

Abstract
Introduction:
Type 1 diabetes (T1D) is an autoimmune disease in which pro-inflammatory and cytotoxic signaling drive the death of the insulin-producing β cells. This complex signaling is regulated in part by fatty acids and their bioproducts, making them excellent therapeutic targets.
Areas covered:
We provide an overview of the fatty acid actions on β cells by discussing how they can cause lipotoxicity or regulate inflammatory response during insulitis. We also discuss how diet can affect the availability of fatty acids and disease development. Finally, we discuss development avenues that need further exploration.
Expert opinion:
Fatty acids, such as hydroxyl fatty acids, ω-3 fatty acids and their downstream products, are druggable candidates that promote protective signaling. Inhibitors and antagonists of enzymes and receptors of arachidonic acid and free fatty acids, along with their derived metabolites, which cause pro-inflammatory and cytotoxic responses, have the potential to be developed as therapeutic targets also. Further, because diet is the main source of fatty acid intake in humans, balancing protective and pro-inflammatory/cytotoxic fatty acid levels through dietary therapy may have beneficial effects, delaying T1D progression. Therefore, therapeutic interventions targeting fatty acid signaling hold potential as avenues to treat T1D.
Keywords: type 1 diabetes, cell signaling, lipid mediators, saturated fatty acids, ω-6 fatty acids, ω-3 fatty acids, β-cell death, β-cell protection, therapeutic targets
Graphical Abstract

1. Introduction
Type 1 diabetes (T1D) is a chronic metabolic condition that is estimated to affect 8.4 million people worldwide [1]. It is caused by autoimmune destruction of the insulin-producing β cells of the islets of Langerhans in the pancreas [2]. This islet autoimmune (IA) response, also known as insulitis, is characterized by islet inflammation and the appearance of circulating autoantibodies against β-cell proteins [3]. Many systemic and cellular components have been associated with T1D development including immune cell signaling, ER stress and lipids. Although lipids are recognized as structural entities of cellular membranes and energy storage molecules, they act as inflammatory mediators in T1D development [4]. This is supported by the recent findings of a pro-inflammatory lipid signature in children at high-risk for developing T1D and in the T1D non-obese diabetic (NOD) mouse model at the disease onset [5]. Lipids are also T1D biomarkers [6–8]. For example, triacylglycerol and phosphatidylcholine levels are reduced in plasma prior to the onset of T1D [6] along with lipoprotein-associated proteins that transport them [9]. There is also an increase in circulating free fatty acids (FFAs) [8], suspected to have a pathological/negative effect in islets during T1D development. Besides the FFAs, many inflammatory mediators are fatty acids or oxidized products of fatty acids, making them excellent targets for therapeutic development. In fact, fatty acid signaling is the target for many inflammatory medicines, including popular over-the-counter drugs such as aspirin and acetaminophen [10,11].
Structurally, fatty acids are carboxylic acids of different aliphatic hydrocarbon chain lengths (Figure 1) [12]. These aliphatic chains can be further modified by methylation, desaturation, oxidation and acylation [12], extending their repertoire of biological roles. Structurally fatty acid modifications are as important as the aliphatic chain length in determining the fatty acid physicochemical proprieties and biological role. Methylation of fatty acids, for example, leads to chain branching and can add stereoisomerism [13]. Beyond methylation, fatty acids can be hydroxylated and further acylated into fatty acid esters of hydroxy fatty acids (FAHFAs), such as the palmitic acid esters of hydroxy stearic acids (PAHSAs). Aliphatic chains can also undergo desaturation, a modification that adds double bonds to the aliphatic fatty acid chains by enzymes named desaturases. This modification of fatty acid chains has a major impact on their physicochemical properties. Double bonds result in sterically distinct trans or cis configurations (Figure 1) [12]. Whereas trans double bonds impart a linear structure to fatty acids, cis double bonds induce curvature and can increase the fluidity of membranes [12]. Unsaturated fatty acids are named according to the double bond position relative to the carboxyl end (Δ), or to the methyl terminus (ω) (Figure 1) [12]. Fatty acid desaturases add double bonds in the fatty acid’s carbon chain, and they are named based on the position of the Δ carbon undergoing dehydrogenation. For instance, the fatty acid Δ-9 desaturase adds a double bond between the C9 and C10 from the carboxyl end (Figure 1) [14]. Fatty acids can also be classified based on the position of the double bound. For example, ω-3 and ω-6 fatty acids are important regulators of the immune response. These fatty acids are prone to both non-enzymatic and enzymatic oxidation. Oxidized products of ω-6 fatty acids, such as pro-inflammatory leukotrienes [15] are considered pharmacological targets for treating inflammation. There are also anti-inflammatory fatty acids, such as ω-3 fatty acids, their derivates, and FAHFAs. In this review, we provide insights to explore fatty acids, their receptors, and related enzymes as potential targets for T1D treatment/prevention, describing their roles and mechanisms in the context of toxic and beneficial effects on β cells. We focused this article on T1D because despite T2D having a similar condition of inability of controlling the blood glucose levels, their mechanisms of development are completely different. Furthermore, fatty acids as potential targets for T2D therapies have been reviewed in numerous publications [16–19].
Figure 1 – Structure and nomenclature of fatty acids.

The figure shows examples of A) saturated, B) branched and, D) unsaturated fatty acids. An example of desaturation reaction is shown in D). Double bonds are named based on their position relative to the Δ carbon or classes of unsaturated fatty acids are named based on the position of the ω carbon. “E” or “Z” is designated to specify the trans and cis stereochemistry, respectively. Abbreviations: CoA, Coenzyme A; FAHFA, fatty acid esters of hydroxy fatty acid.
2. Polyunsaturated fatty acids (PUFAs) and the regulation of inflammatory response
In insulitis, bioactive lipids are generated in β cells by activation of phospholipases A2 (PLA2s), including the inducible calcium-independent PLA2 beta (iPLA2β) [20]. The PLA2s hydrolyze the stereospecific numbering (sn)-2 substituent of membrane glycerophospholipids to generate FFAs and lysophospholipids (Figure 2). The resulting FFAs can be metabolized into pro- or anti-inflammatory lipid mediators [5,21,22]. Reducing the expression of iPLA2β significantly decreases the production of pro-inflammatory lipids and insulitis, leading to preservation of β-cell mass and reduced T1D onset in NOD mice. Treating mice with the reversible iPLA2β inhibitor FKGK18 also shows a similar effect [21]. A prominent fatty acid released by iPLA2β is the ω-6 fatty acid arachidonic acid (AA), which upon hydrolysis can be oxidized by cyclooxygenases (COX), lipoxygenases (LOX) and the superfamily of cytochrome P450 (CYP450) to generate oxidized pro-inflammatory lipids named eicosanoids [23] (Figure 2). Eicosanoids induce immune responses [24] and reduce inflammation-resolving processes [25,26]. For instance, pro-inflammatory eicosanoids induce macrophage phagocytosis, adhesion, and apoptosis, and further amplify eicosanoid release, and promote β-cell death in T1D development [2,27,28].
Figure 2. Arachidonic acid pathway.

The pathway shows examples of bioactive products of arachidonic acid oxidation along with their binding receptors. Receptors are colored based on their pro- or anti-inflammatory activity or both. Agonists (blue), inhibitors/antagonists (red), and drug candidates (underlined) are highlighted. Abbreviations: ALX, lipoxin A4 receptor; COX, cyclooxygenase; CYP, cytochrome P450; CysLT1, cysteinyl leukotriene receptor 1; CysLT2, cysteinyl leukotriene receptor 2; DHET, dihydroxyeicosatrienoic acid; DP, prostaglandin D2 receptor; EET, epoxyeicosatrienoic acid; EP1, prostaglandin E2 receptor 1; EP2, prostaglandin E2 receptor 2; EP3, prostaglandin E2 receptor 3; EP4, prostaglandin E2 receptor 4; FP, prostaglandin E2 receptor; FPR2, formyl peptide receptor 2; HETE, hydroxyeicosatetraenoic acid; IP, protacylcin receptor; iPLA2β; Calcium-independent phospholipase A2β; LT, leukotriene; LTB4R; leukotriene B4 receptor; LTB4R2, leukotriene B4 receptor 2; LOX, lipoxygenase; NOX, NADPH oxidase; PG, prostraglandin; TP, thromboxane A2 receptor.
The LOX enzymes oxidize AA to produce leukotrienes and hydroxyeicosatetraenoic acids (HETEs) (Figure 2) [29]. In the context of T1D, these products trigger inflammation and attract immune cells to induce β-cell death [20]. The LOX products induce p38 kinase, reactive oxygen species (ROS)-generating nicotinamide adenine dinucleotide phosphate (NAPDH) oxidase (NOX), pro-inflammatory cytokine production, migration of leukocytes, and β-cell death [30–35]. Inhibition of NOX-1 by the chemical inhibitors ML171 or GKT137831 reduces diabetes onset in NOD mice [36] (Figure 4). 12-LOX is detectable in islet cells in recent-onset individuals [37]. Global 12-Lox-null NOD mice have reduced T1D incidence by downregulating macrophage production of the pro-inflammatory cytokines IL-1β, TNF-α and IFN-γ [38]. 12-Lox deletion specific in NOD mouse β cells reduces insulitis and results in almost complete protection from T1D [39]. Similar findings were observed with macrophage-specific deletion of the 12-Lox [40]. Chemical inhibitors of 12/15-LOX ML127, ML355 and ML351, also afford protection by reducing oxidative stress and dysglycemia [41,42].
