

Abstract
The synthesis of heretofore unknown γ-spirobutenolides has been achieved via an m-CPBA-mediated oxidation of β-furyl amides. The reaction employs a tethered amide, ostensibly a poorly reactive carbonyl, as a nontraditional nucleophile resulting in spirolactone formation and concurrent amide cleavage. The transformation exhibits functional group tolerance and compatibility with complex compounds. In situ 1H NMR spectroscopic studies reveal the identities of key intermediates in the oxidation–spirolactonization–oxidation cascade, suggesting a plausible mechanistic pathway. The distinct diastereofaces of the electrophilic butenolide product may be used for diastereoselective cycloaddition and conjugate addition reactions.
Graphical Abstract

Spirobutenolides and spirobutyrolactones represent common motifs in both natural products and bioactive compounds (Scheme 1a).1–3 Surprisingly, the structure resulting from the fusion of these two substructures—a spiro-γ-butenolide-γ-butyrolactone (Scheme 1b)—has remained underexplored, with the exception of several benzofused examples.4,5 Given the growing interest in spirocycles for drug discovery6,7 and agricultural applications,8 it was with excitement that we observed the formation of spiro-γ-butenolide-γ-butyrolactone 2a upon attempted Baeyer–Villager oxidation of aryl ketone 1a (Scheme 1b). Although furan oxidation by peracids is well established,9,10 efficient engagement of the amide was not initially anticipated. This transformation bears some resemblance to the m-CPBA-mediated synthesis of spirobutenolide substructures previously reported by Robertson and co-workers,11 where a tethered alcohol was used as the nucleophile.
Scheme 1.

(a) Spirobutenolide and Spirolactone Motifs. (b) Upon Treatment with m-CPBA, Exclusive Formation of a γ-Spirobutenolide 2a was Unexpectedly Observed. (c) Current Work Utilizing Ostensibly Poorly Reactive Amides As Tethered Nucleophiles
Recognizing the possible utility of the observed transformation and the potential advantages of β-furyl amides as synthetic precursors to spirobutenolide substructures, not the least of which are the former’s stability and potential to be carried through numerous synthetic manipulations, we herein disclose a method developed based upon this original, serendipitous result (Scheme 1c). A brief screen of reaction conditions revealed 2.5 equiv of meta-chloroperbenzoic acid (m-CPBA) were sufficient, rather than the 8.0 equiv used in the initial discovery. With that modification, the scope of the discovered transformation was explored (Scheme 2).
