

Abstract
Targeted protein degradation (TPD) involving chimeric molecules has emerged as one of the most promising therapeutic modalities in recent years. Among various reported TPD strategies, proteolysis targeting chimeras (PROTACs) stand out as a significant breakthrough in small molecule drug discovery and have garnered the most attention to date. However, PROTAC is mainly capable of depleting intracellular proteins. Given that many important therapeutic targets such as cytokines, growth factors and numerous receptors are extracellular secreted and membrane proteins, there is interest in the development of novel strategies to degrade these protein categories. We review advances in this emerging area and provide insights to enhance the development of novel TPDs targeting extracellular proteins.
Keywords: LYTAC, PROTAC, Degradation, Lysosome, Extracellular
Targeted protein degradation – a new paradigm for drug discovery
Cells constantly synthesize and degrade proteins. The turnover of protein is essential for maintaining cellular protein homeostasis. While most intracellular proteins in eukaryotic cells are degraded through ubiquitin-proteasome system (UPS, see glossary)[1,2], both intra- and extracellular proteins can be degraded by the lysosomal system through autophagy and endocytosis[3,4]. Various targeted protein degradation (TPD) strategies using chimeric molecules that leverage these cellular degradation systems have been developed[5–7]. These hetero-bifunctional chimeric molecules have a moiety on one end that binds to the protein of interest (POI), while the moiety on the other end binds to an effector to form a ternary complex that can direct the degradation of the POI in either the proteasome or lysosome. Over the past two decades, proteolysis targeting chimera (PROTAC) received the most attention among different TPD strategies[5–7]. PROTACs have a POI-binding ligand linked using a suitable linker with an E3 ligase ligand, which directs the POI for ubiquitination by E3 ubiquitin ligase and route it to proteasome for degradation.
TPD by chimeric molecules is a transformative paradigm for drug discovery[5–7]. TPD modality such as PROTAC promises to overcome a major limitation of traditional therapeutics, which generally need to bind to the functional site of the protein target in order to alter its function. Despite the advantages of TPD modality such as PROTACs (TextBox 1), the cytosolic localization of the UPS restricts the PROTAC technology to cell-permeable small molecule binders and protein targets with cytosolic domains, limiting its utilization for the therapeutic degradation of extracellular secreted and some membrane proteins. Extracellular secreted and membrane proteins, compose around 40% of the human proteome, and are also therapeutic targets in various diseases including cancer and autoimmune illnesses[8]. Cell permeable small molecule PROTACs have been developed to target membrane proteins with ligandable cytosolic domains (TextBox 1). Antibody-based strategies such as sweeping antibodies and Seldegs have also been developed for the degradation of extracellular proteins albeit with limited utilities[9–11].
Textbox 1; PROTACs.
Advantages of PROTACs
TPD paves the way for targeting proteins that lack accessible active sites for the binding of small molecules. The chimeric molecules for TPD only require a binder to the protein target and this binder does not need to be functional as the degradation modality enables the complete removal of proteins from a cellular environment. TPD modality allows the flexibility to use ligands that can bind anywhere on the protein to overcome resistance resulting from mutations at certain functional sites. Indeed, we have witnessed the tremendous success of PROTAC technology in the last few years because of its potential to target proteins with scaffolding functions that cannot be easily blocked or overcome the drug resistance caused by mutations[62].
Degradation of membrane proteins by recruiting intracellular E3 ligases
PROTACs can also be used for the degradation of certain membrane proteins, but not secreted proteins. In all previous strategies, the degradation of extracellular secreted and membrane proteins is realized by binding to the extracellular domain of the protein target, which is the focus of this review. It is also possible to degrade membrane proteins by PROTACs, if a cell permeable small molecule ligand is available for the intracellular domains of the membrane targets[63]. However, the presence of hydrophobic transmembrane domains and their positioning within the lipid bilayer can present challenges for PROTAC-mediated internalization of the membrane targets. Additionally, the current repertoire of reported PROTACs predominantly recruit cytosolic von hippel-lindau (VHL) or cereblon (CRBN) E3 ligases, which need to be recruited to the appropriate location for the ubiquitination of membrane proteins and the resulting ubiquitinated membrane proteins need to be disengaged from the cell membrane for proteasomal degradation. Despite these potential challenges, there are a number of successful cases that deserve to be highlighted. PROTACs for membrane proteins such as anaplastic lymphoma kinase (ALK)[64–73], EGFR[74–86], fibroblast growth factor receptor (FGFR)[63,87–89], FMS-like tyrosine kinase 3 (FLT-3)[90–92], G protein-coupled receptors (GPCRs)[93,94] and PD-L1[95–97] have been reported for effective degradation of membrane proteins. It is expected that the discovery of new pairs of “hijackable” E3 ligases and their cell permeable small molecule ligands, especially membrane E3 ligases/ligands, can further expand the scope of degradable membrane proteins by PROTACs (Figure 4).
Recently, studies on the development of more effective strategies to degrade both extracellular secreted and membrane proteins are increasing. These innovative strategies such as lysosome targeting chimeras (LYTACs)[12], which can bind to and redirect plasma membrane-associated or secreted proteins to lysosomes, have greatly expanded the potential of TPD to other extracellular targets. Other degraders of extracellular proteins such as molecular degraders of extracellular proteins (MoDEs)[13], dendronized DNA chimeras (DENTACs)[14], integrin-facilitated lysosomal degradation (IFLD)[15], cytokine receptor-targeting chimeras (KineTAC)[16], covalent nanobody-based PROTAC strategy (termed as GlueTAC)[17], antibody-based PROTACs (AbTACs)[18], proteolysis-targeting antibodies (PROTABs)[19], R-spondin chimera (ROTACs)[20], and signal-mediated lysosome-targeting chimeras (SignalTACs)[21] show great promise. In all of these strategies, the bifunctional degraders bind to the POI outside of the cells. In most cases, the effectors are cell-surface lysosome targeting receptors (LTRs). Other effectors such as intracellular lysosome sorting sequence (LSS) recognition proteins and membrane E3 ubiquitin ligases have also been employed as effectors. In this review we critically assess and highlight recently developed extracellular protein degraders that use cell-surface LTR mechanism of action (MoA) and cell-surface LTR independent MoA to inform on challenges and opportunities for future research (Table 1).
Table 1.
Comparison of therapeutic strategies for the degradation of extracellular secreted and membrane proteins based on different effectors
| CI-M6PR | Conjugation of POI binders and LTR binders (e.g. antibody, small molecule, aptamer) | APOE4 EGFR, PD-L1, and CD71 | Ubiquitous | Targets both extracellular and membrane proteins in a broad range of cells | poor delivery and poor tissue permeability for most antibody-based degraders; poor in-vivo stability of DNA-based degraders | Endogenous kinetics of protein trafficking and turnover; the amount of POI; the inherent susceptibility to lysosomal transportation by endocytosis; relative ratio of POI and LTR | [12,30,36,38] |
| ASGPR | EGFR; HER2; MIF; α-DNP; integrins | Primarily hepatocytes | Liver cell-specificity; targets both extracellular and membrane proteins | [13,43–45,47–49] | |||
| MGL1 | hIAPP | multiple types | Unique mimic of α-helical peptides; resistant to hydrolysis | [50] | |||
| Integrin | PD-L1 | multiple types | Potential tumor tissue-selectivity | [15] | |||
| SR | NCL; EGFR | multiple types | High expression and wide distribution of SRs. Inherent chemical programmability and tunability of dendritic DNAs | [14] | |||
| Cytokine receptor | genetically encoded bispecific antibodies | PD-L1; HER2; EGFR | multiple types | Modular and easy preparation | Potential immunogenicity | Binding affinity and the choice of epitope of KineTAC to cytokine receptors and POI; construction design and ratio of effector to POI | [16] |
| LSS recognition proteins | Nanobody and cationic CPP-LSS | PD-L1 | Ubiquitous | Independent of E3 ligase or receptor | Short serum half-life, potential toxicity associated with cationic CPP, no tissue-selectivity | Binding affinity of covalent nanobody to POI; lysosomal sorting efficiency of LSS | [17,21] |
| Membrane E3 ligases | recombinant bispecific IgGs / RSPO2 furin domains fused to POI binders | PD-L1; HER2; EGFR; IGF1R; FZD5; | Depend on E3 | Modular and easy preparation (antibodies); many E3s are accessible | Limited available E3 ligands; potentially immunogenic (AbTAC/PROTAB); short serum half-life (ROTAC) | Binding affinity and epitope of each arm; orientation, and valency; cell-surface levels of POI; ratio of POI and E3; endocytosis kinetics upon antibody binding; turnover rate of the POI, and the specific E3 ligase | [18–20,57,58] |
| Intracellular E3 ligases | Cell permeable small molecules | ALK; EGFR; FGFR; FLT-3; GPCRs; PD-L1 | Depend on E3 | Potentially orally available small molecule (PROTACs); many E3s are accessible | Limited available E3 ligands; extensive optimization | Length and type of linker; binding affinity of the ligands to E3 and POI; turnover rate of the POI | [63–97] |
Degradation of extracellular proteins by recruiting cell surface lysosome-targeting receptor (LTR)
Most degraders of extracellular proteins are created by conjugating ligands of the lysosome targeting receptors (LTRs) on the cell surface with ligands that can bind to the extracellular POI. Research from the drug delivery field, particularly receptor-mediated drug delivery, inspired this extracellular protein degrader development strategy. Cell surface carbohydrate binding proteins or lectins including cation-independent mannose-6-phosphate receptor (CI-M6PR) and asialoglycoprotein receptor (ASGPR), both currently utilized for TPD, were previously employed for the delivery of therapies: CI-M6PR was used for the delivery of recombinant hydrolases to treat lysosomal storage diseases[22], while ASGPR was successfully exploited for the delivery of siRNAs to the liver in clinical settings[23–25]. Studies on the utilization of other LTRs such as macrophage galactose type lectin, integrins, scavenger receptors and cytokine receptors in TPD development are growing and have revealed their potential for degradation of specific extracellular proteins.
