

SUMMARY
Ferroptosis is a form of regulated cell death with roles in degenerative diseases and cancer. Excessive iron-catalyzed peroxidation of membrane phospholipids, especially those containing the polyunsaturated fatty acid arachidonic acid (AA), is central in driving ferroptosis. Here, we reveal that an understudied Golgi-resident scaffold protein, MMD, promotes susceptibility to ferroptosis in ovarian and renal carcinoma cells in an ACSL4- and MBOAT7-dependent manner. Mechanistically, MMD physically interacts with both ACSL4 and MBOAT7, two enzymes that catalyze sequential steps to incorporate AA in phosphatidylinositol (PI) lipids. Thus, MMD increases the flux of AA into PI, resulting in heightened cellular levels of AA-PI and other AA-containing phospholipid species. This molecular mechanism points to a pro-ferroptotic role for MBOAT7 and AA-PI, with potential therapeutic implications, and reveals that MMD is an important regulator of cellular lipid metabolism.
In brief
Phadnis et al. show that an understudied membrane scaffold protein, MMD, interacts with ACSL4 and MBOAT7 to promote incorporation of arachidonic acid into phosphatidylinositol (PI). This MMD-potentiated ACSL4-MBOAT7 pathway drives ferroptosis susceptibility in ovarian and renal carcinoma cells and highlights a potential role for PI phospholipids in ferroptotic cell death.
Graphical abstract
INTRODUCTION
Ferroptosis is a form of non-apoptotic cell death that results from excessive iron-catalyzed peroxidation of membrane phospholipids.1,2 Ferroptosis may contribute to cell death in degenerative diseases and acute injury of the kidney, liver, heart, and brain.3–6 Moreover, ferroptosis inducers can selectively target aggressive, mesenchymal-enriched and therapy-resistant cancer cells.7,8 Mounting evidence suggests that ferroptosis may also be induced in cancer cells by CD8+ T cells.9,10 Hence, insights into molecular mechanisms underlying ferroptosis susceptibility may translate to therapeutic interventions for various diseases.
The ferroptosis-susceptible cell state is characterized by abundant polyunsaturated fatty acid (PUFA)-phospholipids, such as phosphatidylethanolamine (PE) and phosphatidylcho-line (PC) containing arachidonic acid (AA; 20:4) or adrenic acid (22:4), which serve as critical substrates of peroxidation.11 Several lipid metabolic enzymes, including acyl-coenzyme A (CoA) synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and AGPS, contribute to PUFA-phospholipid synthesis and remodeling, thereby shaping this cell state.12,13 Lipid peroxides are generated when bis-allylic hydrogens on the PUFA chains of PUFA-phospholipids are abstracted by lipoxygenase enzymes or reactive oxygen species (ROS) generated by iron-dependent Fenton chemistry.14 Lipid peroxidation is counteracted by several repair systems, especially the system xc−/glutathione/glutathione peroxidase 4 (GPX4), ferroptosis suppressor protein 1 (FSP1)/CoQ10, and GCH1/BH4 pathways.15–19 Upon inhibition of one or more of these pathways in cells poised for ferroptosis, a radical chain reaction ensues that leads to ferroptotic cell death.2
We previously reported the findings of two independently performed, genome-wide CRISPR-Cas9 screens in ovarian and renal cancer cells, which revealed genes encoding proteins that promote ferroptosis susceptibility.12,20 At the intersection of these two screens were 12 candidate genes, the loss of which reduced ferroptosis sensitivity; these included the ACSL4, LPCAT3, and AGPS genes, as well as the monocyte-to-macrophage differentiation-related (MMD) gene.
MMD, also termed PAQR11, is a poorly studied member of the progestin and adipoQ receptor (PAQR) family and functions as an integral endomembrane scaffold protein to promote vesicle trafficking, mitogenic signaling, and metastatic outgrowth, a setting in which ferroptosis induction may offer therapeutic utility.21–24 It was unclear how the known molecular functions of MMD might contribute to increased ferroptosis susceptibility. Here, we reveal that MMD scaffolds sequentially acting, membrane-embedded metabolic enzymes to increase levels of phosphatidylinositol lipids containing AA, thereby promoting ferroptosis susceptibility in ovarian and renal carcinoma cells.
RESULTS
MMD promotes susceptibility to ferroptosis
MMD was one of only 12 putative pro-ferroptotic genes identified in common by two previously published CRISPR-Cas9 ferroptosis suppressor screens. These independently conducted screens were performed in the OVCAR-8 (human high-grade serous ovarian carcinoma) and 786-O (human clear cell renal cell carcinoma) cell lines (Figures 1A and S1A).12,20 Given that ovarian and renal cancers are highly aggressive and generally show heightened sensitivity to ferroptosis inducers,8 we investigated the role of MMD in modulating ferroptosis susceptibility in these cell lines.
Figure 1. MMD promotes susceptibility to ferroptosis.

See also Figure S1.
(A) Venn diagram showing the 12 putative pro-ferroptosis genes identified in two previously published CRISPR screens. Data from Zou et al12
(B) Schematic of some key processes and proteins in ferroptosis; ferroptosis-inducing drugs in red.
(C) Viability of OVCAR-8 control (NT sg) and MMD KO cells in response to indicated concentrations of ferroptosis inducers.
(D) Viability of 786-O control (NT sg) and MMD KO cells in response to indicated concentrations of ferroptosis inducers.
(E) Immunoblot of membrane proteins in OVCAR-8 NT sg cells, MMD KO cells transduced with empty vector (MMD KO + EV), and MMD KO cells transduced withvector containing HA-MMD (MMD KO + MMD). COXIV was used as a loading control.
(F) RT-qPCR in OVCAR-8 MMD KO + EV and MMD KO + MMD cells, normalized to NT sg.
(G) Viability of OVCAR-8 NT sg, MMD KO + EV, and MMD KO + MMD cells in response to indicated concentrations of ferroptosis inducers.
(H) Immunoblot of membrane proteins in OVCAR-8 cells transduced with EV or vector containing HA-MMD (MMD). COXIV was used as a loading control.
(I) RT-qPCR in OVCAR-8 MMD OE cells, normalized to EV.
(K) Viability of OVCAR-8 EV and MMD OE cells in response to indicated concentrations of ferroptosis inducers.
(K) Viability of OVCAR-8 MMD OE cells in response to ML210 when treated concurrently with Fer-1, Z-VAD-FMK, Nec-1, or no drug (untreated).
(L) Boxplot in which each point represents the Z-scored Pearson correlation coefficient of expression of MMD with area under the viability curve for a distinct cytotoxic drug (481 drugs total) across 860 cancer cell lines. Boxplot interquartile range was set to ~0.75. Ferroptosis inducers with relatively low Z scores identified in red. Data from the Cancer Therapeutics Response Portal.
For cell viability curves, data points or bar graphs show mean ± SD of n = 3 biological replicates; for RT-qPCR graphs, individual data points are shown on a bar graph representing mean ± SD of n = 3 biological replicates; all experimental data are representative of three independent experiments. Statistical analyses by Student’s t test; ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.
Using CRISPR-Cas9, we established clonal derivatives of the OVCAR-8 and 786-O cell lines with genetic deletions at the MMD locus (Figure S1B) and resulting loss of MMD protein (Figure S1C). Indeed, when compared to control cells expressing a non-targeting control sgRNA (NT sg), MMD knockout (KO) cells were less sensitive to a spectrum of ferroptosis inducers that act via distinct biochemical mechanisms; among these were the covalent GPX4 inhibitors RSL3 and ML210, the system xc_ inhibitor erastin (ERA), and the GPX4-depleting compound FIN5616,19,25,26 (Figures 1B–1D, S1D, and S1E). Consistent with the observed cell viability responses, lipid peroxidation upon ML210 treatment was attenuated in MMD KO cells compared to control cells (Figures S1F and S1G).
