

SUMMARY
Cre/loxP technology has revolutionized genetic studies and allowed for spatial and temporal control of gene expression in specific cell types. Microglial biology has particularly benefited because microglia historically have been difficult to transduce with virus or electroporation methods for gene delivery. Here, we investigate five of the most widely available microglial inducible Cre lines. We demonstrate varying degrees of recombination efficiency, cell-type specificity, and spontaneous recombination, depending on the Cre line and inter-loxP distance. We also establish best practice guidelines and protocols to measure recombination efficiency, particularly in microglia. There is increasing evidence that microglia are key regulators of neural circuits and major drivers of a broad range of neurological diseases. Reliable manipulation of their function in vivo is of utmost importance. Identifying caveats and benefits of all tools and implementing the most rigorous protocols are crucial to the growth of the field and the development of microglia-based therapeutics.
In brief
Microglia are key regulators of neural circuits and neuroinflammation. Tools to specifically manipulate their function in vivo are critical. Faust et al. show that recombination efficiency and specificity differ when comparing different microglial CreER mouse lines. They then provide methods and best practices for using these mice going forward.
Graphical Abstract
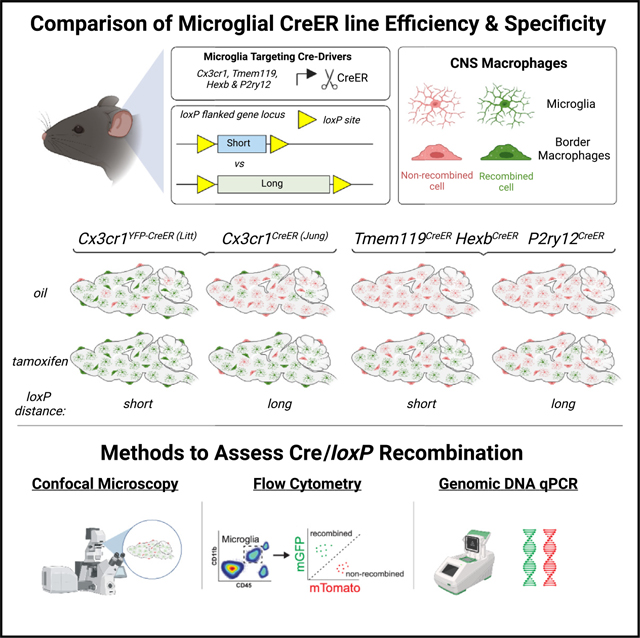
INTRODUCTION
Microglia are resident central nervous system (CNS) macrophages now appreciated to play key roles in regulation of synaptic connectivity and modulation of neural activity.1,2 Microglia are also increasingly recognized as important cellular drivers of brain diseases, ranging from neurodegenerative diseases such as Alzheimer’s disease to neuropsychiatric disorders such as schizophrenia.3,4 This includes studies that have identified many microglia-enriched genes as key risk factors,5 which are likely contributing to the underlying etiology of disease. Thus, methods to study microglial gene function in a cell-type-specific way are of utmost importance. Although recent work has provided evidence of viral targeting of microglia for genetic manipulation,6 this still remains a challenge in the field. In addition, electroporation for gene delivery has been ineffective, and Tet-On/Off systems require experiments to be carried out in the presence of immunosuppressive drugs. As a result, the Cre/loxP system has been the primary method to genetically manipulate microglia in vivo, and a number of microglial Cre lines have been made in recent years. Here, we provide a comprehensive comparison of some of the most widely available tamoxifen (TAM)-inducible microglial Cre lines and a guide for best practices to ensure efficient and specific recombination.
When the Cre protein is fused to a mutated estrogen-binding receptor domain (ER), the fusion CreER protein is retained in the cytoplasm and prevents the Cre-mediated recombination of loxP sequences in the genomic DNA (gDNA).7 Once the CreER binds 4-hydroxytamoxifen (4-OHT), the active metabolite of TAM, the CreER translocates to the nucleus and recombination can occur. Recombination efficiency depends on several factors, such as the level of CreER expression, the amount of TAM that enters the cell, the accessibility of the loxP locus, and the inter-loxP distance of the floxed alleles.8 These variables necessitate validation of Cre/loxP recombination in each experimental paradigm.9 The most widely used CreER lines to target microglia have leveraged the promoter for the gene encoding the fractalkine receptor (CX3CR1).10–12 Compared with other recently generated lines such as Sall1CreER13 or Cd11bCre,14 there is less recombination in other brain or immune cell types. Cx3cr1 is also expressed by other peripheral immune cells, such as circulating, bone marrow-derived monocytes. However, the majority of these peripheral cells are short lived and turn over within 4 weeks because of ongoing, lifelong hematopoiesis.10–12 Thus, 4 weeks after TAM administration, recombination is largely restricted to microglia and other long-lived macrophages (e.g., perivascular macrophages) in Cx3cr1CreER mouse lines.15 Although these mice have led to significant advancements in the field as a more-specific microglial genetic tool, there are some limitations. First, these are knockin lines in which CreER was knocked into the Cx3cr1 locus in a way that ablates endogenous Cx3cr1 gene expression. Although most studies use heterozygotes to avoid knocking out Cx3cr1, these heterozygous mice still lack a copy of the Cx3cr1 gene. In addition, the need for peripheral macrophage turnover after 4 weeks following TAM administration limits studies during the first few weeks of post-natal development. There are also specific CX3CR1+ populations of longer-lived tissue-resident macrophages in the CNS that, similar to microglia, still remain recombined after 4 weeks following TAM administration.15 In certain cases, it has also been shown that Cx3cr1CreER mouse lines can induce loxP recombination in microglia in the absence of TAM.16–18 Also, TAM administration in early postnatal Cx3cr1YFP–CreER (Litt) pups can induce an interferon-responsive population of microglia later in development.19 These limitations have led to the recent development of new inducible Cre lines that more specifically target microglia vs. other macrophage populations, including Tmem119CreER,20 HexbCreER,21 and P2ry12CreER mice.22 Another recent development is the generation of a non-inducible, highly specific microglial split-Cre line for experiments where high recombination efficiency and temporal regulation of gene recombination are not required.23
The effectiveness of CreER lines depends on several factors, such as (1) the cell-type specificity of CreER expression, (2) the expression level of CreER, (3) the degree of off-target effects in both the presence and absence of TAM, and (4) the degree of spontaneous recombination in the absence of TAM. Therefore, several quality-control experiments should be performed for every study. In the current study, we performed a systematic comparison of five of the most widely available inducible microglial CreER lines: Tmem119CreER, HexbCreER, P2ry12CreER, and two Cx3cr1CreER lines. Together, our results demonstrate that recombination efficiency and cell-type specificity vary across these CreER lines. Our data further support that recombination efficiency can be boosted by breeding the CreER to homozygosity for some lines and using floxed mouse lines with shorter inter-loxP distances. Care should also be taken because some CreER lines induce a higher degree of spontaneous recombination, which also depends on the inter-loxP distance. We finally provide guidelines, quality-control measures, and methods for future use of the Cre/loxP system in microglia, which can be extended to other cell types.
RESULTS
Recombination efficiency varies across microglial CreER lines
We initially compared recombination across the Cx3cr1YFP–CreER (Litt)(JAX #021160),10 Cx3cr1CreER (Jung) (JAX #020940),11 Tmem119CreER (JAX #031820),20 HexbCreER,21and P2ry12CreER (JAX #034727)22 mouse lines using the Rosa26mTmG reporter line24 (Figure 1A). This reporter line expresses loxP-(membrane tdTomato [mTomato])-STOP-loxP-(membrane GFP [mGFP]) under the Rosa26 promoter (Figure 1B). For these initial experiments, we used mice heterozygous for the Rosa26mTmG allele such that only one allele will be recombined for a given cell. At baseline, all cells express mTomato, but in cells that undergo Cre/loxP recombination, the mTomato and stop codon are removed, leading to mGFP expression. Mice were injected with TAM daily at post-natal day 28 (P28) through P31. At P56, half of each brain was used for fluorescence-activated cell sorting (FACS), and the other half of the brain was used for immunofluorescence microscopy. For FACS, brains were mechanically dissociated, subjected to Percoll gradient, and immunostained for CD11b and CD45. Live microglia (DAPINegative) were sorted by FACS as CD11bHigh CD45Mid (Figure S1A). We then assessed recombination by evaluating the percentage of microglia that were non-recombined (mTomato+) vs. recombined (mGFP+) across the CreER lines (Figures 1C and S1B). For the Cx3cr1YFP–CreER (Litt) line, in which all microglia constitutively express YFP, recombined cells (mGFP+YFP+) were distinguished from non-recombined cells (mTomato+ YFP+) by both the increased intensity of mGFP and the loss of mTomato (Figure S1B). The percentage of recombined mGFP+ microglia was highest in heterozygous P56 Cx3cr1YFP–CreER/+ (Litt) (96.56% ± 0.72%) and Cx3cr1CreER/+ (Jung) (92.56% ± 2.03%) mice, which had nearly 100% of cells recombined (Figures 1D, 1E, and 1I). In comparison, using the same TAM paradigm, recombination was less efficient in Tmem119CreER/+ (49.9% ± 3.55%), HexbCreER/+ (33.80% ± 2.80%), and P2ry12CreER/+(24.9% ± 2.58%) mice (Figures 1F–1I), which is consistent with independent observations by another group.25 However, if the CreER was bred to homozygosity, recombination efficiency was increased in the Tmem119CreER/CreER (66.1% ± 6.47%) and HexbCreER/CreER (62.40% ± 4.61%) mice (Figures S2A–S2H). Although not directly tested, the P2ry12CreER mice were engineered in a similar way and would likely yield similar results. One remaining important consideration for experiments in homozygous animals is whether the expression of the gene driving the CreER is impacted. Unlike the knockin Cx3cr1CreER lines, the Tmem119CreER, HexbCreER, and P2ry12CreER mice were engineered to retain native gene expression for the gene driving CreER. Still, we checked the protein encoded by the genes driving the CreER for all of these mice. Importantly, we found that in the Tmem119CreER and HexbCreER mice, TMEM119 protein and HEXB enzymatic activity were reduced, respectively, in heterozygous and homozygous CreER mice compared with wild-type mice (Figures S2I–S2K). In contrast, in P2ry12CreER mice, no change in P2RY12 levels were observed (Figures 2A, 2C, 2E, 2G, 2I, and S3A–S3F), which is consistent with previous reports.22 Thus, the efficiency of recombination can be increased in Tmem119CreER, HexbCreER, and P2ry12CreER mouse lines by breeding the CreER to homozygosity, but there is some decrease in native protein levels or activity that should be taken into consideration for the Tmem119CreER and HexbCreER lines.
Figure 1. Recombination efficiency varies across microglial CreER lines.
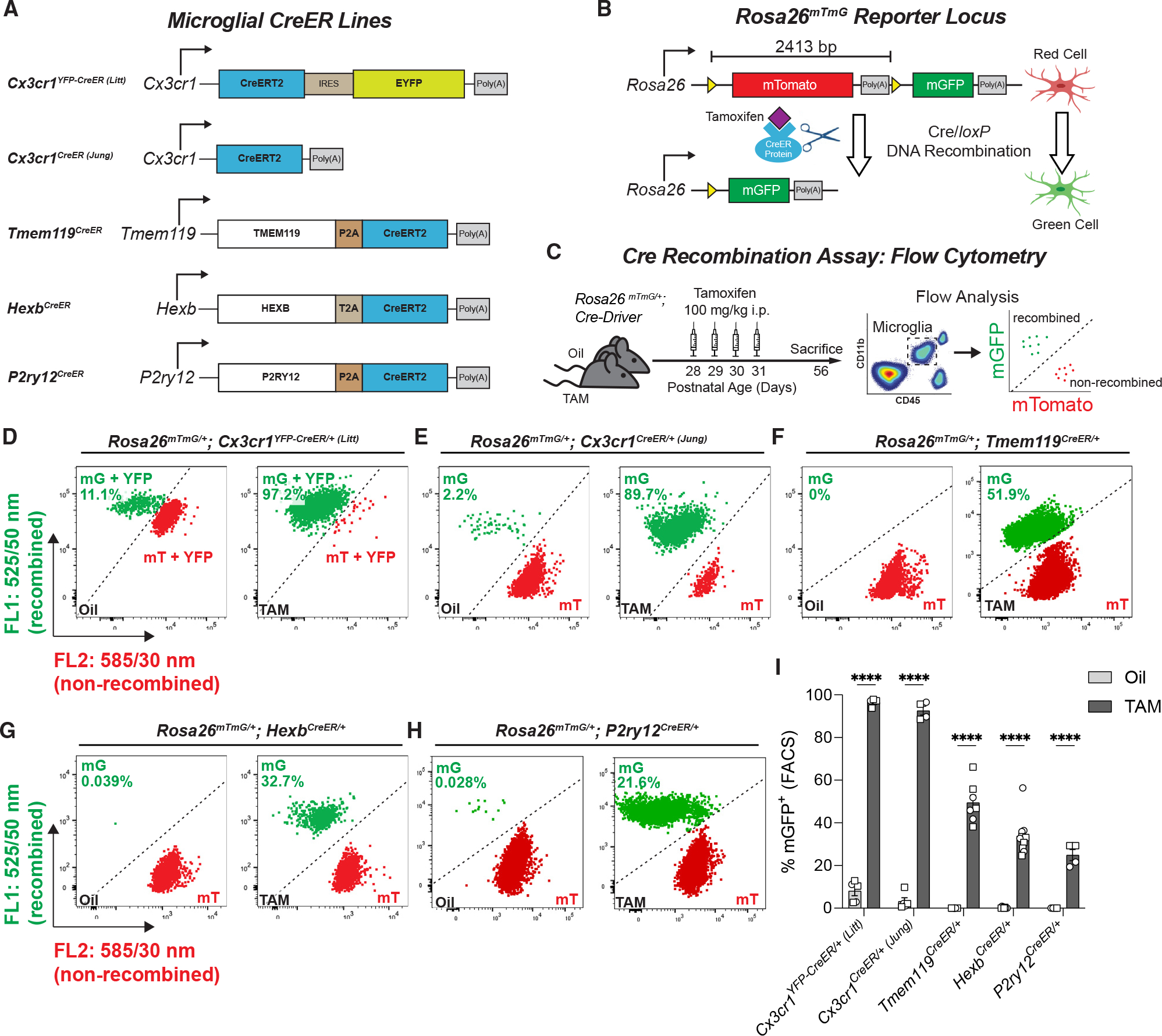
(A) Diagram of transgenes used to express CreER in Cx3cr1YFP–CreER (Litt), Cx3cr1CreER (Jung), Tmem119CreER, HexbCreER, and P2ry12CreER mice.
