

Abstract
Chimeric antigen receptor (CAR) T cell therapy targeting CD19 has achieved tremendous success treating B-cell malignancies, however some patients fail to respond due to poor autologous T cell fitness. To improve response rates, we investigated whether disruption of the co-inhibitory receptors CTLA-4 or PD-1 could restore CART function. CRISPR-Cas9-mediated deletion of CTLA4 in preclinical models of leukemia and myeloma improved CART cell proliferation and anti-tumor efficacy. Importantly, this effect was specific to CTLA4, and not seen upon deletion of CTLA4 and/or PDCD1 in CART cells. Mechanistically, CTLA-4 deficiency permitted unopposed CD28 signaling and maintenance of CAR expression on the T cell surface under conditions of high antigen load. In clinical studies, deletion of CTLA4 rescued the function of T cells from leukemia patients that previously failed CART cell treatment. Thus, selective deletion of CTLA4 reinvigorates dysfunctional CLL patient T cells, providing a strategy for increasing patient responses to CART cell therapy.
Keywords: CAR T cells, Acute Lymphoblastic Leukemia (ALL), Chronic Lymphocytic Leukemia (CLL), CRISPR/Cas9, Cancer Immunotherapy, Checkpoint blockade, PD-1, CTLA-4, resistance, T cell exhaustion
eTOC Blurb
CD19-directed CAR T cell therapy is an effective treatment for B cell malignancies, but some patients fail to respond. Agarwal et. al. demonstrate that deletion of CTLA4 enhances anti-tumor efficacy and surface CAR expression in models of leukemia and lymphoma and in CLL patient CART cells. Interestingly, deletion of PDCD1 or of PDCD1 and CTLA4 did not promote antitumor efficacy of CART19 cells.
Graphical Abstract:
Introduction
CD19-directed CAR T cell (CART19) therapy has impressive clinical efficacy in patients with B cell malignancies.1,2 For instance, pediatric and young adult patients with relapsed/refractory (R/R) acute lymphoblastic leukemia (ALL) achieve an initial complete response rate of 80–96%2 and 50% of patients experience event-free survival 1 year after CART19 therapy.3–5 For ALL, the primary mechanism of failure is relapse due to antigen escape.6 CAR T cell products are also now approved for non-Hodgkin’s lymphoma (NHL), including large B-cell lymphoma (LBCL), follicular lymphoma (FL), and mantle cell lymphoma (MCL).7 In NHL, complete response rates vary from 4% to 60% depending on histology and/or product with about 30–50% of patients maintaining response rates long term.8 In contrast, complete response rates are 30–45% for patients with R/R chronic lymphocytic leukemia (CLL),9 but relapse is unusual after CAR T therapy for CLL and durable remissions rates are 20–30%; the primary mechanism of failure is lack of initial response.10 Thus, despite the tremendous success with CART19 treatment, many patients with hematological malignancies need improved therapies.
Failure of patients to respond to CART19 therapy is linked to T cell dysfunction which can be intrinsic to the starting T cell population or acquired as a result of exposure to cognate antigen, the tumor microenvironment (TME), patient disease activity and previous treatments.11,12 Transcriptomic profiling of autologous T cells from CLL patients reveals that sustained remissions associate with elevated frequencies of CD27+CD45RO− CD8+ T cells with memory-like characteristics, whereas non-responder T cells exhibit increased expression of genes associated with effector differentiation, glycolysis, exhaustion and apoptosis.13 In CLL, T cell dysfunction is characterized by increased expression of immune checkpoints like programmed death 1 (PD-1) and/or cytotoxic T lymphocyte-associated antigen 4 (CTLA-4).12–15 Further, elevated levels of PD-1 and/ or CTLA-4 expression at peak CAR T cell expansion in vivo correlate with poor CLL patient responses to CART19 therapy.13,16 In contrast, CART19 products enriched for central memory17 T cells exhibit promising results in R/R NHL patients.18
Immune checkpoint blockade (ICB) with antibodies against PD-1, PD-L1 and CTLA-4 has dramatically improved clinical outcomes for select groups of patients with advanced cancer. 19 For example, patients with advanced melanoma historically had dismal 5-year survival rates, however with the advent of ICB these patients achieve 5-year overall survival rates of 52% when treated with nivolumab (anti-PD-1 antibody)-plus-ipilimumab (anti-CTLA-4 antibody), 44% with nivolumab alone, and 26% with ipilimumab alone.17,20–22 As in endogenous tumor-infiltrating lymphocytes (TILs), increased expression of immune checkpoints on CAR T cells can limit the strength and duration of their anti-tumor response. In pre-clinical studies, antibody blockade of PD-1 improved CAR T cell-mediated anti-tumor response.23,24 Further, the efficacy of IL-13Ra2-directed CAR T cells was enhanced in mouse models when combined with CTLA-4 blockade or a self-secreted anti-CTLA-4 mini-body.25 In the clinical setting, mesothelin (MSLN)-directed CAR T cell therapy followed by administration of the anti-PD-1 agent pembrolizumab induced partial response in patients with malignant pleural disease.26 Similarly, pembrolizumab was well tolerated by patients with (R/R) large B cell lymphoma (LBCL) who relapsed after CART19 therapy, and 3 patients (25%) had complete or partial responses.27 Further, PD-1 inhibition augmented CART19 therapy in a subset of pediatric and young adult patients with R/R B lymphoblastic malignancies.28
Immune checkpoint signaling can also be disrupted in T cells using CRISPR/Cas9 technology. In this instance, CAR T cells are manufactured from patient T cells with CRISPR-Cas9-mediated disruption of the checkpoint before product administration and only infused T cells contain the deletion. Such CAR T cells may exhibit different efficacy and toxicity profiles than what has been observed for ICB, which is systemically administered to endogenous T cells. There is conflicting preclinical evidence as to whether PD-1 suppresses T cell activity through CD28 and/or TCR signaling29,30 and how CRISPR-Cas9-mediated disruption of PD-1 signaling impacts CART cell tumor clearance in preclinical mouse models of cancer.31–33 CTLA-4-deficient cytotoxic T-lymphocytes (CTLs) exhibit improved anti-tumor activity against bladder and colon cancer in xenograft mouse models.34,35 Furthermore, decreased expression of PD-1 and/or CTLA-4 increases cytotoxicity in CSPG4 CAR T cells against melanoma.36 First in-human pilot studies demonstrate the feasibility and safety of CRISPR-Cas9-mediated checkpoint gene-editing of human T cells in patients with advanced refractory cancer.37–39
Here, we tested whether single or dual CRISPR/Cas9 mediated deletion of PDCD1 and CTLA4 could prevent CAR T cell dysfunction, characterized by gradual loss of effector function and cytokine expression, as well as sustained expression of inhibitory receptors.40,41 We found that CTLA-4-deficient CART19 cells exibited enhanced anti-tumor activity in both an in vitro model of CAR T dysfunction and in xenograft mouse models, unlike PD-1-deficient CART19 cells and CART19 cells deficient for both PD-1 and CTLA-4. We provide mechanistic evidence to support these findings; CTLA-4 disrupted CART19 cells display prolonged surface CAR expression, increased proliferation and enhanced CART19 effector function supported by unopposed CD28 co-stimulation which is lost in CART19 cells deficient for both PD-1 and CTLA-4. Importantly, when CTLA4 is disrupted in nonresponding (NR) CLL patient T cells, these modified T cells enhance the efficacy of CLL CART19 compared to the unedited (WT) CART19 NR product. These findings nominate CTLA-4 deletion as a promising strategy to overcome T cell dysfunction and increase the efficacy of CAR T cell therapy.
Results
CRISPR/Cas9 deletion of CTLA4 in CART19 cells results in superior effector function in vitro.
To evaluate whether single or dual CRISPR/Cas9 mediated deletion of PD-1 and CTLA-4 checkpoint receptors could improve CART19 therapy we manufactured CART19 cells from WT and PDCD1 and/or CTLA4 deleted normal donor (ND) human T cells42 (Figure S1A), using a humanized CD19 binding scFv and CD8a hinge and transmembrane domains fused to 4–1BB and CD3z cytoplasmic signaling domains.2,43 Deletion efficiency was greater than 90% for PD-1-deficient, CTLA-4-deficient, and PD-1 and CTLA-4-deficient T cells (Figure 1A, Figure S1B–C). The on-target editing efficiency of PDCD1, and CTLA4 was above 99% (Figure 1B). Although most mutations were on-target, rare off-target mutations were identified but predicted to not have negative consequences in T cells (Figure S1D). PD-1- and/or CTLA-4-deficient CART19 cells showed similar proliferative capacity, memory phenotype, levels of inhibitory receptors, and surface CAR expression compared to WT CART19 cells during CAR T cell expansion (Figure 1C–E, Figure S1E). Likewise, WT and edited CART19 cells showed equivalent levels of cytotoxic activity, degranulation capacity and intracellular levels of TNFα, IFNγ, GM-CSF and IL-2 production (Figure 1F, Figure S1F–G).
Figure 1. CD19 BBz CAR T cells exhibit comparable effector function and cytokine secretion after single stimulation with target cells irrespective of deletion of PD-1 and/or CTLA-4.
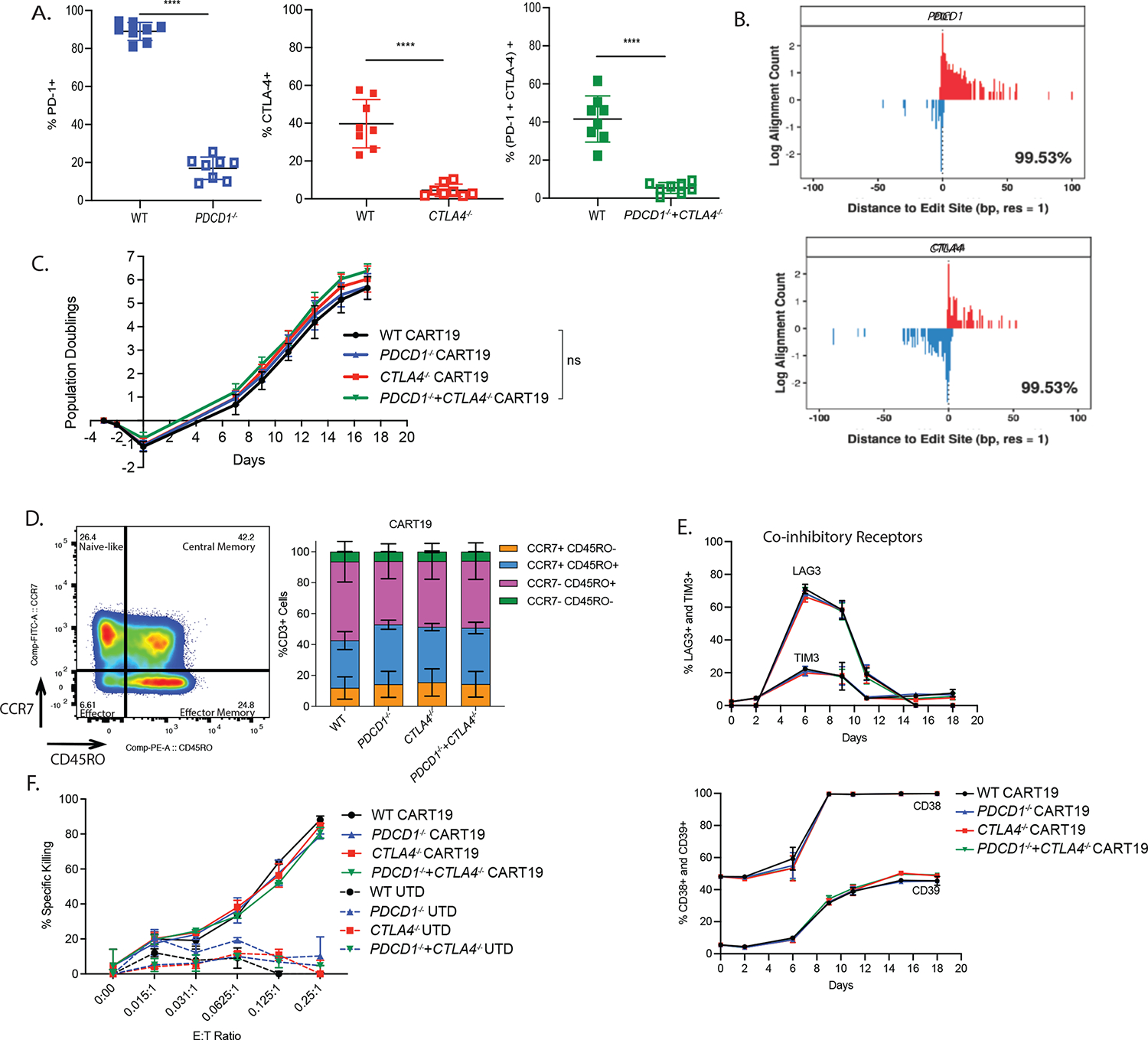
A. PD-1 and/or CTLA-4 deletion efficiency as detected by flow cytometry on day 4 of T cell expansion in ND’s (n=8 donors). Error bars indicate mean±standard deviation. Not significant (ns) P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤0.001, **** P ≤ 0.0001 by unpaired t-test.
B. Distribution of inferred positions of cleavage and dsODN incorporation at an on-target locus using iGUIDE-seq. Incorporation in different strand orientations is shown on the positive (red) and negative (blue) y axis. The percentage in the bottom right corner is an estimate of the number of incorporations associated with the on-target site (based on pileups) captured within the allowed window of 100bp. The PDCD1 sgRNA binds the positive strand and the CTLA4 sgRNA binds the negative strand.
C. Population doublings of edited CAR T cells during the expansion in ND’s (n=3 ND’s). Error bars indicate mean±standard error of the mean (SEM). Not significant (ns) P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤0.001, **** P ≤ 0.0001, by repeated measure two-way ANOVA with Bonferroni correction for multiple comparisons.
D. Representative flow plots (left, n=1 ND) and quantification (right, n=3 ND’s) showing memory phenotype of PD-1 and/or CTLA-4 disrupted CART19 cells. Memory phenotype populations are defined as: Naïve-like (CCR7+ CD45RO−), Central Memory (CM; CCR7+ CD45RO+), Effector memory (EM; CCR7− CD45RO+), Effector (EMRA; CCR7− CD45RO−). Error bars indicate mean±SEM in each memory sub-population.
E. Co-inhibitory receptor expression (LAG3, TIM3, CD38 and CD39) assessed by flow cytometry on PD-1 and/or CTLA-4 disrupted CART19 cells during the expansion (n=1 ND). Error bars indicate mean±SD from technical replicates.
F. Cytotoxicity of WT and edited CART19 cells tested in a 24 hr luciferase-based assay with NALM6 as targets. Different E:T ratios are shown (n=2 ND’s). Error bars indicate mean±SD.
See also Figure S1.
To determine whether deletion of PDCD1 and/or CTLA4 could prevent or delay CAR T dysfunction, we developed an in vitro stress test using chronic antigen exposure (CAE).41 In this assay CART19 cells were repeatedly stimulated with CD19-expressing NALM6 at a high tumor to T cell ratio, leading to dysfunction in the WT CART19 cells (Figure 2A). Of note, NALM6 cells do not express CD80, CD86 or PDL-1, allowing the study of cell-intrinsic mechanisms of resistance (Figure S2A). Two hallmarks of T cell exhaustion are loss of proliferation and cytotoxicity and both phenotypes are recapitulated in our in vitro model (Figure S2B–C). Importantly, CTLA-4-deficient CART19 cells show increased doubling capacity and enhanced anti-tumor efficacy, compared to PD-1-deficient CART19 cells, and CART19 cells deficient for both PD-1 and CTLA-4 (Figure 2B–C; Figure S2D). CTLA-4-deficient CART19 exhibited a similar phenotype when tested against NALM6 expressing the B7 ligand, CD80 (Figure S2E–F). Surprisingly, at the end of the stress test, CTLA-4-deficient CART19 cells have a higher proportion of central memory T (TCM) cells and decreased early apoptotic cells than WT and other edited CART19 cells, consistent with their increased functionality (Figure 2D; Figure S2G). Further upon stress testing, CTLA-4-deficient CART19 cells secreted increased levels of GM-CSF, IL-13, and CCL5 compared to WT, PD-1-deficient CART19 cells, and CART19 cells deficient for both PD-1 and CTLA-4 (Figure 2E–F). IL-13 (Th2 cytokine) was originally isolated as a CD28-dependent gene44 and is produced in response to CD28 co-stimulation in memory T cells.45 CCL5 has been implicated in sustaining CD8 T cell responses during chronic viral infection.46 Further, upon CAE stress, both CTLA-4-deficient BBz and CD28z CART19 cells exhibited higher doubling capacity, and tumor clearance than their WT counterparts (Figure 2G–H, Figure S2H). Thus, deletion of CTLA-4 in CART19 cells improves anti-tumor efficacy for both BBz and CD28z CART19 cells.
Figure 2. Deletion of CTLA4 endows CART19 cells with superior in vitro effector function under stress conditions.
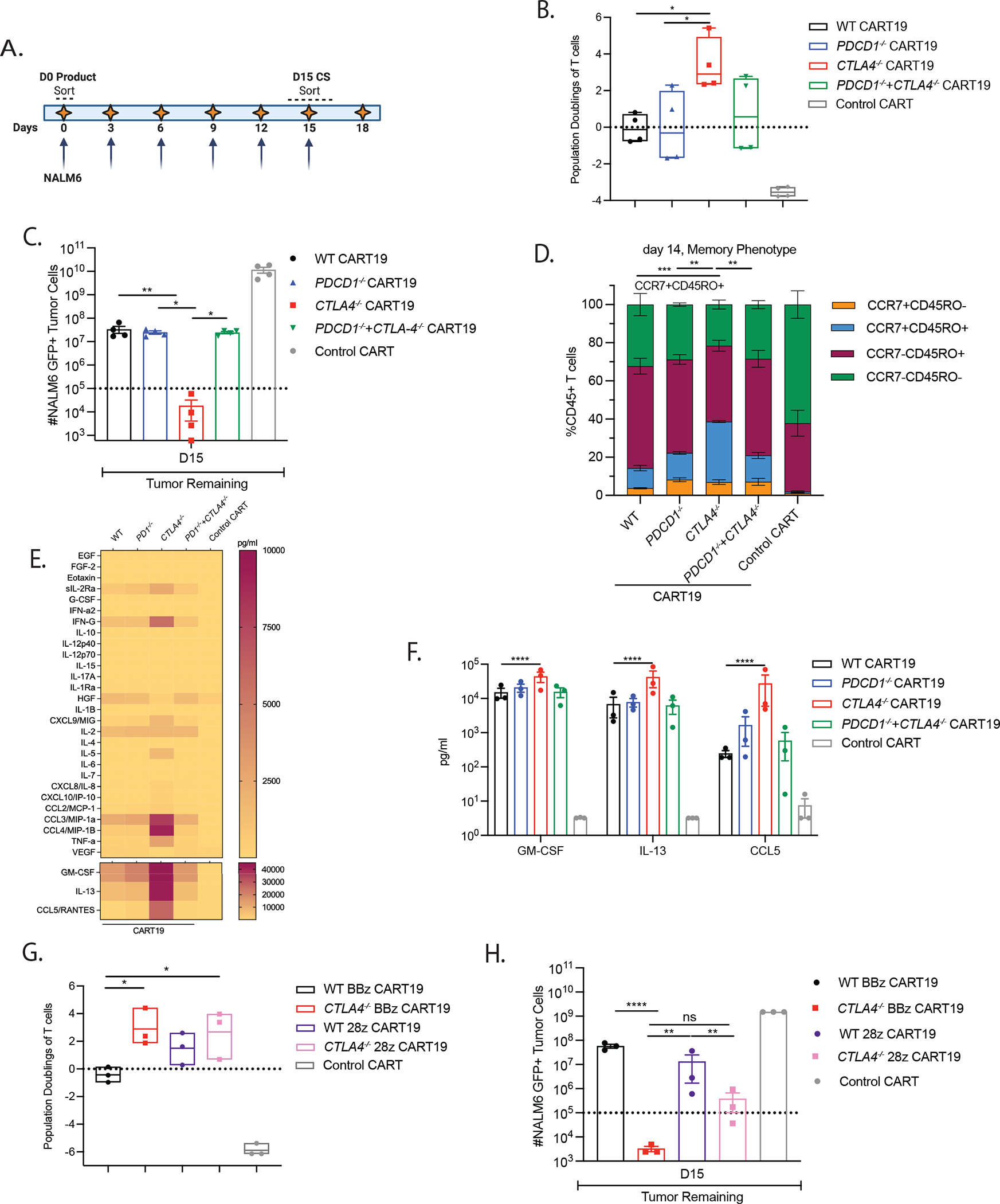
A. Experimental design of CAR T cell dysfunction in an in vitro CAE stress test model in which 2.5e5 CART19 cells (Day 0 product) are repeatedly stimulated with 1e6 NALM6 cells every 3 −4 days. Arrows represent each round of re-stimulation with fresh NALM6 target cells. Day 0 product and Day 15 continuously stimulated CART19 cells are sorted for transcriptional analysis.