To confirm that these effects could be attributed specifically to loss of MMD function, we expressed an sgRNA-resistant, HA-tagged MMD cDNA in MMD KO cells (MMD KO + MMD; Figures 1E, 1F, and S1H). Re-expression of MMD partially reverted the sensitivity of MMD KO cells to all four ferroptosis inducers (Figure 1G). Moreover, forced overexpression (OE) of HA-MMD in OVCAR-8 or 786-O parental cells (Figure 1H, 1I, S1H, and S1I) resulted in hypersensitivity to ferroptosis inducers (Figures 1J and S1J). Further supporting the conclusion that this increased cell death was due to ferroptosis, the viability of ML210-treated OVCAR-8 MMD OE cells could be rescued by co-treatment with the ferroptosis inhibitor ferrostatin-1 (Fer-1) but not with the apoptosis inhibitor Z-VAD-FMK or necroptosis inhibitor necrostatin-1 (Nec-1) (Figure 1K). In contrast to MMD, CRISPR-Cas9 editing at the closely related MMD2 locus (Figures S1K and S1L) did not affect susceptibility to RSL3 or ML210 (Figure S1M), indicating that the ferroptosis-resistant phenotype is specific to loss of MMD. Together, these data verified that MMD supports ferroptosis sensitivity in OVCAR-8 and 786-O cells.
We also noted that, in a survey of 800+ cancer cell lines profiled in the Cancer Therapeutics Response Portal, high expression of MMD preferentially correlates with sensitivity to four well-established ferroptosis inducers vs. 400+ other cytotoxic compounds27–29 (Figure 1L). Given that MMD was originally identified as an mRNA upregulated during monocyte-to-macrophage differentiation,22 we also investigated whether monocytes and macrophages display differential sensitivity to ferroptosis. Correlating with increased MMD expression, in vitro-differentiated human macrophages or PMA-differentiated THP-1 macrophage-like cells were more sensitive to ML210 than primary human monocytes from unmatched individuals or undifferentiated THP-1 monocyte-like cells, respectively (Figures S1N–S1Q). Collectively, these results indicate that MMD is a bona fide enabler of ferroptosis susceptibility in ovarian and renal cancer cells and suggest that its role in ferroptosis may be generalizable to other cell types as well.
MMD increases cellular levels of polyunsaturated phospholipids
We next pursued the mechanism by which MMD promotes ferroptosis susceptibility. To begin, we compared levels of known ferroptosis regulators in OVCAR-8 and 786-O control vs. MMD KO cells, reasoning that any factors influenced by MMD should be altered consistently in both cell line models. Upon MMD KO, we observed no consistent changes in intracellular levels of reduced or oxidized glutathione (GSH and GSSG, respectively) as measured by polar metabolomics (Figure S2A) or in protein levels of ACSL4, GPX4, or FSP1 (Figures S2B and S2C).
Given that membrane lipid composition is a key determinant of ferroptosis sensitivity, we performed a lipidomics analysis of OVCAR-8 and 786-O MMD KO and control cells and assessed effects of MMD KO in an unbiased manner. We identified 17 phospholipid species whose relative abundance was significantly reduced (p < 0.05) upon MMD KO in both OVCAR-8 and 786-O cells (Figure S2D). Of these, 15 were PUFA-phospholipids and eight were similar to oxidized phospholipids characterized by others as potential pro-ferroptotic death signals (see Table S1 of Kagan et al.11) (Figures 2A and 2B), suggesting that some of these lipid species may mediate MMD’s pro-ferroptotic functions.
Figure 2. MMD increases cellular levels of polyunsaturated phospholipids.

See also Figure S2.
(A) Heatmap showing the 21 lipid species whose relative abundance is consistently (same direction) and significantly (p < 0.05 by Student’s t test) altered in both OVCAR-8 and 786-O cells upon MMD KO. Species are annotated as containing AA (20:4), belonging to the PI subclass, and/or as similar (same head group and PUFA chain) to ferroptosis-relevant species identified in Kagan et al.11
(B) Fold changes of species downregulated upon MMD KO from part (A) that were also identified in Kagan et al. Abundances in OVCAR-8 or 786-O MMD KO cells were normalized to those of the corresponding NT sg cell line. Individual data points are shown on a bar graph representing mean ± SD of n = 3 biological replicates.
(C) Distribution of PUFA chains of species from (A) that were PUFA-phospholipids downregulated upon MMD KO.
(D) Pie charts, by phospholipid subclass, showing proportions of lipid species containing AA that were identified in (A) (blue or red) vs. those that did not consistently change upon MMD KO in OVCAR-8 and 786-O cells (gray).
(E) Immunoblot in OVCAR-8 NT sg and ACSL4 KO cells. GAPDH was used as a loading control. Data are representative of two independent experiments.
(F) Viability of OVCAR-8 NT sg, MMD KO, and ACSL4 KO cells in response to indicated concentrations of BSA-conjugated palmitate. Viabilities were normalized to the 750 μM BSA-control condition.
Data points show mean ± SD of n = 3 biological replicates and data are representative of three independent experiments. All lipidomics plots in this figure are analyses of a single lipidomics experiment.
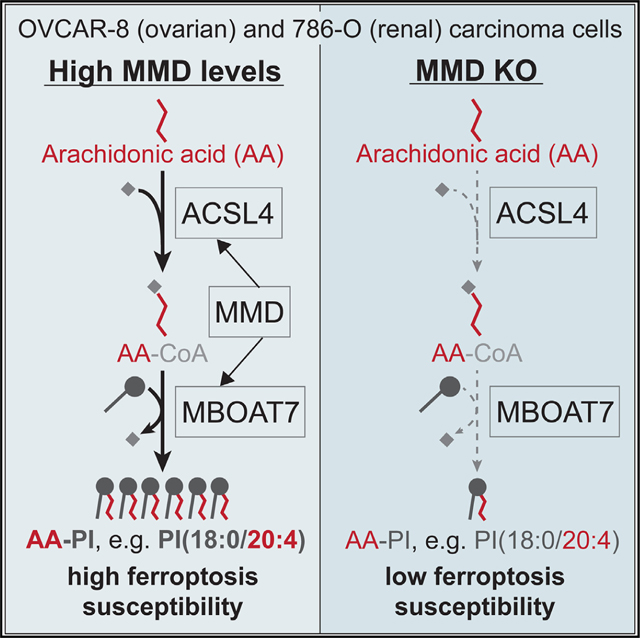
Noting that many of these 17 phospholipids had a phosphatidylinositol (PI) head group and/or contained AA (20:4) (Figure 2A), we further analyzed their head group and fatty acyl composition. In fact, AA was present in nearly half of the 15 PUFA-phospholipids that were consistently downregulated upon MMD KO (Figure 2C). Surprisingly, four out of seven PI species containing AA were consistently downregulated in MMD KO cells, compared to only one out of 15 AA-PE and two out of 11 AA-PC species (Figure 2D). Thus, MMD has the broadest effect on AA-containing species in the PI class.
To assess the cell-biological relevance of lower levels of PUFA-phospholipids in MMD KO cells, we treated them with high doses of exogenous palmitate, which preferentially kills cells with elevated saturated-to-unsaturated phospholipid ratios, such as ACSL4 KO cells.30,31 Indeed, we found that MMD KO cells exhibited increased sensitivity to palmitate-induced cell death, although ACSL4 KO cells were even more sensitive, consistent with expected broad reductions in lipid unsaturation upon ACSL4 KO31 (Figures 2E and 2F). Collectively, these results support the notion that MMD substantially increases cellular levels of PUFA-phospholipids, especially AA-PI, which may render cells more sensitive to induced ferroptosis.