(B) Diagram of the Rosa26mTmG allele and corresponding cellular fluorescence before and after Cre/loxP DNA recombination. loxP sites are indicated by yellow triangles.
(C) Diagram of experimental protocol used to assess tamoxifen (TAM)-induced Cre/loxP recombination of Rosa26mTmG/+ in microglia by flow cytometry.
(D–H) Representative flow cytometry results show the percentage of mGFP+ (mG+) and mTomato+ (mT+) microglia from individual animals from each group.
(I) Quantification of the percentage of recombined mGFP+ microglia in microglial CreER lines showed increased recombination in TAM vs. oil for all five CreER lines (Student’s t tests: Cx3cr1YFP–CreER/+ (Litt):n = 5 oil, 5 TAM mice; Cx3cr1CreER/+ (Jung):n = 5 oil, 4 TAM mice; Tmem119CreER/+:n = 8 oil, 7 TAM mice; HexbCreER/+: n = 9 oil, 10 TAM mice; P2ry12CreER/+: n = 4 oil, 4 TAM mice; ****p < 0.0001). All data are presented as mean ± SEM. Individual data points indicate males (squares) and females (circles).
See also Figures S1 and S2 and Table S1.
Figure 2. Spatial distribution of Rosa26mTmG recombination in microglial CreER lines.
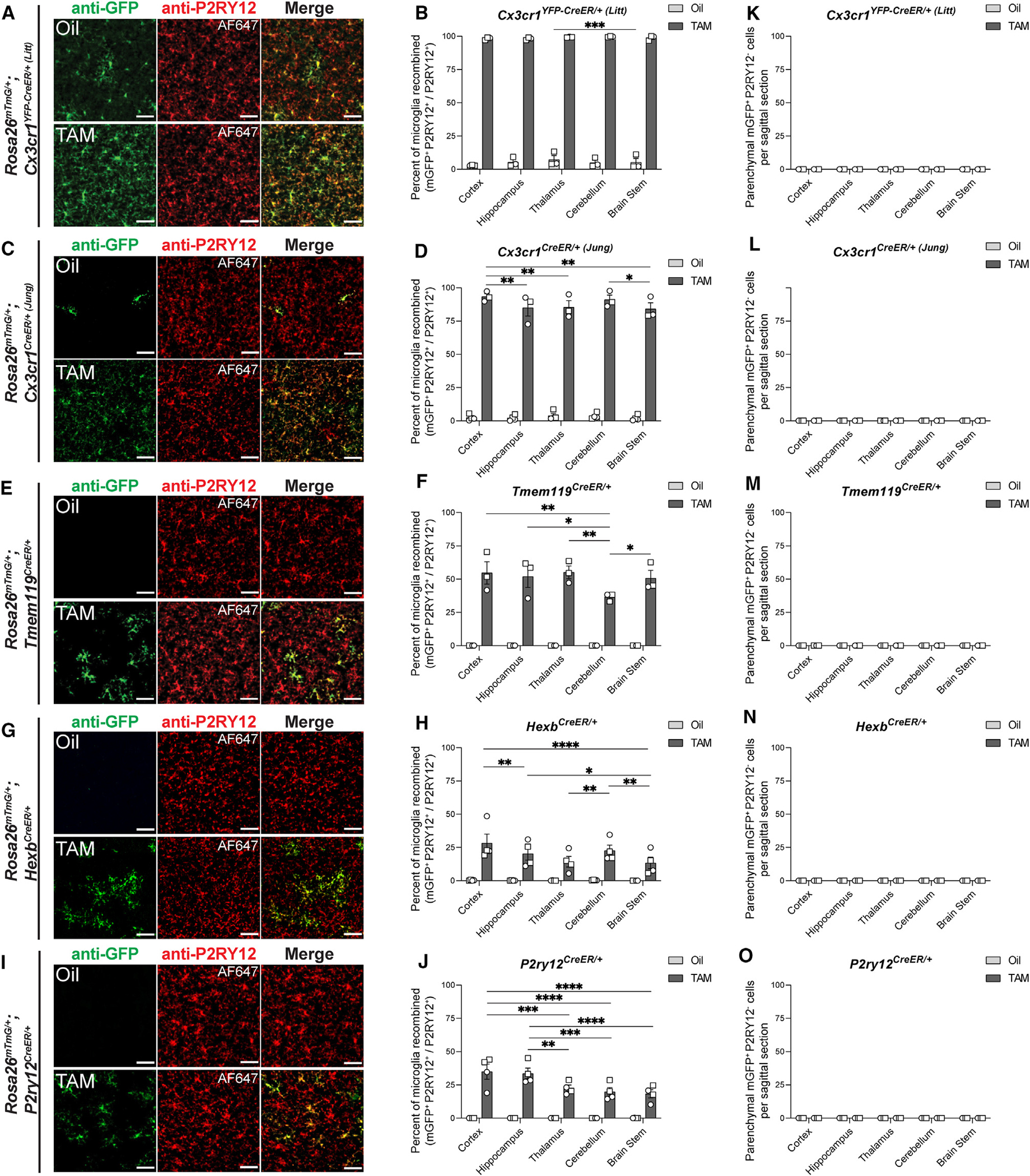
(A, C, E, G, and I) Representative immunofluorescent images of brain sections from right hemispheres of oil- and TAM-injected mice used for flow cytometry analysis in Figure 1. Sections were immunolabeled for anti-P2RY12 (Alexa Fluor 647 [AF647] pseudo-colored red) to identify microglia and anti-GFP (green) to identify recombined cells. Scale bars, 50 μm.
(B, D, F, H, and J) Quantifications of the percentage of P2RY12+ microglia that are mGFP+ in the cortex, hippocampus, thalamus, cerebellum, and brainstem of each CreER line after exposure to oil or TAM (B: Cx3cr1YFP–CreER (Litt), n = 3, 3 mice; D: Cx3cr1CreER (Jung), n = 4, 3 mice; H: HexbCreER, n = 4, 4 mice; J: P2ry12CreER, n = 4, 4 mice; two-way repeated-measures ANOVA with Tukey’s post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
(K–O) Quantifications of the number of P2RY12− cells per section that are mGFP+ in the cortex, hippocampus, thalamus, cerebellum, and brainstem of each CreER line after exposure to oil or TAM (K: Cx3cr1YFP–CreER (Litt), n = 3, 3 mice; L: Cx3cr1CreER (Jung), n = 4, 3 mice; M: Tmem119CreER, n = 4, 3 mice; N: HexbCreER: n = 4, 4 mice; O: P2ry12CreER: n = 4, 4 mice; two-way repeated measures ANOVA with Tukey’s post hoc test).
All data are presented as mean ± SEM. Individual data points indicate males (squares) and females (circles). See also Figure S3.
Regional and cell-specific recombination in microglial CreER lines
We next confirmed our flow cytometry results and assessed microglial specificity and regional differences in recombination by immunofluorescence microscopy in fixed hemi-brain sections from the same animals used for flow cytometry (Figure 1). These experiments confirmed results obtained by flow cytometry and further showed recombination occurs across all brain regions analyzed (Figures 2A–2J and S3A–S3F). There were some significant differences in recombination between brain regions within a given CreER line, but these differences were relatively subtle and much smaller in magnitude than the differences in recombination when comparing across CreER lines. Note, the Cx3cr1YFP–CreER/+ (Litt) line includes a YFP reporter. The fluorescent YFP signal is largely in the soma of microglia, and anti-GFP immunolabeling of mGFP+YFP+ microglia can be distinguished from mGFP−YFP+ microglia by fluorescence intensity and more intense membrane and process labeling (Figure S3G). In the process of assessing recombination across brain regions, we further identified that both Cx3cr1CreER lines induced recombination in anti-LYVE1+ brain border macrophages (Figures 3A and 3B), and that the Tmem119CreER line induced recombination in a small subset of brain-border-associated anti-ALDH1A2/anti-S100A6-positive fibroblasts (Figures 3C–3E and S3H). There was no recombination detected in border-associated cells in the HexbCreER or P2ry12CreER lines (Figure 3). When analyses were restricted to the brain parenchyma (excluding brain borders), recombination was restricted to microglia in all five CreER lines (Figures 2K–2O). Thus, this immunofluorescence method is particularly useful for determining whether recombination is restricted to microglia vs. other CNS cell types in intact tissue, and it is advantageous for detecting whether recombination is evenly distributed across the tissue. Although most efficient, the recombination is the least specific in the Cx3cr1CreER lines.
Figure 3. Cell-type specificity of microglial CreER lines.
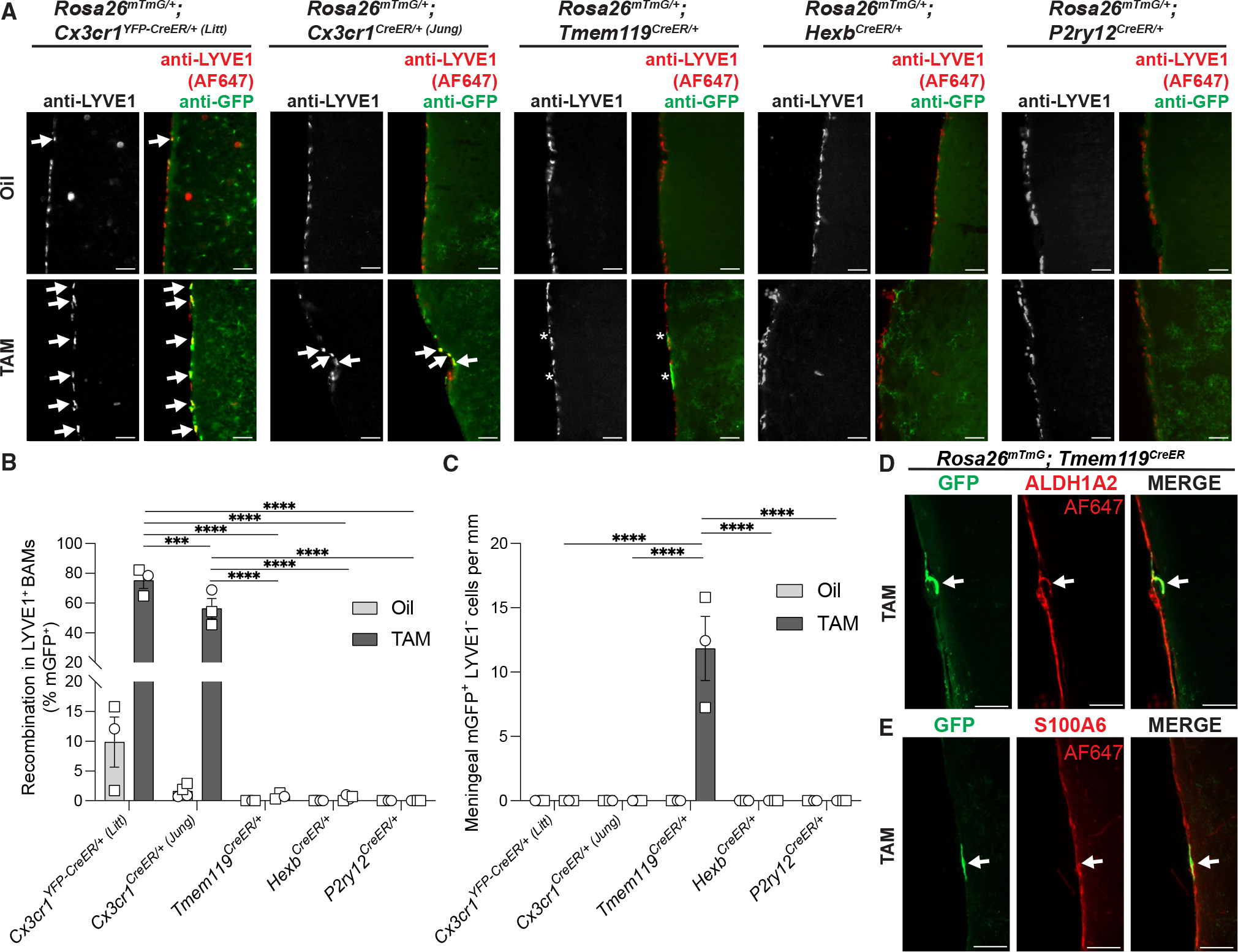
(A) Representative immunofluorescent images of brain sections from right hemispheres of oil- and TAM-injected Rosa26mTmG/+;Cre-DriverCreER/+ mice used for flow cytometry analysis in Figure 1. Sections were immunolabeled for anti-LYVE1 (AF647 pseudo-colored red) to identify border-associated macrophages and anti-GFP (green) to identify recombined cells. Arrows indicate mGFP+ border-associated macrophages. Asterisks indicate mGFP+ cells along the meninges immunonegative for anti-LYVE1. Scale bars, 50 μm.
