

Abstract
Diffuse large B cell lymphoma (DLBCL) is the most common histological subtype of non-Hodgkin B cell lymphoma (NHL), and manifest highly heterogeneous genetic/phenotypic characteristics as well as variable responses to conventional immunochemotherapy (1). Genetic profiling of DLBCL patients has revealed highly recurrent mutations of epigenetic regulator genes such as CREBBP, KMT2D, EZH2 and TET2. These mutations drive malignant transformation by through aberrant epigenetic programming of B-cells and may influence clinical outcomes (2–4). These and other chromatin modifier genes also play critical roles in normal B-cells, as they undergo the various phenotypic transitions characteristic of the humoral immune response. Many of these functions have to do with impairing immune surveillance and may critically mediate resistance to immunotherapies. In this review, we describe how epigenetic dysfunction induces lymphomagenesis and discuss ways for implementing precision epigenetic therapies to reverse these immune resistant phenotypes.
Keywords: Diffuse large B cell lymphoma (DLBCL), Epigenetics, CREBBP, EP300, KMT2D, EZH2, TET2, epigenetic heterogeneity
Dysfunction of genes involved in epigenetic regulation drives lymphomagenesis
Mutations of epigenetic regulator genes are frequently found in germinal center-like DLBCL (GCB-DLBCL) or follicular lymphomas (FL), along with translocation and/or activating mutations of BCL2(2, 3, 5–7). These lymphomas reflect the transcriptional profiles and many biological hallmarks of B-cells transiting the germinal center (GC) reaction, which are transient structures that form during the T-cell dependent humoral immune response. GC formation is dependent on the transcriptional repressor protein BCL6, which silences hundreds of gene promoters to facilitate the proliferative and DNA damage tolerant phenotype of GC B-cells(8). BCL6 also attenuates expression of antigen presenting genes and other immune signaling mediators(9–12). BCL6 mediates these effects through interaction with a series of histone modifying enzymes(4), and is opposed by epigenetic regulatory proteins that induce gene activation.
CREBBP and EP300
CREBBP and EP300 are responsible for histone acetylation (H3K27Ac) of enhancers controlling genes essential for exit of B-cells from the GC reaction and terminal differentiation towards the plasma cell fate (4). Loss-of-function mutations in CREBBP and EP300 are common in GCB-DLBCL and associated with reduced acetyltransferase activity(13, 14). Crebbp-deficient mice develop B cell lymphoma in cooperation with Bcl2 and that Crebbp loss induces focal depletion of H3K27Ac at enhancers (10, 11, 15). CREBBP-regulated enhancers are normally repressed by the BCL6 transcriptional repressor through recruitment of SMRT and NCOR corepressors during GC reaction(9, 10). SMRT/NCOR complexes contain HDAC3 which is required to allosterically induces its deacetylation of H3K27(9). CREBBP loss of function results in unopposed repression of gene enhancers, most notably those controlling expression of MHC class II genes, through HDAC3(10, 16). Accordingly, HDAC3 loss of function in DLBCL cells with or without CREBBP mutation restores enhancer H3K27Ac and BCL6-SMRT target gene expression, suggesting that HDAC3-targeted therapy can be a precision approach for CREBBP-mutated B cell lymphoma(10, 12). In addition, a recent study identified that CREBBP and EP300 partially compensate for each other through shared transcriptional targets, and EP300 loss confers a synthetic lethality effect in CREBBP-deficient B cells(17). These findings also suggest that EP300 can be a target for CREBBP-mutated B cell lymphoma.
KMT2D
Loss-of-function mutations of KMT2D are also common in GCB-DLBCL and FL. KMT2D forms part of the COMPASS epigenetic modifier complex that regulates gene enhancer functions, and mediates H3K4 mono-methylation(18). Kmt2d-deficiency induces GC hyperplasia in GCB cells and impairs B cell differentiation and class switching in mouse models, as well as accelerating B cell lymphomagenesis in cooperation with Bcl2(19, 20). During the GC reaction, KMT2D target enhancers are demethylated by the histone demethylase LSD1, which is recruited to these sites by BCL6. LSD1 deletion accordingly impairs GC formation and prevents BCL6-driven lymphomagenesis, and LSD1 knockdown has anti-proliferative effects in DLBCL cell lines (21). However, blockade of LSD1 enzymatic activity is insufficient to cause this effect, which is also mediated through the LSD1 tower domain that recruits the COREST complex(21), suggesting that perhaps LSD1 degrader molecules might be more appropriate for precision therapy of KMT2D mutant lymphomas.