MMD interacts with consecutively acting, membrane-embedded lipid metabolic enzymes ACSL-4 and MBOAT7
MMD is a poorly studied Golgi-resident scaffold protein with no known enzymatic activity of its own,23 so its effect on levels of PUFA-phospholipids was intriguing. To define the mechanism linking MMD to lipid metabolism, we explored a published proximity labeling dataset that identified hundreds of interactors of MMD in H1299 human lung carcinoma cells.32 At the intersection of this dataset and the list of hits from both CRISPR screens was a single protein: the 79-kDa membrane-embedded isoform of ACSL4 (henceforth memACSL4), a PUFA-metabolizing enzyme with an established pro-ferroptotic function13 (Figure 3A).
Figure 3. MMD interacts with consecutively acting, membrane-embedded lipid metabolic enzymes ACSL4 and MBOAT7.

See also Figure S3.
(A) Venn diagram showing the intersection of the two published CRISPR screens with a published proximity labeling (BioID) dataset identifying potential interactors of MMD in H1299 cells. Numbers in circles represent total hits in each dataset.
(B) Ordered Pearson correlation coefficients of the effects of loss of MMD (left), ACSL4 (middle), or MBOAT7 (right) with that of loss of every other gene in the genome across all cancer cell lines in DepMap (22Q2). See Table S1 for full ranked lists.
(C) Schematic of the consecutive reactions that ACSL4 and MBOAT7 catalyze to generate PI(18:0/20:4) and other AA-PI.
(D) Volcano plot of hits with average log2fold change ≥1.5 from previously published 786-O CRISPR screen, highlighting MMD, ACSL4, and MBOAT7. Data from Zou et al.12
(E) Schematic of MaMTH. Cub, C-terminal half of ubiquitin; Nub, N-terminal half of ubiquitin; TF, transcription factor; DUBs, deubiquitinase enzymes. Bait andprey are tagged such that the Cub and Nub respectively are cytosol facing. When the bait and prey interact, Cub and Nub are reconstituted as pseudoubiquitin, allowing DUBs to recognize pseudoubiquitin and cleave off the TF. The TF is now free to enter the nucleus and promote expression of the luciferase reporter.
(F) Luciferase activity (relative light units [RLU]) indicating MaMTH reporter activity in HEK293 cells expressing C-terminally tagged MBOAT7 (MBOAT7-C) bait and N-terminally tagged MMD (MMD-N) prey or N-terminally tagged PEX7 (PEX7-N) prey. Bars show mean ± SD of n = 3 biological replicates.
(G) Luciferase activity (RLU) indicating MaMTH reporter activity in OVCAR-8 parental cells. Left: cells expressing MBOAT7-C bait and MMD-N prey, C-terminally tagged membrane-embedded ACSL4 (memACSL4-C) prey, or PEX7-N prey. Right: cells expressing memACSL4-C bait and MMD-N prey or PEX7-N prey. Bars show mean ± SD of n = 3 biological replicates.
(H) Relative MaMTH reporter activity in OVCAR-8 NT sg or MMD KO cells transfected with MBOAT7-C bait and memACSL4-C prey. Normalized signal is expressed as a percentage of signal generated by the positive control (signal in cells transfected with the luciferase-driving TF) minus the signal generated by the negative control (N-terminally tagged PEX7 prey) in each cell line. Bars show mean ± SD of n = 5 biological replicates.
All experimental figure panels are representative of three independent experiments. Statistical analyses by Student’s t test; ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.
To further explore whether this potential protein-protein interaction was relevant for MMD’s molecular functions, we mined the DepMap database for codependencies of MMD across over 1,000 cancer cell lines.33,34 Such codependencies can be used to generate hypotheses about functional similarity or shared membership in a protein complex.35 Strikingly, out of 17,385 ranked genes, MMD’s top codependencies included not only ACSL4 (ranked #67) but also membrane-bound O-acyltransferase family member 7 (MBOAT7, ranked #7) (Figure 3B). MMD, ACSL4, and MBOAT7 reciprocally ranked within the top 0.75% of one another’s codependencies, suggesting a functional link (Figure 3B; Table S1).
These observations were noteworthy because ACSL4 and MBOAT7 catalyze sequential reactions to generate AA-PI lipids—the main group of phospholipids upregulated by MMD (Figure 2D)—in membranes of the endoplasmic reticulum (ER) and Golgi36,37 (Figure 3C). Specifically, ACSL4 converts long-chain PUFAs, especially AA, to fatty acyl-CoA thioesters that serve, in turn, as key intermediates in lipid metabolism.38,39 MBOAT7 is a Lands cycle enzyme40 that preferentially esterifies the resulting AA-CoA into lyso-PI, leading to enrichment of AA at the sn2 position of PI.41–43 While MBOAT7 had not been previously implicated in ferroptosis, it was identified in one of our ferroptosis suppressor CRISPR screens (Figure 3D). Accordingly, we hypothesized that MMD physically interacts with both ACSL4 and MBOAT7, either successively or simultaneously, to direct biosynthetic flux of AA toward AA-PI, which in turn heightens ferroptosis sensitivity.
To begin to test this hypothesis, we sought to determine whether MMD could interact with MBOAT7 using the mammalian membrane two-hybrid (MaMTH) assay.44–50 Adapting the yeast two-hybrid assay concept to mammalian cells, MaMTH involves expression of a membrane protein ‘‘bait’’ tagged with the C-terminal half of ubiquitin linked to a transcription factor and a protein ‘‘prey’’ tagged with the N-terminal half of ubiquitin. In principle, any physical interaction of bait and prey should reconstitute a pseudoubiquitin, which is cleaved by deubiquitinases, freeing the transcription factor to drive expression of luciferase (schematized in Figure 3E).44 As we found, in HEK293 human embryonic kidney cells, co-expression of MBOAT7 bait and MMD prey generated higher luciferase signals compared to that of MBOAT7 bait and a negative control peroxisomal protein prey, PEX7 (Figure 3F). This provided proof of concept that MMD and MBOAT7 can physically interact in cells.
We next conducted additional MaMTH experiments in OVCAR-8 cells to confirm that interactions between MMD, MBOAT7, and ACSL4 can occur in our ovarian carcinoma cell model. Indeed, expression of MBOAT7 bait with MMD prey or membrane-embedded ACSL4 prey (memACSL4, the isoform of ACSL4 identified in the published proximity labeling dataset described in Figure 3A) generated luciferase signals above negative control levels, indicating that MBOAT7 can physically interact with both MMD and memACSL4 in OVCAR-8 cells (Figure 3G). Furthermore, expression of memACSL4 bait with MMD prey demonstrated that memACSL4 can interact with MMD in OVCAR-8 cells (Figure 3G), establishing the potential for pairwise interactions to occur among all three of these proteins. Corroborating the interaction data, immunofluorescence analyses showed that ectopically expressed HA-MMD localized to the Golgi and ER based on its colocalization with the Golgi-resident protein GM130 and the ER-resident protein calnexin respectively (Figure S3). We also observed some spatial overlap between HA-MMD and endogenous ACSL4, and HAMMD and ectopically expressed FLAG-MBOAT7, especially in perinuclear regions of the ER and/or Golgi (Figure S3). Overall, these interaction data support a model in which MMD, MBOAT7, and ACSL4 all interact with one another at cellular endomembranes.
Finally, to test the hypothesis that MMD potentiates interactions between ACSL4 and MBOAT7, we conducted MaMTH experiments in OVCAR-8 NT sg and MMD KO cells. Compared to control cells, MMD KO cells showed reduced, but persistent, interactions between MBOAT7 bait and memACSL4 prey (Figure 3H). Thus, MMD is not required for, but enhances, interactions between memACSL4 and MBOAT7. Taken together, these results demonstrate that MMD can interact with and affect interactions between two sequentially acting lipid metabolic enzymes that incorporate AA into PI lipids.