(B) Quantification of the percentage of recombined mGFP+ border-associated macrophages shows increased recombination in TAM-injected Cx3cr1CreER lines compared with Tmem119CreER, HexbCreER, and P2ry12CreER lines (two-way ANOVA with Tukey’s post hoc test; n = 3 oil Cx3cr1YFP–CreER (Litt), 3 TAM Cx3cr1YFP–CreER (Litt), 4 oil Cx3cr1CreER (Jung), 3 TAM Cx3cr1CreER (Jung), 4 oil Tmem119CreER, 3 TAM Tmem119CreER, 4 oil HexbCreER, 4 TAM HexbCreER, 4 oil P2yr12CreER, and 4 TAM P2ry12CreER mice; ***p < 0.001, ****p < 0.0001).
(C) Quantification of the frequency of recombined mGFP+ cells immunonegative for anti-LYVE1 along the meningeal border shows increased recombination in Tmem119CreER mice (two-way ANOVA with Tukey’s post hoc test; n = 3 oil Cx3cr1YFP–CreER (Litt), 3 TAM Cx3cr1YFP–CreER (Litt), 4 oil Cx3cr1CreER (Jung), 3 TAM Cx3cr1CreER (Jung), 4 oil Tmem119CreER, 3 TAM Tmem119CreER, 4 oil HexbCreER, 4 TAM HexbCreER, 4 oil P2yr12CreER, and 4 TAM P2ry12CreER mice; ****p < 0.0001).
(D and E) Representative immunofluorescent images of brain sections from right hemispheres of oil- and TAM-injected Rosa26mTmG;Tmem119CreER mice used for flow cytometry analysis. Sections were immunolabeled for anti-GFP (green) to identify recombined cells and (D) anti-ALDH1A2 (AF647 pseudo-colored red) to identify arachnoid meningeal fibroblasts or (E) anti-S100A6 (AF647 pseudo-colored red) to identify pial meningeal fibroblasts. Arrows indicate mGFP+ cells along the meninges. Scale bars, 50 μm.
All data are presented as mean ± SEM. Individual data points indicate males (squares) and females (circles).
Off-target effects of TAM in microglial CreER mice
In addition to regional and cell-specificity analyses, we also used immunofluorescence analyses to assess off-target effects of TAM on microglia. Similar to a recent publication,19 we observed that TAM injection in neonates caused persistent downregulation of the homeostatic microglial marker P2RY12 in subpopulations of microglia in Cx3cr1YFP–CreER/+ (Litt) mice (Figures 4A–4C and 4G). This was not observed in another microglial CreER line Tmem119CreER/+. It also did not occur in Cx3cr1EGFP/+ mice, which demonstrates this effect was not due to loss of one allele of Cx3cr1. Indeed, recently published work demonstrated that this change in homeostatic microglial phenotype is likely due to DNA damage resulting from cleavage of pseudo-loxP sites,19 which requires both high levels of Cre activity and TAM to induce CreER translocation to the nucleus. Unlike TAM administration in neonates, TAM administration at P28 did not induce gross changes in P2RY12 in Cx3cr1YFP–CreER/+ (Litt) mice (Figures 4D–4G, S3A, S3B, and S3I). Similar results were obtained for other microglial CreER lines (Figures S3C–S3F and S3I). To further investigate whether P28 TAM administration itself causes more subtle changes in microglial gene expression, we performed bulk RNA sequencing (RNA-seq) on microglia isolated from Cx3cr1YFP–CreER/+ (Litt) mice (Figures 4H and 4I), as well as other CreER lines (Figure S4). In all cases, TAM exposure had little effect on microglial gene expression, demonstrating that P28–P31 TAM exposure does not significantly affect the microglial transcriptome.
Figure 4. Off-target effects of TAM in Cx3cr1YFP–CreER (Litt) mice.
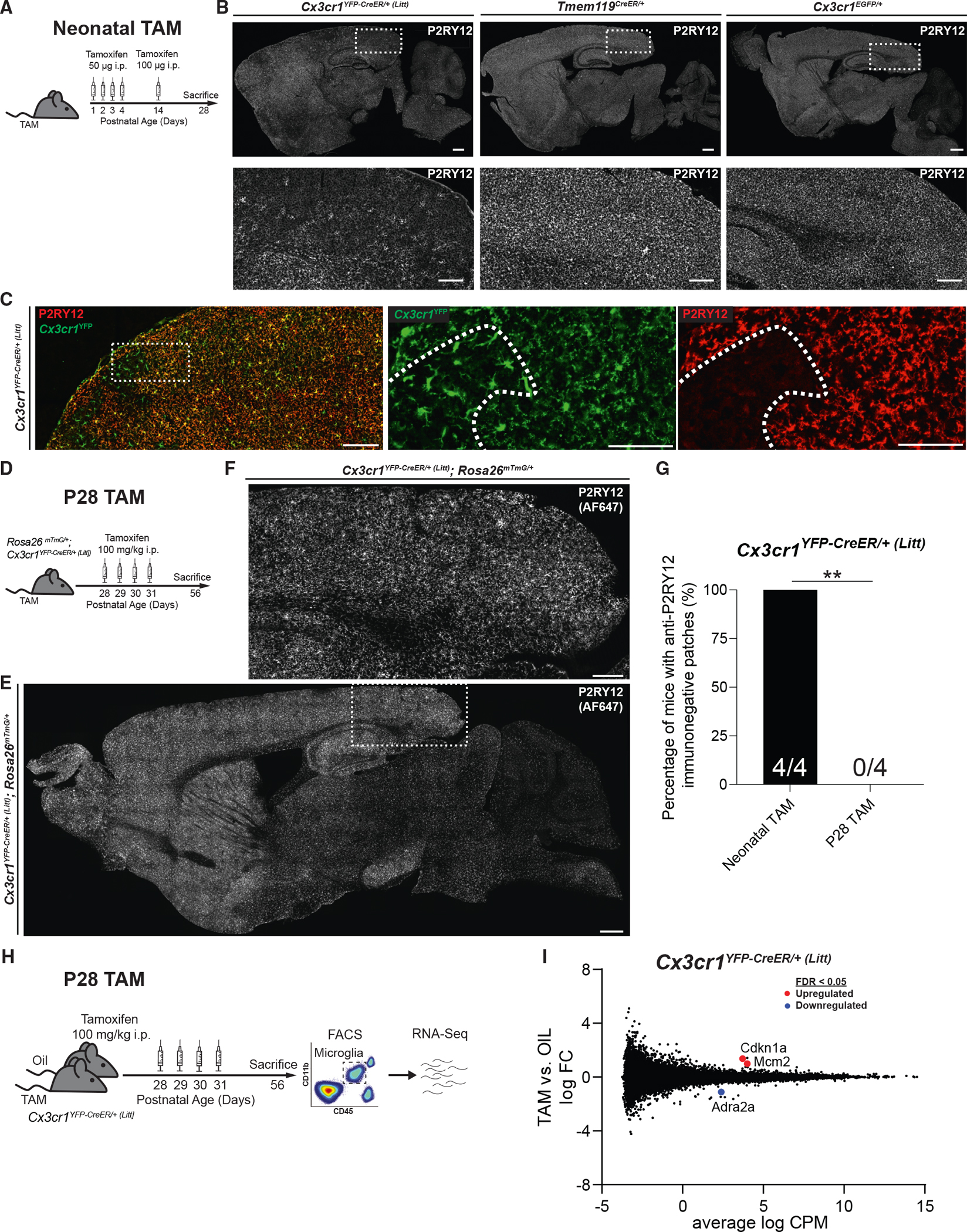
(A) Timeline of neonatal TAM injection for images in (B) and (C).
(B) Fluorescent images of brain sections from Cx3cr1YFP–CreER/+ (Litt), Tmem119CreER/+, and Cx3cr1EGFP/+ mice injected with TAM neonatally, immunolabeled with anti-P2RY12. Large regions of the cortex are devoid of anti-P2RY12 immunofluorescence in Cx3cr1YFP–CreER/+ (Litt) mice, but not Tmem119CreER/+ or Cx3cr1EGFP/+ mice. Scale bars, 500 mm; 200 μm (insets).
(C) Fluorescent images of brain sections from Cx3cr1YFP–CreER/+ (Litt) mice injected with TAM neonatally, immunolabeled with anti-P2RY12 (red) and anti-GFP to identify Cx3cr1YFP+ cells (green). Regions of the cortex devoid of anti-P2RY12 immunofluorescence (dotted outline in inset) contain Cx3cr1YFP+ cells with a morphology characteristic of reactive microglia. Scale bars, 500 μm; 100 μm (insets).
(D) Diagram of experimental protocol used to assess the effect of TAM injection at P28 on anti-P2RY12 immunofluorescence in Cx3cr1YFP–CreER/+ (Litt);Rosa26mTmG/+ mice.
(E and F) Fluorescent images of brain sections from Cx3cr1YFP–CreER/+ (Litt);Rosa26mTmG/+ mice used for flow cytometry analysis in Figure 1, immunolabeled with anti-P2RY12 (imaged using AF647). No patches are devoid of anti-P2RY12 immunofluorescence. Scale bars, 500 μm (E); 200 μm (F).
(G) Quantification of the percentage of mice with patches devoid of anti-P2RY12 immunofluorescence shows higher prevalence in Cx3cr1YFP–CreER/+ (Litt) mice injected with TAM as neonates vs. Cx3cr1YFP–CreER/+ (Litt) mice injected with TAM at P28 (chi-squared test: n = 4, 4 mice; **p < 0.01).
(H) Diagram of experimental protocol used to perform RNA sequencing on Cx3cr1YFP–CreER/+ (Litt) mice injected with TAM or oil at P28.
(I) Smear plot of TAM-vs. oil-injected Cx3cr1YFP–CreER/+ (Litt) mice depicting log fold change (FC) on the y axis against log counts per million (CPM) on the x axis (n = 4, 5 mice). Differentially expressed genes with false discovery rate (FDR) < 0.05 are annotated in red (upregulated by TAM) or blue (downregulated by TAM).
See also Figure S4.
Spontaneous recombination in microglial CreER lines
Besides recombination in other long-lived macrophages and off-target effects in Cx3cr1CreER mouse lines, we also observed spontaneous recombination in the absence of TAM administration by flow cytometry and immunofluorescence microscopy (Figures 1I, 2A–2D, and 5). This spontaneous recombination occurred in both the Cx3cr1YFP–CreER/+ (Litt) and Cx3cr1CreER/+ (Jung) mice in oil and non-injected controls for these lines (Cx3cr1YFP–CreER/+ (Litt) = 8.02% ± 2.15%; Cx3cr1CreER/+ (Jung) = 3.19% ± 1.68%). In contrast, spontaneous recombination was negligible in Tmem119CreER, HexbCreER, and P2ry12CreER mice (Figures 1I, 2E–2J, and 5). To further investigate the spontaneous recombination in Cx3cr1YFP–CreER/+ (Litt);Rosa26mTmG/+ mice, we assessed recombination at younger (1 month) and older (6 months) ages. In Cx3cr1YFP–CreER/+ (Litt);Rosa26mTmG/+ mice, we observed recombination (mGFP+) in 9.7% ± 0.2% of cortical IBA1+ microglia at 1 month, which increased to 21.8% ± 3.9% of microglia by 6 months of age (Figure S5). This is in comparison with 6-month-old Tmem119CreER/+;Rosa26mTmG/+ mice, which displayed no spontaneous recombination. These data are consistent with previous reports of TAM-independent, spontaneous recombination in Cx3cr1YFP–CreER/+ (Litt) mice.16–18
Figure 5. Spontaneous recombination of Rosa26mTmG in microglial CreER lines.