Figure 4 – Free fatty acid pathway.
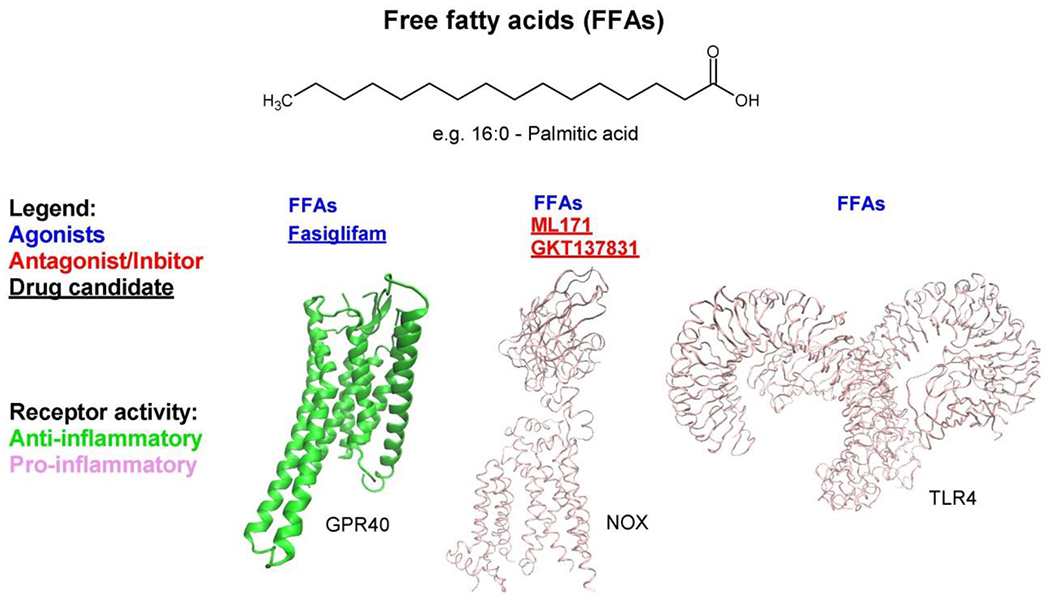
The pathway shows an example of bioactive lipid, palmitic acid, along with binding receptors and an enzyme. Receptors and enzymes are colored based on their pro-inflammatory activity. Agonists (blue), inhibitors (red), and drug candidates (underlined) are highlighted. Abbreviations: FFAs, free fatty acids; GPR40, G-protein coupled receptor 40; NOX, NADPH oxidase; TLR4, Toll-like receptor 4.
The pro-inflammatory lipids dihydroxyeicosatrienoic acids (DHETs) are generated by metabolism of AA by CYP450/soluble epoxide hydrolase (sEH) enzymes (Figure 2). Oxidation of polyunsaturated fatty acids by CYP450 enzymes also generates epoxy fatty acids (EpFAs), designated epoxyeicosatrienoic acids (EETs) (Figure 2), that are anti-inflammatory and lead to resolution of inflammation [43]. However, sEH converts EETs into pro-inflammatory DHETs (Figure 2) [44]. Increases in DHETs are accompanied by decreases in EETs, which together serve to amplify a pro-inflammatory landscape [45]. Inhibitors of sEH, such as t-AUCB [46] and TPPU [47], reduce mice islets apoptosis or dysfunction. Another sEH inhibitor, GSK2256294, has been proposed to target diabetes [48].
The COX enzymes catalyze the formation of cyclic carbon rings on lipids to generate prostaglandins (PG) and thromboxanes (Figure 2). For instance, the pro-inflammatory prostaglandin E2 (PGE2) reduces cellular debris clearance [49] and apoptotic defects in macrophages and dendritic cells of NOD mice [50] in insulitis [51]. Additional PGs of potential relevance in T1D are the F-series prostaglandins (PGF), specifically PGF2α and 8-iso-PGF2α. The PGF2α is generated in almost all tissues, and it signals through PGF2α receptors (FPRs), being a biomarker of inflammation [52,53]. Clinical studies reveal that PGF2α is the most predominant PG formed at inflammatory sites [54]. Binding of prostaglandin F2α (PGF2α) to FPRs triggers multiple pathways, including NF-κB, and leads to the production of chemokines and pro-inflammatory cytokines [55]. Inflammation and oxidative stress pathways are integral to events that lead to T1D-associated β-cell death [56,57], and decreases in PGF2α and 8-iso-PGF2α signaling are associated with reduced inflammation and oxidative stress and consequential amelioration of inflammatory pathologies [55,58]. Taken together, PGF2α and 8-iso-PGF2α signaling could play important roles in T1D development [5,21,41]. The COX-2 inhibitors, NS-398 and SC-236, prevent IL-1β inhibition of insulin secretion by blocking PGE2 synthesis [59]. Moreover, the PGE2 receptor EP4 antagonist grapiprant reduced insulitis in NOD mice [60].
In response to inflammation, various resolution processes also evolve. A component of these is an increased production of resolving bioactive lipids that are derived from metabolism of PUFAs: AA (lipoxins), and the ω-3 fatty acids eicosapentaenoic acid - EPA (E-series resolvins) and docosahexaenoic acid - DHA (D-series resolvins, maresins, and protectins) (Figures 2 and 3). Collectively, they are referred to as specialized resolving lipids mediators [61,62], and they can also be explored as therapeutic targets. Specialized resolving lipids mediators counter-regulate the early initiators (PGs and LTs) of acute inflammation, leading to inhibition of proinflammatory cytokines and upregulation of anti-inflammatory cytokines (e.g., IL-10) [63]. For instance, they participate in the switching from pro-inflammatory phenotype M1 state of macrophages to anti-inflammatory phenotype M2 [23]. In humans, PGE2 induces LOX-class switch from LTB4 to lipoxins, which represents a stop signal for polymorphonuclear cell recruitment and initiation of a resolution phase that promotes an anti-inflammatory macrophage phenotype and function (i.e., phagocytosis) [62]. Selexipag (Synonyms: NS-304; ACT-293987) an oral prodrug deriving in PGI2 receptor agonist [64], improves β-cell function in NOD mice [65]. Resolvins, in particular, exhibit a bi-pronged mode of effectiveness in reducing inflammation and promoting resolution, as evidenced by clearance of debris (i.e., antigens) from inflamed sites [66] in several inflammatory disorders [67]. Resolvins have strong therapeutic potential since they are natural lipids, potent at nanomolar levels, and devoid of causing systemic toxicity. To date, six D-series resolvins (RD1-6, RE1-4) have been reported to be highly potent in vivo and are at various phases of clinical trials due it anti-inflammatory effects [68].
Figure 3 – ω-3 fatty acid pathway.
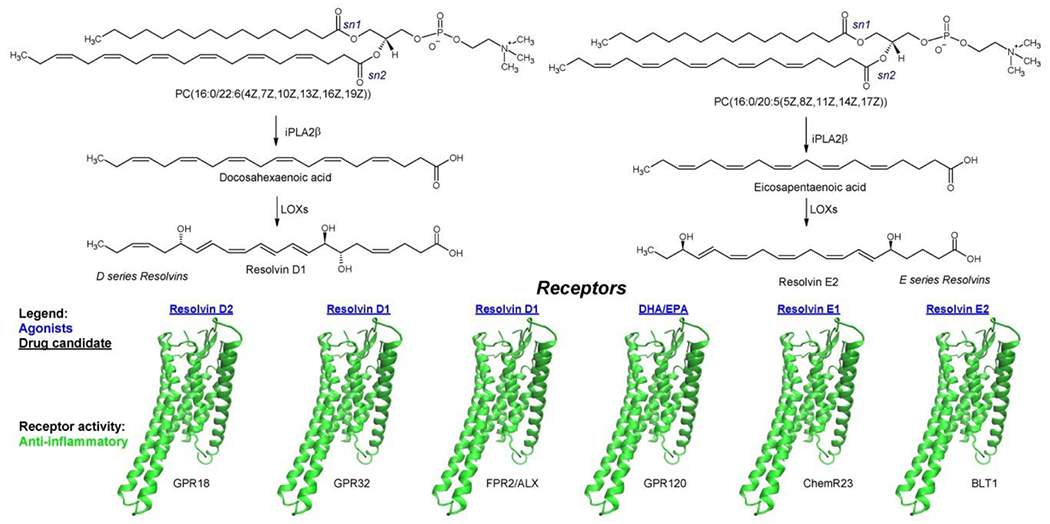
The pathway shows examples of bioactive lipids of ω-3 fatty acid pathway along with their binding receptors. Agonists (blue), and drug candidates (underlined) are highlighted. Receptors are colored based on their pro-inflammatory activity. Abbreviations: ALX, lipoxin A4 receptor; BLT1, leukotriene B4 receptor 1; ChemR23, Chemerin-like receptor 1; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; FPR2, formyl peptide receptor 2; GPR18, G-protein coupled receptor 18; GPR32, G-protein coupled receptor 32; GPR120, G-protein coupled receptor 120; iPLA2β; Calcium-independent phospholipase A2β; LOX, lipoxygenase.