B. Population doublings of WT and CTLA-4-deficient BBz CART19 cells at the end of the CAE stress test quantified by using counting beads-based flow cytometry gated on CD45+ T cells (n=4 ND’s).
C. Total tumor burden remaining at the end of the CAE stress test quantified by using counting beads-based flow cytometry gated on GFP+ NALM6 target cells (n=4 ND’s).
D. Flow cytometry characterization of CCR7+ CD45RO−, CCR7+ CD45RO+, CCR7− CD45RO+ and CCR7− CD45RO− subsets of WT and CTLA-4-deficient CART19 cells measured on day 14 of the CAE stress test using flow cytometry (n=2 ND’s).
E. Heatmap showing the levels of cytokine secretion in the supernatant collected on day 15 in WT and CTLA-4-deficient CART19 cells detected using a 31-plex Luminex assay (n=3 ND’s).
F. Concentration of the cytokines that are differentially secreted in the supernatant on day 15 between WT and CTLA-4-deficient CART19 cells detected using a 31-plex Luminex assay (n=3 ND’s).
G. Population doublings of BBz and 28z CART19 cells in the in vitro CAE stress test quantified by using counting beads-based flow cytometry gated on CD45+ T cells (n=3 ND’s).
H. Total tumor burden remaining at the end of the CAE stress test quantified by using counting beads-based flow cytometry gated on GFP+ NALM6 target cells (n=3 ND’s).
Error bars indicate mean±SEM in each memory population. ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤0.001, **** P ≤ 0.0001, by ordinary one-way ANOVA or repeated measure two-way ANOVA with Bonferroni correction for multiple comparisons. See also Figure S2.
CTLA4 deletion improves T cell fitness in CAR T cells obtained from patients with leukemia.
To determine whether CTLA4 deletion can invigorate dysfunctional T cells isolated from CLL patients, WT and CTLA-4-deficient CART19 cells were manufactured from complete responding (CR)-01, partial responding (PR)-01, non-responding (NR)-01 and NR-02 patients (Figure S3A). Consistent with findings from ND’s, higher TCM cells were observed at the end of expansion in NR-01 and NR-02 CTLA-4-deficient CART19 cells (Figure S3B–C). CTLA-4-deficient CART19 cells derived from a CLL patient (NR-01) showed enhanced proliferation, persistence and cytolytic activity compared to WT CART19 at day 18 of stress (Figure 3A–B, top). Similar, but less pronounced differences in proliferation and killing capacity were observed for CTLA-4-deficient PR-01 CART19 cells compared to WT PR-01 CART19 cells, likely because the starting PR-01 T cells were more functional than the starting NR-01 T cells (Figure 3A–B, bottom).13 Additionally, CTLA-4-deficient NR-01 and PR-01 CART19 cells expressed higher levels of granzyme B+ and Ki67, greater production of CD107a, and increased levels of intracellular cytokines IL-2 and TNFα compared to WT NR-01, PR-01 CART19 cells, confirming their superior performance (Figure 3C–E; Figure S3D). In agreement with results observed with ND’s, GM-CSF, IL-13 and CCL4 are secreted at increased levels by CTLA-4-deficient compared to WT NR-01 and PR-01 CART19 cells (Figure 3F). It is worth noting that, in addition to IL-13, production of IFNγ, and IL-5 are also secreted upon CD28 co-stimulation45 and consistent with this, we observed enhanced secretion in CTLA-4-deficient CAR T cells after stress testing.
Figure 3. Deletion of CTLA-4 in T cells from non-responding CLL patients enables CART19 cells to clear tumor under stress-test conditions.
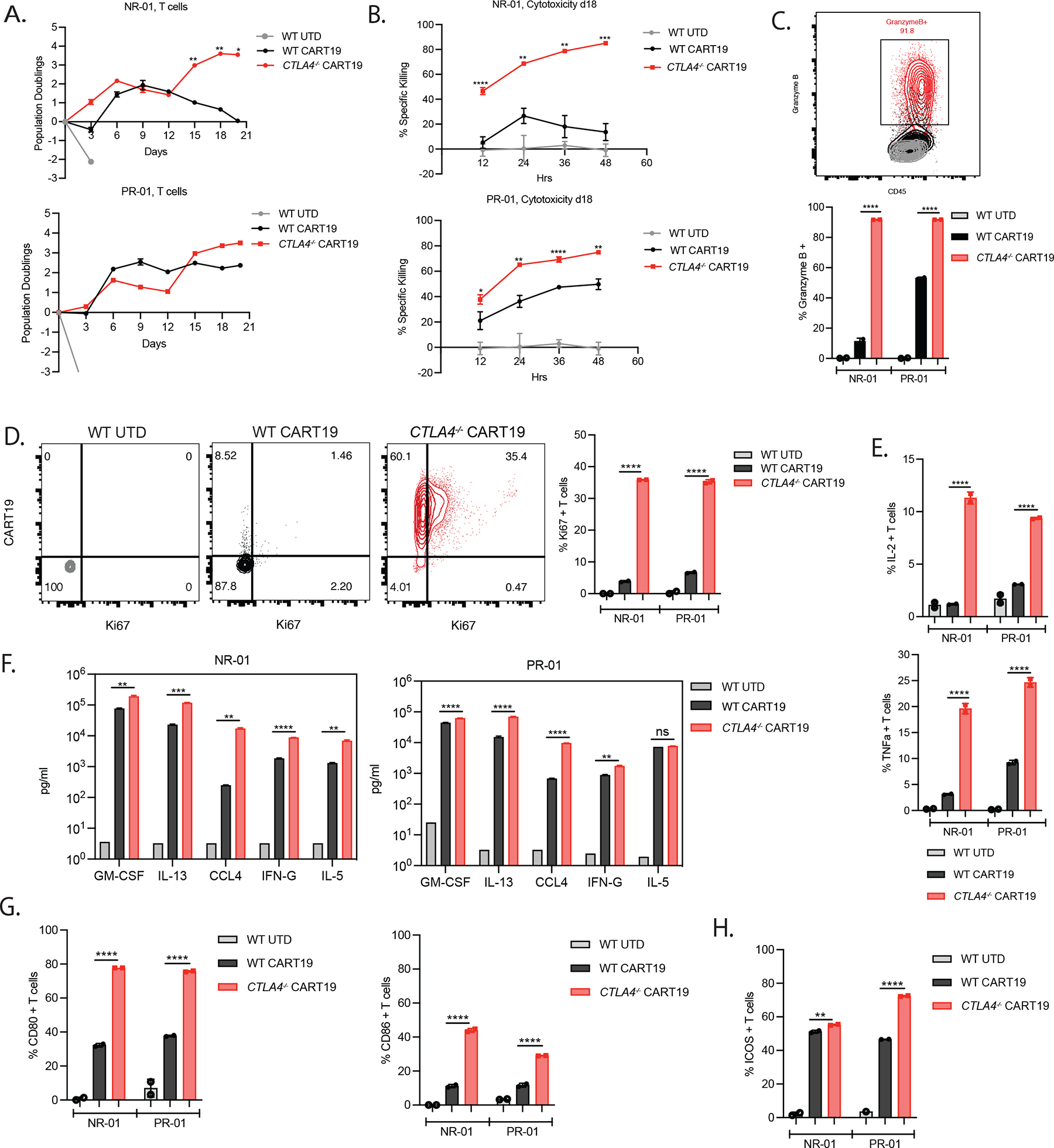
A. Population doublings of UTD T cells, WT, and CTLA-4-deficient CART19 cells during the in vitro CAE stress test quantified assessed by counting beads-based flow cytometry gated on CD45+ T cells. NR-01 (top) and PR-01 (bottom) CLL patients are shown.
B. Cytotoxicity assessment of surface CAR-normalized day 18 CAE CD45+ sorted WT, and CTLA-4-deficient CART19 and UTD T control cells from NR-01 and PR-01 CLL patients against fresh NALM6 target cells at a 1:2 E:T ratio.
C. Representative flow plot (top, NR-01) and quantification (bottom, NR-01 and PR-01) of granzyme B production by UTD T cells, WT and CTLA-4-deficient CART19 cells on day 18 of CAE stress test.
D. Representative flow plots (left, NR-01) and quantification (right, NR-01 and PR-01) of Ki67 production by UTD T cells, WT and CTLA-4-deficient CART19 cells on day 18 of CAE stress test.
E. Quantification of IL-2 (top) and TNFa (bottom) intracellular production levels in NR-01 and PR-01 CLL patients measured using flow cytometry on day 18 of the CAE stress test.
F. Concentration of cytokines in the supernatant from UTD T cells, WT, and CTLA-4-deficient CART19 cells from NR-01 and PR-01 CLL patients detected using 31-plex Luminex assay on day 18 of stress testing.
G. Frequency of CD45+ CD80+ and CD45+ CD86+ T cells from NR-01 and PR-01 CLL patients detected using flow cytometry on day 18 of the CAE stress test.
H. Frequency of CD45+ICOS+ T cells from NR-01 and PR-01 CLL patients assessed by flow cytometry on day 18 of the CAE stress test.
Error bars indicate mean±SD from technical replicates in one patient. NR-01 and PR-01 are shown separately. ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤0.001, **** P ≤ 0.0001, by multiple unpaired t-tests with Bonferroni-Dunn correction for multiple comparisons. See also Figure S3.
We then hypothesized that the increased functionality of CTLA-4-deficient CAR T cells might be due to the removal of CD28 signaling inhibition. To investigate this, we measured levels of B7 ligands and observed increased binding of antibodies to surface CD80 and CD86 protein in CTLA-4-deficient compared to WT CART19 cells after stress testing (Figure 3G).47,48 Additionally, ICOS expression is upregulated in CTLA-4-deficient CART19 cells (Figure 3H). It is noteworthy that CTLA-4 blockade has been shown to induce the expansion of tumor infiltrating Th1-like CD4+ T cells and that expansion of ICOS+ CD4+ T cells following ipilimumab therapy has been associated with an increased likelihood of clinical benefit.21,49
CTLA-4 deletion prevents loss of surface CAR expression in CAR T cells.
CAR T cells lose surface CAR expression during stress testing, leading to CAR dysfunction.16,41,50 Further, reduced expression of CAR on the surface is observed in clinical samples obtained from pre-infusion nonresponding (NR) CLL patient samples and peritoneal/ pleural fluid samples isolated from ovarian cancer patients treated with CAR T cells directed against MSLN.41 These data, along with the observation that CTLA-4-deficient CART19 cells show increased anti-tumor efficacy in the CAR dysfunction model, prompted us to assay the relative levels of surface CAR expression among the WT and CART19 edited groups. We determined that CTLA-4-deficient CART19 cells maintain surface CAR expression through the progression of the stress test, in contrast to WT, PD-1-deficient CART19 cells and CART19 cells deficient for both PD-1 and CTLA-4 (Figure 4A–B). Furthermore, CTLA-4-deficient CART19 cells show enhanced cytolytic activity that is associated with reduced tumor burden at day 15, unlike PD-1-deficient CART19 cells, and CART19 cells deficient for both PD-1 and CTLA-4 (Figure 4C). Likewise, after repeated stimulation, WT CART19 cells in both NR and PR CLL patients exhibit a reduction in the percentage of CAR expression on the surface and inability to clear tumor, whereas CTLA-4-deficient CAR T cells maintain surface CAR expression (Figure 4D, Figure S4A) and display dramatically improved cytolytic activity resulting in reduced tumor burden (Figure 4E). However, when co-cultured with high amounts of tumor, CTLA-4-deficient CART19 cells are unable to eradicate tumor and lose surface CAR expression (Figure 4F–J, Figure S4B). At either E:T ratio, CTLA-4-deficient CART19 do not show significantly enhanced surface TCR (CD3e) expression compared to WT CART19 cells (Figure S4C). These data suggest that CTLA-4-deficient CAR T cells maintain increased surface CAR expression during stress test due, at least in part, to increased tumor clearance.
Figure 4. CTLA-4 deficiency promotes surface CAR expression under stress-test conditions.
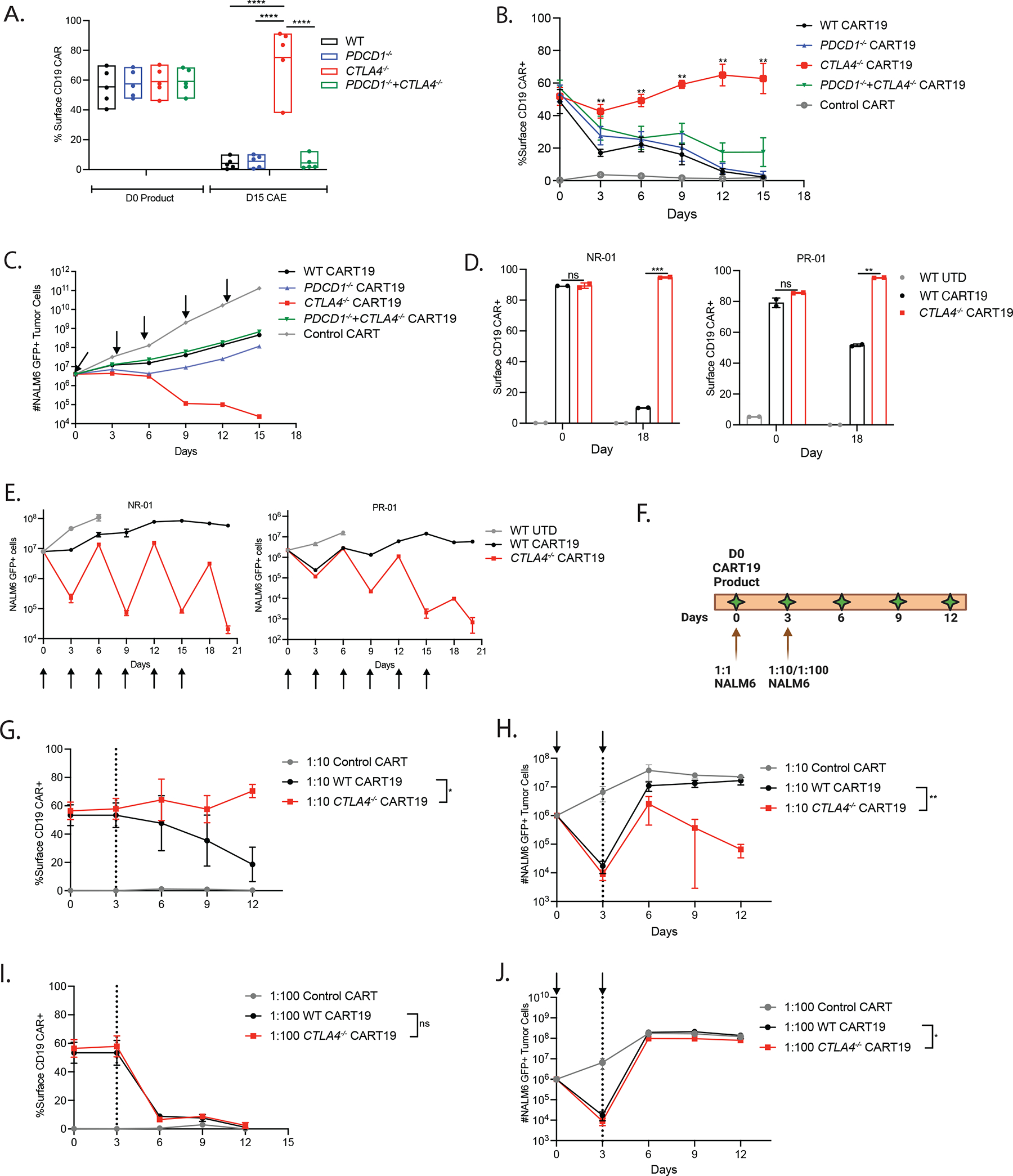
A. Quantification of surface CD19 CAR expression of WT and edited CART19 cells at day 0 and day 15 of the CAE stress test (n=5 ND’s). CAR detection was performed using an anti-idiotype antibody conjugated to a fluorophore.
B. Level of surface CD19 CAR expression for WT and edited CART19 cells during the stress test (n=4 ND’s).
C. Tumor burden in assay with WT and edited CART19 cells during the in vitro stress test model quantified by using counting beads-based flow cytometry gated on GFP+ NALM6 target cells. Arrows indicate addition of new NALM6 cells in each round of restimulation during CAE stress test.
D. Quantification of surface CAR expression by UTD, WT, and CTLA-4-deficient CART19 cells in T cells from CLL patients measured using flow cytometry on day 0 and day 18 of CAE stress testing.
E. Tumor burden in assay with UTD T cells, WT, and CTLA-4-deficient CART19 cells during the in vitro stress test model quantified by using counting beads-based flow cytometry gated on GFP+ NALM6 targets. NR-01 (left) and PR-01 (right) CLL patients are shown. Arrows show each round of restimulation with the NALM6 targets.
F. Experimental design showing cocultures with CART19 cells and NALM6 at E:T ratio of 1:1 on day 0 followed by either 1:10 or 1:100 E:T ratio on day 3.
G. Level of surface CD19 CAR expression for WT and CTLA-4-deficient CART19 cells at 1:10 E:T ratio (n=3 ND’s).
H. Tumor burden in WT and CTLA-4-deficient CART19 cell groups at 1:10 E:T ratio quantified by using counting beads-based flow cytometry gated on GFP+ NALM6 target cells (n=3 ND’s). Arrows indicate addition of NALM6 cells at 1:1 E:T ratio followed by 1:10 E:T ratio.
I. Level of surface CD19 CAR expression for WT and CTLA-4-deficient CART19 cells at 1:100 E:T ratio (n=3 ND’s).
J. Tumor burden in assay with WT and CTLA-4-deficient CART19 cell groups at 1:100 E:T ratio quantified by using counting beads-based flow cytometry gated on GFP+ NALM6 target cells (n=3 ND’s). Arrows indicate addition of NALM6 cells at 1:1 E:T ratio followed by 1:100 E:T ratio.
Error bars indicate mean±SEM. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤0.001, **** P ≤ 0.0001, by multiple unpaired t-tests or repeated measure two-way ANOVA with Bonferroni correction for multiple comparisons. See also Figure S4A–C.
In mice, post thymic deletion of CTLA4 in T cells can lead to lymphoproliferation and infiltration into organs such as the pancreas.51,52 To determine if deletion of CTLA4 in CAR T cells results in lymphoproliferation or GVHD, we injected high doses of WT and CTLA-4-deficient CART19 cells (Figure S4D). Additionally, we assessed the weight and survival of the mice, and measured the degree of GVHD as an initial surrogate of lymphoproliferation (Figure S4E–G). Moreover, at regular intervals, we tracked the expansion of T cells in the peripheral blood (Figure S4H–I). We observed that 1 of 16 mice in the CTLA-4-deficient cohort showed signs of GVHD and high levels of T cell expansion in the peripheral blood, spleen, lung, and liver, ultimately leading to death (Figure S4F, I) and 2 of 16 mice in the WT CART19 cell group died of unknown causes without lymphoproliferation (Figure S4F). Thus, CTLA-4 deletion in CAR T cells did not lead to uncontrolled lymphoproliferation in this model in 15/16 mice.