Cation-independent mannose-6-phosphate receptor (CI-M6PR) as the LTR
The application of CI-M6PR, a ubiquitously expressed cell-surface lysosomal shuttling receptor, for the degradation of extracellular secreted and membrane protein was initiated by the Bertozzi lab in 2020[12]. The ligand of CI-M6PR, mannose-6-phosphate (M6P) or its derivatives, can be conjugated to therapeutic drugs to leverage its lysosomal shuttling function to deliver drugs intracellularly. Besides its use for delivering recombinant hydrolases for lysosomal enzyme replacement therapy[26], various molecules, such as peptides and proteins, have been covalently linked to modified M6P to leverage the CI-M6PR shuttling mechanism for drug delivery[27–29] . The Bertozzi lab used an antibody fused to chemically synthesized glycopeptide ligands that are agonists of CI-M6PR. They referred to this conjugate as LYTACs. The antibody can bind to the extracellular domain of POI, while the glycopeptide ligand bears multiple units of mannose-6-phosphonate (M6Pn) motif which recruits CI-M6PR effectively enabling the internalization and delivery of POI to lysosomes for subsequent proteolysis. The CI-M6PR is dissociated from the POI in the endosome and recycled to the cell surface (Figure 1). In this proof-of-concept work, several proteins including secreted apolipoprotein E4 , and membrane proteins epidermal growth factor receptor (EGFR) , CD71 and programmed death-ligand 1 (PD-L1), which are important targets for the treatment of Alzheimer’s disease and cancers, were successfully degraded using the M6Pn-based LYTACs[12].
Figure 1. Schematic illustration of degraders that recruit various lysosome targeting receptors (LTRs).
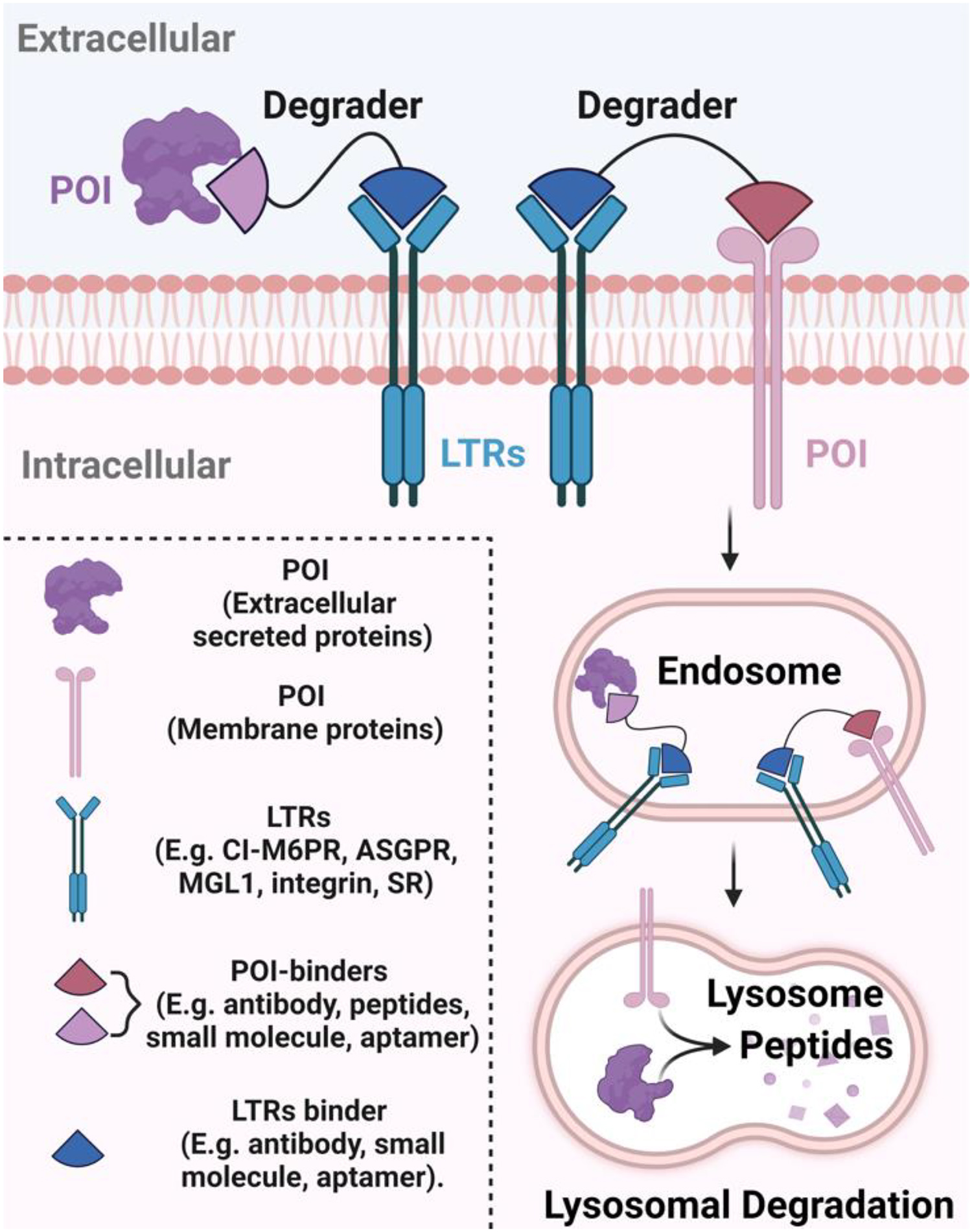
Bifunctional degraders simultaneously bind to the protein of interest (POI) and LTR, forming a ternary complex. This receptor-ligand interaction initiates the internalization of the entire complex via endocytosis pathways. Once inside the endosome, the LTR dissociates from its ligand and recycles back to the cell surface, while the degrader and a part of the POI are likely directed to the lysosome for degradation afterward. The binder of POI and LTR can be antibody, peptide, small molecule, and aptamer. (Created with BioRender.com)
The polymeric glycopeptide ligands used in the Bertozzi study are prepared by co-polymerization, which produced a heterogeneous mixture with variable lengths, diverse numbers of M6Pn moieties (e.g. 20–40), and undefined distances among the M6Pn units[12], because the required number of M6Pn motifs and the distance between the M6Pn moieties are not well understood. It is obvious that only portions of the polymeric ligands can bind to the receptor. A structurally well-defined CI-M6PR ligands will yield more effective LYTACs. Recently, the Tang lab systemically investigated the structure-activity relationship (SAR) of the CI-M6PR ligands for TPD[30]. They prepared structurally well-defined CI-M6PR glycopeptide ligands using a library of oligopeptides with multiple units of cysteine residues (e.g. 1, 2, 3, 4, and 6) and a highly efficient thiol-ene reaction to couple the alkene-containing M6Pn to the cysteine residues on the oligopeptides. They demonstrated that LYTACs bearing glycopeptides with four units of M6Pn could efficiently promote the degradation of therapeutically relevant EGFR in cancer cells.