MMD increases ferroptosis susceptibility in an ACSL4and MBOAT7-dependent manner
Our working model predicts that MMD should increase ferroptosis sensitivity only in the presence of ACSL4 and MBOAT7. To test this hypothesis, we used CRISPR-Cas9 to deplete either ACSL4 or MBOAT7 in control (empty vector [EV]) and MMD OE cells (Figures 4A and 4C). Depletion of either ACSL4 or MBOAT7 alone rendered control cells resistant to ferroptosis, while MMD OE alone sensitized control cells to ferroptosis (Figures 4B and 4D). Importantly, however, MMD OE failed to increase ferroptosis susceptibility in cells lacking either ACSL4 or MBOAT7 (Figures 4B and 4D). Moreover, clonal MMD KO cells with additional population-level depletion of MBOAT7 were as resistant to ferroptosis as MMD KO cells, suggesting no additive genetic effects (Figures S4A and S4B).
Figure 4. MMD increases ferroptosis susceptibility in an ACSL4- and MBOAT7-dependent manner.

See also Figure S4.
(A) Immunoblot of membrane proteins in OVCAR-8 EV cells transduced with a CRISPR vector containing NT sg (EV + NT sg) or ACSL4 sg (EV + ACSL4 sg) and inOVCAR-8 MMD OE cells transduced with a CRISPR vector containing NT sg (MMD + NT sg) or ACSL4 sg (MMD + ACSL4 sg). Overexpressed MMD was HA tagged. COXIV was used as a loading control.
(B) Viability of cell lines from (A) in response to indicated concentrations of ferroptosis inducers.
(C) Immunoblot in OVCAR-8 EV cells transduced with a CRISPR vector containing NT sg (EV + NT sg) or MBOAT7 sg (EV + MBOAT7 sg) and in OVCAR-8 MMD OE cells transduced with a CRISPR vector containing NT sg (MMD + NT sg) or MBOAT7 sg (MMD + MBOAT7 sg). Overexpressed MMD was HA tagged. The MMD polyclonal antibody was used and images are displayed with both short exposure (SE) and long exposure (LE) times. COXIV was used as a loading control.
(D) Viability of cell lines from (C) in response to indicated concentrations of ferroptosis inducers.
Data points plotted are mean ± SD of n = 3 biological replicates, and experimental figure panels are representative of three independent experiments.
Since MBOAT7 belongs to the same family of proteins as LPCAT3, the enzyme known to promote ferroptosis sensitivity downstream of ACSL4 by esterifying PUFA-CoA into PC, PE, and phosphatidylserine (PS),13,51,52 we tested whether LPCAT3 may be functionally related to MMD as are ACSL4 and MBOAT7. In contrast to MBOAT7, when we used CRISPR-Cas9 to edit the LPCAT3 locus in control and MMD OE cells, we found that MMD OE could still sensitize LPCAT3 sg cells to ferroptosis (Figures S4C–S4E). Together, these tests of genetic epistasis underscore the idea that MMD collaborates specifically with ACSL4 and MBOAT7 to increase ferroptosis susceptibility.
As a complementary approach, we tested whether ectopically expressed memACSL4 and MBOAT7 could resensitize MMD KO cells to ferroptosis. Interestingly, OE of HA-tagged memACSL4 or concurrent OE of HA-memACSL4 and FLAG-tagged MBOAT7, but not OE of FLAG-MBOAT7 alone, could partially restore the ferroptosis susceptibility of MMD KO cells (Figures S4F and S4G). Taken together, these data show that, in the absence of MMD, OE of MBOAT7 (at least to the level achieved) is not sufficient on its own to increase ferroptosis susceptibility. In contrast, OE of memACSL4 can overcome some of the ferroptosis resistance caused by loss of MMD, consistent with the continued presence of LPCAT3 and other downstream enzymes that can utilize ACSL4-catalyzed fatty acids to generate pro-ferroptotic PUFA-phospholipids. These results suggest that MBOAT7, but not memACSL4, requires MMD to perform its pro-ferroptotic function, demonstrating a tighter link between MMD and MBOAT7 function than between MMD and ACSL4 function.
Since our data also implicated MBOAT7 as a regulator of ferroptosis susceptibility, we tested its effects in other cell lines. In 786-O renal carcinoma cells, Huh7 hepatoma cells, and HT-1080 fibrosarcoma cells, MBOAT7 depletion (Figures S4H, S4J, and S4L) reduced sensitivity to ML210-induced ferroptosis (Figures S4I, S4K, and S4M), supporting a generalizable role for MBOAT7 in influencing ferroptosis sensitivity.
MMD directs the flux of AA into PI in an MBOAT7-dependent manner and generates putative proferroptotic lipids
To gain greater mechanistic insight, we tested the hypothesis that MMD affects flux through the ACSL4-MBOAT7 PI remodeling pathway. We incubated EV + NT sg (control), MMD KO + EV, and MMD KO + MMD cells with radiolabeled AA and quantified incorporation into PI, PE, PC, PS, phosphatidylglycerol (PG), and phosphatidic acid (PA) lipids 15 and 30 min later. Here we found that MMD KO cells incorporated AA into PI at a slower rate than control cells, and this was partially reversed upon restoration of MMD expression (Figure 5A), consistent with partial reversion of ferroptosis susceptibility in MMD KO + MMD cells (Figure 1H). In sharp contrast, AA incorporation into any other tested phospholipid subclass did not recapitulate this pattern (Figure S5A). Hence, MMD acutely affects incorporation of AA into PI, but not other phospholipid subclasses. Combined with our lipidomics data showing that the presence of MMD increases relative cellular levels of PI(18:0/20:4) (Figure 5C, top left panel), the major PI species in cells,53 this indicates that MMD increases the flux of AA into PI.
Figure 5. MMD directs the flux of AA into PI in an MBOAT7-dependent manner and generates putative pro-ferroptotic lipids.

See also Figure S5.
(A) Time course of [3H]AA content in PI phospholipids, normalized to total protein, in OVCAR-8 EV + NT sg (control), MMD KO + EV, and MMD KO + MMD cells. Data are from one independent experiment and show mean ± SD of n = 3 biological replicates.
(B) Time course of [3H]AA content in PI phospholipids, normalized to total protein, in OVCAR-8 EV + NT sg (control), MMD OE + NT sg, EV + MBOAT7 sg, and MMD OE + MBOAT7 sg cells (same cells as from Figure 4C). This experiment was conducted once, independently of the experiment in (A). Data show mean ± SD of n = 3 biological replicates.
(C) Barplots showing relative abundance of four out of six candidate pro-ferroptotic species identified by unbiased analysis of lipidomics data (see Figure S5B). Fold changes were normalized to the corresponding control cell line (NT sg for MMD KO + EV and MMD KO + MMD; EV + NT sg for MMD + NT sg, EV + MBOAT7 sg, and MMD + MBOAT7 sg). Individual data points are shown on a bar graph representing mean ± SD of n = 3 biological replicates. Statistical analyses by ordinary one-way ANOVA with Dunnett’s multiple comparisons test.
(D) Heatmap of candidate pro-ferroptotic species downstream of MMD and MBOAT7 based on unbiased analysis of another lipidomics dataset (see Figure S5C). Species are annotated as containing AA (20:4) or belonging to the PI subclass.
(E) Pie charts, by phospholipid subclass, showing proportions of lipid species containing AA that were consistently upregulated (red) or consistently down-regulated (blue) upon MMD or MBOAT7 depletion vs. those that did not consistently change (gray). Based on same lipidomics dataset analyzed in (D).