CTLA-4 deletion in CAR T cells results in enhanced anti-tumor efficacy in pre-B-cell acute leukemia and myeloma xenograft models.
To determine if the results from our in vitro models of CAR T-dysfunction correlated with in vivo efficacy, we tested the anti-tumor activity of WT and PD-1-deficient and/or CTLA-4-deficient CART19 cells in an acute lymphoblastic leukemia (ALL) model in NOD-SCID-IL-2Rnull (NSG) mice (Figure 5A). CTLA-4-deficient CART19 cells exhibited enhanced tumor control (Figure 5B, Figure S5A), improved survival (Figure 5C) and greater T cell expansion as compared to WT, and PD-1-deficient CART19 cells and CART19 cells deficient for both PD-1 and CTLA-4 (Figure 5D) but were no longer detected in the peripheral blood after tumor clearance (Figure S5B). Therefore, we subsequently focused on the CTLA-4-deficient group.
Figure 5. Deletion of CTLA4 in CART19 cells manufactured from patient T cells enhances anti-tumor efficacy in xenograft models of acute leukemia.
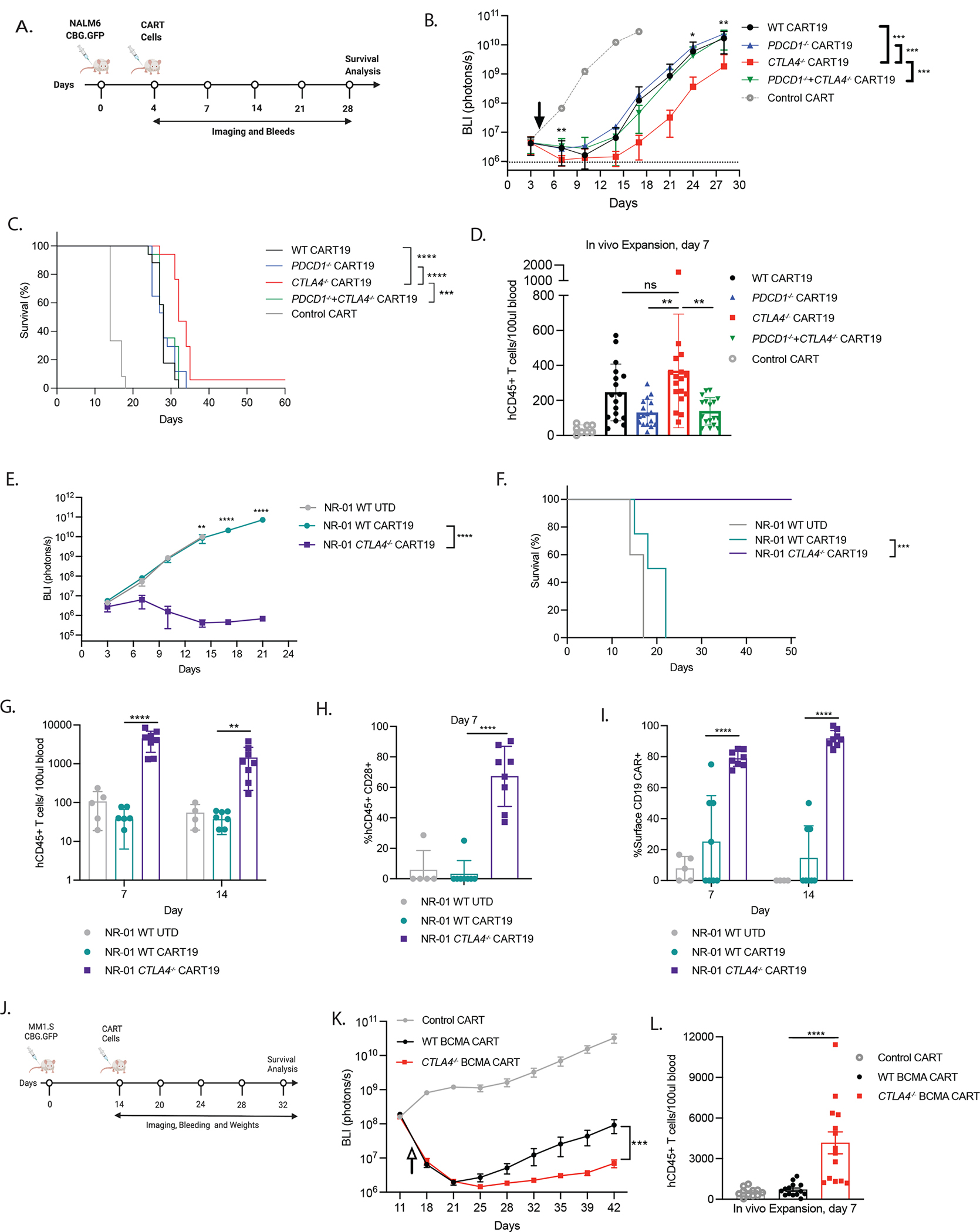
A. Timeline showing experimental design of the ALL-xenograft model. NOD-SCID-IL2rγ−/− (NSG) mice were intravenously injected with NALM6 CGB-GFP. Established NALM6 liquid tumors were treated with WT and respective edited CART19 cells manufactured from ND or CLL patients’ T cells. Mice were imaged every 3–4 days; weighed and retro-orbitally bled every three days.
B. Kinetics of tumor burden assessed by bioluminescence from two independent experiments (n=17 mice).
C. Kaplan-Meier survival curves from two independent experiments (n=17 mice).
D. Frequency of CD3+CD45+ ND T cells in the NSG mice. Peripheral blood was collected 7 days post adoptive transfer of CAR T cells and counted using Trucount based flow cytometry (n=17 mice).
E. Kinetics of tumor bioluminescence for NALM6-bearing mice treated with UTD, and CART19 WT and CART19 CTLA-4-deficient T cells derived from NR-01 CLL patient (n=8 mice).
F. Kaplan-Meier survival curves for mice treated with UTD, and CART19 WT and CART19 CTLA-4-deficient T cells derived from NR-01 CLL patients (n=8 mice).
G. Frequency of CD3+CD45+ human T cells in the NSG mice peripheral blood collected 7 and 14 days post adoptive transfer of NR-01 CLL patient CAR T cells counted using Trucount-based flow cytometry (n=8 mice).
H. Surface protein expression of CD28 on CD3+CD45+ human T cells NSG mice in the peripheral blood collected 7 days post adoptive transfer of NR-01 CLL patient CAR T cells measured using flow cytometry (n=8 mice).
I. Surface protein expression of CD19 CAR on CD3+CD45+ human T cells in the NSG mice peripheral blood collected 7 and 14 days post-adoptive transfer of NR-01 CLL patient CAR T cells measured using flow cytometry (n=8 mice).
J. Timeline showing experimental design of the MM-xenograft model. NOD-SCID-IL2rγ−/− (NSG) mice were intravenously injected with MM1.S CGB-GFP. Established MM1.S liquid tumors were treated with high or low dose WT and CTLA-4-deficient BCMA CAR T cells manufactured from ND. Mice were imaged every 3–4 days; weighed and retro-orbitally bled every week.
K. Kinetics of tumor burden in NSG mice treated with WT and CTLA-4-deficient BCMA CAR T cells assessed by bioluminescence from two independent experiments (n=11 mice).
L. Frequency of CD45+ ND T cells in the NSG mice treated with WT and CTLA-4-deficient BCMA CAR T cells. Peripheral blood was collected 7 days post adoptive transfer of CAR T cells and counted using counting beads-based flow cytometry (n=14 mice).
** P < 0.01 *** P < 0.001, **** P < 0.0001, ns not significant by repeated measure two-way ANOVA with Bonferroni correction for multiple comparisons or ordinary one-way ANOVA. For Kaplan Meier Curves, Log-rank (Mantel Cox) test was performed. Error bars indicate mean±SEM from two individual experiments. See also Figure S4D–I and S5.
We next assessed whether deletion of CTLA4 in T cells isolated from CLL patients could enhance anti-leukemic efficacy of CAR T cells using an in vivo mouse model. Indeed, deletion of CTLA4 in NR-01 CART19 cells led to superior tumor clearance (Figure 5E) and significantly enhanced survival compared to WT CAR T cells (Figure 5F), with no signs of GVHD. Patient NR-02 exhibited limited peak CAR T expansion in the blood in our previous clinical trial (Figure S3A), which is known to be correlated with a poor response to CART19 therapy in pediatric and young adult ALL.13 Intriguingly, deletion of CTLA4 in NR-02 patient T cells enabled a partial rescue of NR-02 CART19 anti-tumor activity and improved survival compared to WT CAR T cells (Figure S5C–D). Both WT and CTLA-4-deficient CR-01 CART19 cells demonstrated tumor clearance and survival (Figure S5E–F). This is anticipated because the baseline T cell populations in responding patients cleared leukemia target cells and did not have a defect in T cell fitness, exhibiting high levels of peak CAR T cell expansion in the patient.13
We further explored our hypothesis that deletion of CTLA4 would augment CD28 signaling given CTLA-4 is known to bind with higher affinity than CD28 to the B7 ligands, CD80 and CD86.53 In comparison to WT, CTLA-4-deficient NR-01 and NR-02 CAR T cells showed increased T cell expansion in the blood at days 7 and 14 (Figure 5G, Figure S5G). Moreover, CD28 surface expression was elevated at day 7 post T cell engraftment on the CAR T cells, offering strong support for this theory (Figure 5H, Figure S5H). Surface expression of the scFv CAR was also maintained at day 7 and day 14 in both CTLA-4-deficient NR-01, NR02 CART19 cells (Figure 5I; Figure S5I), providing in vivo confirmation of our in vitro observation that CTLA-4-deficient CAR T cells maintain surface CAR expression under conditions of repeated exposure to the cognate antigen. Together these data provide a strong correlation between CTLA-4 deficiency, increased CD28 signaling, maintenance of surface CAR expression and anti-tumor efficacy.
Further, we tested the anti-tumor activity of WT and CTLA-4-deficient BCMA CAR T cells using xenograft mouse models of multiple myeloma (MM) (Figure 5J). NSG mice treated with CTLA-4-deficient BCMA CAR T cells exhibited enhanced tumor control (Figure 5K), improved survival (Figure S5J), and displayed greater in vivo T cell expansion at day 7 (Figure 5L) as compared to WT BCMA CAR T cells. This data confirms the superior proliferation and anti-tumor efficacy of CTLA-4-deficient CAR T cells in two clinically validated CAR targets.
Transcriptional dynamics of CTLA-4-deficient CAR T cells.
To examine the molecular pathways driving increased anti-tumor efficacy of CTLA-4-deficient CAR T cells, we performed single cell RNA sequencing (scRNA-seq) analyses on day 0 and day 15 WT and CTLA4-deleted CAR T cells after CAE. The UMAP clusters and top marker genes in each cluster are shown in heatmaps (Figure 6A; S6A). We identify 3 clusters at day 0: terminally effector (red); proliferating (green), and effector memory (blue) T cells. Three additional clusters are identified at day 15: dysfunctional (yellow), terminally exhausted (pink), and stressed and dying (purple) T cells. To gain insight into how deletion of CTLA4 impacts the broad distribution of T cells, we plotted the relative number of cells in each cluster by bar graph (Figure 6B). Interestingly at day 15, CTLA4-deleted CART19 cells contain a greater percentage of cells in the proliferation cluster and a lower percentage of cells in the terminally effector and the stressed and dying clusters compared to WT CART19 cells. These data provide insights on how CTLA-4-deficient CAR T cells outperform WT CAR T cells.
Figure 6. Transcriptional dynamics of CTLA4-deleted CART19 cells.
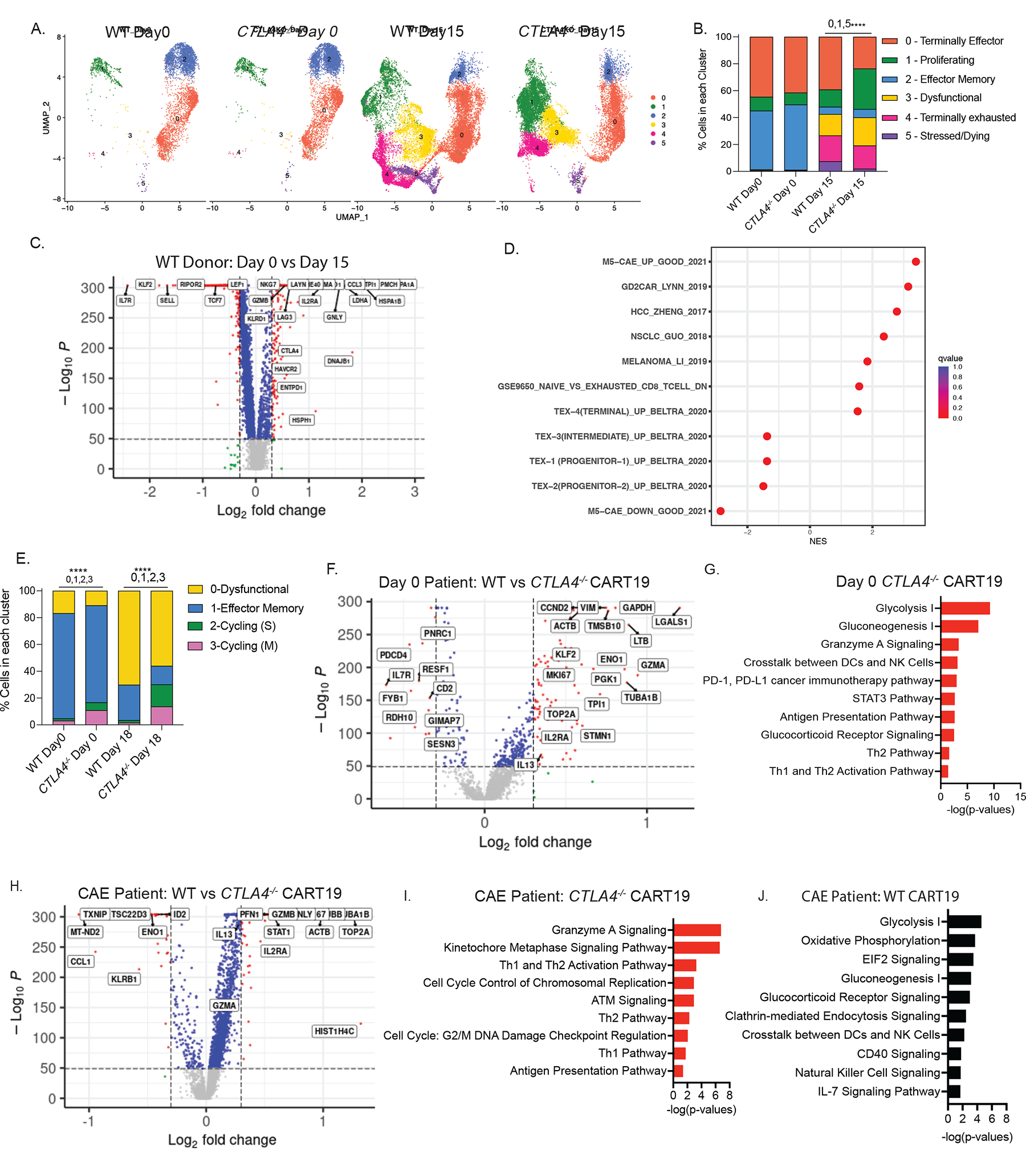
A. UMAP projection of scRNA-seq data on day 0 and on day 15 of stress testing of WT and CTLA-4-deficient CART19 cells, determined by Seurat v.4.1. Each dot corresponds to one individual cell. A total of 5 clusters were identified and color-coded (n=2 ND’s).
B. Bar plot showing percentage of cells in each cluster in WT and CTLA4-deleted CART19 cells on day 0 and day 15 (n=2 ND’s). Fisher’s exact test was used. Top 3 genes in each cluster are cluster 0 (CCL5, GZMK, KLRK1), cluster 1 (HIST1H4C, TOP2A, TUBA1B), cluster 2 (IL7R, KLF2, SELL), cluster 3 (CCL3, GNLY, GZMB), cluster 4 (PMCH, HSPA1A, HSPA1B), cluster 5 (BIRC3, MALAT1, HSP90AA1).
C. Volcano plot identifying the DEGs between day 0 (left) vs day 15 (right) WT CART19 cells. Genes upregulated in the day 15 WT CART19 are indicated on the right side. Red dots indicate genes with p < 1e-50 and log2FC >0.3 (n=2 ND’s). The x axis represents the log fold change; the y axis represents the log10 adjusted p values. A two-sided Wilcoxon rank sum test was used.
D. GSE analysis showing the enrichment of publicly available dysfunction and exhaustion datasets when considering all DEGs between day 15 WT CART19 CAE stress test vs day 0 WT CART19 product.
E. Bar plot showing percentage of cells in each cluster in WT and CTLA-4-deleted CART19 cells on day 0 and day 18 from NR-01 and PR-01 CLL patients. Fisher’s exact test was used. Top 3 genes in each cluster are cluster 0 (PMCH, GNLY, GZMB), cluster 1 (LTB, FTL, RPS28), cluster 2 (HIST1H4C, TOP2A, TUBA1B), cluster 3 (HIST1H1B, HMGN2, LGALS1).
F. Volcano plot identifying the DEGs between day 0 WT CART19 cells (left) and day 0 CTLA-4-deficient CART19 cells (right) from NR-01 and PR-01 CLL patients. Red dots indicate genes with log10 adjusted p values < 1e-50 and log2FC >0.3.). The x axis represents the log fold change; the y axis represents the log10 adjusted p values. A two-sided Wilcoxon rank sum test was used.
G. IPA of pathways upregulated at day 0 CTLA-4-deficient CART19 compared to day 0 WT CART19 cells shown as bar graphs. Top pathways were determined by using a significant p-value of <0.05 and pathways with a negative z-score were removed from analysis.
H. Volcano plot identifying the DEGs between WT CART19 cells (left) and CTLA-4-deficient CART19 cells (right) after day 18 of CAE stress from NR-01 and PR-01 CLL patients. Red dots indicate genes with p < 1e-50 and log2FC >0.3. The x axis represents the log fold change; the y axis represents the log10 adjusted p values. A two-sided Wilcoxon rank sum test was used.
I. IPA of pathways upregulated at day 18 CTLA-4-deficient CART19 compared to day 0 CTLA-4-deficient CART19 cells shown as bar graphs. Top pathways were determined by using a significant p-value of <0.05 and pathways with a negative z-score were removed from analysis.
J. IPA of pathways upregulated at day 18 WT CART19 compared to day 0 WT CART19 cells shown as bar graphs. Top pathways were determined by using a significant p-value of <0.05 and pathways with a negative z-score were removed from analysis. See also Figure S6 and Table S1–5.
