaling occurs and redox-dependent secondary messages may profoundly impact distant events. In addition, this where the more active “plus” ends of actin filaments link to the focal adhesions that control cytoskeletal shape and membrane extension. This work provides a novel framework for Nox1-dependent inflammatory signaling whereby targeted cytoplasmic delivery of ROS can alter vasomotor function and contribute to vascular disease. Anion channel inhibition may provide a novel approach to treatment of vascular inflammation.
Figure 10. TNFR1/Nox1-dependent regulation of VSMC contractility.
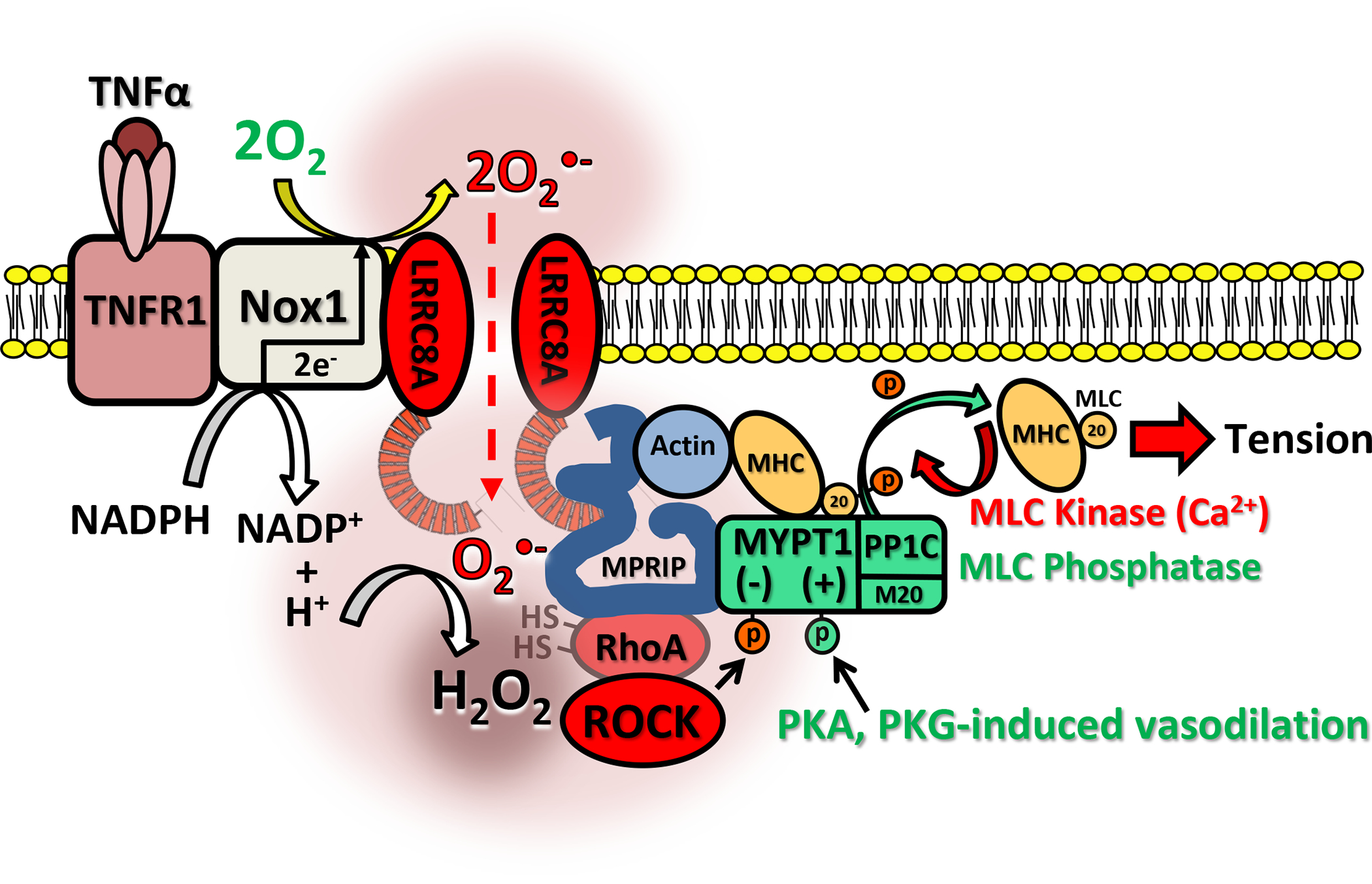
TNFR1 associates with Nox1 and LRRC8A at the plasma membrane. TNFα activates Nox1, resulting in NADPH conversion to NADP+ + H+ and 2 electrons are passed across the membrane and donated to O2 to produce extracellular O2−•. Local membrane depolarization by Nox1 facilitates O2−• influx through LRRC8A into a proton rich environment which may facilitate localized H2O2 production, contributing to creation of an oxidized nanodomain. These oxidants promote redox cycling of RhoA, causing ROCK activation and inhibitory phosphorylation of MYPT1. This reduces myosin light chain phosphatase (MLCP = MYPT1 + PP1C + M20) activity, increasing phosphorylation of the 20kD subunit of MLC and enhancing the calcium sensitivity of MLC which promotes contraction. MPRIP is also oxidized which may impact complex assembly. Loss or inhibition of VRACs inhibits Nox1 activity and prevents O2−• influx, reducing cytoplasmic oxidation, RhoA activation and ROCK phosphorylation of MYPT1. This enhances the ability of PKA and PKG to phosphorylate MYPT1, activate MLCP, and dephosphorylate MLC, thus reducing calcium sensitivity and potentiating VSMC relaxation.
Supplementary Material
Acknowledgements
The live cell images using Zeiss LSM880 Airyscan confocal microscope were performed through the use of the Vanderbilt Cell Imaging Shared Resource.
Grants
This work was supported by the NIH via R35 GM138191 to RJS and via R01 HL160975 and R01 DK132948 to FSL.
Footnotes
Disclosures
None declared.
Data availability statement
Additional data and materials will be individually shared upon reasonable request to the corresponding author.
References
- 1.Puszkarska A, Niklas A, Gluszek J, Lipski D, and Niklas K (2019) The concentration of tumor necrosis factor in the blood serum and in the urine and selected early organ damages in patients with primary systemic arterial hypertension. Medicine (Baltimore) 98, e15773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbaro NR, Fontana V, Modolo R, De Faria AP, Sabbatini AR, Fonseca FH, Anhe GF, and Moreno H (2015) Increased arterial stiffness in resistant hypertension is associated with inflammatory biomarkers. Blood Press 24, 7–13 [DOI] [PubMed] [Google Scholar]
- 3.Ruiz-Ortega M, Esteban V, Ruperez M, Sanchez-Lopez E, Rodriguez-Vita J, Carvajal G, and Egido J (2006) Renal and vascular hypertension-induced inflammation: role of angiotensin II. Curr Opin Nephrol Hypertens 15, 159–166 [DOI] [PubMed] [Google Scholar]
- 4.Pioli MR, and de Faria AP (2019) Pro-inflammatory Cytokines and Resistant Hypertension: Potential for Novel Treatments? Curr Hypertens Rep 21, 95. [DOI] [PubMed] [Google Scholar]