The LYTACs used in the studies by Bertozzi and Tang were generated by non-specific labelling of lysine residues on the antibodies with bicyclononyne-N-hydroxysuccinimidyl ester (BCN-NHS) or dibenzo cyclooctyne-N-hydroxysuccinimidyl ester (DBCO-NHS), and subsequent conjugation of the antibody to azide-terminated M6Pn glycopolypeptides via copper-free strain-promoted Click reaction[12][30]. Although the degradation of the therapeutic targets were successful using LYTACs generated by this method, the heterogeneity of antibody glycoform can modulate the efficacy of LYTACs, as evident in antibody only therapeutics, which could also pose a challenge in LYTAC development[31,32]. Endoglycosidases Endo-S, Endo-S2, and their mutants have previously been shown to be useful in the preparation of a homogeneous antibody glycoform[31–35]. These enzymes can convert diverse N-glycans on a mixture of antibodies to a product bearing a GlcNAc residue with or without the Fuc-residue. The resulting product can then be used as the substrate for a series of glycosyl transferases to attach oligosaccharides with an oxazoline motif to the antibody. Recently, the Wang lab combined the chemoenzymatic method and Click chemistry to prepare a homogenous antibody conjugate with structurally defined glycans containing multiple mannose-6-phosphate (M6P) motifs[36]. They demonstrated that these homogenous antibody conjugates could be used to effectively degrade human epidermal growth factor receptor-2 (HER2) and EGFR, important targets for cancer treatment[36].
Because monomeric M6P, or its analogues such as M6Pn, has relatively weak binding to CI-M6PR, multiple units with appropriate linkers are required for efficient binding[12][30]. Alternative binders with increased binding affinity, such as aptamers, may overcome this limitation. Aptamers are oligonucleotides possessing unique tertiary structures that enable them to bind proteins effectively and specifically. Aptamers can also be used as strong binders of the POI. The lower molecular weight of aptamers compared to antibodies, can provide several benefits in therapeutic applications such as better tissue penetration[37]. In addition, aptamers of LTR or POI can be readily identified by systematic evolution of ligands by exponential enrichment (SELEX) and other methods. Han lab recently prepared bispecific aptamer chimeras that can bind to both CI-M6PR and POI on the membrane[38]. They demonstrated that these bispecific aptamer chimeras were efficient degraders of membrane proteins such as Met and protein tyrosine kinase 7 (PTK-7) by recruiting CI-M6PR. More recently, Hamada’s lab reported bispecific DNA-aptamer-based LYTAC for the degradation of HER2 by recruiting CI-M6PR[39]. Despite the potential advantages, the development of aptamer-based therapeutics need to overcome a number of challenges such as the stability and durability.
Asialoglycoprotein receptor (ASGPR) as the LTR
Tissue-selective degraders could be highly significant to increase therapeutic index and reduce unwanted side effect. ASGPR is primarily and highly expressed on hepatocytes[40]. ASGPR and its ligand triantennary N-acetylgalactosamine (tri-GalNAc) system has been extensively used in delivering various drugs to the liver[41,42]. Recently, research groups of Bertozzi, Spiegel and Tang exploited this unique ligand/receptor pair to generate ASGPR-recruiting LYTACs for the selective degradation of extracellular protein targets in liver[13,43,44]. Similar to CI-M6PR-based LYTACs, ASGPR-triGalNAc interaction triggers the internalization of the extracellular POI through receptor-mediated endocytosis, further inducing the degradation of the targets in the lysosome. This degradation mechanism was termed molecular degraders of extracellular proteins through ASGPR (MoDE-A) -by Spiegel’s lab. The Spiegel lab focused on the development of bifunctional small molecules for the depletion of cytokines (e.g. MIF) and antibodies (α-DNP antibody)[13], while Bertozzi and Tang focused on using antibody or peptide conjugates for the degradation of extracellular proteins, such as EGFR, HER2 and integrins[43,44].
Because of issues associated with heterogeneous antibodies with different glycoforms, Wang lab also used the same chemoenzymatic method to develop homogenous antibody conjugates that recruit ASGPR for the degradation of EGFR and PCSK9, as the representative membrane and secreted POI, respectively[45]. They attached different ASGPR ligands including natural bi- and triantennary N-glycans as well as the synthetic tri-GalNAc ligands to the antibody and observed some interesting SARs. Both the ligands and linker can significantly impact the binding to ASGPR and ASGPR-mediated degradation of PCSK9. While synthetic tri-GalNAc-antibody conjugates showed a “hook effect”[46] for binding to ASGPR and degradation of PCSK9, antibody conjugates with the natural N-glycans did not. This observation clearly indicates that optimization of ligand and linker are important for the development of degraders of extracellular proteins.
As discussed before, aptamers have the following advantages: 1) they have well-defined molecular structure; 2) they have lower molecular weight than antibodies; and 3) aptamer binders for different target proteins can be quickly identified. Zhu lab recently reported that cell-specific aptamer-based LYTACs could induce the degradation of extracellular protein platelet-derived growth factor (PDGF) and membrane protein PTK7 via ASGPR by attaching tri-GalNAc ligand to aptamer binders[47]. Further chemical modifications that can improve the stability of the aptamers are necessary for in-vivo studies and future therapeutic development.
For most LYTACs, we need to covalently link the binder of POI and the binder of LTR. A self-assembled ASGPR-recruiting nano-LYTAC, which may simplify the preparation of LYTACs, was reported by the labs of Dai, Huang, and Ma[48]. In this study, they prepared amphiphilic peptide-modified GalNAc, which could self-assemble into nanospheres to bind to ASGPR. The antibody that can bind to the POI, such as CD24, is conjugated to the amino groups on the peptides of the nanosphere. The resulting nano-LYTAC could efficiently degrade CD24 on the membrane of liver cell lines and affect the CD24/Siglect-10 signaling pathway for immunotherapy. In addition, the internal hydrophobic environment of the nanosphere provided the space for loading of other anti-cancer drugs, such as glucose oxidase (GOx) enzyme, which promotes the glucose utilization as a starvation therapy for the treatment of cancer. They observed synergistic anti-cancer effects in-vitro and in-vivo by combining GOx with the nano-LYTAC targeting CD24. The nano-LYTAC provides accurate targeting for the starvation therapy.
The degradation activity of most LYTACs were only evaluated in cellular assays. There are very few studies on the in-vivo structure activity relationship of LYTACs. Recently, Novartis reported the in-vivo activity of a series of ASGPR-recruiting LYTACs that can accelerate the clearance of secreted proprotein convertase subtilisin/kexin type 9 (PCSK9), which has the potential for the treatment of hyperlipidemia[49]. They employed both antibody and cyclic peptides as the binders of PCSK9 and their resulting heterobifunctional molecules showed great in-vivo degradation activity.
Macrophage galactose type lectin 1 (MGL1) as the LTR
In addition to liver, LTRs that are expressed in other types of cells such as macrophage are of great interest for cell- and tissue-specific degraders. Qu lab reported a metallohelix-based degrader that is composed of tri-GalNAc and chiral metallohelix binding human islet amyloid polypeptide (hIAPP)[50]. Chiral metallohelices are mimics of α-helical peptides and they were shown to bind to amyloid proteins. Metallohelices have the advantage of being resistant to enzymatic hydrolysis[51]. It was shown that the degrader recruited MGL1 to mediate the endocytosis of hIAPP and subsequent lysosomal degradation. MGL1 is an essential transmembrane C-type lectin receptor expressed in macrophage and has high affinity to lactose and its various monosaccharide derivatives[52]. However, the tri-GalNAc ligand also has high affinity to ASGPR on the liver. Not surprisingly, the tri-GalNAc functionalized metallohelix also facilitated the clearance of hIAPP in liver cells through ASGPR.
Integrin as the LTR
Integrins, also referred to as adhesion or junction proteins, were shown to be effective LTRs recently. Integrins are transmembrane receptors that facilitate the interaction between cells and their external environment. Some integrins, like αvβ3, are overexpressed in various types of tumor cells. This characteristic has been leveraged for receptor-mediated, cancer-selective drug delivery[53]. For instance, the recognition sequence of integrin αvβ3, Arg-Gly-Asp (RGD), has been utilized as a ligand to selectively deliver drugs into tumor cells[54]. The research labs of Fang, Chen, and Li reported a new approach known as integrin-facilitated lysosomal degradation (IFLD) for the degradation of PD-L1[15]. The degrader is composed of a small molecule ligand for PD-L1, a cyclic RGD-containing peptide, and a linker uniting the two. The PD-L1 degraders that employ integrin αvβ3 demonstrate a significant anti-tumor effect in mice. Although IFLD has the potential to selectively degrade PD-L1 in tumor tissue over normal tissue based on the expression profile of certain integrins, the tumor tissue-selective degradation was not confirmed experimentally. It’s also worth noting that the RGD sequence can bind to multiple members of integrins, which may lower the tissue specificity.