To identify the dysfunctional gene signature from our in vitro stress test model of dysfunction, we determined differentially expressed genes (DEGs) between day 0 and day 15 after CAE. We observe upregulation of exhaustion genes such as CTLA4, HAVCR2, LAG3, KLRD1, IL2RA, ENTPD1, GNLY, LAYN, CCL3, CCL4, TNFRSF18, and DUSP4; genes all previously identified in our solid tumor model of anti-MSLN CAR T cell dysfunction (Figure 6C, Table S1).41 To further explore the overlap between our in vitro dysfunction model and publicly available T cell exhaustion and dysfunction datasets, we performed GSEA. We confirmed significant positive enrichment of seven exhaustion and dysfunctional datasets: upregulated genes in anti-MSLN CAR,41 anti-GD2 CAR,54 hepatocellular carcinoma,55 non-small cell lung cancer,56 Melanoma,57 GSEA9650 exhaustion dataset,58 terminally exhaustion;59 and negative enrichment of intermediate, progenitor 1, and progenitor 2 exhausted T cells,59 downregulated genes in anti-MSLN CAR41 in our in vitro dysfunctional model (Figure 6D, Figure S6B). These data, together with observed reductions in proliferation, cytotoxicity and surface CAR expression at day 15 cells after stress, provide evidence that our in vitro dysfunction CAR T cell model aligns with previous models of T cell exhaustion and dysfunction in endogenous T cells.40,60,61
We next extended these analyses to CLL patient samples, performing scRNA-seq analyses on WT and CTLA4-deleted CAR T cells on day 0 and day 18 of CAE stress. Based on top marker gene expression, we identify the following UMAP T cell clusters; dysfunctional (yellow); effector memory (blue); cycling in S phase (green); and cycling in M phase (pink) T cells (Figure S6C, D). Both proliferation clusters are virtually absent in WT cells and prominent in CTLA4-deleted CAR T cells, supporting the observation that deletion of CTLA4 increases expansion of CLL patient CAR T cells both at day 0 and day 18 of stress (Figure 6E). Further, an increased percentage of dysfunctional cells are present in WT compared to CTLA4-deleted CLL CAR T cells at both day 0 and day 18 of stress testing, reinforcing the notion that CTLA-4 disruption increases CAR T cell fitness at baseline and after CAE.
To gain insight into how CTLA4 deletion can increase the fitness of CLL CAR T cell patient products we identified day 0 DEGs between WT, and CTLA4-deleted NR-01 and PR-01 patient CAR T cells (Figure 6F, Table S2). Ingenuity pathway analysis (IPA)62 on this gene set reveals RNAs encoding glycolytic enzymes including ENO1, PGK1 and GAPDH and upregulation of the glycolysis pathway is enriched in day 0 CTLA4-deleted CLL CAR T cells, aligning with the increased percentage of proliferating cells (Figure 6G, Table S3). In agreement with the increased efficacy of CTLA-4-deficient day 0 CAR T cells, upregulation of granzyme A signaling and STAT3 signaling pathways are also observed.
Uninhibited CD28 co-stimulation enhances the efficacy of CTLA-4-deficient CAR T cells.
To understand mechanistically how CTLA4 deletion contributes to the increased anti-tumor efficacy of CAR T cells from leukemia patients after stress testing, we identified and then compared DEGs between WT and CTLA4-deficient cells from ND and CLL CART cells and observed significant alignment (NES=2.19; FDR=0.00) (Figure 6H, S6E–F; Table S4). In accordance, there are many commonly upregulated pathways in CTLA4-deleted ND and CLL CAE cells (Figure 6I–J, 7A; Figure S6G–H,7A; Table S5). Glycolysis is the only commonly down regulated pathway (Figure 7A). Further, we provide biological data that supports the observed upregulation of many of these pathways. For instance, CTLA-4-deficient ND and CLL CAR T cells display enhanced in vitro and in vivo antitumor cytotoxicity (Figure 2C; Figure 3B; Figure 5E) in agreement with upregulation of granzyme A signaling and granzyme B signaling pathways (Figure 3C). Likewise, pathways involved in cell cycle regulation are also represented in the top upregulated pathways in both ND and CLL CTLA-4-deficient cells after stress testing (Figure 6I, 7A; Figure S6G), correlating with the increase in percentage of proliferating cells expressing elevated levels of cell cycle genes (Figure 6B, E, F; Figure S6A, D). Interestingly, G2/M DNA damage checkpoint regulation pathways are also upregulated after stress testing in CTLA-4-deficient cells manufactured from ND and CLL patient T cells (Figure 7A). This suggests that deleting the immune checkpoint CTLA-4 in CAR T cells promotes T cell expansion and further, that this is accompanied by the induction of DNA damage. In agreement with this, CTLA-4 has been demonstrated to facilitate DNA-induced apoptosis by binding to the ATM inhibitor, protein phosphatase 2A (PP2A).63 A number of genes involved in cellular redox homeostasis and in the protection of cells from oxidative stress (TXNRD1, TXNRD2, TXN) were upregulated in CTLA-4-deficient cells during CAE (Figure 6H,S6E; Table S4).
Figure 7. Uninhibited CD28 co-stimulation enhances the anti-tumor efficacy of CTLA-4-deficient CART19 cells.
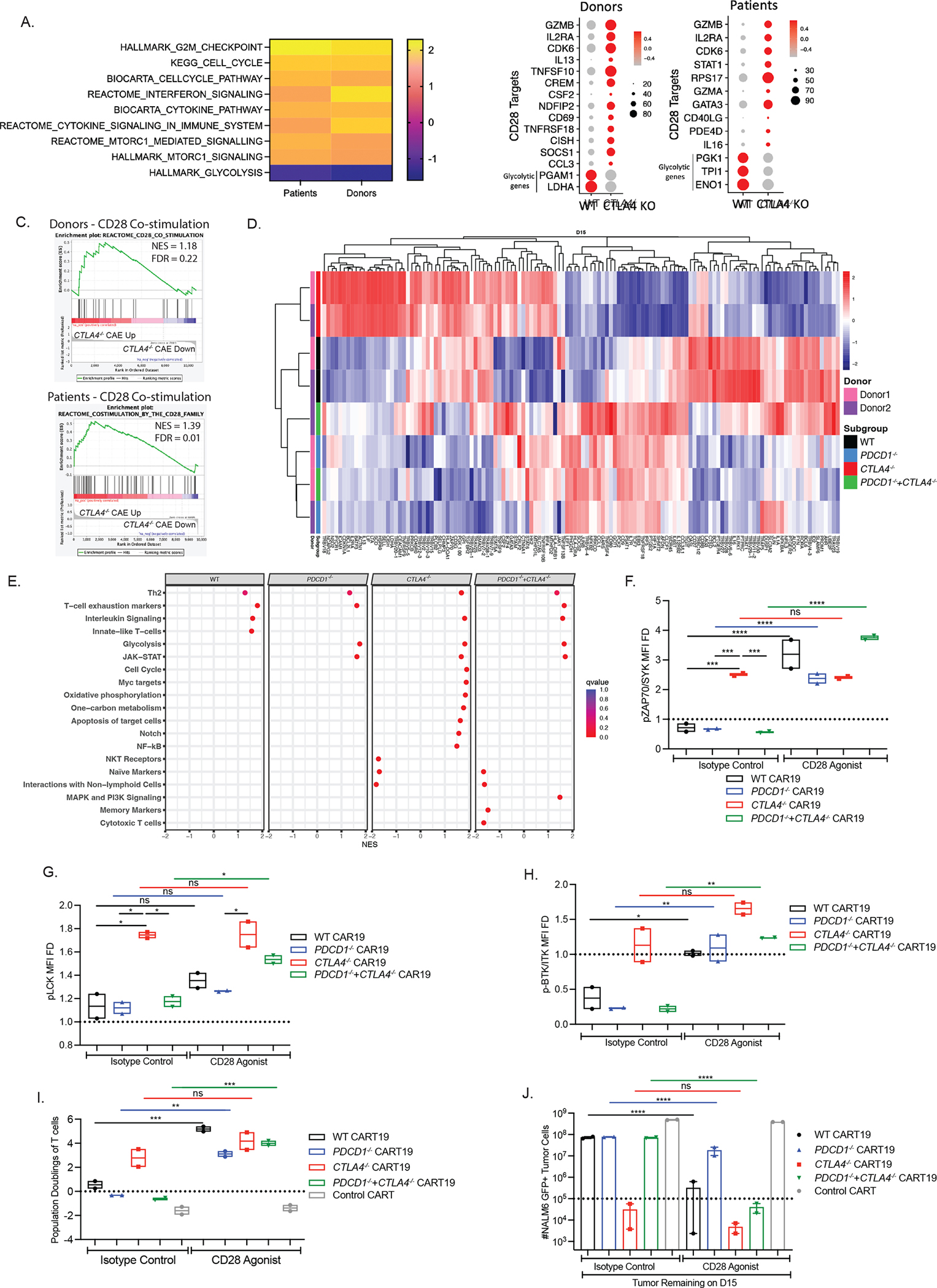
A. GSE analysis of common pathways upregulated in CTLA4-deleted CART19 cells in ND and CLL patients showing normalized enrichment scores (NES).
B. Downstream targets of CD28 in CTLA-4-deficient CART19 cells after stress testing as predicted by IPA in both ND and CLL patients. Genes that are differentially expressed in stress tested WT and CTLA-4-deficient CART19 cells compared to day 0 WT and CTLA-4-deficient CART19 cells. Glycolytic genes are shown as controls.
C. GSE analysis showing positive enrichment of CD28 signaling in genes upregulated after stress testing CTLA4-deleted CART19 compared to WT CART19 cells from donors shown on the top; and genes upregulated at CAE CTLA4-deleted CART19 compared to CAE WT CART19 cells from CLL patients shown at the bottom (n=2 ND’s).
D. Heatmap depicting the normalized expression of top 130 DEGs between day 15 of the CAE stress test vs day 0 product in WT CART19 cells and PDCD1- and/ or CTLA4-deleted CART19 cells using nCounter-based readout (n=2 ND’s).
E. Dotplot of the NES scored by GSEA showing the significantly enriched pathways when considering all DEGs between day 15 CAE stress test vs day 0 product in each group using nCounter-based readout (n=2 ND’s).
F. MFI FD of phospho-ZAP70 (normalized to unstimulated T cells) in WT and edited BBz CART19 cells in the presence or absence of CD28 agonist at the end of the CAE stress test quantified by flow cytometry gated on CD45+ T cells (n=2 ND’s).
G. MFI FD of phospho-LCK (normalized to unstimulated T cells) in WT and edited BBz CART19 cells in the presence or absence of CD28 agonist at the end of the CAE stress test quantified by flow cytometry gated on CD45+ T cells (n=2 ND’s).
H. MFI FD of phospho-BTK/ITK (normalized to unstimulated T cells) in WT and edited BBz CART19 cells in the presence or absence of CD28 agonist at the end of the CAE stress test quantified by flow cytometry gated on CD45+ T cells (n=2 ND’s).
I. Population doublings of WT and edited BBz CART19 cells in the presence of CD28 agonist at the end of the CAE stress test quantified by using counting beads-based flow cytometry gated on CD45+ T cells (n=2 ND’s).
J. Total tumor burden remaining presence of CD28 agonist at the end of the CAE stress test quantified by using counting beads-based flow cytometry gated on GFP+ NALM6 target cells (n=2 ND’s).
Error bars indicate mean±SEM. ns P > 0.05, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤0.001, **** P ≤ 0.0001, by ordinary one-way ANOVA with Bonferroni correction for multiple comparisons. See also Figure S7 and Table S6–7.
Expression of downstream targets of CD28 in WT and CTLA-4-deficient ND and CLL CAR T cells is illustrated by dot plots (Figure 7B, Table S6). CTLA4-deficient CAR T cells showed upregulation of GZMB, IL2RA, and CDK6 contributing to superior proliferation, effector function and cell cycle regulation (Figure S7B). In contrast, WT ND and CLL CAR T cells display upregulation of TSC22D3 which inhibits IL2/IL2R expression and NF-kB, resulting in reduced proliferation (Figure S7B). Further, GSEA demonstrates an enhancement of CD28 co-stimulation in ND (NES=1.18; FDR=0.22) and CLL patient (NES=1.39; FDR=0.01) CTLA-4-deficient compared to WT CART19 CAE cells (Figure 7C). The enhanced CD28 co-stimulation signature occurs in the absence of transcriptional changes in CD80, CD86, CD28, ICOS or an increase in CAR T transcript levels with CTLA-4 depletion (Figure S7C–D). Further, CD274 (PDL-1) and PDCD1LG2 (PDL-2) did not show any changes in transcriptional levels (Figure S7C). The enhancement in CD28 co-stimulation in CTLA-4-deficient cells observed after stress testing is supported by our xenograft mouse studies where increased CD28 cell surface expression is detected in the peripheral blood CAR T cells of mice treated with CTLA-4-deficient compared to WT CLL CAR T cells (Figure 5H, 5SH). Based on these data we propose that CTLA4 deletion in CAR T cells results in unopposed and enhanced CD28 signaling, thereby maintaining CAR co-stimulation, and reduced CAR dysfunction, which together leads to superior CAR T cell effector function, increased proliferation and maintained surface CAR expression.
To investigate why CART19 cells deficient for both PD-1 and CTLA-4 do not display increased anti-tumor efficacy under stress conditions, we compared DEGs before and after CAE stress (Table S7). A high degree of overlap in the gene signatures and hierarchical clustering is visually apparent in the heatmap between PDCD1-deleted CART19 cells and CART19 cells deleted for both PDCD1 and CTLA4(Figure 7D, Table S7). In contrast, the WT and CTLA4-deleted CART19 cells each exhibit unique gene signatures with minimal overlap to the other groups. Genes associated with effector function (STAT6, STAT5, TNFRSF9, IL2RA, BATF3), proliferation (NCAPG2, CDKN1A, BCL2L1), cytokine response (IL13, LTA, CSF2, STAM, CRLF2), central memory (SELL, CD45RO) and kinases such as ZAP70 and PRKCB involved in T cell activation and co-stimulation (Figure 7D) were upregulated in CTLA4-deleted CAR T cells as compared to the other groups, consistent with the increased central memory phenotype (Figure 2D), proliferation and anti-tumor efficacy in CTLA-4-deficient CART19 cells on day 15 of CAE stress (Figure 2B–C). Likewise differential pathway analysis showed that CTLA4-deleted CAR T cells were enriched in pathways associated with increased proliferation and cytotoxicity (Figure 7E). In contrast, WT, PDCD1-deleted CART19 cells, and PDCD1- and CTLA4-deleted CART19 cells positive enrichment of the T cell exhaustion pathway (Figure 7E), unlike CTLA4-deleted CART19 cells. Of note, BATF3 overexpression has recently been shown to prevent exhaustion in mouse CD8 T cells.64 Thus, deletion of PDCD1 on CTLA4-deleted CART19 cells negates the increases in proliferation, cytotoxic capacity, cytokine production, and memory phenotype (Figure 2B–F) observed in CTLA-4-deficient CAR T cells after stress testing.
We hypothesize that deletion of CTLA4 enables unopposed CD28 signaling, resulting in superior function, and that this phenotype is lost with deletion of PDCD1 expression in CTLA4-deleted CART19 T cells. We assessed CD28 signaling by measuring the phosphorylation of overlapping downstream targets of CD28 and CAR signaling. CTLA-4-deficiency resulted in potent phosphorylation of ZAP70 and LCK compared to WT and PD-1-deficient CART19 cells and CART19 cells deficient in both PD-1 and CTLA-4 after CAE stress (Figure 7F–G, Figure S7E–F). Interestingly, in the presence of CD28 agonist, WT, PD-1-deficient CART19 cells and CART19 cells deficient for both PD-1 and CTLA-4 exhibited an increase in phosphorylation of ZAP70, LCK and BTK/ITK resulting in a similar pattern of phosphorylation exhibited by CTLA-4-deficient CART19 cells (Figure 7F–H, Figure S7E–G). Whereas CTLA-4-deficient CART19 cells did not show a significant increase in phosphorylation in the presence of CD28 agonist (Figure 7F–H, Figure S7E–G), consistent with already augmented CD28 co-stimulation. Next, we investigated the impact of CD28 signaling in the CAE stress model and observed that addition of CD28 agonistic antibody results in more than a log fold increase in proliferation in WT CART19 cells after 6 days of stress (Figure S7H). During the CAE stress test, addition of CD28 agonistic antibody results in increased population doublings, enhanced tumor clearance, and increased surface CAR expression on day 15 of CAE stress in WT and PD-1-deficient CART19 cells and CART19 cells deficient for both PD-1 and CTLA-4 (Figure 7I, J, Figure S7I), but does not provide additional benefit to CTLA-4-deficient CART19 cells. Instead, in CTLA-4-deficient CART19 cells population doublings, anti-tumor activity and surface CAR expression remained at the high level observed without CD28 agonistic antibody treatment (Figure 7I–J, Figure S7I). This data aligns with our in vitro and in vivo models and transcriptomic data and provides evidence that relieving inhibition of CD28 signaling under stress conditions via CTLA4 deletion increases the proliferative capacity, anti-tumor activity, and surface CAR expression of CART19 cells.
Discussion
CAR T cell therapies targeting B-cell malignancies have shown remarkable responses, leading to long lasting disease remission.65 However, not all leukemia and lymphoma patients experience durable benefit. In this study, we ask whether combining CART19 cells with checkpoint depletion could improve the response rates of this therapy, potentially broadening the number of patients that can benefit. We examined whether CRISPR-Cas9 mediated deletion of PDCD1 and/ or CTLA4 could increase the anti-tumor efficacy of CART19, in both in vitro and in vivo models of CART dysfunction. Indeed, our findings show that CTLA-4-deficient CART19 cells, compared to PD-1-deficient CART19 cells and CART19 cells deficient for both PD-1 and CTLA-4, exhibit higher proliferative capacity, and superior anti-tumor efficacy, which enables prolonged surface CAR expression due to reduced antigen load. Further, we demonstrate that dysfunctional CART19 CLL patient cells can be invigorated by CTLA-4-deficiency in the starting T cell population. Therefore, deletion of CTLA-4 in CART19 cell products has the potential to improve clinical efficacy in a subset of CLL patients that do not respond to currently available treatments. This technology may also increase the feasibility of manufacturing CAR T cell products from heavily pretreated patient T cells.
We observed superior responses in vitro and in vivo with CTLA-4-deficient CART19 cells wherein CTLA4 is deleted in the starting T cell population used for CAR T manufacturing as opposed to systemic delivery of an immune checkpoint inhibitor. CTLA-4 is known to compete with CD28 for binding to its ligands, CD80 and CD86.53 Thus, eliminating CTLA-4 permits uninterrupted CD28 co-stimulation. In support of this, after repeated stimulation of CTLA-4-deficient CAR T cells we detect increased levels of CD28 protein and elevated levels of targets downstream of CD28 signaling that positively mediate T cell effector function, without noting transcriptional changes in CD80 or CD86 by scRNA-seq. In agreement, shRNA-mediated knockdown of CTLA-4 has been noted to enhance the anti-tumor efficacy of a first-generation CAR T cells endowed with CD80 signaling 19z1-CD80.66 Furthermore, CTLA-4 has been shown to interact with CD80 and CD86 via transendocytosis, leading to their degradation inside the cell and impaired co-stimulation via CD28.48,67,68 This has been demonstrated to occur in both a cell-intrinsic and extrinsic manner for CD80.47,48,68 Thus, it is possible that eliminating CTLA-4 may not only permit uninterrupted CD28 co-stimulation but also may prevent the loss of B7-H1/PD-L1 ligands on the cell surface.69
Another key mechanism leading to the superior function of CTLA-4-deficient CAR T is maintenance of CAR expression on the surface for longer durations, promoting cognate-antigen specific cytotoxicity. Our data suggests that, at least in part, the increase in anti-tumor efficacy of CTLA-4-disrupted CART19 cells leads to reduced tumor load and prevents CAR overstimulation, resulting in improved fitness of CART19 cells and increased surface CAR expression in a feed forward circuit. However, under conditions of high tumor burden CTLA-4-deficient CAR T cells are no longer able to clear tumor and lose surface CAR expression. This is consistent with previous data demonstrating that transient rest can enhance CAR T cell efficacy by preventing or reversing CAR T dysfunction and restoring antigen stress-induced loss of surface CAR expression.62,70
Homozygous deficiency of CTLA4 in mice leads to a lymphoproliferative disorder and destruction of major organs by 2–3 weeks of age.71,72 Additionally, deletion of CTLA4 in mice post-thymic development causes lymphoproliferation, however it is not fatal.52,73 In humans, inherited heterozygous mutations in CTLA4 lead to CTLA4 haploinsufficiency with autoimmune infiltration (CHAI) characterized by dysregulation of regulatory T cells, hyperactivation of effector cells, and loss of circulating B cells.74,75 However, the consequences of CTLA4 deletion in post-thymic human T cells are not well understood. Moreover, it is important to note, that unlike genetic disorders or anti-CTLA-4 ICB, only T cells in the infused CAR T cell product exhibit CTLA4 deletion and endogenous T cells remain CTLA4 replete. Importantly, endogenous regulatory T (Treg) cells play an important role in regulating autoimmunity.76 We observed a low incidence of GVHD and lymphoproliferation after injecting an excess of WT or CTLA-4-deficient CART19 cells into NSG mice. Further, all NSG CD19-positive tumor bearing mice injected with CTLA-4-deficient CART19 cells exhibited contraction of CART19 cells after tumor clearance. Although encouraging, more studies are needed to understand the persistence and toxicity related to deletion of CTLA4 in CAR T cells in an immunocompetent setting before this therapy is translated to the clinic.