EZH2
EZH2 is an enzymatic subunit of polycomb repressive complex 2 (PRC2) which represses gene expression by catalyzing histone 3 lysine 27 trimethylation (H3K27me3) at gene promoters (22). Gain-of-function mutations of EZH2 are frequently observed in GCB-DLBCL and FL(2, 23). EZH2 plays an essential role during the GC reaction through repression of plasma cell differentiation and cell cycle checkpoint genes in cooperation with BCL6(24–27). >90% of lymphoma EZH2 mutations affect the Y646 residue within its catalytic SET domain(23). Mice engineered to express Ezh2Y646 in GC B-cells manifest accelerated lymphomagenesis in cooperation with Bcl2 or BCL6(24, 25). Expression of mutant Ezh2 in GC B-cells impairs T-cell immune surveillance of GC B-cells, resulting in GC hyperplasia and an altered immune microenvironment(28). Pharmacological inhibition of EZH2 is highly effective for the Ezh2-mutated B cell lymphomas both in mouse models and patients with EZH2 mutant lymphoma(24, 25, 29). Collectively, somatic mutations of CREBBP, EP300, EZH2 and KMT2D are associated with the newly defined EZB/cluster 3 class of DLBCLs(2, 3, 30), and correspond to a subset of GCB-DLBCL patients.
TET2
Approximately 10% of DLBCLs feature loss of function mutations of TET2, which defines a unique class of GCB-DLBCLs, distinct from those with CREBBP, EP300, EZH2 and KMT2D mutations(30). Tet2 deficiency yields a preneoplastic phenotype in mice including GC hyperplasia, impaired class switch recombination, and impaired plasma cell differentiation(31). Gene expression profiling of TET2-deficient DLBCL showed repression of immune synapse genes partially overlapping with those affected by CREBBP mutants. This was linked to loss of gene enhancer 5’ hydroxymethylation and impaired H3K27Ac and induced sensitivity to HDAC3 knockdown, similar to the case of CREBBP mutations(31). TET2 mutant GC B-cells and DLBCLs also featured gene repression due to aberrant promoter hypermethylation(32). Hence combinations of HDAC3 inhibitors with DNA methyltransferase inhibitors might be required to restore proper epigenetic programming to TET2 mutant lymphoma cells. Taken together, disruption of epigenetic programming is a hallmark of GCB-DLBCL and FL, due to alteration of regulatory marks controlling gene enhancers and promoters. These defects can be restored by using compounds specific to these mechanisms as a form of precision therapy for these patients, including the opportunity to revert the characteristic loss of immune surveillance observed in GCB-DLBCL and FLs (Figure 1).
Figure 1.