Scavenger receptors (SRs) as the LTRs
Cell surface scavenger receptors (SRs) were employed as LTRs recently. SRs represent a diverse family of cell surface receptors that are expressed in many tissues and cell types including macrophages, endothelial cells, and immune cells[55]. Degraders that recruit SRs have the potential to degrade extracellular proteins in a broad range of tissues and cells. Because certain SRs can bind to polyanionic DNAs and being internalized via endocytosis, they have been exploited in drug delivery and lysosome-targeted delivery[56]. Inspired by this delivery strategy, the labs of Li and Zhang reported dendronized DNA chimera (DENTAC) by linking a POI binder with a dendritic DNA, which acts as a ligand for SRs[14]. They demonstrated that DENTACs could successfully induce the degradation of membrane proteins such as nucleolin (NCL) and EGFR through SR-mediate internalization and lysosomal degradation. Similar to aptamers, the development of DNA-based therapeutics will face many aspects of challenges such as the stability and durability.
Cytokine receptor as the LTR
Finally, cytokine receptors were exploited recently as LTRs for the development of fully genetically encoded degraders, which are relatively straightforward to prepare than LYTACs employing antibody conjugates. Well’s lab developed recombinant cytokine-antibody fusion proteins as cytokine receptor-targeting chimeras (KineTACs)[16] for the degradation of extracellular secreted and membrane proteins. KineTACs are consisted of a cytokine arm and a target-binding arm, which can induce protein internalization and lysosomal delivery by forming a ternary complex with the extracellular POI and the cytokine receptor (Figure 2). The versatility and generalizability of KineTACs are demonstrated by fusing C-X-C (cysteine-any amino acid-cysteine) motif of chemokine ligand (CXCL)12, a ligand of receptor CXCR7, to antibodies that can bind to various POIs to degrade membrane proteins such as PD-L1, HER2, EGFR, and PD-1, and secreted proteins such as VEGF and TNF-α. Other cytokines such as CXCL11, viral macrophage inflammatory protein-II (vMIP-II) and interleukin-2 (IL-2), can also be employed for the development of KineTACs, which may provide a broad range of tissue-specific degraders. In addition to the choice of cytokine/cytokine receptor pair, they showed that binding affinity, the epitope of antibodies and the relative expression level of receptors were also important factors of the degradation efficiency.
Figure 2. Schematic illustration of KineTAC mediated degradation.
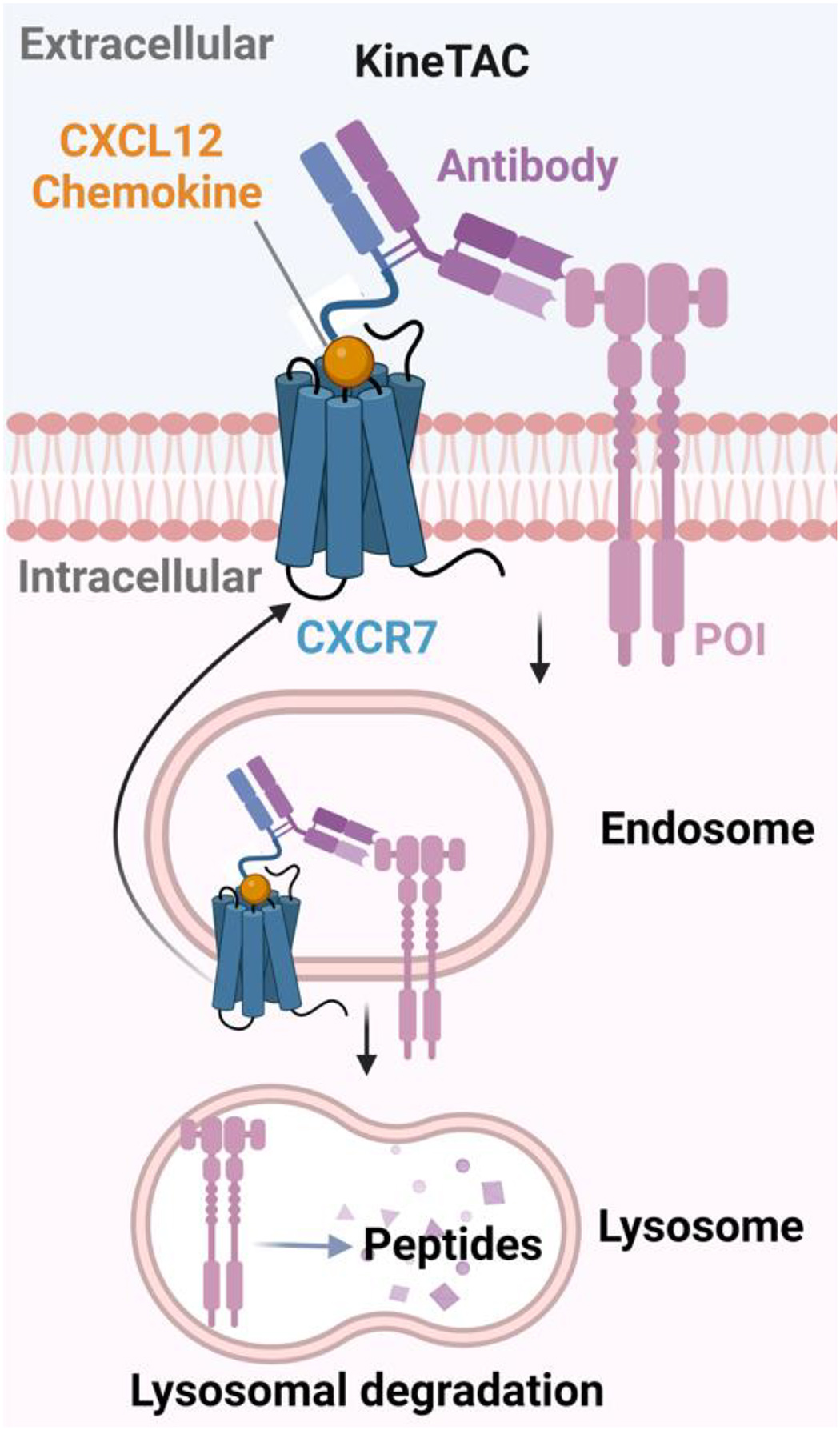
The KineTACs are fully genetically encoded bispecific antibodies with one arm being cytokine that can bind to the cytokine receptor and the other arm binding to the POI. The ligand-receptor interaction promotes the endocytosis pathway and subsequently the POI is degraded in the lysosome. (Created with BioRender.com)
Degradation of membrane proteins by recruiting intracellular lysosome sorting sequence recognition proteins
GlueTACs[17] were developed as cell surface LTR-independent degraders, which may work for cells that do not express suitable LTRs. They are composed of a nanobody that can covalently bind to POI, a cationic cell-penetrating peptide (CPP) with nine arginine residues, and a previously reported lysosome-sorting sequence (LSS) NPGY[54]. Nanobodies are smaller than antibodies and they often have lower binding affinity. A proximity reactive uncanonical amino acid was introduced to site-specifically react with PD-L1. In this way, the nanobody can covalently bind to cell-surface proteins such as PD-L1 to form a stable complex. A cationic CPP and LSS are incorporated into the GlueTAC to promote the internalization and delivery of the protein target to lysosome, respectively (Figure 3). The GlueTAC strategy relies on the cationic CPP instead of cell surface LTR-mediated endocytosis for efficient internalization. However, the cationic CPP lacks specificity in cellular uptake and may lead to on-target off-tissue side effects. In addition, cationic CPP may lead to membrane disruption and cause toxicity. Various methods are being investigated to address the toxicity issues associated with cationic CPPs while preserving their cellular uptake capabilities.
Figure 3. Schematic illustration of GlueTAC-mediated degradation.