Scheme 2. Substrate Scope of Oxidative Spirocyclizationc.
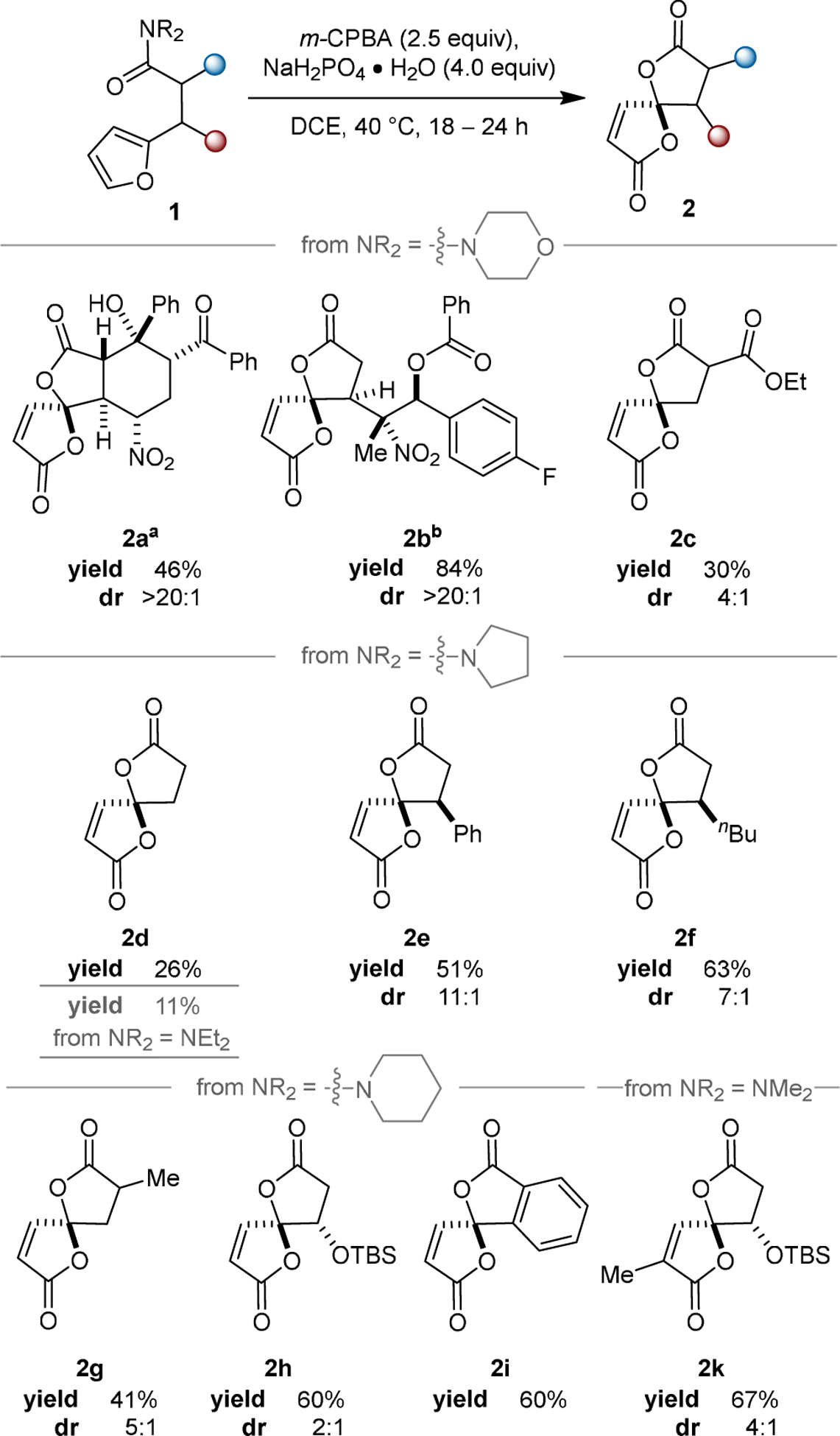
aReaction performed with 8.0 equiv m-CPBA and 10.0 equiv NaH2PO4·H2O on a 0.13 mmol scale. bReaction performed with 8.0 equiv m-CPBA and 10.0 equiv NaH2PO4·H2O on 0.40 and 0.06 mmol scales. The reported data are the average of these two trials. cAll reactions were performed on a 0.30 mmol scale unless otherwise noted. All yields are isolated yields. Diastereoselectivity was determined by analysis of 1H NMR spectra of crude products.
Several amides were tested as nucleophiles: morpholine (toward 2a–2c), pyrrolidine (toward 2d–2f), piperidine (toward 2g–2i), and N,N-dimethyl (toward 2k) amides were all used successfully. Functional group tolerance was probed with polyfunctional compounds (2a and 2b), which proved to be suitable substrates for the transformation and afforded the corresponding γ-spirobutenolides in 46% and 84% yields, respectively, with complete retention of functionality and stereocomplexity. The limited solubility of 1a and 1b necessitated more dilute reaction conditions, and excess m-CPBA was required to achieve efficient transformations for these examples. Subjecting a β-ester amide (1c) to the reaction conditions resulted in 30% isolated yield of spirocycle 2c. The low yield may be partially due to the reduced nucleophilicity of the amide in this compound; however, no amide-containing spirocyclic byproducts were observed in the crude product. Exclusive formation of 2c suggests that the amide is uniquely suited for participation in this transformation over other carbonyl groups such as esters. A simple β-furylamide afforded novel spirobutenolide 2d in low yield (26%) despite full consumption of the starting material. In situ hydrolysis of bis(lactone) 2d to a diacid product (vide infra) likely contributed to the poor performance of this substrate. The lack of substitution on the backbone of 1d may also contribute to its poor performancȩ as more highly substituted substrates may benefit from the Thorpe–Ingold effect during multiple points of the mechanism.12 Spirocycle 2d could also be prepared in 11% yield from the acyclic N,N-diethylamide, suggesting that both cyclic and acyclic amides are capable participants in the spirocyclization.
