

Summary
The tumor suppressor BRCA1-BARD1 is a RING-type Ubiquitin E3 ligase that modifies nucleosomal histone and other substrates. The importance of BRCA1-BARD1 E3 activity in tumor suppression remains highly controversial, mainly stemming from studying mutant ligase-deficient BRCA1-BARD1 species that we show here still retain significant ligase activity. Using full-length BRCA1-BARD1, we establish robust BRCA1-BARD1-mediated ubiquitylation with specificity, uncover multiple modes of activity modulation, and construct a truly ligase-null variant and a variant specifically impaired in targeting nucleosomal histones. Cells expressing either of these BRCA1-BARD1 separation-of-function alleles are hypersensitive to DNA-damaging agents. Furthermore, we demonstrate that BRCA1-BARD1 ligase is not only required for DNA resection during homology-directed-repair (HDR), but also contributes to later stages for HDR completion. Altogether, our findings reveal crucial, previously unrecognized roles of BRCA1-BARD1 ligase activity in genome repair via HDR, settle prior controversies regarding BRCA1-BARD1 ligase functions, and catalyze new efforts to uncover substrates related to tumor suppression.
Keywords: BRCA1-BARD1, Ubiquitin E3 ligase, DNA damage response, Homology-Directed Repair (HDR), DNA end resection, later stages for HDR completion
Graphical Abstract

In brief
While unveiling the intricate regulatory mechanisms of BRCA1-BARD1 ubiquitylation, Wang & Li et al. identify a truly ligase-null variant and another variant specifically impaired in targeting nucleosomal histones, and elucidate the pivotal roles of BRCA1-BARD1 ligase in not only early DNA end resection but also subsequent stages in homology-directed repair.
Introduction
Mutations in BRCA1 (Breast Cancer 1), the first familial breast cancer susceptibility locus identified from genetic linkage analysis1, also cause familial ovarian cancer and various types of sporadic cancers2. BRCA1 forms an obligate heterodimer with BARD1 (BRCA1-associated RING domain 1) to interact with many protein factors that function in diverse biological processes3–16, including DNA double-strand break (DSB) removal by homology-directed repair (HDR)5,6,15,17. It is generally accepted that the HDR role of BRCA1-BARD1 is crucial for its tumor suppression function4,16–18. Both subunits of the BRCA1-BARD1 complex include N-terminal RING and C-terminal BRCT domains separated by long disordered regions. The RING domains govern BRCA1-BARD1 heterodimer formation and endow the complex with its only known enzymatic activity, ubiquitin E3 ligase activity19,20. Though the BRCA1 RING interacts directly with E2~Ub conjugates (e.g., UBE2D3~Ub), heterodimerization with BARD1 enhances E3 ligase activity, and both subunits participate in substrate recognition21,22. Substrates linked to HDR include phosphor-CtIP (pCtIP) that functions in DNA end resection and nucleosomal histone H2A on which BRCA1-BARD1 specifically promotes mono-ubiquitylation of residues (K125, K127 and K129) in its C-terminal tail23–25.
The BRCA1 RING domain constitutes a mutational hotspot26,27. Highly penetrant mutations affect the E2~Ub binding surface and can also impair association with BARD1 (e.g., C61G), making assignment of loss-of-function and pathogenicity difficult. Thus, the role of BRCA1-BARD1 ligase activity in HDR, tumor suppression, and other biological processes has yet to be rigorously established and remains controversial28–33. Much of the debate and the prevailing belief that E3 ligase activity is dispensable stems from use of two designed missense mutations28–33, BRCA1 I26A and BARD1 R99E, shown to disrupt substrate-independent E3-ligase activity (i.e., auto-ubiquitylation) of truncated BRCA1-BARD1 constructs that harbor the RING domains in vitro30,34. Our ability to generate full-length BRCA1-BARD1 complex17 and bona fide substrates23,33, along with recent structures revealing insights into nucleosomal substrate recognition24,35, prompted us to reassess the effects of BRCA1/BARD1 RING mutations in reconstituted systems of protein ubiquitylation. We find that full-length BRCA1I26A-BARD1 and BRCA1-BARD1R99E retain substantial ligase activity toward all substrates tested. We provide contrasting evidence that a triple BRCA1 mutant, I26A/L63A/K65A36–38, ablates ligase activity and report a BARD1 mutant that is selectively impaired for nucleosomal substrate ubiquitylation. These separation-of-function mutants allow us to demonstrate involvement of BRCA1-BARD1 E3 ligase activity in multiple key stages of HDR. Notably, ligase-dead BRCA1-BARD1 exhibits an HDR deficiency similar to the well-established cancer-causing BRCA1 mutation C61G, implying a link between loss of BRCA1-BARD1 E3 ligase activity and tumor formation.
Results
Specificity and regulation of BRCA1-BARD1 ubiquitin E3 ligase
We reconstituted BRCA1-BARD1-dependent ubiquitin conjugation reactions using highly purified BRCA1-BARD1 and multiple E2s identified before21 with the nucleosome core particle (NCP) and other known interactors including phosphorylated CtIP (pCtIP) and RAD51 as potential substrates17,22–25,35,39,40 (Figure 1A and S1A), and found that BRCA1-BARD1 ubiquitylates histone H2A, H3, and pCtIP significantly, but not H2B and RAD51 (Figure 1B,C and S2A, B, D, E). Also, H2A ubiquitylation became greatly attenuated in the reaction with BRCA1-BARD1, but not with another RING-type E3 RING1B/BMI141, when BRCA1-BARD1 specific sites K125/127/129 in H2A of NCP substrates are mutated to Arg (i.e., 3KR) (Figure S2C). Notably, the ubiquitylation efficiency varied between different E2s (i.e., UBE2D3 and UBE2W are most efficient for H2A and H3 ubiquitylation, respectively, while UBE2B is dead for both) (Figure 1B, C). These results highlight the substrate, site and E2 specificity of BRCA1-BARD1-mediated ubiquitylation in vitro. By testing a series of BRCA1-BARD1 heterodimers with truncations in one or the other subunit (Figure S1B–D), we noticed that deletion of the C-terminal tandem BRCT domain of BARD1 impaired the ability of BRCA1-BARD1 to ubiquitylate both histone H2A and pCtIP, while the tandem BRCT domain of BRCA1 is essential for pCtIP39,40 but dispensable for H2A ubiquitination (Figure 1A and S2D, E), indicating distinct domain requirements for each substrate.
Figure 1. Specificity and regulation of BRCA1-BARD1 mediated ubiquitylation in vitro. (See also Figure S1–4).

A. Domain organization of BRCA1 and BARD1. The domain names are indicated below the cartoons (RING, really interesting new gene; CC, coiled-coil; BRCT, BRCA1 C-terminal; ANK, Ankyrin repeat domain). The amino acid boundaries of exon 11 are labeled above. The findings that auto-ubiquitylation (Auto-ub) promotes the E3 ligase activity towards substrates (NCP/pCtIP) (black arrows), that polypeptides derived from the BRCA1 region encoded by exon 11 inhibits E3 activity (red block), and that NCP/pCtIP substrates require distinct domains of BRCA1-BARD1 for efficient ubiquitylation (black arrows) are summarized below. Double-direction cyan arrows show the known interactions between E3 and E2~Ub/NCP/pCtIP. Symbol: Ubyn, ubiquitylation.
B. Representative nucleosome ubiquitylation assays monitoring H2A-Ub efficiency of BRCA1-BARD1 in conjunction with different E2s. UBE2B that does not bind BRCA1-BARD1 was included as a negative control. Symbol: BC1-BD1, BRCA1-BARD1. n=3 independent experiments.
C. Representative nucleosome ubiquitylation assays monitoring H3-Ub efficiency of BRCA1-BARD1 in conjunction with different E2s. UBE2B that does not bind BRCA1-BARD1 was included as a negative control. Symbol: BC1-BD1, BRCA1-BARD1. n=3 independent experiments.
D. Quantification of nucleosome ubiquitylation assays monitoring H2A-Ub efficiency of BRCA1Δ11q -BARD1 in conjunction with indicated amount of BSA and fragments of BRCA1 and BARD1 (BRCA1200–500, BRCA1500–894, BRCA936–1316, BARD11123–270, BARD11216–425 and BARD1425–565). Error bars, SEM (n=3). Representative gels are shown in Figure S3A, C.
E. Quantification of nucleosome ubiquitylation assays monitoring H2A-Ub efficiency of auto-ubiquitylation form or native form of BRCA1-BARD1. Error bars, SEM (n=4). Representative gel is shown in Figure S4A.
F. Quantification of nucleosome ubiquitylation assays monitoring H3-Ub efficiency of BRCA1-BARD1 in conjunction with unmodified, K13ub or K15ub form of nucleosome. Error bars, SEM (n=3–5). Representative gel is shown Figure S4C.
BARD1-BRCA1Δ11q, which harbors a clinically relevant allele missing ~1100 residues (amino acid residues 263–1365 encoded by exon 11) of BRCA1, showed more robust ligase activity with NCP and pCtIP than the wild-type counterpart (Figure S2D–F). This suggests that regions within exon 11 may exert an inhibitory effect on the E3 ligase activity of BRCA1-BARD1. Consistent with this hypothesis, BRCA11–112-BARD1 displayed more rapid H2A ubiquitylation than BRCA1-BARD1(Figure S2F), polypeptides derived from the BRCA1 region encoded by exon 11, namely, BRCA1200–500, BRCA1500–894, and BRCA1936–1316, significantly inhibited H2A and pCtIP ubiquitylation catalyzed by BRCA1Δ11q-BARD1 with different inhibitory regions for two substrates (Figure 1D, S1E and S3A,B), and little or no inhibition of H2A ubiquitylation occurred when any of three BARD1-derived polypeptides (BARD1124−270, BARD1216−425 and BARD1425−565) was used in the reactions with BRCA1Δ11q-BARD1 (Figure1D, S1E and S3C). Next, we examined the three BRCA1 exon 11 fragments in H2A ubiquitylation reactions with the E3 RING1B-BMI1. In this case, the fragments had no detectable effect, allowing us to rule out inhibition through any of the ubiquitylation machinery commonly present in the above assays (Figure S3D). The BRCA1 exon 11 fragments display modest inhibition in assays containing BRCA11–304-BARD126–142 (Figure S3E). Finally, while BRCA1500–894 exhibited robust binding to NCP, neither of the two fragments (BRCA1200–500 and BRCA1936–1316) that strongly inhibited H2A ubiquitylation appears to bind nucleosomes (Figure S3F). Having ruled out components of the ubiquitylation machinery and binding to the substrate itself, the results support the premise that internal domain-domain interactions modulate BRCA1-BARD1 E3 ligase activity in a substrate-selective manner, as reported for autoinhibition mechanism in other E3s like RING1B-BMI142.
We found that the auto-ubiquitylated form of BRCA1-BARD1 made by pre-incubation with UBE2D3 exhibits higher ligase activity than native BRCA1-BARD1 (Figure 1E and S4A, B), and that H3 ubiquitylation was greatly enhanced when H2A in the NCP substrate was modified with mono-ubiquitylation at K13/K15 (Figure 1F and S4C), presumably because the C-terminal domains of BARD1 bind to these modified H2A-containing nucleosomes with high affinity33,35. Altogether, we conclude that BRCA1-BARD1 ligase activity can be modulated up and down towards its NCP histone substrates and towards other bona fide substrates (e.g., pCtIP) through mechanisms that include intramolecular domain interactions and post-translational modifications.
Construction of a E3 ligase truly dead mutant of BRCA1-BARD1
To establish the biological significance of the BRCA1-BARD1 E3 ligase activity, we endeavored to isolate a variant of BRCA1-BARD1 that is devoid of E3 ligase activity but possesses all other known attributes, namely, retention of the BRCA1-BARD1 heterodimeric structure, DNA binding, RAD51 interaction, and enhancement of the recombinase activity of RAD515,17,43,44. Our previous published NMR analysis showed that multiple RING domain residues, including I26, L51, L63, K65, N66, and I68 are affected upon addition of various E2 enzymes (Figure S5A)34. These observations provide a rationale for results from a recent high-throughput analysis that showed several BRCA1 RING domain missense mutants (e.g., I26N, I26T, N66Y, I68K, and I68R) as non-functional and pathogenic45. Accordingly, we constructed 13 BRCA1 mutants in which the aforementioned RING domain residues were changed individually and in combination to assess the mutations impact on BRCA1-BARD1 complex formation and on E3 ligase activity. We found that the triple BRCA1I26A, L63A, K65A mutant36–38 (Figure 2A), is the best candidate as it retains the ability to heterodimerize with BARD1 when co-expressed with the latter in bacterial and insect cells but is devoid of E2 interaction and ligase activity as revealed by testing with NCP as substrate in the context of BRCA1-BARD1 truncation variants (Figure S1F and S5B–E). This is consistent with the fact that this triple mutant not only targets the BRCA1-E2 binding interface, but also alters a key residue (BRCA1K65) required for allosteric activation of bound E2~Ub conjugates and promotion of Ubiquitin (Ub) transfer to substrates34. We also verified by co-immunoprecipitation that BRCA1I26A, L63A, K65A interacts normally with BARD1 in lysates of 293T cells and HeLa cells, with and without prior DNA damaging treatment of cells (Figure S5F, G). Using full-length BRCA1I26A, L63A, K65A-BARD1 and BRCA1I26A-BARD1 complexes purified from insect cells (Figure S1G), we found that both BRCA1I26A, L63A, K65A-BARD1 and BRCA1I26A-BARD1 mutant complexes are just as proficient as the wild-type counterpart in DNA binding, RAD51 interaction, and promoting D-loop formation in conjunction with RAD5115,17 (Figure S6A–E). Notably, we showed that BRCA1I26A-BARD1, previously reported to be E3 deficient28,29,31, possesses robust ubiquitylation activity, whereas BRCA1I26A, L63A, K65A-BARD1 is almost completely devoid of ligase activity with all substrates and all E2s tested (Figure 2B and S6F–I). As well, another reported E3 ligase-deficient mutant, BARD1R99E-BRCA130,32,33 retains a significant level of ubiquitylation activity with unmodified NCP or pCtIP as substrates and it is almost as active as wild-type BRCA1-BARD1 in reactions with H2AK13ub form of NCP (Figure 2B and S6F–I).