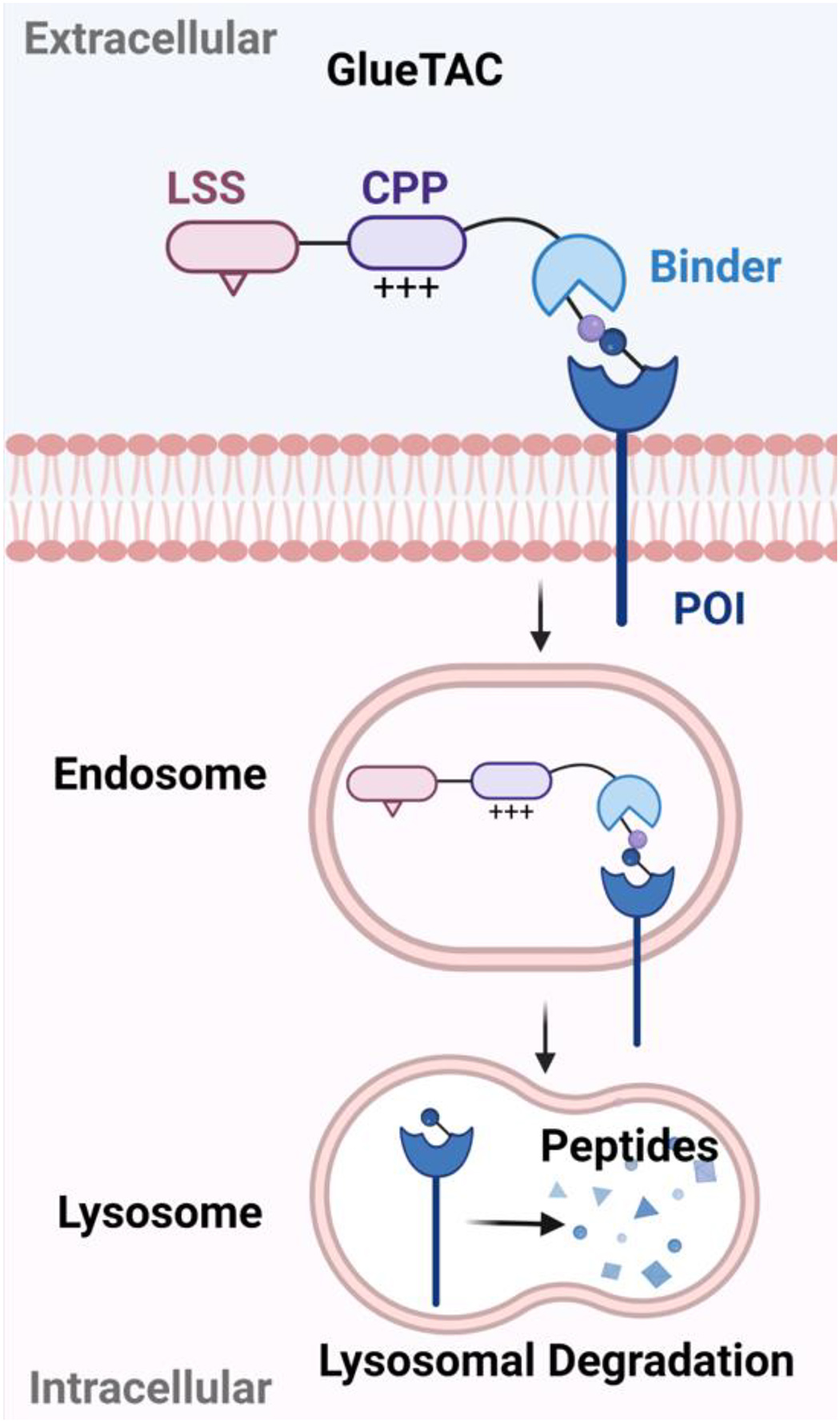
GlueTACs are composed of covalent nanobody binder of the POI, cationic cell-penetrating peptides (CPP) and lysosome sorting sequence (LSS). The cationic CPP and LSS promote the internalization and delivery of the POI to lysosome for degradation. (Created with BioRender.com)
Recently, a similar technology, termed as SignalTACs[21], was reported by employing a LSS (SFHDDSDEDLLHI) that is more effective than NPGY. The SignalTACs are constructed by genetically fusing the above LSS and a cationic CPP that is composed of four arginine and one lysine residues to antibodies of the target protein. Membrane proteins such as EGFR, HER2, PD-L1, CD20 and CD71 were degraded effectively using this technology.
Degradation of membrane proteins by recruiting membrane-bound E3 ligases
All previously discussed degraders utilized cell surface LTRs or LSS for lysosome targeting. Alternative mechanisms may offer more opportunities for developing tissue-specific degraders. The Well’s lab first utilized the membrane-associated E3 ligase, ring finger protein 43 (RNF43), to induce the degradation of the cell-surface proteins. They developed fully recombinant bispecific antibodies termed antibody-based PROTACs or AbTACs that can recruit membrane-bound E3 ligase RNF43 via its N-terminal glycprotein D (gD) for the degradation of cell-surface protein PD-L1[18]. AbTACs represent a novel strategy by inducing the proximity outside the cells using designed biologics to promote the ubiquitination reaction and subsequent degradation inside the cells. Interestingly, the degradation occurred mainly in lysosome instead of proteasome. Subsequently, the Well’s lab explored various factors that can affect degradation efficiency including epitope, affinity, orientation, and valency. Higher expression level of E3 ligase facilitates the AbTAC-induced degradation. The epitopes for both E3 ligase and POI play an important role in determining degradation efficiency of AbTAC. The affinity between AbTAC and E3 ligase or POI, while less sensitive than the epitope, also contributes to the degradation efficiency, especially for the binding between AbTAC and the POI. A threshold exists, meaning only moderate binding affinity is required to reach Dmax. Moreover, dual-binding IgG increases the potency of target protein degradation and the spacing of the binding arm and the format of the antibody affects the degradation efficiency as well[57].
Among more than 600 E3 ubiquitin ligase complexes, there are many membrane-associated E3 ligases[58]. Recently, Genentech reported their proteolysis-targeting antibody (PROTAB) platform by employing several membrane E3 ligases to degrade a series of membrane targets[19]. The MoA of PROTAB is the same as AbTAC. The scope of the PROTAB platform was demonstrated by the degradation of insulin-like growth factor 1 receptor (IGF1R), HER2, PD-L1 and frizzled class receptor 5 (FZD5). The initial membrane E3 ligases employed in the PROTAB platform are RNF43 or zinc- and ring finger 3 (ZNRF3), two closely related negative regulators of Wnt signaling pathway. Moreover, they showed that the degradation induced by ZNRF3 can be specific to colorectal cancer[19]. To further expand the scope of PROTAB, they identified 38 putative membrane E3 ligases defined by the presence of a signal peptide, transmembrane domains and prediction or reports from literature. Many of them are tissue-specific. They further demonstrated that some of these cell-surface E3 ligases (e.g. RNF128, RNF130, RNF133, RNF149, and RNF150) could be recruited by the corresponding bispecific antibodies to degrade therapeutically relevant membrane targets such as FZD5, HER2 and PD-L1[19]. Interestingly, they found that both proteasome and lysosome are involved in the PROTAB-mediated degradation. Further exploration showed that antibody epitope, format and affinity could all impact the efficiency of degradation[19].
Degraders with lower molecular weight may provide better tissue penetration than the large bispecific antibodies used for AbTACs and PROTABs. Recently, the Niehrs lab reported the development of bispecific WNT- and BMP-signaling-disabled R-spondin (RSPO) chimera (ROTACs), which are smaller than antibodies, for the degradation of membrane proteins[20]. RSPOs belong to a family of secreted stem cell growth factors and they are ligands for two cell-surface E3 ubiquitin ligases, RNF43 and its homolog ZNRF3. ROTACs are composed of the RSPO2 furin domains, which is known for its specific recognition of ZNRF3 and RNF43, as well as a high-affinity ligand of POI. They demonstrated the utility of ROTACs for the lysosomal degradation of PD-L1 in three melanoma cell lines. Mutations introduced to RSPO prevent its activities on BMP inhibition and Wnt activation, ensuring PD-L1 degradation caused by ROTACs are Wnt and BMP signaling independent. Although ROTACs are much smaller than AbTACs and PROTABs, which is advantageous for penetration and delivery, it has a shorter serum half-life and is limited to specific cell lines expressing ZNRF3 or RNF43.
Concluding Remarks and Future Perspectives
We categorized the approaches for the degradation of extracellular membrane and secreted proteins by the effectors employed in the process, 1) LTRs such as CI-M6PR, ASGPR, MGL1, integrins, SR, and cytokine receptors; 2) LTR-independent mechanisms such as LSS binding proteins and membrane E3 ligases; 3) PROTACs that bind to the intracellular domains of membrane proteins and recruit cytosolic E3 ligases. Within each approach, different types of binders of the LTR and POI have been explored such as antibodies and small molecules. Antibodies have the advantages of high specificity, low off-target toxicity, and long circulation time, while small molecules have simpler and well-defined structures, better tissue penetration, and are non-immunogenic[59]. Most importantly, small molecules have the potential to be orally available. However, not all therapeutically relevant proteins have well-defined binding sites for small molecules. For secreted proteins, either ASGPR that is expressed on liver or ubiquitously expressed LTRs such as CI-M6PR can be used. The choice of degradation approaches for membrane proteins is largely dependent on the tissue specificity of the LTR. The LTRs used to date are all receptors previously employed for drug delivery. We anticipate that additional receptors used in drug delivery[60] may also be leveraged for the degradation of extracellular membrane and secreted proteins, potentially providing more tissue-selective degraders for a diverse range of pathogenic protein targets. Furthermore, as we identify novel LTRs and their ligands in the future, exciting new possibilities for drug delivery strategies may emerge. Apart from LTRs, a few cell-surface E3 ubiquitin ligases have been harnessed for the degradation of membrane proteins. With approximately 84 estimated cell-surface E3 ligases[60] there exists immense potential for expanding our arsenal of tools to develop degraders for membrane protein targets.