Aryl- and alkyl-substituted spirolactones 2e and 2f were isolated in reasonable yields and diastereomeric ratios (51%, 11:1 and 63%, 7:1, respectively). Methyl-substituted spirolactone 2g was isolated as a 5:1 mixture of separable diastereomers in 41% yield. β-Silyloxy amide afforded 2h in 60% yield, albeit in poor diastereoselectivity (2:1 dr). We were pleased to observe that a methyl-substituted furan could also participate in the spirocyclization reaction, delivering spirobutenolide 2k in 67% yield as a 4:1 mixture of diastereomers. Inverse diastereoselectivity was obtained in 2h and 2k when compared with β-substituted lactones 2e and 2f. Unprotected furfuryl alcohols were not viable substrates for spirolactonization (vide infra). Using a slightly modified procedure (see Supporting Information for details), a 1.2 mmol scale reaction was performed to obtain spirobutenolide 2h in 69% yield with 2.5:1 dr. An ortho-furylbenzamide was also capable of the title transformation, producing benzo-fused butenolide 2i in 60% yield.
Having established the efficacy of the one-pot synthesis of γ-spirobutenolides, we were interested in probing the reaction mechanism. Several examples of β-furyl carbonyl oxidations have been previously reported,13–16 some of which propose spirocyclic intermediates.17 To observe reaction intermediates, a solution of β-furyl amide 1d and the phenanthrene internal standard was prepared in CDCl3 (Figure 1a, t = 0 min). Upon addition of 2.0 equiv m-CPBA, a well-precedented cis-ene-1,4-dicarbonyl (Int-1)18 was observed in 70% yield after 10 min (NMR yield vs internal standard), identifiable by the characteristic aldehyde C–H methine resonance at δ 10.2 ppm (Figure 1a). After the solution was heated at 40 °C for 1 h, hemiacetal Int-2 was observed. Continued heating resulted in bis(lactone) formation and ultimately production of undesired acyclic diacid SP-1, presumably from hydrolysis and isomerization of spirocycle 2d (t = 30 h).19 The in situ 1H NMR spectroscopy experiments provided insights into the low yield observed for 2d during scope exploration: oxidation of Int-2 to 2d appears to be the lowest-yielding step of the cascade.
Figure 1.

Mechanistic investigations of the oxidative cascade reaction. (a) Oxidation–spirolactonization–oxidation cascade observed by in situ 1H NMR spectroscopy. (b) Proposed reaction mechanism consistent with 1H NMR spectroscopy study. (c) Investigations on the reactivity of furfuryl alcohols.
Reactions of β-furylpropionic acid and ethyl β-furylpropionate with m-CPBA showed no evidence of spirolactone formation by 1H NMR spectroscopic analysis, highlighting the unique behavior of the amide (See Supporting Information for details). Additionally, the furan oxidations from the ester and acid derivatives were much less efficient (30% and 27% 1H NMR yields of cis-ene-1,4-dicarbonyl, respectively) when compared to the amide example (70% 1H NMR yield). Allylic and homoallylic amides are capable of directing m-CPBA alkene epoxidations;20 these experiments collectively suggest that the amide is important in directing the initial furan oxidation by m-CPBA.
A plausible mechanism that accounts for the species observed via in situ 1H NMR spectroscopy and the differences in reactivity between carbonyl identities is outlined in Figure 1a, where two separate oxidations with m-CPBA occur in the overall transformation. Based on the unique efficiency of furan oxidation in the presence of a tethered amide, we propose an amide-directed, dearomative epoxidation of the furan ring. The resulting bicyclic intermediate rapidly opens to reveal 1,4-dicarbonyl intermediate Int-1; heating the solution at 40 °C for 1 h elicits nucleophilic activity from the tethered amide in a manner similar to that observed in some lactonization protocols,21–25 and cyclization proceeds in a 5-exo-trig fashion26 to afford hemiacetal Int-2. Oxidation of the hemiacetal Int-2 by m-CPBA affords the γ-spirobutenolide product.
This proposed mechanism suggests that an alternative oxidant might be used to convert lactol Int-2 to spiro bis(lactone) 2d, especially since m-CPBA is an unconventional reagent for lactol → lactone oxidation. A brief screen of alternative oxidants was conducted for this step, but no significant improvements in yield were obtained (see Supporting Information for details).