Figure 2. Identification of a BRCA1-BARD1 truly E3-dead mutant. (See also Figure S5, 6).

A. The BRCA1-BARD1 RING structure showing the locations of BRCA1 residues I26, L63, and K65 and BARD1 R99E with the E2~Ub binding and activation surface indicated by dotted lines (PDB: 7JZV).
B. Representative nucleosome ubiquitylation assays monitoring H2A-Ub efficiency for wild-type or mutant BRCA1-BARD1 in conjunction with UBE2D3 & NCP_H2A (top), UBE2D3+UBE2N-UBE2V2 & NCP_H2A (middle); UBE2D3 & NCP_H2AK13ub (bottom). Quantification shown on the right. Error bars, SEM (n=3).
C. Ub discharge was monitored as the decay of the Ub488~UBE2D3594 FRET signal over time after the addition of wild-type or mutant BRCA1-BARD1 (E3; 100 nM) and 200 nM NCP (substrate). The E3 UBR3 (RING trimer of TRIM5a) was included as a control.
A truly ligase-dead E3 should be incapable of facilitating Ub transfer from its cognate E2s. A FRET-based assay monitoring loss of activated UBE2D3~Ub46 was used to assess the enhancement of Ub discharge by BRCA1-BARD1 and mutants in the presence and absence of NCP. As expected, wild type BRCA1-BARD1 displayed robust enhancement of E2~Ub discharge (Figure 2C and S6J). Importantly, both in the presence or absence of NCP, BRCA1I26A-BARD1 and BRCA1-BARD1R99E clearly retain significant activity in the discharge reaction, while BRCA1I26A, L63A, K65A-BARD1 is devoid of such activity. Altogether, our results provide strong evidence that BRCA1I26A, L63A, K65A-BARD1 represents a true E3 ligase-null form. We therefore refer to it as an E3-dead mutant (“BRCA1E3d-BARD1”).
BRCA1-BARD1 ligase is essential for DNA damage response and DSB repair
To assess the physiological significance of the E3 ligase activity of BRCA1-BARD1, we constructed HeLa-shBRCA1 cell lines that ectopically express similar levels of shBRCA1-resistant HA-tagged BRCA1 species (BRCA1I26Ares, BRCA1E3dres, or BRCA1C61Gres) in which endogenous BRCA1 is depleted by shRNA against BRCA1 upon doxycycline treatment (Figure S7A). Both BRCA1I26Ares and BRCA1E3dres are nuclear (Figure S7B) and form foci upon IR treatment of cells (Figure S7C). While also nuclear, the cancer-mutant BRCA1C61Gres forms fewer and smaller foci as reported47. This highlights an important difference between our E3-defective mutant and pathogenic BRCA1C61Gres as the latter also displays decreased heterodimerization with BARD126,31,48. Cells ectopically expressing BRCA1E3dres or BRCA1C61Gres are hypersensitive to DNA-damaging agents olaparib, camptothecin (CPT), mitomycin C (MMC), and cisplatin, whereas cells expressing BRCA1I26Ares show an intermediate phenotype (Figure 3A), whether BARD1WT is ectopically overexpressed or not. Of note, overexpression of BARD1 partially restored BRCA1 protein level, leading to the moderate effect of olaparib in all cell lines when compared to that without BARD1 overexpression. Furthermore, results from stable BRCA1-deficient MDA-MB-436 cell lines expressing wild-type HA-BRCA1 or mutants (Figure S7D) revealed that, in contrast to BRCA1WTres or BRCA1I26Ares, BRCA1E3dres is defective in supporting cell survival upon treatment with olaparib (Figure 3B). Altogether, the data provide strong evidence that BRCA1-BARD1 E3 ligase activity plays a crucial role in the DNA damage response.
Figure 3. BRCA1-BARD1 E3 ligase activity is required for cell survival following DNA damage (see also Figure S7).

A. Clonogenic survival of HeLa-shBRCA1 cells stably expressing wild-type or mutant HA-BRCA1 upon treatment with olaparib, CPT, MMC or cisplatin. Error bars, SEM (n=3–6). Symbol: EV, empty vector; OE BARD1, with ectopic expression of Flag-BARD1.
B. Clonogenic survival of MDA-MB-436 cells stably expressing wild-type or mutant HA-BRCA1 without or with ectopic expression of Flag-BARD1 upon treatment with olaparib. Error bars, SEM (n=3–4). Symbol: EV, empty vector; OE BARD1, with ectopic expression of Flag-BARD1.
Three reporter assays were used to identify the type of DSB repair process for which BRCA1-BARD1 E3 activity is required, evaluating homologous recombination (HR), single-strand annealing (SSA), and non-homologous end joining (NHEJ)49. As expected, endogenous BRCA1 knockdown by small interfering RNA (siRNA) impaired HR and SSA but had no significant effect on NHEJ (Figure 4A, B and S7E, F). Ectopic expression of BRCA1WTres in BRCA1-deficient cells substantially restored both HR and SSA, while ectopic expression of BRCA1E3dres resulted in significantly less complementation (Figure 4A, B and S7F). Notably, BRCA1I26Ares showed only a partial defect, while BRCA1C61Gres was more impaired in repair activity than BRCA1E3dres. Thus, we surmise that the BRCA1-BARD1 ligase activity is essential for DSB repair via HR and SSA.
Figure 4. BRCA1-BARD1 E3 activity is critical for efficient HDR and early DNA end resection of HDR. (See also Figure S8, 9).

A. Schematic of HR assay using the DR-GFP reporter (top). Quantification of HR assay results (bottom) from DR-U2OS cells transiently expressing wild-type or mutant HA-BRCA1 upon treatment with siRNA against BRCA1 or control siRNA (siCtrl). Error bars, SD (n=3–4). Symbol: EV, empty vector.
B Schematic of the SSA assay using the SA-GFP reporter (top). Quantification of SSA assay results (bottom) from examining SA-U2OS cells transient expressing wild-type or mutants of HA-BRCA1 upon their treatment with siRNA against BRCA1 or control siRNA (siCtrl). Error bars, SD (n=2–4). Symbol: EV, empty vector.
C. Quantification of cells with >5 RAD51 foci four hours after exposure to 6 Gy irradiation in HeLa-shBRCA1 cells stably expressing wild-type or mutant HA-BRCA1, where endogenous BRCA1 was depleted by doxycycline incubation. Mean values ± SEM of at least three independent experiments are shown.
D. Quantification of cells with >10 RPA foci four hours after exposure to 6 Gy irradiation in HeLa-shBRCA1 cells stably expressing wild-type or mutant HA-BRCA1, where endogenous BRCA1 was depleted by doxycycline incubation. Mean values ± SEM of at least three independent experiments are shown.
E. Quantification of cells with >10 BrdU foci four hours after exposure to 6 Gy irradiation in HeLa-shBRCA1 cells stably expressing wild-type or mutant HA-BRCA1, where endogenous BRCA1 was depleted by doxycycline incubation. Mean values ± SEM of at least three independent experiments are shown.
Statistical significance was assessed by two-tailed unpaired Student’s t-test and two-way ANOVA. ns: not significant, *P ⩽ 0.05, **P ⩽ 0.01, ***P⩽0.001, and ****P⩽0.0001 were considered significant.
BRCA1-BARD1 promotes DNA end resection via its ligase activity
Both HR and SSA require extensive resection of DNA ends, suggesting that ligase activity could be indispensable for proper end resection. We examined ionizing radiation (IR)-induced foci formation of Replication Protein A (RPA), a ubiquitous ssDNA-binding protein that serves as a reliable marker of resected DNA ends. Moreover, we examined RAD51 foci as an additional readout of DNA end resection. As expected, knockdown of endogenous BRCA1 by doxycycline treatment diminished RPA and RAD51 foci formation (Figure S8A–C). In cells depleted of endogenous BRCA1 but expressing BRCA1E3dres or BRCA1C61Gres, significantly fewer RPA and RAD51 foci were formed compared to cells expressing BRCA1WTres or BRCA1I26Ares at 4h after IR (Figure 4C, D and S8D, E). Notably, γH2AX foci formation was identical in cells expressing either wild type BRCA1 or any of the three BRCA1 mutants (Figure S8D), indicating DSB formation is not affected by any of the BRCA1 mutations. Together, these results and those that indicated that both HR and SSA are impaired by the BRCA1E3d mutation strongly suggest that successful execution of DNA resection is reliant on the BRCA1-BARD1 E3 ligase activity. For more direct evidence to support our premise, we labeled resection tracks in genomic DNA using 5-bromo-2’-deoxyuridine (BrdU) that can be detected by anti-BrdU antibody under non-denaturing conditions50. We found that cells with BRCA1WTres generate more ssDNA/BrdU foci in response to IR treatment than cells expressing BRCA1E3dres or BRCA1C61Gres (Figure 4E and S8F). Consistent with this observation, fewer CtIP foci40 and proximity ligation assay (PLA) signals of BrdU-CtIP interaction51 were detected in cells expressing empty vector or BRCA1E3dres, compared with wild type BRCA1 cells (Figure S9A, B). Of note, cells expressing wild type or mutant BRCA1 exhibited similar cell cycle progression profiles (Figure S7G). Thus, we conclude that BRCA1-BARD1 ligase activity is needed for the successful recruitment of CtIP and execution of DNA end resection.
Late steps for HDR completion require BRCA1-BARD1 ligase
Assembly and disassembly of the RAD51 presynaptic filament are essential HDR steps following DNA end resection. In cells expressing BRCA1WTres, RAD51 foci peaked at approximately 4 h after IR and then declined dramatically. In contrast, RAD51 foci in cells expressing BRCA1E3dres accumulated at a slow pace (peak at 8 h) and disappeared more slowly (Figure S8G). Comparing BRCA1WTres and BRCA1E3dres cells, RPA foci decreased in a similar manner after peaking (Figure S8G). These observations suggest a possible role of BRCA1-BARD1 E3 ligase in HDR stages downstream of DNA end resection. Thus, we measured the decline of γH2AX phosphorylation over time post-IR as a readout of DSB repair kinetics. Peak γH2AX levels were observed 2 h post-IR and rapidly decreased to baseline by 3h in cells with BRCA1WTres, whereas levels remained high even after 8 h post-IR in cells expressing BRCA1E3dres (Figure 5A). Importantly, we found that BRCA1E3dres cells remain hypersensitive to olaparib in comparison with BRCA1WTres cells upon 53BP1 depletion (Figure 5B and S9C), which restores DNA end resection capacity in BRCA1E3dres cells (Figure 5B, C and S9C–E) due to its established role in restricting DNA end resection52. Furthermore, we observed a marked persistence of RAD51 foci and γH2AX levels in cells expressing BRCA1E3dres after release from high-dose cisplatin or CPT treatment (Figure 5D and S10A–D). Using a neutral comet assay, we found that BRCA1E3dres cells exhibit similar levels of DSBs as BRCA1WTres cells right after 4 μM cisplatin treatment, but retain high levels of unrepaired DSBs even after 48 h post-cisplatin while DNA breaks in cells with BRCA1WTres decline significantly (Figure 5E and S10E). These data support the premise that the BRCA1-BARD1 E3 activity promotes the completion of HR beyond DNA end resection.
Figure 5. BRCA1-BARD1 E3 activity is critical for late stages beyond DNA end resection of HDR. (See also Figure S9, 10).

A. Western blot to detect γ-H2AX (α-Tubulin as the loading control) at various times (0, 2, 3, 4 and 8h) after 2Gy irradiation.