The rapid advancements in the field of targeted protein degradation hold tremendous promise for revolutionizing drug discovery and therapeutic interventions. As we gain a deeper understanding of the intricacies of cellular pathways and protein regulation, the potential applications of LYTACs and other innovative strategies will continue to expand. Future research efforts will focus on refining the design and selectivity of degraders, enhancing their delivery efficiency and drug-like properties especially oral bioavailability, and exploring novel ligands and receptors for more precise and potent protein degradation. Additionally, exploring the combination of degradation modalities with other therapeutic strategies such as irradiation and understanding of their detailed mechanisms will be pivotal in advancing this field[61]. The translation of the emerging TPD technologies from preclinical studies to clinical applications is the ultimate testimony for the success. With the collective efforts of researchers, the development of effective and safe targeted protein degraders has the potential to significantly impact the treatment landscape for a wide range of diseases, offering novel therapeutic options and improved outcomes for patients with unmet medical needs.
Figure 4. Schematic illustration of PROTAC, PROTAB/AbTAC, and ROTAC-mediated degradation.
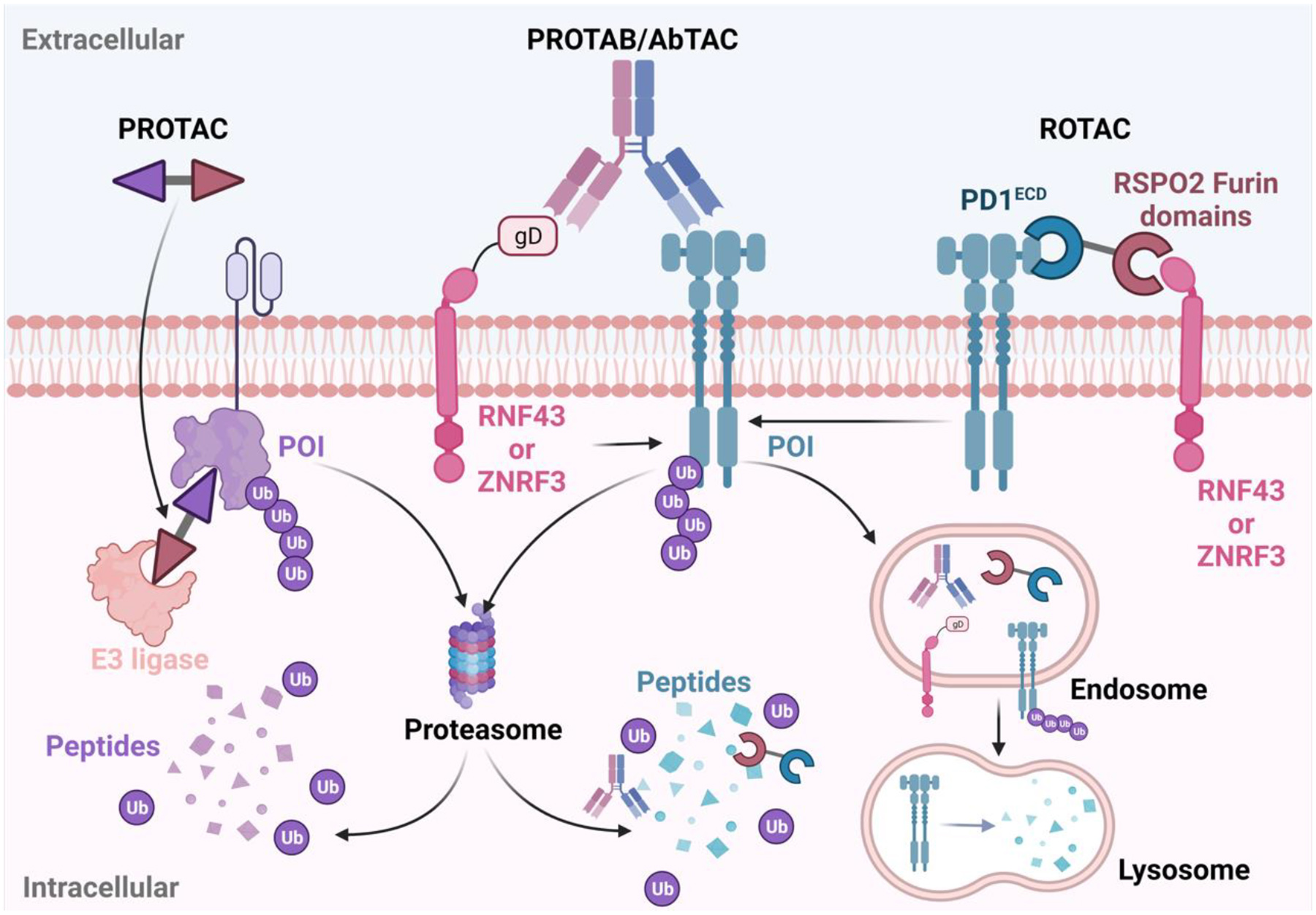
PROTACs for membrane protein of interest (POI) are comprised of a binder of the intracellular domain of the POI and a binder of the E3 ubiquitin ligase, and a linker between the two binders. PROTAB and AbTAC are fully genetically encoded bispecific antibodies that can bind to the membrane POI and a membrane E3 ubiquitin ligase (RNF43 or ZNRF3). ROTAC uses mutated R-spondin furin domain as the binder of RNF43 or ZNRF3. The formation of POI-degrader-E3 complex ternary complex promotes the ubiquitination of the POI and subsequent proteasomal or lysosomal degradation. (Created with BioRender.com)
Outstanding questions.
What factors should be considered when choosing suitable degradation approaches for the extracellular proteins?
What are the potential future directions for the degradation of extracellular proteins?
What elements are used by the degraders to route extracellular proteins into cells own intrinsic degradation pathway?
What are the factors contributing to the degrader efficiency and need to be considered when designing the degrader?
What are the advantages and limitations for small molecule and antibody-based degrader?
Highlights.
Targeted protein degradation (TPD), represented by proteolysis targeting chimeras (PROTACs) and lysosome targeting chimeras (LYTACs), is a major breakthrough in drug discovery.
LYTACs, designed to degrade extracellular proteins, provide a solution to the significant limitation of PROTACs, which are restricted to degrading intracellular proteins.
New strategies that use various effectors to degrade extracellular proteins are promising, potentially signaling a new era in targeted protein degradation (TPD).
A comprehensive understanding of the new approaches for the degradation of extracellular proteins could accelerate the advancements in clinical applications.
Acknowledgement
We thank the financial support from the University of Wisconsin- Madison Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation (WARF) through a UW2020 award and National Institutes of Health under the award number R35 GM148266. Y.Z. was supported by Novo Nordisk through the NovoSTAR program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
- Autophagy
Autophagy is an evolutionarily conserved process wherein the cells transports the unwanted components to lysosomes for degradation and recycling. It is responsible for disposing of damaged organelles, misfolded proteins, and other macromolecules through the lysosomal pathway.
- Cytokine
Cytokines are small proteins involved in cell communication within the immune system and other biological processes. They are produced by various cells, including immune cells and non-immune cells.
- Endocytosis
Endocytosis involves the internalization of entities ranging from large particles like bacteria to the uptake of fluids or macromolecules within small vesicles.
- Hook effect
It is a phenomenon often observed for both PROTAC and LYTAC bifunctional degraders, where the degradation effect decreases with increasing concentration of the degrader past a certain peak point. It is due to the formation of more binary complexes and less productive ternary complexes at high concentration of degraders.
- Lysosome
Lysosomes are membrane-bound organelles found in eukaryotic cells. They contain a diverse array of hydrolytic enzymes, including proteases, nucleases, lipases, and glycosidases. It degrades many types of proteins, protein aggregates, and damaged organelles.
- PROTACs
PROTACs are heterobifunctional molecules comprising of a binder of the targeted protein and a binder of the E3 ligase, and a linker between the two binders.
- Ternary complex
The simultaneous binding of a target protein and an E3 ligase mediated by PROTAC promotes the formation of ternary complexes known as Target-PROTAC-E3 Ligase complexes. Similar types of ternary complexes can be formed by the simultaneous binding of a target protein and a lysosome targeting receptor mediated by the lysosome targeting degrader.
- Ubiquitin-proteasome system
It is a tightly regulated mechanism responsible for intracellular protein degradation and turnover. It involves the activation of ubiquitin by the ubiquitin activating enzyme (E1), followed by its transfer to ubiquitin conjugases (E2). Ultimately, the substrate is conjugated to ubiquitin by a ubiquitin ligase (E3). The polyubiquitinated protein is then recognized by the 26S proteasome, leading to its final degradation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
W. Tang is the cofounder and shareholder of Chimergen Therapeutic Inc. Y. Zhou, Y. Zhao and W. Tang are inventors of patents related to this manuscript.