The α-substituted ketone Int-1 would be the key for understanding the origin of diastereoselectivity in a kinetically controlled spirolactonization. Felkin–Anh analysis27,28 would suggest that sterically bulkier α-keto groups would result in higher diastereoselectivity during carbonyl addition; this trend is observed (dr for 2b > 2e > 2f) and is useful for the qualitative prediction of diastereoselectivity. For silyl ether substrates 2h and 2k where diastereoselectivity is opposite that which would be predicted by Felkin–Anh analysis, other factors may be controlling the diastereoselectivity of spirolactonization. To probe whether interconversion between diastereomers was taking place under the reaction conditions, diastereopure spirocycle 2e was subjected to catalytic m-chlorobenzoic acid (m-CBA) in CDCl3. Heating the sample at 40 °C for 20 h resulted in partial diastereomerization (2e:epi-2e = 21:1) and product hydrolysis (see Supporting Information for details). This experiment suggests that product thermodynamic stability contributes, at least in part, to the observed diastereoselectivity of some of the oxidative spirocyclization. This experiment also demonstrates that acidic conditions promote product hydrolysis, perhaps explaining why slightly higher yields were obtained in reactions with the added phosphonate base.
Previous reports have used organic peroxides to perform the Achmatowicz rearrangement, which converts furfuryl alcohols to dihydropyranone hemiacetals.29,30 To probe the current method’s compatibility with furfuryl alcohols as well as to understand the relative rates of the two possible reaction pathways, β-furyl amide 1j was subjected to the standard reaction conditions (Figure 1b). Spirobutenolide 2j was not observed, but the dihydropyranone Achmatowicz-type product 3a was isolated as a 1:1.6 mixture of diastereomers in 60% yield. These results suggest that the 6-exo-trig cyclization of the Achmatowicz rearrangement is faster than the 5-exo-trig cyclization required to obtain the spirobutenolide product. The Achmatowicz predominance can reasonably be attributed to the higher nucleophilicity of the alcohol as compared to the amide.31 As demonstrated with silyl ethers 2h and 2k, the Achmatowicz-type reactivity can be arrested by the simple protection of furfuryl alcohol groups.