The ω-3 fatty acids induce anti-inflammatory responses by activating the G-protein coupled receptor GPR120 (also known as free fatty acid receptor 4 – FFA4) (Figure 3) [69,70], making this receptor an excellent target for drug discovery. β-cell death caused by infiltration of autoreactive CD8+ T cells can be prevented by restoring the T helper (Th)1/Th2 ratio balance administrating ω-3 fatty acids or resolvins, which reduce the population of Th1 cells and increase the populations of Th2 and regulatory T cells [71]. The ω-3 fatty acids also attenuate the inflammatory state of macrophages by reducing the activation of the inflammasome and production of nitric oxide [72,73]. Moreover, diet rich in ω-3 fatty acids restores the gut barrier integrity inducing immune homeostasis in NOD mice by reducing intestinal inflammation [73].
Agonists of GPR120 showed to improve glucose tolerance in diet-induced obese mice [74]. However, no clinical tests have been conducted so far. The GPR120 agonist ‘compound A’ [75], reduces the unfolded protein response (UPR) by attenuating ER stress and improving the survival and function of β cells exposed to an environment of proinflammatory cytokines [75,76]. As ‘compound A’, TUG-891 and GSK137647A are both GPR120 agonists and presented antidiabetic activity [77]. Like GPR120, resolvin receptors are also excellent targets for drug development. A compounds screening has been performed for resolvin D1 receptor (DRV1/GPR32), leading to the identification of anti-inflammatory molecules [78].
3. Effects of free fatty acids (FFAs) on β cells
Saturated FFAs have been shown to have both beneficial and detrimental effects on β-cell function. For example, in early studies in rats, depletion of intra-islet FFAs levels impaired glucose-stimulated insulin secretion, whereas their restoration led to recovery [79]. These beneficial effects, likely mediated through FFA receptor 1 (GPR40) and FFA metabolism (Figure 4) [80,81], are thought to represent physiologic roles for FFAs in the context of lipid homeostasis. By contrast, excess FFAs can promote inflammation, oxidative stress, and β-cell death [82–86]. Although better characterized in the context of type 2 diabetes (T2D), growing lines of evidence, though indirect, suggest that FFA-mediated lipotoxicity may play a role in T1D development [87]. This includes observations of (1) there is an increase in circulating levels of FFAs in individuals with IA during the pre-diabetic period frequently characterized by reduced β cell function [84,85]; (2) palmitic acid-mediated induction in human islets of several pro-inflammatory cytokines (IL-1β, TNF-α, IL-6, IL-8) and chemokines (CXCL1 and CCL2) [8], which are associated with T1D development [3]; (3) FFAs induce NF-κB and COX-2 via TLR4 in murine macrophages (Figure 4) [88], another pathway that induces the expression of pro-inflammatory cytokines [89]; (4) FFAs decreases the expression of the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2) in β cells, thereby increasing their susceptibility to apoptosis [90–92]; and (5) FFAs increases ROS production in β cells via activation of NOX [80,93] (Figure 4).
Medium- to long-chain FFAs promote both insulin secretion and reduce insulin production, leading to short-term hyperinsulinemia and long-term β-cell exhaustion. Medium to long chain FFAs acutely activate G protein-coupled receptor 40 (GPR40) in β cells and stimulate insulin secretion via protein kinase PKD1 signaling [94]. They also activate the mammalian target of rapamycin (mTOR) pathway via L-type Ca2+ channels to promote cytokine and chemokine expression, as part of the unfolded protein response [95]. Longer term, however, multiple detrimental effects of FFAs have been observed in β cells. These include promotion of an M1-like state of macrophage inflammation [96], increasing β-cell ROS, inducing mitochondrial dysfunction [97,98], promoting endoplasmic reticulum stress (with attendant reductions in insulin processing, alterations in calcium homeostasis, and reductions in pancreatic and duodenal homeobox protein-1 levels) [94,99], and altering autophagy [100]. Targets to reduce lipotoxicity-related dysfunction of β cells has been recently reviewed [86], but the approaches are largely related to their downstream effects on β-cell stress and signaling pathways (macrophage inflammation, ROS overproduction, endoplasmic reticulum stress and autophagy). Although these potential therapies (e.g. GLP-1 analogs, pioglitazone, ER chaperones, LOX inhibitors, verapamil, NOX inhibitors) have not been approved for use in human T1D, several have shown potential benefit in preclinical models of T1D, including pioglitazone [101] and LOX [41] and NOX inhibitors [102–104]. Still others are in clinical trials, such as ER chaperone TUDCA (ClinicalTrials.gov Identifier: NCT02218619) and verapamil [105].
4. Fatty acid esters of hydroxy fatty acids (FAHFAs)
FAHFAs were first reported in 2014 by Barbara Kahn’s group as molecules that improve glucose tolerance, β-cell function and reduce inflammation [106]. FAHFAs are synthesized by trans-esterification of hydroxylated fatty acyl chains of triacylglycerols by the adipose triglyceride lipase ATGL, which is further released from the glycerol moiety (Figure 5) [107]. FAHFAs can vary the acylation position and the composition of both fatty acid chains, resulting in a variety of structures with different potencies and activities [108]. More recently, palmitic acid ester of hydroxy stearic acid (PAHSA) delayed T1D onset in NOD mice [109,110]. Mechanistically, FAHFAs have been proposed to activate GPR120 and GPR40 (Figure 5). FAHFAs binding to GPR120 reduce ER stress and attenuate ERK1/2 and JNK1/2 MAP kinases-mediated signaling [109,110]. This leads to reductions in cytokine-mediated β-cell apoptosis and necrosis, thus preserving β-cell mass. FAHFAs also induce glucagon-like peptide 1 (GLP-1), a powerful insulin secretagogue. Therefore, as drug targets for T1D, FAHFAs can reduce β-cell death and improve insulin secretion.
Figure 5 – Fatty acid ester of hydroxy fatty acid (FAHFA) pathway.

The pathway shows the formation of FAHFAs along with their binding receptors. Agonists (blue), and drug candidates (underlined) are highlighted. Receptors are colored based on their pro-inflammatory activity. Abbreviations: FAHFA, fatty acid ester of hydroxy fatty acid; GPR40, G-protein coupled receptor 40; GPR120, G-protein coupled receptor 120.
As anti-diabetic and anti-inflammatory compounds, FAHFAs are considered excellent targets for drug development [110]. Therapeutic levels of PAHSA in circulation can be achieved through oral administration in pharmacological levels [111]. Another approach would be to target the signaling network of these molecules. For instance, the GPR40 agonist fasiglifam (TAK-875) had a clinical trial for treating T2D, which could also be effective in countering T1D [112]. However, its development into a therapeutic was interrupted because of liver toxicity concerns [74]. Another GPR40 agonist, SCO-267, stimulated insulin and GLP1 secretion, improving glucose tolerance in rats [113]. The GPR40 agonist rosiglitazone, an approved drug for T2D, was shown to preserve β-cell function in recent onset T1D but failed to promote glycemic control when the disease was already established [114,115]. Agonists for the other FAHFA receptor, GPR120, have also been studied as discussed above for ω-3 fatty acids. However, despite FAHFAs and ω-3 fatty acids targeting the same receptor more studies are need for determine if they somehow interact or cooperate in inducing protective signaling to the pancreatic islets.
5. The impact of dietary fatty acids on T1D development
The role of fatty acids in the development of islet autoimmunity and T1D has been investigated in a few large prospective cohorts of children with increased genetic risk of T1D, i.e., DAISY, DIPP, TEDDY, and TRIGR studies. Total longitudinal fat intake (adjusted for energy) during the first 6 years of life was associated with decreased risk of islet autoimmunity and T1D [116]. A child’s higher intake or status of ω-3 fatty acids was relatively consistently associated with decreased risk of islet autoimmunity prospectively, particularly in young children [117], but not with progression from islet autoimmunity to T1D [118]. On the other hand, both inverse [27] and direct [119], associations between serum/erythrocyte levels of saturated fatty acids with islet autoimmunity were shown in infants aged 3 to 6 months while associations observed in 1 to 6-year-old children were direct [27] [118–121]. Maternal fatty acid intake or status during pregnancy or lactation has not been associated with childhood T1D endpoints [122,123].
5.1. PUFAs.
Child’s higher intake of total and ω-3 fatty acids to decreases in up to 55% in risk for developing islet autoimmunity in prospective cohort studies [116,117]. Similarly, dietary DHA and EPA have been shown to reduce the onset of T1D in 60% of NOD mice [71]. In the DAISY study, ω-3 fatty acid content in erythrocytes showed a protective association with islet autoimmunity development over a 6-year follow-up [117], while in the TEDDY study erythrocyte EPA and DHA content at 3 months of age was protectively associated with IA risk [119]. In the DIPP study, DHA was associated with protection against IA at 3 months of age in non-breastfed children. Furthermore, EPA at 3 and 6 months and DHA at 6 months showed protective associations with primary insulin autoimmunity [27]. In the DIPP study, α-linoleic acid showed a direct association with islet autoimmunity at 3 months of age, whereas in the TEDDY study an inverse association was seen but only in non-breastfed children [27,119]. In the TRIGR study, only a weak protective association was detected between cord blood DPA with IA [120]. The most important dietary sources of ω-3 fatty acids are fish and vegetable oils, although they are also formed endogenously from α-linoleic acid. A child’s fish intake has also been protectively associated with islet autoimmunity [124]. Taken together, the evidence that ω-3 fatty acids may protect from IA is relatively strong, supported by recent oxylipin findings [125]. The ω-3 PUFAs influences the immune system activation and development, maturation of gut microbiota, permeability, barrier function, inflammatory responses, viral infections, and immune response [126,127].