- 5.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, and Griendling KK (2001) Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88, 888–894. [DOI] [PubMed] [Google Scholar]
- 6.Lamb FS, Choi H, Miller MR, and Stark RJ (2020) TNFalpha and Reactive Oxygen Signaling in Vascular Smooth Muscle Cells in Hypertension and Atherosclerosis. American journal of hypertension [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao W, Zhang D, Li Q, Liu Y, Jing S, Cui J, Xu W, Li S, Liu J, and Yu B (2017) Biomechanical Stretch Induces Inflammation, Proliferation, and Migration by Activating NFAT5 in Arterial Smooth Muscle Cells. Inflammation 40, 2129–2136 [DOI] [PubMed] [Google Scholar]
- 8.Birukov KG (2009) Cyclic stretch, reactive oxygen species, and vascular remodeling. Antioxid Redox Signal 11, 1651–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mironova GY, Mazumdar N, Hashad AM, El-Lakany MA, and Welsh DG (2022) Defining a role of NADPH oxidase in myogenic tone development. Microcirculation 29, e12756. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, and Pagano PJ (2017) Microvascular NADPH oxidase in health and disease. Free Radic Biol Med 109, 33–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu XQ, and Zhang L (2022) Oxidative Regulation of Vascular Ca(v)1.2 Channels Triggers Vascular Dysfunction in Hypertension-Related Disorders. Antioxidants (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, and Nakano T (2003) Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res 92, 411–418 [DOI] [PubMed] [Google Scholar]
- 13.Yazdanpanah B, Wiegmann K, Tchikov V, Krut O, Pongratz C, Schramm M, Kleinridders A, Wunderlich T, Kashkar H, Utermohlen O, Bruning JC, Schutze S, and Kronke M (2009) Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 460, 1159–1163 [DOI] [PubMed] [Google Scholar]
- 14.Choi H, Stark RJ, Raja BS, Dikalova A, and Lamb FS (2019) Apoptosis signal-regulating kinase 1 activation by Nox1-derived oxidants is required for TNFalpha receptor endocytosis. Am J Physiol Heart Circ Physiol 316, H1528–H1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi H, Ettinger N, Rohrbough J, Dikalova A, Nguyen HN, and Lamb FS (2016) LRRC8A channels support TNFalpha-induced superoxide production by Nox1 which is required for receptor endocytosis. Free Radic Biol Med 101, 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi H, Rohrbough JC, Nguyen HN, Dikalova A, and Lamb FS (2021) Oxidant-resistant LRRC8A/C anion channels support superoxide production by NADPH oxidase 1. J Physiol 599, 3013–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai JM, Syyong H, Navarro-Dorado J, Redondo S, Alonso M, van Breemen C, and Tejerina T (2010) A comparative study of alpha-adrenergic receptor mediated Ca(2+) signals and contraction in intact human and mouse vascular smooth muscle. Eur J Pharmacol 629, 82–88 [DOI] [PubMed] [Google Scholar]
- 18.Kendrick DJ, Mishra RC, John CM, Zhu HL, and Braun AP (2021) Effects of Pharmacological Inhibitors of NADPH Oxidase on Myogenic Contractility and Evoked Vasoactive Responses in Rat Resistance Arteries. Front Physiol 12, 752366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somlyo AP, and Somlyo AV (1994) Signal transduction and regulation in smooth muscle. Nature 372, 231–236 [DOI] [PubMed] [Google Scholar]