B. Clonogenic survival upon treatment with olaparib of HeLa-shBRCA1 cells stably expressing empty vector, wild-type or E3d mutant of HA-BRCA1, where endogenous BRCA1 and 53BP1 were depleted by doxycycline incubation and siRNA transfection, respectively. Error bars, SEM (n=4). Symbol: EV, empty vector. **** P⩽0.0001, by two-way ANOVA.
C. Quantification of cells with >10 RPA foci four hours after exposure to 6 Gy irradiation in (right) HeLa-shBRCA1 cells stably expressing wild-type or E3d mutant of HA-BRCA1, where endogenous BRCA1 and 53BP1 were depleted by doxycycline incubation and siRNA transfection, respectively. Error bars, SEM (n=3). ****p<0.0001, by Student’s t-test.
D. Representative micrographs of RAD51 foci (red) in the nucleus of HeLa-shBRCA1 cells stably expressing wild-type or E3d mutant of HA-BRCA1 at 24h, 34h and 48h after 24h exposure of 4 μM cisplatin. Blue: DAPI. Quantification of cells with >5 RAD51 foci at various time points from the release of cisplatin exposure. Mean values ± SEM of at least three independent experiments are shown. Scale bar: 10 μm.
E. Quantification of Olive tail moment from the neutral comet assay of HeLa-shBRCA1 cells stably expressing wild-type or E3d mutant HA-BRCA1, where endogenous BRCA1 was depleted by doxycycline incubation. One hundred representative comets were analyzed in ImageJ and plotted in GraphPad PRISM. **p<0.01, ****p<0.0001, by Student’s t-test.
Statistical significance was assessed by two-tailed unpaired Student’s t-test and two-way ANOVA. ns: not significant, *P ⩽ 0.05, **P ⩽ 0.01, ***P⩽0.001, and ****P⩽0.0001 were considered significant.
Role of histone ubiquitylation by BRCA1-BARD1 in DNA end resection
Although histone H2A is a well-established ubiquitylation substrate of BRCA1-BARD1 and it has been reported that H2A-ubiquitin fusion polypeptide recruits the chromatin remodeler SMARCAD1 for 53BP1 repositioning30, the biological significance for this modification is still unclear due to conflicting reports involving R99E mutant of BARD130,32,33. As we also detected H3 ubiquitylation by BRCA1-BARD1, we wanted to further identify a nucleosome-binding mutant of BRCA1-BARD1 that is specifically defective in both H2A and H3 ubiquitylation for testing in cells. Our endeavor was guided by recently solved structures of nucleosome-BRCA1-BARD124,35, wherein BRCA1 Arg71 side chain was seen to insert into a pocket in the nucleosome acidic patch and BARD1 Trp91 and Pro89 were found to engage the nucleosome H2B/H4 cleft24,35. Accordingly, we generated mutants of P89A-W91A of BARD1 (BARD1PW) and R71G of BRCA1 (BRCA1R71G) (Figure 6A) and expressed BRCA1-BARD1PW, BRCA1R71G-BARD1, and BRCA1R71G-BARD1PW complexes in insect cells. We purified these mutant complexes using the same procedure developed for the wild-type counterpart (Figure S1H). Importantly, all three mutant complexes (BRCA1-BARD1PW, BRCA1R71G-BARD1, and BRCA1R71G-BARD1PW) behaved like their wide-type counterpart in RAD51 interaction, DNA binding, and D-loop formation (Figure S11A–C). Based on H2A/H3 ubiquitylation and E2 discharge measurement with UBE2D3 as E2 and NCP as substrate, we found that all three mutants are functionally impaired, to varying degrees, in both assay systems, with the R71G-PW double mutant being the most impaired (Figure S11D–F), followed by R71G (Figure S11D–F), then PW (Figure 6B, C and S11D–F). However, when 50 mM Lysine substrate was used in the E2 discharge assay, we found that only BRCA1-BARD1PW possesses the same intrinsic E3 ligase activity as the wild type complex (Figure 6D and S11F), whereas BRCA1R71G-BARD1 is significantly impaired in this regard (Figure S11F). Likewise, BRCA1-BARD1PW, but not BRCA1R71G-BARD1, has the same ability as wild-type BRCA1-BARD1 to ubiquitylate other substrates, including pCtIP and BRCA1-BARD1 itself (Figure S11G, H). Of note, BRCA1R71G has been shown to be HDR proficient and the pathogenicity of the founder mutation R71G could stem from a splicing defect38,53–55. We therefore left BRCA1 NCP-contacting residues (e.g., R71) intact to avoid possible disruption of E2 binding and intrinsic ligase activity known to occur with mutations in adjacent residues (e.g., L63, K65) (Figure 6A). This left BRCA1-BARD1PW as the most suitable species to address the biological role of nucleosome binding and ubiquitylation by the tumor suppressor complex. We made stable cell lines (HeLa-shBARD1), where endogenous BARD1 can be efficiently depleted by doxycycline treatment (Figure 6E). As expected, knock down of BARD1 hypersensitized the cells to olaparib, cisplatin and CPT (Figure 6F and S12A). Importantly, cells ectopically expressing BARD1WTres were more resistant to DNA damaging agents than those expressing BARD1PWres (Figure 6E, F and S12A). Accordingly, we found that BARD1PWres is less capable in restoring HR efficiency in BARD1-depleted cells (Figure 6G and S12B), and BARD1PWres cells exhibit lower numbers of RPA, BrdU and RAD51 foci than BARD1WTres cells (Figure 6H, I and S12C–E). Of note, upon 53BP1 depletion, cells expressing BARD1PWres became as resistant to olaparib as the WT BARD1 counterpart (Figure S12F). Together with our E3d mutant data, these results support a multifaceted and essential role for BRCA1-BARD1 Ub ligase activity in HDR and highlight a specific contribution of histone ubiquitylation by BRCA1-BARD1 in the DNA end resection step of HDR.
Figure 6. Role of the BARD1-nucleosome complex in histone ubiquitylation and in cell survival. (See also Figure S11, 12).
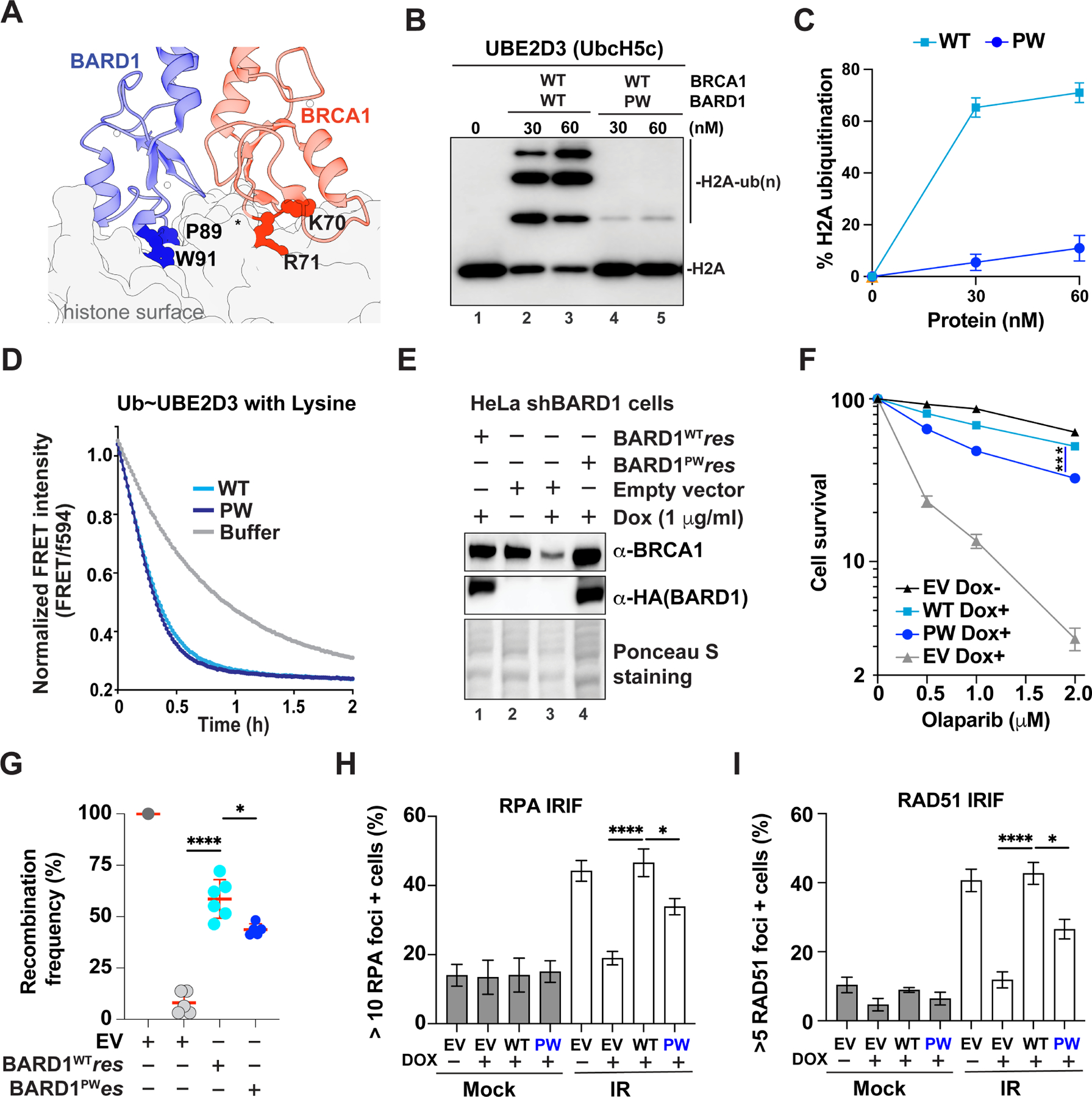
A. The BRCA1-BARD1 RING-histone interface showing the locations of BRCA1 K70R71 and BARD1 P89W91 relative to the histone surface (PDB: 7JZV).
B. Representative nucleosome ubiquitylation assays monitoring H2A-Ub efficiency of wild-type or PW (P89AW91A mutant of BARD1) of BRCA1-BARD1.
C. Quantification of b. Error bars, SEM (n=4).
D. Ub discharge was monitored by the decay of the Ub488~UBE2D3594 FRET signal over time after the addition of wild-type or PW mutant BRCA1-BARD1 (E3; 100 nM) and 50 mM lysine (substrate).
E. Western blot analysis to detect HA-BARD1 and BRCA1 from HeLa-shBARD1 cells stably expressing wild-type or PW mutant of HA-BARD1, where endogenous BARD1 was depleted by doxycycline incubation.
F. Clonogenic survival of HeLa-shBARD1 cells stably expressing wild-type or PW mutants of HA-BARD1 upon treatment with olaparib. Error bars, SEM (n=3–6). Symbol: EV, empty vector. *** P⩽0.001, by two-way ANOVA.
G. Quantification of HR assay results from DR-GFP U2OS cells transiently expressing wild-type or mutant HA-BARD1 upon treatment with siRNA against BARD1 or control siRNA (siCtrl). Error bars, SD (n=5–6). Symbol: EV, empty vector. *p<0.05, **** P⩽0.0001, by Student’s t-test.
H. Quantification of cells with >10 RPA foci four hours after exposure to 6 Gy irradiation in HeLa- shBARD1 cells stably expressing wild-type or mutant HA-BARD1, where endogenous BARD1 was depleted by doxycycline incubation. Mean values ± SEM of at least three independent experiments are shown. Symbol: EV, empty vector. *p<0.05, **** P⩽0.0001, by Student’s t-test.
I. Quantification of cells with >5 RAD51 foci four hours after exposure to 6 Gy irradiation in HeLa-shBAD1 cells stably expressing wild-type or mutant HA-BARD1, where endogenous BARD1 was depleted by doxycycline incubation. Mean values ± SEM of at least three independent experiments are shown. Symbol: EV, empty vector. *p<0.05, **** P⩽0.0001, by Student’s t-test.
Statistical significance was assessed by two-tailed unpaired Student’s t-test and two-way ANOVA. ns: not significant, *P ≤ 0.05, **P ⩽ 0.01, ***P⩽0.001, and ****P⩽0.0001 were considered significant.