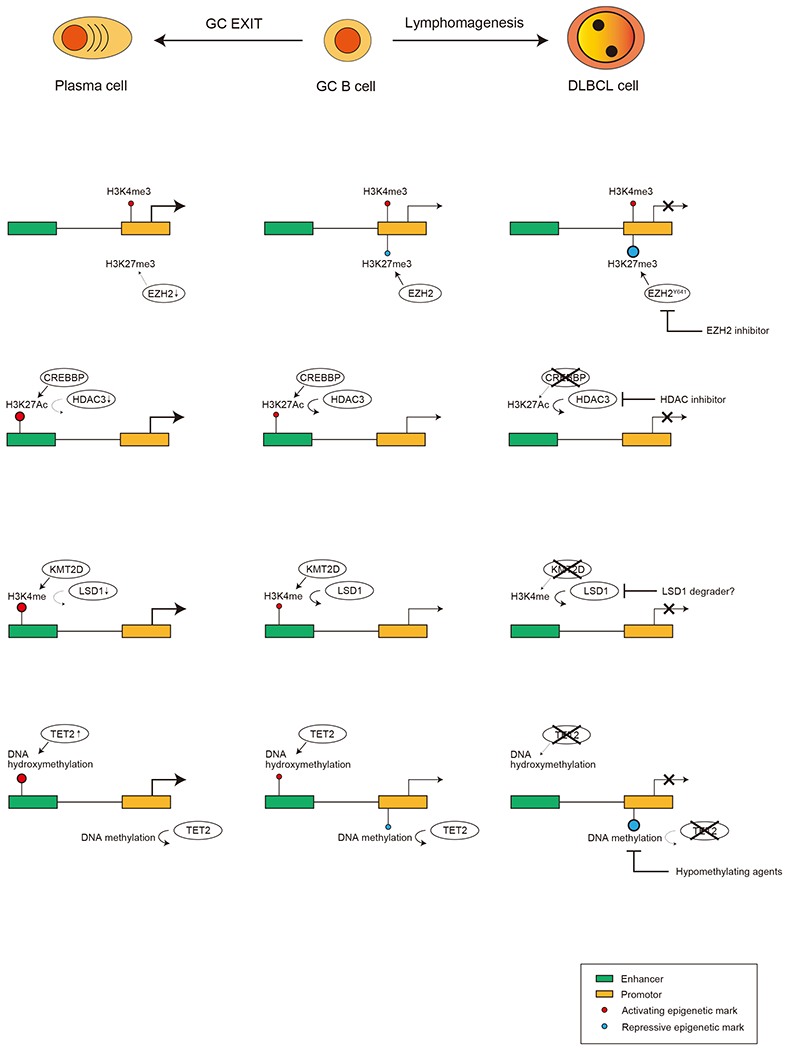
Mechanisms of lymphomagenesis initiated by epigenetic dysfunctions and therapeutic targets.
EZH2 catalyzes H3K27me3 at gene promotors and represses gene expression. EZH2 target genes in GC B-cells are also usually marked with H3K4me3, an active histone mark at promotors, and those genes are called bivalent genes. EZH2 is activated in GCB cells and plays an important role to maintain GC reaction by repressing B cell differentiation associated genes and cell cycle checkpoint genes. Once GC reaction is terminated, EZH2 is downregulated and bivalent genes are reactivated, which induces plasma cell differentiation. EZH2 mutations in DLBCL are usually gain-of-function mutations, and enable GC B-cells to become less dependent on T-cell help. EZH2 inhibitors are a rational approach to target lymphomas with EZH2 mutations. CREBBP and EP300 catalyze H3K27Ac at enhancers and activate genes involved in differentiation and exit from the GC reaction. HDAC3 is responsible for deacetylation of H3K27 and antagonizes CREBBP and EP300. Loss-of-function mutations of CREBBP and EP300 results in H3K27Ac loss at enhancers and are associated with GCB-DLBCL initiation. Since HDAC3 acts in an unopposed manner in those case, HDAC3 inhibitors can specifically effective in GCB-DLBCL with CREBBP and possibly EP300 mutations. KMT2D is responsible for H3K4me1, another active histone mark at enhancers, and is also important to terminate GC reaction. Since LSD1 antagonizes the function of KMT2D, inhibition of LSD1 may be effective in in GCB-DLBCL with loss-of-function mutations of KMT2D. TET2 mediated DNA hydroxymethylation at enhancers is also important to activate B cell differentiation associated genes. TET2 loss causes inactivation of those target genes and lymphomagenesis. Hypomethylating agents such as azacytidine or decitabine may help to overcome the effects of TET2 insufficiency.
Mechanisms of escape from immune surveillance and resistance to conventional therapy initiated by epigenetic dysfunctions.
