

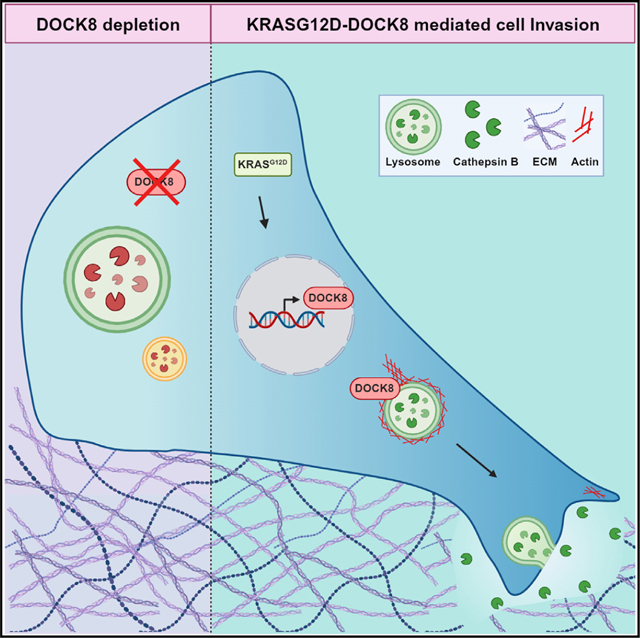
SUMMARY
Amplified lysosome activity is a hallmark of pancreatic ductal adenocarcinoma (PDAC) orchestrated by oncogenic KRAS that mediates tumor growth and metastasis, though the mechanisms underlying this phenomenon remain unclear. Using comparative proteomics, we found that oncogenic KRAS significantly enriches levels of the guanine nucleotide exchange factor (GEF) dedicator of cytokinesis 8 (DOCK8) on lysosomes. Surprisingly, DOCK8 is aberrantly expressed in a subset of PDAC, where it promotes cell invasion in vitro and in vivo. DOCK8 associates with lysosomes and regulates lysosomal morphology and motility, with loss of DOCK8 leading to increased lysosome size. DOCK8 promotes actin polymerization at the surface of lysosomes while also increasing the proteolytic activity of the lysosomal protease cathepsin B. Critically, depletion of DOCK8 significantly reduces cathepsin-dependent extracellular matrix degradation and impairs the invasive capacity of PDAC cells. These findings implicate ectopic expression of DOCK8 as a key driver of KRAS-driven lysosomal regulation and invasion in pancreatic cancer cells.
In brief
Gutierrez Ruiz et al. analyzed the lysosomal proteome of pancreatic cancer cells expressing oncogenic KRAS. They identified the GEF DOCK8 as being aberrantly expressed in pancreatic cancer cells, where it controls lysosome morphology. DOCK8 regulates lysosomal actin, which impacts trafficking and secretion of lysosomal cathepsins to promote tumor cell invasion.
Graphical Abstract
INTRODUCTION
Lysosomes are acidic, degrative organelles that are implicated in nearly every hallmark of cancer.1–5 Pancreatic ductal adenocarcinoma (PDAC), the third leading cause of cancer death in the US, exhibits enhanced lysosome biogenesis and activity, which contributes to tumor progression by multiple mechanisms including amino acid homeostasis, survival signaling, and immune evasion.2,6–10 Lysosomes also contribute to invasion, such as through the degradation of E-cadherin and the secretion of lysosomal hydrolases for invasive degradation and remodeling of the extracellular matrix (ECM).7,11,12 In particular, the lysosomal protease cathepsin B is upregulated in PDAC and enhances pancreatic cancer progression and metastasis.8,11 However, the mechanisms regulating these tumor-promoting actions are not well defined.
Most PDAC tumors are driven by mutations in the oncogene KRAS,13–15 which amplify the lysosome-targeted nutrient scavenging processes of macropinocytosis and autophagy.16–18 The molecular mechanisms by which oncogenic KRAS regulates lysosomal action to promote tumor invasion and metastasis remain unclear. KRAS activation can promote invasive ECM degradation in part through actin-based invadopodia and matrix metalloproteinase secretion.19–23 Invadopodia are regulated by the small GTPase Cdc42, which is activated by guanine nucleotide exchange factors (GEFs).24,25 Aberrant activation of GEFs in PDAC increases Rac1/Cdc42-mediated invasive migration and potentiates metastasis.26–28 However, pro-invasive functions for GEFs beyond leading-edge actin dynamics and invadopodia remain elusive.
We hypothesized that oncogenic KRAS regulates lysosome function to support PDAC tumor progression and metastasis. In this study, we investigated how oncogenic KRAS regulates the lysosomal proteome using mass spectrometry-based proteomic profiling of lysosomes isolated from PDAC cells. Surprisingly, we identified that a Rac/Cdc42 GEF, dedicator of cytokinesis 8 (DOCK8), is enriched on lysosomes isolated from oncogenic KRAS-expressing cells. DOCK8 is primarily expressed in immune cells, where it regulates immune synapse formation29,30 and immune cell migration via Rac1 and Cdc42.31 Here, we identify that DOCK8 is ectopically expressed in a subset of PDACs, and this aberrant expression promotes tumor growth and invasion. Surprisingly, this ectopically expressed GEF localizes in part to tumor cell lysosomes, where it plays a central role in metastatic invasion by controlling both actin-based lysosome morphology and cathepsin B-dependent ECM degradation. Together, these findings implicate DOCK8 as a mediator of KRAS-driven lysosomal regulation and metastatic invasion in pancreatic tumor cells.
RESULTS
Lysosome proteomic profiling identifies enrichment of DOCK8 at lysosomes in pancreatic cancer cells
Oncogenic mutations in KRAS are key drivers of PDAC. As lysosome biogenesis and activity are amplified in PDAC, we sought to identify changes to the lysosomal proteome regulated by KRAS. To this end, we performed comparative proteomics on isolated lysosomes from cells expressing wild-type (WT) or oncogenic KRAS using a PDAC cell line derived from a mouse with doxycycline-inducible oncogenic KRAS G12D (iK-RAS).13,32 The lysosomal transmembrane protein TMEM192–3xHA was stably expressed in iKRAS cells to label lysosomes (Figures S1A and S1B), and lysosomes were isolated by immunoprecipitation using anti-HA magnetic beads33 from cells treated with or without doxycycline, thus expressing KRASG12D or KRASWT, respectively. Lysosome integrity and purity were evaluated by immunoblotting of lysosomal (TMEM192-HA, LAMP2A, cathepsin B) and non-lysosomal markers (COXIV, GM130), as well as lysosome-associated proteins (mTOR, Rab7) (Figure S1C). Isolated lysosomes were analyzed using mass spectrometry and comparative proteomic analysis. Overall, 52 proteins were significantly enriched at lysosomes from cells expressing oncogenic KRASG12D cells versus KRASWT, while 138 proteins were decreased (Figure 1A). Surprisingly, we identified the GEF DOCK8 as enriched by 2.4-fold on lysosomes isolated from iKRASG12D cells compared with iKRASWT cells (Figure 1A). This was unexpected, as DOCK8 is primarily expressed in the immune system, has not been reported to be expressed in PDAC, and is not known to associate with lysosomes. DOCK8 was of particular interest as it is required for interstitial migration of dendritic cells.34 In considering novel regulators of pancreatic cancer progression and metastasis, we hypothesized that DOCK8 might support the highly similar process of invasive migration by tumor cells.
Figure 1. Lysosome proteomics reveal that DOCK8 is regulated by oncogenic KRASG12D.

(A) Comparative proteomics of lysosomes isolated from iKRASG12D cells ± doxycycline (KRASG12D versus WT), showing enrichment of 57 proteins (right of blue line) and decrease of 138 proteins (left of blue line) on lysosomes from KRASG12D-expressing cells. Red point: DOCK8.
(B) DOCK8 associates with lysosomes. Immunoblotting of whole-cell lysates (WCLs) and isolated lysosomes from control iKRASG12D, or iKRASG12D-TMEM192-HA cells ± doxycycline, indicating enrichment of DOCK8 in cells expressing KRASG12D. LAMP1 and cathepsin B: lysosomal positive controls; TOM20, GM130, and GAPDH: negative controls.
(C) Quantitation of DOCK8 immunoblotting on isolated lysosomes from KRASG12D normalized to WT cells.
(D) Immunofluorescence of iKRASG12D-TMEM192-HA cells + doxycycline showing DOCK8-YFP (green) localization to TMEM192-HA (red) lysosomes.
(E) DOCK8 protein schematic, including DHR1 (green) and DHR2 (blue) domains, and a truncated mutant containing the DRH2 domain with HA tag (DOCK8-DHR2-HA).
(F and G) Immunofluorescence of iKRASG12D cells + doxycycline (F) and Panc04.03 cells (G) showing DOCK8-DHR2-HA (red) colocalization with LAMP1 (green) lysosomes.
(H) Oncogenic KRASG12D increases DOCK8 expression. Immunoblotting of DOCK8 in cell lysates from iKRASG12D cells ± doxycycline. RAW 264.7 macrophages: positive control.
(I) Quantitation of DOCK8 immunoblotting normalized to actin, compared to KRASWT cells.
(J) Relative DOCK8 mRNA levels by qRT-PCR from iKRASG12D cells ± doxycycline, normalized to RAW 264.7 macrophages.
(K) Isolated lysosomes were treated with the indicated concentrations of Proteinase K. Intraluminal proteins cathepsin B and LAMP1 (intraluminal epitope) are protected from degradation, whereas proteins at the lysosomal surface (DOCK8, mTOR, Rab7) are sensitive to digestion. Scale bars, 10 μm.
Graphs: mean ± SEM of at least three independent biological replicates. Student’s t test: *p < 0.05; **p < 0.01; ***p < 0.001; ****p ≤ 0.0001.
See also Figure S1.
DOCK8 enrichment on lysosomes was validated by immunoblotting purified lysosome fractions for DOCK8 in iKRAS cells, as well as in Panc04.03 cells (Figures 1B, 1C, and S1D). Further, while DOCK8 is primarily cytoplasmic, a YFP-tagged-DOCK8 construct could be detected at the membrane of lysosomes, defined by costaining for TMEM192-HA, in iKRAS cells (Figure 1D), as well as by LysoTracker labeling in iKRAS and Panc04.03 cells (Figures S1E and S1F). In addition, we transfected a truncated version of DOCK8, comprised of the DHR2 (GEF) domain tagged with HA, and immunofluorescence staining of LAMP1, and HA detected this mutant at the membrane of lysosomes in iKRAS and Panc04.03 cells, suggesting that this domain is sufficient for the recruitment of DOCK8 to lysosomes (Figures 1E–1G). Together, both biochemical and immunocytochemical methods indicate a surprising lysosomal association for the GEF DOCK8 in PDAC cells.
We first assessed if KRASG12D affected total DOCK8 expression. Immunoblotting revealed that DOCK8 protein levels were substantially increased by 11-fold in doxycycline-induced iK-RASG12D cells (Figures 1H and 1I). Quantitative RT-PCR indicated a 4.5-fold increase in DOCK8 transcript levels in oncogenic iKRASG12D cells (Figure 1J). As induction of KRASG12D results in increased KRAS protein levels (Figures S1G and S1H), we tested if increasing levels of WT KRAS were sufficient to elevate DOCK8 expression. However, overexpression of HA-tagged WT KRAS did not enhance expression of DOCK8 (Figures S1I and S1J). This suggests that oncogenic KRAS, and not the WT form, promotes DOCK8 expression in PDAC cells, which can enhance its association with lysosomes.
The biochemical and morphological association of DOCK8 with lysosomes described above does not distinguish if this GEF is a luminal protein, is associated with the lysosome surface, or is a cargo destined for degradation. Proteinase K digestion of isolated lysosomes was used to discriminate between lysosomal surface proteins, which would be sensitive to Proteinase K digestion, as compared with luminal proteins, which would be more protected from degradation. Lysosome-associated DOCK8 was extremely sensitive to Proteinase K, as were known lysosome interacting proteins mTOR and Rab7, indicating DOCK8 is associating with the lysosomal surface (Figure 1K). To test if DOCK8 is degraded by lysosomes, lysosome degradative activity was inhibited using bafilomycin A1, and DOCK8 protein levels were assessed by immunoblotting. Lysosome inhibition did not increase total DOCK8 levels compared to DMSO control (Figure S1K), suggesting that DOCK8 recruitment to lysosomes is not a protein clearance mechanism but rather that DOCK8 may act at the lysosomal surface.
DOCK8 is aberrantly expressed in PDAC cells and promotes tumor growth and invasion
DOCK8 signaling has been studied extensively in immune cells; however, its expression and role in other cell types remain poorly defined. We first evaluated the expression of DOCK8 in pancreatic cancers. Gene expression analysis of normal pancreas from the Genotype-Tissue Expression (GTEx) project and tumor samples from The Cancer Genome Atlas (TCGA) Program revealed that DOCK8 expression is elevated in PDAC (Figure 2A). However, as high expression of DOCK8 in infiltrating immune cells could confound these data, we analyzed DOCK8 expression using RNA sequencing (RNA-seq) data from PDAC patient-derived organoids (PDOs) and cell lines. DOCK8 was expressed in 20% of PDAC PDOs generated by both the Mayo Clinic and Crown Bioscience (Figures 2B and 2C).35 Transcript analysis from the Cancer Cell Line Encyclopedia identified that 21 out of 41 (51%) PDAC cell lines expressed DOCK8, which was validated by immunoblotting of a subset of cell lines (Figures 2D and 2E). DOCK8 protein was detected in two murine cell lines (iKRAS,13 mKPC, which is derived from the KPC genetic mouse model of pancreatic cancer (PDX-1 Cre, KrasG12D/+, p53T172H/+36) and four human cell lines (6741-P, L3.6, Panc04.03, and Capan1, consistent with transcriptomic data from CCLE) (Figure 2E). Although our data suggest that DOCK8 expression is enhanced by KRASG12D, not all cell lines with oncogenic KRAS mutations express DOCK8, suggesting additional regulatory mechanisms of DOCK8 expression. Together, these data identify aberrant expression of the GEF DOCK8 in a subset of PDAC tumor cells.
Figure 2. DOCK8 is ectopically expressed in a subset of pancreatic cancer tumors and is required for cancer cell invasion in vivo and in vitro.

(A–D) Relative DOCK8 mRNA expression in (A) normal, healthy pancreas (GTEx) and pancreatic cancer tumors (TCGA), patient-derived organoids (PDOs) generated at (B) the Mayo Clinic or (C) Crown Bioscience, and (D) pancreatic cancer cell lines (Cancer Cell Line Encyclopedia).
(E) Immunoblotting of DOCK8 in pancreatic cancer cell lines. RAW 264.7 macrophages: positive control; non-cancerous HPDE (human pancreatic ductal epithelial) cells: negative control.
(F) Immunoblotting of DOCK8 in three independent control or DOCK8 CRISPR knockout mKPC cell clones. GAPDH: loading control.
(G) Control and DOCK8 knockout average tumor weight (mg) from syngeneic orthotopic tumors in male (blue squares) and female (red triangles) mice.
(H) Representative images of control or DOCK8 knockout metastases and H&E staining in male mice. Scale bar, 50 μm.
(I and J) Mean number of metastatic tumors at indicated sites in (I) male and (J) female mice.
(K) Total number of metastases per mouse, male (blue squares) and female (red triangles).
(G, I, J, and K) Graphs: mean ± SEM of n = 3 male and 3 female mice for control clone 1 and DOCK8 knockout (KO) clone 2, n = 5 each control clone 3, and n = 2 for DOCK8 KO clone 1.
(L) Lysosome activity is required for tumor cell invasion. Quantitation of Transwell invasion assay of 6741-P and mKPC cells treated with DMSO or bafilomycin A1 (1 μM).