Discussion
Since the initial demonstration that mutation of BRCA1 Ile26 to Ala impairs the auto-ubiquitylation of heterodimeric RING-RING BRCA1 and BARD1 fragments34, this mutant has been regarded as a “E3 ligase-dead” mutant and used in over 200 publications to date. Notably, examination of the BRCA1I26A mutant in mouse models led to the conclusion that BRCA1-BARD1 E3 ligase activity is dispensable for HDR and tumor suppression19,20, a conclusion that has shaped the field and its focus. However, there is profound dissonance between this conclusion and the fact that the BRCA1 RING domain, which comprises only 6% of the protein, harbors ~60% of known pathogenic missense mutations27. Further characterization of the structure and function of the BRCA1-BARD1 ligase revealed that it functions with multiple E2s21 and via a critical allosteric linchpin residue36, and that both BRCA1 and BARD1 subunits are required to bind substrates as well as to localize the complex at sites of HDR15,17,24,33,35,47. Herein, we have shown that, contrary to previous assumptions, full-length BRCA1I26A-BARD1 retains substantial E3 ligase activity toward both NCP or pCtIP substrates even at concentrations as low as 60 nM (Figure 2B) and is almost as active as wild-type BRCA1-BARD1 when the physiological substrates, i.e., H2AK13/K15ub form of NCP, are used in the reaction. In contrast, a mechanism-based triple mutant appears devoid of E3 ligase activity with multiple substrates and multiple E2s. By all metrics we used to measure the cellular response to DNA damage, the BRCA1E3d-BARD1 mutant showed much more severe deficiencies than ligase-competent BRCA1I26A-BARD1. Whether monitoring the response to IR treatment, DNA damaging agents, or PARP inhibitors, the BRCA1I26A-BARD1 showed only a partial defect, whereas in most assays the BRCA1E3d-BARD1 mutant more closely matched that of the known cancer-predisposing mutant BRCA1C61G. Even the inherent ability to repair DNA breaks in untreated cells expressing BRCA1E3d-BARD1 is substantially diminished (Figure S10F).
The BRCA1E3d-BARD1 complex provides a valuable tool to reassess the importance of BRCA1-BARD1 E3 ligase activity for its functions and can be used to parse various steps of the HDR pathways. Herein, we demonstrated that BRCA1-BARD1 E3 ligase activity enhances DNA end resection for DSB repair by both the HR and SSA pathways. In subsequent steps, the absence of ligase activity delays both the formation and resolution of RAD51 foci and results in the persistence of γH2AX and unrepaired DSBs showing that the kinetics of DSB removal are significantly altered (Figure 5A, D, E, S8G and S10A–D). 53BP1 deletion fully rescues DNA end resection defect (Figure 5C and S9C–E), but not DNA damage sensitivity of cells expressing BRCA1E3d (Figure 5B), highlighting the role of BRCA1-BARD1 E3 ligase in later stages (e.g., assembly and disassembly of the RAD51 nucleoprotein) in HDR. Complementing these experiments, we have developed a BARD1 mutant that is specifically impaired in the recognition of nucleosomal histones. BARD1PW prevents docking of the RING-RING domains on the nucleosome surface and suppresses BRCA1-BARD1-dependent histone ubiquitylation while leaving other functional attributes intact. We have shown that lack of histone (e.g., H2A and H3) ubiquitylation by the PW BARD1 mutant exerts a significant impact on DNA end resection during HDR (Figure 6H, I and S12C–E), presumably due to a defect in recruitment of downstream readers to antagonize 53BP1 (Figure S12F), and overall cell survival in the presence of cisplatin, CPT, and PARP inhibitors (Figure 6F and S12A). Altogether, our results answer the long-standing questions of whether and how BRCA1-BARD1 E3 ligase activity is involved in HDR and also encourage further studies to reveal substates involved especially in late steps of HDR and to establish their molecular functions.
A model for potential roles of BRCA1-BARD1 E3 ligase involving its substrates (e.g., histones) in HDR is presented here (Figure 7). Our findings open up ways to define how BRCA1-BARD1 E3 ligase activity affects its DNA damage repair functions, other biological roles (e.g., heterochromatin formation and gene silencing), and ultimately, its tumor suppressor activity. Interestingly, our work present evidence that the ubiquitin E3 ligase of BRCA1-BARD1 can be tuned down or up by internal domains (e.g., exon 11) and post-translational modification status (e.g., ubiquitylation), in addition to the regulation by the post-translational modification status of substrates (e.g., K13/K15ub form of H2A in NCP) and the paired E2s (e.g., UBE2D3 for H2A vs UBE2W for H3) (Figure 1A). This implies that mutations in the domains beyond RING of BRCA1/BARD1 may also affect the ability of BRCA1-BARD1 to ubiquitylate key substrates (e.g., C-terminal domains of BARD1 and exon 11 domains of BRCA1 for ubiquitylating NCP/pCtIP) and thereby contribute to tumor development and therapeutic response in mutation carriers. The discovery of multiple BRCA1-BARD1 E3 ligase regulatory features, in conjunction with the E3-deficient mutants, provides a framework to address major questions that remain despite decades of research on BRCA1-BARD1 and cancer mutations in the domains not limited to RING with regards to its only known enzymatic function and will help guide future efforts to develop specific BRCA1-BARD1 E3 ligase inhibitors towards substrates as possible cancer therapeutics. Additionally, our discovery that previously presumed ligase-dead BRCA1-BARD1 mutants actually retain ligase activity in the context of full-length complex with bona-fide substrates demands reinterpretation of scores of studies that have used these mutants over decades.
Figure 7. Model for BRCA1-BARD1 ubiquitin E3 ligase functions in DSB repair by HDR.

Our work demonstrates 1) critical roles for BRCA1-BARD1 E3 ligase in key stages of HDR, including DNA end resection (green arrow) and potential late steps for HDR completion (red arrows) via ubiquitylation substrates (to be identified), and 2) that histone (as one key substrate) ubiquitylation by BRCA1-BARD1 is important for DNA end resection therein. Symbol: S, Substrate; Ub, Ubiquitin (either monoubiquitylation or polyubiquitylation).
Limitations of the study
Our study demonstrates that BRCA1-BARD1 ubiquitin E3 ligase activity not only plays a critical role in DNA end resection during HDR, but also contributes to later stages for HDR completion. One limitation of the current study stems from the inability to detect histone ubiquitylation by BRCA1-BARD1 in cells due to the lack of antibodies that are specific to this modification. Secondly, we do not fully define how the BRCA1-BARD1 E3 ligase participates in different stages of HDR by identifying the full spectrum of substrates and establishing their biological roles. Thirdly, we cannot conclude rigorously that BRCA1-BARD1 E3 ligase is required for tumor suppression, as we have not tested the ligase-dead version of BRCA1-BARD1 in a mouse model.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Weixing Zhao (zhaow2@uthscsa.edu).
Materials availability
Plasmids, recombinant proteins, DNA substrates and cell lines are available without restriction upon requests, which should be directed to the lead contact, Weixing Zhao (zhaow2@uthscsa.edu).
Data and code availability
Confocal fluorescence microscopy images and western blot have been deposited at Mendeley: https://doi:10.17632/9s37k6s8jr.1 and is publicly available as of the date of publication. DOI is listed in the key resources table. All data reported in this publication will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-VSV-G epitope antibody | Sigma | Cat# V4888 |
| Anti-H3 antibody | Cell Signaling Technology | Cat# 4499 |
| Anti-H2B antibody | Cell Signaling Technology | Cat# 2934 |
| Anti-FLAG M2-HRP | Sigma | Cat# A8592 |
| Anti-RAD51 | Cell Signaling Technology | Cat# 18875 |
| Anti-RPA34 | Sigma | Cat# MABE285 |
| Anti-His-HRP | Sigma | Cat# A7058 |
| Anti-GAPDH | Cell Signaling Technology | Cat# 2118S |
| Anti-β-Tubulin | Cell Signaling Technology | Cat# 2128S |
| Anti-HA | Cell Signaling Technology | Cat# 3724S |
| Anti-BRCA1 | Santa Cruz Biotech | Cat# SC6954 |
| Anti-BARD1 | Abcam | Cat# ab50984 |
| Anti-Lamin B1 | Santa Cruz Biotech | Cat# SC374015 |
| Anti-53BP1 | Sigma | Cat# MAB3802 |
| Anti-Actin | Abcam | Cat# ab3280 |
| Anti-γH2AX | Sigma | Cat# 05636 |
| Anti-©H2AX | Cell Signaling Technology | at# 9718S |
| Anti-CtIP | Active Motif | Cat# 61141; RRID: AB_2714164 |
| Anti-BrdU | BD Biosciences | Cat# 347580; RRID: AB_400326 |
| Anti-CtIP | Invitrogen | Cat# PA5–84133; RRID: AB_2791285 |
| Chemicals, peptides, and recombinant proteins | ||
| Olaparib | Selleckchem | Cat#HY-10162; CAS: 763113–22-0 |
| Camptothecin | Sigma | Cat#208925; CAS: 7689–03-4 |
| Cisplatin | Selleckchem | Cat#P4394; CAS: 15663–27-1 |
| Mitomycin C | Sigma | Cat#HY-13316; CAS: 50–07-7 |
| Doxycycline | Sigma | Cat# D3447 |
| BRCA1-BARD1 (WT or mutants) | In this study | N/A |
| BRCA1 fragments (BRCA1200–500, BRCA1500–894, and BRCA1936–1316) | In this study | N/A |
| BARD1 fragments (BARD1124–270, BARD1216–425, and BARD1425–565 | In this study | N/A |
| 15N-labeled UBE2D3 | In this study | N/A |
| 15N-labeled BRCA11–112-BARD26–142 | In this study | N/A |
| E1 (UBA1) | In this study | N/A |
| E2s (UBE2D3, UBE2W, UBE2E3, UBE2E2, UBE2E1, UBE2N, UBE2V2, UBE2B) | In this study | N/A |
| Ubiquitin | In this study | N/A |
| RING1B1–159/BMI11–109 | In this study | N/A |
| Histone octamer | In this study | N/A |
| phosphorylated CtIP | In this study | N/A |
| RAD51 | In this study | N/A |
| AlexaFluor488-Ubiquitin | In this study | N/A |
| AlexaFluor594-UBE2D3 | In this study | N/A |
| Critical commercial assays | ||
| Duolink PLA kit | Sigma | Cat# DUO92101 |
| Q5 Site-Directed Mutagenesis Kit | New England BioLabs | Cat# E0554 |
| Mammalian β-Galactosidase Kit | Thermo Fisher Scientific | Cat# T1029 |
| Deposited data | ||
| Original images and blots | This manuscript | Mendeley data: doi:10.17632/9s37k6s8jr.1 |
| Experimental models: Cell lines | ||
| HeLa | ATCC | CCL-2 |
| U2OS (DR, SA, EJ5) | Jeremy Stark Lab | N/A |
| MDA-MB-436 | Bing Xia Lab | N/A |
| Oligonucleotides | ||
| Control siRNA (UAGCCGGUAGACUUAGGUCUG), | Qiagen | N/A |
| BRCA1 siRNA (AAGCUCCUCUCACUCUUCAGU) | Qiagen | N/A |
| TP53BP1 siRNA | Thermo Fisher Scientific | Cat# s14313 |
| Recombinant DNA | ||
| pFB-BRCA1 | In this study | N/A |
| pFB-BRCA1 I26A | In this study | N/A |
| pFB-BRCA1 E3d | In this study | N/A |
| pFB-BRCA1 R71G | In this study | N/A |
| pFB-BARD1 | In this study | N/A |
| pFB-BARD1-P89A/W91A | In this study | N/A |
| pcDNA5/FRT-BRCA1 | In this study | N/A |
| pcDNA5/FRT-BRCA1 I26A | In this study | N/A |
| pcDNA5/FRT-BRCA1 E3d | In this study | N/A |
| pcDNA5/FRT-BRCA1 C61G | In this study | N/A |
| pcDNA5/FRT-BARD1 | In this study | N/A |
| pcDNA5/FRT-BARD1 P89A/W91A | In this study | N/A |
| pTRIP-Puro-P2A-BRCA1 | In this study | N/A |
| pTRIP-Puro-P2A-BRCA1 I26A | In this study | N/A |
| pTRIP-Puro-P2A-BRCA1 E3d | In this study | N/A |
| pTRIP-Blasticidin-P2A-BARD1 | In this study | N/A |
| Software and algorithms | ||
| GraphPad Prism 9 | Graphpad | https://www.graphpad.com/ |
| Image J | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Image lab | Bio-Rad | https://www.bio-rad.com |
| Celleste 5 image analysis | Invitrogen | https://www.thermofisher.com/ |
| Casplab software | Casplab | https://www.casplab.com/ |
| Matlab | MathWorks | https://www.mathworks.com/ |
| FlowJo software | FlowJo | https://www.Flowjo.com |
| Adobe Illustrator 2020 | Adobe | https://www.adobe.com |
| Adobe Photoshop 2023 | Adobe | https://www.adobe.com |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Bacterial strains
DH5a E. coli strain (genotype: F– endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG purB20 φ80dlacZΔM15 Δ(lacZYA-argF) U169, hsdR17(rK–mK+), λ–) and BL21(DE3) E. coli strain (genotype: B F– ompT gal dcm lon hsdSB(rB–mB–) λ (DE3 [lacI lacUV5-T7p07 ind1 sam7 nin5]) [malB+]K-12(λS)) was transformed with protein expression plasmids (key resources table) and grown in Luria Broth at 37°C in the presence of ampicillin (100 mg/l), Kanamycin (50 mg/l) or Kanamycin (50 mg/l) + chloramphenicol (34 mg/l) with constant agitation (180 rpm). MAX Efficiency™ DH10Bac Competent Cells (genotype: F-mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ-rpsL nupG/pMON14272/pMON7124) was transformed with the baculovirus vectors (key resources table) and grown in Luria Broth plate at 37°C in the presence of 50 mg/l kanamycin, 7 mg/l gentamicin and 10 mg/l tetracycline with constant agitation (180 rpm).