References
- 1.Ciechanover A and Schwartz AL (1998) The ubiquitin-proteasome pathway: The complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. U. S. A 95, 2727–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bedford L et al. (2011) Ubiquitin-like protein conjugation and the ubiquitin–proteasome system as drug targets. Nat. Rev. Drug Discov 10, 29–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamb CA et al. (2013) The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol 14, 759–774 [DOI] [PubMed] [Google Scholar]
- 4.Yim WW-Y and Mizushima N (2020) Lysosome biology in autophagy. Cell Discov. 6, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai AC and Crews CM (2017) Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov 16, 101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerry CJ and Schreiber SL (2020) Unifying principles of bifunctional, proximity-inducing small molecules. Nat. Chem. Biol 16, 369–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deshaies RJ (2020) Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 580, 329–338 [DOI] [PubMed] [Google Scholar]
- 8.Uhlén M et al. (2015) Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 9.Ward ES and Ober RJ (2018) Targeting FcRn to Generate Antibody-Based Therapeutics. Trends Pharmacol. Sci 39, 892–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klaus T and Deshmukh S (2021) pH-responsive antibodies for therapeutic applications. J. Biomed. Sci 28, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn G et al. (2021) Degradation from the outside in: Targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell Chem. Biol 28, 1072–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banik SM et al. (2020) Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caianiello DF et al. (2021) Bifunctional small molecules that mediate the degradation of extracellular proteins. Nat. Chem. Biol 17, 947–953 [DOI] [PubMed] [Google Scholar]
- 14.Zhu C et al. (2023) Dendronized DNA Chimeras Harness Scavenger Receptors To Degrade Cell Membrane Proteins. Angew. Chem. Int. Ed 62, e202300694. [DOI] [PubMed] [Google Scholar]
- 15.Zheng J et al. (2022) Bifunctional Compounds as Molecular Degraders for Integrin-Facilitated Targeted Protein Degradation. J. Am. Chem. Soc 144, 21831–21836 [DOI] [PubMed] [Google Scholar]
- 16.Pance K et al. (2023) Modular cytokine receptor-targeting chimeras for targeted degradation of cell surface and extracellular proteins. Nat. Biotechnol 41, 273–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H et al. (2021) Covalently Engineered Nanobody Chimeras for Targeted Membrane Protein Degradation. J. Am. Chem. Soc 143, 16377–16382 [DOI] [PubMed] [Google Scholar]
- 18.Cotton AD et al. (2021) Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc 143, 593–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marei H et al. (2022) Antibody targeting of E3 ubiquitin ligases for receptor degradation. Nature 610, 182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun R et al. (2023) ROTACs leverage signaling-incompetent R-spondin for targeted protein degradation. Cell Chem. Biol DOI: 10.1016/j.chembiol.2023.05.010 [DOI] [PubMed] [Google Scholar]
- 21.Yu J et al. (2023) Harnessing the Lysosomal Sorting Signals of the Cation-Independent Mannose-6-Phosphate Receptor for Targeted Degradation of Membrane Proteins. J. Am. Chem. Soc DOI: 10.1021/jacs.3c07687 [DOI] [PubMed] [Google Scholar]
- 22.Gary-Bobo M et al. (2007) Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr. Med. Chem 14, 2945–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimmermann TS et al. (2017) Clinical Proof of Concept for a Novel Hepatocyte-Targeting GalNAc-siRNA Conjugate. Mol. Ther 25, 71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuda S et al. (2015) siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem. Biol 10, 1181–1187 [DOI] [PubMed] [Google Scholar]
- 25.Kanasty R et al. (2013) Delivery materials for siRNA therapeutics. Nat. Mater 12, 967–977 [DOI] [PubMed] [Google Scholar]
- 26.Dalle Vedove E et al. (2018) Mannose and Mannose-6-Phosphate Receptor–Targeted Drug Delivery Systems and Their Application in Cancer Therapy. Adv. Healthc. Mater 7, 1701398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crucianelli E et al. (2014) Liposomes containing mannose-6-phosphate-cholesteryl conjugates for lysosome-specific delivery. RSC Adv. 4, 58204–58207 [Google Scholar]
- 28.Das S et al. (2016) Controlled Synthesis of End-Functionalized Mannose-6-phosphate Glycopolypeptides for Lysosome Targeting. ACS Macro Lett. 5, 809–813 [DOI] [PubMed] [Google Scholar]
- 29.Hyun JY et al. (2018) A Glycoengineered Enzyme with Multiple Mannose-6-Phosphates Is Internalized into Diseased Cells to Restore Its Activity in Lysosomes. Cell Chem. Biol 25, 1255–1267.e8 [DOI] [PubMed] [Google Scholar]
- 30.Stevens CM et al. (2023) Development of Oligomeric Mannose-6-phosphonate Conjugates for Targeted Protein Degradation. ACS Med. Chem. Lett DOI: 10.1021/acsmedchemlett.2c00479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L-X et al. (2019) Glycoengineering of Antibodies for Modulating Functions. Annu. Rev. Biochem 88, 433–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boune S et al. (2020) Principles of N-Linked Glycosylation Variations of IgG-Based Therapeutics: Pharmacokinetic and Functional Considerations. Antibodies 9, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang W et al. (2012) Chemoenzymatic Glycoengineering of Intact IgG Antibodies for Gain of Functions. J. Am. Chem. Soc 134, 12308–12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li T et al. (2016) Glycosynthase Mutants of Endoglycosidase S2 Show Potent Transglycosylation Activity and Remarkably Relaxed Substrate Specificity for Antibody Glycosylation Remodeling *. J. Biol. Chem 291, 16508–16518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang F et al. (2017) Chemoenzymatic synthesis of glycoengineered IgG antibodies and glycosite-specific antibody–drug conjugates. Nat. Protoc 12, 1702–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X et al. (2022) Site-Specific Chemoenzymatic Conjugation of High-Affinity M6P Glycan Ligands to Antibodies for Targeted Protein Degradation. ACS Chem. Biol 17, 3013–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keefe AD et al. (2010) Aptamers as therapeutics. Nat. Rev. Drug Discov 9, 537–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miao Y et al. (2021) Bispecific Aptamer Chimeras Enable Targeted Protein Degradation on Cell Membranes. Angew. Chem. Int. Ed 60, 11267–11271 [DOI] [PubMed] [Google Scholar]
- 39.Hamada K et al. (2023) Development of a bispecific DNA-aptamer-based lysosome-targeting chimera for HER2 protein degradation. Cell Rep. Phys. Sci 4, 101296 [Google Scholar]
- 40.Spiess M (1990) The asialoglycoprotein receptor: a model for endocytic transport receptors. Biochemistry 29, 10009–10018 [DOI] [PubMed] [Google Scholar]
- 41.Huang Y (2017) Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Mol. Ther. - Nucleic Acids 6, 116–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glazier DA et al. (2020) Chemical Synthesis and Biological Application of Modified Oligonucleotides. Bioconjug. Chem 31, 1213–1233 [DOI] [PubMed] [Google Scholar]
- 43.Ahn G et al. (2021) LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol 17, 937–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Y et al. (2021) Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Cent. Sci 7, 499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Donahue TC et al. (2023) Synthetic Site-Specific Antibody–Ligand Conjugates Promote Asialoglycoprotein Receptor-Mediated Degradation of Extracellular Human PCSK9. ACS Chem. Biol 18, 1611–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Douglass EF Jr. et al. (2013) A Comprehensive Mathematical Model for Three-Body Binding Equilibria. J. Am. Chem. Soc 135, 6092–6099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Y et al. (2023) Aptamer-LYTACs for Targeted Degradation of Extracellular and Membrane Proteins. Angew. Chem. Int. Ed 62, e202218106 [DOI] [PubMed] [Google Scholar]
- 48.Wang K et al. Nano-LYTACs for Degradation of Membrane Proteins and Inhibition of CD24/Siglec-10 Signaling Pathway. Adv. Sci n/a, 2300288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bagdanoff JT et al. (2023) Clearance of plasma PCSK9 via the asialoglycoprotein receptor mediated by heterobifunctional ligands. Cell Chem. Biol 30, 97–109.e9 [DOI] [PubMed] [Google Scholar]
- 50.Liu Z et al. (2023) Enantioselective Degrader for Elimination of Extracellular Aggregation-Prone Proteins hIAPP Associated with Type 2 Diabetes. ACS Nano DOI: 10.1021/acsnano.2c11476 [DOI] [PubMed] [Google Scholar]