- 20.Turner SR, Chappellaz M, Popowich B, Wooldridge AA, Haystead TAJ, Cole WC, and MacDonald JA (2019) Smoothelin-like 1 deletion enhances myogenic reactivity of mesenteric arteries with alterations in PKC and myosin phosphatase signaling. Sci Rep 9, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobe B, and Kajava AV (2001) The leucine-rich repeat as a protein recognition motif. Current opinion in structural biology 11, 725–732 [DOI] [PubMed] [Google Scholar]
- 22.Surks HK, Riddick N, and Ohtani K (2005) M-RIP targets myosin phosphatase to stress fibers to regulate myosin light chain phosphorylation in vascular smooth muscle cells. J Biol Chem 280, 42543–42551 [DOI] [PubMed] [Google Scholar]
- 23.Surks HK, Richards CT, and Mendelsohn ME (2003) Myosin phosphatase-Rho interacting protein. A new member of the myosin phosphatase complex that directly binds RhoA. J Biol Chem 278, 51484–51493 [DOI] [PubMed] [Google Scholar]
- 24.Mulder J, Poland M, Gebbink MF, Calafat J, Moolenaar WH, and Kranenburg O (2003) p116Rip is a novel filamentous actin-binding protein. J Biol Chem 278, 27216–27223 [DOI] [PubMed] [Google Scholar]
- 25.Riddick N, Ohtani K, and Surks HK (2008) Targeting by myosin phosphatase-RhoA interacting protein mediates RhoA/ROCK regulation of myosin phosphatase. J Cell Biochem 103, 1158–1170 [DOI] [PubMed] [Google Scholar]
- 26.Stark RJ, Nguyen HN, Bacon MK, Rohrbough JC, Choi H, and Lamb FS (2022) Chloride Channel-3 (ClC-3) Modifies the Trafficking of Leucine-Rich Repeat-Containing 8A (LRRC8A) Anion Channels. J Membr Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee E, and Stafford WF 3rd. (2015) Interaction of Myosin Phosphatase Target Subunit (MYPT1) with Myosin Phosphatase-RhoA Interacting Protein (MRIP): A Role of Glutamic Acids in the Interaction. PLoS One 10, e0139875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shinohara M, Shang WH, Kubodera M, Harada S, Mitsushita J, Kato M, Miyazaki H, Sumimoto H, and Kamata T (2007) Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J Biol Chem 282, 17640–17648 [DOI] [PubMed] [Google Scholar]
- 29.Resta TC, Broughton BR, and Jernigan NL (2010) Reactive oxygen species and RhoA signaling in vascular smooth muscle: role in chronic hypoxia-induced pulmonary hypertension. Adv Exp Med Biol 661, 355–373 [DOI] [PubMed] [Google Scholar]
- 30.Rivera J, Sobey CG, Walduck AK, and Drummond GR (2010) Nox isoforms in vascular pathophysiology: insights from transgenic and knockout mouse models. Redox Rep 15, 50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edwards G, Feletou M, Gardener MJ, Thollon C, Vanhoutte PM, and Weston AH (1999) Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br J Pharmacol 128, 1788–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benfenati V, Caprini M, Nicchia GP, Rossi A, Dovizio M, Cervetto C, Nobile M, and Ferroni S (2009) Carbenoxolone inhibits volume-regulated anion conductance in cultured rat cortical astroglia. Channels (Austin) 3, 323–336 [DOI] [PubMed] [Google Scholar]