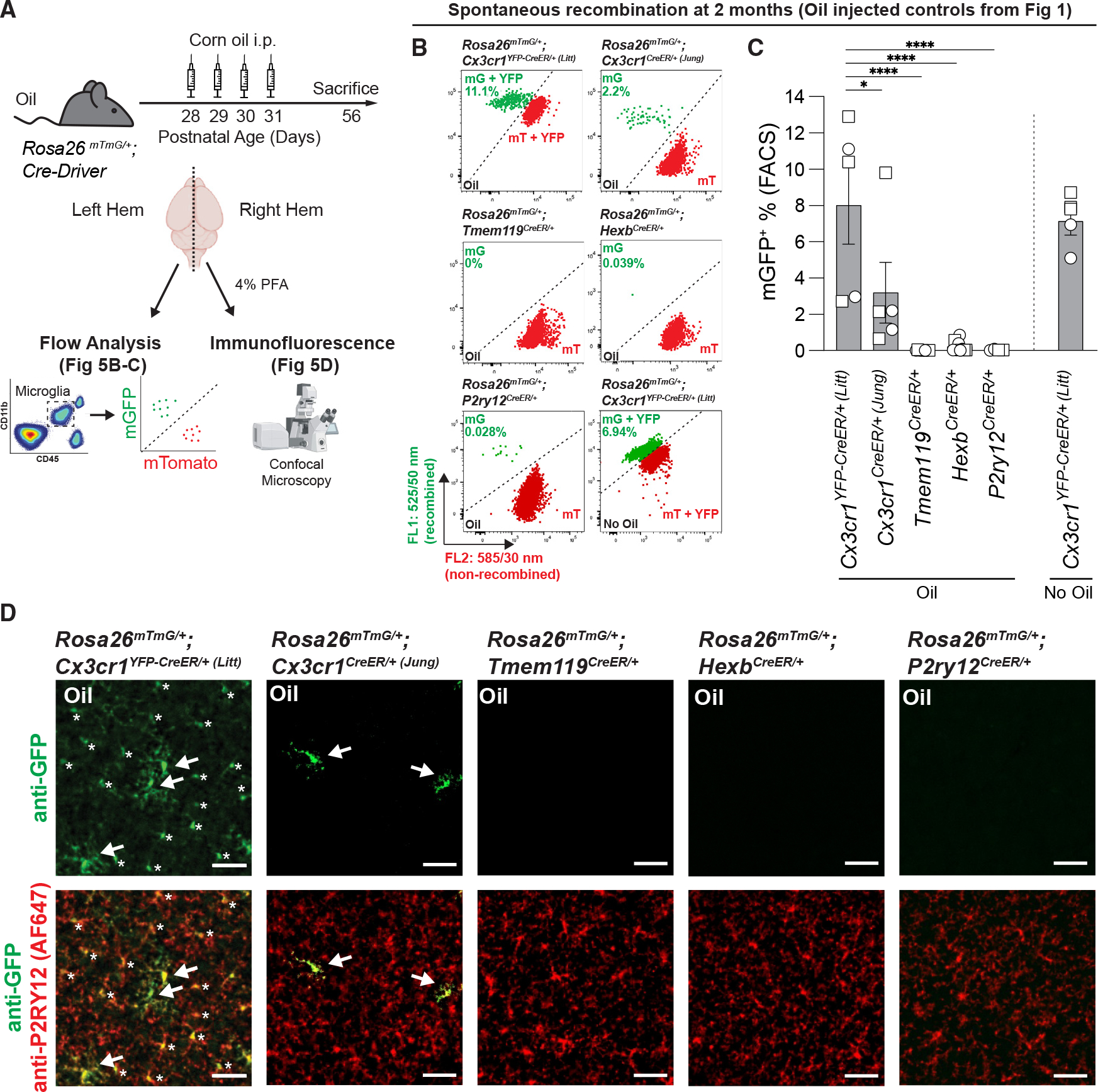
(A) Diagram of experimental protocol used to assess spontaneous Cre/loxP recombination of Rosa26mTmG/+ in microglia by flow cytometry and immunofluorescence.
(B) Representative flow cytometry results show the percentage of recombined mGFP+ (mG) and mTomato+ (mT) microglia from individual animals from each group from Figure 1 and uninjected (no oil) Cx3cr1YFP–CreER/+ (Litt) mice.
(C) Quantification of the percentage of recombined mGFP+ microglia shows increased spontaneous recombination of the Rosa26mTmG allele in the Cx3cr1YFP–CreER (Litt) line compared with the Cx3cr1CreER (Jung), Tmem119CreER, HexbCreER, and P2ry12CreER lines (one-way ANOVA with Tukey’s post hoc test; n = 5 Cx3cr1YFP–CreER/+ (Litt), 5 Cx3cr1CreER/+ (Jung), 8 Tmem119CreER/+, 9 HexbCreER/+, and 4 P2ry12CreER/+ mice; *p < 0.05, ****p < 0.0001).
(D) Representative immunofluorescent images of brain sections from right hemispheres of oil-injected mice used for flow cytometry analysis in (B) and (C). Sections were immunolabeled for anti-P2RY12 (AF647 pseudo-colored red) to identify microglia and anti-GFP (green) to identify recombined cells. The number of recombined mGFP+ microglia (white arrows) matches the results observed by flow cytometry. In the Cx3cr1YFP–CreER/+ (Litt) line, the soma of non-recombined microglia are also immunolabeled by anti-GFP because of the constitutive expression of YFP (asterisks), but it can be distinguished from recombined mGFP+ microglia by fluorescence intensity and membrane labeling. Scale bars, 50 μm.
All data are presented as mean ± SEM. Individual data points indicate males (squares) and females (circles). See also Figure S5.
Inter-loxP distance is a determinant of recombination efficiency in microglial CreER lines
After more thoroughly characterizing recombination efficiency and phenotypic differences among the different microglial CreER lines, we sought parameters that could modify this recombination efficiency. First, we identified that increasing the dose of Cre enzyme could increase efficiency (Figure S2). Second, because previous work has shown a strong correlation between inter-loxP distance and recombination rates,8,18 we next assessed the impact of inter-loxP distance on recombination. For these experiments, we compared recombination of the shorter inter-loxP distance Rosa26Ai9 allele (871 bp inter-loxP distance) with the longer inter-loxP distance Rosa26mTmG allele (2.4 kb inter-loxP distance) when crossed to either a highly efficient microglial CreER line (Cx3cr1YFP–CreER (Litt)) or a less efficient microglial CreER line (Tmem119CreER) (Figures 6A and 6B). Mice were injected with 100 mg/kg TAM intraperitoneally (i.p.) or corn oil from P28 to P31. Microglia were then assessed and collected by FACS at P56, microglial gDNA was isolated, and recombination was measured by flow cytometry and endpoint PCR (Figure 6B). As predicted, the Cx3cr1YFP–CreER (Litt) line recombined efficiently when crossed to either of the floxed lines, independent of inter-loxP distance (Figures 6C–6E). These mice also displayed spontaneous recombination events in the absence of TAM, which was further increased when the mice were crossed with the allele with the shorter inter-loxP distance (Rosa26Ai9). In contrast, minimal spontaneous recombination was observed in the Tmem119CreER line (Figures 6F–6H). Also, the Tmem119CreER line induced more efficient recombination of the floxed allele with a shorter 871 bp inter-loxP distance (Rosa26Ai9) compared with a floxed allele with a longer 2.4 kb inter-loxP distance (Rosa26mTmG) (Figures 6F–6H). Similar results were obtained in the P2ry12CreER line by another group.25 Endpoint PCR was also sufficient to identify highly efficient recombination and spontaneous recombination events (Figures 6I–6K). The increased efficiency of the Tmem119CreER line when crossed to lines with shorter inter-loxP distances was further confirmed using a Becn1Flox line (600 bp inter-loxP distance) and a C1qaFlox line (1,156 bp inter-loxP distance) (Figure S6). These findings are also consistent with previously published work showing nearly 100% recombination at the protein level in Tmem119CreER/+;C1qaFlox/Flox mice.26
Figure 6. Inter-loxP distance is a determinant of recombination efficiency in microglial CreER lines.
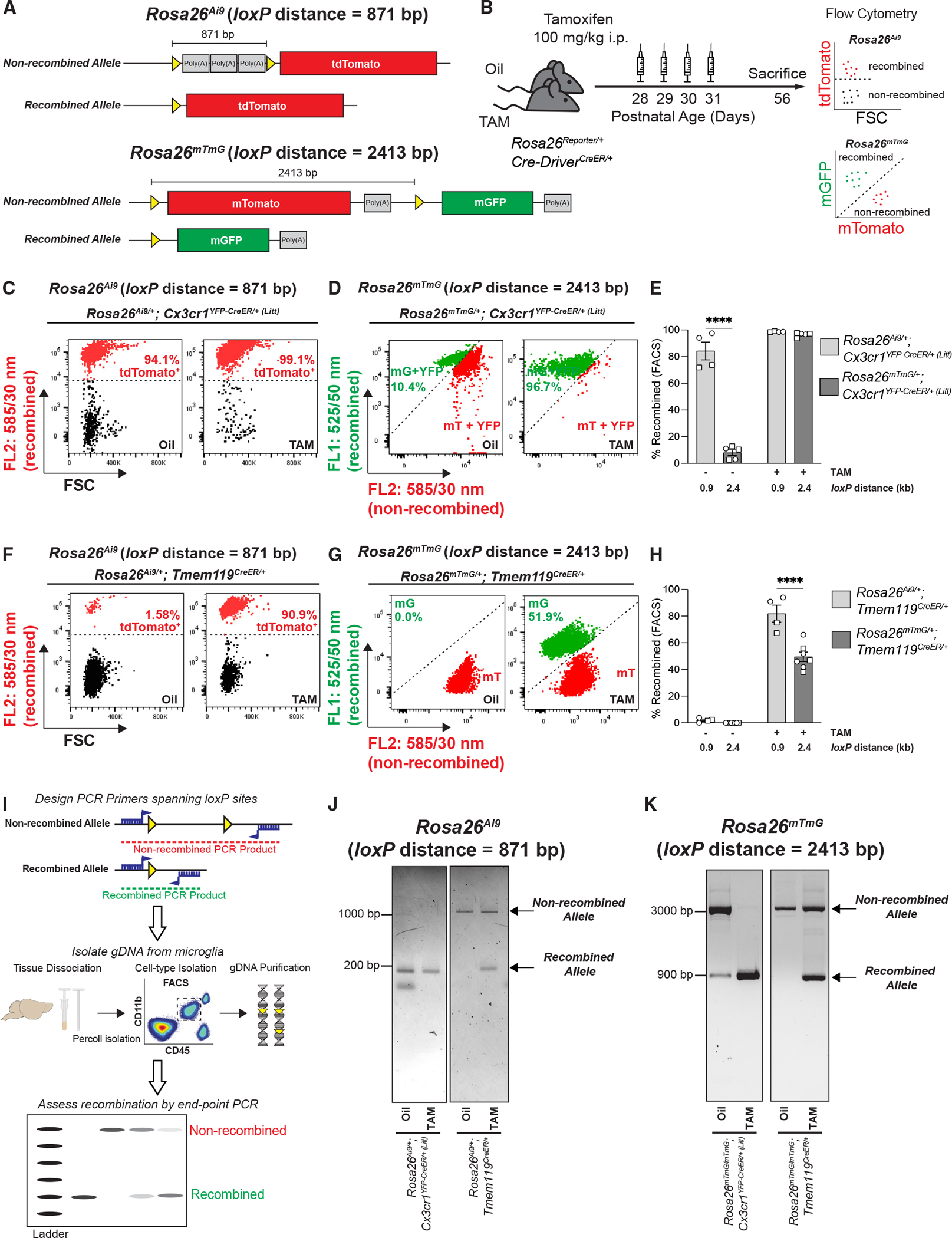
(A) Diagram of the Rosa26Ai9 allele and Rosa26mTmG allele before and after Cre/loxP recombination shows the locations of the loxP sites (yellow triangles) and the inter-loxP distance.
(B) Diagram of experimental protocol used to assess TAM-induced Cre/loxP recombination of Rosa26Ai9 and Rosa26mTmG in microglia by flow cytometry.
(C, D, F, and G) Representative flow cytometry results from individual oil- and TAM-injected Cx3cr1YFP–CreER/+ (Litt) and Tmem119CreER/+ mice expressing Rosa26Ai9/+ are compared with flow cytometry results of Rosa26mTmG/+ from Figure 1. Recombined Rosa26Ai9 microglia are tdTomato+, whereas recombined Rosa26mTmG microglia are mGFP+ (mG) and non-recombined Rosa26mTmG microglia are mTomato+ (mT).
(E and H) Quantifications of recombination of Rosa26Ai9 and Rosa26mTmG in microglia from oil- and TAM-injected mice show significantly more recombination of the 0.9 kb Rosa26Ai9 floxed allele vs. the 2.4 kb Rosa26mTmG floxed allele in (E) Cx3cr1YFP–CreER/+ (Litt) mice (two-way ANOVA with Sidak’s post hoc test; n = 4 Rosa26Ai9 oil, 5 Rosa26mTmG oil, 4 Rosa26Ai9 TAM, and 5 Rosa26mTmG mice; ****p < 0.0001) and (H) Tmem119CreER/+ mice (two-way ANOVA with Sidak’s post hoc test; n = 4 Rosa26Ai9 oil, 8 Rosa26mTmG oil, 4 Rosa26Ai9 TAM, and 7 Rosa26mTmG mice; ****p < 0.0001).