Derivatizations of the γ-spirobutenolide products were investigated (Figure 2). Spirobutenolide 2e readily participated in a Zn(II)-catalyzed Diels–Alder cycloaddition with cyclopentadiene to afford tetracycle 4a in 67% isolated yield as an 8.5:1 mixture of separable diastereomers. The relative configuration of the major diastereomer was established by X-ray crystallography, revealing the major stereoisomer to be the endo product resulting from the diene approach syn to the oxygen of the saturated lactone. Rhodium(I)-catalyzed arylation of spirocycle 2e proceeded with high diastereoselectivity (>20:1 dr), and spirolactone 4b was isolated in 64% yield. The relative stereochemistry of bis(lactone) 4b was established by X-ray crystallography; the observed diastereomer results from addition syn relative to the oxygen in the butyrolactone of spirocycle 2e (see Supporting Information for discussion of related literature examples).11
Figure 2.
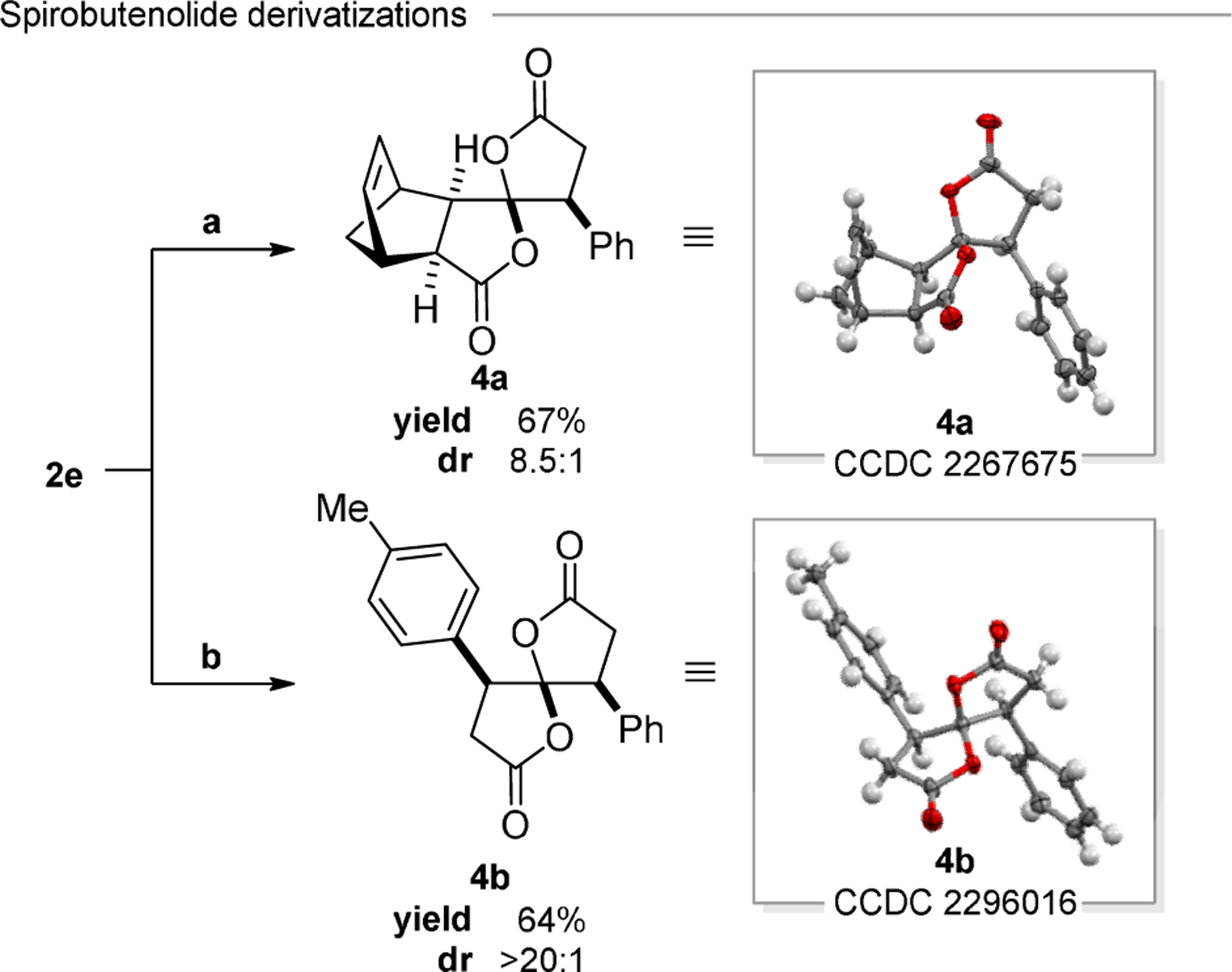
(a) ZnCl2 (0.70 equiv), cyclopentadiene (12.0 equiv), DCM (0.65 M), rt, 18 h and (b) [RhCl(COD)]2 (0.5 mol % Rh), ptolueneboronic acid (3.0 equiv), CsF (5.0 equiv), Proton Sponge (1.0 equiv), 1,4-dioxane (70 mM), 50 °C, 24 h. X-ray structures of 4a and 4b are shown as 50% thermal ellipsoids.
In conclusion, a method for the synthesis of γ-spirobutenolides from β-furyl amides using m-CPBA has been disclosed. The operationally simple, one-pot procedure provides access to previously unknown spirocyclic substructures. Additionally, this method represents a novel oxidative method for the cleavage of ostensibly inert amide groups under relatively mild conditions. The reported method expands the chemical space, that may be accessed from biomass-derived starting materials such as furfural and may enable broader exploration of γ-spirobutenolide architectures.
Supplementary Material
ACKNOWLEDGMENTS
The project described was supported by award R35 GM 118055 from the National Institute of General Medical Sciences and NCATS U01 TR002625 and by a National Science Foundation Graduate Research Fellowship to K.M.K. under Grant No. DGE-2040435. We thank Dr. B. Ehrmann and D. Weatherspoon (UNC Chemistry Mass Spectrometry Core Laboratory) for their assistance with mass spectrometry analysis and Dr. M. ter Horst (UNC Chemistry NMR Core Laboratory) for assistance with NMR spectroscopy. X-ray crystallography (compound 4a and 4b) was performed by E. T. Crawford (UNC Chapel Hill) with assistance from Dr. C. H. Chen (UNC Chapel Hill) on instrumentation acquired under the NSF MRI program under Grant No. CHE-2117287. We are grateful to P. de Jesús Cruz (UNC Chapel Hill) for the initial synthesis and characterization of compound 2b.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.3c03100.