Insect cell lines
Spodoptera frugiperda (Sf9) cells were manipulated according to the instructions accompanying Bac-to-bac system (Life technologies). Sf9 insect cells were grown in Sf-900 III serum free media (Gibco) and seeded at 1 ×106 cells/ml for virus amplification. Trichoplusia ni High Five (BTI-TN-5B1–4) insect cells were grown in HYQ SFX INSECT (Cytiva) and infected with a multiplicity of infection equal to 5 for 40–44 hours for protein production. Both Sf9 cells and High Five cells were grown at 27°C with 140 rpm agitation.
Mammalian cell lines
HEK293T (ATCC), HeLa (ATCC), U2OS (DR, SA, EJ5) clones were gifts from Jeremy Stark) and MDA-MB-436 (gift from Bing Xia) cells were grown in Dulbecco’s modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum (Sigma), 100 μg/ml streptomycin, and 100 U/ml penicillin (Sigma) at 37°C with 5% CO2. The chemicals used in this study to treat cells are listed in key resources table.
METHOD DETAILS
Plasmids construction
pFastbac-Flag-BRCA1(from Jeffrey Parvin) and (pFastBac-Twin-StrepTagII-BARD1; gift from Andrew Deans, Addgene plasmid #137166)56 were used for BRCA1 and BARD1 expression in insect cells. pCOT7n-BRCA11–304, pCOT7n-BRCA11–112 and pET28-BARD126–142 were used for BRCA1-BARD1 RING heterodimer expression in bacterial cells. pcDNA5/FRT-HA-BRCA1 (from Jeffrey Parvin)38, pTRIP-Puro-P2A-HA-BRCA1, pTRIP-Blasticidin-P2A-Flag-BARD1 and pcDNA5/FRT-Flag-BARD1 were used and modified for BRCA1 and BARD1 expression in mammalian cells. QuikChange site-directed mutagenesis was used to construct the mutant variants of BRCA1 and BARD1 in full length or truncation forms, namely, I26A, C61G, L63A+K65A, N66Y+I68R, L63A+K65A+N66Y+I68R, I26A+L63A+K65A (AAA or 3A or E3d), I26A+N66Y+I68R, I26A+L63A+K65A +N66Y+I68R, L51A, I26A+L51A+L63A+K65A, I26S+L63A+K65A (SAA), I26N+L63A+K65A (NAA), I26T+L63A+K65A (TAA), R71G of BRCA1; P89AW91A (PW) and R99E of BARD1. The fragment of BRCA1200–500, BRCA1500–894, BRCA1936–1316, BARD1124–270, BARD1216–425, BARD1425–565 and BARD1124–777 into pRSFDuet-His-Sumo-Flag vector for protein expression in bacterial cells. The sequences of the primers used are available upon request. The plasmids for expressing proteins of ubiquitylation machinery, including human E1 (UBA1), E2s (UBE2D3, UBE2W, UBE2E3, UBE2E2, UBE2E1, UBE2N, UBE2V2/MMS2), Ubiquitin, Histone (pET3a-H2B-1K, pET3a-H4, pET3a-H3.2-C110A, and pMCSG7-His-TEV-VSVG-H2A) were described before21,34. pFastbac-Flag-CtIP was a gift from Patrick Sung, plasmids of UBE2B (#31408) and UBE2C (#15779) were from Addgene.
Protein purification
Purification of BRCA1-BARD1 from insect cells
The bacmid production in E. coli strain DH10Bac, baculoviruses generation and amplification in SF9 cells, and protein expression in Hi5 cells were done as described17. All purification steps were carried out at 0°C to 4°C. To prepare extract, the frozen cell pellet (8 g, from 600 ml culture) was thawed and suspended in 50 ml of cell breakage buffer A (50 mM Tris-HCl, pH 7.5, 450 mM KCl, 1 mM EDTA, 0.01% Igepal-CA630, 1 mM 2-mercaptoethanol (βME), 10% glycerol, 5 mM MgCl2, 2 mM ATP and the following protease inhibitors: aprotinin, chymostatin, leupeptin, and pepstatin A at 3 μg/ml each, 1 mM PMSF, 0.9 mg/ml benzamidine hydrochloride) for cell lysis using 10% strength sonication (Microtip, Branson Digital Sonifier) for 3 min (2 sec pause on, 5 sec pause off). The lysate was cleared by centrifugation at 40,000 × g for 30 min, filtered through 0.45 mm PES filter (Thermofisher, FB12566509), and loaded onto 1ml StrepTrap XT Chromatography Column (Cytiva, 09920071) at 0.5 ml/min. After the column was washed with 50 ml buffer B (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 0.5 mM EDTA, 0.01% Igepal-CA630, 1 mM bME, 10% glycerol, 5 mM MgCl2, 2 mM ATP, the following protease inhibitors: aprotinin, chymostatin, leupeptin, and pepstatin A at 0.03 μg/ml each, 1 mM PMSF, 0.9 mg/ml benzamidine hydrochloride), before the bound proteins were eluted 10 ml of buffer B containing 50 mM biotin at 0.5 ml/min. The eluates were mixed with 32 ml buffer C (25 mM Tris-HCl, pH 7.5, 10% glycerol, 0.5 mM EDTA, 0.01% Igepal CA630, 1 mM DTT) before being further fractionated in a 1 ml HiTrap SP HP Sepharose column (GE Healthcare) using a 12 ml gradient of 75–500 mM KCl in buffer C. The peak fractions were pooled, divided into 10 μl portions, frozen in liquid nitrogen, and stored at −80°C. The mutant forms of BRCA1-BARD1 were expressed and purified using the same procedures.
Purification BRCA1 and BARD1 Fragments and BRCA11–304-BARD126–142 from E. coli.
Either fragment of BRCA1200−500, BRCA1500–894, BRCA1936–1316, BARD1124–270, BARD1216–425, and BARD1425–565 in pRSFDuet-His-Sumo-Flag or pCOT7n-BRCA11–304 and pET28-BARD126–142 was introduced into E. coli BL21 (DE3) cells. An overnight culture derived from a single colony in 50 ml LB medium grown at 37°C was used to inoculate 2 l fresh LB medium. Expression was induced by the addition of 0.4 mM IPTG when the cell density had reached OD600=0.8, and cells were harvested after a 3-h incubation at 37°C. All the subsequent steps were carried out at 0–4°C. The cell pellet (8 g) was suspended in 50 ml buffer D (25 mM Tris-HCl pH 7.5, 10% glycerol, 150 mM KCl, 1mM DTT, 0.01% Igepal-CA630, 10 mM imidazole) containing the protease inhibitors (aprotinin, chymostatin, leupeptin, and pepstatin A at 0.3 μg/ml each, and 1 mM PMSF) and cell lysate was prepared by sonication. After centrifugation (100,000 × g for 90 min), the clarified lysate was incubated with 2 ml Ni-NTA resin (Ni Sepharose™ 6 fast flow, Cytiva, 17531803) for 1h. The resin was transferred to a glass column (1.5 × 15 cm), washed with 20 ml buffer E (25 mM Tris-HCl pH 7.5, 10% glycerol, 1 M KCl, 1 mM DTT, 0.01% Igepal-CA630, 10 mM imidazole) and then 50 ml D, before being eluted 8 times with 2 ml of 200 mM imidazole in buffer D. The eluates were mixed with 32 ml buffer F (25 mM Tris-HCl pH 7.5, 10% glycerol, 0.01% Igepal-CA630, 1 mM DTT) before being further fractionated in a 1 ml HiTrap SP HP Sepharose column (GE Healthcare) using a 12 ml gradient of 50–500 mM KCl in buffer F. The eluates were pooled and concentrated in Centricon-10K concentrator (Amicon) to 0.5 ml before being further fractionated in a Superdex 200 10/300 GL column (GE Healthcare) with 24 ml of buffer F containing 300 mM KCl. The peak fractions were pooled, concentrated to ~100 μl, divided into 5 μl portions, frozen in liquid nitrogen, and stored at −80°C.
Purification of Histone
The individual expression vector for H2A, H2B, H3 and H4 was transformed into E. coli BL21(DE3) cells, cultured at 37°C in TB medium (Yeast extract, 24 g; Tryptone, 20 g; Glycerol, 4 mL; Phosphate buffer (0.17 M KH2PO4, 0.72 M K2HPO4), 100 ml) to OD=0.5–0.6, and incubated with 0.4 mM IPTG IPTG for 3 hours at 37°C before harvest. Histone octamers were purified using the one-pot refolding protocol as described previously24,57. Briefly, the mixed pellet with 1:1:1:1 of H2A:H2B:H3:H4 was suspended in buffer G (50 mM Tris-HCl pH 7.5 and 100 mM NaCl) and cell lysate was prepared by sonication and centrifuged at 40,000 × g, 4°C for 30 min. The supernatant was discarded and the inclusion bodies were resolved in buffer H (10 M urea, 20 mM acetate, pH 5.2, and 10 mM DTT) at room temperature by shaking 2 h gently. After insoluble debris was removed by centrifugation, the supernatant containing histone octamers were refolded by dialysis in refolding buffer I (20 mM Tris-HCl pH 8, 2 M NaCl and 2 mM β-mercaptoethanol) at 4°C for overnight, followed by centrifugation at 40,000 × g, 4°C for 20 min. The clarified supernatant was incubated with Ni-NTA resin for 2hs, and then histone octamers were captured on resin according to the manufacturer’s instructions of Ni-NTA (Cytiva). The 6×His-tag on H2A was removed using TEV protease (produced in-house) overnight at 4 °C in refolding buffer I. The eluates were concentrated in Centricon-10K concentrator (Amicon) to 0.5 ml before being further fractionated in a Superdex® 200 Increase 10/300 GL column (GE Healthcare) with 24 ml of buffer I. The octamer fractions were pooled, concentrated to ~100 μl, divided into 10 μl portions, frozen in liquid nitrogen, and stored at −80°C.
Other recombinant proteins
Ubiquitylation machinery including human E1 (UBA1), E2s (UBE2D3, UBE2W, UBE2E3, UBE2E2, UBE2E1, UBE2N, UBE2V2, UBE2B), Ubiquitin, RING1B1–159/BMI11–109, phosphorylated CtIP and RAD51 were purified to near homogeneity using our previously described procedures21,34,58,59.
Purification of 15N-labeled UBE2D3 and BRCA1-BARD1 RING heterodimer from E. coli.
The expression and purification of 15N-labeled Ube2D3 and wild-type or E3d of BRCA11–112/BARD126–140 for use in NMR experiments were as previously described21,34.
Nucleosome reconstruction
Mono-nucleosome core particles (NCPs) were reconstituted by the standard salt dialysis method by mixing histone octamers with 147 bp ‘601’ DNA in high salt buffer (25 mM Tris-HCl pH 7.5, 2 M NaCl, 0.1 mM EDTA, 1 mM DTT) in a mini dialysis tube (ThermoFisher, 87734) floating in 100 ml high salt buffer. Low-salt buffer (25 mM Tris-HCl pH7.5, 0.1 mM EDTA, 1 mM DTT) was pumped in over ~24 hours in a cold room with stirring. A final dialysis was performed into NCP storage buffer (25 mM Tris-HCl pH 7.5, 100 mM NaCl, 0.1 mM EDTA, 1 mM DTT). For ubiquitin G76C and dichloroacetone crosslinking of H2A-Ub, H2A K13C, H2A K15C and His-ubiquitin G76C were purified and cross-linked with 1,3-dichloroacetone (DCA) as previously described60.
Ubiquitylation assay in vitro
The standard condition for ubiquitylation assay was carried out with indicated amount of BRCA1-BARD1 and mutants, 50 nM UBA1, 0.5 μM E2, 180 nM nucleosome core particles (NCPs) or X nM pCtIP and 10 μM ubiquitin in the buffer J (25 mM Tris-HCl pH 7.5, 0.01% Igepal-CA630, 0.5 mM DTT, 100 μg/ml BSA and 100 mM KCl, 2.5 mM MgCl2, 2.5 mM ATP). The mixtures were incubated at 37°C for 30 min or the temperature & time indicated and then added 1:1 of 2x SDS-PAGE load buffer before being processed for Western Blotting, where VSV-G epitope antibody (Sigma, V4888), H3 antibody (CST, 4499) and H2B antibody (CST, 2934) were used. To isolate auto-ubiquitylation form of BRCA1-BARD1, a 100 μl standard reaction of BRCA1-BARD1 with or without with ATP/MgCl2 was incubated at 37°C for 15 min, and then 30 μl prewashed Flag resin was added for shaking at 4°C. After the supernatant was removed, the resin was washed for 3 times with buffer J and aliquoted into 3 tubes for following reactions with 180 nM NCP at 37°C for 1, 2, 5 min respectively.