To verify that the impact of MMD on AA incorporation into PI is dependent on MBOAT7, we conducted a similar experiment in EV + NT sg, MMD + NT sg, EV + MBOAT7 sg, and MMD + MBOAT7 sg cells. Correlating with the ferroptosis susceptibility phenotypes, MMD OE alone dramatically increased the rate of incorporation of AA into PI, and MBOAT7 depletion alone decreased this rate, consistent with previous work54 (Figure 5B). However, MMD OE in MBOAT7-depleted cells had minimal effect on the rate of incorporation of AA into PI (Figure 5B). Steady-state levels of PI(18:0/20:4) in these cell lines were concordant with their relative rates of AA incorporation into PI (Figure 5C, top left panel), indicating that MBOAT7 mediates the effects of MMD on AA flux into PI. These results directly demonstrate that MMD potentiates flux through the pathway converting AA into AA-PI, and further implies that any effects of MMD on AA-containing lipids outside the PI subclass are likely indirect.
In search of putative pro-ferroptotic lipids directly or indirectly generated by MMD and MBOAT7, we performed further unbiased lipidomics analyses to identify lipids whose steady-state levels in cell lines correlated with their ferroptosis susceptibility phenotypes (Figure S5B). Our pipeline identified six lipids, four of which were PI, PE, PC, and DG (diacylglycerol) with fatty acyl composition (18:0/20:4) (Figure 5C). Levels of each of these lipids decreased upon MMD KO, increased upon MMD OE in control or MMD KO cells, decreased upon MBOAT7-depletion, and were not significantly altered upon MMD OE in MBOAT7depleted cells. A similar unbiased analysis of an independent lipidomics experiment (Figure S5C) reinforced the potent effect of MMD or MBOAT7 depletion on several AA-PI phospholipids, as well as on DG(18:0/20:4) and PE(18:0/20:4) (Figures 5D and 5E). Further, levels of PI(18:0/20:4), DG(18:0/20:4), PE(18:0/20:4), and PC(18:0/20:4) were similar between MMD KO, MBOAT7-depleted, and MMD/MBOAT7 double-knockout cells from Figure S4A, indicating that MMD and MBOAT7 act in the same pathway to increase levels of these lipids (Figure S5D). We also verified that levels of these four lipids were not consistently altered upon MMD2 depletion (Figure S5E). Together, these observations highlight the possibility that AA-PI species generate AA-DG and other AA-phospholipids, for example via the PI kinase/phospholipase C (PLC)55 and Kennedy pathways56 (Figure S5F). These AA-phospholipids likely drive the ferroptosis susceptibility of OVCAR-8 cells downstream of MMD and MBOAT7.
DISCUSSION
Highly aggressive cancer cells exhibiting a mesenchymal gene signature, including drug-tolerant persister cells, epithelial-mesenchymal transition-induced cancer cells, sarcomas, and ovarian and renal carcinomas, are difficult to eliminate therapeutically but are often vulnerable to ferroptosis.7,8 In the present study, we sought to investigate the mechanisms underlying such elevated ferroptosis susceptibility, both to offer insights into the biochemical mechanisms underlying ferroptosis and to guide future efforts to develop ferroptosis-inducing cancer therapies.
In this study, we uncovered a role for the understudied Golgi scaffold protein, MMD, in modulating lipid metabolism and ferroptosis susceptibility. More specifically, we demonstrated that MMD interacts with sequential lipid remodeling enzymes ACSL4 and MBOAT7 and increases the relative cellular abundance of AA-phospholipids, likely derived from AA-PI, thereby heightening ferroptosis susceptibility. Previous reports suggested that membrane-embedded ACSL3 and ACSL4 promote metabolic flux of AA toward PI via MBOAT736,57; however, there was no mechanistic explanation indicating precisely how the ACSL4-catalyzed intermediate, AA-CoA, would preferentially and efficiently encounter MBOAT7 in the crowded intracellular milieu. To explain these previous observations, our data support a model in which MMD interacts with both memACSL4 and MBOAT7 to enable rapid incorporation of AA into PI phospholipids. While the precise mechanism by which MMD promotes flux through the ACSL4-MBOAT7 pathway remains to be clarified, we propose two, not mutually exclusive, possibilities: (1) that MMD interacts simultaneously with memACSL4 and MBOAT7, thereby serving as a structural scaffold that enhances spatial proximity between the two enzymes and enables channeling of metabolites (which is achieved in other pathways by direct interactions between sequentially acting enzymes themselves58,59), or (2) that MMD interacts sequentially with memACSL4 and then MBOAT7, carrying MBOAT7 substrate(s) in a lipid binding pocket and/or allosterically enhancing the enzymatic activity of MBOAT7. Regardless of the specific model, this work highlights the importance of integral membrane scaffold proteins in regulating cellular lipid metabolism.
Our work also reveals additional contributors to the ferroptosis-susceptible cell state. LPCAT3, also known as MBOAT5, is well established as an enzyme that promotes ferroptosis sensitivity downstreamofACSL4.13 Our results illustrate that MMD promotes ferroptosis susceptibility through MBOAT7, a protein in the same class of enzymes as LPCAT3 that has not been implicated in ferroptosis. The continued presence of MBOAT7 may explain the relatively modest pro-ferroptotic effect of LPCAT3 compared to ACSL4 that others have observed.13 Conversely, the presence of LPCAT3 may explain why OE of ACSL4 but not MBOAT7 increases ferroptosis sensitivity in the absence of MMD. Stated differently, MBOAT7 may represent an alternative to LPCAT3 in the synthesis of PUFA-phospholipids downstream of ACSL4, with a preference for PI rather than PC, PE, and PS. It is interesting to note that, during revision of this manuscript, an independent publication identified two other members of the MBOAT family of proteins (MBOAT1/LPEAT1 and MBOAT2/LPCAT4) as suppressors of ferroptosis.60 Collectively, there is rapidly accumulating evidence that enzymes involved in the phospholipid-remodeling Lands cycle are critical gatekeepers of ferroptotic cell death.
The role of PI phospholipids in determining ferroptosis susceptibility warrants further investigation. While AA-PE and AAPC species are well-established pro-ferroptotic substrates of lipid peroxidation,11,61 the PI subclass constitutes a relatively minor proportion of cellular phospholipids53 and has not yet been implicated in ferroptosis. Nevertheless, in a previous redox phospholipidomics analysis, oxidized PI(18:0/20:4) showed a significant, greater than 3-fold increase in ferroptotic vs. control cells and demonstrated the highest correlation coefficient with cell death out of 17 species characterized as potential ferroptotic death signals.11 Other published datasets also report PI(18:0/20:4) oxidation in ferroptosis.62–64
In light of our present observations and those of others, we propose at least three, not mutually exclusive, mechanisms by which AA-PI species may promote ferroptosis sensitivity: (1) by themselves serving as direct substrates of lipid peroxidation, (2) by being remodeled to AA-PE or AA-PC species via the PI kinase/PLC and Kennedy pathways, and (3) by potentiating pro-ferroptotic signaling via PLC/protein kinase C (PKC) signaling and generating AA-DG in the process. This signalling mechanism is especially provocative, as recent work showed that PKCβII is recruited to membranes upon lipid peroxidation by an unknown mechanism and, once activated, phosphorylates ACSL4 to increase its activity.65 Classically, PKCβII is activated by interaction with DG generated from PLC signaling in the plasma membrane.66 Thus, it is tempting to speculate that oxidized AA-PI metabolized to AA-DG recruits PKCβII, which enhances ACSL4 activity and sets up a positive feedback loop to promote ferroptotic cell death. This cascade may be dampened in MMD KO cells given their reduced abundance of AA-PI and AA-DG.
Intriguingly, a recent report demonstrated that CD8+ T cell-derived interferon gamma (IFNγ) induces ferroptosis in cancer cells in the presence of exogenous AA and does so in an ACSL4-dependent manner.9 Upon concurrent IFNγ and AA treatment, cancer cells actively incorporated AA into a variety of phospholipids, especially PI.9 Future work should investigate whether MMD and MBOAT7 are involved in this dynamic remodeling toward AA-PI and drive T cell-mediated ferroptosis of cancer cells in vivo. If so, activating the MMD-potentiated ACSL4-MBOAT7 axis may enhance tumor cell killing via ferroptosis during checkpoint immunotherapy.