Regarding ω-6 fatty acids, contrasting associations of AA intake and status with islet autoimmunity have been observed [116,117]. The same applies to conjugated linoleic acid levels, which have shown both inverse and direct associations with islet autoimmunity [119].
5.2. Monounsaturated and saturated fatty acids.
The impacts of monounsaturated and saturated fatty acid (MUFA) intake [116] or status [27] in relation to T1D-related endpoints have also been examined. Intake of MUFA during the first 6 years of life was inversely associated with islet autoimmunity [116]. In contrast erythrocyte oleic acid at 3 months of age and nervonic acid content at 1-6 years of age was directly associated with islet autoimmunity [119]. In infancy, serum palmitoleic and cis-vaccenic acid levels were inversely associated with the risk of IA and primary insulin autoimmunity [27].
In infancy, levels of the saturated fatty acid pentadecanoic acid were associated with decreased islet autoimmunity risk [27] and palmitic acid with both decreased and increased islet autoimmunity risk [27,119]. In childhood, some even- (myristic, stearic) and odd-chain (pentadecanoic, heptadecanoic) saturated fatty acid levels showed direct associations with islet autoimmunity, but not with T1D endpoints [119–121]. Odd chain saturated fatty acids pentadecanoic acid and heptadecanoic acid and conjugated linoleic acid are known biomarkers of dairy or human milk intake (although other factors like fiber intake also influence their formation) [27,128,129]. There is some indication that breastmilk may protect from IA, whereas cow’s milk consumption has been consistently linked to increased IA and T1D risk [130].
One phenomenon that should be kept in mind is the endogenous formation of saturated fatty acids in the liver from shorter-chain fatty acids, and by de novo lipogenesis [131]. Their contributions to T1D endpoints are largely unknown [119]. The major even-chain SFAs [palmitic (16:0), stearic (18:0)], and MUFAs [oleic (18:1(9Z)) and nervonic (24:1(15Z)) acids] levels in erythrocytes were associated with an increased risk of islet autoimmunity. Furthermore, for a multiple islet autoimmunity endpoint, stearic (18:0) and cis vaccenic acid (18:1(11Z)) showed increased risk [119].
Overall, dietary fatty acids are strongly associated with islet autoimmunity and the development of T1D and can be targeted to promote a protective effect.
6. Conclusion.
In this article, we summarize lipids, receptors and enzymes that are relevant targets for both IA and T1D development. There is a crescent number of drug candidates that target fatty acid signaling. As the mechanistic knowledge of the complex fatty acid signaling network increases, it will open opportunities for identifying new druggable targets. Screening large libraries of small chemical compounds can speed up this process. The coming years will offer new therapies focused on T1D fatty acid-related pathways targets to prevent both IA and T1D. The effectiveness improvement of these putative treatments will likely require years and will be developed as complementing therapies to the existing insulin-based treatments. The success of this approach will also rely on sustained investments and research on testing different therapeutic candidates in cells, tissues, animals, and humans.
7. Expert opinion
T1D therapies targeting fatty acid signaling are in different stages of development, from initial discovery to in vivo testing, to early stages of clinical trials. Extensive knowledge obtained in these pathways to treat other autoimmunity, inflammatory, and even infectious diseases can contribute to speeding up the process of drug development. For instance, using a pharmacological targets database [132], we identified 763 drugs that target fatty acid signaling pathways, out of which only 42 target-specific drugs are approved by the U.S. Food & Drug Administration (FDA), for a variety of diseases and conditions Table 1, opening opportunities for repurposing them for treating T1D. Anyway, these drugs were not approved to treat T1D by FDA. In addition, in Table 2 we report a list of 18 compounds, targeting fatty acid-mediated pathways relevant in both IA and T1D that were tested in rodent and human-derived diabetes models out of which 2 are FDA approved: rosiglitazone and selexipag to treat T2D and high pulmonary pressure, respectively. Although compounds listed here can potentially improve the T1D therapies, scaling to clinical trials needs further investigation. Below we give insights about how these pharmaceutical compounds can be developed into therapies.
TABLE 1 – List of drugs targeting fatty acid-mediated signaling.
Contains a list of drugs targeting fatty acid signaling pathways associated with T1D from the curated database of International Union of Basic and Clinical Pharmacology (IUPHAR)/ British Pharmacological Society (BPS) Guide to PHARMACOLOGY (https://www.guidetopharmacology.org/) [132]. Columns (from left to right) indicate: 1) the type of target (enzyme or receptor), 2) the abbreviated name of the target, 3) the total number of drugs identified for the specific target, 4) the number of drugs identified to interact primarily with the indicated target, 5) the number of drugs known to interact primarily with the specified target approved by the FDA and 6) the drug names of column 5.
| Type of target | Target | Drugs identified | Target-specific drugs | FDA approved1 target-specific drugs | FDA approved1 target-specific drug names |
|---|---|---|---|---|---|
| Receptor | Leukotriene receptor B1 | 21 | 0 | 0 | |
| Receptor | Chemerin receptor 23 | 8 | 0 | 0 | |
| Enzyme | Cyclooxygenase-2 | 44 | 28 | 23 | lumiracoxib, benzquinamide, valdecoxib, flurbiprofen, diclofenac, celecoxib, meclofenamic acid, carprofen, meloxicam, rofecoxib, nimesulide, ketoprofen, etoricoxib, ibuprofen, aspirin, naproxen, ketorolac, suprofen, mefenamic acid, oxaprozin, etodolac, piroxicam, phenylbutazone |
| Receptor | Cysteinyl leukotriene receptor 1 | 30 | 6 | 6 | zafirlukast, montelukast, pranlukast, pranlukast, zafirlukast, montelukast |
| Receptor | Cysteinyl leukotriene receptor 2 | 29 | 0 | 0 | |
| Receptor | Prostaglandin D2 receptor 1 | 38 | 0 | 0 | |
| Receptor | Prostaglandin D2 receptor 2 | 45 | 4 | 0 | |
| Receptor | Prostaglandin E receptor 1 | 50 | 3 | 3 | PGE2, PGE1, PGI2, |
| Receptor | Prostaglandin E receptor 2 | 59 | 2 | 2 | PGE1, PGE2 |
| Receptor | Prostaglandin E receptor 3 | 84 | 1 | 1 | misoprostol (methyl ester) |
| Receptor | Prostaglandin E receptor 4 | 83 | 1 | 0 | |
| Receptor | Prostaglandin F receptor | 37 | 5 | 4 | latanoprost (free acid form), latanoprost (isopropyl ester), bimatoprost, tafluprost |
| Receptor | Formyl peptide receptor 2 | 57 | 0 | 0 | |
| Receptor | G protein-coupled receptor 120 | 13 | 1 | 0 | |
| Receptor | G protein-coupled receptor 18 | 9 | 0 | 0 | |
| Receptor | G protein-coupled receptor 32 | 4 | 0 | 0 | |
| Receptor | G protein-coupled receptor 40 | 19 | 2 | 0 | |
| Receptor | G protein-coupled receptor 12 | 2 | 0 | 0 | |
| Enzyme | 12-lipoxygenase | 1 | 1 | 0 | |
| Enzyme | 15-lipoxygenase | 4 | 1 | 0 | |
| Receptor | Leukotriene B4 receptor 2 | 14 | 0 | 0 | |
| Enzyme | NADPH oxidase 1 | 4 | 3 | 0 | |
| Enzyme | Prostaglandin I2 | 41 | 4 | 3 | treprostinil, iloprost, treprostinil |
| Enzyme | Phospholipases A2 | 3 | 0 | 0 | |
| Enzyme | Soluble epoxide hydrolase | 14 | 2 | 0 | |
| Receptor | Thromboxane A2 receptor | 50 | 0 | 0 |
Target-specific drugs approved by FDA for a variety of diseases. None of them is approved for T1D.
Table 2 – List of fatty acid signaling drugs tested in diabetic models.
Contains a list of compounds targeting fatty acid signaling pathways discussed in the text, as a potential treatment for T1D. Columns (from left to right) indicate: 1) the compound name found in bibliography or common drug for the compound; 2) the abbreviated name of the specific target; 3) the mechanism of action reported for the specific target; 3) the diabetic experimental models where the compound has been tested in; 3) FDA approval information in humans to treat not-T1D pathologies (https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm); 4) references for the mentioned diabetic experimental models.
| Compound name | Target | Type | Experimental models | FDA approved1 | Reference |
|---|---|---|---|---|---|
| Fasiglifam | G protein-coupled receptor 40 | Agonist | Human having T2D | No | [111,112] |
| FKGK18 | Ca2+-Independent Phospholipase A2β | Inhibitor | NOD mice | No | [21] |
| GKT137831 | NADPH Oxidase 1 | Inhibitor | NOD mice & human with T1D | No | [36,137,138] |
| Grapiprant | receptor EP4 | Agonist | NOD mice | No | [60] |
| GSK137647A | G protein-coupled receptor 120 | Agonist | Diet-induced obesity mice model | No | [77] |
| GSK2256294 | Soluble epoxide hydrolase | Inhibitor | Mice islets | No | [48] |
| ML127 | 12/15-lipoxygenase | Inhibitor | NOD mice, human islets & mice islets | No | [41,42] |
| ML171 | NADPH Oxidase 1 | Inhibitor | NOD mice | No | [36] |
| ML351 | 12/15-lipoxygenase | Inhibitor | NOD mice, human Islets & mice islets | No | [41,42] |
| ML355 | 12/15-lipoxygenase | Inhibitor | NOD mice, human islets, human Islets from T2D donors & mice islets | No | [41,42] |
| NS-398 | Cyclooxygenase-2 | Inhibitor | Rat Islet & human Islets | No | [59,133] |
| Rosiglitazone | G protein-coupled receptor 40 | Agonist | Human with T1D | Yes | [114,115] |
| SC-236 | Cyclooxygenase-2 | Inhibitor | Rat pancreatic islet | No | [59] |
| SCO-267 | G protein-coupled receptor 40 | Agonist | N-STZ-1.5 Rats | No | [113] |
| Selexipag | Prostaglandin I2 receptor | Agonist | NOD mice | Yes | [64,65,140] |
| t-AUCB | Soluble epoxide hydrolase | Inhibitor | Mice islets | No | [46] |
| TPPU | Soluble epoxide hydrolase | Inhibitor | Mice islets | No | [47] |
| TUG-891 | G protein-coupled receptor 120 | Agonist | Diet-induced obesity mice model | No | [77] |
Target-specific drugs approved by FDA for a variety of diseases. None of them is approved for T1D.