Experimental details, materials, methods, characterization data, NMR spectra for all compounds, chromatograms for chiral separations, and information on X-ray diffraction experiments (PDF)
Accession Codes
CCDC 2267675 and 2296016 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.orglett.3c03100
Contributor Information
Katelyn M. Kitzinger, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States
Jeffrey S. Johnson, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
REFERENCES
- (1).Yadav P; Pratap R; Ji Ram V Natural and Synthetic Spirobutenolides and Spirobutyrolactones. Asian J. Org. Chem 2020, 9 (10), 1377–1409. [Google Scholar]
- (2).Quintavalla A Spirolactones: Recent Advances in Natural Products, Bioactive Compounds and Synthetic Strategies. Curr. Med. Chem 2018, 25 (8), 917–962. [DOI] [PubMed] [Google Scholar]
- (3).Thirupathi B; Mandal S Strategies for the construction of γ-spirocyclic butenolides in natural product synthesis. Org. Biomol. Chem 2020, 18, 5287–5314. [DOI] [PubMed] [Google Scholar]
- (4).Bayat M; Imanieh H; Farjam MH Triphenylphosphine-Promoted Synthesis of Spiroketals from Phthalic Anhydride with Dialkyl Acetylenedicarboxylates. Synth. Commun 2010, 40 (16), 2475–2482. [Google Scholar]
- (5).Marsili A; Scartoni V; Morelli I; Pierangeli P Nitrogen Heterocycles. Part 6. Conversion of 3-(α-Bromobenzylidene)-2-Phenethylphthalimidine into Pyrrole and Benzazepine Derivatives; Base-Catalysed Rearrangement and Photolysis of 3-(α-Bromobenzylidene)Phthalimidine. J. Chem. Soc. Perkin Trans 1 1977, No. 9, 959–965. [Google Scholar]
- (6).Hiesinger K; Dar’in D; Proschak E; Krasavin M Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem 2021, 64 (1), 150–183. [DOI] [PubMed] [Google Scholar]
- (7).Bora D; Kaushal A; Shankaraiah N Anticancer Potential of Spirocompounds in Medicinal Chemistry: A Pentennial Expedition. Eur. J. Med. Chem 2021, 215, No. 113263. [DOI] [PubMed] [Google Scholar]
- (8).Yu L; Dai A; Zhang W; Liao A; Guo S; Wu J Spiro Derivatives in the Discovery of New Pesticides: A Research Review. J. Agric. Food Chem 2022, 70 (35), 10693–10707. [DOI] [PubMed] [Google Scholar]
- (9).Lefebvre Y Oxidation of Furans - I. Synthesis of 6-Hydroxy-2HPyran-3(6H)-Ones. Tetrahedron Lett 1972, 13 (2), 133–136. [Google Scholar]
- (10).Laliberte R; Medawar G; Lefebvre Y Oxidation of Furans. 2. Synthesis and Biological Properties of 6-Hydroxy-2H-Pyran-3(6H)-Ones and Derivatives. J. Med. Chem 1973, 16 (10), 1084–1089. [DOI] [PubMed] [Google Scholar]
- (11).Robertson J; Meo P; Dallimore JWP; Doyle BM; Hoarau C Stereoselective Synthesis of the Lituarine Tricyclic Spiroacetal. Org. Lett 2004, 6 (21), 3861–3863. [DOI] [PubMed] [Google Scholar]
- (12).Beesley RM; Ingold CK; Thorpe JF CXIX.—The Formation and Stability of Spiro-Compounds. Part I. Spiro-Compounds from Cyclohexane. J. Chem. Soc. Trans 1915, 107 (0), 1080–1106. [Google Scholar]
- (13).Choi J; Laird JM; Salomon RG An Efficient Synthesis of γ-Hydroxy-α,β-Unsaturated Aldehydic Esters of 2-Lysophosphatidylcholine. Bioorg. Med. Chem 2011, 19 (1), 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shcherbakov RO; Eshmemet’eva DA; Merkushev AA; Trushkov IV; Uchuskin MG Transformation of 3-(Furan-2-Yl)-1,3-Di(Het)Arylpropan-1-Ones to Prop-2-En-1-Ones via Oxidative Furan Dearomatization/2-Ene-1,4,7-Triones Cyclization. Molecules 2021, 26 (9), 2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hoogewijs K; Deceuninck A; Madder A Aromatic Capping Surprisingly Stabilizes Furan Moieties in Peptides against Acidic Degradation. Org. Biomol. Chem 2012, 10 (20), 3999. [DOI] [PubMed] [Google Scholar]