- 33.Beleznai TZ, Yarova PL, Yuill KH, and Dora KA (2011) Smooth muscle Ca(2+)-activated and voltage-gated K+ channels modulate conducted dilation in rat isolated small mesenteric arteries. Microcirculation 18, 487–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka Y, Koike K, and Toro L (2004) MaxiK channel roles in blood vessel relaxations induced by endothelium-derived relaxing factors and their molecular mechanisms. J Smooth Muscle Res 40, 125–153 [DOI] [PubMed] [Google Scholar]
- 35.Brandes RP, Weissmann N, and Schroder K (2014) Nox family NADPH oxidases in mechano-transduction: mechanisms and consequences. Antioxid Redox Signal 20, 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bleeke T, Zhang H, Madamanchi N, Patterson C, and Faber JE (2004) Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ Res 94, 37–45 [DOI] [PubMed] [Google Scholar]
- 37.Ratz PH, Berg KM, Urban NH, and Miner AS (2005) Regulation of smooth muscle calcium sensitivity: KCl as a calcium-sensitizing stimulus. Am J Physiol Cell Physiol 288, C769–783 [DOI] [PubMed] [Google Scholar]
- 38.Gong MC, Fujihara H, Somlyo AV, and Somlyo AP (1997) Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. J Biol Chem 272, 10704–10709 [DOI] [PubMed] [Google Scholar]
- 39.Lai EY, Wellstein A, Welch WJ, and Wilcox CS (2011) Superoxide modulates myogenic contractions of mouse afferent arterioles. Hypertension 58, 650–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chennupati R, Wirth A, Favre J, Li R, Bonnavion R, Jin YJ, Wietelmann A, Schweda F, Wettschureck N, Henrion D, and Offermanns S (2019) Myogenic vasoconstriction requires G12/G13 and LARG to maintain local and systemic vascular resistance. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alghanem AF, Abello J, Maurer JM, Kumar A, Ta CM, Gunasekar SK, Fatima U, Kang C, Xie L, Adeola O, Riker M, Elliot-Hudson M, Minerath RA, Grueter CE, Mullins RF, Stratman AN, and Sah R (2021) The SWELL1-LRRC8 complex regulates endothelial AKT-eNOS signaling and vascular function. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Collins C, Kiosses WB, Murray AM, Joshi M, Shepherd TR, Fuentes EJ, and Tzima E (2013) A novel pathway spatiotemporally activates Rac1 and redox signaling in response to fluid shear stress. J Cell Biol 201, 863–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huo C, Liu Y, Li X, Xu R, Jia X, Hou L, and Wang X (2021) LRRC8A contributes to angiotensin II-induced cardiac hypertrophy by interacting with NADPH oxidases via the C-terminal leucine-rich repeat domain. Free Radic Biol Med 165, 191–202 [DOI] [PubMed] [Google Scholar]
- 44.Pak VV, Ezerina D, Lyublinskaya OG, Pedre B, Tyurin-Kuzmin PA, Mishina NM, Thauvin M, Young D, Wahni K, Martinez Gache SA, Demidovich AD, Ermakova YG, Maslova YD, Shokhina AG, Eroglu E, Bilan DS, Bogeski I, Michel T, Vriz S, Messens J, and Belousov VV (2020) Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab 31, 642–653 e646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balta E, Kramer J, and Samstag Y (2020) Redox Regulation of the Actin Cytoskeleton in Cell Migration and Adhesion: On the Way to a Spatiotemporal View. Front Cell Dev Biol 8, 