Interestingly, our findings do not fully align with results from dual checkpoint blockade in which PD-1 and CTLA-4 blockade or PD-1 blockade alone lead to significantly longer progression free survival for melanoma and select other indications as opposed to CTLA-4 blockade alone.20 In contrast, genetic deletion of PDCD1 in the starting T cell population does not enhance CART19 response compared to WT CART19 cells under CAE stress. Given that NALM6 cells lack PD-L1/2 ligands it is perhaps not surprising that cell-intrinsic deletion of PD-1 did not endow CAR T cells with increased function.31,32 Not all cancer patients respond to anti-PD-1, anti-PD-L1 and/or anti-CTLA-4 checkpoint therapy and the reasons for this variable response are under intense investigation in numerous laboratories and clinical trials.77,78 Similarly, the benefits of deleting PDCD1 and/or CTLA4 in CAR T cell therapy are likely context dependent, including tumor ligand expression. In some cases, deletion of PDCD1 provides benefit to CART cell function31,32,79 and in other scenarios its presence regulates optimal anti-tumor activity, engraftment, and memory formation.80–82 A complete mechanistic understanding of this will require further examination.
However, unexpectedly, we observe that PDCD1 deletion negates the increased proliferation, enhanced surface CAR expression, memory phenotype, and improved anti-tumor efficacy observed in CTLA-4-deficient CART19 cells under CAE stress. Nevertheless, if CAR T cells deficient for both PD-1 and CTLA-4 are treated with a CD28 agonistic antibody during CAE stress the dominant negative phenotype acquired by deletion of PDCD1 is lost and instead PD-1- and CTLA-4-deficient-CART19 cells express the superior phenotype endowed by deletion of CTLA4. Concomitant 4–1BB and CD28 signaling has been demonstrated to enhance CAR T cell function83,84 and our data suggest that maintenance of CD28 signaling during continuous antigen stimulation enhances CAR T cell function. It is important to note that these CAR T cells also express PD-L1 and PD-L2 (Figure S7C).85 Further, PD-L1 not only binds PD-1 in a cell extrinsic manner, but also, like CTLA-4, has been shown to bind CD80 in a cell-intrinsic fashion.86 These cis interactions disrupt the PD-1/PDL-1 axis but not CTLA-4/CD28 binding to CD80 in a cell-extrinsic and/or cell-intrinsic fashion in APC-sparse tissues.87,88
In summary, we investigated whether single or dual CRISPR-Cas9-mediated disruption of immune checkpoint receptors in T cells could enhance the efficacy of CAR T cell therapy. Surprisingly we demonstrate that CTLA-4-deficient CART19 cells, unlike WT and PD-1-deficient CART19 cells and CART19 cells deficient for both PD-1 and CTLA-4, have superior anti-tumor efficacy under stress-test conditions that otherwise lead to CAR dysfunction. Importantly we confirm the clinical relevance of these results, demonstrating that deletion of CTLA4 can bolster the fitness of human T cells and enable the manufacture of potent CART19 cell products from CLL patient T cells that failed. Further we provide mechanistic insights, revealing that CTLA-4-deficiency permits unopposed CD28 signaling and prevents or delays CAR internalization. In conclusion, deletion of CTLA4 can invigorate patient T cells, providing a strategy for increasing durable responses to CAR T cell therapy.
Limitations of Study
Further work is needed to understand why deletion of PDCD1 has a dominant negative effect on CTLA4-deleted CART19 cells in a CAE setting. The effects of PDCD1 deletion may be context-dependent and determined by multiple factors such as immunosuppression on T cells mediated by PDL-1/2 expression, the degree of PD-1 signaling required for proper CAR T cell activation, and the level and timing of PDCD1 depletion, where partial deletion of PDCD1 could be more beneficial than complete deficiency in some cases. We hypothesize that these factors may mechanistically contribute to the absence of enhanced CD28 signaling with depletion of both CTLA4 and PDCD1. The decrease in memory markers and cytotoxic T cell pathways observed in CART19 cells deleted for both PDCD1 and CTLA4, compared to WT, and single PDCD1- or CTLA4-deleted CART19 cells supports this theory and warrants future investigation.
STAR METHODS
RESOURCE AVAILBILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Carl H. June (cjune@upenn.edu)
Material Availability
The study did not generate new unique reagents.
Data and code availability
All associated raw genomics data from scRNA-seq experiments have been deposited at database of genotypes and phenotypes (dbGaP). They are available upon request if access is granted as of the date of publication. Accession number is listed in the key resource table.
This paper does not report original code.
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-human CD45 | Biolegend | Cat# 304032 |
| anti-human CD45 | Biolegend | Cat# 304028 |
| anti-human CD3 | Biolegend | Cat# 317322 |
| anti-human CD8 | Biolegend | Cat# 301066 |
| anti-human CD8 | Biolegend | Cat# 301016 |
| anti-human CD8 | BD Pharmingen | Cat# 560179 |
| anti-human CD4 | Biolegend | Cat# 317440 |
| anti-human CD4 | Biolegend | Cat# 317414 |
| anti-human CD28 | Biolegend | Cat# 302926 |
| anti-human CD38 | eBioscience | Cat# 25-0388-42 |
| anti-human CD39 | Biolegend | Cat# 328228 |
| anti-human PD-1 | Biolegend | Cat# 329928 |
| anti-human PD-1 | Biolegend | Cat# 329918 |
| anti-human TIM3 | eBioscience | Cat# 46-3109-42 |
| anti-human LAG3 | eBioscience | Cat# 11-2239-42 |
| anti-human CTLA-4 | Invitrogen | Cat# 12-159-42 |
| anti-human CD45RO | Biolegend | Cat# 304244 |
| anti-human CD45RO | BD Pharmingen | Cat# 555493 |
| anti-human CCR7 | BD Pharmingen | Cat# 130-126-090 |
| anti-human CD107a | Biolegend | Cat# 328618 |
| anti-human IL-2 | Invitrogen | Cat# 46-7029-42 |
| anti-human TNFa | Biolegend | Cat# 502928 |
| anti-human GM-CSF | BD Horizon | Cat# 562930 |
| anti-human IFNg | Biolegend | Cat# 506507 |
| anti-human Granzyme B | BD Horizon | Cat# 562462 |
| anti-human Ki67 | Biolegend | Cat# 350526 |
| anti-human CD80 | Biolegend | Cat# 305230 |
| anti-human CD86 | Biolegend | Cat# 305442 |
| anti-human ICOS | Biolegend | Cat# 313536 |
| anti-mouse CD45 | Biolegend | Cat# 103116 |
| anti-mouse FMC63 scFv MAb | Bioswan | Cat# 200106 |
| anti-mouse FMC63 scFv MAb | Bioswan | Cat# 200102 |
| BCMA CAR Detection | Miltenyi Biotec | Cat# 130-126-090 |
| BTK/ITK (Tyr551, Tyr511) Monoclonal Antibody (M4G3LN) | Thermo Fisher | Cat#12-9015-42 |
| Phospho-LCK (Tyr505) monoclonal antibody (SRRCHA) | Thermo Fisher | Cat#50-9076-42 |
| Phospho-ZAP70/Syk (Tyr319, Tyr352) Monoclonal Antibody (n3kobu5) | Thermo Fisher | Cat#12-9006-42 |
| mouse IgG2a, κ Isotype control Antibody | Biolegend | Cat# 400269 |
| mouse IgG1, κ Isotype control Antibody | Biolegend | Cat# 400126 |
| mouse IgG1, κ Isotype control Antibody | Biolegend | Cat# 400168 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | C7373-03 |
| Biological samples | ||
| T lymphocytes from human healthy donors | UPenn Human Immunology Core | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Live/Dead Aqua | ThermoFisher | Cat# L34957 |
| Zombie NIR (Biolegend) Fixable Viability Kits | Biolegend | Cat# 423106 |
| Apotracker™ Green | Biolegend | Cat# 427403 |
| Annexin V with 7AAD | Biolegend | Cat# 640930 |
| Alt-R® Cas9 Electroporation Enhancer, 10 nmol | Integrated DNA Technologies | Cat# 1075916 |
| SpyFi Cas9 | Aldeveron | Cat# 9214 |
| P3 Primary cell 4D-nucleofactor X Kit L | Lonza | Cat# V4XP-3024 |
| OpTmizer T Cell Expansion SFM | Gibco | Cat# A1048501 |
| Human AB Serum | GeminiBio | Cat#100-512 |
| Recombinant Human IL-7 | Peprotech | Cat#200-07 |
| Recombinant Human IL-15 | Peprotech | Cat#200-15 |
| DNAse I roche | Sigma | Cat#10104159001 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#1166500 |
| DNase I from bovine pancreas | Sigma | Cat# 11284932001 |
| EDTA (0.5 M), pH 8.0, RNase-free | Thermo Fisher | Cat# AM9261 |
| PE Streptavidin | BD Pharmingen | Cat# 554061 |
| APC Streptavidin | Biolegend | Cat# 405235 |
| Golgi Stop protein transport inhibitor (containing monensin | Thermo Fisher | Cat# BD554724 |
| Cell stimulation cocktail (PMA/Ionomycin) | Thermo Fisher | Cat#00497093 |
| Critical commercial assays | ||
| RNA Clean & Concentrator™-5 | ZYMO | Cat#R1016 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69504 |
| NovaSeq6000 S2-100 cycle kit | Illumina | Cat#20028316 |
| Chromium Next GEM Single Cell 3' GEM, Library & Gel Bead Kit v3.1 | 10X Genomics | Cat#1000121 |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | Cat#1000120 |
| Chromium Next GEM Single Cell 3’ Library Construction Kit v3.1 | 10X Genomics | Cat#1000157 |
| Single Index Kit T Set A | 10X Genomics | Cat#1000213 |
| XT_HS_CART Panel_CSO | Nanostring | Cat# 115000343 |
| Master Kit-12 rxns | Nanostring | Cat# 100052 |
| DynaBeads CD3x28 (Human) | ThermoFisher | Cat# 11131D |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | Cat# 74609.50 |
| QIAGEN Plasmid Plus Maxi Kit | QIAGEN | Cat# 12963 |
| Fixation Medium (Medium A) | Invitrogen | Cat# GAS001S100 |
| Permeabilization Medium (Medium B) | Invitrogen | Cat# GAS002S100 |
| CountBright Absolute Counting Beads, (ThermoFisher) | Thermo Fisher | Cat# C36950 |
| LongAmp™ Taq 2X Master Mix | New England Biolabs | Cat# M0287S |
| Vacuum Filter/Storage Systems | Corning | Cat# 430770 |
| Dead cell removal kit | Miltenyi Biotec | Cat# 130-090-101 |
| CliniMACS CD4 GMP Microbeads | Miltenyi Biotec | Cat# 170-076-702 |
| CliniMACS CD4 GMP Microbeads | Miltenyi Biotec | Cat# 170-076-703 |
| LOVO Pouch Disposable Kit | Fenwal | Cat# X6R4907 |
| LOVO Processing kit w/ Acess | Fenwal | Cat# X6R4909 |
| Deposited data | ||
| Raw and analyzed data | This paper | dbGAP: phs001707.v3 |
| Experimental models: Cell lines | ||
| Human (female) HEK293T | ATCC | CRL-11268 |
| Human (female) NALM6 | ATCC | CRL-3273 |
| Human (female) MM.1S | ATCC | CRL-2974 |
| Experimental models: Organisms/strains | ||
| NOD/scid/IL2rγ−/− (NSG) | Jackson Laboratory | Cat# 5557 |
| Oligonucleotides | ||
| PD-1 sgRNA: 5’ GGCCAGGATGGTTCTTAGGT3’ | Integrated DNA Technologies | (Ren et al, 2017) |
| CTLA-4 sgRNA: 5’ TATGCCCAGGTAGTATGG3’ | Integrated DNA Technologies | N/A |
| PD-1.PCR.F (gDNA): 5’ AGTTTCCCTTCCGCTCACCTC3’ | Genewiz | N/A |
| PD-1.PCR.R (gDNA): 5’ ACTAACCTTGGCTTTACGACGT3’ | Genewiz | N/A |
| PD-1.Seq.R (PCR): 5’ TGGCAGCCCAGGGGTC3’ | Genewiz | N/A |
| CTLA-4.PCR.F (gDNA): 5’GGACATGGGGGAAGTGTGAC3’ | Genewiz | N/A |
| CTLA-4.PCR.R (gDNA): 5’AGGTTTACTTTTAGGACTGTGGACA3’ | Genewiz | N/A |
| CTLA-4.Seq.F (PCR): 5’ TGTTCTTCCTGCCACAACCA3’ | Genewiz | N/A |
| CTLA-4.Seq.R (PCR): 5’ GACACCTGTTGCATTGCAGTC3’ | Genewiz | N/A |
| Recombinant DNA | ||
| pTRPE CD19BBz | This paper | N/A |
| pTRPE BCMABBz | This paper | N/A |
| Software and algorithms | ||
| R version 4.1.0 | CRAN | https://cran.r-project.org/ |
| Seurat_4.1.0 | (Butler et al., 2018; Stuart et al., 2019) | https://satijalab.org/seurat/ |
| Cell Ranger v4.0 | 10X Genomics | https://www.10xgenomics.com/ |
| sctransform_0.3.3 | (Hafemeister and Satija, 2019) | https://cran.r-project.org/web/packages/sctransform/index.html |
| DESeq2 1.38.3 | Love et al, 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| EdgeR 3.40.2 | Robinson et al, 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| FlowJo™ v10.8 Software | BD Life Sciences | https://www.flowjo.com |
| Ingenuity Pathway Analysis Software | QIAGEN | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Other | ||
| Benchling sgRNA designer tool (https://www.benchling.com, [2020]) | Benchling | https://benchling.com/ |
| BioRender illustration design tool | BioRender | https://biorender.com/ |
| TIDE: Tracking of Indels by DEcompositionTool | TIDE | http://shinyapps.datacurators.nl/tide/ |
| Gene Set enrichment Analysis Software | GSEA | https://www.gsea-msigdb.org/gsea/index.jsp |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
NALM6, MM.1S and HEK293T cell lines were obtained from American Type Culture Collection (ATCC) and were cultured in standard culture media (RPMI 1640 + 10% FBS, 1% penicillin/streptomycin (50IU/ml), 1% HEPES, 1% Glutamax; R10 media) (Gibco, Life Technologies) at 37°C in 5% CO2. NALM6- click beetle green (CBG) and green fluorescent protein (GFP) expressing cells, NALM6 CBG GFP CD80 cells, and MM.1S CBG GFP expressing cells were generated by lentiviral transduction for cell killing assays and in vivo studies. All cell lines were authenticated by the University of Arizona Genetics Core and were tested for the presence of mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza) every 6 months.
Mice
Animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Six- to eight-week-old male NOD/scid/IL2rg/ (NSG) were procured from Jackson Laboratories and bred in the vivarium at the University of Pennsylvania in pathogen-free conditions. Mice were maintained under pathogen free conditions. Mice were randomly assigned to experimental groups based on their individual tumor burden to ensure that the tumor burden remained consistent across all experimental conditions prior to CAR T infusion.
Human Samples
Healthy donor primary T lymphocytes were provided by the University of Pennsylvania Human Immunology Core. Samples are de- identified for compliance with HIPAA rules. Donor sex and age is shown below: ND500 (female, age 24), ND538 (male, 50), ND503 (female, 24), ND534 (male, 25), ND410 (female, 54, 59), ND520 (male, 46), ND451 (female, 30), ND572 (male, 35), ND561 (male, 26), ND052 (male, 56), ND502 (female, 54).
Frozen apheresis samples were collected from CLL patients who enrolled in clinical trials of single-agent CTL019 therapy. Patient’s enrolled in this trial were adults aged 18 years or older with CD19+ CLL with relapsed or persistent disease after at least two previous treatment regimens. All patients gave informed consent in accordance with the Declaration of Helsinki. This study was registered at ClinicalTrials.gov (identifier NCT01029366).
METHOD DETAILS
General Cell Culture
NALM6, MM.1S and HEK293T cell lines were obtained from American Type Culture Collection (ATCC) and were cultured in standard culture media (RPMI 1640 + 10% FBS, 1% penicillin/streptomycin (50IU/ml), 1% HEPES, 1% Glutamax; R10 media) (Gibco, Life Technologies) at 37°C in 5% CO2. NALM6- click beetle green (CBG) and green fluorescent protein (GFP) expressing cells, NALM6 CBG GFP CD80 cells, and MM.1S CBG GFP expressing cells were generated by lentiviral transduction for cell killing assays and in vivo studies. The cell lines were selected by sorting on FACS Aria Sorter (BD Biosciences) Sorter and monitored for growth and stable expression of ligands for 8 weeks. All cell lines were authenticated by the University of Arizona Genetics Core and were tested for the presence of mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza) every 6 months.
Lentiviral vector production
Lentiviral vector production was performed as previously described 89. Briefly, replication defective lentiviral vectors were generated by transient transfection of HEK293T cells (ATCC ACS-4500) using Lipofectamine 2000 (ThermoFisher Scientific, Cat#11668500). Approximately 6e6 cells were plated in T150 culture vessels in standard culture media and incubated overnight at 37°C. 18–24h later, cells were transfected using a combination of Lipofectamine 2000 (96μL, Invitrogen), pTRP gag/pol (Lot# RR13SEP19A) (18μg), pTRP RSV-Rev (Lot# RR13SEP19B-3) (18μg), pTRP VSVG (Lot# RR13SEP19C) (7μg) packaging plasmids and 15μg of expression plasmid (CD19bbz scFv cloned in pTRPE). Lipofectamine and plasmid DNA were diluted in 3mL Opti-MEM media (Gibco, Life Technologies) prior to transferring into lentiviral production flasks. At both 24 and 48hrs following transfection, culture media was isolated and concentrated using high-speed ultracentrifugation (25,000g for 2.5 hours or 8000g O/N). To generate the lentiviral stocks, the resulting concentrated lentivirus batches were resuspended in cold R10 media and stored at −80°C.