- (16).Halila S; Velasco T; Clercq PD; Madder A Fine-Tuning Furan Toxicity: Fast and Quantitative DNA Interchain Cross-Link Formation upon Selective Oxidation of a Furan Containing Oligonucleotide. Chem. Commun 2005, No. 7, 936. [DOI] [PubMed] [Google Scholar]
- (17).Yu H; Zhong W; He T; Gu W; Yin B An Entry to Polysubstituted Furans via the Oxidative Ring Opening of Furan Ring Employing NBS as an Oxidant. Tetrahedron Lett 2013, 54 (10), 1256–1260. [Google Scholar]
- (18).Piancatelli G; D’Auria M; D’Onofrio F Synthesis of 1,4-Dicarbonyl Compounds and Cyclopentenones from Furans. Synthesis 1994, 09, 867–889. [Google Scholar]
- (19).Barton P; Page MI The Esterase Catalysed Resolution of Lactones and Spirodilactone. J. Chem. Soc., Perkin Trans 1993, No. 12, 2317. [Google Scholar]
- (20).Kocovsky P; Stary I Steric Control of Epoxidation by Carbamate and Amide Groups. Evidence for the Carbonyl-Directed Epoxidation. J. Org. Chem 1990, 55 (10), 3236–3243. [Google Scholar]
- (21).Blot V; Reboul V; Metzner P Asymmetric Induction of the Iodolactonization Reaction of α-Sulfurated γ-Unsaturated Amides. J. Org. Chem 2004, 69 (4), 1196–1201. [DOI] [PubMed] [Google Scholar]
- (22).Robin S; Rousseau G Electrophilic Cyclization of Unsaturated Amides. Tetrahedron 1998, 54 (45), 13681–13736. [Google Scholar]
- (23).Rozners E; Liu Y Toward Amide-Linked RNA Mimics: Total Synthesis of 3’-C Branched Uridine Azido Acid via an Ene–Iodolactonization Approach. Org. Lett 2003, 5 (2), 181–184. [DOI] [PubMed] [Google Scholar]
- (24).Najdi S; Reichlin D; Kurth MJ Enantioselective Route to.Gamma.-Butyrolactones: Chiral Auxiliary Mediated Amide Alkylation and Iodolactonization. J. Org. Chem 1990, 55 (26), 6241–6244. [Google Scholar]
- (25).Oderinde MS; Hunter HN; Bremner SW; Organ MG Iodolactonization: Synthesis, Stereocontrol, and Compatibility Studies. Eur. J. Org. Chem 2012, 2012 (1), 175–182. [Google Scholar]
- (26).Claveau E; Noirjean E; Bouyssou P; Coudert G; Gillaizeau I Access to Novel Bicyclic Fused γ-Butyrolactone Using [3,3]Sigmatropic Rearrangement and Acid-Lactonization Sequence as Key Transformation. Tetrahedron Lett 2010, 51 (23), 3130–3133. [Google Scholar]
- (27).Anh NT; Eisenstein O Theoretical Interpretation of 1–2 Asymmetric Induction. The Importance of Antiperiplanarity. Nouv. J. Chim 1977, 1, 61–70. [Google Scholar]
- (28).Paddon-Row MN; Rondan NG; Houk KN Staggered Models for Asymmetric Induction: Attack Trajectories and Conformations of Allylic Bonds from Ab Initio Transition Structures of Addition Reactions. J. Am. Chem. Soc 1982, 104 (25), 7162–7166. [Google Scholar]
- (29).Ren J; Tong R Asymmetric Total Synthesis of (+)-Didemniserinolipid B via Achmatowicz Rearrangement/Bicycloketalization. J. Org. Chem 2014, 79 (15), 6987–6995. [DOI] [PubMed] [Google Scholar]
- (30).Li Z; Leung T-F; Tong R Total Syntheses of (±)-Musellarins A–C. Chem. Commun 2014, 50 (75), 10990–10993. [DOI] [PubMed] [Google Scholar]
- (31).Robertson J; North C; Sadig JER Asymmetric Synthesis of the C(6–18) Bis(Tetrahydropyran)Spiroacetal Fragment of the Lituarines. Tetrahedron 2011, 67 (27–28), 5011–5023. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.