(M–R) Mean ± SEM of three biological replicates. Immunoblotting of DOCK8 and quantitation of Transwell invasion following DOCK8 knockdown in (M) iKRAS, (N) Panc04.03, (O) mKPC, (P) 6741-P, (Q) Capan-1, and (R) mKPC control and DOCK8 KO clones 1–3.
(S) Immunoblotting of Panc1 cells stably expressing FLAG, FLAG-DOCK8, or FLAG-DOCK8-V1985A, and quantitation of Transwell cell invasion.
(L–S) NTsi, Nontargeting siRNA. Mean ± SEM from at least three independent biological replicates. p values were calculated using a Student’s t test or one-way ANOVA with the Tukey’s post hoc test (G, K, and R). *p < 0.05; **p < 0.01; ***p < 0.001; ****p ≤ 0.0001.
See also Figures S2–S4.
We next sought to determine if DOCK8 played a role in PDAC tumor progression. CRISPR-Cas9 was used to knock out DOCK8 in mKPC cells (Figures 2F and S2A), which are derived from the KPC mouse model of pancreatic cancer and express high levels of DOCK8 (Figure 2E).36 Two control (clones 1 and 3) or DOCK8 knockout (clones 1 and 2) mKPC cells were orthotopically injected into the pancreas of syngeneic C57BL/6 male and female mice. After 3 weeks, tumor size and intraperitoneal dissemination were measured upon necropsy. There was a mixed effect on tumor size in the DOCK8 knockout clones, as tumor size was slightly reduced in knockout clone 1 but increased in knockout clone 2 compared with the control clones (Figures 2G and S2B–S2D). Histological analysis of the primary tumors indicated that DOCK8 knockout clone 1 formed low-grade tumors, whereas clone 2 formed high-grade tumors, which is likely related to enhanced tumor growth (Figures 2H, S2B, and S2C). The most striking difference was the marked reduction in tumor cell dissemination and colonization (Figures 2H–2K, S2B, and S2C). Even within 3 weeks, invasive lesions could be detected in the control tumors in this aggressive model of PDAC, particularly along the vasculature of the intestinal mesentery, as we and others have observed previously (Figures 2H–2J and S2C).37–39 Remarkably, DOCK8 knockout profoundly reduced invasive dissemination by 80% even in the presence of a large primary tumor (Figures 2G, 2K, and S2C). These in vivo findings suggest that the ectopically expressed DOCK8 GEF plays a role in PDAC tumor growth and metastatic invasion.
DOCK8 promotes pancreatic tumor cell invasion and survival
From these initial findings, we pursued cell biological and biochemical studies to understand the role of DOCK8 in PDAC using multiple cell lines including established human cell lines (Panc04.03, L3.6, Capan1, PANC1), cells isolated from PDAC patient-derived xenografts (6741-P),40,41 and two mouse PDAC cell lines described above, iKRASG12D 13 and cells derived from the KPC genetic mouse model of pancreatic cancer (PDX-1 Cre, KrasG12D/+, p53T172H/+).36
Given the association of DOCK8 with lysosomes (Figure 1) and the significant effect of DOCK8 on intraperitoneal dissemination in vivo (Figures 2H–2K), we sought to determine if DOCK8 might affect lysosome-mediated tumor cell invasion. Lysosomal proteolytic activity has been implicated in tumor growth and invasion, though the mechanisms remain incompletely defined.5,42–44 Indeed, treatment of PDAC cells with the lysosomal inhibitor bafilomycin A1 decreased tumor cell invasion by 50%–60% (Figure 2L), highlighting the importance of lysosome function in tumor cell invasion.
Therefore, we tested if DOCK8 promotes migratory invasion by PDAC cells. To this end, we used small interfering RNA (siRNA) to knock down DOCK8 in multiple PDAC cell lines and assessed Transwell cell invasion and cell migration in a wound-healing assay. Both siRNA pools (DOCK8siP) and two individual siRNAs (DOCK8si1 and DOCK8si2) for both mouse and human DOCK8 efficiently reduced DOCK8 mRNA and protein levels (Figures S3A and S3B) and significantly decreased Transwell cell invasion by 50%–85% (Figures 2M–2Q and S3C). Transwell invasion was measured by scoring the cells on the top and bottom of the Transwell filter and calculating the percentage of invasion, which minimizes any indirect effects of viability or proliferation. In addition, using three CRISPR-Cas9 control clones and three DOCK8 knockout clones, we found that knockout of DOCK8 also reduced Transwell invasion by 80% (Figure 2R). Interestingly, 2D migration was not significantly reduced by DOCK8 knockdown or knockout, as measured by wound-healing assays (Figures S3D–S3G). This is an important distinction, as it suggests that DOCK8 promotes PDAC invasion in the presence of an ECM but not necessarily in the context of 2D migration. This is consistent with the role of DOCK8 as an important regulator of interstitial immune cell migration through the ECM.34,45–47
In immune cells, the GEF activity of DOCK8 is crucial for cell migration by activating CDC42 during immune responses.34 To determine whether the GEF activity of DOCK8 is required to mediate invasion in PDAC cells, a lentivirus was used to stably express FLAG-DOCK8-WT, or the DOCK8 GEF-inactive mutant FLAG-DOCK8-V1985A,48 in either Panc1 cells, which do not express DOCK8, or mKPC cells, which do express DOCK8. Expression of DOCK8-WT increased invasion in Panc1 cells by nearly 2-fold, whereas the GEF inactive mutant DOCK8-V1985A reduced invasion by 50% (Figure 2S). Similar to the knockdown and knockout experiments, overexpression of DOCK8 in Panc1 cells had no effect on 2D migration (Figures S3H and S3I). In mKPC cells, which already express endogenous DOCK8, overexpression of DOCK8-WT did not further increase invasion, but overexpression of DOCK8-V1985A did reduce invasion by nearly 50% (Figures S3J and S3K). Epithelial-to-mesenchymal transition (EMT) is a hallmark of cancer that contributes to the invasive capacity of cancer cells.49 Thus, we assessed whether DOCK8 depletion induces changes to the EMT program in PDAC. However, DOCK8 knockdown had no significant effect on protein levels of EMT markers E-cadherin, N-cadherin, or vimentin, suggesting that the decreased invasion observed upon DOCK8 depletion is not due to defects in EMT (Figures S3L–S3N). Together, these data indicate that ectopic expression of DOCK8 in PDAC promotes tumor cell invasion in a GEF-dependent manner.
Given the mixed effects of DOCK8 on tumor size in vivo, we investigated the role of DOCK8 in cell viability in vitro. In contrast to invasion, DOCK8 expression had a modest effect on cell viability. Crystal violet staining and MTT assays showed that knockdown of DOCK8 had no effect on the viability of 6741-P cells but that it did reduce cell numbers in L3.6 cells (Figures S4A–S4F). In mKPC CRISPR-mediated knockout cells, the viability/proliferation of the control clones was highly variable, but there was not a significant decrease in crystal violet staining of DOCK8 knockout clones (Figures S4G and S4H). In addition, knockout of DOCK8 did not cause a significant change in cell-cycle distribution measured by propidium iodide staining (Figure S4I) and did not induce apoptosis as measured by immunoblotting for cleaved caspase-3 (Figures S4J and S4K). Overexpression of WT or GEF-mutant DOCK8-V1985A had no effect on viability in PANC1 cells, which do not express DOCK8 (Figures S4L and S4M). This suggests that while DOCK8 regulates viability only in some contexts, it plays a consistent and important role in regulating PDAC invasion. Thus, in this study, we focused on how DOCK8 impacts lysosome-mediated tumor cell invasion.
DOCK8 regulates lysosome morphology, motility, and actin nucleation
As we uncovered an unexpected association of DOCK8 with lysosomes (Figure 1A), we next defined its contribution to lysosome morphology, motility, and function. DOCK8 was depleted by siRNA in iKRASG12D, 6741-P, mKPC, and Panc04.03 cells followed by immunofluorescence staining for the lysosome marker LAMP1. There was a significant enlargement of a subpopulation of lysosomes upon depletion of DOCK8. The mean lysosome area was increased by over 20% in the DOCK8 knockdown cells (Figures 3A–3C and S5A–S5J), but more strikingly, there was a 4.5-fold increase in lysosomes with an area greater than 2 μm2 (Figure S5K). Similarly, CRISPR-mediated knockout of DOCK8 caused a marked 35%–55% increase in the mean lysosome size in all three clonal cell lines (Figures 3E and 3F). The number of lysosomes per cell was not consistently affected across cell lines (Figures 3D, S5D, S5G, and S5J). Ultrastructural analysis supported these findings, as transmission electron microscopy of 6741-P and L3.6 DOCK8 knockdown cells also revealed the presence of markedly enlarged lysosomes compared with control cells (Figures 3G, S5L, and S5M), as did labeling of lysosomes with LysoTracker, which labels acidic compartments, in mKPC and 6741-P cells (Figures S5N–S5Q).
Figure 3. DOCK8 regulates lysosome morphology via Cdc42 activity.
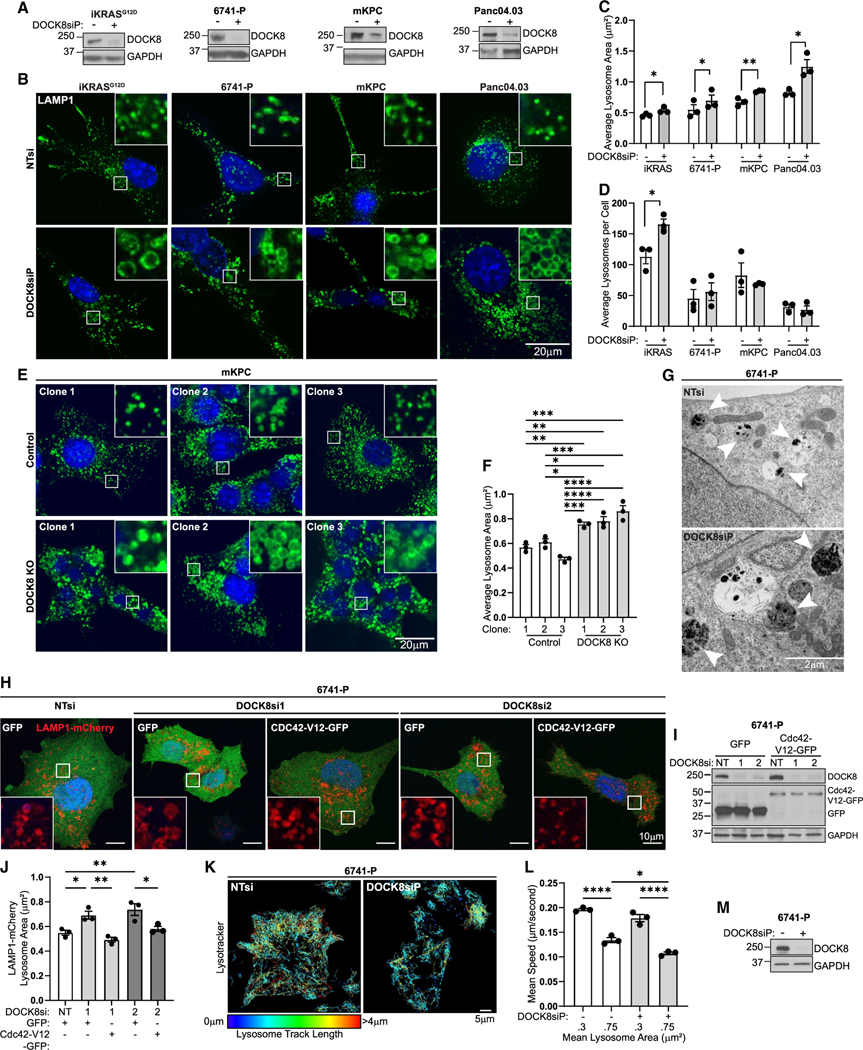
(A) Confirmation of DOCK8 knockdown by immunoblotting. GAPDH: loading control.
(B) Immunofluorescence of LAMP1 (green) and nuclei (Hoechst, blue) in cells (iKRASG12D, 6741-P, mKPC, Panc04.03) following DOCK8 knockdown. Scale bar, 20 μm.
(C and D) Quantitation of (C) average lysosome area (μm2) and (D) average number of lysosomes per cell. (iKRASG12D NT control n = 168 cells, DOCK8 knockdown n = 137; 6741-P NT control n = 307, DOCK8 knockdown n = 241; mKPC NT control n = 183, DOCK8 knockdown n = 173; Panc04.03 NT control n = 152, DOCK8 knockdown n = 168).
(E) Immunofluorescence of LAMP1 (green) and nuclei (Hoechst, blue) in DOCK8 KO mKPC cells. Scale bar, 20 μm.
(F) Quantitation of average lysosome area (μm2) (control clone 1 n = 120 total cells; control clone 2 n = 168; control clone 3 n = 141; DOCK8 KO clone 1 n = 156; DOCK8 KO clone 2 n = 140; DOCK8 KO clone 3 n = 198).
(G) Transmission electron microscopy of DOCK8 knockdown 6741-P cells compared to non-targeting control. White arrows: lysosome compartments, which are enlarged in DOCK8 knockdown cells. Scale bar, 2 μm.
(H) Overexpression of LAMP1-mCherry and GFP or active Cdc42-V12-GFP in 6741-P following DOCK8 knockdown with two individual siRNAs. Scale bar, 10 μm.
(I) Immunoblotting confirming DOCK8 knockdown and expression of GFP or Cdc42-V12-GFP. GAPDH: loading control.
(J) Quantitation of average lysosome area (μm2) (NT control GFP n = 52 cells; DOCK8si 1 GFP n = 63; DOCK8si 1 Cdc42-V12-GFP n = 66; DOCK8si 2 GFP n = 58; DOCK8si 2 Cdc42-V12-GFP n = 57).
(K) Track displacement of lysosomes over 5 min in 6741-P cells treated with LysoTracker (NT control = 147 total cells, DOCK8si = 158). Tracks are pseudocolored to represent track length. Scale bar, 5 μm.
(L) Quantitation of lysosome mean speed (of cells in K).
(M) Immunoblotting confirming DOCK8 knockdown in 6741-P cells. GAPDH: loading control.
Graphs: mean ± SEM, at least three independent biological replicates. p values were calculated using a Student’s t test or one-way ANOVA with the Tukey’s post hoc test (F). *p < 0.05; **p < 0.01; ***p < 0.001; ****p ≤ 0.0001.
See also Figure S5.
To test if the effects on lysosome size required DOCK8’s known role in activating Cdc42, we tested if expression of constitutively active Cdc42 could rescue lysosome size in the DOCK8 knockdown cells. Indeed, cotransfection of GFP Cdc42-G12V reduced the size of the lysosomes enlarged by knockdown of DOCK8 (Figures 3H–3J). This suggests that DOCK8 regulates lysosome size through activating Cdc42. Importantly, expression of oncogenic KRAS alone in iKRAS cells did not alter lysosome size (Figures S5R and S5S), though the number of lysosomes per cell was decreased by nearly 25% and the LysoTracker signal was increased by 20% (Figures S5T–S5V), indicating alterations in the lysosomal compartment. Together, these findings suggest that DOCK8 may restrict lysosome size through modulating Cdc42, which may have implications for lysosome function.2,50,51
Lysosomes are highly motile organelles, with their trafficking and positioning within the cell being crucial for their activity. To test if enlarged lysosomes in DOCK8-depleted cells exhibit altered motility, lysosomes were labeled with LysoTracker, which marks small acidic organelles. Live-cell imaging and particle tracking were performed over 10 min and revealed that lysosomes in DOCK8-depleted 6741-P cells were less motile in comparison to control cells, with decreases in mean speed by 25% (Figures 3K–3M; Videos S1 and S2). The decreased motility was particularly evident in the enlarged subpopulation of lysosomes, which is enriched in the DOCK8 knockdown cells (Figure 3L). As an additional approach, L3.6 cells were transfected with LAMP1-mCherry to label lysosomes. Particle tracking analysis of LAMP1-mCherry-labeled lysosomes in L3.6 DOCK8 knockdown cells also revealed a substantial decrease in lysosomal mean speed and maximum distance traveled by nearly 50% (Figures S5W–S5Z; Videos S3 and S4). Live-cell imaging of iKRASG12D transfected with LAMP1-GFP also showed a reduction in lysosome motility and enlarged lysosomes upon DOCK8 depletion (Videos S5 and S6). Together, these results suggest that DOCK8 promotes lysosomal motility and dynamics.