(I) Diagram of protocol to assess Cre/loxP recombination in microglia by endpoint PCR of genomic DNA (gDNA).
(J and K) Gel images of endpoint PCR products from microglial gDNA isolated by florescence-activated cell sorting (FACS) from oil- and TAM-injected Cx3cr1YFP–CreER/+ (Litt) and Tmem119CreER/+ mice expressing Rosa26Ai9/+ or Rosa26mTmG/+.
All data are presented as mean ± SEM. Individual data points indicate males (squares) and females (circles). See also Figure S6.
All experiments in Rosa26mTmG mice described above were performed in mice heterozygous for the floxed allele. When we also analyzed microglia from Cx3cr1YFP–CreER/+ (Litt) and Tmem119CreER/+ mice homozygous for the Rosa26mTmG allele by flow cytometry, we noticed three populations of microglia for each line (Figures S7A–S7C): (1) non-recombined microglia (mTomato+/mTomato+), (2) microglia with a single allele recombined (mGFP+/mTomato+), and (3) microglia with two recombined alleles (mGFP+/mGFP+). These results indicated that recombination of the two Rosa26mTmG alleles in homozygous Rosa26mTmG/mTmG microglia can occur independently, which is consistent with previous reports of independent recombination events in individual cells.27,28 Using the assumption of two alleles per cell, we calculated the theoretical maximum and minimum number of fully recombined cells that would be possible from the recombination of a given number of alleles (Figures S7D and S7E). As a test of these assumptions, we compared the percentage of doubly recombined Rosa26mTmG/mTmG microglia (mGFP+/mGFP+) detected in each sample by flow cytometry vs. the percentage of recombined Rosa26mTmG alleles (mGFP+) inferred from the relative numbers of singly and doubly recombined microglia. We found that the number of doubly recombined cells fit along a curve proportional to the square of the Rosa26mTmG allelic recombination rate (Figure S7E; r2 = 0.999), which can be used to better predict the allelic recombination rate following PCR-based methods.
Together, these data show that inter-loxP distance can influence the recombination efficiency of a given microglial CreER line. The Cx3cr1YFP–CreER (Litt) line is highly efficient at inducing recombination independent of inter-loxP distance but at the expense of spontaneous recombination. Conversely, the Tmem119CreER line is significantly less efficient when crossed to floxed lines with longer inter-loxP distances, but this line has a high likelihood of achieving specific and efficient recombination if inter-loxP distances are less than ~1.2 kb. Finally, endpoint PCR from gDNA can be used to detect spontaneous recombination events, and it can be used to approximate recombination efficiency, particularly if recombination is highly efficient.
A qPCR protocol to quantitatively assess recombination in microglia
Endpoint PCR is not highly quantitative and can offer only an approximation of recombination efficiency. Southern blot is a classic and highly quantitative technique, but it requires large amounts (~5–10 μg) of gDNA.29 We therefore implemented a quantitative measurement of recombination efficiency using qPCR of gDNA (Figure 7A). For a given floxed mouse line, loxP sites are first mapped and then three qPCR primer pairs are designed: one outside the loxP region as a control, a second overlapping a single loxP site to measure non-recombined DNA, and a third spanning the loxP-flanked region to measure recombined DNA. qPCR conditions are then optimized for the primers, microglia are isolated by FACS, and gDNA is purified for qPCR. We used Cx3cr1YFP–CreER (Litt);Rosa26mTmG/+ mice as proof of concept to establish this protocol because we also had a well-established FACS protocol for this line. First, we tested the protocol in cultured microglia. To induce different degrees of recombination, we treated primary mixed glia cultures prepared from Cx3cr1YFP–CreER (Litt);Rosa26mTmG/+ mice with increasing concentrations of 4-OHT or vehicle (ethanol) (Figure S7F). Microglia were then isolated from the mixed glia and re-plated. Endogenous fluorescence within the purified microglia cultures confirmed recombination by an increase in mGFP and loss of mTomato in 4-OHT-treated cultures (Figure S7G). A subset of technical replicates was analyzed by flow cytometry to quantify the percentages of microglia that were mGFP+ (recombined) or mTomato+ (non-recombined) (Figure S7H). Another subset of technical replicates was collected and analyzed by qPCR (Figure S7I). As expected, 4-OHT-treated cultures recombined more efficiently in microglia than vehicle-treated cells, and the recombination efficiency measured by qPCR was highly correlated with the recombination efficiency calculated by flow cytometry (Figure S7J; r2 = 0.954). These data suggest that the qPCR-based method can be used to accurately quantify recombination efficiency.
Figure 7. A quantitative PCR (qPCR) protocol to quantitatively assess recombination in microglia.
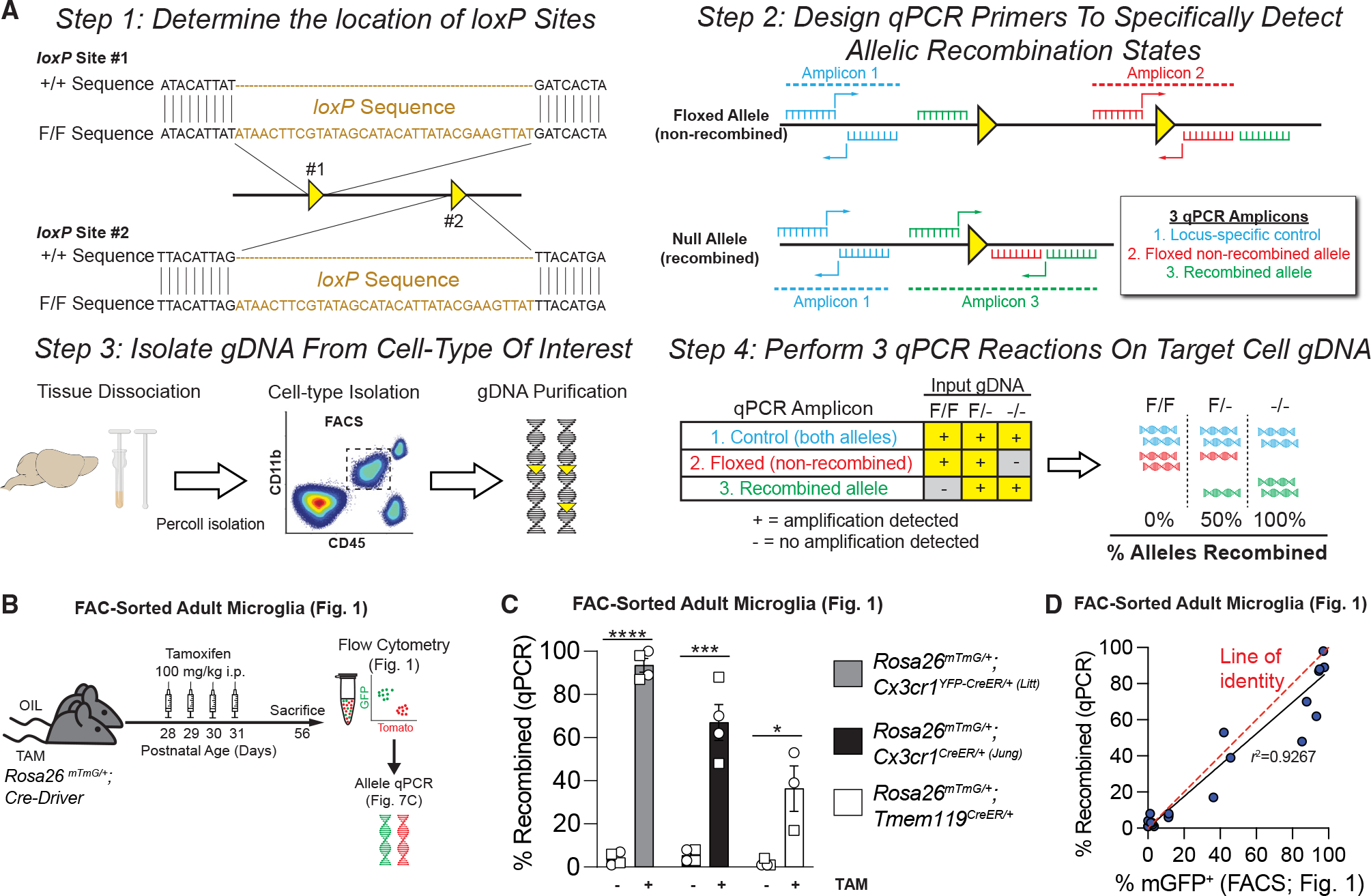
(A) Diagram of protocol used to quantify Cre/loxP recombination of microglial gDNA by qPCR.
(B) Diagram of experiment to assess Cre/loxP recombination in mice injected with TAM or oil.
(C) Quantification of the percentage of recombined gDNA by qPCR shows increased recombination in TAM vs. oil for all three CreER lines (Rosa26mTmG/+;Cx3cr1YFP–CreER/+ (Litt): Student’s t test, n = 4 oil, 4 TAM mice; Cx3cr1CreER/+ (Jung): Student’s t test, n = 4 oil, 4 TAM mice; Tmem119CreER/+: Student’s t test, n = 4,oil, 3 TAM mice; *p < 0.05, ***p < 0.001, ****p < 0.0001).
(D) Graph of percent recombination of Rosa26mTmG in microglia in mice injected with TAM or oil as measured by flow cytometry analysis (see also Figure 1) vs. the recombination rate as measured by qPCR of microglial gDNA isolated by FACS. Data points fit to a linear curve (black line; r2 = 0.9267), closely aligned with the line of identity (red dashed line), indicating that qPCR provides a linear, quantitative measurement of Rosa26mTmG recombination in in vivo samples.
Data in (C) are presented as mean ± SEM. Individual data points in (C) indicate males (squares) and females (circles). See also Figure S7.
After initial validation in vitro, we next tested the qPCR protocol on in vivo microglia acutely isolated by FACS from Rosa26mTmG/+; Cx3cr1YFP–CreER/+ (Litt), Rosa26mTmG/+; Cx3cr1CreER/+ (Jung), or Rosa26mTmG/+; Tmem119CreER/+ mice (Figure 7B). Similar to in vitro experiments, the measurements of Cre/loxP recombination by qPCR from gDNA strongly correlated with recombination efficiency calculated by flow cytometry for higher efficiency (Cx3cr1YFP–CreER/+ (Litt) and Cx3cr1CreER/+ (Jung)) and lower efficiency (Tmem119CreER/+) microglial CreER lines (Figures 7C, 7D, and 1I; r2 = 0.9267). Of note, the experiments described above were conducted in mice heterozygous for the floxed Rosa26mTmG allele. In mice homozygous for a floxed allele, the calculation described in Figures S7D–S7E is needed to estimate the number of singly and doubly recombined cells.
Together, these results provide important proof of concept that using gDNA isolated from microglia in vitro or in vivo, combined with qPCR to detect the floxed allele cassette, is a quantitative method for measuring recombination efficiency. This is particularly useful when antibodies are not available to test recombination by FACS or immunofluorescence microscopy. Predictions of cells with doubly or singly recombined alleles can be further estimated (Figures S7D and S7E). Still, assessment at the mRNA level and, even better, at the protein level by flow cytometry or immunofluorescence remain the best ways to validate effective and cell-type-specific gene ablation.
DISCUSSION
We have performed an in-depth comparison of the most widely available CreER lines that target microglia (Table S1). The Cx3cr1CreER lines more efficiently recombine compared with Tmem119CreER, HexbCreER, and P2ry12CreER lines, but the Cx3cr1CreER lines display significant spontaneous recombination events, and the recombination is less specific for microglia. The Cx3cr1CreER lines also express 50% less Cx3cr1, and the Cx3cr1YFP–CreER/+ (Litt) line displays abnormal microglial phenotypes if TAM is administered to neonates. For lower efficiency microglial CreER lines, recombination is equivalent to Cx3cr1CreER lines when crossed to floxed mice with inter-loxP distances <1.2 kb and also can be boosted by using mice homozygous for CreER. To further aid in assessing recombination of different floxed alleles that are not easily measured by flow cytometry or immunofluorescence microscopy, we describe a protocol to measure recombination of gDNA purified from microglia by endpoint PCR and more quantitatively by qPCR.