Mechanistically, histone modifier gene dysfunction in EZB-DLBCLs induces loss of immune recognition molecule expressions on lymphoma cells, which results in immune escape and resistance to immunotherapy. Accordingly, GCB-DLBCLs and FLs manifest poor response to checkpoint inhibitor therapy, and moderate response to CAR-T cell therapy. Loss-of-function mutations of CREBBP result in downregulation of MHC-II, CIITA and CD74 on lymphoma cells through the reduction of H3K27Ac at the respective enhancers and promoters, leading to the escape from CD4 T cell mediated anti-tumor immunity(10–12, 33). T-cells from Primary human FLs with CREBBP mutations manifested impaired activation when placed in mixing studies with their respective lymphoma cells(16). However, lymphoma infiltrating T-cells could be induced to kill lymphomas that had been pre-incubated with specific HDAC3 inhibitors(12). Treatment of syngeneic lymphomas with HDAC3 inhibitors in vivo induced CD4 and CD8 cell infiltration, killing of B-cells, and significantly enhanced the anti-lymphoma activity of PD-L1 blockade(12).
Gain-of-function mutations of EZH2 are also associated with downregulation of both MHC-I and II, and which results in the decreased infiltration of T cells into tumor microenvironment(34). Ezh2 mutation reduces the expression of surface proteins required for the interaction with Tfh cells such as SLAM and ICAM-1, and B cells acquire independency on Tfh cells(28). Expression of these antigen presentation genes is restored by EZH2 inhibitors in vitro, and treatment of syngenic Ezh2 mutant lymphomas with EZH2 inhibitors in vivo recruited activated CD4 and CD8 cells into the tumors, suggesting that these compounds may also enhance T cell mediated anti-tumor immunity(34). Kmt2d mutation is associated with loss of response to CD40 signaling, (19). Notably, CD40 agonist antibodies have been used as therapeutic agents for DLBCL(35), but KMT2D mutations would be expected to confer resistance to such agents(19). As noted earlier it is possible that such effects might be rescued through LSD1 loss of function although this has not yet been explored in the literature.
In addition to the histone modifier gene mutations, aberrant DNA methylation status is associated with poor survival in patients with DLBCL(36). Most notably, hypermethylation and repression of SMAD1 can induce chemotherapy resistance in DLBCL, and SMAD1 de-methylation by hypomethylating agents restores the chemosensitivity in human patients treated with these drugs(37, 38). Finally, the 2016 WHO classification defined as high grade B cell lymphoma (HGBL) those harboring MYC and BCL2 and/or BCL6 translocations, with significantly inferior prognosis(39). Recent studies revealed that HGBL usually derive from EZB/C3 type GCB-DLBCLs with mutations in the aforementioned histone modifier genes(30, 40, 41). Although it is logical that lymphomas with very strong proliferative drive due to MYC and resistance to apoptosis due to BCL6 might be more chemotherapy resistant, it is not yet clear mechanistically exactly how this plays out in the context of these EZB/C3 epigenetically perturbed cases. Overall, disruption of epigenetic programming is associated with resistance to conventional chemotherapy and immunotherapy (Figure 2), providing a strong rationale for use of specifically targeted epigenetic therapies to overcome these mechanisms.