Lysosome size and motility are regulated in part by polymerization of actin at their surface.52,53 As a Cdc42 GEF, DOCK8 could potentially regulate lysosome size and motility via actin dynamics at the lysosomal surface. To test for DOCK8-mediated changes in lysosome-associated actin, siRNA was used to deplete DOCK8 in human 6741-P and mouse mKPC cells, and immunofluorescence staining for actin and LAMP1 was performed. Interestingly, DOCK8 knockdown significantly decreased actin associated with lysosomes, with overlap between actin and LAMP1 reduced by approximately 50%, in addition to a reduction in cytosolic actin (Figures 4A–4C, S6A, and S6B), though cortical actin remained unaffected. To test if this represented a defect in actin nucleation on the lysosomal surface, cells were permeabilized and incubated with rhodamine-G-actin for 15 min, which is incorporated into sites of nucleating actin, and were quantified by fluorescence microscopy. Newly nucleated actin was detected on LAMP1-GFP-positive lysosomes in non-targeting treated control cells but was reduced by 30% in DOCK8-depleted cells (Figures S6C–S6E). As reduced lysosomal actin can result in both impaired motility and lysosomal enlargement,54 these data suggest a role for DOCK8 in regulating actin levels at the lysosome surface that, in turn, regulates the size, motility, and perhaps function.
Figure 4. DOCK8 regulates lysosomal actin and cathepsin B activity.
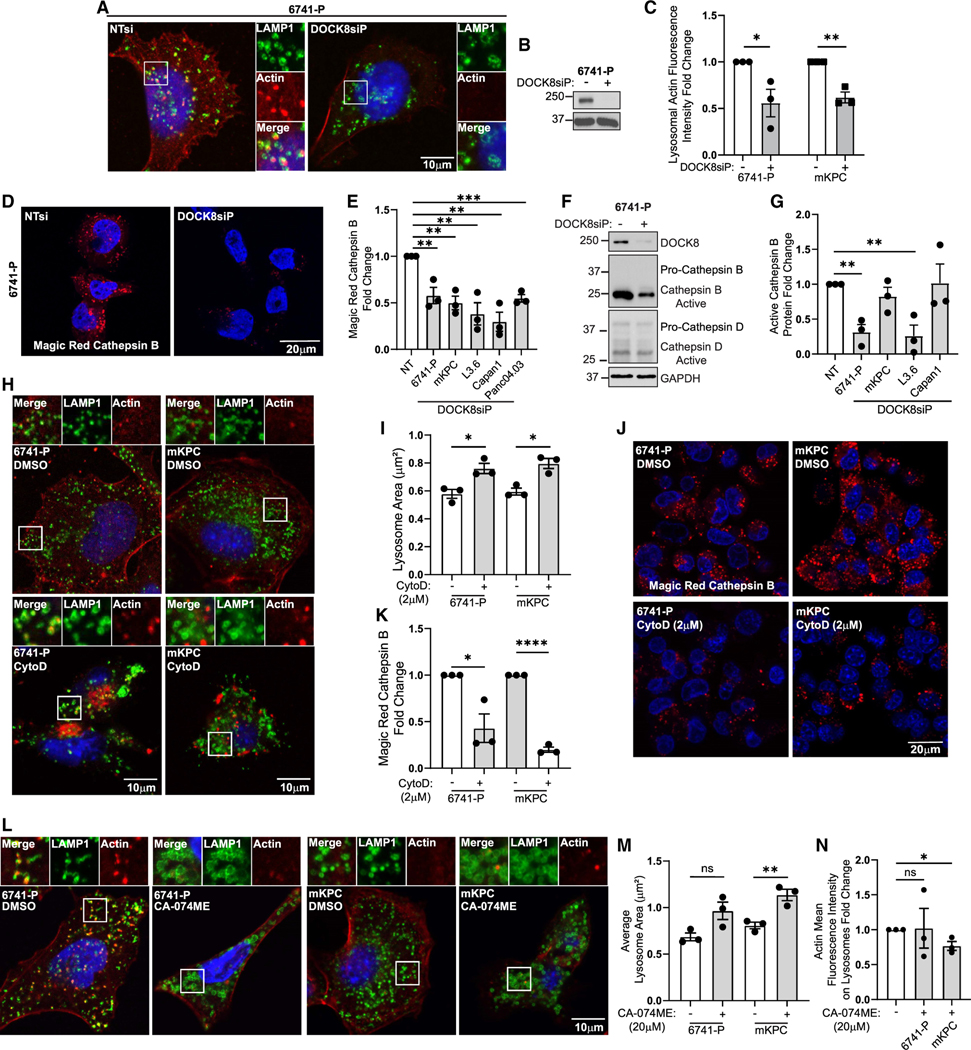
(A) Immunofluorescence of LAMP1 (green), actin (red), and nuclei (Hoechst, blue) in 6741-P cells.
(B) Immunoblotting confirming DOCK8 knockdown. GAPDH: loading control.
(C) Quantitation of actin fluorescence intensity at lysosomes in 6741-P and mKPC DOCK8 knockdown cells, normalized to cortical actin. Control cells = 1 for each cell type (6741-P, NT n = 147 cells, DOCK8 KD n = 141; mKPC, NT n = 115, DOCK8 KD n = 129).
(D) DOCK8 knockdown reduces cathepsin B activity. Magic Red cathepsin fluorescence indicating active cathepsin B in 6741-P cells following DOCK8 knockdown.
(E) Quantitation of Magic Red cathepsin B following DOCK8 knockdown in the indicated cells, normalized to non-targeting control for each cell line (6741-P, NT n = 173 cells, DOCK8 KD n = 195; mKPC, NT n = 217, DOCK8 KD n = 228; L3.6, NT n = 231, DOCK8 KD n = 221; Capan1, NT n = 163, DOCK8 KD n = 190; Panc04.03, NT n = 268, DOCK8 KD n = 240).
(F) Immunoblotting of 6741-P cell lysates showing cathepsins B and D.
(G) Quantitation of immunoblots for cathepsin B protein levels in DOCK8 knockdown cells relative to NT control cells (control set to 1 for each cell line). GAPDH: loading control.
(H) Immunofluorescence of LAMP1 (green) and actin (red) in 6741-P and mKPC cells treated with the actin inhibitor cytochalasin D (2 μM) for 5 h.
(I) Quantitation of mean lysosome area (mm2) shows that F-actin disassembly increases lysosome size (6741-P, DMSO n = 173 cells, cytochalasin D [CytoD] n = 182; mKPC, DMSO n = 158, CytoD n = 169).
(J) Magic Red cathepsin B fluorescence indicating active cathepsin B in 6741-P and mKPC cells treated with DMSO or CytoD (2 μM) for 5 h.
(K) Quantitation of Magic Red cathepsin B mean fluorescence intensity shows that F-actin disassembly inhibits cathepsin B activity (6741-P, DMSO n = 234 cells, CytoD n = 184; mKPC, DMSO n = 236, CytoD n = 291). DMSO condition set to 1 for each cell type.
(L) Immunofluorescence of LAMP1 (green) and actin (red) in 6741-P or mKPC cells treated with DMSO or the cathepsin B inhibitor CA-074ME (20 μM, overnight).
(M) Quantitation of average lysosome area (μm2).
(N) Actin mean fluorescence intensity (actin, red) at the lysosome membrane (6741-P, DMSO n = 124 cells, CA-074ME n = 122; mKPC, DMSO n = 192, CA-074ME n = 205). DMSO-treated cells were set to 1 for each cell line.
Scale bars, 10 μm (A, H, and L) and 20 μm (D and J). Graphs: mean ± SEM of three independent biological replicates. ns, p > 0.05; *p ≤ 0.05; **p < 0.01; ***p < 0.001; ****p ≤ 0.0001.
See also Figures S6 and S7.
DOCK8 is required for invasive migration via cathepsin B-mediated ECM degradation
As lysosome morphology and motility depend upon DOCK8 expression, it was important to test if DOCK8 affected lysosome function by measuring lysosomal cargo degradation. First, the EGF receptor, which is degraded by lysosomes following stimulation with high levels of EGF, was degraded at equal rates following EGF treatment in both control and DOCK8-depleted cells (Figures S6F–S6H), suggesting no defect in lysosomal degradation. Second, we assessed the fluorescence of DQ-BSA, which is internalized by macropinocytosis and fluoresces upon cleavage in the acidic lysosome. Following DOCK8 knockdown, 6741-P cells were incubated with DQ-BSA for 1 h followed by a 3 h chase. However, there was no change in DQ-BSA fluorescence when normalized to a second macropinocytic cargo, tetramethylrhodamine (TMR)-dextran10 (Figures S6I–S6L). Third, lysosome acidification was assessed by measuring the fluorescence intensity of cells treated with LysoTracker, which labels acidic organelles. Both mKPC and 6741-P cells showed no defects in LysoTracker fluorescence intensity upon DOCK8 knockdown, indicating no gross dysregulation of lysosome pH (Figures S5P and S6M–S6O). These data suggest that DOCK8 does not globally regulate the degradative function of lysosomes.
We next measured the effects of DOCK8 on the activity of cathepsins, which are proteases that are enzymatically activated in the low pH within the lysosome lumen. To this end, control or DOCK8 knockdown 6741-P, mKPC, L3.6, Capan1, and Panc04.03 cells were incubated with Magic Red cathepsin B, which is a cathepsin B substrate that fluoresces upon cleavage. Importantly, cathepsin B activity was dramatically reduced by 40%–70% upon DOCK8 depletion, using both siRNA pools and individual siRNAs (Figures 4D, 4E, and S7A–S7D). Similarly, CRISPR-mediated knockout of DOCK8 also reduced cathepsin B activity (Figures S7E and S7F). Cathepsin B levels were not significantly or consistently altered at the transcriptional or protein levels, suggesting differences in the regulation of its activity rather than its expression (Figures 4F, 4G, and S7G–S7J). This finding revealed a link between DOCK8 and the activity of the lysosomal protease cathepsin B, which has been defined as essential for pancreatic cancer development and progression.8
As DOCK8 regulates lysosomal size, actin, and cathepsin B, we investigated the relationship among these effects by first testing if the DOCK8-dependent inhibition of actin impacted lysosome size and cathepsin activity. Indeed, F-actin disassembly by cytochalasin D also increased lysosome size by 30% (Figures 4H and 4I) and reduced cathepsin B activity by 60%–80% (Figures 4J and 4K), mimicking DOCK8 depletion. Conversely, we tested if cathepsin B activity regulates lysosome size and actin association. While inhibition of cathepsin B with CA-074ME increased lysosome size by 40%, it did not significantly affect the amount of lysosome-associated actin (Figures 4L–4N). These data suggest that DOCK8-dependent actin polymerization at the lysosome impacts cathepsin B activity and lysosome size.
In addition to degrading intracellular cargo, lysosomal cathepsins can also be secreted to degrade extracellular substrates including ECM. Given the defects in lysosome motility, invasion, and cathepsin B activation, we hypothesized that the activation or the secretion of lysosomal cathepsins more broadly might be defective in the absence of DOCK8. Using a Protease Array, we assessed conditioned medium from 6741-P cells and found that siRNA-mediated knockdown of DOCK8 reduced the secretion of multiple lysosomal cathepsins, including cathepsin B, into the medium (Figures 5A, 5B, and S7K). In contrast, other lysosomal and non-lysosomal secreted proteases were unaffected. Thus, these data implicate a role for DOCK8 in the activation and secretion of a subset of lysosomal cathepsins.
Figure 5. DOCK8 regulates lysosomal protease secretion and cathepsin B-mediated ECM degradation.

(A) Proteases from conditioned media of DOCK8 knockdown 6741-P cells using a Protease Array. Green bars: lysosomal proteases; two independent replicates.
(B) Immunoblotting confirming DOCK8 knockdown. GAPDH: loading control.
(C) DOCK8 is required for ECM degradation. Images of cleaved DQ-collagen (green) in 6741-P cells either overlayed with DQ-collagen/Matrigel (top) or seeded on a DQ-collagen substrate with gelatin (bottom, green puncta).
(D) DOCK8 immunoblotting confirmed knockdown in 6741-P and mKPC cells. Quantitation of DQ-collagen mean fluorescence intensity in Matrigel-embedded model in DOCK8 knockdown cells relative to NT control (set to 1 for each cell line) (6741-P, NT n = 1007 cells, DOCK8 KD n = 1399; mKPC, NT n = 969, DOCK8 KD n = 939).
(E) DQ-collagen/gelatin substrate relative fluorescence intensity in DOCK8 knockdown cells versus NT control (set to 1 for each cell line) (6741-P, NT n = 114 cells, DOCK8 KD n = 91; mKPC, NT n = 187, DOCK8 KD n = 190). Nuclei: Hoechst, blue.
(F) Cathepsin B is necessary and sufficient for ECM degradation. Immunoblotting verifying DOCK8 knockdown and overexpression of cathepsin B-mCherry in 6741-P cells.
(G) Images of degraded DQ-collagen fluorescence (green) in DOCK8 knockdown 6741-P cells transfected with mCherry vector or cathepsin B-mCherry (red) and nuclei (Hoechst, blue).
(H) Quantitation of DQ-collagen/gelatin mean fluorescence intensity in 6741-P DOCK8 knockdown and NT control cells expressing mCherry or cathepsin B-mCherry relative to NT-mCherry control cells (set to 1) (6741-P, NT-mCherry n = 67 cells; NT-cathepsin B-mCherry n = 47; DOCK8 KD-mCherry n = 53; DOCK8 KD-cathepsin B-mCherry n = 44).
(I) Manders’s coefficient measuring cathepsin B-mCherry overlap with degraded DQ-collagen.
(J) Quantitation of 6741-P and mKPC Transwell cell invasion upon cathepsin B inhibition. Nuclei: Hoechst, blue.
Only mCherry vector- or cathepsin B-mCherry-positive cells were scored (H and J). Scale bars, 20 μm. Graphs: mean ± SEM of three independent biological replicates. ns, p > 0.05; *p ≤ 0.05; **p < 0.01; ***p < 0.001; ****p ≤ 0.0001.
See also Figure S7.
As secreted cathepsins can promote stromal remodeling during tumor cell invasion,6,8,55–57 we next tested if the lysosome-regulatory function of DOCK8 contributed to its role in tumor cell invasion. We assessed the ability of PDAC cells to degradDistinct forms of the actin cross-linking proteine DQ-collagen, an ECM protein that fluoresces upon its proteolytic cleavage. DOCK8 was knocked down in 6741-P cells that were overlaid with an ECM comprised of Matrigel/DQ-collagen or that were plated on top of gelatin/DQ-collagen for more constrained degradation of the ECM that accumulates in and marks lysosomes. The control cells exhibited high levels of DQ-collagen fluorescence, indicating proteolytic ECM cleavage. Importantly, in both ECM environments, the DQ-collagen fluorescence intensity was substantially reduced upon DOCK8 knockdown (Figures 5C–5E and S7L), indicating that DOCK8 promotes ECM degradation. To confirm that degradation of ECM is dependent upon cathepsin activity, 6741-P cells were treated with the cathepsin B inhibitor CA-074ME (20 μM) overnight, which significantly reduced the degradation of DQ-collagen (Figures S7M and S7N). To test if cathepsin B is both necessary and sufficient for DOCK8-dependent ECM degradation, we overexpressed cathepsin B-mCherry in 6741-P DOCK8 knockdown cells and assessed ECM degradation after 48 h by measuring the intracellular accumulation of cleaved DQ-collagen in lysosomes. Importantly, overexpression of cathepsin B-mCherry rescued DQ-collagen degradation in the absence of DOCK8, consistent with DOCK8 regulating ECM degradation through cathepsin B activity (Figures 5F–5H). Interestingly, we noticed a significant fraction of cathepsin B-mCherry in the DOCK8 knockdown cells that did not colocalize with cleaved DQ-collagen, which marks lysosomes. A Manders’s overlap coefficient indicated that in control cells, 85% of the cathepsin B-mCherry colocalized with DQ-collagen-positive lysosomes but that only 60% localized to these structures in the DOCK8 knockdown cells (Figure 5I). This suggests the presence of extra-lysosomal cathepsin B that perhaps has been misdirected into other membranous organelles, indicating defective trafficking and routing of cathepsin B in the absence of DOCK8.
