

Summary:
In this issue, Maganti and colleagues described an epigenetic link between reduced abundance of Polycomb-related protein MTF2 and chemotherapy resistance in refractory acute myeloid leukemia. MTF2 deficiency impaired expression of the PRC2 complex and deposition of H3K27me3 at many genes, including the key target gene MDM2, leading to increased MDM2 expression that in turn depleted p53 and thereby conferred chemoresistance.
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy that arises from uncontrolled proliferation of myeloid cells. Up to 40% of patients with de novo AML are refractory to standard induction chemotherapy (1). Current risk stratification biomarkers do not reliably predict for refractory AML. Hence, new biomarkers and treatment strategies are needed to improve clinical outcomes.
Aberrant epigenetic programming has emerged as a hallmark of AML and is present in virtually all patients (2). Unlike genetic lesions, leukemic epigenetic marks are potentially reversible through pharmacologic intervention. Recent data indicate that aberrant epigenetic programming and epigenetic modifiers contribute to chemotherapy resistance. For example, aberrant cytosine methylation patterning has been linked to chemotherapy resistance in hematologic malignancies, and clinical trials are currently testing whether DNA methyltransferase inhibitors administered prior to chemotherapy can overcome this effect (3, 4). In AML, loss-of-function of the histone modifier EZH2, which is part of the polycomb repressive complex 2 (PRC2), was suggested to mediate multidrug resistance (5). The PRC2 core complex is formed by EED, RbAp46/48, SUZ12, and one of the two histone methyltransferases EZH1 or EZH2 that catalyze methylation of histone H3 at lysine 27, resulting in formation of H3K27me3, a repressive chromatin mark (6). MTF2 interacts with PRC2 and directs it to specific gene loci for H3K27 trimethylation (7).
Herein, Maganti and colleagues (8) identify decreased levels of MTF2 as a biomarker for chemoresistance in refractory AML. The authors analyzed global H3K27me3 levels in CD34+CD38− cells (enriched with leukemia stem cells) derived from 32 patients with AML and observed that they segregated into those exhibiting basal levels of H3K27me3 similar to normal hematopoietic CD34+CD38− cells and others with markedly reduced H3K27me3 levels. Patients with reduced H3K27me3 had a 3-fold shorter survival time than patients with normal H3K27me3 abundance. Notably, H3K27me3 levels were more highly correlated with MTF2 transcription than EZH2 or SUZ12. Higher MTF2 expression was associated with longer survival in a multivariate analysis among favorable, but not intermediate-risk, cytogenetics.
MTF2 levels were reduced in the H3K27me3-low patients, and the authors observed that this was associated with aberrant MTF2 gene promoter hypermethylation as compared with AML with normal MTF2 levels (Fig. 1A). The authors validated that MTF2 deficiency reduced H3K27me3 levels and created an H3K27me3 “landscape” resembling those of refractory AML. MTF2 knockdown led to reduced expression of PRC2 components (EZH1, EZH2, and SUZ12) through posttranslational regulation because mRNA levels were unchanged. MTF2 knockdown in both Lin−CD34+ normal hematopoietic cells and AML samples enhanced chemoresistance to cytarabine and daunorubicin. Chemotherapy-surviving MTF2-deficient cells manifested less DNA damage and greater cell proliferation (PCNA+ cells; Fig. 1B and C). On the other hand, rescue of MTF2 with lentiviral expression in MTF2-deficient AML reestablished global H3K27me3 levels indicating that MTF2 directly influences the H3K27me3 level (Fig. 1B). Restoration of MTF2 also sensitized AML cells to cytarabine and daunorubicin, leading to increased cell death. Hence, MTF2 expression rendered refractory AML cells chemosensitive, whereas loss of MTF2 enables drug resistance in chemosensitive immature hematopoietic cells (Fig. 1B and C).
Figure 1.