7.1. Inhibitors of pro-inflammatory enzymes and antagonists of pro-inflammatory receptors.
Most of the enzymatic drug targets in fatty acid signaling for T1D belong to the AA-signaling pathway. The phospholipase iPLA2β inhibitor FKGK18 has been successfully tested in NOD mice [21]. However, no clinical trials have been conducted so far. The case is similar for the lipooxygenases 12-LOX and 15-LOX inhibitors, ML127 and ML351. The COX-2 inhibitors NS-398 and SC-236 have been mainly tested in rat and human islets [59,133]. In addition, there are 27 FDA-approved drugs targeting COX-2, including the over-the-counter drugs acetaminophen, aspirin, and ibuprofen, opening opportunities to test them as potential targets for T1D therapies. Acetaminophen intake was addressed in the TEDDY study, but no association of the drug was found with the onset of IA [134]. However, to be effective, these drugs might need to be administrated in multiple doses during IA and tested in a clinical trial rather than an observational study. Most of the clinical trials with COX-2-targeting drugs evaluate their efficacy and safety towards improving hyperglycemia-induced complications, such as thrombosis and renal disease. The sEH inhibitors t-AUCB [46] and TPPU [47] have been only tested in mice. However, there are currently two approved drugs targeting sEH, the anti-asthmatic drug Zafirlukast and the anti-inflammatory drug Oxaprozin [135]. None of them have been tested against insulitis and could be candidates for T1D therapy. Zafirlukast induces insulin secretion by triggering calcium signaling, which is independent on the glucose levels [136], and might be an undesired side effect if this compound is developed into a drug. Regarding NOX inhibitors, GKT137831 has currently being evaluated in clinical trials for treating diabetic kidney disease [137,138], rather than directly targeting the β-cell survival or function.
Antagonists of PG receptors are also candidates for T1D therapies. For instance, the PGE2 receptor EP4 antagonist grapiprant reduced insulitis in NOD mice, but it’s efficacy in preventing T1D development has not been tested [60].
7.2. Agonists of anti-inflammatory receptors.
Anti-inflammatory receptors that bind to fatty acids and their bioactive products represent excellent drug targets. The fatty acids and their active bioproducts themselves can be considered as drug candidates. For instance, GPR120 is activated by both protective ω-3 fatty acids and FAHFAs. Research in FAHFA has only been conducted in vitro and in animals [80,106–110]. However, ω-3 fatty acids are extensively studied in clinical trials, with over 1500 studies currently registered in ClinicalTrials.gov. There are currently 11 ω-3 fatty acid clinical trial studies registered for T1D. One concluded study followed for one year the supplement of ω-3 fatty acids by individuals with recent onset of T1D, which showed a reduced demand for insulin in the ω-3 supplemented group [139]. As mentioned above, ω-3 fatty acids can reduce the risk of developing IA [117]. FPR2/ALX, GPR18, GPR32, ChemR23 and BLT1 are receptors for lipoxins and resolvins. There are currently, 23 and 30 studies registered in ClinicalTrials.gov for lipoxins and resolvins, respectively. Selexipag, the antagonist of PIG2 receptor, an approved drug for pulmonary arterial hypertension [140], preserves β-cell function in NOD mice [65]. However, it still needs to be systematically tested for T1D. Even though none of these studies are in T1D, the results can bring important information regarding safety and efficacy in other autoimmune or inflammatory disease. Despite that FAHFAs, ω-3 fatty acids, resolvins, and lipoxins being active themselves and have potential to be developed into drugs, there is still a value for screening new compounds that are more stable. FAHFAs can be degraded by lipases, whereas ω-3 fatty acids, resolvins and lipoxins can be oxidized into inactive byproducts.
7.3. Diet as a potential intervention.
In terms of targeting dietary fatty acids for T1D therapies, changes in diet tending, such as reducing FFAs intake may have an impact by regulating metabolic processes that induces increased levels of circulating FFAs [141]. Diet accompanied by exercise and changing in lifestyles can further decrease the levels of FFAs [141]. Diets rich in ω-3 PUFAs can reduce the risk of developing islet autoimmunity, i.e., preclinical T1D [117]. While fatty acids themselves are not considered as drugs, ω-3-rich fish-oil or from other sources can be taken as supplements [142], or by eating foods rich in these fatty acids, such as fish from cold waters and nuts.
Article highlights.
Fatty acids are key mediators in islets inflammation, autoimmune response, and β-cell death in T1D development.
Dietary fatty acids can alter the risk of developing T1D.
Fatty acid signaling-targeting compounds and dietary supplements are potential T1D therapeutic targets.
FDA-approved drugs have potential to be repurposed for T1D therapies.
Funding
This work was supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grants U01 DK127786 (to ES Nakayasu), U01 DK127786 (to RG Mirmira, TO Metz. and S Ramanadham), R01 DK060581 (to R.G.M), and R01 DK126444 (to S Ramanadham). ES Nakayasu was supported by a Catalyst Award from the Human Islet Research Network. S Ramanadham was also supported by UAB-DRC P&F, CDIB Department, and UAB-Comprehensive Diabetes Center.
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants, or patents received or pending, or royalties.
Footnotes
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Gregory GA, Robinson TIG, Linklater SE, et al. Global incidence, prevalence, and mortality of type 1 diabetes in 2021 with projection to 2040: a modelling study. Lancet Diabetes Endocrinol. 2022. Oct;10(10):741–760. [DOI] [PubMed] [Google Scholar]
- 2.DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet. 2018. Jun 16;391(10138):2449–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009. Apr;5(4):219–26. [DOI] [PubMed] [Google Scholar]
- 4.Lei X, Zhang S, Emani B, et al. A link between endoplasmic reticulum stress-induced beta-cell apoptosis and the group VIA Ca2+-independent phospholipase A2 (iPLA2beta). Diabetes Obes Metab. 2010. Oct;12 Suppl 2(0 2):93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson AJ, Stephenson DJ, Bone RN, et al. Lipid mediators and biomarkers associated with type 1 diabetes development. JCI Insight. 2020. Aug 20;5(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamichhane S, Ahonen L, Dyrlund TS, et al. Dynamics of Plasma Lipidome in Progression to Islet Autoimmunity and Type 1 Diabetes - Type 1 Diabetes Prediction and Prevention Study (DIPP). Sci Rep. 2018. Jul 13;8(1):10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamichhane S, Ahonen L, Dyrlund TS, et al. A longitudinal plasma lipidomics dataset from children who developed islet autoimmunity and type 1 diabetes. Sci Data. 2018. Nov 13;5:180250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Webb-Robertson BM, Bramer LM, Stanfill BA, et al. Prediction of the development of islet autoantibodies through integration of environmental, genetic, and metabolic markers. J Diabetes. 2021. Feb;13(2):143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sarkar S, Elliott EC, Henry HR, et al. Systematic review of type 1 diabetes biomarkers reveals regulation in circulating proteins related to complement, lipid metabolism, and immune response. medRxiv. 2023:2023.02.21.23286132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004. Sep;56(3):387–437. [DOI] [PubMed] [Google Scholar]
- 11.Esh CJ, Chrismas BCR, Mauger AR, et al. Pharmacological hypotheses: Is acetaminophen selective in its cyclooxygenase inhibition? Pharmacol Res Perspect. 2021. Aug;9(4):e00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rustan AC, Drevon CA. Fatty Acids: Structures and Properties. eLS2005. [Google Scholar]
- 13.Fievez V, Colman E, Castro-Montoya JM, et al. Milk odd- and branched-chain fatty acids as biomarkers of rumen function—An update. Animal Feed Science and Technology. 2012. 2012/02/28/;172(1):51–65. [Google Scholar]
- 14.Nachtschatt M, Okada S, Speight R. Integral Membrane Fatty Acid Desaturases: A Review of Biochemical, Structural, and Biotechnological Advances [ 10.1002/ejlt.202000181]. European Journal of Lipid Science and Technology. 2020. 2020/12/01;122(12):2000181. [DOI] [Google Scholar]
- 15.Dobrian AD, Morris MA, Taylor-Fishwick DA, et al. Role of the 12-lipoxygenase pathway in diabetes pathogenesis and complications. Pharmacol Ther. 2019. Mar;195:100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gendaszewska-Darmach E, Drzazga A, Koziolkiewicz M. Targeting GPCRs Activated by Fatty Acid-Derived Lipids in Type 2 Diabetes. Trends Mol Med. 2019. Oct;25(10):915–929. [DOI] [PubMed] [Google Scholar]
- 17.Mancini AD, Poitout V. GPR40 agonists for the treatment of type 2 diabetes: life after ‘TAKing’ a hit. Diabetes Obes Metab. 2015. Jul;17(7):622–9. [DOI] [PubMed] [Google Scholar]
- 18.Sekiguchi H, Kasubuchi M, Hasegawa S, et al. A novel antidiabetic therapy: free fatty acid receptors as potential drug target. Curr Diabetes Rev. 2015;11(2):107–15. [DOI] [PubMed] [Google Scholar]