Finally, inhibition of cathepsin B reduced Transwell cell invasion by 70%–80% (Figure 5J), indicating that cathepsin B is required for tumor cell invasion. However, cathepsin B-mCherry overexpression alone was not sufficient to restore invasion in the DOCK8 knockdown cells (Figures 5F and S7O). This is likely because the secretion of additional lysosomal proteases was impacted by DOCK8 and may also be due to localization defects of cathepsin B (Figures 5G and 5I). Together, these observations suggest that DOCK8 promotes tumor cell invasion by supporting the activation and secretion of lysosomal cathepsins, at least in part through lysosome-associated actin dynamics and proper sorting or trafficking of lysosomal proteases.
DISCUSSION
In this study, we provide evidence linking the Cdc42 GEF DOCK8 to lysosome-mediated tumor cell invasion and identify it as a potential target in pancreatic cancer. These findings indicate that lysosomes are key players in PDAC cell invasion and that oncogenic KRAS remodels the lysosomal proteome to promote tumor growth and invasion at least in part through regulation of DOCK8.
DOCK8 is primarily expressed and studied in immune cells, where it plays key roles in cell migration, activation of the immune response, and T cell survival.30,58 DOCK8 has been suggested to be elevated in some cancers; however, this was attributed to immune cell infiltrates.59 Further, DOCK8 mutation causes a rare immunodeficiency that affects immune cell numbers and migration and increases cancer risk.34,60 DOCK8 is essential for migration of natural killer (NK) cells, dendritic cells, and T cells and induces T cell infiltration into the central nervous system via activation of Cdc42-mediated cell migration61 and for migration through constricted regions of ECM.34 Intriguingly, immune cell infiltration and tumor cell invasion share common characteristics, including remodeling of the actin cytoskeleton, ECM degradation, and cytokine secretion, that allow cells to invade and migrate through constricted spaces. Similar to DOCK8, aberrant expression of an additional immune-specific GEF, Vav1, in PDAC also drives metastatic invasion.26–28 Thus, aberrant expression of immune-regulatory GEFs in PDAC may confer invasive abilities in tumor cells comparable to their roles in immune cells.
Interestingly, depletion of DOCK8 decreased the secretion of a subset of lysosomal cathepsins, reduced cathepsin B activity, and diminished tumor cell invasion. Cathepsin B, a cysteine protease, is widely expressed across tissues, is required for lysosome recycling and degradation of cargo, and is upregulated in cancer malignancies.6,8,12,57,62 Extra-lysosomal cathepsin B substrates go beyond ECM components and include E-cadherin, insulin-like growth factor-1 (IGF-I), glucagon, and perilipin 1.63–65 High expression of cathepsin B in PDAC correlates with a poor prognosis.66 In addition, a genetic mouse model demonstrated that depletion of cathepsin B extends survival time and decreases metastasis. Similarly, tumor growth was upregulated in a flank injection model in mice injected with cells expressing cathepsin B, compared to control cells.8 Together, these studies link cathepsin B to PDAC outcomes.
DOCK8 GEF function activates Rac1 and Cdc42 and links Cdc42 to WIP and Talin to drive actin nucleation in immune cells.34,45,48 In migratory cancer cells, GEF action is well studied in actin polymerization in the invasive leading edge. Indeed, actin dynamics are a main driver of invasion and migration through leading-edge dynamics and mechanotransduction, and treatment with inhibitors of actin polymerization including cytochalasins inhibits tumor growth and invasion.67,68 While DOCK8 likely impacts actin in multiple subcellular regions, the data presented here suggest that DOCK8 also promotes actin dynamics at lysosomes. DOCK8 may drive actin-based lysosome motility by inducing actin nucleation and actin comet formation at lysosomal membranes or by actin-myosin-dependent lysosome movement.69,70 Further, lysosomal actin may control membrane dynamics and modulate lysosome fission, fusion, and tubulation, which may regulate lysosome size.54,71,72 Indeed, disruption of F-actin leads to enlarged lysosomes (Figures 4H and 4I). In addition, the Arp2/3 activator Wiskott-Aldrich syndrome protein and SCAR homolog (WASH) is required for lysosome maturation and recycling of cathepsins.73 Thus, it is possible that DOCK8-mediated Cdc42 activation leads to actin-regulated lysosome maturation and cathepsin recycling. The pathway by which specific cathepsins are trafficked and recycled are not comprehensively known.74–79 We propose that DOCK8-dependent actin regulation may affect the trafficking, lysosomal targeting, and activation of the subset of cathepsins identified in Figure 5A. Further studies are required to elucidate how DOCK8 selectively regulates a subset of cathepsins including cathepsin B. Together, these data suggest that KRAS-dependent upregulation of DOCK8 in PDAC enhances its association with lysosomes and promotes actin nucleation to maintain lysosome homeostasis. Of note, while we could readily detect DOCK8 on lysosomes using biochemical means, the localization of DOCK8 was detectable by microscopy on a subset of lysosomes and in a subset of cells, while the majority of DOCK8 appeared localized in the cytoplasm. This suggests that DOCK8’s lysosomal association may be dynamic or regulated by secondary factors, or that its detection may be limited by technical factors.
While oncogenic KRAS enhanced expression of DOCK8 at the transcript and protein levels, not all PDAC cell lines with oncogenic KRAS mutations express DOCK8, suggesting a more complex regulatory mechanism of DOCK8 expression. It will be interesting to define the mechanisms regulating DOCK8 expression in PDAC cells and to determine if a specific subset of tumor cells elevate DOCK8 levels. It is important to note that tumor cells that do not express DOCK8 may still undergo invasion and metastasis, likely due to the aberrant expression of a different GEF.26,27 For example, the GEF Vav1, which is an activator of Rho GTPases Cdc42 and Rac1, is also ectopically expressed in a subset of PDACs.26 Thus, it is possible that different PDAC tumors use different GEFs to drive cell invasion. Indeed, PDAC cells express a variety of Cdc42 GEFs including VAV1, VAV2, FGD1, and B-PIX, among others,28 and many cell lines have unique patterns of GEF expression. Yet, tumors with upregulated DOCK8 expression become dependent on it for growth and invasion, making it an essential factor for tumor progression and metastasis and thus a potential therapeutic target.
Our data revealed that depletion of DOCK8 results in a substantial reduction in tumor cell invasion in vitro and in vivo, with moderate and mixed effects on cell viability. This was surprising given the modest effects of DOCK8 on cell proliferation in vitro (Figure S4H) and suggests both clonal variation and that DOCK8 may interact with factors in the tumor microenvironment to promote proliferation in some contexts. Invasion was significantly reduced in both cases, though for clone 1 in vivo, this may be attributed to reduced tumor size. DOCK8-dependent cathepsin B regulation may impact tumor growth, as cathepsin B promotes tumor growth and metastasis in multiple cancers including PDAC.8,56,80,81 In addition, the contribution of the tumor microenvironment, particularly immune cell infiltration or the dense ECM, could also contribute to impaired tumor growth. To this point, DOCK8-deficient T cells undergo cytothripsis (cell shattering) when migrating though ECM.82 Thus, loss of DOCK8 may sensitize tumor cells to a harsh, desmoplastic PDAC microenvironment. Future studies will shed light on how DOCK8 expression in pancreatic tumor cells interacts with the tumor microenvironment to promote tumor growth.
In summary, these results reveal a mechanism by which KRAS regulates lysosome-dependent tumor cell invasion through DOCK8 in vitro, as well as tumor metastasis in vivo. These findings highlight the importance of DOCK8 as a potential therapeutic target to abrogate PDAC progression and metastasis.
Limitations of the study
Pancreatic tumors are quite heterogeneous, which makes it difficult to determine the subpopulations of tumors or tumor cells in which DOCK8 expression promotes tumor growth and progression. This is further complicated by the expression of DOCK8 in immune cells, the limitations of antibodies for staining of endogenous DOCK8, and the mixed effects of DOCK8 on tumor growth. While we cannot rule out the effects of DOCK8 on regulating tumor growth in vivo, the inhibition of dissemination in vivo, even in the presence of a large primary tumor, is consistent with the in vitro data demonstrating that loss of DOCK8 consistently reduces tumor cell invasion.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents should be directed to and will be fulfilled by the lead contact, Gina Razidlo, PhD. (Razidlo.Gina@mayo.edu).
Materials availability
All the materials generated in this study are accessible upon request.
Data and code availability
Comparative lysosome proteomic data has been deposited in MassIVE: https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp. Dataset accession is MSV000091036. Western blot images and CRISPR KO sanger sequencing have been deposited at Mendeley Data: https://doi.org/10.17632/zmwy6bgfg8.1. RNA sequencing data from pancreatic cancer organoids have been deposited in Gene Expression Omnibus: GSE185335.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
Animals were maintained under specific pathogen-free conditions. Wild type C57BL/mice were purchased from the Jackson Laboratory. Female and male mice at 8 weeks of age were used for experiments. The laboratory animal facility of Mayo Clinic has been accredited by AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care International) and animal studies were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Mayo Clinic.
Cell lines
mKPC, 6741-P, Capan1, Panc1, DanG, CFPAC, MIA PaCa2, HPAFII, HuPT3, and HEK293T cells were cultured in DMEM (Corning, 10-013-CV). L3.6 cells were cultured in DMEM with no phenol red (Corning, 17-205-CV). Panc04.03, and BxPC3 were cultured in RPMI (Corning, 10-040-CV). iKRASG12D cells were cultured in RPMI supplemented with 1 μg/mL doxycycline hyclate (Sigma, D9891) or 72-h doxycycline withdrawal to revert cells to WT KRAS. RAW 264.7 macrophages were cultured in DMEM. Media was supplemented with 10% FBS and Penicillin/Streptomycin (Gibco, 15140-122). HPDE cells were cultured in keratinocyte-SFM containing epidermal growth factor, bovine pituitary extract, and Pen/Strep (Gibco, 15140-122). Cell lines were obtained from ATCC or kindly provided by Dr. David Tuveson (mKPC T4-2D; Cold Spring Harbor Laboratory, Cold Spring Harbor, NY),36 Dr. Marina Pasca di Magliano (iKRASG12D, 4292; University of Michigan, Ann Arbor, MI),13,32 Dr. Daniel Billadeau (HPDE; CFPAC, RAW 264.7 Mayo Clinic, Rochester, MN), or 6741-P are human patient–derived xenograft (PDX) cells isolated from pancreatic tumors implanted in nude mice41 and were provided by the Mayo Clinic SPORE in Pancreatic Cancer. Cell lines were authenticated by IDEXX BioAnalytics. Authentication of the PDX-derived 6741 cell line showed high similarity with PaTu8898, and thus are denoted as 6741-P. Cells were tested for Mycoplasma by PCR (Southern Biotech) and Hoechst staining. Cells were treated with DMSO (Sigma, D2438-10ML). Bafilomycin A1 (Cayman Chemical, 11038), CA-074ME (Cayman Chemical, 18469), Cytochalasin D (Cayman Chemical, 11330), Staurosporine (Cayman Chemical, 81590).
METHOD DETAILS
Plasmids, transfections and generation of stable cell lines
Lipofectamine 2000 (Invitrogen, 11668019) was used for plasmid transfections. pCI2-DOCK8-YFP, pCI2-DOCK8-V1985A-YFP, and pLenti6.3. F-MCS were kindly provided by Dr. Daniel Billadeau. Human cathepsin B was provided by Dr. Hyeryun Choe (Addgene plasmid #11249)84 and was cloned into pmCherry-N1 (Clontech, 632523). Briefly, pmCherry-N1 and pLenti6.3. F-MCS vectors were digested with XhoI (New England BioLabs, R0146S) and BamHI (New England Biolabs, R3136S) for 1 h at 37°C. Human cathepsin B was PCR amplified with primers: Fwd 5′- CCGCTAGCGCTACCGGACTCAGATCTCGAGATGTGGCAGCTCTG GGCCTCC-3′ and Rev 5′- GCTCACCATGGTGGCGACCGGTGGATCCCGGGCGGGGGCCACCTGGCTGG-3’. Full length DOCK8 was PCR amplified in two parts using primers: Fwd1 5′-GATTACAAGGATGACGACGATAAGGGATCCATGGCCACTCTGCC GAGCGC-3′, Rev1 5′ GAGAAGGTCATACAAGAAGAAAGCCAGGCTGATGTTCATCTTTTCCGCCT-3′, Fwd2 5′-AGCCTGGCTTTCTT CTTGTATGACCTTCTCTCCCTCATGGATCGGGGCTT-3’. Rev2 5′- CGATCATTACTAACCGGTACGCGTCACTCGAGTTAGCTGCC CTGTGACAACTGG-3’. DOCK8-DHR2-HA was cloned into pcDNA3.1-HA provided by Dr. Oskar Laur (Addgene plasmid #128034). In brief pcDNA3.1-HA was digested with BamHI BamHI (New England Biolabs, R3136S) and EcoRV (New England Biolabs, R0195S) for 1 h at 37°C. The DHR2 domain of DOCK8 was PCR amplified using pCI2-DOCK8-YFP as template with primers: Fwd: 5′-ATTACGCTGGTACCGAGCTCCTCCGGAGGTTCATGTACACCACCCCGTTCACC-3′ and Rev: 5′ CGGCCGCCACTGTGCTGGAT TTAGCTGCCCTGTGACAACTGGGTTTCACATTTCC-3’. Q5 High-Fidelity DNA Polymerase (New England BioLabs, M0491S) was used for PCR amplification. Digested vectors and PCR products were gel purified using Illustra GFX 96 PCR purification kit (Cytiva, 28-9034-71). Human cathepsin B was cloned into pmCherry-N1, and DOCK8 or DOCK8-V1985A (GEF inactive) were cloned into the pLenti6.3. F-MCS lentiviral vector, and and DOCK8-DHR2-HA was cloned into pcDNA3.1-HA using NEBuilder HiFi DNA Assembly (New England BioLabs, E2621L). pEGFP-N1-LAMP1 was previously described.85 pEGFP-N1 (Clontech, 6085-1). pBabe-Kras Wt was provided by Dr. Channing Der (Addgene plasmid #75282). Tmem192-3xHA was a gift from Dr. David Sabatini (Addgene plasmid #102930).33 To generate stable cells, lentivirus was produced by cotransfecting HEK293T cells with pLJC5-Tmem192-3xHA, pLenti6.3. F-MCS, pLenti6.3. F-MCS-DOCK8, or pLenti6.3. F-MCS-DOCK8-V1985A with VSV-G-pseudotyped viral packaging plasmids (provided by Dr. Y. Ikeda, Mayo Clinic). After 72 h, conditioned media containing lentivirus was collected and passed through 0.45mm filters. Cells were transduced with lentivirus for 72 h in the presence of 10mg/ml polybrene (Millipore, TR1003-G). Cells were then selected with puromycin (Gibco, A1113—03) or blasticidin (Gibco, A11139-03) for 72 h to obtain stable cells. The stable cell lines were tested for the presence of replication-competent lentivirus using an ELISA for lentiviral p24 (QuickTiter Lentivirus Titer Kit, Cell Biolabs, Inc, VPK-107-T), and the signal was below the limit of detection. siGENOME Mouse DOCK8 (Dharmacon, M-026106-01-0005) or siGENOME Human DOCK8 (Dharmacon, M-056235-01-0005) siRNA SMARTpools (DOCK8siP), mouse DOCK8si1 (Dharmacon, D-056235-01-0010), mouse DOCK8si2 (Dharmacon, D-056235-04-0010), Human DOCK8si1 (Dharmacon, D-026106-02-0010), Human DOCK8si2 (Dharmacon, D-026106-04-0010) individual siRNA, and On-Target Plus nontargeting siRNA control (Dharmacon, D-001810) were used for knockdowns. siRNA was transfected using RNAiMAX (Invitrogen, 13778-150).