DNA substrates and DNA binding assay
DNA bubble and double-stranded DNA were assembled from oligonucleotides 1 (5’-Cy5-TTATATCCTTTACTTTGAATTCTATGTTTAACCTTTTACTTATTTTGTATTAGCCGGAC CTTATTTCAATTATGTTCAT-3’) and oligo-nucleotides 2 (5’-ATGAACATAATTGAAATAA GGATCCACTCTACATGCTCACACACTCGAACTCATGATAGAATTCAAAGTAAAGATATAA-3’), and from oligonucleotides 1 and oligonucleotides 3 (5’-ATGAACATAATTGAAATAA GGATCCGGCTAATACAAAATAAGTAAAAGGTTAAACATAGAATTCAAAGTAAAGGATATAA-3’) respectively; the asterisk identifies the oligonucleotide that was Cy5-labeled at its 5´ ends in each substrate. These DNA substrates (2 or 5 nM each) were incubated with wild-type or the specified mutant form of BRCA1-BARD1 at 4°C in 10 μl buffer K (25 mM Tris-HCl, pH 7.5, 90 mM KCl, 1 mM DTT, and 100 μg/ml BSA) for 20 min. After the addition of loading buffer (50% glycerol, 20 mM Tris-HCl, pH 7.4, 0.5 mM EDTA, 0.05% orange G), the reaction mixtures were resolved by 5% native polyacrylamide gel electrophoresis in TAE buffer (30 mM Tris-acetate, pH 7.4 and 0.5 mM EDTA) at 4°C. The DNA species were visualized by the Cy5 channel of the ChemiDoc MP Imaging System and the data was quantified with the Image Lab software (Biorad).
Affinity pulldown
RAD51 (1 μM) was incubated with 0.1 μM of Flag-BRCA1-BARD1 (WT or mutants) at 4°C for 30 min in 30 μl buffer L (25 mM Tris-HCl pH 7.5, 10% Glycerol, 0.5 mM EDTA, 0.05% Igepal CA630, 1 mM 2-mercaptoethanol and 150 mM KCl). Then, the reaction mixture was mixed with 10 μl anti-Flag M2 affinity resin at 4°C for 30 min to capture protein complexes through the Flag tag on BRCA1. After washing the resin three times with 200 μl buffer L, bound proteins were eluted with 20 μl 2% SDS at 37°C for 5 min. The supernatant (S), last wash (W) and SDS eluate (E), 8 μl each, were analyzed by SDS-PAGE and Coomassie blue staining.
D-loop assay
This was conducted as described61,62. Briefly, the Cy5-labeled 90-mer oligonucleotide (5’-Cy5AAATCAATCTAAAGTATATATGAGTAAACTTGGTCTGACAGTTACCAATGCTTA ATCAGTGAGGCACCTATCTCAGCGATCTGTCTATTT-3’; 0.7 μM nucleotides) was incubated with RAD51 (0.3 μM) at 37°C for 5 min in buffer K containing 1 mM MgCl2 and 2 mM AMP-PNP. Following the incorporation of the indicated concentration (30, 60 and 90 nM) of BRCA1-BARD1 and a 5-min incubation at 37°C, the D-loop reaction was initiated by adding pBluescript SK replicative form I DNA (15 μM base pairs) and was incubated at 37°C for 7 min. The molar ratio of the 90-mer to pBluescript plasmid in the reactions was 1.6 to 1. The reaction was terminated by adding an equal volume of 1% SDS containing 1 mg/ml proteinase K, 2.5 μl loading buffer, and a 5-min incubation at 37°C. The deproteinized reaction mixtures were resolved by electrophoresis in a 1% agarose gel, which was directly imaged with the Cy5 channel of the ChemiDoc MP Imaging System. The data was analyzed with the Image Lab software.
FRET E2~Ub discharge assay
Ubiquitin and UBE2D3 were fluorescently labeled at the N-terminus with AlexaFluor488 and AlexaFluor594, respectively46. Ub488 and UBE2D3594 were conjugated in buffer M (50 mM Na2HPO4/NaH2PO4 pH 7.4, 150 mM NaCl, 5 mM MgSO4, 2 mg/ml BSA, and 5 mM ATP). The Ub~2D3 conjugates were subsequently purified and flash frozen for long-term storage at 1 μM (E3 Bioscience LLC). To measure the Ub488~2D3594 discharge reaction, a 20 μL volume of 200 nM BRCA1-BARD1 or mutants and 200 nM NCP in buffer N (50 mM sodium phosphate pH 7.4, 100 mM NaCl, 5 mM MgSO4, 1 mM TCEP and 2 mg/ml BSA) was mixed with an equivalent volume of 200 nM Ub488~2D3594 conjugate. Fluorescence intensity measurements were performed in 384 well plates on a Synergy 2 microplate reader. For Ub488 measurements, we used 485/20 nm excitation and 530/20 nm emission filters and for 2D594, we used 530/25 nm excitation and 590/20 nm emission filters. For the FRET signal, we used 485/20 nm excitation and 590/20 nm emission filters. Data for Ub~2D discharge reactions were processed and analyzed using MATLAB.
NMR spectroscopy
NMR experiments were performed on Bruker 500-MHz Avance II spectrometer and analyzed as described34. 1H,15N- transverse relaxation optimized spectroscopy (TROSY) spectra were acquired using 1024 complex points in the 1H dimension and 100 complex points in the 15N dimension. NMR samples were prepared in buffer containing 25 mM Na-phosphate, 150 mM NaCl, and 10% D2O at pH7.0. Spectra of 235 μM 15N-labeled BRCA11–112-BARD126–142 complexes (wild-type, IBRCA1I26A-BARD1, or BRCA1E3d-BARD1) were collected at 35°C. Titrations of 15N-labeled UBE2D3 binding to unlabeled wild-type or E3d of BRCA11–112-BARD126–142 were performed by acquiring a series of 1H,15N-TROSY spectra at 25°C starting with 225 μM 15N-enriched samples in the absence and after sequential additions of unlabeled binding partner(s). Data were processed using NMRPipe/NMRDraw and visualized with NMRView.
Mammalian cell culture and transfection
HEK293T (ATCC), HeLa (ATCC), U2OS (DR, SA, EJ5) clones were gifts from Jeremy Stark) and MDA-MB-436 (gift from Bing Xia) cells were grown in Dulbecco’s modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum (Sigma), 100 μg/ml streptomycin, and 100 U/ml penicillin (Sigma). The cells were tested for mycoplasma contamination by Bionique testing labs (http://www.bionique.com/). Control siRNA (UAGCCGGUAGACUUAGGUCUG), BRCA1 siRNA (AAGCUCCUCUCACUCUUCAGU) oligonucleotides were purchased from Qiagen. TP53BP1 siRNA (s14313) was purchased from Ambion-Thermo Fisher Scientific. Dharmacon™ TRIPZ™ lentiviral shRNAs against BRCA1 (V2THS) and BARD1 (RHS4696) were purchased from Horizon-PerkinElmer. Transfection of siRNA, mammalian expression vectors, shRNA and pCMV-I-SceI-3xNLS was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. To create a FLP-in version of HeLa, we stably integrated a flippase recognition target (FRT) sequence into the cells by using the pFRT/lacZeo plasmid (Thermo Fisher Scientific). We tested Zeocin-resistant clones that had a single integration site detected by Southern blot for high-activity integration sites by using the mammalian β-galactosidase activity assay (Gal-Screen T1029, Thermo Fisher Scientific). Clonal expansion of the selected colony established the HeLa-FRT cell line. To generate stable HeLa-FRT shBRCA1/BARD1 cells, TRIPZ™ lentiviral shRNAs against BRCA1 or BARD1 were transfected with their respective plasmids and individual clones were selected with 2 μg/ml puromycin. To generate HeLa-FRT shBRCA1/BARD1 and MDA-MB-436 cell lines expressing HA-BRCA1/BARD1 or their mutants, cells were transfected with their respective plasmids and individual clones were selected with 200 μg/ml hygromycin, 2 μg/ml blastcidin or 2 μg/ml puromycin.
Co-immunoprecipitation analysis
HeLa cells grown on 10 cm cell culture dishes were treated with or without 10 μM olaparib overnight prior to harvest. Following two washes with PBS (phosphate buffered saline), cells were scraped off and transferred to Eppendorf tubes. Whole cell lysate was prepared by adding 1 ml of lysis buffer (300 mM NaCl, 1.0% Triton X-100, 5 mM EDTA, 2 mM NaVO4, 2 mM Na4O7P2, 0.02% NaN3, and 50 mM Tris-HCl, pH7.4) with protease inhibitors (Roche complete protease inhibitor tablet) to cell pellets. Following a 10s sonication, the cell extract was cleared by centrifugation at 13000 × g for 20 min at 4°C. The supernatant fraction (2 mg protein in total) was incubated with DNase I (20U) for 15 min at 4°C. Then, 30 μl of anti-Flag resin (Sigma) or 20ul Pierce™ Anti-HA Magnetic Beads (Thermo Scientific™, 88837) was added, followed by incubation at 4°C overnight. After washing the resin 4 times with lysis buffer, bound proteins were eluted with 30 μl SDS gel loading buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 0.1% bromophenol blue, 10% glycerol, 10% 2-mercaptoethanol) and the eluates were subject to Western blot analysis with anti-Flag and anti-RAD51 antibodies.
Immunoblot analysis
Protein was extracted from cells harvested two days after transfection with the indicated siRNAs or three days after 1 μg/ml doxycycline treatment using NETN buffer (20 mM Tris-HCl pH 8, 420 mM NaCl, 1 mM EDTA, 0.5% Igepal CA630, 1 mM DTT, and Roche Protease Inhibitor Cocktail). Blots (20–50 mg of total protein) were probed with the following antibodies: HA (3724S, Cell Signaling; 1:1000), BRCA1(SC6954, Santa Cruz; 1:500), BARD1(ab50984, Abcam; 1:1000), Tubulin (2128S, Cell Signaling; 1:2000), GAPDH (2118S, Cell Signaling; 1:1500), Lamin B1 (SC374015, Santa Cruz, 1:500), 53BP1(MAB3802, Millipore Sigma; 1:1000), Phospho-Histone H2A.X (9718S, Cell Signaling; 1:2000), Flag M2-HRP (Sigma, A8592), and Actin (Abcam, ab3280) according to the instructions provided by the manufacturers. If needed, the blots were incubated with HRP-conjugated secondary antibodies (Pierce 31450 for rabbit anti-mouse IgG-HRP; Sigma A6154 for goat anti-rabbit IgG-HRP; Santa Cruz Biotech SC2032 for goat anti-rat IgG-HRP) before visualization of protein signals using the ECL max kit (Biorad).
DNA repair reporter assays (HR, SSA and NHEJ)
The DR-U2OS cell line containing a single integrated copy of the DR-GFP reporter was used63,64. Exponentially growing cells were seeded in 6-well plates at 2 × 105 cells per well prior to reverse transfection with 2 μl siRNA (20 μM) and 3 μl Lipofectamine™ RNAiMAX. One day after siRNA transfection, cells were transfected with 1.25 μg HA-BRCA1 or HA-BARD1 and 0.75 μg I-SceI expression vector (pCBASce) and 3 μl Lipofectamine™ 2000. HR proficiency was determined by counting the fraction of GFP-positive cells using a BD FACS Calibur S at 72 h post I-SceI transfection. The results were derived from 3 to 5 transfections of at least 3 independent experiments. Same procedures were followed in EJ5-U2OS (for NHEJ) and SA-U2OS (for SSA) cell lines.
Immunofluorescence microscopy and image analysis
Cells were subjected to the treatment of 6 Gy X- or γ-irradiation (IR) or 4 μM Cisplatin for 24 hours followed by 3 washes with PBS, before being pre-extracted and fixed at different time points post-treatment. Pre-extraction was performed on ice for 10 min with cold cytoskeleton buffer (10 mM PIPES pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 0.5% Triton X-100) followed by 10 min with cytoskeleton stripping buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mm MgCl2, 1% Tween 40 (v/v), 0.5% sodium deoxycholate). Next, cells were fixed with 4% paraformaldehyde on ice for 20 min, washed with PBS, and permeabilized for 10 min with 0.5% Triton X-100 in PBS. After being blocked in blocking buffer (0.2% Triton-X, 5% Goat serum, 2% BSA in PBS) on ice for 20 min, cells were incubated with primary antibodies in blocking buffer overnight at 4°C in a humid chamber. The following primary antibodies were used HA (3724S, Cell Signaling; 1:500) to quantify HA-BRCA1 wild-type and mutants, RAD51(8875S, Cell Signaling; 1:500), γH2AX (05636, Millipore), RPA (MABE285, Millipore, 1:500), CtIP (61141, Active Motif) and BrdU (347580, BD Biosciences). Pre-extraction steps were omitted for the experiment with RAD51 and γH2AX foci. Then, cells were washed in PBS 3 times, incubated with Alexa Fluor or FITC conjugated secondary antibodies (Jackson Immunno Research Labs) for 2 h at room temperature, and stained with DAPI for another 15 min, before slides were mounted using antifade mounting media (Cell signaling 9071). Images were captured using a Nikon Swept Field fluorescence microscope or an EVOS M5000 microscope. The average number of RAD51 and RPA foci per nucleus and percentage of cells that were positive for these foci positive were determined after scoring at least 100 nuclei. Images were generated in the Core Optical Imaging Facility which is supported by UTHSCSA, NIH-NCI P30 CA54174 (CTRC at UTHSCSA).