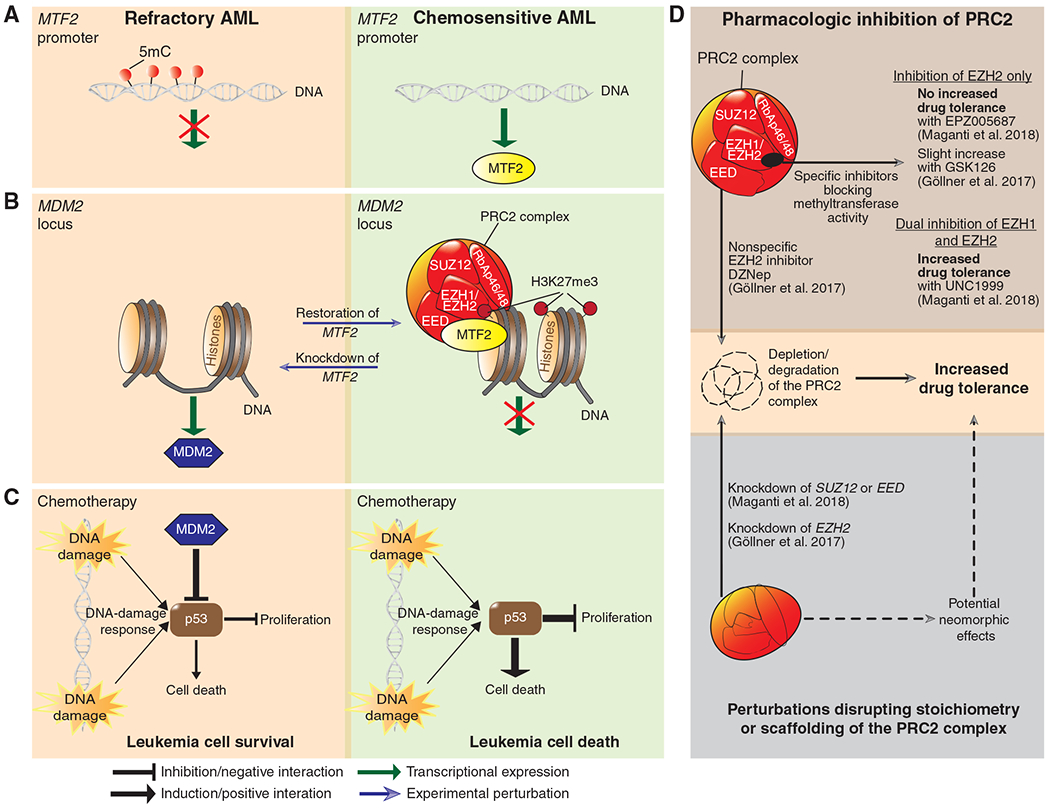
Epigenetic regulation of MTF2–PRC2 involved in chemoresistance in AML. A, The MTF2 promoter is hypermethylated (red) in MTF2-deficient AMLs that are significantly more unresponsive/resistant to induction therapy without undergoing complete remission (defined as refractory AML). In contrast, chemosensitive AML contains unmethylated CpG islands at the MTF2 promoter and expresses basal (normal) MTF2 mRNA levels. B, MTF2 mediates silencing of MDM2 in chemosensitive AML, whereas refractory, MTF2-deficient AMLs exhibit abundant levels of MDM2. MTF2 knockdown results in posttranslational reduction of the PRC2 core proteins EZH1, EZH2, and SUZ12 with concomitant reduction of H3K27me3, linked to increased chemoresistance (blue arrow). Conversely, rescue of MTF2 through ectopic expression in MTF2-deficient leukemia cells restores H3K27me3 levels that are associated with reduced chemoresistance. C, PRC2-mediated silencing of the MDM2 locus renders leukemia cells sensitive to chemotherapy (cytarabine and daunorubicin) through activation of the p53 pathway in response to DNA damage. On the other hand, refractory AML cells resist chemotherapy-induced DNA damage through MDM2-mediated depletion of p53. Inhibition of MDM2 (Nutlin-3a or MI-773) renders refractory cells sensitive to chemotherapy. D, Top plot shows that inhibition of EZH2 with specific SAM-competitive inhibitors (EPZ005687, GSK126) confers non-to minimal increase in drug tolerance (for cytarabine and daunorubicin) in contrast to dual inhibition of EZH1 and EZH2 (UNC1999). Inhibition with the SAH inhibitor DZNep (used as an EZH2 inhibitor) also affects other PRC core components leading to depletion/degradation of the whole PRC complex. DZNep treatment increases drug tolerance in AML. Bottom plot shows that knockdown of EED, SUZ12, or EZH2 induced drug tolerance, presumably through perturbation that can affect scaffolding or stoichiometry of the PRC2 complex. This can either cause depletion/degradation of the complex or potentially induce divergent functions by compensating proteins (e.g., EZH2 vs. EZH1). AML carrying frameshift mutations in EZH2 possibly perturb the stoichiometry of the PRC2 complex as well. The role of EZH2 missense mutations is unknown in AML.