Immunoblotting
Cells were lysed in ice-cold NP-40 lysis buffer (20mM Tris-HCl, pH8, 137mM NaCl, 10% glycerol, 1% IGEPAL, 2mM EDTA, pH8) supplemented with Complete Protease Inhibitor Cocktail (Roche, 11873580001) and PhosSTOP (Roche, 4906845001). Protein concentration was measured by BCA protein assay (Thermo Fisher Scientific, 23225). Equal amounts of protein were resolved on SDS-PAGE gels and transferred to PVDF (Millipore, IPVH00010) membrane. Membranes were probed with primary antibodies: Abcam: DOCK8 (ab227529), LAMP2A (ab18528). Cell Signaling: Flag (8146S), HA (2367S), mCherry (43590S), cathepsin B (31718S), TOM20 (42406S), GM130 (12480S), GAPDH (5174S), phospho-ERK 1/2 (4377S), ERK 1/2 (9102S), mTOR (2983), COXIV (11967S), EGFR (2232S), p62 (5114), LC3B (2775), RAB7 (9367S), Cleaved Caspase 3 (9661S), Caspase 3 (9662S). LAMP1 (DHSB, 1D4B). Sigma: Actin (A2066), KRAS (WH0003845M1), Santa Cruz: cathepsin D (sc-377124), cathepsin L (sc-390367), GFP (Roche, 11814460001). Secondary horseradish peroxidase-conjugated antibodies (Invitrogen), HyBlot CL film (LabForce, 1141J52), and SuperSignal West Pico PLUS (Thermo Fisher Scientific, 34579) were used to detect immunoreactive signals. Band densitometry was quantitated with ImageJ. Full Western blot images have been deposited at Mendeley Data (https://doi.org/10.17632/zmwy6bgfg8.1).
Protease secretion detection
Nontargeting control or DOCK8siP were transfected into 6741-P cells. 48 post-knockdown, cells were washed three times with HBSS (Corning, 21-021-CV), and incubated overnight in serum free DMEM. After 24 h, conditioned media was collected and centrifuged for 5 min at 1,200 RPM at 4°C. The supernatant was collected and Proteome Profiler Human Protease Array Kit (R&D Systems, ARY021B) was used to detect secreted proteases following the manufacturer’s protocol. A map of the Protease secretion array is provided in Mendeley Data under supplementary data.
Immunofluorescence microscopy
For LAMP1 and actin immunofluorescence staining, cells were fixed with methanol at −20°C for 15 min. For immunofluorescence staining of transwell filters and others, cells were fixed with formaldehyde fixing buffer (0.1M PIPES, pH6.9, 1Mm EGTA, 3mm MgSO4, 2.5% formaldehyde), and permeabilized with 0.1% Triton X-100 (Sigma, 93443) for 2 min. Fixed cells were incubated in blocking buffer (5% goat serum, 5% glycerol, 0.04% Na-azide, ph7.2) for 1 h at 37°C. Cells were probed with primary antibodies: LAMP1 (Santa Cruz, sc-20011) for Human cells), LAMP1 (DSHB, 1D4B) for murine cells, HA (Cell Signaling, 23676), actin (Sigma, A2066). Secondary antibodies were from Invitrogen: Anti-Mouse-Alexa Fluor 488 (A11029), Anti-Rat-Alexa Fluor 488 (A11006), Anti-Rabbit-Alexa Fluor 594 (A11037). Phalloidin-FITC was from Sigma (P5282). Nuclei were stained with Hoechst 33342 (Invitrogen, H3570). Coverslips were mounted on ProLong Gold antifade mountant (Invitrogen, P36934). Images were acquired on a Zeiss LSM-980 or a Zeiss LSM-780 confocal microscope with a 63x or 403 oil objective lens, respectively, and Zen software. Quantitation of lysosome area and actin mean fluorescence intensity was done using ImageJ. In brief, the Auto Local Threshold function was used to threshold lysosomes, then the following functions were performed in order: convert to mask, fill holes, watershed. The Analyze Particles function was used to measure lysosome area. For actin quantitation at lysosomes, a further step was performed. A 0.4μm band around thresholded lysosomes was drawn using the function ‘‘Make Band’’, then all ROI were selected, and actin mean fluorescence intensity was measured. Cortical actin was measured and used to normalize lysosomal actin. Manders’ overlap coefficient was measured using the JaCoP plug-in in ImageJ86.
Actin polymerization assay
Cells were transfected with GFP-Lamp1, following DOCK8 knockdown, and plated onto coverslips. The next day cells were cultured in serum free media for 3 h. 12 μM Rhodamine-labeled G-actin (Cytoskeleton, AR05) was reconstituted in G-actin buffer (5 mM Tris-HCl, 0.2 mM CaCl2, 0.2 mM ATP, 1 mM DTT, pH 8.0). Cells were permeabilized with Buffer C (138 mM KCl, 10mM PIPES, 0.1 mM ATP, 3 mM EGTA, 4 mM MgCl2, 1% BSA, 0.025% saponin, pH 6.9) containing 1μM Rhodamine-labeled G-actin for 15 min at room temperature. Cells were fixed with 3% formaldehyde fixation buffer (0.1M PIPES, pH6.9, 1mM EGTA, 3mM MgSO4, 2.5% formaldehyde) for 20 min and gently washed with PBS. Nuclei were stained with Hoechst 33342 (Invitrogen, H3570) and coverslips were mounted onto glass slides with Prolong Gold (Invitrogen; #P36934). Images were acquired using a Zeiss LSM 980 confocal microscope with Zeiss Zen software. Quantitation of polymerized Rhodamine-G-actin mean fluorescence intensity associated with lysosomes was done using ImageJ as described above.
Migration and transwell invasion and assays
For transwell invasion assays, 3 × 105 cells were seeded in the upper chamber of 8μm pore Matrigel-coated transwell filters (Costar, 3428) in 0.1% FBS media and allowed to invade for 20 h toward the lower chamber containing media supplemented with 10% FBS. Filters were cut and fixed with 2.5% formaldehyde followed by nuclei staining with Hoechst 33342 on the top and bottom of the filters (Invitrogen, H3570) and mounted on slides with ProLong Gold antifade mountant (Thermo Fisher Scientific, P36930). The filters were imaged by fluorescence microscopy to distinguish the cells on the top (uninvaded) and bottom (invaded) of the filter. Transwell invasion percent was calculated by quantitating the nuclei on the top and bottom of the filter (at least 500 cells were scored per each of three independent biological replicates) and dividing the number of nuclei on the bottom of the filter by the total number of nuclei. For Cathepsin B-mCherry overexpression experiments, only cells expressing mCherry or Cathepsin B-mCherry were scored. Cells were pretreated with 1μM Bafilomycin A1 (Cayman Chemical, 11038) for 5 h, or 20μM CA-074ME (Cayman Chemical, 18469) overnight, before and during transwell invasion assays. For wound healing assays, cells were plated into a monolayer in each well of an Ibidi Culture-Insert 2 well system (Ibidi, 80209) after knockdown of DOCK8. The next day the ibidi chambers were removed, cells were carefully washed with HBSS and allowed to migrate in fresh culture media. Three brightfield images per insert were taken at t = 0 and t = 8h. Migration distances were quantitated using ImageJ by drawing the border on the leading edge of the migrating cells and measuring the area.
Lysosome immunoprecipitation (LysoIP)
Lysosomes were isolated as previously described33 with minor modifications. In brief, 4 × 106 iKRASG12D cells stably expressing TMEM192-HA, compared to no TMEM192-HA as a negative control, were seeded on five 15cm dishes per condition and cultured with 1 μg/mL doxycycline hyclate (Sigma, D9891) or following doxycycline withdrawal for 72 h. Panc 04.03 cells stably expressing TMEM192-HA, or not expressing as a negative control, were plated similarly. Cells were washed 3 times with PBS (Corning, 21-040-CV), scraped in 1mL of ice-cold KPBS buffer (136mM KCl, 10mM KH2PO4, 2mM EDTA, pH 7.25) supplemented with Complete EDTA free Protease inhibitor. Cells were centrifuged at 1000xG for 2 min at 4°C, resuspended in 1mL ice-cold KPBS, and homogenized by 30 strokes with a 2mL homogenizer. Cell homogenate was centrifuged at 1000xG for 2 min at 4°C, 100μL of supernatant was collected (whole cell lysate), and the remaining supernatant was incubated with 100μL of Pierce Anti-HA magnetic beads (Thermo Fisher Scientific, 88837) for 30 min on an end-over-end rotator at 40°C. Beads were collected on the side of the tube with a magnet and washed five times with KPBS, and five times with KPBS+ buffer (170mM KCl, 10mM KH2PO4, 2mM EDTA, pH 7.25) supplemented with Complete EDTA free Protease inhibitor, followed by two gentle washes with KPBS+ on the vortex. The beads were collected with a magnet, the supernatant was discarded, and the beads were resuspended in 100μL of KPBS buffer. The purity of isolated lysosomes was validated by immunoblotting of lysosome markers (TMEM192-HA, LAMP2A, cathepsin B), lysosome binding proteins (mTOR, Rab7), and non-lysosome markers (GM130, Golgi; COXVI, TOM20, mitochondria).
For proteinase K degradation of extraluminal lysosome-associated proteins, isolated lysosomes bound to anti-HA magnetic beads were resuspended in ice-cold proteinase K buffer, (33.3 mM HEPES, 1mM CaCl4, pH 7.4) and treated with increasing concentrations of Proteinase K (0, 0.1, 0.25, 0.5 μg/mL) for 15 min on ice. Digestion was terminated with a final concentration of 1mM AEBSF (Sigma, SBR00015). Equal volumes of samples were used for immunoblotting.
Lysosome comparative mass spectrometry analysis
Isolated lysosomes from three independent biological replicates were resolved in an SDS-PAGE gel followed by staining with Bio-Safe Coomassie G-250 (BioRad, 1610786) and submitted for mass spectrometry analysis at the Mayo Clinic Proteomics Core. For iKRAS 4292 cells, sample groups included no TMEM192-HA control, TMEM192-HA + doxycycline (KRASG12D), or TMEM192-HA without doxycycline (KRAS WT).
In-gel tryptic digestions were performed by excising gel lanes and mincing them into 1 mm cubes. Gel pieces were suspended in 200 mM Tris pH 8.2 prior to destaining with 40% acetonitrile/50 mM Tris pH 8.2, and dehydration with 100% acetonitrile. These three steps were repeated until color was removed from gel pieces. Destaining was followed by reduction with 50 mM TCEP for 30 min at 60°C, dehydration with acetonitrile, alkylation with 25 mM iodoacetamide/50 mM Tris pH 8.2 for 30 min at room temperature in the dark, and dehydration with acetonitrile prior to trypsin digestion. Proteins were in-gel typsin digest with 80 μL of 0.002 μg/μL trypsin (Promega Corporation, Madison, WI) prepared in 25 mM Tris pH 8.2/0.0002% Zwittergen 3–16 and incubated overnight at 37°C. Peptide extraction was accomplished by adding 4% trifluoroacetic acid and acetonitrile to the digestion solution and incubated at room temperature for 40 min. Liquid was saved. A second extraction of acetonitrile was added to the gel pieces for 10 min and saved with the initial extraction. Extractions were dried and stored at −20°C.
Tryptic peptides were suspended in sample buffer (0.2% formic acid/0.1% TFA/0.002% zwittergent 3–16). A portion of the sample was analyzed by nano-flow liquid chromatography electrospray tandem mass spectrometry (nanoLC-ESI-MS/MS) using a Thermo Scientific Q-Exactive Mass Spectrometer (Thermo Fisher Scientific, Bremen, Germany) coupled to a Thermo Ultimate 3000 RSLCnano HPLC system. The digest peptide mixture was loaded onto a 330 nL Halo 2.7 ES-C18 trap (Optimize Technologies, Oregon City, OR). Chromatography was performed using a 2%–40% gradient of solvent B over 90 min where solvent A is (98% water/2% acetonitrile/0.2% formic acid) and solvent B is (80% acetonitrile/10% isopropanol/10% water/0.2% formic acid). Peptides were eluted at a flow rate of 400 nL/min from the trap through a PicoFrit (New Objective, Woburn, MA) 100 μm × 33 cm column hand packed with Agilent Poroshell 120 EC C18 packing (Agilent Technologies, Santa Clara, CA). Q-Exactive mass spectrometer was set to acquire an ms1 survey scans from 340 to 1600 m/z at resolution 70,000 (at 200 m/z) with an AGC target of 3e6 ions and a maximum ion inject time of 60 msec. Survey scans were followed by HCD MS/MS scans on the top 15 ions at resolution 17,500 with an AGC target of 2e5 ions, and maximum ion inject time of 60 msec. The isolation window set at 2.5 m/z with a 0.3 m/z offset. Dynamic exclusion placed selected ions on an exclusion list for 40 s.
Analysis was performed using MaxQuant and Perseus software (both from Max Planck Institute of Biochemistry, Martinsried, Germany). MaxQuant software, version 1.6.7.0, was used to extract, time align, and database search (Uniprot mouse, 2019_05 release date) chromatographic extracted peptide peaks generated from mass spectrometry files.87,88 Label-free relative quantitation parameters within the MaxQuant software were used to generate normalized protein intensities89 reported in the proteins group table. Perseus software, version 1.6.7.0, was used to perform differential expression of identified proteins.90 Briefly, protein intensities were log2 transformed, missing values were imputed (0.3 width, 1.8 downshift), students T test was performed in which an estimation of difference was calculated, and p values and q-values were reported. Protein groups with a p value of ≤0.05, and a log2 difference of at least ±1 were considered as differentially enriched on lysosomes.
The proteomic data have been deposited in MassIVE (Welcome to MassIVE (ucsd.edu)) through ProteomeXchange (http://www.proteomexchange.org/). Dataset accession is MSV000091036.
Extracellular matrix degradation assay
Extracellular matrix degradation was measured following two similar approaches. In the first approach, cells were embedded in matrix composed of 75μg/ml of DQ-Collagen Type I (Invitrogen, D12060) and 5 mg/ml Matrigel (Corning, 356231). In the second approach, cells were plated on coverslips coated with matrix composed of 75μg/ml of DQ-Collagen Type I (Invitrogen, D12060) and 0.2% gelatin (Sigma, G815′−100G). Matrix degradation was carried out for 48h. Cells from the first approached were imaged live, whereas cells from second approach were fixed with 2.5% formaldehyde fixing buffer as described above. Nuclei were stained with Hoechst 33342 (Invitrogen, H3570). The mean fluorescence intensity of cleaved DQ-Collagen was quantitated with ImageJ. In brief, the Auto Local Threshold function was used to threshold cleaved DQ-collagen I fluorescent spots, then the following functions were performed in order: convert to mask, watershed. The Analyze Particles function was used to measure DQ-Collagen I mean fluorescence intensity. For cathepsin B-mCherry overexpression experiments, only cells expressing mCherry or Cathepsin B-mCherry were scored.