Selecting CreER lines for assessment of microglia
Care should be taken to select the appropriate CreER line for each study. Cx3cr1CreER lines appear to have the strongest Cre activity compared with Tmem119CreER, HexbCreER, and P2ry12CreER lines. These Cx3cr1CreER lines also allow for the most efficient and homogeneous targeting of all microglia independent of inter-loxP distance. In contrast, the Tmem119CreER, HexbCreER, and P2ry12CreER lines offer distinct advantages. First, although the Tmem119CreER line does target a small subset of brain-border fibroblasts, the Tmem119CreER, HexbCreER, and P2ry12CreER lines do not target border-associated macrophages in the CNS.15,20,21,30 Another group had similar results and also showed that the Cx3cr1CreER and P2ry12CreER lines recombined to a greater degree in splenic macrophages compared with the Tmem119CreER line.25 Second, there is significantly less spontaneous recombination in the Tmem119CreER, HexbCreER, and P2ry12CreER lines compared with Cx3cr1CreER lines. Third, the Cx3cr1YFP–CreER/+ (Litt) line displays changes in microglial homeostasis if TAM is administered in neonates. Thus, for developmental experiments, other microglial CreER lines or a non-inducible microglial split-Cre line should be considered.23
The endogenous protein levels in the Tmem119CreER and HexbCreER lines are an additional consideration; that is, although the Tmem119CreER and HexbCreER lines were engineered to retain native gene expression, we still found that there were reduced levels of TMEM119 protein and HEXB enzymatic activity in the Tmem119CreER and HexbCreER lines, respectively. In contrast, despite being made in a similar fashion, P2RY12 levels were retained in the P2ry12CreER mice.22 All of the exons for Tmem119, Hexb, and P2ry12 are maintained in the respective CreER lines, so the reductions in protein level and enzymatic activity are unexpected and the mechanism remains unclear. Insertion of the CreER sequence before the stop codon of these genes could potentially affect epigenetic regulation of mRNA expression, the stability and degradation of the mRNA, or its translation among other possible mechanisms. There could also be incomplete cleavage of the T2A/P2A protein sequences, which could affect functional protein levels. It is unclear to what degree the reductions in TMEM119 and HEXB truly alter microglial phenotypes. For example, mice lacking HEXB enzymatic activity suffer neurodegeneration,31 which is not apparent in heterozygous or homozygous HexbCreER mice.13 Thus, the reduced amount of HEXB enzymatic activity that occurs in the HexbCreER line appears to still be sufficient to preserve function.
Also, the Tmem119CreER, HexbCreER, and P2ry12CreER lines are less efficient in recombining, particularly when crossed to floxed mice with longer inter-loxP distances. For these CreER lines, breeding to homozygosity for CreER can boost recombination efficiency. In addition, the inter-loxP distance of the floxed line can be used to help predict an anticipated recombination efficiency while designing experiments. For at least the Tmem119CreER line, we found that three different floxed alleles with shorter inter-loxP distances (<1.2 kb) have more efficient recombination vs. a longer >2-kb floxed allele. However, it should be noted that other factors can also influence recombination efficiency, such as chromatin accessibility. Still, we recommend determining the inter-loxP distance of the floxed allele (if not publicly listed) when designing a study prior to selecting the CreER line.
Selecting appropriate controls for Cre/loxP experiments
When performing Cre/loxP experiments, the following should be considered: (1) the impact of TAM exposure (dose and timing of administration), (2) spontaneous Cre/loxP recombination, (3) theendogenous expression of the gene driving CreER, and (4) off-target effects of CreER activation. Oil-injected Cre-DriverCreER/+; Floxed AlleleFlox/Flox mice are recommended to assess spontaneous recombination. If spontaneous recombination is low, one should ideally use TAM-injected Cre-DriverCreER/+;Floxed Allele+/+ mice as controls for TAM-injected Cre-DriverCreER/+; Floxed AlleleFlox/Flox mice. We note that TAM had minimal impact on microglial gene expression 4 weeks after exposure in adolescent mice; however, it is best practice to control for organism-wide effects of TAM.32,33 If spontaneous recombination is high, oil-injected Cre-DriverCreER/+;Floxed AlleleFlox/Flox mice should be included to control for the effects of spontaneous recombination on important phenotypes. However, oil-injected Cre-DriverCreER/+;Floxed AlleleFlox/Flox mice controls do not control for off-target effects of TAM or off-target effects of CreER activation, so TAM-injected Cre-DriverCreER/+;Floxed Allele+/+ mice are also recommended as controls. A high rate of spontaneous recombination is also concerning if parents express both the Cre-Driver and the floxed allele. For example, ablation of a gene in microglia in the parents could affect parental behavior and/or pregnancy. Oil-injected Cre-DriverCreER/+; Floxed AlleleFlox/Flox and/or TAM-injected Cre-DriverCreER/+; Floxed Allele+/+ littermate controls could be used to control for any effects of spontaneous CreER activation in the parents. Also, if endogenous expression of genes driving the CreER are reduced, as we observed in the CreER lines in this study, a comparison with mice negative for the CreER and expressing Floxed AlleleFlox/Flox should be considered.
Protocols for assessing recombination in microglia
Lastly, we provide protocols to assess recombination efficiency in microglia by flow cytometry, immunofluorescence microscopy, endpoint PCR of gDNA, and qPCR of gDNA. Because Cre/loxP recombination is dependent on a variety of factors (e.g., inter-loxP distance or chromatin accessibility of a floxed allele), using a floxed reporter line as a proxy for recombination can be highly misleading. Methods that measure bulk levels of RNA or protein in microglia (e.g., RNA qPCR or western blot) are useful, but they are most robust for molecules expressed at higher levels, they rely on high-quality RNA probes or antibodies, and they are not sufficient to detect individual recombination events. For these reasons, recombination is most reliably assessed at the protein level in microglia by flow cytometry or immunofluorescence microscopy in tissue. These approaches are preferred because each cell can be individually classified as recombined or non-recombined at the protein level. Single-molecule fluorescence in situ hybridization (smFISH) is also an option, although levels of RNA and protein are not always well correlated. Still, both immunofluorescence microscopy and smFISH allow for the assessment of recombination in all cell types in the tissue of interest. The drawback of these methods is that they rely on the availability of high-quality, often-cost-prohibitive RNA probes or antibodies. For these reasons, we also include protocols for endpoint PCR of microglial gDNA for assessment of spontaneous recombination events and qPCR from microglialgDNA as a more quantitative measure. Still, it should be noted that if a floxed gene is necessary for cell survival, these assessments of recombination will not include dead or dying homozygous cells, and the analysis will be strongly biased toward heterozygous cells.
In summary, as interest in microglia continues to grow, common guidelines for the effective use of widely used genetic tools such as Cre/loxP are critical to achieve reliable, reproducible results. Cre/loxP technology is of particular importance for molecular studies designed to elucidate the microglia-specific functions of individual genes associated with increased risk of brain diseases such as Alzheimer’s disease and schizophrenia. This study provides the pros and cons of different CreER lines, offers guidance on how to properly select the appropriate controls, and recommends best practice protocols for accurately measuring Cre/loxP recombination in microglia. Although the specific CreER lines and isolation methods will differ for other cell types, the guidelines and protocols outlined in this study are also informative for the general use of Cre/loxP technology across all other cell types.
Limitations of the study
To efficiently compare between different CreER lines, this study primarily focused on recombination of the Rosa26mTmG allele in 8-week-old C57Bl/6J mice injected for 4 days with 100 mg/kg TAM i.p. at 4 weeks of age. The absolute level of recombination will depend on the specific floxed allele targeted, the age of the mice, and the method of TAM administration. Still, the relative levels of recombination between different CreER lines should remain largely consistent. Indeed, our results are consistent with independent observations by another group that targeted the Rosa26YFP and Rosa26Ai9 alleles by a different TAM paradigm, administering 180 mg/kg TAM by oral gavage for 5 days at 8–12 weeks of age.25 However, it is important to note that manipulations that affect the expression levels of Cx3cr1, Tmem119, Hexb, or P2ry12 could affect the relative strength and specificity of different CreER lines. For example, neonatal mice, aged mice, and models of inflammation and disease that exhibit reactive microglial phenotypes are likely to show the greatest differences from healthy 4-week-old wild-type mice. The TAM we used is near the maximum tolerated dose, but higher dosages could induce off-target effects beyond what we observed in this study. Lastly, although we identified reduced expression of TMEM119 and HEXB in their respective CreER lines, the exact mechanism by which they are reduced remains unclear.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Dorothy Schafer (dorothy.schafer@umassmed.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cx3cr1YFP–CreER(Litt) (Stock# 021160, B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ), Cx3cr1CreER (Jung) (Stock# 020940, B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Jung/J), Tmem119CreER (Stock# 031820, C57BL/6-Tmem119em1(cre/ERT2)Gfng/J), P2ry12CreER (Stock# 034727, B6(129S6)-P2ry12em1(icre/ERT2)Tda/J), Rosa26mTmG (Stock# 007676, B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J), Cx3cr1EGFP (Stock# 005582, B6.129P2(Cg)-Cx3cr1tm1Litt/J), Rosa26Ai9 (Stock# 007909, B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J), C1qaFlox (Stock# 031261, B6(SJL)-C1qatm1c(EUCOMM)Wtsi/TennJ), and Becn1Flox (Stock# 028794, STOCK Becn1tm1.1Yue/J) mice were obtained from Jackson Laboratories (Bar Harbor, ME). HexbCreER (Hexbem2(cre/ERT2)Mp) mice were directly obtained from Dr. Marco Prinz. All mice were maintained on a C57Bl/6J background. Animals were group housed after weaning and maintained on a 12 h light/dark cycle with food and water provided ad libitum. All animals were healthy and were not immune compromised. Littermates of the same sex were randomly assigned to experimental groups. Equal numbers of males and females were used for all experiments except for RNA Sequencing for which only males were used. Circles (females) and squares (males) are used to designate to the sex of the individual data points throughout the figures. The hexosaminidase enzymatic activity was performed in 4 week old mice. Unless otherwise noted, all other experiments were performed in 8 week old mice. All animal experiments were performed in accordance with Animal Care and Use Committees (IACUC) and under NIH guidelines for proper animal use and welfare.
METHOD DETAILS
Tamoxifen injection
To induce CreER activity in adults, animals were injected once daily i.p. with 100 mg/kg tamoxifen (Sigma T5648) dissolved 20 mg/mL in corn oil (Sigma C8267) on postnatal days 28, 29, 30, and 31. Neonatal animals were injected once daily with 1 mg/mL tamoxifen on days P1-P4 (50 μg) and P14 (100 μg).
Microglia isolation from adult mouse brain
Microglia were isolated following a published protocol35 with some modifications. All tubes used to spin cells during this protocol were precoated for at least 1 h with 1% bovine serum albumin (BSA; Sigma A2153) diluted in ultrapure water to improve cell yields and viability. Centrifuges and tools were all prechilled to 4°C or on ice. When collecting samples for RNA, standard RNAse free conditions were followed. Mice for gDNA sample collection were anesthetized and transcardially perfused using ice-cold, Ca2+-free, Mg2+-free Hank’s balanced salt solution (HBSS; GIBCO 14175095) and the brains were quickly dissected and placed in HBSS on ice. Mice used for RNA sample collection were sacrificed with CO2 and the brains were removed and placed in HBSS on ice. For mice where one hemisphere was used for immunohistochemistry, brains were hemisected along the sagittal midline, the right half was transferred to a 4% paraformaldehyde (PFA; Electron Microscopy Sciences 15711) phosphate buffered saline (PBS) solution for immunohistochemistry and the left half placed in HBSS for microglial isolation. For microglial isolation, the cerebellum and brainstem were removed and the remaining brain minced using a razor, taking care not to carve plastic fragments from the dish. Samples were then dounce homogenized (2mL homogenizer; DWK Life Sciences (Kimble) 885300–0002) in 1.5 mL ice-cold HBSS 30 times each with the loose (A) and tight (B) pestles while simultaneously rotating the pestle. The cell suspension was then transferred through a pre-wet (with HBSS) 70 μm cell strainer (Miltenyi 130–098-462) into prechilled, BSA-coated 50 mL tubes. Cell suspensions were then spun down at 300 × g for 10 min in a centrifuge set to 4°C. For microglial enrichment and myelin removal, a 40% Percoll solution was prepared from Percoll (Sigma GE17–0891-01), 10X HBSS, and water for a final 1X HBSS concentration then pH adjusted to 7.3 with dilute HCl. Cell pellets were resuspended in 2 mL 40% Percoll then transferred to a pre-chilled, BSA-coated 15 mL tube. The 50 mL tube was rinsed with up to an additional 7 mL 40% Percoll and transferred to the 15 mL tube. The final volume for each sample was adjusted to 10 mL total by adding Percoll to the 10 mL line. The samples were then spun for 30 min at 500 × g with full acceleration and braking. The myelin at the top of the solution was removed using vacuum suction, and then the rest of the Percoll solution was removed. The cell pellet was resuspended in 1–2 mL ice-cold HBSS and transferred to a new pre-chilled, BSA-coated 15 mL tube. An additional 6 mL of HBSS was used to rinse the Percoll spin tube to ensure complete transfer of cells to the new tube. The final volume was adjusted to 10 mL with HBSS and spun again for 10 min at 300 × g at 4°C. All samples were then resuspended in 300 μL of ice-cold FACS buffer (0.5% BSA, 1 mM EDTA, in 1x PBS, Sterile Filtered) then transferred to a pre-chilled, BSA-coated 1.5 mL tube. Several additional rinses with 300 μL per transfer were done to ensure complete transfer of cells from the 15 mL tube to the 1.5 mL tube. The samples were again spun for 10 min at 300 × g at 4°C and resuspended in ice-cold FACS buffer containing APC anti-CD11b (eBioscience clone M1/70) and PerCP-Cy5.5 anti-CD45 (eBioscience clone 30-F11) antibodies at a 1:50 dilution for 15 min on ice. Samples were then washed in 1 mL of ice-cold FACS buffer and spun for 10 min at 300 × g at 4°C and then resuspended in 300–500 μL of ice-cold FACS buffer supplemented with DAPI (Miltenyi 130–111-570, 1:100 dilution). 10,000–50,000 microglia (DAPI− CD11b+ CD45Mid) were sorted per sample on a SONY MA800 or SONY MA900 into 1.5 mL tubes containing either 200 μL FACS buffer for gDNA samples or 300 μL TRIzol LS reagent (Invitrogen 10296028) for RNA samples. For samples from Rosa26mTmG mice and Rosa26Ai9 mice, GFP (488 nm laser; FL1 525/50 nm) and TdTomato (561 nm laser; FL2 585/30 nm) fluorescent signal was also collected for further analysis using FlowJo v10.80 Software (BD Life Sciences). The sample and collection tubes were maintained at 4°C and each sample was kept on ice before and after the sort. Each sample took approximately 10–20 min to sort. After the sort, gDNA samples were spun at 10,000 x g for 10 min and the supernatant was discarded. Cell-pellet-containing tubes were kept at −20°C for short term storage until all samples were collected for batch processing (see genomic DNA isolation). The RNA samples (TRIzol tubes) were stored at −80°C until all samples were collected for batch processing (see total RNA isolation).