- 19.Watterson KR, Hudson BD, Ulven T, et al. Treatment of type 2 diabetes by free Fatty Acid receptor agonists. Front Endocrinol (Lausanne). 2014;5:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo P, Wang MH. Eicosanoids, beta-cell function, and diabetes. Prostaglandins & other lipid mediators. 2011. Aug;95(1–4):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bone RN, Gai Y, Magrioti V, et al. Inhibition of Ca2+-independent phospholipase A2beta (iPLA2beta) ameliorates islet infiltration and incidence of diabetes in NOD mice. Diabetes. 2015. Feb;64(2):541–54. ** This work demonstrated that the inhibition of Ca2+-independent phospholipase A2beta reduces the onset of type 1 diabtes in mice
- 22.Lei X, Bone RN, Ali T, et al. Evidence of contribution of iPLA2beta-mediated events during islet beta-cell apoptosis due to proinflammatory cytokines suggests a role for iPLA2beta in T1D development. Endocrinology. 2014. Sep;155(9):3352–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norris PC, Dennis EA. A lipidomic perspective on inflammatory macrophage eicosanoid signaling. Adv Biol Regul. 2014. Jan;54:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imai Y, Dobrian AD, Morris MA, et al. Lipids and immunoinflammatory pathways of beta cell destruction. Diabetologia. 2016. Apr;59(4):673–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez M, Clare-Salzler M. Eicosanoid imbalance in the NOD mouse is related to a dysregulation in soluble epoxide hydrolase and 15-PGDH expression. Ann N Y Acad Sci. 2006. Oct;1079:130–4. [DOI] [PubMed] [Google Scholar]
- 26.Luther JM, Brown NJ. Epoxyeicosatrienoic acids and glucose homeostasis in mice and men. Prostaglandins & other lipid mediators. 2016. Sep;125:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niinisto S, Takkinen HM, Erlund I, et al. Fatty acid status in infancy is associated with the risk of type 1 diabetes-associated autoimmunity. Diabetologia. 2017. Jul;60(7):1223–1233. [DOI] [PubMed] [Google Scholar]
- 28.Zaitseva L, Vaisburd M, Shaposhnikova G, et al. Role of eicosanoids in regulation of macrophage phagocytic functions by platelet-activating factor during endotoxic shock. Bulletin of experimental biology and medicine. 2000. Sep;130(9):879–81. [DOI] [PubMed] [Google Scholar]
- 29.Dobrian AD, Lieb DC, Cole BK, et al. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res. 2011. Jan;50(1):115–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma K, Nunemaker CS, Wu R, et al. 12-Lipoxygenase Products Reduce Insulin Secretion and {beta}-Cell Viability in Human Islets. J Clin Endocrinol Metab. 2010. Feb;95(2):887–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weaver JR, Holman TR, Imai Y, et al. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol Cell Endocrinol. 2012. Jul 6;358(1):88–95. [DOI] [PubMed] [Google Scholar]
- 32.Wen Y, Gu J, Vandenhoff GE, et al. Role of 12/15-lipoxygenase in the expression of MCP-1 in mouse macrophages. Am J Physiol Heart Circ Physiol. 2008. Apr;294(4):H1933–8. [DOI] [PubMed] [Google Scholar]
- 33.McDuffie M, Maybee NA, Keller SR, et al. Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes. 2008. Jan;57(1):199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prasad KM, Thimmalapura PR, Woode EA, et al. Evidence that increased 12-lipoxygenase expression impairs pancreatic beta cell function and viability. Biochem Biophys Res Commun. 2003. Aug 29;308(3):427–32. [DOI] [PubMed] [Google Scholar]
- 35.Zhou RH, Pesant S, Cohn HI, et al. Enhanced sterol response element-binding protein in postintervention restenotic blood vessels plays an important role in vascular smooth muscle proliferation. Life Sci. 2008. Jan 16;82(3–4):174–81. [DOI] [PubMed] [Google Scholar]
- 36. David Taylor AP, Glenn Lindsey, Orr Kara S, Tersey Sarah A and Taylor-Fishwick David A. Small molecule inhibition of NOX-1 reduces diabetes conversion in NOD mice. Integrative Molecular Medicine. 2019;6. ** This paper demonstrated that the inhibition of NOX-1 reduces the onset of type 1 diabtes in mice
- 37.Grzesik WJ, Nadler JL, Machida Y, et al. Expression pattern of 12-lipoxygenase in human islets with type 1 diabetes and type 2 diabetes. J Clin Endocrinol Metab. 2015. Mar;100(3):E387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Green-Mitchell SM, Tersey SA, Cole BK, et al. Deletion of 12/15-lipoxygenase alters macrophage and islet function in NOD-Alox15(null) mice, leading to protection against type 1 diabetes development. PLoS One. 2013;8(2):e56763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pineros AR, Kulkarni A, Gao H, et al. Proinflammatory signaling in islet beta cells propagates invasion of pathogenic immune cells in autoimmune diabetes. Cell Rep. 2022. Jun 28;39(13):111011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulkarni A, Pineros AR, Walsh MA, et al. 12-Lipoxygenase governs the innate immune pathogenesis of islet inflammation and autoimmune diabetes. JCI Insight. 2021;6(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hernandez-Perez M, Chopra G, Fine J, et al. Inhibition of 12/15-Lipoxygenase Protects Against beta-Cell Oxidative Stress and Glycemic Deterioration in Mouse Models of Type 1 Diabetes. Diabetes. 2017. Nov;66(11):2875–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma K, Xiao A, Park SH, et al. 12-Lipoxygenase Inhibitor Improves Functions of Cytokine-Treated Human Islets and Type 2 Diabetic Islets. J Clin Endocrinol Metab. 2017. Aug 1;102(8):2789–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Z, He Z, Wang DW. CYP450 Epoxygenase Metabolites, Epoxyeicosatrienoic Acids, as Novel Anti-Inflammatory Mediators. Molecules. 2022. Jun 16;27(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho HJ, Switzer CH, Kamynina A, et al. Complex interrelationships between nitro-alkene-dependent inhibition of soluble epoxide hydrolase, inflammation and tumor growth. Redox Biol. 2020. Jan;29:101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McReynolds C, Morisseau C, Wagner K, et al. Epoxy fatty acids are promising targets for treatment of pain, cardiovascular disease and other indications characterized by mitochondrial dysfunction, endoplasmic stress and inflammation. Adv Exp Med Biol. 2020;1274:71–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo P, Chang HH, Zhou Y, et al. Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther. 2010. Aug;334(2):430–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koike S, Hsu MF, Bettaieb A, et al. Genetic deficiency or pharmacological inhibition of soluble epoxide hydrolase ameliorates high fat diet-induced pancreatic beta-cell dysfunction and loss. Free Radic Biol Med. 2021. Aug 20;172:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lazaar AL, Yang L, Boardley RL, et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol. 2016. May;81(5):971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim R, Emi M, Tanabe K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol Ther. 2005. Sep;4(9):924–33. [DOI] [PubMed] [Google Scholar]
- 50.Atkinson MA. Mechanisms underlying the loss of self tolerance in NOD mice. Res Immunol. 1997. Jun;148(5):301–6. [DOI] [PubMed] [Google Scholar]
- 51.Ganapathy V, Gurlo T, Jarstadmarken HO, et al. Regulation of TCR-induced IFN-gamma release from islet-reactive non-obese diabetic CD8(+) T cells by prostaglandin E(2) receptor signaling. Int Immunol. 2000. Jun;12(6):851–60. [DOI] [PubMed] [Google Scholar]
- 52.Basu S Novel cyclooxygenase-catalyzed bioactive prostaglandin F2alpha from physiology to new principles in inflammation. Med Res Rev. 2007. Jul;27(4):435–68. [DOI] [PubMed] [Google Scholar]
- 53.Oyesola OO, Tait Wojno ED. Prostaglandin regulation of type 2 inflammation: From basic biology to therapeutic interventions. Eur J Immunol. 2021. Oct;51(10):2399–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scher JU, Pillinger MH. The anti-inflammatory effects of prostaglandins. J Investig Med. 2009. Aug;57(6):703–8. [DOI] [PubMed] [Google Scholar]
- 55.Xu C, Liu W, You X, et al. PGF2alpha modulates the output of chemokines and pro-inflammatory cytokines in myometrial cells from term pregnant women through divergent signaling pathways. Mol Hum Reprod. 2015. Jul;21(7):603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clark M, Kroger CJ, Tisch RM. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front Immunol. 2017;8:1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dominguez C, Ruiz E, Gussinye M, et al. Oxidative stress at onset and in early stages of type 1 diabetes in children and adolescents. Diabetes Care. 1998. Oct;21(10):1736–42. [DOI] [PubMed] [Google Scholar]
- 58.Nemzer BV, Rodriguez LC, Hammond L, et al. Acute reduction of serum 8-iso-PGF2-alpha and advanced oxidation protein products in vivo by a polyphenol-rich beverage; a pilot clinical study with phytochemical and in vitro antioxidant characterization. Nutr J. 2011. Jun 15;10:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tran PO, Gleason CE, Poitout V, et al. Prostaglandin E(2) mediates inhibition of insulin secretion by interleukin-1beta. The Journal of biological chemistry. 1999. Oct 29;274(44):31245–8. [DOI] [PubMed] [Google Scholar]
- 60.Rahman MJ, Rodrigues KB, Quiel JA, et al. Restoration of the type I IFN-IL-1 balance through targeted blockade of PTGER4 inhibits autoimmunity in NOD mice. JCI Insight. 2018. Feb 8;3(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014. Jun 5;510(7503):92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018. Jul 2;128(7):2657–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2017. Apr;31(4):1273–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuwano K, Hashino A, Asaki T, et al. 2-[4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy]-N-(methylsulfonyl)acetamide (NS-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther. 2007. Sep;322(3):1181–8. [DOI] [PubMed] [Google Scholar]