MMD may also be dynamically upregulated to increase ferroptosis susceptibility in settings beyond cancer, based on our findings in this work in primary human monocytes and macrophages and THP-1 monocyte- and macrophage-like cells. Previously published data showed that, upon PMA differentiation of THP-1 cells, PI(18:0/20:4) is significantly upregulated,67 which is consistent with our findings that increased MMD levels in cancer cells potentiate ACSL4-MBOAT7 axis activity and sensitivity to ferroptosis. However, if MMD underlies the ferroptosis susceptibility of macrophages as well as cancer cells, any MMD-potentiating targeted therapies in cancer may have complex and even conflicting effects on the tumor microenvironment.
In summary, our work demonstrates that MMD interacts with key enzymes in AA-PI synthesis, clarifying its mechanistic role in ferroptosis and opening many avenues for future investigation in the fields of cell death, cancer biology, signal transduction, and molecular metabolism. Modulating MMD, and in turn the ACSL4-MBOAT7 axis, may also offer therapeutic opportunities to enhance or inhibit ferroptosis in relevant disease contexts.
Limitations of the study
While our work defines a clear role for MMD in promoting ferroptosis sensitivity in OVCAR-8 and 786-O cells, it remains to be determined whether this role generalizes to other cancer cell lines and non-cancer contexts, as well as in vivo. Due to technical challenges with co-immunoprecipitation of membrane proteins, we performed MaMTH assays to demonstrate that ectopic tagged forms of MMD, memACSL4, and MBOAT7 can interact with one another in cells. Future work should investigate whether the endogenous proteins interact physically and whether they interact directly with one another. Elucidating the structural basis for these physical interactions may reveal the precise mechanisms by which MMD promotes flux through the ACSL4-MBOAT7 pathway. Finally, while our analyses narrowed down potential causative pro-ferroptotic lipid species, it remains unclear whether AA-PI species can directly promote ferroptosis independently of AA-PE and AA-PC phospholipids.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Whitney S. Henry (whenry@wi.mit.edu).
Materials availability
All new materials and reagents generated in this study are available from the lead contact upon request.
Data and code availability
Raw metabolomics data have been deposited at Metabolights and are publicly available as of the date of publication. The accession number is listed in the key resources table. Spreadsheets related to the four lipidomics datasets and one polar metabolomics dataset reported in this study have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-HA | Cell Signaling Technology | Cat#3724; RRID: AB_1549585 |
| Rabbit polyclonal anti-MMD | Cell Signaling Technology | N/A |
| Rabbit monoclonal anti-MMD | Cell Signaling Technology | Cat#20226 |
| Rabbit polyclonal anti-FSP1 | Cell Signaling Technology | Cat#24972S |
| Rabbit polyclonal anti-ACSL4 | Invitrogen | Cat#PA527137; RRID: AB_2544613 |
| Rabbit monoclonal anti-GPX4 | abcam | Cat#ab125066; RRID: AB_10973901 |
| Rat monoclonal anti-MBOAT7 | Dr. Nozomu Kono (The University of Tokyo, Tokyo, Japan); Lee et al.41 | N/A |
| Mouse monoclonal anti-COXIV | Cell Signaling Technology | Cat#11967; RRID: AB_2797784 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat#2118; RRID: AB_561053 |
| Mouse monoclonal anti-CANX | Invitrogen | Cat#MA3–027; RRID: AB_2069043 |
| Mouse monoclonal anti-HA-Tag Alexa Fluor 647 Conjugate |
Cell Signaling Technology | Cat#3444S; RRID: AB_10693329 |
| Rabbit monoclonal anti-GM130 | Cell Signaling Technology | Cat#12480; RRID: AB_2797933 |
| Rabbit monoclonal Anti-DYKDDDDK (FLAG) | Cell Signaling Technology | Cat#14793S; RRID: AB_2572291 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| One Shot™ MAX Efficiency™ DH5α-T1R competent cells | Invitrogen | 12297016 |
|
| ||
| Biological samples | ||
|
| ||
| Discarded human blood products | Crimson Core Specimen Bank (Brigham & Women’s Hospital, Boston, MA, USA) | https://crimson-core.partners.org/samples/ |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RSL3 | SelleckChem | Cat#S8155 |
| ML210 | Sigma-Aldrich | Cat#SML0521 |
| Erastin | SelleckChem | Cat#S7242 |
| FIN56 | SelleckChem | Cat#S8254 |
| Ferrostatin-1 | Sigma-Aldrich | Cat#SML0583 |
| Z-VAD-FMK | SelleckChem | Cat#S7023 |
| Necrostatin-1 | SelleckChem | Cat#S8037 |
| BSA-control | Cayman Chemical | Cat#29556 |
| BSA-palmitate | Cayman Chemical | Cat#29558 |
| PMA | Sigma-Aldrich | Cat#P8139 |
| Recombinant human M-CSF | Peprotech | Cat#300–25 |
| 3H-Arachidonic acid | Perkin Elmer | Cat#NET298Z050UC |
|
| ||
| Critical commercial assays | ||
|
| ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat#G7571 |
| BODIPY™ 581/591 C11 (Lipid Peroxidation Sensor) | Invitrogen | Cat#D3861 |
|
| ||
| Deposited data | ||
|
| ||
| Lipidomics dataset #1 (OVCAR-8 and 786-O NT sg vs. MMD KO cells) and associated code | This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169857 |
| Lipidomics dataset #3 (OVCAR-8 NT sg, MMD KO, EV + NT sg, and EV + MBOAT7 sg cells) and associated code |
This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169908 |
| Lipidomics dataset #4 (OVCAR-8 NT sg, MMD2 sg bulk, MMD2 KO1, MMD2 KO2, NT sg + NT sg, NT sg + MBOAT7 sg, MMD KO + NT sg, and MMD KO + MBOAT7 sg cells) and associated code | This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169917 |
| Polar metabolomics dataset (OVCAR-8 and 786-O NT sg vs. MMD KO cells) | This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169924 |
| Code for calculating all ranked codependencies of MMD, ACSL4, and MBOAT7 in DepMap | This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169937 |
| All raw mass spectrometry (metabolomics) data | This paper; Metabolights | Metabolights: MTBLS8280 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| OVCAR-8 parental | Dr. Joan Brugge (Harvard Medical School, Boston, MA, USA) | RRID: CVCL_1629 |
| OVCAR-8 NT sg (puro) | This paper | N/A |
| OVCAR-8 NT sg (blast) | This paper | N/A |
| OVCAR-8 MMD KO (puro) | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-EF1a-IRES-blast] EV | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-EF1a-IRES-blast] MMD (HA-MMD) |
This paper | N/A |
| OVCAR-8 [pLJM1] EV | This paper | N/A |
| OVCAR-8 [pLJM1] MMD OE (a.k.a. MMD, HA-MMD) | This paper | N/A |
| OVCAR-8 HA-MMD + [pRH115] FLAG-MBOAT7 | This paper | N/A |
| OVCAR-8 MMD2 sg1 (puro) | This paper | N/A |
| OVCAR-8 MMD2 KO1 (puro) | This paper | N/A |
| OVCAR-8 MMD2 KO2 (puro) | This paper | N/A |
| OVCAR-8 ACSL4 sg (blast) | This paper | N/A |
| OVCAR-8 [pLJM1] EV + NT sg (blast) | This paper | N/A |
| OVCAR-8 [pLJM1] EV + ACSL4 sg (blast) | This paper | N/A |
| OVCAR-8 MMD + NT sg (blast) | This paper | N/A |
| OVCAR-8 MMD + ACSL4 sg (blast) | This paper | N/A |
| OVCAR8 [pLJM1] EV + MBOAT7 sg (blast) | This paper | N/A |
| OVCAR-8 MMD + MBOAT7 sg (blast) | This paper | N/A |
| OVCAR-8 [pLJM1] EV + LPCAT3 sg (blast) | This paper | N/A |
| OVCAR-8 MMD + LPCAT3 sg (blast) | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-mCherry] ACSL4-HA | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-mCherry] FLAG-MBOAT7 | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-mCherry] ACSL4-HA-P2A-FLAG-MBOAT7 | This paper | N/A |