Clonogenic survival assay
HeLa cell lines stably expressing HA-BRCA1 wild-type or mutants were pretreated with doxycycline for 3 days or longer as described above. 400 cells/well were seeded into 12-well plates, treated with indicated amount of MMC (Sigma), olaparib (Selleckchem), camptothecin (Sigma) or cisplatin (Selleckchem) in regular growth medium for 11–12 days. Cells were fixed with methanol and stained with 0.5% crystal violet in methanol before colonies were counted. Clonogenic survival was determined for a given concentration of cells that were plated by dividing the number of colonies on each treated plate by the number of colonies on the untreated plate, taking the plating efficiency of untreated cells into account.
Preparation of cytoplasmic and nuclear extracts
The REAP method for the preparation of cytoplasmic and nuclear extracts was followed65. Briefly, HeLa cells from 10-cm dishes were washed with ice-cold phosphate buffer saline (PBS) pH 7.4, collected by centrifugation, resuspended in 900 ml of ice-cold PBS with 0.1% NP40 and protease inhibitors, and triturated 5 times using a p1000 micropipette. The lysed cell suspension was centrifuged for supernatant (this is the cytoplasmic fraction) and the pelleted nuclei was washed once with PBS with 0.1% NP40 and lysed by NETN buffer with protease inhibitors to yield the nuclear extract fraction. The cytoplasmic and nuclear fractions, 20 μg each, were analyzed by immunoblotting for their content of BRCA1, BARD1, Tubulin and Lamin B1.
Single Cell Neutral or Alkaline Gel Electrophoresis (Comet Assay)
Hela-shBRCA1 cells stable expressing HA-BRCA1 wild-type or E3d were pretreated with doxycycline for 3 days before indicated drug treatment, then cell samples were harvested at various time points and subjected to comet assay. Briefly, single cell suspensions were seeded in low melting agarose (20×103 cells/ml) on pre-coated frosted microscope slides. Neutral or alkaline lysis (1 hour) and unwinding (30 min) were performed prior to electrophoresis in neutral or alkaline running buffer as described66. Samples were dehydrated in 100% ethanol, and dried in a 50-degrees oven for 30 mins and sequentially at room temperature for another 24 hours. Then the slides were hydrated in water for 30 mins prior to staining in SYBR Gold (Invitrogen Cat# S11494, Carlsbad, CA) for 15 minutes. Images were captured using an EVOS M5000 microscope. Olive Tail Moment was determined using Casplab software (casplab.com). At least 100 representative comets were measured for each treatment group.
Analysis of cell cycle profiles
For cell cycle profile analysis, 2×106 cells exponentially growing cells were washed with cold PBS, collected by centrifugation, and then fixed in 10 ml of ice-cold 70% ethanol/PBS for at least 2 days at 4°C. The fixed cells were washed sequentially with 1 ml each of 30% ethanol/PBS and 0.05% BSA/PBS at 4°C. Cells were incubated in the staining solution (40 μg/ml RNase A and 30 μg/ml propidium iodide in PBS) for 15 min at 37°C in the dark. The stained cells were fractionated in a BD FACS Calibur S instrument and analyzed by the FlowJo software.
PLA (Proximity Ligation Assay)
To analyze colocalization of BrdU/CtIP in HeLa-shBRCA1 cells expressing wild-type or E3d mutant HA-BRCA1, where endogenous BRCA1 was depleted by doxycycline treatment (1 μg/ml, 72 h), proximity ligase assays (PLAs) were carried out as per manufacturer’s protocol (Duolink PLA kit; DUO92101; Sigma-Aldrich). Cells were cultured with glass bottom dishes (SKU#801002; NEST). To chase resection after DSB, BrdU (10 μM) was added to the cells for 24 h before cells were exposed to X-ray (6 Gy) and cultured for 2h. The cells were washed with PBS and pre-permeabilized with 0.5% Triton X-100 in PBS, before being fixed with 1% of formaldehyde/PBS for 15 min. The fixed cells were washed twice with PBS and blocked with Duolink Blocking buffer for 1 h at 37°C with humidity. Next, the cells were incubated with anti-BrdU (347580; clone B44; BD) and anti-CtIP (PA5–84133; Invitrogen) antibodies in Duolink antibody diluent at 4°C overnight. In Situ PLA probes (anti-mouse plus and anti-rabbit minus) were diluted 1:5 in Duolink antibody diluent and incubated to detected anti-BrdU (mouse) and anti-CtIP (rabbit) antibodies for 1 h at 37°C. After being washed with 1x Wash buffer A solution three times for 5 min each, dishes were incubated at 37°C for 30 min with 1x Duolink ligation buffer solution and then washed twice with 1x Wash buffer A solution for 5 min each. For amplification signals, 1x Amplification mix solution was prepared as per manufacturer’s instructions and added into dishes for incubation at 37°C for 100 min in the dark. Lastly, dishes were washed with 1x Wash buffer B solution three times for 5 min each and 0.01× diluted Wash buffer B solution once for 5 min before being mounted with emulsion oil including DAPI and applied to the confocal microscope (FV3000; Olympus). Foci formation was analyzed using ImageJ (1.53a version; NIH) software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Each experiment was repeated three times or more. The statistical analysis was performed using Prism 9 (GraphPad Software, Inc., La Jolla, CA; http://www.graphpad.com/quickcalcs/ttest1.cfm) on the data from at least three independent experiments, as specified. Statistical significance was assessed by two-tailed unpaired Student’s t-test and two-way ANOVA. *P ≤ 0.05, ** P ≤ 0.01, *** P≤0.001, and **** P≤0.0001 were considered significant. The statistical details are shown in the figures and figure legends.
Supplementary Material
Highlights:
Provide insights into BRCA1-BARD1 regulation and identify a truly inactive mutant
BRCA1-BARD1 E3 ligase activity is critical for DNA end resection
BRCA1-BARD1 E3 ligase contributes to later steps of homologous recombination
Histone ubiquitylation by BRCA1-BARD1 plays an important role in DNA end resection
Acknowledgments:
We are grateful to Jeffery Parvin, Neil Johnson, Jeremy Stark, and Daniel Durocher for reagents and technical advice, and to Patrick Sung, Trisha Davis, and Reuben Harris for helpful discussions and critical reading of the manuscript. This study was supported by a V Scholar Cancer Research Grant, a Young Investigator Award from Max and Minnie Tomerlin Voelcker Fund, the Cancer Prevention and Research Institute of Texas (RP210102), and NIH research grants R01GM141091 and RSG-22-721675-01-DMC awarded to W.Z. Additional funding was from NIH research grants including R50CA265315 (Y. K.), R01CA244212 (A.A.H), R01CA246807 (S.B.), R01NS106173 (G.S.), R01AI136697 (D.N.I.), R01CA268641 (T.J.C and W.Z.) and R01CA260834 (R.E.K. and P.S.B.). R.E.K. is the Edmond H. Fischer/Washington Research Foundation Endowed Chair in Biochemistry and S.B. is the Mays Family Foundation Distinguished Chair in Oncology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests:
Authors except DNI declare that they have no competing interests. DNI is a co-founder and a shareholder of E3 Bioscience LLC, a commercial entity that manufactures FRET-active E2~Ub conjugates used in this study.
Data and materials availability:
All data, code, and materials used in the analysis will be made available upon request.
References
- 1.Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, and King MC (1990). Linkage of early-onset familial breast cancer to chromosome 17q21. Science 250, 1684–1689. 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- 2.Petrucelli N, Daly MB, and Pal T (1998). BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In GeneReviews((R)), Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, and Amemiya A, eds. [updated 2022 May 26]. [Google Scholar]
- 3.Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, et al. (2015). BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell 57, 636–647. 10.1016/j.molcel.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silver DP, and Livingston DM (2012). Mechanisms of BRCA1 tumor suppression. Cancer Discov 2, 679–684. 10.1158/2159-8290.CD-12-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, and Livingston DM (1997). Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88, 265–275. 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 6.Moynahan ME, Chiu JW, Koller BH, and Jasin M (1999). Brca1 controls homology-directed DNA repair. Mol Cell 4, 511–518. 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 7.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, and Scully R (2014). BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559. 10.1038/nature13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savage KI, Gorski JJ, Barros EM, Irwin GW, Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ, McCluggage WG, et al. (2014). Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol Cell 54, 445–459. 10.1016/j.molcel.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawai S, and Amano A (2012). BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J Cell Biol 197, 201–208. 10.1083/jcb.201110008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleiman FE, and Manley JL (1999). Functional interaction of BRCA1-associated BARD1 with polyadenylation factor CstF-50. Science 285, 1576–1579 10.1126/science.285.5433.1576. [DOI] [PubMed] [Google Scholar]
- 13.Kleiman FE, and Manley JL (2001). The BARD1-CstF-50 interaction links mRNA 3’ end formation to DNA damage and tumor suppression. Cell 104, 743–753. 10.1016/s0092-8674(01)00270-7 [DOI] [PubMed] [Google Scholar]
- 14.Deng CX (2006). BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 34, 1416–1426. 10.1093/nar/gkl010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao W, Wiese C, Kwon Y, Hromas R, and Sung P (2019). The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu Rev Biochem 88, 221–245. 10.1146/annurev-biochem-013118-111058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tarsounas M, and Sung P (2020). The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat Rev Mol Cell Biol 21, 284–299. 10.1038/s41580-020-0218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao W, Steinfeld JB, Liang F, Chen X, Maranon DG, Jian Ma C, Kwon Y, Rao T, Wang W, Sheng C, et al. (2017). BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 550, 360–365. 10.1038/nature24060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Densham RM, and Morris JR (2019). Moving Mountains-The BRCA1 Promotion of DNA Resection. Front Mol Biosci 6, 79. 10.3389/fmolb.2019.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, and Weissman AM (1999). RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci U S A 96, 11364–11369. 10.1073/pnas.96.20.11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brzovic PS, Rajagopal P, Hoyt DW, King MC, and Klevit RE (2001). Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol 8, 833–837. 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 21.Christensen DE, Brzovic PS, and Klevit RE (2007). E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol 14, 941–948. 10.1038/nsmb1295. [DOI] [PubMed] [Google Scholar]
- 22.Witus SR, Stewart MD, and Klevit RE (2021). The BRCA1/BARD1 ubiquitin ligase and its substrates. Biochem J 478, 3467–3483. 10.1042/BCJ20200864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalb R, Mallery DL, Larkin C, Huang JT, and Hiom K (2014). BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep 8, 999–1005. 10.1016/j.celrep.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witus SR, Burrell AL, Farrell DP, Kang J, Wang M, Hansen JM, Pravat A, Tuttle LM, Stewart MD, Brzovic PS, et al. (2021). BRCA1/BARD1 site-specific ubiquitylation of nucleosomal H2A is directed by BARD1. Nat Struct Mol Biol 28, 268–277. 10.1038/s41594-020-00556-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witus SR, Zhao W, Brzovic PS, and Klevit RE (2022). BRCA1/BARD1 is a nucleosome reader and writer. Trends Biochem Sci 47, 582–595. 10.1016/j.tibs.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruffner H, Joazeiro CA, Hemmati D, Hunter T, and Verma IM (2001). Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci U S A 98, 5134–5139. 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fowler DM, Parvin JD, Shendure J, and Fields S (2015). Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics 200, 413–422. 10.1534/genetics.115.175802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reid LJ, Shakya R, Modi AP, Lokshin M, Cheng JT, Jasin M, Baer R, and Ludwig T (2008). E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc Natl Acad Sci U S A 105, 20876–20881. 10.1073/pnas.0811203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, deRooij DG, Hirsch S, Ravi K, Hicks JB, et al. (2011). BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334, 525–528. 10.1126/science.1209909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, Daza-Martin M, Fletcher A, Blair-Reid S, Beesley J, Johal B, et al. (2016). Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol 23, 647–655. 10.1038/nsmb.3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, Ganesan S, and Xia B (2017). Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. Elife 6. 10.7554/eLife.21350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura K, Saredi G, Becker JR, Foster BM, Nguyen NV, Beyer TE, Cesa LC, Faull PA, Lukauskas S, Frimurer T, et al. (2019). H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat Cell Biol 21, 311–318. 10.1038/s41556-019-0282-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becker JR, Clifford G, Bonnet C, Groth A, Wilson MD, and Chapman JR (2021). BARD1 reads H2A lysine 15 ubiquitination to direct homologous recombination. Nature 596, 433–437. 10.1038/s41586-021-03776-w. [DOI] [PubMed] [Google Scholar]