The authors next tested whether loss of PRC2 core proteins phenocopied MTF2-mediated chemoresistance. They observed that dual inhibition of EZH1 and EZH2 (using 2 μM UNC1999) conferred drug resistance, whereas EZH2 inhibition alone (using 2 μM EPZ005687) was not sufficient (Fig. 1D) . These findings are somewhat different from the study by Göllner and colleagues (5), who reported that EZH2 inhibition alone was sufficient to confer drug resistance in AML. One factor that might contribute to this discrepancy is that Maganti and colleagues used a potent EZH2 selective inhibitor (EPZ005687), whereas Göllner and colleagues primarily utilized 3-deazaneplanocin (DZNep), a compound with pleiotropic effects. DZNep inhibits S-adenosyl-L-homocysteine (SAH) hydrolase and promotes accumulation of SAH, a byproduct of S-adenosyl methionine (SAM)–dependent methylation. SAM is used as a methyl donor for many methyltransferases (including EZH1 and EZH2) and converted to SAH, in which elevated SAH levels cause by-product inhibition of the methylation reactions (9). Additionally, DZNep depletes several PRC2 protein components besides EZH2, thus also affecting the PRC2 complex (ref. 9; Fig. 1D). Indeed, Göllner and colleagues showed that the direct EZH2 selective GSK126 mediated a 2- to 10-fold less drug-resistant phenotype than DZNep in AML cell lines, indicating distinct drug-dependent effects. Still, GSK126 treatment increased drug tolerance, whereas EPZ005687 did not in the report by Maganti and colleagues (Fig. 1D). It is unclear whether these dissimilarities can be attributed to differences between GSK126 and EPZ005687. Maganti and colleagues suggested that AMLs were not sensitized by EZH2 inhibition, because EZH1 compensated for loss of EZH2. However, their flow cytometry measurements indicated that H3K27me3 levels were markedly reduced after EZH2 inhibition alone, although not as much as with the dual EZH1/2 inhibitor. These results suggest either a threshold effect of H3K27me3 or locus-specific effects of EZH1 versus EZH2. Göllner and colleagues also showed that inhibition of CDK1 (which can trigger EZH2 degradation), HSP90, or proteasomal degradation led to increased EZH2 levels that were associated with higher drug sensitivity (5), although it is not possible to distinguish whether this drug-sensitizing effect was caused by EZH2 upregulation or the pleiotropic effects of these drugs. In contrast, it is also worth noting that an additional study (10) reported that dual inhibition of EZH1/2 potently impaired the growth of MLL-rearranged leukemia cell lines, suggesting that decreased H3K27me3 levels are detrimental in these leukemias (Göllner and colleagues also studied MLL-rearranged cell lines).
Of note, Göllner and colleagues demonstrated that EZH2 knockdown increased drug resistance. Knockdown of EZH2 may have a greater impact than enzymatic inhibition (e.g., EPZ005687), because RNAi will reduce the protein levels and thus the scaffolding function of EZH2. This could perturb the assembly of the whole PRC2 complex instead of affecting only the enzymatic activity with an inhibitor (Fig. 1D). Supporting this notion, Maganti and colleagues showed that knockdown of SUZ12 or EED also increased drug resistance, suggesting that perturbation of the whole PRC2 complex can influence drug tolerance (Fig. 1D). The extent to which knockdown of EZH1 and/or EZH2 affected drug resistance was not reported. Given that a subset of AML cases carry EZH2 frameshift mutations, which would likely disrupt PRC2 stoichiometry, it is possible that these could lead to chemotherapy resistance through similar mechanisms.
Exploring in greater depth potential mechanisms through which MTF2 reduction mediates chemoresistance, Maganti and colleagues discovered that the MDM2 locus is a target of H3K27me3 by the MTF2-PRC2 complex (ref. 8; Fig. 1B). MDM2 encodes an E3-ubiquitin ligase that depletes the tumor suppressor p53, and low p53 level increased drug resistance in AML (refs. 5, 8; Fig. 1C). Inhibition of MDM2 (Nutlin-3a and MI-773) activated p53 targets and mediated drug resistance without affecting PRC2 targets such as HOXA9 and HOXB7. In contrast, Göllner and colleagues reported that EZH2-mediated drug resistance is less dependent on p53 because TP53 knockdown did not increase drug resistance to the same extent as EZH2 knockdown in an AML cell line. They observed association of chemotherapy resistance with expression of HOXA9 and HOXB7 in an EZH2-low chemoresistant cell line (5). HOX gene expression is linked to stem cell features such as increased expression of drug efflux pumps, which led Göllner and colleagues to link the chemotherapy-resistant phenotype to increased expression of the MRP1 transporter (ABCC1). Yet, MRP1 is able to transport anthracyclines but not cytarabine and hence cannot fully address the drug-resistant phenotype. Increased expression of HOXA9 and HOXB7 was associated with poor survival in patients with AML, and knockdown of HOXA9 and HOXB7 in AML cell lines reduced drug resistance (5). It is unclear, however, whether HOX gene knockdown affects more chemoresistant AML or whether it creates this effect due to a more general impairment in leukemia cell survival and proliferation irrespective of drug resistance.