Primary human T cells
Normal donor healthy human T cells were procured through the University of Pennsylvania Human Immunology Core. Autologous peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteer donors. CD4 and CD8 T cells were isolated using CD4 (Catalog# 15062, Stem Cell Technologies) and CD8 (Catalog# 15063, Stem Cell Technologies) selection kits. CD4 and CD8 cells were combined at a 1:1 ratio and incubated overnight at 3e6/ml with 5ng/ml IL-7 and IL-15 (Peprotech). They were then modified using CRISPR cas9 technology, detailed below, and maintained at 30°C for 2 days.42 They were then activated using Dynabeads CD3/CD28 CTS™ (Thermo fisher) at a 3:1 bead-to-cell ratio in RPMI 1640, 10% FBS, 1% penicillin (50IU/ml), 1% HEPES, and 1% Glutamax. The murine CD19 CAR plasmid vector was used for transduction approximately 24hrs post bead stimulation at MOI of 3. Beads were removed on day 7 of stimulation, and cells were monitored daily using the Multisizer 4 Coulter Counter (Beckman Coulter) until growth kinetics and cell size demonstrated that they had rested from stimulation. T cells were grown for 8–10 days in the presence of 5ng/ml of IL-7 and IL-15 and maintained at 8e5 cells/ml prior to cryopreservation. Prior to all experiments, T cells were thawed and rested at 37°C for 16 hours. All experiments were conducted using the second generation CD19 CAR composed of a mouse CD19-binding (FMC63) scFv and CD8α hinge and transmembrane domains fused to 4–1BB and CD3-ζ cytoplasmic signaling domains expressed under control of the EF-1α promoter, unless otherwise noted. Untransduced (UTD) T cells were as controls for alloreactivity.
Patient CAR T cell manufacturing
We generated WT and CTLA-4-deficient CART19 cells using banked apheresis samples from previously reported90 responding (CR)-01, partially responding (PR)-01, non-responding (NR)-01, and NR-02 CLL patients (Figure S3A). Given the limited amounts of available patient apheresis material and the increased in vitro and in vivo efficacy observed in the ND CTLA-4 disrupted CART19 group, we chose to focus on WT and CTLA-4-deficient CART19 cells. Frozen banked apheresis patient products were thawed and washed using the LOVO Cell Processing System (Fresenius Kabi). CliniMACS CD4 and CD8 MicroBeads (Miltenyi Biotec) were used to positively select for T cells from the apheresis material using the CliniMACS machine (Miltenyi Biotec). The CD4 and CD8 T cells were subjected to CRISPR editing and T cell expansion as described above and used for in vitro and in vivo assays as described.
CRISPR/Cas9-guide design, genomic engineering
CRISPR single guide RNA (sgRNA) targeting CTLA-4 were designed using software integrated into Benchling (2017) and chemically modified and synthesized by Integrated DNA Technologies (IDT). PDCD1 sgRNA sequence was taken from Ren et al 31. One of six sgRNA targeting CTLA-4 was selected for further experiments after validation for highest deletion efficiency. Gene deletion, T cell activation, transduction, expansion, and knockout validation of PDCD1 deletion, CTLA4 deletion, and PDCD1 and CTLA4 deletion CART19 cells were performed following an optimized protocol previously described 42. Briefly, primary T cells were electroporated using the Lonza 4D-Nucleofector Core/X Unit and the P3 Primary Cell 4-D Kit (Lonza). For Cas9 and sgRNA delivery, the ribonucleoprotein (RNP) complex was formed by incubating 10μg of Spy Fi Cas9 (Aldeveron) with 5μg of sgRNA (IDT) for 10 mins at room temperature (RT). Cells were spun down at 300×g for 5mins and resuspended at a concentration of 5–10e6 cells/100μL in the specified buffer (P3 Solution with Supplement). The RNP complex, 100μL of resuspended cells, and 4μL of 100μM IDT Electroporation Enhancer (a non-homologous ssDNA oligonucleotide) were combined and electroporated in a cuvette. Pulse codes EH115 were used for primary T cells. After electroporation, the cells were incubated in standard media at a 5e6 cells/mL at 30°C for 48h in 12-well plates, then cultured at 37°C for the duration of experimental procedures.
Genomic DNA extraction, Sanger sequencing, indel detection
From each screening culture, 3e6 – 5e6 were flash frozen as dry cell pellets. At time of DNA extraction, cell pellets were thawed and resuspended in 200ul Phosphate Buffer Saline (PBS). Genomic DNA from electroporated cells was isolated using the Qiagen DNeasy Blood & Tissue Kit (Cat#69504) and 200–300ng DNA was PCR amplified using LongAmp™ Taq 2X Master Mix (NEB) and 10μM forward and reverse primers flanking the region of interest. Primers were designed such that the amplicon was at a target size of 600–700bp. PCR products were gel purified and sequenced, and trace files were analyzed using Desktop Genetics software (tide.deskgen.com, Desktop Genetics) to determine deletion efficiency. TIDE (Tracking of Indels by DEcomposition) analysis was used to detect deletion efficiency at the genomic level.91 R2 values were calculated, reflecting goodness of fit after non-negative linear modeling by TIDE software. PCR primers and sequencing primers were designed to detect each target locus. Analysis of gene editing efficiency was assessed by Sanger sequencing. The following sequences were used for generation and deletion efficiency validation: PD-1 sgRNA #1: 5’ GGCCAGGATGGTTCTTAGGT3’, CTLA-4 sgRNA #2: 5’ TATGCCCAGGTAGTATGG3’; Primers for target sequence amplification and Sanger sequencing: PD-1.PCR.F 5’ AGTTTCCCTTCCGCTCACCTC3’, PD-1.PCR.R 5’ ACTAACCTTGGCTTTACGACGT3’, PD-1.Seq.R 5’ TGGCAGCCCAGGGGTC3’; CTLA-4.PCR.F 5’GGACATGGGGGAAGTGTGAC3’, CTLA-4.PCR.R 5’AGGTTTACTTTTAGGACTGTGGACA3’, CTLA-4.Seq.F 5’ TGTTCTTCCTGCCACAACCA3’, and CTLA-4.Seq.R 5’ GACACCTGTTGCATTGCAGTC3’.
iGUIDE
Library preparation, DNA sequencing, and analysis. Libraries were prepared as described in the associated protocol for iGUIDE.92 Genomic DNA from samples was purified and randomly fragmented by ultrasonication. Adapters were ligated to end-repaired DNA, and targeted DNA was amplified through a nested-PCR from the incorporated dsODN to the ligated adapter sequence. Amplicons were purified and sequenced on an Illumina MiSeq with 300 cycle v2 reagent kits. Output sequence data was analyzed using the iGUIDE pipeline. iGUIDE standard operating procedure (SOP) for carrying out iGUIDE is detailed in Nobles et al., 2019.92 For the CTLA-4 sgRNA, there were low-abundance mutations in CAMK1G and PLA2R1; however, deletion of these genes is not expected to have negative consequences because they are not reported to be expressed in T cells. For the PDCD1 sgRNA, off-target edits were identified within the transcriptional unit of AKAP7 (Figure S1D).
Bioluminescence-based cytotoxicity assays
NALM6 CBG GFP target cells were used, and cell survival was measured using bioluminescent quantification. Target and effector cells were co-cultured at different effector: target ratios. After 24 hrs, D-luciferin potassium salt (Perkin-Elmer) was added to cell cultures at a final concentration of 15μg/mL and incubated at 37°C for 10min. This was repeated at either 12hr or 24 hr intervals. Bioluminescent signal was detected using a BioTek Synergy H4 imager, and the signal was analyzed using BioTek Gen5 software. Percent specific lysis was calculated using a control of target cells without effectors. Triplicate wells were averaged and percent lysis was calculated from the data with the following formula: % specific lysis = 100×(tumor only − test sample lysis)/(tumor only − maximum lysis).
Degranulation Assays and Intracellular Cytokine Staining
5e4 surface CAR+ T cells (day 0 CD19BBz product or day 15/18 CAE) were cocultured with 5e4 NALM6 cells or left in R10 media in 48 well plates in the presence of Golgi Stop protein transport inhibitor (containing monensin) (Catalog# BD554724) purchased from Thermo Fisher. Cell stimulation cocktail (PMA/Ionomycin) (Catalog #00497093) was used as a positive control purchased from Thermo Fisher. After 6–8 hours, the cells were harvested for flow cytometry staining. The cells were stained with CD107a antibody (Catalog#328618) fromBiolegend, anti-human antibodies to intracellular cytokines IL-2 (Catalog# 46–7029-42) purchased from Invitrogen, IFNg (Catalog# 506507) and TNFa (Catalog# 502948) purchased from Biolegend, and GM-CSF (Catalog# 562930) purchased from BD Horizon. Samples were acquired on an LSRII Fortessa Cytometer. All data analysis was performed using FlowJo 9.0 software (FlowJo, LLC).
Chronic antigen exposure (CAE) stress test assay
CART19 cells (50–70% transduction efficiency) were thawed and rested at 2e6 cells/ml in T75 flasks with R10 media. After 24 hours, the T cell number (CD45+) was calculated and 2.5e5 T cells/well were mixed with 1e6 NALM6 cells/well for an E:T ratio of 1:4 per well. After 2–3 days, 200ul of co-culture was used to count the absolute number of T cells and NALM6 tumor cells by flow cytometry using Countbright absolute counting beads (Thermo Fisher). At each timepoint, cell suspension was spun down and the supernatant (conditioned media) was collected and filtered with a 0.45μm filter (Corning). The cells were resuspended in media containing equal amounts of conditioned media and fresh R10. Based on counts, co-cultures were re-fed with new NALM6 tumor cells at the same E:T ratio of 1:4 per well. Cultures were maintained at a concentration of 1e6 tumor cells/ml. This process was repeated for 15–18 days. At the end of the assay, supernatant was collected and analyzed for cytokines as described below. Additionally, CD45+ T cells were sorted for genomics analysis. Flow Cytometry analysis was performed on Day 0, 7 and 15 for T cell immunophenotyping. To test whether CAE and surface CAR expression are directly correlated we exposed CTLA-4-deficient CART19 cells to very high levels of tumor burden i.e., 1:100 E:T ratio and used 1:10 E:T ratio as control (Figure 4F). For CD28 agonist antibody experiments, CD28 Agonist antibody (#16–029-95) or isotype control (#14–4301-82) purchased from Thermo Fisher were pre-coated at 37C for 2h on 6 well plates at 5ug/ml. The plates were then washed and used for CAE stress test. For all assays, NALM6 cells do not express HER2, allowing the use of HER2-CAR as a specificity control (Control CART).
Flow cytometry and sorting
For flow cytometry and sorting assays of the CAE, CART19 cells were stained in fluorescence-activated cell sorting (FACS) buffer consisting of PBS (Gibco), 0.5% bovine serum albumin (BSA) (GEMINI), 2 mM EDTA (Invitrogen), and 100mg/ml DNase (Roche). CountBright™ Absolute Counting Beads (ThermoFisher) were used as an internal standard according to the manufacturer’s instructions to calculate absolute cell counts in cell suspensions.
Antibodies specific for human CD45 (304032/304028, clone HI30), CD3 (317322, OKT3), CD4 (317414/317440, OKT4), CD8(301066/301016), CD28 (302926, CD28.2), CD39(328228), PD-1 (329928/329918, EH12.2H7), CD45RO (304244, UCHL1), Ki67 (350526), CD80 (305230), CD86 (305442), ICOS (313536), and anti-mouse CD45 (103116) were purchased from BioLegend. Antibodies specific for human CD8 (560179, SK1), CD45RO (555493), and CCR7 (130–126-090, 150503) were purchased from BD Pharmingen. Anti-human CTLA-4 antibody (12–1529-42, 14D3) was purchased from Invitrogen. Anti-human CD38 antibody (25–0388-42), LAG3 (11–2239-42), TIM3 (46–3109-42) was purchased from eBioscience. CD19 CAR was detected using an anti-idiotype antibody, specifically binding to the anti-CD19 scFv that was derived from the FMC63, conjugated to PE provided by Novartis Pharmaceuticals, or purchased from CytoArt (Catalog # 200107/200106/200102, Antibody Registration Number AB_2857947). If not conjugated to a fluorochrome, AffiniPure F(ab’)₂ Fragment Goat Anti-Human IgG AF647 secondary antibody was used purchased from Jackson Immunoresearch. Cell viability was measured using Live/Dead Aqua fixable staining kit (Life Technologies) according to the manufacturer’s instructions. Intracellular staining was performed with the Medium A/ Medium B Buffer set (Invitrogen) according to the manufacturer’s instructions. CFSE Proliferation Assay was performed using the CFSE Cell Proliferation Kit (Catalog # C34570) purchased from Thermo Fisher according to manufacturer instructions. Samples were acquired on an LSRII Fortessa Cytometer (BD Biosciences) or FACSymphony™ A5 SE Cell Analyzer (BD Biosciences). All data analysis was performed using FlowJo 9.0 software (FlowJo, LLC). Sorting assays were performed using a FACS Aria Cytometer (BD Bioscience).
Cytokine and cytolytic molecule quantification
Human cytokine quantification was performed using a custom 31-plex Luminex panel (EMD Millipore) containing the following analytes: EGF, FGF-2, Eotaxin, sIL-2Ra, G-CSF, GM-CSF, IFN-α2, IFN-γ, IL-10, IL-12P40, IL-12P70, IL-13, IL-15, IL-17A, IL-1RA, HGF, IL-1β, CXCL9/MIG, IL-2, IL-4, IL-5, IL-6, IL-7, CXCL8/IL-8, CXCL10/IP-10, CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, RANTES, TNF-α, VEGF. Cell culture supernatants, on day 15 of long term restimulation assays, were flash frozen on dry ice, and thawed at time of cytokine analysis. Assays were established per manufacturer recommendations. Data were acquired on a FlexMAP 3D quantification instrument, and analysis was done using xPONENT software. Data quality was determined by ensuring the standard curve for each analyte had a 5P R2 value > 0.95 with or without minor fitting using xPONENT software. To pass assay technical quality control, the results for two controls in the kit were required to be within the 95% confidence interval provided by the vendor for >25 of the tested analytes.
Phospho-Flow Signaling Assay
CAE stress test assay was sampled on day 15 to test phospho-flow. The assay was terminated by fixation with 1.5% paraformaldehyde (PFA) for 15 min at room temperature with agitation. Cells were washed and permeabilized with ice-cold 100% methanol for 60 min on ice. Cells were then washed with FACS buffer and stained for 1h at 4°C in the dark with BTK/ITK (Tyr551, Tyr511) Monoclonal Antibody (M4G3LN) (Cat#12–9015-42), Phospho-LCK (Tyr505) monoclonal antibody (SRRCHA) (Cat#50–9076-42), and Phospho-ZAP70/Syk (Tyr319, Tyr352) Monoclonal Antibody (n3kobu5) (Cat#12–9006-42)purchased from Thermo Fisher Cells were washed twice and acquired on a FACSymphony™ A5 SE Cell Analyzer (BD Biosciences). All data analysis was performed using FlowJo 9.0 software (FlowJo, LLC). Data represent the median fluorescence intensity (MFI) fold change (FD) compared to unstimulated T cells.
Nanostring nCounter technology
Day 0 product and day15 CAE stress test were sorted on CD45+ T cells from two donors. Cell pellets were frozen at −80C in lysis buffer and total RNA was extracted using the Low input RNeasy mini kit purchased from Qiagen. RNA expression was analyzed using the nCounter human CART characterization panel (#XT-CSO-HCART1–12) purchased from Nanostring Technologies. Briefly, 3’ mRNA gene expression profiling was generated from 100 ng of total RNA. The concentration of the total RNA was assessed using NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and the quality using Agilent 4200 TapeStation (Agilent Technologies, Santa Clara, CA). Hybridization between target mRNA and reporter-capture probe pairs using nCounter CAR-T Characterization Panel (NanoString Technologies, Seattle, WA) was performed at 65°C for 18 hours using Mastercycler Pro S Thermal Cycler (Eppendorf, Framingham, MA). Post hybridization processing was carried out on a fully automated nCounter Prep Station (NanoString Technologies, Seattle, WA) liquid-handling robot using the High Sensitivity setting. The probe/target complexes were immobilized on the nCounter cartridge, which was then placed in nCounter Digital Analyzer (NanoString Technologies, Seattle, WA) for image acquisition and data processing as per the manufacturer’s protocol with FOV set to 555. The expression level of a gene was measured by counting the number of times the specific barcode for that gene was detected. The barcode counts were then tabulated in a comma-separated value (CSV) format. The raw digital count of expression was exported from nSolver v3.0 software for downstream analysis.
Nanostring nCounter technology data analysis
Raw counts and data quality control occurred within the nCounter digital analyzer software, nSolver, provided by Nanostring Technologies. Gene expression was normalized using the default median size factor method. Differential expression analysis was performed using EdgeR v3.40.293 package using TMM normalization and glmQLFit function and filtered on FDR<0.05 and absolute value log2 fold change >95th percentile among all the different groups. For heatmap visualization, top 130 significant DEG were chosen, normalized using VST94, plotted using the pheatmap R package and scaled by row. Gene set enrichment analysis was performed with nCounter CART cell panel derived gene-sets using the clusterProfiler v4.6.2 and visualized using the ggplot2 v3.4.1 R packages.
Mice study approval and experiments
The University of Pennsylvania Institutional Animal Care and Use Committee approved all animal experiments, and all animal procedures were performed in the animal facility at the University of Pennsylvania in accordance with Federal and Institutional Animal Care and Use Committee requirements. Male and female 6–8-week-old NOD-SCID-IL2rγ−/− (NSG) mice were purchased from Jackson laboratories or bred in-house in the vivarium at the University of Pennsylvania and maintained in pathogen free conditions. All experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. Mice were maintained in dedicated BSL-2 animal barrier spaces. Age and sex-matched animals were randomly assigned to experimental groups; no gender-specific influences were detected in the experimental results. For ALL xenograft models, animals were injected via the tail vein with 0.5e6 WT NALM6 cells in 200ul sterile PBS. On day 4 after tumor delivery, 3e6 T cells (control or CAR19+ T cells) were injected via tail vein in 200ul sterile PBS. For MM xenograft models, animals were injected via the tail vein with 2.5e6 MM.1S cells in 200ul sterile PBS. NALM6 and MM.1S cells do not express HER2, allowing the use of HER2-CAR as a specificity control. On day 14 after tumor delivery, 1.5e6 T cells (control or BCMA CAR+ T cells) were injected via tail vein in 200ul sterile PBS. Mice were evaluated by bi-weekly bioluminescent imaging, weekly weight measurements and serial retro-orbital bleeding. Disease burdens were monitored over time every 3–4 days using the Xenogen IVIS-200 spectrum camera. Data was analyzed using Living Image version 4.4 (Caliper LifeSciences, PerkinElmer). Peripheral blood was obtained by retro-orbital bleeding and cell numbers of each subset (hCD45, CD4, CD8) were quantified using TruCount tubes (BD Biosciences) or countBright™ Absolute Counting Beads (ThermoFisher) using flow cytometry. Animals were monitored for signs of lymphoproliferation, and overt toxicity as evidenced by >10% loss in body weight, loss of fur, diarrhea, conjunctivitis and disease-related hind limb paralysis. All experiments were performed in a randomized fashion.
For xenogeneic GVHD studies, 16 NSG mice per cohort (T cells manufactured from two ND’s) were injected with 10e6 T cells, intravenously. GVHD severity was assessed by a scoring system that incorporates four clinical parameters: weight loss, posture (hunching), mobility and anemia as previously reported.95 Each parameter received a score of 0 (minimum) to 2 (maximum). Mice were assessed for GVHD score twice weekly, weighed weekly and monitored daily during the experiments. Mice reaching a GVHD score of 6/8 were sacrificed. Peripheral blood was obtained by retro-orbital bleeding and cell numbers of each human CD45+ subset was quantified using TruCount tubes (BD Biosciences). Mice were followed up for 6 months for survival analysis.
scRNA-seq library prep and sequencing
For donor ND520 and ND052 and patient NR-01 and PR-01, cells were sorted at the end of the assay and were subjected to single-cell RNA-seq library prep. We followed the protocol by 10x Genomics to perform scRNA-seq (scRNA-seq). scRNA-seq libraries were generated using a Chromium Single-Cell 3’ Library and Gel Bead Kit v3.1 (10x Genomics) according to the manufacturer’s protocol. Briefly, 20,000 CD45+ T cells were sorted by flow cytometry and washed with ice cold PBS + 0.04% BSA. After washing, cells were partitioned into single cell droplets using the 10x Genomics Chromium Controller as per manufacturer’s protocol. Following reverse transcription, the emulsion of single cell droplets was disrupted, and barcoded complementary DNA was isolated and amplified by PCR for 10 cycles. After fragmentation, end repair, and poly A tailing, sample indexes were added and amplified following the manufacturer’s protocol. Size of the final libraries was determined using BioAnalyzer 2100 (Agilent) and final loading concentration was determined using the KAPA Library qPCR Quantification kit for Illumina sequencing platforms (Roche). Sequencing was completed on an Illumina NovaSeq 6000 platform (Illumina) with a 100-cycle kit with parameters Read 1: 28, Read 2: 91, Index 1: 8, Index 2: 0. Twelve samples were sequenced per flow cell to achieve coverage of at least 20,000 reads/single cell.