| OVCAR-8 NT sg (puro) + NT sg (blast) | This paper | N/A |
| OVCAR-8 NT sg (puro) + MBOAT7 sg (blast) | This paper | N/A |
| OVCAR-8 MMD KO (puro) + NT sg (blast) | This paper | N/A |
| OVCAR-8 MMD KO (puro) + MBOAT7 sg (blast) | This paper | N/A |
| 786-O parental | Broad Institute Biological Samples Platform (Broad Institute, Cambridge, MA, USA) | RRID: CVCL_1051 |
| 786-O NT sg (blast) | This paper | N/A |
| 786-O MMD KO (blast) | This paper | N/A |
| 786-O [pLJM1] MMD OE (puro) | This paper | N/A |
| 786-O MBOAT7 sg (blast) | This paper | N/A |
| Huh7 WT | Dr. J. Mark Brown (Cleveland Clinic, Cleveland, OH, USA); Helsley et al.68 | N/A |
| Huh7 MBOAT7 KO | Dr. J. Mark Brown (Cleveland Clinic, Cleveland, OH, USA); Helsley et al.68 | N/A |
| HT-1080 parental | Dr. Robert A. Weinberg (Whitehead Institute for Biomedical Research, Cambridge, MA, USA) | RRID: CVCL_0317 |
| HT-1080 NT sg (blast) | This paper | N/A |
| HT-1080 MBOAT7 sg (blast) | This paper | N/A |
| THP-1 | Dr. Jaime Cheah (Koch Institute for Integrative Cancer Research, Cambridge, MA, USA) | RRID: CVCL_0006 |
| HEK293T | ATCC | Cat#CRL-3216; RRID: CVCL_0063 |
| OVCAR-8 parental MaMTH Gaussia princeps luciferase reporter line (B0755) | This paper | N/A |
| OVCAR-8 NT sg MaMTH Gaussia princeps luciferase reporter line (B0781) | This paper | N/A |
| OVCAR-8 MMD KO MaMTH Gaussia princeps luciferase reporter line (B0772) | This paper | N/A |
| HEK293 | Thermo-Fisher | Cat#R78007; RRID: CVCL_0045 |
| HEK293 MaMTH Gaussia princeps luciferase reporter line | Dr. Igor Stagljar (University of Toronto, Toronto, Canada); Saraon et al.49 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| MMD qPCR primer (Fwd, 5’→3′): CCGCTACAAGCCAACTTGCTAT | This paper | N/A |
| MMD qPCR primer (Rev, 5’→3′): AGTCCCATTCCATAAATCCATG | This paper | N/A |
| ACTB qPCR primer (Fwd, 5’→3′): CACCATTGGCAATGAGCGGTTC | This paper | N/A |
| ACTB qPCR primer (Rev, 5’→3′): AGGTCTTTGCGGATGTCCACGT | This paper | N/A |
| sgRNA target sequences, see Table S2 | This paper | N/A |
| CRISPR Sanger sequencing primers, see Table S2 | This paper | N/A |
| MaMTH plasmid cloning primers, see Table S3 | This paper | N/A |
| MaMTH plasmid sequencing primers, see Table S3 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Vector for CRISPR cloning: lentiCRISPRv2-puro | Dr. Feng Zhang (Broad Institute, Cambridge, MA, USA); Sanjana et al.69 | Addgene #52961; RRID: Addgene_52961 |
| Vector for CRISPR cloning: lentiCRISPRv2-blast | Dr. Mohan Babu (University of Regina, Regina, Canada) | Addgene #83480; RRID: Addgene_83480 |
| Vector for cDNA expression: pLJM1 | Dr. Joshua Mendell (UT Southwestern, Dallas, TX, USA); Golden et al.70 | Addgene #91980; RRID: Addgene_91980 |
| Vector for cDNA expression: pLV-EF1a-IRES-blast | Dr. Tobias Meyer (Stanford University School of Medicine, Stanford, CA, USA); Hayer et al.71 | Addgene #85133; RRID: Addgene_85133 |
| Vector for FLAG-MBOAT7 cloning: pRH115 | Dr. Tobiloba Oni (Whitehead Institute for Biomedical Research, Cambridge, MA, USA) | N/A |
| MMD cDNA | Horizon Discovery Mammalian Gene Collection | Cat#BC026324 |
| MBOAT7 cDNA | Horizon Discovery Mammalian Gene Collection | Cat#BC003164 |
| memACSL4 cDNA | GenScript | Cat#OHu23492, transcript variant 2, mRNA |
| pLV-mCherry-EF1a-memACSL4-HA | VectorBuilder, custom made for this paper | N/A |
| pLV-mCherry-EF1a-FLAG-MBOAT7 | VectorBuilder, custom made for this paper | N/A |
| pLV-mCherry-EF1a-memACSL4-HA:P2A:FLAG-MBOAT7 | VectorBuilder, custom made for this paper | N/A |
| lentiCRISPRv2-puro_NT sg | This paper | N/A |
| lentiCRISPRv2-blast_NT sg | This paper | N/A |
| lentiCRISPRv2-puro_MMDsg10 | This paper | N/A |
| lentiCRISPRv2-blast_MMDsg8 | This paper | N/A |
| lentiCRISPRv2-puro_MMD2sg1 | This paper | N/A |
| lentiCRISPRv2-blast_ACSL4sg | This paper | N/A |
| lentiCRISPRv2-blast_MBOAT7sg3 | This paper | N/A |
| lentiCRISPRv2-blast_LPCAT3sg3 | This paper | N/A |
| pLV-EF1a-IRES-blast_HA-MMD_sgResistant | This paper | N/A |
| pLJM1_HA-MMD | This paper | N/A |
| pRH115_FLAG-MBOAT7 | This paper | N/A |
| Gateway cloning entry vector: pDONR221 | Dr. Tsukasa Shibue (Broad Institute, Cambridge, MA, USA) | N/A |
| MaMTH N-terminally tagged MMD prey: A1245 MMD | This paper | N/A |
| MaMTH C-terminally tagged memACSL4 prey: A1379 memACSL4 | This paper | N/A |
| MaMTH C-terminally tagged MBOAT7 bait: A1160 MBOAT7 | This paper | N/A |
| MaMTH C-terminally tagged memACSL4 bait: A1160 memACSL4 | This paper | N/A |
| MaMTH N-terminally tagged PEX7 prey: A1245 PEX7 | Dr. Igor Stagljar (University of Toronto, Toronto, Canada); Saraon et al.50 | N/A |
| MaMTH 5xGAL4 UAS Gaussia princeps reporter plasmid containing hygromycin resistance cassette (A1687) | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ApE Version 3.1.3 | Davis et al.72 | https://jorgensen.biology.utah.edu/wayned/ape/ |
| SnapGene Version 6.2.1 | Dotmatics | www.snapgene.com |
| GraphPad Prism Version 9.5.1 | Dotmatics | www.graphpad.com |
| RStudio Version 2022.07.0 + 548 | RStudio Team73 | www.rstudio.com |
| R Version 4.2.1 | R Core Team74 | https://www.R-project.org |
| ICE Analysis Tool Version 3.0 | Synthego | https://ice.synthego.com/#/ |
| Imaris Version 10.0 | Oxford Instruments | https://imaris.oxinst.com/ |
| LipidSearch Version 4.2.27 | Thermo Fisher Scientific, Mitsui Knowledge Industries75,76 | N/A |
| TraceFinder | Thermo Fisher Scientific | N/A |
| Progenesis QI | Nonlinear Dynamics | https://www.nonlinear.com/progenesis/qi/ |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines and culture conditions
OVCAR-8 cells (human female high-grade serous ovarian carcinoma cells) were obtained from J. Brugge (Harvard Medical School) and cultured in 1:1 MCDB105 and Medium 199 with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). 786-O cells (human male clear cell renal cell carcinoma cells) were obtained from the Broad Institute Biological Samples Platform and cultured in RPMI with 10% FBS and 1% P/S. Huh7 cells (human male hepatocellular carcinoma cells), both wild-type and MBOAT7 KO, were obtained from J. M. Brown (Cleveland Clinic) and cultured in DMEM with 10% FBS, 1% L-glutamine, 1% non-essential amino acids, and 1% P/S. HT-1080 cells (human male fibrosarcoma cells) were obtained from the Weinberg Lab Cell Bank and cultured in DMEM with 10% FBS and 1% P/S. HEK293T cells (human female immortalized embryonic kidney cells) were obtained from the American Type Culture Collection (ATCC) and were cultured in DMEM with 10% FBS. HEK293 cells (human female immortalized embryonic kidney cells) were obtained from Thermo Fisher (Flp-In 293 TREx cells) and were cultured in DMEM with 10% FBS and 1% P/S. THP-1 cells (human male acute monocytic leukemia cells) were obtained from Jaime Cheah (Koch Institute for Integrative Cancer Research) and cultured in RPMI with 10% heat-inactivated FBS and 1% P/S. To induce differentiation to macrophage-like cells, THP-1 cells were incubated in media containing 100 nM phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich) for 48 h, followed by media without PMA for 24 h. All cells were cultured in a humidified incubator at 37°C with 5% CO2. OVCAR-8 and 786-O cells were verified to be free of mycoplasma contamination and were STR profiled by the Duke University DNA analysis facility.