Immunostaining & confocal microscopy
Brains from transcardially perfused mice were fixed in 4% PFA (electron microscopy sciences) 1X PBS solution. Brains were kept at 4°C overnight then rinsed with PBS before being transferred to 30% sucrose 1X PBS solution for dehydration. 40 μm thick sagittal sections were cut on a vibratome (Leica) in a bath containing PBS. Sections were transferred to individual wells of 24-well plates for staining. When necessary, photobleaching of endogenous fluorophores was performed by incubating sections overnight in Liberate Antibody Binding Solution (Polyscience, Inc. 24310), followed by 3 washes in 0.1M phosphate buffer (PB) for 5 min each. The sections were then blocked for 1 h using PBTGS (0.1M phosphate buffer, 5% normal goat serum (NGS; Sigma G9023), 0.3% Triton X-100 (Sigma X100)). Sections were incubated overnight at room temperature (RT) with primary antibodies diluted in PBTGS. Primary antibodies used: chicken anti-GFP (1:1000, Abcam ab13970), rabbit anti-P2RY12 (1:1000, anaspec AS-55043A), rabbit anti-IBA1 (1:500, Wako 019–19741), rabbit anti-LYVE1 (1:200, Abcam ab14917), rat anti-PECAM1 (1:100, BioLegend 102401), rabbit anti-ALDH1A2 (1:200, Millipore Sigma HPA010022), rabbit anti-S100A6 (1:1000, Novus NBP1–89388), guinea pig anti-TMEM119 (1:500, Synaptic Systems 400 004). Sections were washed 3x in 0.1M PB for 5 min each. Sections were incubated for 2–3 h at RT with secondary antibodies diluted in PBTGS. Secondary antibodies used: goat anti-chicken-AlexaFluor488 (1:1000, Life Technologies), goat anti-guinea pig-AlexaFluor488 (1:1000, Life Technologies), goat anti-rabbit-AlexaFluor594 (1:1000, Life Technologies), goat anti-rat-AlexaFluor594 (1:1000, Life Technologies), and goat anti-rabbit-AlexaFluor647 (1:1000, Life Technologies). Sections were washed 3x in 0.1M PB for 5 min each. Next, sections were mounted onto a glass slide then allowed to dry. Fluoroshield mounting media containing DAPI (Sigma F6057) was applied to the section and a #1.5 coverslip was added. The slide was sealed with nail polish along the edge.
Confocal images were acquired at 10x, 20x, or 40x on a Zeiss Observer microscope (Zeiss; Oberkochen, Germany) equipped with 405 nm, 488 nm, 555 nm, and 639 nm lasers and Zen black acquisition software (Zeiss; Oberkochen, Germany). For quantification of mGFP recombination in IBA1+ cells at 1 month vs. 6 months of age, 20x confocal images were acquired of 3 sagittal sections per animal. To assess mGFP+ recombination in IBA1+ cells, at least 100 IBA1+ cells within the cortex were quantified in each section using FIJI software.34 To assess anti-TMEM119 mean fluorescence intensity, 40x confocal images were acquired within the cortex of 3 sections per animal using identical acquisition settings. To quantify anti-TMEM19 mean fluorescence intensity, images were back-ground subtracted and thresholded to the anti-TMEM119+ area, and then the mean gray value was measured using FIJI software.
For quantification of mGFP recombination in anti-P2RY12+ microglia, parenchymal anti-P2RY12− cells, anti-LYVE1+ border-associated macrophages, or anti-LYVE1− meningeal cells, 20x widefield images of 3 immmunostained sagittal sections per animal were acquired on a TissueFAXS SL slide loader (Tissue Gnostics; Vienna, Austria). To assess mGFP+ recombination in anti-P2RY12+ cells, at least 100 anti-P2RY12+ cells were quantified per region in each section using FIJI software. To assess mGFP+ recombination in parenchymal anti-P2RY12− cells, the entire brain region within each section was quantified. To assess mGFP+ recombination in anti-LYVE1+ cells, at least 100 anti-LYVE1+ cells were quantified per section. To assess mGFP+ recombination in anti-LYVE1− meningeal cells, mGFP+ anti-LYVE1− cells were counted along the border of the section where anti-LYVE1+ cells could be detected, for a length of at least 2.5 mm per section. All data represents the average of 3 sections per animal.
Hexosaminidase assay
For the hexosaminidase assay, an established protocol was followed.36 In brief, FACS sorted microglia were lysed with KPBS (136 mM KCl, 10 mM KH2PO4, pH 7.25) containing 1% Triton X-100 for 10 min on ice. In a total volume of 40 μL per reaction, the cell lysates were incubated at 37°C in a 10 mM sodium citrate buffer (pH 4.2) containing 2 mM 4-methylumbelliferyl-2-acetamido-2-deoxy-β-D-glucopyranoside (MUG). The reaction was stopped by adding 5 volumes of a 0.2 M glycine/0.2 M Na2CO3 solution. The amount of liberated 4-methylumbelliferone was determined fluorometrically at an emission wavelength of 440 nm after excitation at 365 nm.
Total RNA isolation
Total RNA was purified from FACS sorted microglia. Samples were kept on ice unless otherwise stated. Samples were retrieved from the −80°C freezer and allowed to thaw on ice, then transferred a 2 mL Phase Lock Gel tube (VWR 10847–802). We collected ~50,000 microglia, which had a sort volume of ~140 μL, into 300 μL TRIzol LS. To reach the ideal TRIzol:sample ratio of 3:1, we adjusted the volume of TRIzol to 420 μL by adding 120 μL TRIzol LS to the Phase Lock Gel tube. The samples were then incubated at RT for 5 min. Next, 110 μL of 49:1 chloroform:isoamyl alcohol (Sigma 25668) was added to each tube and then the tube was shaken vigorously. Samples were incubated again at RT for 5 min with repeated shaking. Next, samples were spun at 13,000 × g for 10 min at 4°C. Next, an additional 110 μL of chloroform: isoamyl alcohol was added to the tube and then the tube was shaken vigorously. The samples were then spun again at 13,000 × g for 10 min at 4°C. After the spin, 300 μL of the aqueous volume from above the gel was transferred to a clean 1.5 mL tube. Next, 5 μL glycoblue (Invitrogen AM9516), and 30 μL 3 M Sodium Acetate pH 5.5 (Ambion AM9740) were added to the tube and mixed with gently tapping to ensure fully mixed. Next, 300 μL isopropanol was added and the tubes were mixed by inverting 10 times. The samples were stored at −80°C for at least one night. The next day, samples were retrieved from the −80°C freezer and spun for 30 min at 13,000 and 4°C. The supernatant was discarded and 1 mL of ice-cold 75% ethanol was added to wash the pellet. With careful pipetting, the pellet was dislodged from the bottom of the tube. Samples were spun again at 13,000 × g at 4°C for 10 min then rinsed with ice-cold 75% ethanol for a second time. Next the samples were spun for the final time at 13,000 × g at 4°C for 10 min. After the spin, the supernatant was discarded and the tubes were placed at RT with the caps open to air-dry for 10–15 min. Dried pellets were resuspended in 20 μL nuclease-free ultrapure water. Samples were then stored at −80°C until shipped for quality control and further processing for RNA-seq. Samples were prepared in batches containing all tamoxifen and oil injected samples from a particular CreER line.
RNA sequencing
RNA integrity was assessed on an Agilent 2100 Bioanalyzer and samples with RIN <7 were excluded. Libraries were prepared from the remaining samples using SMART-Seq v4 + Nextera XT and then sequenced on NovaSeq 6000 (Illumina) with 2 × 100 bp paired end reads. Quality control (QC) was performed on base qualities and nucleotide composition of sequences, to identify problems in library preparation or sequencing. Short reads were aligned using STAR (v2.4.0) to the mouse genome (mm10). Average input read counts were 87.3M per sample and average percentage of uniquely aligned reads was 87.5%. Read counts for mouse refSeq genes were generated by HT-seq (v0.6.1).37 Low count transcripts were filtered, and count data were normalized using the method of trimmed mean of M-values (TMM). Differentially expressed genes (FDR < 0.05) were then identified using the Bioconductor package EdgeR.38
Genomic DNA isolation
To purify genomic DNA (gDNA) from microglia, cells were lysed with 50 μL Bradley Lysis Buffer (10 mM Tris-HCl, 10 mM EDTA, 0.5% SDS, 10 mM NaCl). Samples were incubated at 55°C for 3 h then returned to RT. Next 2 μL 5M NaCl was added to each tube and mixed with finger flicking. Next 1 μL glycogen (ThermoFisher Scientific R0561) was added to each tube and mixed. Finally, 125 μL 100% ethanol was added to each tube and mixed by inverting the tubes 5–10 times. The samples were then incubated at −20°C for at least 1 h to allow for gDNA precipitation. To pellet the gDNA precipitant, samples were spun at 20,000 × g, 4°C for 30 min. The supernatant was removed, and the pellet was washed with ice-cold 70% ethanol, vortexed on medium speed, then spun again at 15,000 × g for 10 min. This washing step was repeated two additional times for a total of three washes. After the final spin, the supernatant was removed, and the pellet briefly dried using a speed-vac. The pellet was then resuspended in 30 μL ultrapure water, pre-warned to 50°C–55°C. Samples were then incubated at 55°C for at least 30 min to facilitate resuspension. The concentration of DNA was determined using a Nanodrop (Thermo Scientific). Samples were stored at −20°C for short term storage and −80°C for long-term storage.
loxP site mapping
Benchling was used for all primer design (https://www.benchling.com/). Putative loxP site locations were obtained by referencing the original published paper or searching the MGI database (http://www.informatics.jax.org/). The Benchling primer wizard was then used to design three primer pairs for each loxP site with a high probability of flanking the loxP site: typically one primer in the exon and one primer in the intron containing the loxP site. DNA primers were obtained from integrated DNA technology, stored at −20 °C at 100 μM, and used at 20 μM. We tested the primer pairs by running a PCR using wild type (+/+) and homozygous floxed (F/F) gDNA with Apex 2X Taq RED Master Mix with 1.5 mM MgCl2. The PCR products were run out on a 1.5% agarose gel at 100 V (constant voltage) for 30–45 min. A picture of the gel was taken and the best primer pair per loxP site was identified. Next, we repeated the PCR with successful primer pairs in replicate (3–4) to then pool replicates. Next, we ran pooled reactions (20 μL) on a gel using low melting temp agarose. Next, we took pictures of the gel then cut out the +/+ and F/F bands from the gel with a razor under UV light using proper eye protection. Next, we used the Qiagen gel extraction kit (Qiagen) to purify the amplicons following the manufacturers protocol. DNA was eluted from the Qiagen spin column with 30 μL Buffer EB (10 mM Tris-Cl, pH 8.5). A Nanodrop (Thermo Scientific) was used to determine the DNA concentration. Samples were submitted to GeneWiz for Sanger sequencing. Each amplicon was sequenced in both directions using the forward and reverse primers separately. The sequence results were entered into Benchling and using the alignment function, the +/+ and F/F sequences were aligned to locate the loxP sequence (ATAACTTCGTATAGCATACATTATACGAAGTTAT). The sequences directly up- and downstream of the loxP sequence was used to map the coordinates of the loxP in the genome. The distance between the loxP sites in base pairs (bp) was determined in Benchling.
Endpoint PCR
Primer pairs flanking both loxP sites were generated using Benchling. Primer pairs were screened by PCR on non-recombined and recombined DNA. For longer PCR products, Hot Start VeriFi Taq polymerase (PCR Biosystems) was used. Gel electrophoresis was performed on 1.5% agarose gels at 100 V to distinguish PCR products of different sizes. Once the best primer pair was selected (listed in Table S2), PCR was performed on gDNA from FACS microglia samples to estimate the percentage of DNA recombination. Gel images were collected using a Bio-Rad ChemiDoc.