Figure 2.
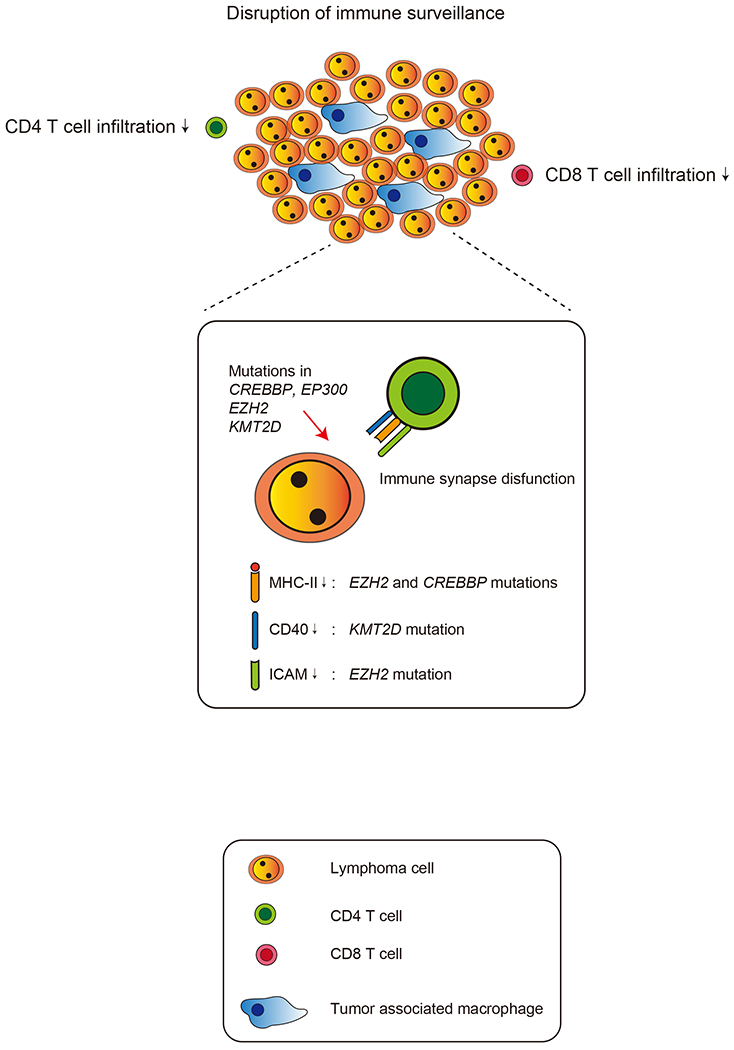
Mechanisms of immune escape induced by mutations in histone modifier genes.
Expressions of MHCs and other surface molecules required for tumor antigen recognition and T cell activation via TCR are downregulated in DLBCL cells with mutations of EZH2, CREBBP, EP300 or KMT2D. Downregulation of those molecules disrupts immune synapse between DLBCL cells and T cells and enables DLBCL cells to escape from immune surveillance. As a result, DLBCL with those mutations shows lower numbers of tumor infiltrating T cells and consists “immune-cold” tumor microenvironment.
Epigenetic heterogeneity and lymphoma progression
Another source of epigenetically encoded therapy resistance in DLBCLs is linked to inherent plasticity and hence heterogeneity of epigenetic marks among populations of lymphoma cells. Indeed, clonal diversity in tumors is known to lead to adverse outcomes, for example due to the presence of therapy-resistant subclones conducive to survival and tolerance of therapeutic agents (42). Recent studies revealed that increased epigenetic heterogeneity based on DNA methylation profiles is associated with inferior clinical outcomes in DLBCL and CLL(36, 43–45). The first study to explore the clinical relevance of DNA methylation heterogeneity in cancer explored cohorts of DLBCL patients and designed a Methylation Heterogeneity score (M-score) to quantify this feature(44). High M-score was shown to be an independent clinical risk factor for outcome(44). Using an independent approach Chambwe et.al. classified DLBCLs based on DNA methylation variability and confirmed higher clinical risk through this orthogonal method(36). Variability in DNA methylation was also linked to outcome in Mantle Cell Lymphomas(46).
Reduced representation bisulfite sequencing (RRBS) in CLL patients analyzed through a novel approach that quantifies disorder of methylation pattern among specific sets of consecutive CpG motifs again showed that heterogeneity is linked to poor outcomes(43). CLL patients also manifested evidence of heterogeneity in histone marks, suggesting additional layers of epigenetic heterogeneity may be functionally relevant in lymphomas(47). RRBS performed in DLBCL cases confirmed that higher levels of epigenetic diversity were linked to risk for relapse(45). Strikingly, there was evidence of selection for specific epigenetic states in relapsed DLBCLs, consistent with the notion of particular epigenetic settings confer a selective advantage among lymphoma cells during treatment. For example, enhancers of therapy-resistance associated genes were aberrantly hypomethylated at relapse(45), and in CLL, epigenetic heterogeneity was linked to variable transcriptional levels of genes further pointing to a relevant functional impact(43).