Lysosome activity assays
Cathepsin B proteolytic activity was assessed in control or DOCK8 knockdown cells, as well as in cells treated with DMSO or 2μM Cytochalasin D (Cayman Chemical, 11330) for 5 h. Cathepsin B proteolytic activity was assessed using Magic Red Cathepsin B Assay (Immunochemistry, 937). Cells were seeded on coverslips at 70% confluency overnight. Coverslips were inverted onto 80μL of 1x Magic Red Cathepsin B in media supplemented with 10% FBS and incubated for 30 min at 37°C covered from light. Coverslips were washed twice with HBSS and cultured for 5 min in media supplemented with 10% FBS and Hoechst 33342 (Invitrogen, H3570) to label nuclei. Coverslips were mounted on 50μL of media on slides and images of live cells were acquired using a Zeiss LSM-980 confocal microscope with a 63× oil objective lens and Zen software. Quantitation of Magic Red mean fluorescence intensity was done using ImageJ. In brief, the Auto Local Threshold function was used to threshold Magic Red Cathepsin B fluorescent spots, then the following functions were performed in order: convert to mask, watershed. The Analyze Particles function was used to measure Magic Red Cathepsin B mean fluorescence intensity, which was normalized to cell number and then to nontargeting control or DMSO (set to one for each of three independent biological replicates).
Changes to lysosome pH were tested in control or DOCK8 knockdown cells seeded on imaging dishes using pH-sensitive LysoTracker. Cells were cultured with 1:20,000 LysoTracker deep red (Invitrogen, L12492) in media with 10% FBS for 1 h at 37°C. Nuclei was labeled using Hoechst 33342 (Invitrogen, H3570). Cells were washed three times with HBSS followed by addition of media with 10% FBS. Images were acquired in live cells using a Zeiss LSM-780 confocal microscope with a 40× oil objective lens and Zen software. Quantitation of LysoTracker mean fluorescence intensity was done using ImageJ following the same methods as for Magic Red Cathepsin B mentioned above.
To measure degradation of the EGF Receptor, control or DOCK8 knockdown cells were cultured with 50 mg/ml EGF (E9644-2MG) for 0, 1.5, 2.5, and 5 h (6741P), 0, 6, 12, and 17 h (L3.6), or 0, 4, 18, 43 h (Panc04.03) at 37°C. Cell lysates were collected, and EGFR protein levels were assessed by immunoblotting as described above. Band densitometry was quantitated with ImageJ. For DQ-BSA assays, control or DOCK8 knockdown cells were cultured overnight in serum-free media, incubated with 0.5 mg/ml DQ-BSA (Invitrogen, D12050) and 1 mg/mL 70 kDa tetramethylrhodamine (TMR)-dextran (Thermo Fisher Scientific, D1818) in serum-free media for 1 h at 37°C. Cells were washed three times with HBSS, cultured in media with 10% FBS for 0 and 3 h, fixed with formaldehyde fixing buffer (0.1M PIPES, pH6.9, 1Mm EGTA, 3mm MgSO4, 2.5% formaldehyde). Nuclei were labeled using Hoechst 33342 (Invitrogen, H3570). Images were acquired using a Zeiss LSM-980 confocal microscope with a 63× oil objective lens and Zeiss Zen software. Quantitation of DQ-BSA and TMR-dextran puncta mean area was performed using ImageJ.
Transcriptomic data analysis
DOCK8 gene expression in pancreatic cancer was assessed by combination of analysis of publicly available databases and RNAseq performed at the Mayo Clinic. Transcriptomic data from normal pancreas (The Genotype-Tissue Expression, (GTEx)) and pancreatic cancer tumors (The Cancer Genome Atlas, TCGA) were analyzed and compared using TNMplot.91 Expression of DOCK8 in pancreatic cancer cells was determined by analysis of RNA seq data from pancreatic cancer PDOs generated by Crown Bioscience (https://www.crownbio.com/databases) and from the Cancer Cell Line Encyclopedia through XenaBrowser. Z score was calculated using gene expression values.
For PDAC PDOs generated at the Mayo Clinic, surgically resected tumor specimens were collected for research with informed consent with approval the Mayo Clinic Internal Review Board and deidentified. Tissues were minced and dissociated using a human Tumor Dissociation Kit (Miltenyi Biotec, 130-095-929) and plated in a 12-well flat bottom culture plate (Corning, 3513) coated with Matrigel (Corning, 354-230) in PaTOM media containing DMEM + Glutamax (Gibco; 10564-011), 0.1% Penicillin-Streptomycin-Amphotericin B Solution, 0.25 μg/mL hydrocortisone (Sigma; H0888), 1% B27 (Gibco; 12587-010), 50 μg/mL L-Ascorbic acid (Sigma; A92902), 20 μg/mL insulin (Sigma; I9278), 100 ng/mL FGF2 (R&D Systems; 233-FB), and 100 nM all-trans retinoic acid (Sigma; R2625). RNA was extracted from the PDAC PDOs using the Qiagen RNeasy Micro Kit (Qiagen; 74004). cDNA libraries were prepared using 200 ng of total RNA according to the manufacturer’s instructions for the TruSeq Stranded mRNA Sample Prep Kit (Illumina). The concentration and size distribution of the completed libraries were determined using an Agilent Bioanalyzer DNA 1000 chip and Qubit fluorometry (Invitrogen). Libraries were sequenced at 6 samples per lane following Illumina’s standard protocol using the Illumina cBot and HiSeq 3000/4000 PE Cluster Kit. The flow cells were sequenced as 100 X 2 paired end reads on an Illumina HiSeq 4000 using HiSeq 3000/4000 sequencing kit and HD 3.4.0.38 collection software. Base-calling was performed using Illumina’s RTA version 2.7.7. RNA-seq FASTQ files were aligned using STAR version 2.7.8a to the human reference genome GRCh38. Gene level counts were obtained using HTseq-count v.0.9.1 and read counts were normalized with R package DESeq2 version 2.11.40.7. Z score was calculated using gene expression values.
The organoid RNA-seq data are deposited and available in the NCBI Gene Expression Omnibus (GEO GSE185335).
qPCR mRNA analysis
iKRASG12D cells were cultured with 1 μg/mL doxycycline hyclate (Sigma, D9891) or 72-h doxycycline withdrawal to revert cells to WT KRAS. Other cell lines were cultured as described above. RNA was isolated using RNeasy Plus Mini Kit (Qiagen, 74134) and reverse transcribed with Super Script III First Strand Kit (Invitrogen, 18080-51). DOCK8 gene expression was quantitated using SYBR green fluorescence on a LightCycler 480 (Roche, 04707516001) and primers for mouse DOCK8: Fwd 5′-TCAGTTTATGTGCGGAGAAGAC-3′, Rev 5′-TGGCAGGGAGCTTAATTTTCAC −3’. Human DOCK8: Fwd 5′-CCGCACAAAGAGATTTTGGA-3′, Rev 5′-TCAGCCTCTGTG GGTAGACA-3’. Mouse cathepsin B: Fwd 5′-TCCTTGATCCTTCTTTCTTGCC-3′, Rev 5′-ACAGTGCCACACAGCTTCTTC-3’. Human cathepsin B: Fwd 5′-TTCTTGCGACTCTTGGGACTTC-3′, Rev 5′-TGACGAGGATGACAGGGAACTA-3’.83 18S: Fwd: 5′-CGCTTCCTT ACCTGGTTGAT-3′, Rev: 5′-GAGCGACCAAAGGAACCATA-3′. Relative mRNA expression was determined by normalizing DOCK8 to 18S using the ΔΔCt method.
Cell viability and cell cycle analysis
Cell viability was assessed in control and DOCK8 knockdown cells. In brief, DOCK8 was knocked down using siRNA, and 48 h later cells were plated on a 96 well plate and allowed to proliferate for three days. MTT assay was performed using CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (Promega, G5421) following the manufacturer’s protocol. For the crystal violet assay, DOCK8 was knocked down, and 48 h later cells were plated in triplicate and allowed to proliferate for three days. Cells were fixed with 25% glutaraldehyde (Sigma, G6257) for 10 min and stained with 12.25mM crystal violet solution in 1:3 methanol and dH20 for 10 min. Cells were washed, dried overnight, and destained for 10 min in 1:1 Ethanol and 200mM sodium citrate solution (Sigma, S-4641). The absorbance was read at 550nm. To measure cell cycle distribution, mKPC CRISPR control or DOCK8 knockout cells were seeded in 6 well plates for 3 days, then when at 80% confluence were trypsinized and resuspended in cell culture medium. The cells were pelleted, washed with PBS, and resuspended in 75% ice-cold ethanol and incubated on ice 30 min. Following washing in PBS, cells were resuspended in 1mL PBS with RNase (60 μg/mL, Sigma, R4642) and propidium iodide (2.5 μg/mL, Sigma, P4864) and incubated on ice for 45 min prior to analysis by flow cytometry in the Mayo Clinic Microscopy and Cell Analysis Core. The cells were analyzed using a BD FACSCanto 2-laser flow cytometer (BD Biosciences) using FACSDiva v8.0.1 software. Cells were collected in the 488C detector with a filter set of 585/42 nm. FlowJo software was used to gate and quantify the peaks for G0/G1, G2/M, and S phase.
CRISPR KO cell lines
DOCK8 was knocked out in mKPC cells using the CRISPR/Cas9 system. CHOPCHOP was used to design the gRNA (Fwd: 5′ CTTCCTTCCTTCCTTCCTTCC-3′ and Rev: 5′-ACACGGGGTGTTTGATTTCC-3′) to target exon 6 of murine DOCK8. gRNA was cloned into lentiCRISPR v2 (a gift from Dr. Feng Zhang, Addgene plasmid #52961) as previously described.92 lentiCRISPR v2-DOCK8-gRNA or control were transfected into mKPC cells. After 48 h, cells were selected with puromycin (Gibco, A11138-03). Single cell clones were isolated and expanded. DOCK8 knockout was validated by immunoblotting and Sanger sequencing. DOCK8-gRNA target region was PCR amplified using primers Fwd 5′-ATAGCTATGTTTGCATTTCC-3′ and Rev 5′-CCCCTCTATTGAAAA GATGCTG-3’. PCR fragments were gel purified and Sanger sequenced using the same primers used for PCR reaction. Control clones 1, 3 and DOCK8 clones 1, 2 were used for in vivo experiments (Sequencing results are deposited in Mendeley Data, https://doi.org/10.17632/zmwy6bgfg8.1).
In vivo tumor growth and metastasis
In vivo experiments were conducted under Institutional Animal Care and Use Committee approval and in accordance with the approved protocol. Pancreatic orthotopic injections were performed on syngeneic 8-week-old female and male C57Bl/6J mice93 (The Jackson Laboratories, 000664). Buprenorphine (0.5 mg/kg) was administered intraperitoneally an hour prior to the procedure. Mice were anesthetized with isoflurane, and 1 × 104 mKPC control clone 1 or 3, or DOCK8 KO clone 1 or 2 cells were resuspended in 30μL of 50% (6.5 mg/ml) Matrigel in sterile PBS94 and were injected into the tail of the pancreas. A power analysis based on previous studies was used to calculate the total of 6 mice per group (n = 3 male and 3 female for each). Mice were observed and weighed daily for 7 days, then every other day for three weeks. Mice were sacrificed by CO2 inhalation compliant with the AVMA three weeks post-surgery. During necropsy, the abdominal cavity as well as chest were inspected. Metastases were detected primarily in the abdominal cavity, intestinal mesentery, spleen, and liver, and were counted. The mean tumor weight and mean number of metastases were calculated. Mice without a primary tumor were excluded from quantitation. Upon necropsy, pancreas, primary and metastatic tumors were resected and weighted. Tissues were washed with sterile PBS and fixed in 4% formaldehyde solution for 24 h at 4°C. Next, tissues were transferred to a 30% sucrose solution (dissolved in sterile PBS) and incubated for 24 h at 4°C. Tissues were embedded and frozen in O.C.T. compound (Tissue-Tek, 4583), and then sectioned and stained with hematoxylin and eosin by the Mayo Clinic Biomaterials and Histomorphometry Core Laboratory. Normal pancreas, primary and metastatic tumors were verified by Mayo Clinic pathologist Dr. Lizhi Zhang. Images of H&E stained tissues were acquired with a Zeiss Axio Scope A1 using 2.5X and 20× objective lenses, and Zeiss Zen software.
Transmission electron microscopy
For standard transmission electron microscopy, samples were prepared as previously described.95 Briefly, cells on carbon-coated coverslips were rinsed in 37°C HBSS and fixed with 37°C primary fixative (100 mM cacodylate, pH 7.4, 60 mM sucrose, 2.5% glutar-aldehyde) for 1 h at room temperature, rinsed three times with washing buffer (100 mM cacodylate, pH 7.4, 200 mM sucrose) then fixed in the secondary fixative (50 mM cacodylate, pH 7.4, 100 mM sucrose, 1% OsO4) for 1 h at room temperature, rinsed three times in water and fixed in 1% uranyl acetate in water for 1 h at room temperature. Samples were then dehydrated in a graded ethanol series, embedded in Quetol 651 (Ted Pella, Redding, CA) and polymerized in a 65C oven overnight. After removal from the oven, the coverslip was removed from the bottom of the sample, the block trimmed down to a trapezoid 1 mm wide at the base, and 100 nm thin sections were cut and viewed on a Jeol 1200 transmission electron microscope (Jeol Ltd, Tokyo, Japan).
Time-lapse live cell microscopy
Cells were cultured on 35 mm Petri dishes with an 18 mm well and #1.5 coverglass (Cell E&G, #GBD00004-200). Imaging was performed on a Zeiss LSM980 + Fast Airyscan2 confocal microscope (Carl Zeiss, Thornwood, NY) using a 63× oil 1.4 NA objective lens with frames acquired every 10 s for 10 min (60 frames total for Lamp1-mCherry tracks) or every 3 s for 5 min (100 frames total for Lysotracker-FR tracks). After acquisition, the files were post processed by the Airyscan module and saved as.czi files.