Single cell RNA-seq data processing
scRNA-seq fastq data was pre-processed using established and custom pipelines. Briefly, raw Illumina data files were subjected to Cell Ranger, which used cellranger mkfastq to wrap Illumina’s bcl2fastq to correctly demultiplex Chromium-prepared sequencing samples and to convert barcode and read data to FASTQfiles. Sequencing data were aligned to the GRCh38 genome, filtered, and then barcodes and unique molecular identifiers were counted using the Cell Ranger v3.1.0 command cellranger count. Data were further analyzed in R using Seurat version 3.1.2.96,97 scRNA-seq was performed in two ND’s and two CLL patient samples in independent experiments. For analysis, the two donors were analyzed together and the CLL patients were analyzed together. Briefly, genes that were not detected in at least 3 cells and cells with >10% for donors and >15% for patients15,98 mitochondrial reads were excluded. Also, cells that expressed < 200 genes or were 3 median absolute deviations higher than the median (outliers) were also excluded. For CLL patient samples, IGCL7 expression (B cell marker) was used to identify a contaminating NALM6 (pre-B cell leukemia cell line) cell cluster and long-noncoding RNAs were used to identify a contaminating CLL tumor cell cluster which were subsequently removed using Seurat’s subset function. Data were normalized using sctransform.99 Clusters and UMAP were generated from the first 10 PCA dimensions using the default parameter settings in Seurat. Sctransform normalized expression was used for the heatmap of marker genes, UMAP feature plots, and dot plots. Marker genes were determined using the FindAllMarkers function in Seurat where at least 25% of the cells must be expressing the gene. Differentially expressed genes were identified using FindAllMarkers function in Seurat where logFC>0.5 and at least 25% of the cells expressing the gene were used as cut-off values. A Wilcoxon rank sum test was used to determine significance between WT and CTLA-4-deficient groups or Day 0 and Day15 DEGs. IPA (Qiagen Inc) was used to determine differential pathways and downstream targets of CD28. For IPA, mitochondrial genes were filtered out and genes with fold change >1.5 were included. Common differential pathways in donors and patients were confirmed using Gene Set Enrichment (GSE) Analysis. Pathways were filtered on Normalized Enrichment Score (NES) > 1 and False Discovery Rate < 0.25. Gene Set Enrichment Analysis was also used to determine the enrichment of (1) exhaustion gene sets in Figure 6D (Reference Gene Sets – Good et al 2021; Lynn et al, 2019; Zheng et al, 2017; Guo et al, 2018; Li et al, 2019; Wherry et al, 2017; Beltra et al, 2020) (2) anti-mesothelin M5CART dysfunctional signature in the DEG’s between day 15 WT and day 0 WT CD19 CAR T cells in Figure 6D and Figure S6B (Reference Gene set - M5 CAR Dysfunctional gene signature from Good et al)41; and (2) d15 CTLA-4-deficient CART19 phenotype from ND’s in the CLL patients’ DEG’s of day 18 CTLA-4-deficient (compared to day 18 WT CART19 cells) in Figure S6F (Reference gene set - All DEG’s between day 15 CTLA-4-deficient vs the day 15 WT CART19 from ND’s).
CART transcript detection in scRNA-seq data
To assess the expression of the CD19 CAR transcript in the scRNA-seq data, the cellranger reference was reindexed (mkref) by adding a single contig for the WPRE+R region sequence (a unique sequence in the CAR plasmid) to the human genome (GRCh38). The gene annotation GTF files was appended with CDS and exon entries spanning the entire sequence and labelled with the gene_id “mCD19CAR”. To analyze expression of CAR and to determine the percent of cells expressing the CAR, we pooled data from two scRNA-seq experiments for both donors (ND520 and ND052) and patients (NR-01 and PR-01) (related to Figure S7D).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism 8. All comparisons between two groups were performed using either a two-tailed unpaired Student’s t-test or Mann-Whitney test, depending on the normality of distribution. Comparisons between more than two groups were performed by multiple unpaired t-tests with Bonferroni-Dunn correction, ordinary one-way analysis of variance or two-way ANOVA with Bonferroni correction for multiple comparisons. Survival data were analyzed using the Log-Rank (Mantel-Cox) test. All in vitro data presented are representative of at least three biological replicates in donors and two biological replicates in CLL patients, unless otherwise stated. scRNAseq was performed in four biological replicates (two donors and two CLL patients). In vivo animal studies were performed using two biological replicates, independently. Pooled data are represented as the mean ± SEM.
ADDITIONAL RESOURCES
Frozen apheresis samples were collected from CLL patients who enrolled in clinical trial of single-agent CTL019 therapy, registered at ClinicalTrials.gov (identifier NCT01029366).
Supplementary Material
Table S1. Full list of DEGs between day 0 and day 15 CAE WT CART19 cells from scRNA-seq experiment, related to Figure 6C.
Table S2. Full list of DEGs between day 0 WT and CTLA4-deleted CART19 product in patients from scRNA-seq experiment, related to Figure 6F.
Table S3. Full list of Ingenuity pathway Analysis pathways enriched in DEGs between day 0 WT and CTLA4-deleted CART19 product from scRNA-seq experiment, related to Figure 6G.
Table S4. Full list of DEGs between day 18 WT and CTLA-4-deleted CART19 cells in patients related to Figure 6H; and day 15 WT and CTLA4-deleted CART19 cells in donors related to Figure S6E from scRNA-seq experiment.
Table S5. Full list of Ingenuity pathway Analysis pathways enriched in DEGs (1) between day 18 and day 0 CTLA4-deleted CART19 (related to Figure 6I); and between day 18 and day 0 WT CART19 cells (related Figure 6J) in patients; and (2) between day 15 and day 0 CTLA4-deleted CART19 (related to Figure S6G); and between day 15 and day 0 WT CART19 cells (related Figure S6H) in donors from scRNA-seq experiment.
Table S6. All CD28 targets predicted by IPA (upstream regulator analysis) using DEGs between day 15 and day 0 CTLA4-deleted CART19 in donors; and between day 18 and day 0 WT CART19 cells in patients, from scRNA-seq experiment, related to Figure 7B.
Table S7. DEGs between day 0 and day 15 CAE CART19 cells in each group from nCounter-based readout, related to Figure 7D, and Figure 7E.
Highlights.
CTLA4 deletion enhances CART efficacy and surface CAR expression under stress.
Deletion of CTLA4 permits unopposed CD28 signaling in CART cells.
CTLA4 deletion rescues the function of nonresponding CLL patient CART cells.
PD-1-deficiency nullifies CD28 signaling enabled by CTLA4 deletion in CART cells.
Acknowledgements
C.H.J, R.M.Y, J.A.F., M.M.D. and D.L.P. are supported by NIH grant P01CA214278 and C.H.J, R.M.Y, J.A.F., M.M.D. and M.A.A. NIH grant U54CA244711. We thank the patients for their participation in the clinical trials from which research samples were obtained. The authors acknowledge the Human Immunology Core, Flow and Cell Sorting Facility, Stem Cell and Xenograft Core at the University of Pennsylvania and the Wistar Genomics Core at The Wistar Institute. The authors would like to thank Carolyn Shaw, and John Scholler for expert technical assistance, and Phillip C. Rommel, and Christopher Roselle for helpful discussions on the manuscript.
Inclusion and Diversity
The authors support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of Interests
A.J.R. is a consultant for Affini-T Therapeutics. M.R. is the scientific founder of viTToria bio and is an inventor of IP managed by Penn and licensed to Novartis, Tmunity, and viTToria. M.R. is part of the SAB of ABClon, viTToria bio and Scailyte and has served as a consultant for GLG, BMS, GSK, nanoString, Sana, Bayer. M.M.D. is a consultant and has received research funding from Tmunity Therapeutics and is a consultant and member of the scientific advisory board for Cellares Corporation. J.A.F. is a member of the Scientific Advisory Boards of Cartography Bio. and Shennon Biotechnologies Inc. and has patents, royalties, other intellectual property. D.L.P. has consulting or advisory Role at Novartis, Kite/Gilead, Incyte, Gerson Lehrman Group, Janssen (Johnson and Johnson), BMS, Bluebird Bio, Angiocrine, Mirror Biologics, Capstan Therapeutics, Instill Bio, Sana Biotherapeutics, Verismo Therapeutics, and research support from Novartis. D.L.P. is a patent inventor for use of CAR T cells in CD19+ malignancies. D.L.P. served as Chair of Board of Directors for National Marrow Donor Program from Oct 2018-Oct 2020. D.L.P. is Associate Editor for Am J Hematology, Wiley and Deputy editor for Transplant and Cell Therapy (ASTCT Journal), Elsevier. D.L.P. spouse has Stock and Other Ownership Interests with Genentech, and Roche from former employment. C.H.J. is an inventor of patents related to the CAR therapy product which is the subject of this paper, as well as other CAR therapy products, and may be eligible to receive a select portion of royalties paid from Kite to the University of Pennsylvania. C.H.J. is a scientific co-founder and holds equity in Capstan Therapeutics, Dispatch Biotherapeutics and Bluewhale Bio. C.H.J. serves on the board of AC Immune and is a scientific advisor to BluesphereBio, Cabaletta, Carisma, Cartography, Cellares, Cellcarta, Celldex, Danaher, Decheng, ImmuneSensor, Kite, Poseida, Verismo, Viracta, and WIRB-Copernicus group. C.H.J., R.M.Y., M.M.D., M.R., and D.L.P. are inventors on patents and/or patent applications licensed to Novartis Institutes of Biomedical Research and Kite and may receive license revenue from such licenses. The remaining authors declare no competing interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. (2014). Efficacy and Toxicity Management of 19–28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. (2018). Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378, 439–448. 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, Leguay T, Bishop MR, Topp MS, Tzachanis D, et al. (2021). KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. The Lancet 398, 491–502. 10.1016/s0140-6736(21)01222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young RM, Engel NW, Uslu U, Wellhausen N, and June CH (2022). Next-Generation CAR T-cell Therapies. Cancer Discov, OF1–OF14. 10.1158/2159-8290.CD-21-1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schultz LM, Baggott C, Prabhu S, Pacenta HL, Phillips CL, Rossoff J, Stefanski HE, Talano JA, Moskop A, Margossian SP, et al. (2022. ). Disease Burden Affects Outcomes in Pediatric and Young Adult B-Cell Lymphoblastic Leukemia After Commercial Tisagenlecleucel: A Pediatric Real-World Chimeric Antigen Receptor Consortium Report. J Clin Oncol. 40, 945–955. 10.1200/JCO.20.03585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruella M, and Muas M (2016). Catch me if you can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput Struct Biotechnol J. 14, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wells DA, Summerlin J, and Halford Z (2022). A Review of CAR T-Cell Therapies Approved for the Treatment of Relapsed and Refractory B-Cell Lymphomas. J Hematol Oncol Pharm 12. [Google Scholar]
- 8.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. (2017). Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 377, 2531–2544. 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siddiqi T, Soumerai JD, Dorritie KA, Stephens DM, Riedell PA, Arnason J, Kipps TJ, Gillenwater HH, Gong L, Yang L, et al. (2022). Phase 1 TRANSCEND CLL 004 study of lisocabtagene maraleucel in patients with relapsed/refractory CLL or SLL. Blood 139, 1794–1806. [DOI] [PubMed] [Google Scholar]
- 10.Frey NV, Gill S, Hexner EO, Schuster SJ, Nasta SD, Loren A, Svoboda J, Stadtmauer E, Landsburg DJ, Mato A, et al. (2020). Long-Term Outcomes From a Randomized Dose Optimization Study of Chimeric Antigen Receptor Modified T Cells in Relapsed Chronic Lymphocytic Leukemia. J Clin Oncol. 38, 2862–2871. 10.1200/JCO.19.03237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sterner RC, and Sterner RM (2021). CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J 11, 69. 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cox MJ, Lucien F, Sakemura R, Boysen JC, Kim Y, Horvei P, Manriquez Roman C, Hansen MJ, Tapper EE, Siegler EL, et al. (2021). Leukemic extracellular vesicles induce chimeric antigen receptor T cell dysfunction in chronic lymphocytic leukemia. Mol Ther 29, 1529–1540. 10.1016/j.ymthe.2020.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang WT, et al. (2018). Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 24, 563–571. 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palma M, Gentilcore G, Heimersson K, Mozaffari F, Nasman-Glaser B, Young E, Rosenquist R, Hansson L, Osterborg A, and Mellstedt H (2017). T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica 102, 562–572. 10.3324/haematol.2016.151100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Bruggen J, Martens A, Fraietta JA, Hofland T, Tonino SH, Eldering E, Levin MD, Siska PJ, Endstra S, Rathmell JC, et al. (2019). Chronic Lymphocytic leukemia cells imapir mitochondrial fitness in CD8+ T cells and impede CAR T cell efficacy. Blood 134, 44–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kong W, Dimitri A, Wang W, Jung IY, Ott CJ, Fasolino M, Wang Y, Kulikovskaya I, Gupta M, Yoder T, et al. (2021). BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia. J Clin Invest 131. 10.1172/JCI145459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. The New England Journal of Medicne 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X, et al. (2016). Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor–modified T cells. Science Tranlational Medicine 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. (2015). Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 373, 23–34. 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, and Allison JP (2017). Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 170, 1120–1133 e1117. 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. (2010). Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. New Eng. J. Med. 363, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serganova I, Moroz E, Cohen I, Moroz M, Mane M, Zurita J, Shenker L, Ponomarev V, and Blasberg R (2017). Enhancement of PSMA-Directed CAR Adoptive Immunotherapy by PD-1/PD-L1 Blockade. Mol Ther Oncolytics 4, 41–54. 10.1016/j.omto.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sang W, Wang X, Geng H, Li T, Li D, Zhang B, Zhou Y, Song X, Sun C, Yan D, et al. (2022). Anti-PD-1 Therapy Enhances the Efficacy of CD30-Directed Chimeric Antigen Receptor T Cell Therapy in Patients With Relapsed/Refractory CD30+ Lymphoma. Front Immunol 13, 858021. 10.3389/fimmu.2022.858021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin Y, Boesteanu AC, Binder ZA, Xu C, Reid RA, Rodriguez JL, Cook DR, Thokala R, Blouch K, McGettigan-Croce B, et al. (2018). Checkpoint Blockade Reverses Anergy in IL-13Ralpha2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol Ther Oncolytics 11, 20–38. 10.1016/j.omto.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adusumilli PS, Zauderer MG, Riviere I, Solomon SB, Rusch VW, O’Cearbhaill RE, Zhu A, Cheema W, Chintala NK, Halton E, et al. (2021). A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov 11, 2748–2763. 10.1158/2159-8290.CD-21-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chong EA, Alanio C, Svoboda J, Nasta SD, Landsburg DJ, Lacey SF, Ruella M, Bhattacharyya S, Wherry EJ, and Schuster SJ (2022). Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood 139, 1026–1038. 10.1182/blood.2021012634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li AM, Hucks GE, Dinofia AM, Seif AE, Teachey DT, Baniewicz D, Callahan C, Fasano C, McBride B, Gonzalez V, et al. (2018). Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 132, 556–556. 10.1182/blood-2018-99-112572. [DOI] [Google Scholar]
- 29.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, and Vale RD (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki IM, and Okazaki T (2019). PD-1 Primarily Targets TCR Signal in the Inhibition of Functional T Cell Activation. Front Immunol 10, 630. 10.3389/fimmu.2019.00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren J, Liu X, Fang C, Jiang S, June CH, and Zhao Y (2017). Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res 23, 2255–2266. 10.1158/1078-0432.CCR-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, and Marson A (2017). CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep 7, 737. 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J, Luo C, Wang Y, Guo Y, Dai H, Tong C, Ti D, Wu Z, and Han W (2019). PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J Immunother Cancer 7, 209. 10.1186/s40425-019-0685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi L, Meng T, Zhao Z, Han J, Zhang W, Gao F, and Cai J (2017). CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene 636, 36–41. 10.1016/j.gene.2017.09.010. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W, Shi L, Zhao Z, Du P, Ye X, Li D, Cai Z, Han J, and Cai J (2019). Disruption of CTLA-4 expression on peripheral blood CD8 + T cell enhances anti-tumor efficacy in bladder cancer. Cancer Chemother Pharmacol 83, 911–920. 10.1007/s00280-019-03800-x. [DOI] [PubMed] [Google Scholar]
- 36.Simon B, Harrer DC, Schuler-Thurner B, Schaft N, Schuler G, Dorrie J, and Uslu U (2018). The siRNA-mediated downregulation of PD-1 alone or simultaneously with CTLA-4 shows enhanced in vitro CAR-T-cell functionality for further clinical development towards the potential use in immunotherapy of melanoma. Exp Dermatol 27, 769–778. 10.1111/exd.13678. [DOI] [PubMed] [Google Scholar]
- 37.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science 367. 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, Huang M, Yi X, Liang M, Wang Y, et al. (2020). Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med 26, 732–740. 10.1038/s41591-020-0840-5. [DOI] [PubMed] [Google Scholar]
- 39.Wellhausen N, Agarwal S, Rommel PC, Gill SI, and June CH (2022). Better living through chemistry: CRISPR/Cas engineered T cells for cancer immunotherapy. Curr Opin Immunol. 74, 76–84. 10.1016/j.coi.2021.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al. (2019). Defining ‘T cell exhaustion’. Nat Rev Immunol 19, 665–674. 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Good CR, Aznar MA, Kuramitsu S, Samareh P, Agarwal S, Donahue G, Ishiyama K, Wellhausen N, Rennels AK, Ma Y, et al. (2021). An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 184, 6081–6100 e6026. 10.1016/j.cell.2021.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agarwal S, Wellhausen N, Levine BL, and June CH (2021). Production of Human CRISPR-Engineered CAR-T Cells. J Vis Exp. 10.3791/62299. [DOI] [PubMed] [Google Scholar]
- 43.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, et al. (2009). Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Molecular Therapy 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minty A, Chalon P, Derocq JM, Dumont X, Guillemot JC, Kaghad M, Labit C, Leplatois P, Liauzun P, and Miloux B (1993). lnterleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature 362, 248–250. 10.1038/362248a0. [DOI] [PubMed] [Google Scholar]