These findings point to the potential significance of identifying mechanisms that can give rise to epigenetic heterogeneity during lymphomagenesis. Along these lines, it was observed that even normal GC B-cells display greater epigenetic heterogeneity than resting B-cells and this was dependent on expression of the enzyme AICDA, which deaminates cytosine residues during immunoglobulin affinity maturation(48, 49). Notably, AICDA expression yielded greater degrees of DNA methylation heterogeneity and a more lethal disease in a murine lymphoma model. Moreover, primary human DLBCLs with the highest levels of AICDA expression also manifested the greatest degree of epigenetic heterogeneity(50). Collectively the data point to epigenetic heterogeneity as a potentially critical source of therapy resistance in DLBCL. It remains to be seen whether pre-treatment with epigenetic therapies such as DNMT inhibitors might reduce epigenetic heterogeneity and hence fitness of lymphoma cell populations in high risk DLBCLs, and in this way contribute to more favorable clinical responses.
Conclusion
Disruption of epigenetic mechanisms is a major theme in lymphomagenesis. Mutations in epigenetic modifiers are associated with immune evasion and likely explain in part resistance to immunotherapies. In addition, aberrant epigenetic programming and epigenetic heterogeneity are also linked to resistance to chemotherapy. Some of these phenotypes can be restored through use of specific and appropriate epigenetic therapy agents. Preclinical studies show promising results for such “precision epigenetic therapies” such as HDAC3 selective inhibitors for the CREBBP mutations, EZH2 inhibitors for the EZH2 mutations and hypomethylating agents for SMAD1 hypermethylated lymphoma. Hence there is a strong rationale for combining these epigenetic targeted drugs with chemotherapy or immunotherapies, although it is critical to do this in an evidence-based manner and addressing specific and well defined disease mechanisms. We contend that approached in this manner, epigenetic combinatorial therapies have the potential to significantly improve outcomes and reduce reliance on toxic therapy modalities for lymphoma patients.
Figure 3.
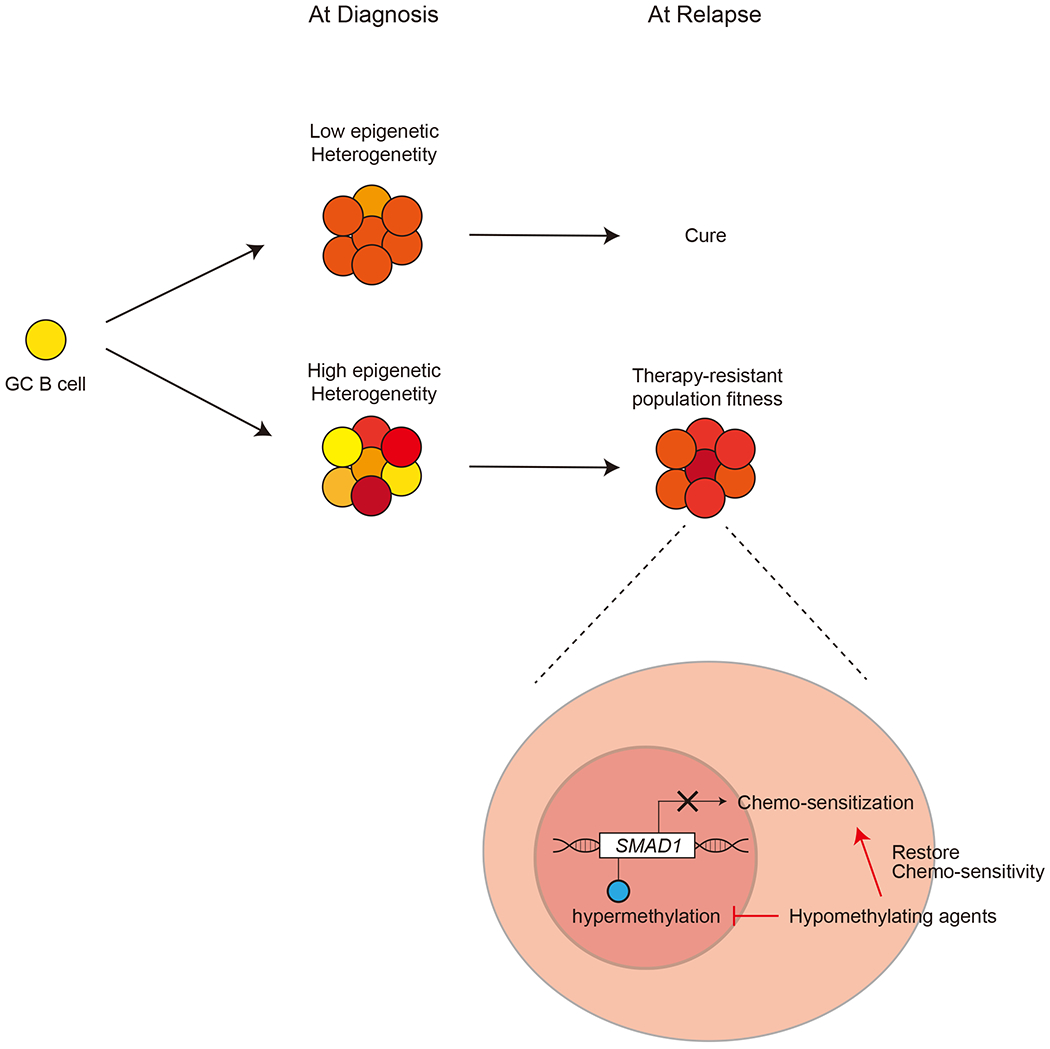
Relationship between DNA methylation heterogeneity and relapse.