Primary microglia cultures
Mixed glia cultures were generated from P1 Rosa26mTmG/+; Cx3cr1YFP–CreER/+ (Litt) mice and grown in advanced DMEM/F12 culture medium supplemented with fetal bovine serum (10%, GIBCO 10437028), pen/strep (GIBCO 15140122), and glutaMax (GIBCO 35050061) at 37°C 5% CO2 for two weeks. Mixed glia cultures were maintained for two weeks with 50% media changes after 5–7 days. CreER activity was induced by addition of 4-hydroxy-tamoxifen (4-OHT, Sigma H7904) dissolved in ethanol to the mixed glia cultures on DIV 13. Three final concentrations were tested: 10nM, 100nM, and 1000nM. Loosely adhered microglia were shaken off at 300 rpm for 3 h at 37°C, 5% CO2 and plated in 24-well plates at a density of 60,000 cells per well 72 h after 4-OHT application. The next day, the media was completely removed and changed to serum-free microglia growth media. Microglia growth media consists of neurobasal media (GIBCO 21103049), insulin (Sigma-Aldrich I6634), sodium pyruvate (GIBCO 11360070), pen/strep, thyroxine (Sigma-Aldrich T6397), glutamax, B27 (GIBCO 17504044), n-acetyl cysteine (Sigma-Aldrich A8199), m-CSF (Shenandoah 200–08), transferrin (Sigma-Aldrich T1147), bovine serum albumin (Sigma-Aldrich A2153), putrescine (Sigma-Aldrich P5780), progesterone (Sigma-Aldrich P8783), and sodium selenite (Sigma-Aldrich S5261). After three days in serum-free conditions, the entire media was changed. Fluorescent images of the primary microglia were acquired 24 h after changing to microglia growth media using an Observer Spinning Disk Confocal microscope equipped with a 40× objective, diode lasers (405 nm, 488 nm, 594 nm, 647 nm) and Zen Blue acquisition software (Zeiss; Oberkochen, Germany). gDNA was also collected 72 h after tamoxifen induction.
Quantitative PCR
Quantitative PCR (qPCR) was performed using primers designed in Benchling (https://www.benchling.com/) to generate specific PCR products for 1) a control region, 2) non-recombined gDNA and 3) recombined gDNA. For each primer pair, a 2-fold dilution series was generated with gDNA and only primer pairs with linear amplification were used for qPCR analyses. Primer stocks were prepared at 100 μM then further diluted to 20 μM working concentration. The final primer concentration in the PCR reaction is 400 nM. Each gDNA sample was tested for the three different qPCR primer pairs (control region, non-recombined allele, recombined allele). Power SYBR master mix (Life technologies 43–676-59) was used and the plates were run using a CFX96 real-time PCR instrument (BioRad). A standard curve for each primer pair was run on recombined or non-recombined gDNA and the equation of the best-fit line was used to extrapolate the abundance of the allele in each sample from the cycle number. Primers used are listed in Table S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 9.5.1. The exact number of samples (n), what n represents, the statistical tests used, and p values for each experiment are noted in the figure legends. For comparisons between two groups, we used Student’s t-test. For comparisons between more than two groups, we used 1-way ANOVA followed by Tukey’s post-hoc test between groups. For comparisons of the distribution of categorical variables between groups, we used a Chi-square test. For 2-way comparisons, we used two-way ANOVA followed by Tukey’s post-hoc test when comparing at least three groups or Sidak’s post-hoc test when comparing two groups. For 2-way repeated measured comparisons, we used 2-way repeated measured ANOVA followed by Tukey’s post-hoc test.
Supplementary Material
Highlights.
Cx3cr1CreER lines efficiently recombine but have more spontaneous recombination
Tmem119CreER, P2ry12CreER, and HexbCreER are less efficient but are microglia specific
Inter-loxP distance is a key determinant of recombination efficiency
Best practices are provided for measuring recombination in microglia
ACKNOWLEDGMENTS
This work was supported by grants from the NIMH (R01MH113743 to D.P.S.; R01 MH118329 to A.S.; F31MH124400 to H.S.), NINDS (R01NS117533 to D.P.S.; R01NS106721 to A.S.), NIA (RF1AG068281 to D.P.S.; RF1AG068558 to A.S.; R01AG072489 to A.S.), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (D.P.S. and R.K.), and Autism Speaks Weatherstone Predoctoral Fellowship 11779 (to P.A.F.). T.M. was supported by AMED (JP20gm6310016 and JP21wm0425001) and JSPS (KAKENHI JP21H02752, JP21H00204, and JP22H05062). M.P. and K.-P.K. were supported by the DFG (TRR167 Project ID: 259373024) and the DFG under Germany’s Excellence Strategy (CIBSS–EXC-2189 Project ID390939984). M.P. was further supported by the Sobek Foundation, the Ernst-Jung Foundation, the Klaus Faber Foundation, the Novo Nordisk Prize, and the German Research Foundation (SFB 992 Project ID 192904750, SFB 1160, SFB 1479 Project ID 441891347, TRR 359 Project ID 491676693, and Gottfried Wilhelm Leibniz Prize). Schematics were created with BioRender.com. Wide-field microscopy images were acquired with help from the SCOPE core at UMass Chan Medical School. FACS experiments were performed with the help of Susanne Pechhold and the UMass Chan Medical School Flow Cytometry Core Facility. We also thank Elisa Vazquez for helpful insights interpreting off-target recombination in the meninges of Tmem119CreER mice and Agnes Luo for useful discussions regarding results with the different CreER lines across our labs.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the genomic datasets. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. We avoided “helicopter science” practices by including the participating local contributors from the region where we conducted the research as authors on the paper.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113031.
REFERENCES
- 1.Wu Y, Dissing-Olesen L, MacVicar BA, and Stevens B (2015). Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 36, 605–613. 10.1016/j.it.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prinz M, Jung S, and Priller J (2019). Microglia Biology: One Century of Evolving Concepts. Cell 179, 292–311. 10.1016/j.cell.2019.08.053. [DOI] [PubMed] [Google Scholar]
- 3.Salter MW, and Stevens B (2017). Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027. 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- 4.Biber K, Möller T, Boddeke E, and Prinz M (2016). Central nervous system myeloid cells as drug targets: current status and translational challenges. Nat. Rev. Drug Discov. 15, 110–124. 10.1038/nrd.2015.14. [DOI] [PubMed] [Google Scholar]
- 5.Hansen DV, Hanson JE, and Sheng M (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. 10.1083/jcb.201709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin R, Zhou Y, Yan T, Wang R, Li H, Wu Z, Zhang X, Zhou X, Zhao F, Zhang L, et al. (2022). Directed evolution of adeno-associated virus for efficient gene delivery to microglia. Nat. Methods 19, 976–985. 10.1038/s41592-022-01547-7. [DOI] [PubMed] [Google Scholar]
- 7.Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, and Chambon P (1996). Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. USA 93, 10887–10890. 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glaser S, Anastassiadis K, and Stewart AF (2005). Current issues in mouse genome engineering. Nat. Genet. 37, 1187–1193. 10.1038/ng1668. [DOI] [PubMed] [Google Scholar]
- 9.McLellan MA, Rosenthal NA, and Pinto AR (2017). Cre-loxP-Mediated Recombination: General Principles and Experimental Considerations. Curr. Protoc. Mouse Biol. 7, 1–12. 10.1002/cpmo.22. [DOI] [PubMed] [Google Scholar]
- 10.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, and Gan WB (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldmann T, Wieghofer P, Muüller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, et al. (2013). A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 16, 1618–1626. 10.1038/nn.3531. [DOI] [PubMed] [Google Scholar]
- 13.Chappell-Maor L, Kolesnikov M, Kim JS, Shemer A, Haimon Z, Grozovski J, Boura-Halfon S, Masuda T, Prinz M, and Jung S (2020). Comparative analysis of CreER transgenic mice for the study of brain macrophages: A case study. Eur. J. Immunol. 50, 353–362. 10.1002/eji.201948342. [DOI] [PubMed] [Google Scholar]
- 14.Shi J, Hua L, Harmer D, Li P, and Ren G (2018). Cre Driver Mice Targeting Macrophages. Methods Mol. Biol. 1784, 263–275. 10.1007/978-1-4939-7837-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805. 10.1038/ni.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, and Tenner AJ (2017). Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflammation 14, 48. 10.1186/s12974-017-0814-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stowell RD, Sipe GO, Dawes RP, Batchelor HN, Lordy KA, Whitelaw BS, Stoessel MB, Bidlack JM, Brown E, Sur M, and Majewska AK (2019). Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat. Neurosci. 22, 1782–1792. 10.1038/s41593-019-0514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Hove H, Antunes ARP, De Vlaminck K, Scheyltjens I, Van Ginderachter JA, and Movahedi K (2020). Identifying the variables that drive tamoxifen-independent CreERT2 recombination: Implications for microglial fate mapping and gene deletions. Eur. J. Immunol. 50, 459–463. 10.1002/eji.201948162. [DOI] [PubMed] [Google Scholar]
- 19.Sahasrabuddhe V, and Ghosh HS (2022). Cx3Cr1-Cre induction leads to microglial activation and IFN-1 signaling caused by DNA damage in early postnatal brain. Cell Rep. 38, 110252. 10.1016/j.celrep.2021.110252. [DOI] [PubMed] [Google Scholar]
- 20.Kaiser T, and Feng G (2019). Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro 6, ENEURO.0448, 18.2019. 10.1523/ENEURO.0448-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masuda T, Amann L, Sankowski R, Staszewski O, Lenz M, D Errico P, Snaidero N, Costa Jordaão MJ, Böttcher C, Kierdorf K, et al. (2020). Novel Hexb-based tools for studying microglia in the CNS. Nat. Immunol. 21, 802–815. 10.1038/s41590-020-0707-4. [DOI] [PubMed] [Google Scholar]
- 22.McKinsey GL, Lizama CO, Keown-Lang AE, Niu A, Santander N, Larpthaveesarp A, Chee E, Gonzalez FF, and Arnold TD (2020). A new genetic strategy for targeting microglia in development and disease. Elife 9, e54590. 10.7554/eLife.54590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JS, Kolesnikov M, Peled-Hajaj S, Scheyltjens I, Xia Y, Trzebanski S, Haimon Z, Shemer A, Lubart A, Van Hove H, et al. (2021). A Binary Cre Transgenic Approach Dissects Microglia and CNS Border-Associated Macrophages. Immunity 54, 176–190.e7. 10.1016/j.immuni.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 25.Bedolla A, McKinsey G, Ware K, Santander N, Arnold T, and Luo Y (2023). Finding the right tool: a comprehensive evaluation of microglial inducible cre mouse models. Preprint at bioRxiv. 10.1101/2023.04.17.536878. [DOI] [Google Scholar]
- 26.Absinta M, Maric D, Gharagozloo M, Garton T, Smith MD, Jin J, Fitzgerald KC, Song A, Liu P, Lin JP, et al. (2021). A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 597, 709–714. 10.1038/s41586-021-03892-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khawaja RR, Agarwal A, Fukaya M, Jeong HK, Gross S, Gonzalez-Fernandez E, Soboloff J, Bergles DE, and Kang SH (2021). GluA2 overexpression in oligodendrocyte progenitors promotes postinjury oligodendrocyte regeneration. Cell Rep. 35, 109147. 10.1016/j.celrep.2021.109147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox BC, Liu Z, Lagarde MMM, and Zuo J (2012). Conditional gene expression in the mouse inner ear using Cre-loxP. J. Assoc. Res. Otolaryngol. 13, 295–322. 10.1007/s10162-012-0324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Southern E (2006). Southern blotting. Nat. Protoc. 1, 518–525. 10.1038/nprot.2006.73. [DOI] [PubMed] [Google Scholar]
- 30.Masuda T, Amann L, Monaco G, Sankowski R, Staszewski O, Krueger M, Del Gaudio F, He L, Paterson N, Nent E, et al. (2022). Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature 604, 740–748. 10.1038/s41586-022-04596-2. [DOI] [PubMed] [Google Scholar]
- 31.Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, McDonald MP, Crawley JN, Sandhoff K, Suzuki K, and Proia RL (1995). Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 11, 170–176. 10.1038/ng1095-170. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Park JW, Ahn IS, Diamante G, Sivakumar N, Arneson D, Yang X, van Veen JE, and Correa SM (2021). Estrogen receptor alpha in the brain mediates tamoxifen-induced changes in physiology in mice. Elife 10, e63333. 10.7554/eLife.63333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donocoff RS, Teteloshvili N, Chung H, Shoulson R, and Creusot RJ (2020). Optimization of tamoxifen-induced Cre activity and its effect on immune cell populations. Sci. Rep. 10, 15244. 10.1038/s41598-020-72179-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wy-soker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2019). Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271.e6. 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wendeler M, and Sandhoff K (2009). Hexosaminidase assays. Glycoconj. J. 26, 945–952. 10.1007/s10719-008-9137-5. [DOI] [PubMed] [Google Scholar]
- 37.Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.