By examining The Cancer Genome Atlas (TCGA) and other patient cohorts, Göllner and colleagues link EZH2 levels to outcome, whereas Maganti and colleagues did not observe this association. This may be due to their consideration of different endpoints. Maganti and colleagues reported an association with overall survival over 10 years among TCGA patients, whereas Göllner and colleagues reported on overall survival up to approximately 3 years. Still, this does not exclude a function for EZH2 in acquired drug resistance. Perhaps more suited for this aspect is the comparison of EZH2 levels in paired AML samples at diagnosis versus relapse, in which 45% of relapsed AML cases exhibited reduced EZH2 levels (5).
Drug resistance in AML can emerge from a variety of sources (e.g., fraction of quiescent cells, genetic background, somatic mutations, and type of drug). Additionally, stochastic epigenetic plasticity might enable cancer cells to survive stress situations such as chemotherapy (11), thus providing subsets of cells with advantageous features. Along these lines, the use by Göllner and colleagues of an MLL-rearranged cell line that was induced to be resistant to tyrosine kinases in vitro might reflect selection of particular epigenetic and/or genetic features that yielded the observed biological associations. In contrast, Maganti and colleagues used patient-derived AML cells for all functional and mechanistic studies, which may represent more a physiologic context. Yet, by and large, both studies point to PRC2 functions as being relevant for chemotherapy resistance.
Acknowledgments
A. Melnick is supported by LLS SCOR 7013-17, NCIR01CA 198089-01, and the Chemotherapy Foundation.
Disclosure of Potential Conflicts of Interest
A. Melnick reports receiving commercial research grants from Janssen, GSK, Roche, and Lilly and is a consultant/advisory board member for Epizyme. No potential conflicts of interest were disclosed by the other author.
REFERENCES
- 1.Thol F, Schlenk RF, Heuser M, Ganser A. How I treat refractory and early relapsed acute myeloid leukemia. Blood 2015;126:319–27. [DOI] [PubMed] [Google Scholar]
- 2.Glass JL, Hassane D, Wouters BJ, Kunimoto H, Avellino R, Garrett-Bakelman FE, et al. Epigenetic identity in AML depends on disruption of nonpromoter regulatory elements and is affected by antagonistic effects of mutations in epigenetic modifiers. Cancer Discov 2017;7:868–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scandura JM, Roboz GJ, Moh M, Morawa E, Brenet F, Bose JR, et al. Phase 1 study of epigenetic priming with decitabine prior to standard induction chemotherapy for patients with AML. Blood 2011;118:1472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clozel T, Yang S, Elstrom RL, Tam W, Martin P, Kormaksson M, et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov 2013;3:1002–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gollner S, Oellerich T, Agrawal-Singh S, Schenk T, Klein HU, Rohde C, et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat Med 2017;23:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chittock EC, Latwiel S, Miller TC, Muller CW. Molecular architecture of polycomb repressive complexes. Biochem Soc Trans 2017;45:193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker E, Chang WY, Hunkapiller J, Cagney G, Garcha K, Torchia J, et al. Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell 2010;6:153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maganti HB, Jrade H, Cafariello C, Manias Rothberg JL, Porter CJ, Yockell-Lelièvre J, et al. Targeting the MTF2–MDM2 axis sensitizes refractory acute myeloid leukemia to chemotherapy. Cancer Discov 2018;8:1376–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pande V, Pocalyko DJ, Dhanak D. Chapter 19 – Emerging Epigenetic Therapies—Lysine Methyltransferase/PRC Complex Inhibitors. In: Gray SG, editor. Epigenetic Cancer Therapy. Boston: Academic Press; 2015. p 427–37. [Google Scholar]
- 10.Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood 2015;125:346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med 2016;22:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]