High DNA methylation heterogeneity is associated with poor prognosis in patients with DLBCL. DLBCL with higher methylation heterogeneity may have more chance to include therapy resistant populations at the diagnosis. SMAD1 hypermethylation is critical to acquire resistance to conventional chemotherapy, and hypomethylating agents may restore chemo-sensitivity in SMAD1 hypermethylated lymphoma.
Acknowledgements:
AM is supported by NCI R35 CA220499, LLS TRP 6572-19, LLS SCOR 7013-17. AM. is also supported by NCI R01 CA198089, The Follicular Lymphoma Consortium and The Chemotherapy Foundation.
Conflict of interest:
AM receives research funding from Janssen and Sanofi. AM consults for Epizyme, Constellation and Jubilant, and is on the advisory board of KDAC.
References
- 1.Al-Hamadani M, Habermann TM, Cerhan JR, Macon WR, Maurer MJ, Go RS. Non-Hodgkin lymphoma subtype distribution, geodemographic patterns, and survival in the US: A longitudinal analysis of the National Cancer Data Base from 1998 to 2011. Am J Hematol. 2015;90(9):790–5. [DOI] [PubMed] [Google Scholar]
- 2.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(5):679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med. 2018;378(15):1396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mlynarczyk C, Fontan L, Melnick A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol Rev. 2019;288(1):214–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell. 2017;171(2):481–94 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109(10):3879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatzi K, Melnick A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med. 2014;20(6):343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatzi K, Jiang Y, Huang C, Garrett-Bakelman F, Gearhart MD, Giannopoulou EG, et al. A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell Rep. 2013;4(3):578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang Y, Ortega-Molina A, Geng H, Ying HY, Hatzi K, Parsa S, et al. CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov. 2017;7(1):38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Vlasevska S, Wells VA, Nataraj S, Holmes AB, Duval R, et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-cell Lymphoma. Cancer Discov. 2017;7(3):322–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mondello P, Tadros S, Teater M, Fontan L, Chang AY, Jain N, et al. Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov. 2020;10(3):440–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471(7337):189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerchietti LC, Hatzi K, Caldas-Lopes E, Yang SN, Figueroa ME, Morin RD, et al. BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. J Clin Invest. 2010;120(12):4569–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Ramirez I, Tadros S, Gonzalez-Herrero I, Martin-Lorenzo A, Rodriguez-Hernandez G, Moore D, et al. Crebbp loss cooperates with Bcl2 overexpression to promote lymphoma in mice. Blood. 2017;129(19):2645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green MR, Kihira S, Liu CL, Nair RV, Salari R, Gentles AJ, et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A. 2015;112(10):E1116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer SN, Scuoppo C, Vlasevska S, Bal E, Holmes AB, Holloman M, et al. Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity. 2019;51(3):535–47 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Froimchuk E, Jang Y, Ge K. Histone H3 lysine 4 methyltransferase KMT2D. Gene. 2017;627:337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21(10):1190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatzi K, Geng H, Doane AS, Meydan C, LaRiviere R, Cardenas M, et al. Histone demethylase LSD1 is required for germinal center formation and BCL6-driven lymphomagenesis. Nat Immunol. 2019;20(1):86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013;20(10):1147–55. [DOI] [PubMed] [Google Scholar]