- 51.Faulkner AD et al. (2014) Asymmetric triplex metallohelices with high and selective activity against cancer cells. Nat. Chem 6, 797–803 [DOI] [PubMed] [Google Scholar]
- 52.Singh SK et al. (2009) Characterization of murine MGL1 and MGL2 C-type lectins: Distinct glycan specificities and tumor binding properties. Mol. Immunol 46, 1240–1249 [DOI] [PubMed] [Google Scholar]
- 53.Kechagia JZ et al. (2019) Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol 20, 457–473 [DOI] [PubMed] [Google Scholar]
- 54.Han Y et al. (2020) Protein labeling approach to improve lysosomal targeting and efficacy of antibody–drug conjugates. Org. Biomol. Chem 18, 3229–3233 [DOI] [PubMed] [Google Scholar]
- 55.Alquraini A and El Khoury J (2020) Scavenger receptors. Curr. Biol 30, R790–R795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kakizawa Y and Kataoka K (2002) Block copolymer micelles for delivery of gene and related compounds. Adv. Drug Deliv. Rev 54, 203–222 [DOI] [PubMed] [Google Scholar]
- 57.Gramespacher JA et al. (2022) Roadmap for Optimizing and Broadening Antibody-Based PROTACs for Degradation of Cell Surface Proteins. ACS Chem. Biol 17, 1259–1268 [DOI] [PubMed] [Google Scholar]
- 58.Chen X et al. (2022) The Role of Membrane-Associated E3 Ubiquitin Ligases in Cancer. Front. Pharmacol 13, 928794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Imai K and Takaoka A (2006) Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 6, 714–727 [DOI] [PubMed] [Google Scholar]
- 60.Srinivasarao M and Low PS (2017) Ligand-Targeted Drug Delivery. Chem. Rev 117, 12133–12164 [DOI] [PubMed] [Google Scholar]
- 61.Dale B et al. (2021) Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 21, 638–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Békés M et al. (2022) PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov 21, 181–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruffilli C et al. (2022) Proteolysis Targeting Chimeras (PROTACs): A Perspective on Integral Membrane Protein Degradation. ACS Pharmacol. Transl. Sci 5, 849–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang CH et al. (2018) Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem. Biophys. Res. Commun 505, 542–547 [DOI] [PubMed] [Google Scholar]
- 65.Sun N et al. (2020) Development of a Brigatinib degrader (SIAIS117) as a potential treatment for ALK positive cancer resistance. Eur. J. Med. Chem 193, 112190. [DOI] [PubMed] [Google Scholar]
- 66.Ren C et al. (2021) Structure-based discovery of SIAIS001 as an oral bioavailability ALK degrader constructed from Alectinib. Eur. J. Med. Chem 217, 113335. [DOI] [PubMed] [Google Scholar]
- 67.Gao Y et al. (2023) Catalytic Degraders Effectively Address Kinase Site Mutations in EML4-ALK Oncogenic Fusions. J. Med. Chem 66, 5524–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie S et al. (2023) Discovery of Norbornene as a Novel Hydrophobic Tag Applied in Protein Degradation. Angew. Chem. Int. Ed 62, e202217246 [DOI] [PubMed] [Google Scholar]
- 69.Yan G et al. (2021) Discovery of a PROTAC targeting ALK with in vivo activity. Eur. J. Med. Chem 212, 113150. [DOI] [PubMed] [Google Scholar]
- 70.Zhang C et al. (2018) Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur. J. Med. Chem 151, 304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie S et al. (2021) Development of Alectinib-Based PROTACs as Novel Potent Degraders of Anaplastic Lymphoma Kinase (ALK). J. Med. Chem 64, 9120–9140 [DOI] [PubMed] [Google Scholar]
- 72.Ren C et al. (2021) Discovery of a Brigatinib Degrader SIAIS164018 with Destroying Metastasis-Related Oncoproteins and a Reshuffling Kinome Profile. J. Med. Chem 64, 9152–9165 [DOI] [PubMed] [Google Scholar]
- 73.Burslem GM et al. (2018) The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol 25, 67–77.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qu X et al. (2021) Effective degradation of EGFRL858R+T790M mutant proteins by CRBN-based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur. J. Med. Chem 218, 113328. [DOI] [PubMed] [Google Scholar]
- 75.Zhang X et al. (2020) Design and synthesis of selective degraders of EGFRL858R/T790M mutant. Eur. J. Med. Chem 192, 112199. [DOI] [PubMed] [Google Scholar]
- 76.Zhao H-Y et al. (2020) Discovery of potent small molecule PROTACs targeting mutant EGFR. Eur. J. Med. Chem 208, 112781. [DOI] [PubMed] [Google Scholar]
- 77.Zhang H et al. (2020) Discovery of potent epidermal growth factor receptor (EGFR) degraders by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem 189, 112061. [DOI] [PubMed] [Google Scholar]
- 78.Khattab RR et al. (2021) Click chemistry based synthesis, cytotoxic activity and molecular docking of novel triazole-thienopyrimidine hybrid glycosides targeting EGFR. J. Enzyme Inhib. Med. Chem 36, 504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang H et al. (2022) Design, Synthesis, and Biological Evaluation of Novel EGFR PROTACs Targeting Del19/T790M/C797S Mutation. ACS Med. Chem. Lett 13, 278–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang W et al. (2022) Discovery of highly potent and selective CRBN-recruiting EGFRL858R/T790M degraders in vivo. Eur. J. Med. Chem 238, 114509. [DOI] [PubMed] [Google Scholar]
- 81.Chang MT et al. (2022) Identifying transcriptional programs underlying cancer drug response with TraCe-seq. Nat. Biotechnol 40, 86–93 [DOI] [PubMed] [Google Scholar]
- 82.Li Q et al. (2022) Design and synthesis of proteolysis targeting chimeras (PROTACs) as an EGFR degrader based on CO-1686. Eur. J. Med. Chem 238, 114455. [DOI] [PubMed] [Google Scholar]
- 83.Shi S et al. (2022) Rational Design for Nitroreductase (NTR)-Responsive Proteolysis Targeting Chimeras (PROTACs) Selectively Targeting Tumor Tissues. J. Med. Chem 65, 5057–5071 [DOI] [PubMed] [Google Scholar]
- 84.O. Aboelez M et al. (2022) Design, synthesis, and molecular docking studies of novel pomalidomide-based PROTACs as potential anti-cancer agents targeting EGFRWT and EGFRT790M. J. Enzyme Inhib. Med. Chem 37, 1196–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rosenberg SC et al. (2023) Ternary complex dissociation kinetics contribute to mutant-selective EGFR degradation. Cell Chem. Biol 30, 175–187.e15 [DOI] [PubMed] [Google Scholar]
- 86.Cheng M et al. (2020) Discovery of Potent and Selective Epidermal Growth Factor Receptor (EGFR) Bifunctional Small-Molecule Degraders. J. Med. Chem 63, 1216–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Du G et al. (2021) Discovery of a Potent Degrader for Fibroblast Growth Factor Receptor 1/2. Angew. Chem. Int. Ed 60, 15905–15911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shibata N et al. (2022) Development of a degrader against oncogenic fusion protein FGFR3-TACC3. Bioorg. Med. Chem. Lett 60, 128584. [DOI] [PubMed] [Google Scholar]
- 89.Guo L et al. (2022) Development of selective FGFR1 degraders using a Rapid synthesis of proteolysis targeting Chimera (Rapid-TAC) platform. Bioorg. Med. Chem. Lett 75, 128982. [DOI] [PubMed] [Google Scholar]
- 90.Řezníčková E et al. (2022) Modulation of FLT3-ITD and CDK9 in acute myeloid leukaemia cells by novel proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem 243, 114792. [DOI] [PubMed] [Google Scholar]
- 91.Cao S et al. (2021) Proteolysis-Targeting Chimera (PROTAC) Modification of Dovitinib Enhances the Antiproliferative Effect against FLT3-ITD-Positive Acute Myeloid Leukemia Cells. J. Med. Chem 64, 16497–16511 [DOI] [PubMed] [Google Scholar]
- 92.Burslem GM et al. (2018) Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc 140, 16428–16432 [DOI] [PubMed] [Google Scholar]
- 93.Huber ME et al. (2022) A Chemical Biology Toolbox Targeting the Intracellular Binding Site of CCR9: Fluorescent Ligands, New Drug Leads and PROTACs. Angew. Chem. Int. Ed 61, e202116782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li Z et al. (2020) First small-molecule PROTACs for G protein-coupled receptors: inducing α1A-adrenergic receptor degradation. Acta Pharm. Sin. B 10, 1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y et al. (2021) In vitro and in vivo degradation of programmed cell death ligand 1 (PD-L1) by a proteolysis targeting chimera (PROTAC). Bioorganic Chem. 111, 104833. [DOI] [PubMed] [Google Scholar]
- 96.Liu Y et al. (2023) Design, synthesis, and evaluation of PD-L1 degraders to enhance T cell killing activity against melanoma. Chin. Chem. Lett 34, 107762 [Google Scholar]
- 97.Cheng B et al. (2020) Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur. J. Med. Chem 199, 112377. [DOI] [PubMed] [Google Scholar]