618261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fiaschi T, Cozzi G, and Chiarugi P (2012) Redox Regulation of Nonmuscle Myosin Heavy Chain during Integrin Engagement. J Signal Transduct 2012, 754964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ko YS, Bae JA, Kim KY, Kim SJ, Sun EG, Lee KH, Kim N, Kang H, Seo YW, Kim H, Chung IJ, and Kim KK (2019) MYO1D binds with kinase domain of the EGFR family to anchor them to plasma membrane before their activation and contributes carcinogenesis. Oncogene 38, 7416–7432 [DOI] [PubMed] [Google Scholar]
- 48.Sandoval YH, Li Y, and Anand-Srivastava MB (2011) Transactivation of epidermal growth factor receptor by enhanced levels of endogenous angiotensin II contributes to the overexpression of Gialpha proteins in vascular smooth muscle cells from SHR. Cell Signal 23, 1716–1726 [DOI] [PubMed] [Google Scholar]
- 49.Aghajanian A, Wittchen ES, Campbell SL, and Burridge K (2009) Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One 4, e8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heo J, Raines KW, Mocanu V, and Campbell SL (2006) Redox regulation of RhoA. Biochemistry 45, 14481–14489 [DOI] [PubMed] [Google Scholar]
- 51.Jin L, Ying Z, and Webb RC (2004) Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol Heart Circ Physiol 287, H1495–1500 [DOI] [PubMed] [Google Scholar]
- 52.Nakatani T, Tsujimoto K, Park J, Jo T, Kimura T, Hayama Y, Konaka H, Morita T, Kato Y, Nishide M, Koyama S, Nada S, Okada M, Takamatsu H, and Kumanogoh A (2021) The lysosomal Ragulator complex plays an essential role in leukocyte trafficking by activating myosin II. Nat Commun 12, 3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koga Y, and Ikebe M (2005) p116Rip decreases myosin II phosphorylation by activating myosin light chain phosphatase and by inactivating RhoA. J Biol Chem 280, 4983–4991 [DOI] [PubMed] [Google Scholar]
- 54.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, and Yabe-Nishimura C (2005) Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation 112, 2677–2685 [DOI] [PubMed] [Google Scholar]
- 55.DeCoursey TE, Morgan D, and Cherny VV (2003) The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature 422, 531–534 [DOI] [PubMed] [Google Scholar]
- 56.Mumbengegwi DR, Li Q, Li C, Bear CE, and Engelhardt JF (2008) Evidence for a superoxide permeability pathway in endosomal membranes. Mol Cell Biol 28, 3700–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hawkins BJ, Madesh M, Kirkpatrick CJ, and Fisher AB (2007) Superoxide flux in endothelial cells via the chloride channel-3 mediates intracellular signaling. Mol Biol Cell 18, 2002–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertelli S, Zuccolini P, Gavazzo P, and Pusch M (2022) Molecular determinants underlying volume-regulated anion channel subunit-dependent oxidation sensitivity. J Physiol 600, 3965–3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morgan D, Cherny VV, Murphy R, Katz BZ, and DeCoursey TE (2005) The pH dependence of NADPH oxidase in human eosinophils. J Physiol 569, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional data and materials will be individually shared upon reasonable request to the corresponding author.