- 65.Batchu SN, Majumder S, Bowskill BB, et al. Prostaglandin I2 Receptor Agonism Preserves beta-Cell Function and Attenuates Albuminuria Through Nephrin-Dependent Mechanisms. Diabetes. 2016. May;65(5):1398–409. [DOI] [PubMed] [Google Scholar]
- 66.Serhan CN, Chiang N, Dalli J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin Immunol. 2015. May;27(3):200–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dalli J, Serhan C. Macrophage Proresolving Mediators-the When and Where. Microbiol Spectr. 2016. Jun;4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chiang N, Serhan CN. Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem. 2020. Sep 23;64(3):443–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010. Sep 3;142(5):687–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mao C, Xiao P, Tao XN, et al. Unsaturated bond recognition leads to biased signal in a fatty acid receptor. Science. 2023. Apr 7;380(6640):eadd6220. [DOI] [PubMed] [Google Scholar]
- 71.Bi X, Li F, Liu S, et al. omega-3 polyunsaturated fatty acids ameliorate type 1 diabetes and autoimmunity. J Clin Invest. 2017. May 1;127(5):1757–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Davanso MR, Crisma AR, Braga TT, et al. Macrophage inflammatory state in Type 1 diabetes: triggered by NLRP3/iNOS pathway and attenuated by docosahexaenoic acid. Clin Sci (Lond). 2021. Jan 15;135(1):19–34. [DOI] [PubMed] [Google Scholar]
- 73.Lo Conte M, Antonini Cencicchio M, Ulaszewska M, et al. A diet enriched in omega-3 PUFA and inulin prevents type 1 diabetes by restoring gut barrier integrity and immune homeostasis in NOD mice. Front Immunol. 2022;13:1089987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang X, Li X, Wei S, et al. Discovery of Novel and Selective G-Protein Coupled Receptor 120 (GPR120) Agonists for the Treatment of Type 2 Diabetes Mellitus. Molecules. 2022. Dec 17;27(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oh DY, Walenta E, Akiyama TE, et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat Med. 2014. Aug;20(8):942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Andreone L, Fuertes F, Setula C, et al. Compound A attenuates proinflammatory cytokine-induced endoplasmic reticulum stress in beta cells and displays beneficial therapeutic effects in a mouse model of autoimmune diabetes. Cell Mol Life Sci. 2022. Nov 12;79(12):587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Azevedo CM, Watterson KR, Wargent ET, et al. Non-Acidic Free Fatty Acid Receptor 4 Agonists with Antidiabetic Activity. J Med Chem. 2016. Oct 13;59(19):8868–8878. [DOI] [PubMed] [Google Scholar]
- 78.Chiang N, Barnaeva E, Hu X, et al. Identification of Chemotype Agonists for Human Resolvin D1 Receptor DRV1 with Pro-Resolving Functions. Cell Chem Biol. 2019. Feb 21;26(2):244–254 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stein DT, Esser V, Stevenson BE, et al. Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. Journal of Clinical Investigation. 1996;97(12):2728–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alquier T, Peyot ML, Latour MG, et al. Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes. 2009. Nov;58(11):2607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nolan CJ, Prentki M. The islet beta-cell: fuel responsive and vulnerable. Trends Endocrinol Metab. 2008. Oct;19(8):285–91. [DOI] [PubMed] [Google Scholar]
- 82.Park EJ, Lee AY, Park S, et al. Multiple pathways are involved in palmitic acid-induced toxicity. Food Chem Toxicol. 2014. May;67:26–34. [DOI] [PubMed] [Google Scholar]
- 83.Ly LD, Xu S, Choi SK, et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med. 2017. Feb 3;49(2):e291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liang H, Zhong Y, Zhou S, et al. Palmitic acid-induced apoptosis in pancreatic beta-cells is increased by liver X receptor agonist and attenuated by eicosapentaenoate. In Vivo. 2011. Sep-Oct;25(5):711–8. [PubMed] [Google Scholar]
- 85.Allagnat F, Cunha D, Moore F, et al. Mcl-1 downregulation by pro-inflammatory cytokines and palmitate is an early event contributing to beta-cell apoptosis. Cell Death Differ. 2011. Feb;18(2):328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lytrivi M, Castell AL, Poitout V, et al. Recent Insights Into Mechanisms of beta-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J Mol Biol. 2020. Mar 6;432(5):1514–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Corkey BE, Kilpatrick LE, Evans-Molina C. Hypothesis: Induction of Autoimmunity in Type 1 Diabetes-A Lipid Focus. Diabetes. 2022. Oct 1;71(10):2067–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Graciano MF, Valle MM, Curi R, et al. Evidence for the involvement of GPR40 and NADPH oxidase in palmitic acid-induced superoxide production and insulin secretion. Islets. 2013. Jul-Aug;5(4):139–48. [DOI] [PubMed] [Google Scholar]
- 89.Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000. Nov;49(11):1939–45. [DOI] [PubMed] [Google Scholar]
- 90.Carlsson C, Borg LA, Welsh N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology. 1999. Aug;140(8):3422–8. [DOI] [PubMed] [Google Scholar]
- 91.Guichard C, Moreau R, Pessayre D, et al. NOX family NADPH oxidases in liver and in pancreatic islets: a role in the metabolic syndrome and diabetes? Biochem Soc Trans. 2008. Oct;36(Pt 5):920–9. [DOI] [PubMed] [Google Scholar]
- 92.Sato Y, Fujimoto S, Mukai E, et al. Palmitate induces reactive oxygen species production and beta-cell dysfunction by activating nicotinamide adenine dinucleotide phosphate oxidase through Src signaling. J Diabetes Investig. 2014. Feb 12;5(1):19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ferdaoussi M, Bergeron V, Zarrouki B, et al. G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia. 2012. Oct;55(10):2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hatanaka M, Maier B, Sims EK, et al. Palmitate induces mRNA translation and increases ER protein load in islet beta-cells via activation of the mammalian target of rapamycin pathway. Diabetes. 2014. Oct;63(10):3404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Igoillo-Esteve M, Marselli L, Cunha DA, et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia. 2010. Jul;53(7):1395–405. [DOI] [PubMed] [Google Scholar]
- 96.Eguchi K, Manabe I, Oishi-Tanaka Y, et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012. Apr 4;15(4):518–33. [DOI] [PubMed] [Google Scholar]
- 97.Barlow J, Affourtit C. Novel insights into pancreatic beta-cell glucolipotoxicity from real-time functional analysis of mitochondrial energy metabolism in INS-1E insulinoma cells. Biochem J. 2013. Dec 15;456(3):417–26. [DOI] [PubMed] [Google Scholar]
- 98.Joseph JW, Koshkin V, Saleh MC, et al. Free fatty acid-induced beta-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem. 2004. Dec 3;279(49):51049–56. [DOI] [PubMed] [Google Scholar]
- 99.Cunha DA, Hekerman P, Ladriere L, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci. 2008. Jul 15;121(Pt 14):2308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008. Oct;8(4):325–32. [DOI] [PubMed] [Google Scholar]
- 101.Maganti AV, Tersey SA, Syed F, et al. Peroxisome Proliferator-activated Receptor-gamma Activation Augments the beta-Cell Unfolded Protein Response and Rescues Early Glycemic Deterioration and beta Cell Death in Non-obese Diabetic Mice. J Biol Chem. 2016. Oct 21;291(43):22524–22533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Elumalai S, Karunakaran U, Moon JS, et al. NADPH Oxidase (NOX) Targeting in Diabetes: A Special Emphasis on Pancreatic beta-Cell Dysfunction. Cells. 2021. Jun 22;10(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Elksnis A, Cen J, Wikstrom P, et al. Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells. Biomedicines. 2021. Dec 8;9(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xiang FL, Lu X, Strutt B, et al. NOX2 deficiency protects against streptozotocin-induced beta-cell destruction and development of diabetes in mice. Diabetes. 2010. Oct;59(10):2603–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Forlenza GP, McVean J, Beck RW, et al. Effect of Verapamil on Pancreatic Beta Cell Function in Newly Diagnosed Pediatric Type 1 Diabetes: A Randomized Clinical Trial. JAMA. 2023. Mar 28;329(12):990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yore MM, Syed I, Moraes-Vieira PM, et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 2014. Oct 9;159(2):318–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Patel R, Santoro A, Hofer P, et al. ATGL is a biosynthetic enzyme for fatty acid esters of hydroxy fatty acids. Nature. 2022. Jun;606(7916):968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aryal P, Syed I, Lee J, et al. Distinct biological activities of isomers from several families of branched fatty acid esters of hydroxy fatty acids (FAHFAs). J Lipid Res. 2021;62:100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rubin de Celis MF, Garcia-Martin R, Syed I, et al. PAHSAs reduce cellular senescence and protect pancreatic beta cells from metabolic stress through regulation of Mdm2/p53. Proc Natl Acad Sci U S A. 2022. Nov 22;119(47):e2206923119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Syed I, Rubin de Celis MF, Mohan JF, et al. PAHSAs attenuate immune responses and promote beta cell survival in autoimmune diabetic mice. J Clin Invest. 2019. Aug 5;129(9):3717–3731. * This work showed that FAHFAs can reduce the onset of type 1 diabetes in mice.