Lysosomal tracking with TrackMate
The.czi files were imported into Fiji v.2.3.0/1.53F96 for analysis with the plugin TrackMate.97 Movie files were analyzed either manually (Lamp1-mCherry, 12 frames analyzed per movie, 3 cells per condition in each of 3 biological replicates) using the manual tracking function or by software automation (Lysotracker-FR movies, 100 frames analyzed). Automatic tracking of the lysotracker movies started with opening the file and identifying lysosomes using the LoG detector (Laplacian of Gaussian) (estimated object diameter: 0.75 (0.3 when indicated), quality threshold: 50.0, pre-process with median filter and sub-pixel localization boxes both checked), no initial thresholding, the LAP tracker (linear assignment problem mathematical framework) was then used to generate tracks (Max distance: 2.5 μm, gap closing allowed: max distance 1.5 μm, and max frame gap of 1 frame) and tracks were then filtered to only count tracks of 5 frames and longer. Colorized tracks representing ‘‘max distance traveled’’ were then generated and pseudocolored with a rainbow heatmap with red colors indicating longer distances and blue indicating shorter distances traveled.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitation of microscopy images and immunoblots were performed using ImageJ. Adjustments of fluorescence intensity for all images were done uniformly in each individual experiment. Data was analyzed and graphed using GraphPad Prism. Graphs represent mean ± SEM. p-values were calculated using a Student’s t-test, or Tukey test following one-way-ANOVA.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit Anti-DOCK8 | Abcam | Cat# ab227529; RRID:AB_2943448 |
| Rabbit Anti-Cathepsin B | Cell Signaling | Cat# 31718S; RRID:AB_2687580 |
| Mouse Anti-HA | Cell Signaling | Cat# 2367S; RRID:AB_10691311 |
| Mouse Anti-Flag | Cell Signaling | Cath# 8146S; RRID:AB_10950495 |
| Rabbit Anti-GAPDH | Cell Signaling | Cat# 5174S; RRID:AB_10622025 |
| Rabbit Anti-phospho-ERK 1/2 | Cell Signaling | Cat# 4377S; RRID:AB_331775 |
| Rabbit Anti-ERK 1/2 | Cell Signaling | Cat# 9102S; RRID:AB_330744 |
| Rat Anti-LAMP1 | DHSB | Cat# 1D4B; RRID:AB_2134500 |
| Rabbit Anti-Actin | Sigma | Cat# A2066; RRID:AB_476693 |
| Mouse Anti-KRAS | Sigma | Cat# WH0003845M1; RRID:AB_1842235 |
| Mouse Anti-GFP | Sigma; Roche | Cat# 11814460001; RRID:AB_390913 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| DMSO | Sigma | Cat# D2438-10ML |
| Bafilomycin A1 | Cayman Chemical | Cat# 11038 |
| CA-074ME | Cayman Chemical | Cat# 18469 |
| Cytochalasin D | Cayman Chemical | Cat# 11330 |
| Staurosporine | Cayman Chemical | Cat# 81590 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Proteome Profiler Human Protease Array | R&D Systems | Cat# ARY021B |
|
| ||
| Deposited Data | ||
|
| ||
| Comparative Lysosome Proteomics | Welcome to MassIVE (ucsd.edu) | MassIVE: MSV000091036 |
| PDAC PDX Organoid RNAseq | Gene Expression Omnibus | GEO: GSE185335 |
| Western blot and CRISPR sequencing data | Mendeley Data | Mendeley: https://doi.org/10.17632/zmwy6bgfg8.1 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| mKPC | Dr. David Tuveson36 | RRID: mKPC T4-2D |
| iKRASG12D | Dr. Marina Pasca di Magliano13,32 | RRID: 4292 iKRASG12D |
| 6741-P | Dr. Daniel Billadeau, Mayo Clinic, Rochester, MN | RRID: 6741-P |
| Panc 0403 | ATCC | Cat# CRL-2555 |
| PANC1 | ATCC | Cat# CRL-1469 |
| DanG | Dr. Daniel Billadeau, Mayo Clinic, Rochester, MN | RRID: NA |
| Capan1 | ATCC | Cat# HTB-79 |
| CFPAC | ATCC | Cat# CRL-1918 |
| MIA PaCa2 | ATCC | Cat# CRL-1420 |
| HPAFII | ATCC | Cat# CRL-1997 |
| L3.6 | Dr. Steven Johnsen, Bosch Institute, Stuttgart, Germany | |
| BxPC3 | ATCC | Cat# CRL-1687 |
| RAW 264.7 | ATCC | Cat# TIB-71 |
| HPDE | Dr. Daniel Billadeau, Mayo Clinic, Rochester, MN | |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| C57Bl/6J mice | The Jackson Laboratories | Cat# 000664 RRID:IMSR_JAX:000664 |
|
| ||
| Oligonucleotides | ||
|
| ||
| 5′- CCGCTAGCGCTACCGGACTCAGATC TCGAGATGTGGCAGCTCTGGGCCTCC-3′ |
This Paper | Human cathepsin B Fwd |
| 5′- GCTCACCATGGTGGCGACCGGTGGA TCCCGGGCGGGGGCCACCTGGCTGG-3′ |
This Paper | Human cathepsin B Rev |
| 5′-ATTACGCTGGTACCGAGCTCC TCCGGAGGTTCATGTACACCACC CCGTTCACC-3′ |
This Paper | DOCK8-DHR2-HA Fwd |
| 5′ CGGCCGCCACTGTGCTGGATTTA GCTGCCCTGTGACAACTGGGTTTCA CATTTCC-3′ |
This Paper | DOCK8-DHR2-HA Rev |
| 5′-TCAGTTTATGTGCGGAGAAGAC-3′ | Primer Bank | qPCR msDOCK8 Fwd |
| 5′-TGGCAGGGAGCTTAATTTTCAC-3′ | Primer Bank | qPCR msDOCK8 Rev |
| 5′-CCGCACAAAGAGATTTTGGA-3′ | Dr. Helen C. Su82 | qPCR hDOCK8 Fwd |
| 5′-TCAGCCTCTGTGGGTAGACA-3′ | Dr. Helen C. Su82 | qPCR hDOCK8 Rev |
| 5′-TTCTTGCGACTCTTGGGACTTC-3′ | Dr. Stephen Shea83 | qPCR hCathpsin B Fwd |
| 5′-TGACGAGGATGACAGGGAACTA-3′ | Dr. Stephen Shea83 | qPCR hCathpsin B Rev |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCI2-DOCK8-YFP | Dr. Daniel Billadeau, | RRID: NA |
| pCI2-DOCK8-V1985A-YFP | Dr. Daniel Billadeau | RRID: NA |
| pLenti6.3. F-MCS | Dr. Daniel Billadeau | RRID: NA |
| pLenti DOCK8 WT | This Paper | RRID:NA |
| pLenti DOCK8 VA | This Paper | RRID:NA |
| pcDNA3.1_hCathepsin B | Dr. Hyeryun Choe | Addgene_11249 |
| pmCherry-N1 | Clontech | Clontech_632523 |
| pcDNA3.1-HA | Dr. Oskar Laur | Addgene_128034 |
| pEGFP-N1 | Clontech | Clontech_6085-1 |
| pBabe-Kras Wt | Dr. Channing Der | Addgene_75282 |
| pLJC5-Tmem192-3xHA | Dr. David Sabatini | Addgene_102930 |
| pEGFP-N1-LAMP1 | Dr. Mark McNiven | RRID:NA |
|
| ||
| Software and Algorithms | ||
|
| ||
| ImageJ | NIH | ImageJ (nih.gov) |
| Zen | ZEISS | ZEISS ZEN Microscopy Software |
| GraphPad Prism | GraphPad by Dotmatics | https://www.graphpad.com |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator |
Highlights.
Lysosomal proteomics of pancreatic cancer cells identified the Cdc42 GEF DOCK8
Expression of DOCK8 promotes tumor cell proliferation and invasion
DOCK8 controls lysosome size and motility by regulating lysosome-associated actin
DOCK8 regulates secretion of lysosomal cathepsins to promote invasion
ACKNOWLEDGMENTS
We acknowledge Dr. Lizhi Zhang for pathology support and the Mayo Clinic Proteomics Core Facility, the Mayo Clinic Cell Analysis Core Facility, and members of the McNiven laboratory for helpful suggestions and technical support. This research was supported by R01 CA104125 (G.L.R. and M.A.M.) and CA265050 (M.E.F.-Z.); Sigma Xi Grants in Aid Award G201903158642619 (O.L.G.-R.); the Mayo Clinic Cancer Center (P30 CA015083); the Mayo Clinic Center for Biomedical Discovery; the Mayo Clinic Center for Signaling in Gastroenterology (P30DK084567); the Mayo Clinic SPORE in Pancreatic Cancer (P50 CA102701); and the Mayo Clinic Graduate School for Biomedical Sciences (O.L.G.-R., R.E.N., N.C.R.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113042.
REFERENCES
- 1.Davidson SM, and Vander Heiden MG (2017). Critical Functions of the Lysosome in Cancer Biology. Annu. Rev. Pharmacol. Toxicol 57, 481–507. 10.1146/annurev-pharmtox-010715-103101. [DOI] [PubMed] [Google Scholar]
- 2.Tang T, Yang Z, Wang D, Yang X, Wang J, Li L, Wen Q, Gao L, Bian X, and Yu S. The Role of Lysosomes in Cancer Development and Progression.Cell Biosci. 2020. Nov 18;10(1):131 10.1186/s13578-020-00489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen RJ, Lyu YJ, Chen YY, Lee YC, Pan MH, Ho YS, and Wang YJ (2021). Chloroquine Potentiates the Anticancer Effect of Pterostilbene on Pancreatic Cancer by Inhibiting Autophagy and Downregulating the RAGE/STAT3 Pathway. Molecules 26, 6741. 10.3390/MOLECULES26216741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao B, Dierichs L, Gu J-N, Trajkovic-Arsic M, Axel Hilger R, Savvatakis K, Vega-Rubin-De-Celis S, Liffers S-T, Peña-Llopis S, Behrens D, et al. (2020). TFEB-mediated lysosomal biogenesis and lysosomal drug sequestration confer resistance to MEK inhibition in pancreatic cancer. Cell Death Discov. 6, 12. 10.1038/s41420-020-0246-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elliott IA, Dann AM, Xu S, Kim SS, Abt ER, Kim W, Poddar S, Moore A, Zhou L, Williams JL, et al. (2019). Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc. Natl. Acad. Sci. USA 116, 6842–6847. 10.1073/PNAS.1812410116/SUPPL_FILE/PNAS.1812410116.SD02.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Podgorski I, Linebaugh BE, Sameni M, Jedeszko C, Bhagat S, Cher ML, and Sloane BF (2005). Bone microenvironment modulates expression and activity of cathepsin B in prostate cancer. Neoplasia 7, 207–223. 10.1593/NEO.04349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dykes SS, Fasanya HO, and Siemann DW (2019). Cathepsin L secretion by host and neoplastic cells potentiates invasion. Oncotarget 10, 5560–5568. 10.18632/oncotarget.27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gopinathan A, DeNicola GM, Frese KK, Cook N, Karreth FA, Mayerle J, Lerch MM, Reinheckel T, and Tuveson DA (2012). Cathepsin B promotes the progression of pancreatic ductal adenocarcinoma in mice. Gut 61, 877–884. 10.1136/GUTJNL-2011-300850. [DOI] [PubMed] [Google Scholar]
- 9.Machado ER, Annunziata I, van de Vlekkert D, Grosveld GC, and d’Azzo A. (2021). Lysosomes and Cancer Progression: A Malignant Liaison. Front. Cell Dev. Biol 9, 373. 10.3389/FCELL.2021.642494/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burton KM, Johnson KM, Krueger EW, Razidlo GL, and McNiven MA (2021). Distinct forms of the actin cross-linking protein α-actinin support macropinosome internalization and trafficking. Mol. Biol. Cell 32, 1393–1407. 10.1091/MBC.E20-12-0755/ASSET/IMAGES/LARGE/MBC-32-1393-G005.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Victor BC, Anbalagan A, Mohamed MM, Sloane BF, and Cavallo-Medved D. (2011). Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res. 13, R115. 10.1186/BCR3058/FIGURES/7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mostafa Mohamed M, and Sloane BF (2006). Cysteine cathepsins: multifunctional enzymes in cancer. Nat. Rev. Cancer 6, 764–775. 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 13.Collins MA, Bednar F, Zhang Y, Brisset JC, Galbá n S, Galbá n CJ, Rakshit S, Flannagan KS, Adsay NV, and Pasca Di Magliano M. (2012). Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest 122, 639–653. 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eser S, Schnieke A, Schneider G, and Saur D. (2014). Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 111, 817–822. 10.1038/bjc.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerr EM, Gaude E, Turrell FK, Frezza C, and Martins CP (2016). Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 531, 110–113. 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’Antonio G et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bryant KL, Mancias JD, Kimmelman AC, and Der CJ (2014). KRAS: feeding pancreatic cancer proliferation. Trends Biochem. Sci 39, 91–100. 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buccione R, Caldieri G, and Ayala I. (2009). Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 28, 137–149. 10.1007/S10555-008-9176-1. [DOI] [PubMed] [Google Scholar]
- 20.Clark ES, Whigham AS, Yarbrough WG, and Weaver AM Cortactin Is an Essential Regulator of Matrix Metalloproteinase Secretion and Extracellular Matrix Degradation in Invadopodia.Cancer Res. 2007. May 1;67(9):4227–4235. doi: 10.1158/0008-5472.CAN-06-3928. 10.1158/0008-5472.CAN-06-3928. [DOI] [PubMed] [Google Scholar]
- 21.Baldassarre M, Ayala I, Beznoussenko G, Giacchetti G, Machesky LM, Luini A, and Buccione R. (2006). Actin dynamics at sites of extracellular matrix degradation. Eur. J. Cell Biol 85, 1217–1231. 10.1016/J.EJCB.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 22.Neel NF, Rossman KL, Martin TD, Hayes TK, Yeh JJ, and Der CJ (2012). The RalB Small GTPase Mediates Formation of Invadopodia through a GTPase-Activating Protein-Independent Function of the RalBP1/RLIP76 Effector. Mol. Cell Biol 32, 1374–1386. 10.1128/MCB.06291-11/ASSET/A0D10192-433E-4EC3-BBE2-5939B0B8D1A7/ASSETS/GRAPHIC/ZMB9991094520009.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eddy RJ, Weidmann MD, Sharma VP, and Condeelis JS (2017). Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol. 27, 595–607. 10.1016/J.TCB.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evers EE, Zondag GC, Malliri A, Price LS, Ten Klooster JP, Van Der Kammen RA, and Collard JG (2000). Rho family proteins in cell adhesion and cell migration. Eur. J. Cancer 36, 1269–1274. 10.1016/S0959-8049(00)00091-5. [DOI] [PubMed] [Google Scholar]
- 25.Murphy DA, and Courtneidge SA (2011). The ‘‘ins’’ and ‘‘outs’’ of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol 12, 413–426. 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernandez-Zapico ME, Gonzalez-Paz NC, Weiss E, Savoy DN, Molina JR, Fonseca R, Smyrk TC, Chari ST, Urrutia R, and Billadeau DD (2005). Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell 7, 39–49. 10.1016/j.ccr.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 27.Razidlo GL, Wang Y, Chen J, Krueger EW, Billadeau DD, and McNiven MA (2013). Dynamin 2 Potentiates Invasive Migration of Pancreatic Tumor Cells through Stabilization of the Rac1 GEF Vav1. Dev. Cell 24, 573–585. 10.1016/j.devcel.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Razidlo GL, Schroeder B, Chen J, Billadeau DD, and McNiven MA (2014). Vav1 as a central regulator of invadopodia assembly. Curr. Biol 24, 86–93. 10.1016/j.cub.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Randall KL, Lambe T, Johnson AL, Treanor B, Kucharska E, Doma-schenz H, Whittle B, Tze LE, Enders A, Crockford TL, et al. (2009). Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat. Immunol 10, 1283–1291. 10.1038/ni.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Randall KL, Chan SSY, Ma CS, Fung I, Mei Y, Yabas M, Tan A, Arkwright PD, Al Suwairi W, Lugo Reyes SO, et al. (2011). DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J. Exp. Med 208, 2305–2320. 10.1084/JEM.20110345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiraishi A, Uruno T, Sanematsu F, Ushijima M, Sakata D, Hara T, and Fukui Y. (2017). DOCK8 Protein Regulates Macrophage Migration through Cdc42 Protein Activation and LRAP35a Protein Interaction * □ S Downloaded from. J. Biol. Chem 292, 2191–2202. 10.1074/jbc.M116.736306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, Galbán CJ, Galbán S, and di Magliano MP (2012). Metastatic Pancreatic Cancer Is Dependent on Oncogenic Kras in Mice. PLoS One 7, e49707. 10.1371/journal.pone.0049707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harada Y, Tanaka Y, Terasawa M, Pieczyk M, Habiro K, Katakai T, Hanawa-Suetsugu K, Kukimoto-Niino M, Nishizaki T, Shirouzu M, et al. (2012). DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood 119, 4451–4461. 10.1182/blood-2012-01-407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hogenson TL, Xie H, Phillips WJ, Toruner MD, Li JJ, Horn IP, Kennedy DJ, Almada LL, Marks DL, Carr RM, et al. (2022). Culture media composition influences patient-derived organoid ability to predict therapeutic responses in gastrointestinal cancers. JCI Insight 7, e158060. 10.1172/jci.insight.158060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boj SF, Hwang CI, Baker LA, Chio IIC, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. (2015). Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 38.Qiang L, Cao H, Chen J, Weller SG, Krueger EW, Zhang L, Razidlo GL, and McNiven MA (2019). Pancreatic tumor cell metastasis is restricted by MT1-MMP binding protein MTCBP-1. J. Cell Biol 218, 317–332. 10.1083/jcb.201802032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su H, Yang F, Fu R, Trinh B, Sun N, Liu J, Kumar A, Baglieri J, Siruno J, Le M, et al. (2022). Collagenolysis-dependent DDR1 signalling dictates pancreatic cancer outcome. Nature 610, 366–372. 10.1038/s41586-022-05169-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pal K, Pletnev AA, Dutta SK, Wang E, Zhao R, Baral A, Yadav VK, Aggarwal S, Krishnaswamy S, Alkharfy KM, et al. (2014). Inhibition of Endoglin-GIPC Interaction Inhibits Pancreatic Cancer Cell Growth. Mol. Cancer Therapeut 13, 2264–2275. 10.1158/1535-7163.MCT-14-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sagar G, Sah RP, Javeed N, Dutta SK, Smyrk TC, Lau JS, Giorgadze N, Tchkonia T, Kirkland JL, Chari ST, et al. (2016). Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut 65, 1165–1174. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu PH, Onodera Y, Giaccia AJ, Le QT, Shimizu S, Shirato H, and Nam JM (2020). Lysosomal trafficking mediated by Arl8b and BORC promotes invasion of cancer cells that survive radiation. Commun. Biol 3, 620–715. 10.1038/s42003-020-01339-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu X, Chen L, Chen Y, Shao Q, and Qin W. (2015). Bafilomycin A1 inhibits the growth and metastatic potential of the BEL-7402 liver cancer and HO-8910 ovarian cancer cell lines and induces alterations in their microRNA expression. Exp. Ther. Med 10, 1829–1834. 10.3892/ETM.2015.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, and Kimmelman AC (2014). Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discov. 4, 905–913. 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shiraishi A, Uruno T, Sanematsu F, Ushijima M, Sakata D, Hara T, and Fukui Y. (2017). DOCK8 protein regulates macrophage migration through Cdc42 protein activation and LRAP35a protein interaction. J. Biol. Chem 292, 2191–2202. 10.1074/jbc.M116.736306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krishnaswamy JK, Singh A, Gowthaman U, Wu R, Gorrepati P, Sales Nascimento M, Gallman A, Liu D, Rhebergen AM, Calabro S, et al. (2015). Coincidental loss of DOCK8 function in NLRP10-deficient and C3H/HeJ mice results in defective dendritic cell migration. Proc. Natl. Acad. Sci. USA 112, 3056–3061. 10.1073/pnas.1501554112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kunimura K, Uruno T, and Fukui Y. (2020). DOCK family proteins: key players in immune surveillance mechanisms. Int. Immunol 32, 5–15. 10.1093/intimm/dxz067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ham H, Guerrier S, Kim J, Schoon RA, Anderson EL, Hamann MJ, Lou Z, and Billadeau DD (2013). DOCK8 Interacts with Talin and WASPto Regulate Natural Killer Cell Cytotoxicity. J Immunol. 190. 10.4049/jimmunol.1202792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joshi VB, Gutierrez Ruiz OL, and Razidlo GL (2023). The Cell Biology of Metastatic Invasion in Pancreatic Cancer: Updates and Mechanistic Insights. Cancers 15, 2169. 10.3390/cancers15072169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huynh C, Roth D, Ward DM, Kaplan J, and Andrews NW (2004). Defective Lysosomal Exocytosis and Plasma Membrane Repair in Chediak-Higashibeige Cells. Proc. Natl. Acad. Sci. USA 101, 16795–16800. 10.1073/pnas.0405905101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Araujo MEG, Liebscher G, Hess MW, and Huber LA (2020). Lysosomal size matters. Traffic 21, 60–75. 10.1111/tra.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taunton J, Rowning BA, Coughlin ML, Wu M, Moon RT, Mitchison TJ, and Larabell CA (2000). Actin-dependent propulsion of endosomes and lysosomes by recruitment of N-WASP. J. Cell Biol 148, 519–530. 10.1083/jcb.148.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vincent C, Maridonneau-Parini I, Le Clainche C, Gounon P, and Labrousse A. (2007). Activation of p61Hck triggers WASp- and Arp2/3-dependent actin-comet tail biogenesis and accelerates lysosomes. J. Biol. Chem 282, 19565–19574. 10.1074/JBC.M701501200. [DOI] [PubMed] [Google Scholar]
- 54.Dai A, Yu L, and Wang HW (2019). WHAMM initiates autolysosome tubulation by promoting actin polymerization on autolysosomes. Nat. Commun 10, 3699–3714. 10.1038/s41467-019-11694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu YJ, Zhang T, Cheng D, Yang J, Chen S, Wang X, Li X, Duan D, Lou H, Zhu L, et al. (2020). Late endosomes promote microglia migration via cytosolic translocation of immature protease cathD. Sci. Adv 6, eaba5783. 10.1126/SCIADV.ABA5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matarrese P, Ascione B, Ciarlo L, Vona R, Leonetti C, Scarsella M, Mileo AM, Catricalà C, Paggi MG, and Malorni W. (2010). Cathepsin B inhibition interferes with metastatic potential of human melanoma: An in vitro and in vivo study. Mol. Cancer 9, 207. 10.1186/1476-4598-9-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guinec N, Dalet-Fumeron V, and Pagano M. (1993). In vitro’’ Study of Basement Membrane Degradation by the Cysteine Proteinases, Cathepsins B, B-Like and L. Digestion of Collagen IV, Laminin, Fibronectin, and Release of Gelatinase Activities from Basement Membrane Fibronectin. Biol. Chem. Hoppe Seyler 374, 1135–1146. 10.1515/BCHM3.1993.374.7-12.1135/HTML. [DOI] [PubMed] [Google Scholar]
- 58.Lambe T, Crawford G, Johnson AL, Crockford TL, Bouriez-Jones T, Smyth AM, Pham THM, Zhang Q, Freeman AF, Cyster JG, et al. (2011). DOCK8 is essential for T-cell survival and the maintenance of CD8 + T-cell memory. Eur. J. Immunol 41, 3423–3435. 10.1002/EJI.201141759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z, Bao Y, Zhou L, Ye Y, Fu W, and Sun C. (2021). DOCK8 Serves as a Prognostic Biomarker and Is Related to Immune Infiltration in Patients With HPV Positive Head and Neck Squamous Cell Carcinoma. Cancer Control 28, 1–10. 10.1177/10732748211011951/ASSET/IMAGES/LARGE/10.1177_10732748211011951-FIG2.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kearney CJ, Randall KL, and Oliaro J. (2017). DOCK8 regulates signal transduction events to control immunity. Cell. Mol. Immunol 14, 406–411. 10.1038/cmi.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu X, Han L, Zhao G, Xue S, Gao Y, Xiao J, Zhang S, Chen P, Wu ZY, Ding J, et al. (2017). LRCH1 interferes with DOCK8-Cdc42–induced T cell migration and ameliorates experimental autoimmune encephalomyelitis. J. Exp. Med 214, 209–226. 10.1084/JEM.20160068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qi X, Man SM, Malireddi RKS, Karki R, Lupfer C, Gurung P, Neale G, Guy CS, Lamkanfi M, and Kanneganti TD (2016). Cathepsin B modulates lysosomal biogenesis and host defense against Francisella novicida infection. J. Exp. Med 213, 2081–2097. 10.1084/JEM.20151938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C, Hanahan D, and Joyce JA (2006). Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 20, 543–556. 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Authier F, Kouach M, and Briand G. (2005). Endosomal proteolysis of insulin-like growth factor-I at its C-terminal D-domain by cathepsin B. FEBS Lett. 579, 4309–4316. 10.1016/J.FEBSLET.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 65.Mizunoe Y, Kobayashi M, Hoshino S, Tagawa R, Itagawa R, Hoshino A, Okita N, Sudo Y, Nakagawa Y, Shimano H, et al. Cathepsin B overexpression induces degradation of perilipin 1 to cause lipid metabolism dysfunction in adipocytes.Sci. Rep 2020. Jan 20;10(1):634 10.1038/s41598-020-57428-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fujimoto T, Tsunedomi R, Matsukuma S, Yoshimura K, Oga A, Fujiwara N, Fujiwara Y, Matsui H, Shindo Y, Tokumitsu Y, et al. (2021). Cathepsin B is highly expressed in pancreatic cancer stem-like cells and is associated with patients’ surgical outcomes. Oncol. Lett 21, 30–39. 10.3892/OL.2020.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gandalovičová A, Fernandes M, Sanz-Moreno V, Brábek J, Heneberg P, Čermák, Petruželka L, Kumar S, Sanz-Moreno V, and Brábek J. (2019). Migrastatics—Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends Cancer 5, 755–756. 10.1016/j.trecan.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gardel ML, Schneider IC, Aratyn-Schaus Y, and Waterman CM (2010). Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol 26, 315–333. 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taunton J, Rowning BA, Coughlin ML, Wu M, Moon RT, Mitchison TJ, and Larabell CA (2000). Actin-dependent Propulsion of Endosomes and Lysosomes by Recruitment of N-WASP 7. J. Cell Biol 148, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cordonnier MN, Dauzonne D, Louvard D, and Coudrier E. (2001). Actin filaments and myosin I alpha cooperate with microtubules for the movement of lysosomes. Mol. Biol. Cell 12, 4013–4029. 10.1091/MBC.12.12.4013/ASSET/IMAGES/LARGE/MK1211695012.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Y, Jiang X, Deng Q, Gao Z, Tang X, Fu R, Hu J, Li Y, Li L, and Gao N. (2019). Downregulation of MYO1C mediated by cepharanthine inhibits autophagosome-lysosome fusion through blockade of the F-actin network. J. Exp. Clin. Cancer Res 38, 457–518. 10.1186/S13046-019-1449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Son S, Baek A, Lee JH, and Kim D-E (2022). Autophagosome-lysosome fusion is facilitated by plectin-stabilized actin and keratin 8 during macroautophagic process. Cell. Mol. Life Sci 79, 95. 10.1007/s00018-022-04144-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.King JS, Gueho A, Hagedorn M, Gopaldass N, Leuba F, Soldati T, and Insall RH (2013). WASH is required for lysosomal recycling and efficient autophagic and phagocytic digestion. Mol. Biol. Cell 24, 2714–2726. 10.1091/MBC.E13-02-0092/ASSET/IMAGES/LARGE/2714FIG8.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.King JS, Gueho A, Hagedorn M, Gopaldass N, Leuba F, Soldati T, and Insall RH (2013). WASH is required for lysosomal recycling and efficient autophagic and phagocytic digestion. Mol. Biol. Cell 24, 2714–2726. 10.1091/mbc.E13-02-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Linke M, Herzog V, and Brix K. (2002). Trafficking of lysosomal cathepsin B-green fuorescent protein to the surface of thyroid epithelial cells involves the endosomal/lysosomal compartment. J. Cell Sci 115, 4877–4889. 10.1242/jcs.00184. [DOI] [PubMed] [Google Scholar]
- 76.Tanaka Y, Tanaka R, Kawabata T, Noguchi Y, and Himeno M. (2000). Lysosomal cysteine protease, cathepsin B, is targeted to lysosomes by the mannose 6-phosphate-independent pathway in rat hepatocytes: Site-specific phosphorylation in oligosaccharides of the proregion. J. Biochem 128, 39–48. 10.1093/oxfordjournals.jbchem.a022728. [DOI] [PubMed] [Google Scholar]
- 77.Otomo T, Schweizer M, Kollmann K, Schumacher V, Muschol N, Tolosa E, Mittrücker HW, and Braulke T. (2015). Mannose 6 phosphorylation of lysosomal enzymes controls B cell functions. J. Cell Biol 208, 171–180. 10.1083/jcb.201407077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Braulke T, and Bonifacino JS (2009). Sorting of lysosomal proteins. Biochim. Biophys. Acta 1793, 605–614. 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 79.Wendland M. (1992). Sorting of Lysosomal Proteins by Mannose 6-Phosphate Receptors. Trends Glycosci. Glycotechnol 4, 200–209. 10.4052/tigg.4.200. [DOI] [Google Scholar]
- 80.Vasiljeva O, Papazoglou A, Krüger A, Brodoefel H, Korovin M, Deussing J, Augustin N, Nielsen BS, Almholt K, Bogyo M, et al. (2006). Tumor Cell–Derived and Macrophage-Derived Cathepsin B Promotes Progression and Lung Metastasis of Mammary Cancer. Cancer Res. 66, 5242–5250. 10.1158/0008-5472.CAN-05-4463. [DOI] [PubMed] [Google Scholar]
- 81.Vasiljeva O, Korovin M, Gajda M, Brodoefel H, Bojič L, Krüger A, Schurigt U, Sevenich L, Turk B, Peters C, and Reinheckel T. (2008). Reduced tumour cell proliferation and delayed development of high-grade mammary carcinomas in cathepsin B-deficient mice. Oncogene 27, 4191–4199. 10.1038/onc.2008.59. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Q, Dove CG, Hor JL, Murdock HM, Strauss-Albee DM, Garcia JA, Mandl JN, Grodick RA, Jing H, Chandler-Brown DB, et al. (2014). DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J. Exp. Med 211, 2549–2566. 10.1084/JEM.20141307/VIDEO-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lohoefer F, Reeps C, Lipp C, Rudelius M, Haertl F, Matevossian E, Zernecke A, Eckstein HH, and Pelisek J. (2014). Quantitative expression and localization of cysteine and aspartic proteases in human abdominal aortic aneurysms. Exp. Mol. Med 46, e95–e95. 10.1038/emm.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang IC, Bosch BJ, Li F, Li W, Lee KH, Ghiran S, Vasilieva N, Dermody TS, Harrison SC, Dormitzer PR, et al. (2006). SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem 281, 3198–3203. 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cao H, Schroeder B, Chen J, Schott MB, and McNiven MA (2016). The endocytic fate of the transferrin receptor is regulated by c-Abl kinase. J. Biol. Chem 291, 16424–16437. 10.1074/JBC.M116.724997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bolte S, and Cordeliè res, F.P. (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc 224, 213–232. 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 87.Cox J, and Mann M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 88.Tyanova S, Temu T, and Cox J. (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 89.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M. (2014). Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. 10.1074/MCP.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 91.Bartha Á, and Gyröffy B. (2021). TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci 22, 2622. 10.3390/IJMS22052622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/SCIENCE.1247005/SUP-PL_FILE/SHALEM.SM.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rozeveld CN, Johnson KM, Zhang L, and Razidlo GL (2020). KRAS controls pancreatic cancer cell lipid metabolism and invasive potential through the lipase HSL. Cancer Res. 80, 4332–4345. 10.1158/0008-5472.CAN-20-1255/654658/AM/KRAS-CONTROLS-PANCREATIC-CANCER-CELL-LIPID. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Erstad DJ, Sojoodi M, Taylor MS, Ghoshal S, Razavi AA, Graham-O’Regan KA, Bardeesy N, Ferrone CR, Lanuti M, Caravan P, et al. (2018). Orthotopic and heterotopic murine models of pancreatic cancer and their different responses to FOLFIRINOX chemotherapy. Dis. Model. Mech 11, dmm034793. 10.1242/DMM.034793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Henley JR, Krueger EW, Oswald BJ, and McNiven MA (1998). Dynamin-mediated Internalization of Caveolae. J. Cell Biol 141, 85–99. 10.1083/JCB.141.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. 10.1016/J.YMETH.2016.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Comparative lysosome proteomic data has been deposited in MassIVE: https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp. Dataset accession is MSV000091036. Western blot images and CRISPR KO sanger sequencing have been deposited at Mendeley Data: https://doi.org/10.17632/zmwy6bgfg8.1. RNA sequencing data from pancreatic cancer organoids have been deposited in Gene Expression Omnibus: GSE185335.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.