- 34.Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D 3rd, Fukuda M, Ohta T, and Klevit R (2003). Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci U S A 100, 5646–5651. 10.1073/pnas.0836054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu Q, Botuyan MV, Zhao D, Cui G, Mer E, and Mer G (2021). Mechanisms of BRCA1-BARD1 nucleosome recognition and ubiquitylation. Nature 596, 438–443. 10.1038/s41586-021-03716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pruneda JN, Littlefield PJ, Soss SE, Nordquist KA, Chazin WJ, Brzovic PS, and Klevit RE (2012). Structure of an E3:E2~Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol Cell 47, 933–942. 10.1016/j.molcel.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart MD, Duncan ED, Coronado E, DaRosa PA, Pruneda JN, Brzovic PS, and Klevit RE (2017). Tuning BRCA1 and BARD1 activity to investigate RING ubiquitin ligase mechanisms. Protein Sci 26, 475–483. 10.1002/pro.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Starita LM, Islam MM, Banerjee T, Adamovich AI, Gullingsrud J, Fields S, Shendure J, and Parvin JD (2018). A Multiplex Homology-Directed DNA Repair Assay Reveals the Impact of More Than 1,000 BRCA1 Missense Substitution Variants on Protein Function. Am J Hum Genet 103, 498–508. 10.1016/j.ajhg.2018.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu X, and Chen J (2004). DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol 24, 9478–9486. 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu X, Fu S, Lai M, Baer R, and Chen J (2006). BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev 20, 1721–1726. 10.1101/gad.1431006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGinty RK, Henrici RC, and Tan S (2014). Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596. 10.1038/nature13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taherbhoy AM, Huang OW, and Cochran AG (2015). BMI1-RING1B is an autoinhibited RING E3 ubiquitin ligase. Nat Commun 6, 7621. 10.1038/ncomms8621. [DOI] [PubMed] [Google Scholar]
- 43.Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, and Baer R (1996). Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet 14, 430–440. 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 44.Paull TT, Cortez D, Bowers B, Elledge SJ, and Gellert M (2001). Direct DNA binding by Brca1. Proc Natl Acad Sci U S A 98, 6086–6091. 10.1073/pnas.111125998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, and Shendure J (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222. 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herkules F, Yu CH, Taylor AB, Dougherty V, Weintraub ST, and Ivanov DN (2022). Structural and functional asymmetry of RING trimerization controls priming and extension events in TRIM5alpha autoubiquitylation. Nat Commun 13, 7104. 10.1038/s41467-022-34920-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krais JJ, Wang Y, Patel P, Basu J, Bernhardy AJ, and Johnson N (2021). RNF168-mediated localization of BARD1 recruits the BRCA1-PALB2 complex to DNA damage. Nat Commun 12, 5016. 10.1038/s41467-021-25346-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S, Klijn C, van der Heijden I, van der Gulden H, Wientjens E, et al. (2011). BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 20, 797–809. 10.1016/j.ccr.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 49.Gunn A, and Stark JM (2012). I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol 920, 379–391. 10.1007/978-1-61779-998-3_27. [DOI] [PubMed] [Google Scholar]
- 50.Mukherjee B, Tomimatsu N, and Burma S (2015). Immunofluorescence-based methods to monitor DNA end resection. Methods Mol Biol 1292, 67–75. 10.1007/978-1-4939-2522-3_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fowler FC, and Tyler JK (2021). A Proximity Ligation Method to Detect Proteins Bound to Single-Stranded DNA after DNA End Resection at DNA Double-Strand Breaks. Methods Protoc 5. 10.3390/mps5010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Panier S, and Boulton SJ (2014). Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15, 7–18. 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 53.Vega A, Campos B, Bressac-De-Paillerets B, Bond PM, Janin N, Douglas FS, Domenech M, Baena M, Pericay C, Alonso C, et al. (2001). The R71G BRCA1 is a founder Spanish mutation and leads to aberrant splicing of the transcript. Hum Mutat 17, 520–521. 10.1002/humu.1136. [DOI] [PubMed] [Google Scholar]
- 54.Ransburgh DJ, Chiba N, Ishioka C, Toland AE, and Parvin JD (2010). Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res 70, 988–995. 10.1158/0008-5472.CAN-09-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Towler WI, Zhang J, Ransburgh DJ, Toland AE, Ishioka C, Chiba N, and Parvin JD (2013). Analysis of BRCA1 variants in double-strand break repair by homologous recombination and single-strand annealing. Hum Mutat 34, 439–445. 10.1002/humu.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan W, Murphy VJ, Charron A, van Twest S, Sharp M, Constantinou A, Parker MW, Crismani W, Bythell-Douglas R, and Deans AJ (2020). Preparation and purification of mono-ubiquitinated proteins using Avi-tagged ubiquitin. PLoS One 15, e0229000. 10.1371/journal.pone.0229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee YT, Gibbons G, Lee SY, Nikolovska-Coleska Z, and Dou Y (2015). One-pot refolding of core histones from bacterial inclusion bodies allows rapid reconstitution of histone octamer. Protein Expr Purif 110, 89–94. 10.1016/j.pep.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sigurdsson S, Trujillo K, Song B, Stratton S, and Sung P (2001). Basis for avid homologous DNA strand exchange by human Rad51 and RPA. J Biol Chem 276, 8798–8806. 10.1074/jbc.M010011200. [DOI] [PubMed] [Google Scholar]
- 59.Anand R, Pinto C, and Cejka P (2018). Methods to Study DNA End Resection I: Recombinant Protein Purification. Methods Enzymol 600, 25–66. 10.1016/bs.mie.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 60.Dos Santos Passos C, Choi YS, Snow CD, Yao T, and Cohen RE (2021). Design of genetically encoded sensors to detect nucleosome ubiquitination in live cells. J Cell Biol 220. 10.1083/jcb.201911130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao W, Saro D, Hammel M, Kwon Y, Xu Y, Rambo RP, Williams GJ, Chi P, Lu L, Pezza RJ, et al. (2014). Mechanistic insights into the role of Hop2-Mnd1 in meiotic homologous DNA pairing. Nucleic Acids Res 42, 906–917. 10.1093/nar/gkt924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao W, and Sung P (2015). Significance of ligand interactions involving Hop2-Mnd1 and the RAD51 and DMC1 recombinases in homologous DNA repair and XX ovarian dysgenesis. Nucleic Acids Res 43, 4055–4066. 10.1093/nar/gkv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, and Jasin M (2005). Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci U S A 102, 1110–1115. 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, and Livingston DM (2006). Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22, 719–729. 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 65.Nabbi A, and Riabowol K (2015). Rapid Isolation of Nuclei from Cells In Vitro. Cold Spring Harb Protoc 2015, 769–772. 10.1101/pdb.prot083733. [DOI] [PubMed] [Google Scholar]
- 66.Olive PL, and Banath JP (2006). The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1, 23–29. 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Confocal fluorescence microscopy images and western blot have been deposited at Mendeley: https://doi:10.17632/9s37k6s8jr.1 and is publicly available as of the date of publication. DOI is listed in the key resources table. All data reported in this publication will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-VSV-G epitope antibody | Sigma | Cat# V4888 |
| Anti-H3 antibody | Cell Signaling Technology | Cat# 4499 |
| Anti-H2B antibody | Cell Signaling Technology | Cat# 2934 |
| Anti-FLAG M2-HRP | Sigma | Cat# A8592 |
| Anti-RAD51 | Cell Signaling Technology | Cat# 18875 |
| Anti-RPA34 | Sigma | Cat# MABE285 |
| Anti-His-HRP | Sigma | Cat# A7058 |
| Anti-GAPDH | Cell Signaling Technology | Cat# 2118S |
| Anti-β-Tubulin | Cell Signaling Technology | Cat# 2128S |
| Anti-HA | Cell Signaling Technology | Cat# 3724S |
| Anti-BRCA1 | Santa Cruz Biotech | Cat# SC6954 |
| Anti-BARD1 | Abcam | Cat# ab50984 |
| Anti-Lamin B1 | Santa Cruz Biotech | Cat# SC374015 |
| Anti-53BP1 | Sigma | Cat# MAB3802 |
| Anti-Actin | Abcam | Cat# ab3280 |
| Anti-γH2AX | Sigma | Cat# 05636 |
| Anti-©H2AX | Cell Signaling Technology | at# 9718S |
| Anti-CtIP | Active Motif | Cat# 61141; RRID: AB_2714164 |
| Anti-BrdU | BD Biosciences | Cat# 347580; RRID: AB_400326 |
| Anti-CtIP | Invitrogen | Cat# PA5–84133; RRID: AB_2791285 |
| Chemicals, peptides, and recombinant proteins | ||
| Olaparib | Selleckchem | Cat#HY-10162; CAS: 763113–22-0 |
| Camptothecin | Sigma | Cat#208925; CAS: 7689–03-4 |
| Cisplatin | Selleckchem | Cat#P4394; CAS: 15663–27-1 |
| Mitomycin C | Sigma | Cat#HY-13316; CAS: 50–07-7 |
| Doxycycline | Sigma | Cat# D3447 |
| BRCA1-BARD1 (WT or mutants) | In this study | N/A |
| BRCA1 fragments (BRCA1200–500, BRCA1500–894, and BRCA1936–1316) | In this study | N/A |
| BARD1 fragments (BARD1124–270, BARD1216–425, and BARD1425–565 | In this study | N/A |
| 15N-labeled UBE2D3 | In this study | N/A |
| 15N-labeled BRCA11–112-BARD26–142 | In this study | N/A |
| E1 (UBA1) | In this study | N/A |
| E2s (UBE2D3, UBE2W, UBE2E3, UBE2E2, UBE2E1, UBE2N, UBE2V2, UBE2B) | In this study | N/A |
| Ubiquitin | In this study | N/A |
| RING1B1–159/BMI11–109 | In this study | N/A |
| Histone octamer | In this study | N/A |
| phosphorylated CtIP | In this study | N/A |
| RAD51 | In this study | N/A |
| AlexaFluor488-Ubiquitin | In this study | N/A |
| AlexaFluor594-UBE2D3 | In this study | N/A |
| Critical commercial assays | ||
| Duolink PLA kit | Sigma | Cat# DUO92101 |
| Q5 Site-Directed Mutagenesis Kit | New England BioLabs | Cat# E0554 |
| Mammalian β-Galactosidase Kit | Thermo Fisher Scientific | Cat# T1029 |
| Deposited data | ||
| Original images and blots | This manuscript | Mendeley data: doi:10.17632/9s37k6s8jr.1 |
| Experimental models: Cell lines | ||
| HeLa | ATCC | CCL-2 |
| U2OS (DR, SA, EJ5) | Jeremy Stark Lab | N/A |
| MDA-MB-436 | Bing Xia Lab | N/A |
| Oligonucleotides | ||
| Control siRNA (UAGCCGGUAGACUUAGGUCUG), | Qiagen | N/A |
| BRCA1 siRNA (AAGCUCCUCUCACUCUUCAGU) | Qiagen | N/A |
| TP53BP1 siRNA | Thermo Fisher Scientific | Cat# s14313 |
| Recombinant DNA | ||
| pFB-BRCA1 | In this study | N/A |
| pFB-BRCA1 I26A | In this study | N/A |
| pFB-BRCA1 E3d | In this study | N/A |
| pFB-BRCA1 R71G | In this study | N/A |
| pFB-BARD1 | In this study | N/A |
| pFB-BARD1-P89A/W91A | In this study | N/A |
| pcDNA5/FRT-BRCA1 | In this study | N/A |
| pcDNA5/FRT-BRCA1 I26A | In this study | N/A |
| pcDNA5/FRT-BRCA1 E3d | In this study | N/A |
| pcDNA5/FRT-BRCA1 C61G | In this study | N/A |
| pcDNA5/FRT-BARD1 | In this study | N/A |
| pcDNA5/FRT-BARD1 P89A/W91A | In this study | N/A |
| pTRIP-Puro-P2A-BRCA1 | In this study | N/A |
| pTRIP-Puro-P2A-BRCA1 I26A | In this study | N/A |
| pTRIP-Puro-P2A-BRCA1 E3d | In this study | N/A |
| pTRIP-Blasticidin-P2A-BARD1 | In this study | N/A |
| Software and algorithms | ||
| GraphPad Prism 9 | Graphpad | https://www.graphpad.com/ |
| Image J | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Image lab | Bio-Rad | https://www.bio-rad.com |
| Celleste 5 image analysis | Invitrogen | https://www.thermofisher.com/ |
| Casplab software | Casplab | https://www.casplab.com/ |
| Matlab | MathWorks | https://www.mathworks.com/ |
| FlowJo software | FlowJo | https://www.Flowjo.com |
| Adobe Illustrator 2020 | Adobe | https://www.adobe.com |
| Adobe Photoshop 2023 | Adobe | https://www.adobe.com |
All data, code, and materials used in the analysis will be made available upon request.