- 23.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23(5):677–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell. 2016;30(2):197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beguelin W, Rivas MA, Calvo Fernandez MT, Teater M, Purwada A, Redmond D, et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun. 2017;8(1):877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest. 2013;123(12):5009–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beguelin W, Teater M, Meydan C, Hoehn KB, Phillip JM, Soshnev AA, et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell. 2020;37(5):655–73 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morschhauser F, Salles GA, Le Gouill S, Radford JA, Mckay P, Cartron G, et al. Phase 2 multi-center study of tazemetostat (EPZ-6438), an inhibitor of enhancer of zeste-homolog 2 (EZH2), in patients with relapsed or refractory B-cell non-Hodgkin lymphoma (NHL). Journal of Clinical Oncology. 2016;34(15). [Google Scholar]
- 30.Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell. 2020;37(4):551–68 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Beguelin W, Fontan L, et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov. 2018;8(12):1632–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosikiewicz W, Chen X, M. DP, Ghamlouch H, Aoufouchi S, Bernard OA, et al. TET2 deficiency reprograms the germinal center B cell epigenome and silences genes linked to lymphomagenesis. Sci Adv. 2020;6:eaay5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashwah H, Schmid CA, Kasser S, Bertram K, Stelling A, Manz MG, et al. Inactivation of CREBBP expands the germinal center B cell compartment, down-regulates MHCII expression and promotes DLBCL growth. Proc Natl Acad Sci U S A. 2017;114(36):9701–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ennishi D, Takata K, Beguelin W, Duns G, Mottok A, Farinha P, et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov. 2019;9(4):546–63. [DOI] [PubMed] [Google Scholar]
- 35.de Vos S, Forero-Torres A, Ansell SM, Kahl B, Cheson BD, Bartlett NL, et al. A phase II study of dacetuzumab (SGN-40) in patients with relapsed diffuse large B-cell lymphoma (DLBCL) and correlative analyses of patient-specific factors. J Hematol Oncol. 2014;7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chambwe N, Kormaksson M, Geng H, De S, Michor F, Johnson NA, et al. Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood. 2014;123(11):1699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clozel T, Yang S, Elstrom RL, Tam W, Martin P, Kormaksson M, et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 2013;3(9):1002–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stelling A, Wu CT, Bertram K, Hashwah H, Theocharides APA, Manz MG, et al. Pharmacological DNA demethylation restores SMAD1 expression and tumor suppressive signaling in diffuse large B-cell lymphoma. Blood Adv. 2019;3(20):3020–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2019;37(3):190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sha C, Barrans S, Cucco F, Bentley MA, Care MA, Cummin T, et al. Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J Clin Oncol. 2019;37(3):202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94. [DOI] [PubMed] [Google Scholar]
- 43.Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26(6):813–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De S, Shaknovich R, Riester M, Elemento O, Geng H, Kormaksson M, et al. Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS Genet. 2013;9(1):e1003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan H, Jiang Y, Boi M, Tabbo F, Redmond D, Nie K, et al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat Commun. 2015;6:6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Queiros AC, Beekman R, Vilarrasa-Blasi R, Duran-Ferrer M, Clot G, Merkel A, et al. Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell. 2016;30(5):806–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pastore A, Gaiti F, Lu SX, Brand RM, Kulm S, Chaligne R, et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat Commun. 2019;10(1):1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood. 2011;118(13):3559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dominguez PM, Teater M, Chambwe N, Kormaksson M, Redmond D, Ishii J, et al. DNA Methylation Dynamics of Germinal Center B Cells Are Mediated by AID. Cell Rep. 2015;12(12):2086–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teater M, Dominguez PM, Redmond D, Chen Z, Ennishi D, Scott DW, et al. AICDA drives epigenetic heterogeneity and accelerates germinal center-derived lymphomagenesis. Nat Commun. 2018;9(1):222. [DOI] [PMC free article] [PubMed] [Google Scholar]