- 111.Kaku K, Enya K, Nakaya R, et al. Efficacy and safety of fasiglifam (TAK-875), a G protein-coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: a randomized, double-blind, placebo-controlled, phase III trial. Diabetes Obes Metab. 2015. Jul;17(7):675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Marcinak JF, Munsaka MS, Watkins PB, et al. Liver Safety of Fasiglifam (TAK-875) in Patients with Type 2 Diabetes: Review of the Global Clinical Trial Experience. Drug Saf. 2018. Jun;41(6):625–640. [DOI] [PubMed] [Google Scholar]
- 113.Furukawa H, Miyamoto Y, Hirata Y, et al. Design and Identification of a GPR40 Full Agonist (SCO-267) Possessing a 2-Carbamoylphenyl Piperidine Moiety. J Med Chem. 2020. Sep 24;63(18):10352–10379. [DOI] [PubMed] [Google Scholar]
- 114. Stone ML, Walker JL, Chisholm D, et al. The addition of rosiglitazone to insulin in adolescents with type 1 diabetes and poor glycaemic control: a randomized-controlled trial. Pediatr Diabetes. 2008. Jul 28;9(4 Pt 1):326–34. * Drug approved by FDA with potential to be repurposed to treat type 1 diabetes.
- 115. Yang Z, Zhou Z, Li X, et al. Rosiglitazone preserves islet beta-cell function of adult-onset latent autoimmune diabetes in 3 years follow-up study. Diabetes Res Clin Pract. 2009. Jan;83(1):54–60. * Drug approved by FDA with potential to be repurposed to treat type 1 diabetes.
- 116.Hakola L, Vuorinen AL, Takkinen HM, et al. Dietary fatty acid intake in childhood and the risk of islet autoimmunity and type 1 diabetes: the DIPP birth cohort study. Eur J Nutr. 2023. Mar;62(2):847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Norris JM, Yin X, Lamb MM, et al. Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes. JAMA. 2007. Sep 26;298(12):1420–8. ** This paper shows the association of dietary omega-3 fatty acid intake with reduction of islet autoimmunity onset in children.
- 118.Miller MR, Yin X, Seifert J, et al. Erythrocyte membrane omega-3 fatty acid levels and omega-3 fatty acid intake are not associated with conversion to type 1 diabetes in children with islet autoimmunity: the Diabetes Autoimmunity Study in the Young (DAISY). Pediatr Diabetes. 2011. Dec;12(8):669–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Niinisto S, Erlund I, Lee HS, et al. Children’s erythrocyte fatty acids are associated with the risk of islet autoimmunity. Sci Rep. 2021. Feb 11;11(1):3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hakola L, Erlund I, Cuthbertson D, et al. Serum fatty acids and risk of developing islet autoimmunity: A nested case-control study within the TRIGR birth cohort. Pediatr Diabetes. 2021. Jun;22(4):577–585. [DOI] [PubMed] [Google Scholar]
- 121.Virtanen SM, Niinisto S, Nevalainen J, et al. Serum fatty acids and risk of advanced beta-cell autoimmunity: a nested case-control study among children with HLA-conferred susceptibility to type I diabetes. Eur J Clin Nutr. 2010. Aug;64(8):792–9. [DOI] [PubMed] [Google Scholar]
- 122.Niinisto S, Takkinen HM, Uusitalo L, et al. Maternal dietary fatty acid intake during pregnancy and the risk of preclinical and clinical type 1 diabetes in the offspring. Br J Nutr. 2014. Mar 14;111(5):895–903. [DOI] [PubMed] [Google Scholar]
- 123.Niinisto S, Takkinen HM, Uusitalo L, et al. Maternal intake of fatty acids and their food sources during lactation and the risk of preclinical and clinical type 1 diabetes in the offspring. Acta Diabetol. 2015. Aug;52(4):763–72. [DOI] [PubMed] [Google Scholar]
- 124.Syrjala E, Nevalainen J, Peltonen J, et al. A Joint Modeling Approach for Childhood Meat, Fish and Egg Consumption and the Risk of Advanced Islet Autoimmunity. Sci Rep. 2019. May 23;9(1):7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Buckner T, Vanderlinden LA, DeFelice BC, et al. The oxylipin profile is associated with development of type 1 diabetes: the Diabetes Autoimmunity Study in the Young (DAISY). Diabetologia. 2021. Aug;64(8):1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hakola L, Oikarinen M, Niinisto S, et al. Serum 25-hydroxyvitamin D and fatty acids in relation to the risk of microbial infections in children: The TRIGR Divia study. Clin Nutr. 2022. Dec;41(12):2729–2739. [DOI] [PubMed] [Google Scholar]
- 127.Radzikowska U, Rinaldi AO, Celebi Sozener Z, et al. The Influence of Dietary Fatty Acids on Immune Responses. Nutrients. 2019. Dec 6;11(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Uusitalo L, Nevalainen J, Salminen I, et al. Fatty acids in serum and diet--a canonical correlation analysis among toddlers. Matern Child Nutr. 2013. Jul;9(3):381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Weitkunat K, Schumann S, Nickel D, et al. Odd-chain fatty acids as a biomarker for dietary fiber intake: a novel pathway for endogenous production from propionate. Am J Clin Nutr. 2017. Jun;105(6):1544–1551. [DOI] [PubMed] [Google Scholar]
- 130.Lampousi AM, Carlsson S, Lofvenborg JE. Dietary factors and risk of islet autoimmunity and type 1 diabetes: a systematic review and meta-analysis. EBioMedicine. 2021. Oct;72:103633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ma W, Wu JH, Wang Q, et al. Prospective association of fatty acids in the de novo lipogenesis pathway with risk of type 2 diabetes: the Cardiovascular Health Study. Am J Clin Nutr. 2015. Jan;101(1):153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Harding SD, Armstrong JF, Faccenda E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2022: curating pharmacology for COVID-19, malaria and antibacterials. Nucleic Acids Res. 2022. Jan 7;50(D1):D1282–D1294. ** Database with curated information about drugs and their targets.
- 133.Heitmeier MR, Kelly CB, Ensor NJ, et al. Role of cyclooxygenase-2 in cytokine-induced beta-cell dysfunction and damage by isolated rat and human islets. The Journal of biological chemistry. 2004. Dec 17;279(51):53145–51. [DOI] [PubMed] [Google Scholar]
- 134.Lundgren M, Steed LJ, Tamura R, et al. Analgesic antipyretic use among young children in the TEDDY study: no association with islet autoimmunity. BMC Pediatr. 2017. May 16;17(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kawai S, Nishida S, Kato M, et al. Comparison of cyclooxygenase-1 and −2 inhibitory activities of various nonsteroidal anti-inflammatory drugs using human platelets and synovial cells. Eur J Pharmacol. 1998. Apr 17;347(1):87–94. [DOI] [PubMed] [Google Scholar]
- 136.Hwang HJ, Park KS, Choi JH, et al. Zafirlukast promotes insulin secretion by increasing calcium influx through L-type calcium channels. J Cell Physiol. 2018. Nov;233(11):8701–8710. [DOI] [PubMed] [Google Scholar]
- 137.De Livera AM, Reutens A, Cooper M, et al. Evaluating the efficacy and safety of GKT137831 in adults with type 1 diabetes and persistently elevated urinary albumin excretion: a statistical analysis plan. Trials. 2020. Jun 3;21(1):459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Reutens AT, Jandeleit-Dahm K, Thomas M, et al. A physician-initiated double-blind, randomised, placebo-controlled, phase 2 study evaluating the efficacy and safety of inhibition of NADPH oxidase with the first-in-class Nox-1/4 inhibitor, GKT137831, in adults with type 1 diabetes and persistently elevated urinary albumin excretion: Protocol and statistical considerations. Contemp Clin Trials. 2020. Mar;90:105892. [DOI] [PubMed] [Google Scholar]
- 139.Cadario F, Pozzi E, Rizzollo S, et al. Vitamin D and omega-3 Supplementations in Mediterranean Diet During the 1st Year of Overt Type 1 Diabetes: A Cohort Study. Nutrients. 2019. Sep 9;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Duggan ST, Keam SJ, Burness CB. Selexipag: A Review in Pulmonary Arterial Hypertension. Am J Cardiovasc Drugs. 2017. Feb;17(1):73–80. * Drug approved by FDA for treating pulmory hypertension with potential to be repurposed to treat T1D
- 141.Henderson GC. Plasma Free Fatty Acid Concentration as a Modifiable Risk Factor for Metabolic Disease. Nutrients. 2021. Jul 28;13(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cholewski M, Tomczykowa M, Tomczyk M. A Comprehensive Review of Chemistry, Sources and Bioavailability of Omega-3 Fatty Acids. Nutrients. 2018. Nov 4;10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]