- 45.Glinos DA, Soskic B, Williams C, Kennedy A, Jostins L, Sansom DM, and Trynka G (2020). Genomic profiling of T-cell activation suggests increased sensitivity of memory T cells to CD28 costimulation. Genes Immun 21, 390–408. 10.1038/s41435-020-00118-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crawford A, Angelosanto JM, Nadwodny KL, Blackburn SD, and Wherry EJ (2011). A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLoS Pathog 7, e1002098. 10.1371/journal.ppat.1002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zenke S, Sica MP, Steinberg F, Braun J, Zink A, Gavrilov A, Hilger A, Arra A, Brunner-Weinzierl M, Elling R, et al. (2022). Differential trafficking of ligands trogocytosed via CD28 versus CTLA4 promotes collective cellular control of co-stimulation. Nat Commun 13, 6459. 10.1038/s41467-022-34156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, H., T. Z., et al. (2011). Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carthon BC, Wolchok JD, Yuan J, Kamat A, Ng Tang DS, Sun J, Ku G, Troncoso P, Logothetis CJ, Allison JP, and Sharma P (2010). Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res 16, 2861–2871. 10.1158/1078-0432.CCR-10-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Qiu S, Chen J, Jiang S, Chen W, Jiang J, Wang F, Si W, Shu Y, Wei P, et al. (2020). Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 53, 456–470 e456. 10.1016/j.immuni.2020.07.011. [DOI] [PubMed] [Google Scholar]
- 51.Ise W, Kohyama M, Nutsch KM, Lee HM, Suri A, Unanue ER, Murphy TL, and Murphy KM (2010). CTLA-4 suppresses the pathogenicity of self antigen-specific T cells by cell-intrinsic and cell-extrinsic mechanisms. Nat Immunol 11, 129–135. 10.1038/ni.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klocke K, Sakaguchi S, Holmdahl R, and Wing K (2016). Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci U S A 113, E2383–2392. 10.1073/pnas.1603892113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walker LS, and Sansom DM (2011). The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol 11, 852–863. 10.1038/nri3108. [DOI] [PubMed] [Google Scholar]
- 54.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, et al. (2019). c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300. 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017). Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356 e1316. 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 56.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 24, 978–985. 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 57.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen J, Blank CU, et al. (2019). Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 176, 775–789 e718. 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684. 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 59.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. (2020). Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841 e828. 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pauken KE, and Wherry EJ (2015). Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36, 265–276. 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thommen DS, and Schumacher TN (2018). T Cell Dysfunction in Cancer. Cancer Cell 33, 547–562. 10.1016/j.ccell.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, Klysz D, et al. (2021). Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372. 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yan Q, Zhang B, Ling X, Zhu B, Mei S, Yang H, Zhang D, Huo J, and Zhao Z (2022). CTLA-4 Facilitates DNA Damage-Induced Apoptosis by Interacting With PP2A. Front Cell Dev Biol 10, 728771. 10.3389/fcell.2022.728771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCutcheon SR, Swartz AM, Brown MC, Barrera A, McRoberts Amador C, Siklenka K, Humayun L, Isaacs JM, Reddy TE, Nair S, et al. (2023). Orthogonal CRISPR screens to identify transcriptional and epigenetic regulators of human CD8 T cell function. bioRxiv. 10.1101/2023.05.01.538906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, Gao P, Bandyopadhyay S, Sun H, Zhao Z, et al. (2022). Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature 602, 503–509. 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Condomines M, Arnason J, Benjamin R, Gunset G, Plotkin J, and Sadelain M (2015). Tumor-Targeted Human T Cells Expressing CD28-Based Chimeric Antigen Receptors Circumvent CTLA-4 Inhibition. PLoS One 10, e0130518. 10.1371/journal.pone.0130518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walker LS, and Sansom DM (2015). Confusing signals: recent progress in CTLA-4 biology. Trends Immunol 36, 63–70. 10.1016/j.it.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kennedy A, Waters E, Rowshanravan B, Hinze C, Williams C, Janman D, Fox TA, Booth C, Pesenacker AM, Halliday N, et al. (2022). Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat Immunol 23, 1365–1378. 10.1038/s41590-022-01289-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Teft WA, Kirchhof MG, and Madrenas J (2006). A molecular perspective of CTLA-4 function. Annu Rev Immunol 24, 65–97. 10.1146/annurev.immunol.24.021605.090535. [DOI] [PubMed] [Google Scholar]
- 70.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC, et al. (2014). Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res 20, 4262–4273. 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khattri R, Auger JA, Griffin MD, Sharpe AH, and JA B. (1999). Lymphoproliferative Disorder in CTLA-4 Knockout Mice Is Characterized by CD28-Regulated Activation of Th2 Responses. The Journal of Immunology 162, 5784–5791. [PubMed] [Google Scholar]
- 72.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, and AH S. (1995). Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547. [DOI] [PubMed] [Google Scholar]
- 73.Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, Arancibia-Carcamo CV, Sobel RA, Rudensky AY, Kuchroo VK, et al. (2015). Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med 212, 1603–1621. 10.1084/jem.20141030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, et al. (2014). Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345, 1623–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schäffer AA, Grüning BA, et al. (2014). Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat med 20, 1410–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walker LSK (2022). The link between circulating follicular helper T cells and autoimmunity. Nat Rev Immunol. 10.1038/s41577-022-00693-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pai CS, Huang JT, Lu X, Simons DM, Park C, Chang A, Tamaki W, Liu E, Roybal KT, Seagal J, et al. (2019). Clonal Deletion of Tumor-Specific T Cells by Interferon-gamma Confers Therapeutic Resistance to Combination Immune Checkpoint Blockade. Immunity 50, 477–492 e478. 10.1016/j.immuni.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J, Goswami S, and Allison JP (2021). The Next Decade of Immune Checkpoint Therapy. Cancer Discov 11, 838–857. 10.1158/2159-8290.CD-20-1680. [DOI] [PubMed] [Google Scholar]
- 79.Dotsch S, Svec M, Schober K, Hammel M, Wanisch A, Gokmen F, Jarosch S, Warmuth L, Barton J, Cicin-Sain L, et al. (2023). Long-term persistence and functionality of adoptively transferred antigen-specific T cells with genetically ablated PD-1 expression. Proc Natl Acad Sci U S A 120, e2200626120. 10.1073/pnas.2200626120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kalinin RS, Ukrainskaya VM, Chumakov SP, Moysenovich AM, Tereshchuk VM, Volkov DV, Pershin DS, Maksimov EG, Zhang H, Maschan MA, et al. (2021). Engineered Removal of PD-1 From the Surface of CD19 CAR-T Cells Results in Increased Activation and Diminished Survival. Front Mol Biosci 8, 745286. 10.3389/fmolb.2021.745286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang OY, Tian L, Yoder T, Xu R, Kulikovskaya I, Gupta M, Melenhorst JJ, Lacey SF, O’Rourke DM, and Binder ZA (2022). PD1 Expression in EGFRvIII-Directed CAR T Cell Infusion Product for Glioblastoma Is Associated with Clinical Response. Front Immunol 13, 872756. 10.3389/fimmu.2022.872756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Odorizzi PM, Pauken KE, Paley MA, Sharpe A, and Wherry EJ (2015). Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med 212, 1125–1137. 10.1084/jem.20142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhong XS, Matsushita M, Plotkin J, Riviere I, and Sadelain M (2010). Chimeric antigen receptors combining 4–1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther 18, 413–420. 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, Eyquem J, Zhao Z, Whitlock BM, Miele MM, et al. (2019). CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 568, 112–116. 10.1038/s41586-019-1054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, Buttar C, Li E, Sundberg B, Salas RD, et al. (2020). PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol 21, 442–454. 10.1038/s41590-020-0620-x. [DOI] [PubMed] [Google Scholar]
- 86.Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, and Okazaki T (2019). Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364, 558–566. [DOI] [PubMed] [Google Scholar]
- 87.Kennedy A, Robinson MA, Hinze C, Waters E, Williams C, Halliday N, Dovedi S, and Sansom DM (2023). The CTLA-4 immune checkpoint protein regulates PD-L1:PD-1 interaction via transendocytosis of its ligand CD80. EMBO J 42, e111556. 10.15252/embj.2022111556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao Y, Caron C, Chan YY, Lee CK, Xu X, Zhang J, Masubuchi T, Wu C, Bui JD, and Hui E (2023). cis-B7:CD28 interactions at invaginated synaptic membranes provide CD28 co-stimulation and promote CD8(+) T cell function and anti-tumor immunity. Immunity. 10.1016/j.immuni.2023.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kutner RH, Zhang XY, and Reiser J (2009). Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 4, 495–505. 10.1038/nprot.2009.22. [DOI] [PubMed] [Google Scholar]
- 90.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V, et al. (2015). Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 7, 303ra139. 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nobles CL, Reddy S, Salas-McKee J, Liu X, June CH, Melenhorst JJ, Davis MM, Zhao Y, and Bushman FD (2019). iGUIDE: an improved pipeline for analyzing CRISPR cleavage specificity. Genome Biol 20, 14. 10.1186/s13059-019-1625-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hannon M, Lechanteur C, Lucas S, Somja J, Seidel L, Belle L, Bruck F, Baudoux E, Giet O, Chantillon AM, et al. (2014). Infusion of clinical-grade enriched regulatory T cells delays experimental xenogeneic graft-versus-host disease. Transfusion 54, 353–363. 10.1111/trf.12279. [DOI] [PubMed] [Google Scholar]
- 96.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Osorio D, and Cai JJ (2021). Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control. Bioinformatics 37, 963–967. 10.1093/bioinformatics/btaa751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Full list of DEGs between day 0 and day 15 CAE WT CART19 cells from scRNA-seq experiment, related to Figure 6C.
Table S2. Full list of DEGs between day 0 WT and CTLA4-deleted CART19 product in patients from scRNA-seq experiment, related to Figure 6F.
Table S3. Full list of Ingenuity pathway Analysis pathways enriched in DEGs between day 0 WT and CTLA4-deleted CART19 product from scRNA-seq experiment, related to Figure 6G.
Table S4. Full list of DEGs between day 18 WT and CTLA-4-deleted CART19 cells in patients related to Figure 6H; and day 15 WT and CTLA4-deleted CART19 cells in donors related to Figure S6E from scRNA-seq experiment.
Table S5. Full list of Ingenuity pathway Analysis pathways enriched in DEGs (1) between day 18 and day 0 CTLA4-deleted CART19 (related to Figure 6I); and between day 18 and day 0 WT CART19 cells (related Figure 6J) in patients; and (2) between day 15 and day 0 CTLA4-deleted CART19 (related to Figure S6G); and between day 15 and day 0 WT CART19 cells (related Figure S6H) in donors from scRNA-seq experiment.
Table S6. All CD28 targets predicted by IPA (upstream regulator analysis) using DEGs between day 15 and day 0 CTLA4-deleted CART19 in donors; and between day 18 and day 0 WT CART19 cells in patients, from scRNA-seq experiment, related to Figure 7B.
Table S7. DEGs between day 0 and day 15 CAE CART19 cells in each group from nCounter-based readout, related to Figure 7D, and Figure 7E.
Data Availability Statement
All associated raw genomics data from scRNA-seq experiments have been deposited at database of genotypes and phenotypes (dbGaP). They are available upon request if access is granted as of the date of publication. Accession number is listed in the key resource table.
This paper does not report original code.
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-human CD45 | Biolegend | Cat# 304032 |
| anti-human CD45 | Biolegend | Cat# 304028 |
| anti-human CD3 | Biolegend | Cat# 317322 |
| anti-human CD8 | Biolegend | Cat# 301066 |
| anti-human CD8 | Biolegend | Cat# 301016 |
| anti-human CD8 | BD Pharmingen | Cat# 560179 |
| anti-human CD4 | Biolegend | Cat# 317440 |
| anti-human CD4 | Biolegend | Cat# 317414 |
| anti-human CD28 | Biolegend | Cat# 302926 |
| anti-human CD38 | eBioscience | Cat# 25-0388-42 |
| anti-human CD39 | Biolegend | Cat# 328228 |
| anti-human PD-1 | Biolegend | Cat# 329928 |
| anti-human PD-1 | Biolegend | Cat# 329918 |
| anti-human TIM3 | eBioscience | Cat# 46-3109-42 |
| anti-human LAG3 | eBioscience | Cat# 11-2239-42 |
| anti-human CTLA-4 | Invitrogen | Cat# 12-159-42 |
| anti-human CD45RO | Biolegend | Cat# 304244 |
| anti-human CD45RO | BD Pharmingen | Cat# 555493 |
| anti-human CCR7 | BD Pharmingen | Cat# 130-126-090 |
| anti-human CD107a | Biolegend | Cat# 328618 |
| anti-human IL-2 | Invitrogen | Cat# 46-7029-42 |
| anti-human TNFa | Biolegend | Cat# 502928 |
| anti-human GM-CSF | BD Horizon | Cat# 562930 |
| anti-human IFNg | Biolegend | Cat# 506507 |
| anti-human Granzyme B | BD Horizon | Cat# 562462 |
| anti-human Ki67 | Biolegend | Cat# 350526 |
| anti-human CD80 | Biolegend | Cat# 305230 |
| anti-human CD86 | Biolegend | Cat# 305442 |
| anti-human ICOS | Biolegend | Cat# 313536 |
| anti-mouse CD45 | Biolegend | Cat# 103116 |
| anti-mouse FMC63 scFv MAb | Bioswan | Cat# 200106 |
| anti-mouse FMC63 scFv MAb | Bioswan | Cat# 200102 |
| BCMA CAR Detection | Miltenyi Biotec | Cat# 130-126-090 |
| BTK/ITK (Tyr551, Tyr511) Monoclonal Antibody (M4G3LN) | Thermo Fisher | Cat#12-9015-42 |
| Phospho-LCK (Tyr505) monoclonal antibody (SRRCHA) | Thermo Fisher | Cat#50-9076-42 |
| Phospho-ZAP70/Syk (Tyr319, Tyr352) Monoclonal Antibody (n3kobu5) | Thermo Fisher | Cat#12-9006-42 |
| mouse IgG2a, κ Isotype control Antibody | Biolegend | Cat# 400269 |
| mouse IgG1, κ Isotype control Antibody | Biolegend | Cat# 400126 |
| mouse IgG1, κ Isotype control Antibody | Biolegend | Cat# 400168 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | C7373-03 |
| Biological samples | ||
| T lymphocytes from human healthy donors | UPenn Human Immunology Core | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Live/Dead Aqua | ThermoFisher | Cat# L34957 |
| Zombie NIR (Biolegend) Fixable Viability Kits | Biolegend | Cat# 423106 |
| Apotracker™ Green | Biolegend | Cat# 427403 |
| Annexin V with 7AAD | Biolegend | Cat# 640930 |
| Alt-R® Cas9 Electroporation Enhancer, 10 nmol | Integrated DNA Technologies | Cat# 1075916 |
| SpyFi Cas9 | Aldeveron | Cat# 9214 |
| P3 Primary cell 4D-nucleofactor X Kit L | Lonza | Cat# V4XP-3024 |
| OpTmizer T Cell Expansion SFM | Gibco | Cat# A1048501 |
| Human AB Serum | GeminiBio | Cat#100-512 |
| Recombinant Human IL-7 | Peprotech | Cat#200-07 |
| Recombinant Human IL-15 | Peprotech | Cat#200-15 |
| DNAse I roche | Sigma | Cat#10104159001 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#1166500 |
| DNase I from bovine pancreas | Sigma | Cat# 11284932001 |
| EDTA (0.5 M), pH 8.0, RNase-free | Thermo Fisher | Cat# AM9261 |
| PE Streptavidin | BD Pharmingen | Cat# 554061 |
| APC Streptavidin | Biolegend | Cat# 405235 |
| Golgi Stop protein transport inhibitor (containing monensin | Thermo Fisher | Cat# BD554724 |
| Cell stimulation cocktail (PMA/Ionomycin) | Thermo Fisher | Cat#00497093 |
| Critical commercial assays | ||
| RNA Clean & Concentrator™-5 | ZYMO | Cat#R1016 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69504 |
| NovaSeq6000 S2-100 cycle kit | Illumina | Cat#20028316 |
| Chromium Next GEM Single Cell 3' GEM, Library & Gel Bead Kit v3.1 | 10X Genomics | Cat#1000121 |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | Cat#1000120 |
| Chromium Next GEM Single Cell 3’ Library Construction Kit v3.1 | 10X Genomics | Cat#1000157 |
| Single Index Kit T Set A | 10X Genomics | Cat#1000213 |
| XT_HS_CART Panel_CSO | Nanostring | Cat# 115000343 |
| Master Kit-12 rxns | Nanostring | Cat# 100052 |
| DynaBeads CD3x28 (Human) | ThermoFisher | Cat# 11131D |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | Cat# 74609.50 |
| QIAGEN Plasmid Plus Maxi Kit | QIAGEN | Cat# 12963 |
| Fixation Medium (Medium A) | Invitrogen | Cat# GAS001S100 |
| Permeabilization Medium (Medium B) | Invitrogen | Cat# GAS002S100 |
| CountBright Absolute Counting Beads, (ThermoFisher) | Thermo Fisher | Cat# C36950 |
| LongAmp™ Taq 2X Master Mix | New England Biolabs | Cat# M0287S |
| Vacuum Filter/Storage Systems | Corning | Cat# 430770 |
| Dead cell removal kit | Miltenyi Biotec | Cat# 130-090-101 |
| CliniMACS CD4 GMP Microbeads | Miltenyi Biotec | Cat# 170-076-702 |
| CliniMACS CD4 GMP Microbeads | Miltenyi Biotec | Cat# 170-076-703 |
| LOVO Pouch Disposable Kit | Fenwal | Cat# X6R4907 |
| LOVO Processing kit w/ Acess | Fenwal | Cat# X6R4909 |
| Deposited data | ||
| Raw and analyzed data | This paper | dbGAP: phs001707.v3 |
| Experimental models: Cell lines | ||
| Human (female) HEK293T | ATCC | CRL-11268 |
| Human (female) NALM6 | ATCC | CRL-3273 |
| Human (female) MM.1S | ATCC | CRL-2974 |
| Experimental models: Organisms/strains | ||
| NOD/scid/IL2rγ−/− (NSG) | Jackson Laboratory | Cat# 5557 |
| Oligonucleotides | ||
| PD-1 sgRNA: 5’ GGCCAGGATGGTTCTTAGGT3’ | Integrated DNA Technologies | (Ren et al, 2017) |
| CTLA-4 sgRNA: 5’ TATGCCCAGGTAGTATGG3’ | Integrated DNA Technologies | N/A |
| PD-1.PCR.F (gDNA): 5’ AGTTTCCCTTCCGCTCACCTC3’ | Genewiz | N/A |
| PD-1.PCR.R (gDNA): 5’ ACTAACCTTGGCTTTACGACGT3’ | Genewiz | N/A |
| PD-1.Seq.R (PCR): 5’ TGGCAGCCCAGGGGTC3’ | Genewiz | N/A |
| CTLA-4.PCR.F (gDNA): 5’GGACATGGGGGAAGTGTGAC3’ | Genewiz | N/A |
| CTLA-4.PCR.R (gDNA): 5’AGGTTTACTTTTAGGACTGTGGACA3’ | Genewiz | N/A |
| CTLA-4.Seq.F (PCR): 5’ TGTTCTTCCTGCCACAACCA3’ | Genewiz | N/A |
| CTLA-4.Seq.R (PCR): 5’ GACACCTGTTGCATTGCAGTC3’ | Genewiz | N/A |
| Recombinant DNA | ||
| pTRPE CD19BBz | This paper | N/A |
| pTRPE BCMABBz | This paper | N/A |
| Software and algorithms | ||
| R version 4.1.0 | CRAN | https://cran.r-project.org/ |
| Seurat_4.1.0 | (Butler et al., 2018; Stuart et al., 2019) | https://satijalab.org/seurat/ |
| Cell Ranger v4.0 | 10X Genomics | https://www.10xgenomics.com/ |
| sctransform_0.3.3 | (Hafemeister and Satija, 2019) | https://cran.r-project.org/web/packages/sctransform/index.html |
| DESeq2 1.38.3 | Love et al, 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| EdgeR 3.40.2 | Robinson et al, 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| FlowJo™ v10.8 Software | BD Life Sciences | https://www.flowjo.com |
| Ingenuity Pathway Analysis Software | QIAGEN | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Other | ||
| Benchling sgRNA designer tool (https://www.benchling.com, [2020]) | Benchling | https://benchling.com/ |
| BioRender illustration design tool | BioRender | https://biorender.com/ |
| TIDE: Tracking of Indels by DEcompositionTool | TIDE | http://shinyapps.datacurators.nl/tide/ |
| Gene Set enrichment Analysis Software | GSEA | https://www.gsea-msigdb.org/gsea/index.jsp |