Primary monocyte culture and differentiation
Discarded blood products were obtained from anonymous human donors and provided by the Crimson Core Specimen Bank (Brigham and Women’s Hospital, Boston, MA). Monocytes were isolated by magnetic labeling with StraightFrom Whole Blood CD14 Microbeads and AutoMacs-directed selection (Miltenyi Biotec). Macrophages were differentiated in vitro from primary monocytes by culturing the monocytes in IMDM with 10% heat-inactivated FBS, 1% P/S, and 20 ng/mL human M-CSF (Peprotech) for 7 days. Macrophages derived from this method were sustained in these media conditions and replated as necessary.
METHOD DETAILS
Lentiviral production and transduction
To produce lentivirus, HEK293T cells were co-transfected with the lentiviral plasmid and VSV-G and psPAX2 packaging plasmids at a 2.1 to 0.2 to 3.7 ratio using polyethylenimine. Lentivirus-containing media was harvested from transfected cells after 48 h, passed through a 0.45 μm filter to exclude cells, and added to target cell lines along with polybrene (10 μg/mL). Media was replaced 24 h after transduction, and selection with puromycin (2 μg/mL or 4 μg/mL) or blasticidin (8 μg/mL) began 48 h after transduction.
Generation of knockout cell lines
CRISPR-Cas9 lentiviral vectors were generated by cloning sgRNAs into BsmBI-linearized lentiCRISPRv2-puro or lentiCRISPRv2-blast using T4 DNA ligase (sgRNA target sequences in Table S2). Target cells were stably transduced with the above lentivirus followed by antibiotic selection as described above. Cells maintained as bulk populations are denoted as “sg” cells, e.g., “NT sg,” or “EV + MBOAT7 sg,” while cells that were single-cell sorted into 96 well plates by fluorescence-activated cell sorting to generate single-cell clones are denoted as “KO” cells, e.g., “MMD KO” and “ACSL4 KO” cells. lentiCRISPRv2-puro was a gift from Feng Zhang (Addgene plasmid #52961; http://n2t.net/addgene:52961; RRID:Addgene_52961).69 lentiCRISPRv2-Blast was a gift from Mohan Babu (Addgene plasmid #83480; http://n2t.net/addgene:83480; RRID:Addgene_83480).
Sanger sequencing of knockout cell lines
Single-cell clones generated from bulk MMD-sg or MMD2-sg populations were screened for knockout of MMD or MMD2 respectively at the genomic DNA level. Genomic DNA was purified from cell pellets using the DNeasy Blood & Tissue Kit (QIAGEN). A region of the MMD or MMD2 locus surrounding the sgRNA target site was amplified using PCR and Sanger sequenced (all primers in Table S2). Single-cell clones used in this paper were homozygous knockouts. Sequencing reads were aligned to the NCBI MMD or MMD2 reference sequence (RefSeq) using ApE72 or SnapGene.
ICE validation of pooled knockout cell lines
Sanger sequencing of bulk MMD2 sg, MBOAT7 sg, or LPCAT3 sg populations enabled assessment of editing at the genomic DNA level using Synthego’s Inference of CRISPR Edits (ICE) tool. First, genomic DNA was purified from cell pellets (of sg-targeted cells as well as the NT sg control) using the PureLink Genomic DNA Mini Kit (Invitrogen). Regions of the target gene locus surrounding the respective sgRNA target sites were amplified using PCR and sequenced using the Sanger method (all primers in Table S2). Subsequently, .ab1 files with mixed peaks in the sg-edited cells were compared to.ab1 files with single peaks in the corresponding control cells within the ICE online tool (https://ice.synthego.com/#/) and the indel score and knockout score were noted.
Generation of overexpression cell lines
MMD cDNA from the Horizon Discovery Mammalian Gene Collection (BC026324) was fused to an HA-tag at its N terminus during PCR and cloned into the lentiviral vector pLJM1 linearized with AgeI (NEB) and EcoRI (NEB) by Gibson assembly (NEB). To re-express MMD in MMD knockout cells, the HA-MMD cDNA sequence was edited by fusion PCR (final sequence) to reduce base-pairing with both MMD-sg8 and MMD-sg10 and cloned into the lentiviral vector pLV-EF1a-IRES-blast by restriction digestion with EcoRI-HF (NEB) and BamHI-HF (NEB). FLAG-MBOAT7 used to generate cells for IF experiments was cloned into the lentiviral vector pRH115-mCherry linearized with XbaI (NEB) and BamHI (NEB) by Gibson assembly (NEB) with a codon-optimized FLAGMBOAT7 gblock (Integrated DNA Technologies). All cDNA inserts that were cloned into vectors were verified by Sanger sequencing. Plasmids (pLV-mCherry-EF1a) for overexpression of memACSL4-HA, FLAG-MBOAT7, or memACSL4-HA-P2A-FLAG-MBOAT7 in MMD KO cells were obtained from VectorBuilder and contained an mCherry selection marker. Overexpression cell lines were generated by stable transduction of lentiviral vectors and antibiotic selection (pLJM1, pLV-EF1a-IRES-blast) or fluorescence-activated cell sorting for mCherry positive cells (pRH115-mCherry and pLV-mCherry-EF1a). pLJM1-Empty was a gift from Joshua Mendell (Addgene plasmid # 91980; http://n2t.net/addgene:91980; RRID:Addgene_91980).70 pLV-EF1a-IRES-Blast was a gift from Tobias Meyer (Addgene plasmid #85133; http://n2t.net/addgene:85133; RRID:Addgene_85133).71 pRH115 was a gift from Tobiloba Oni (Whitehead Institute for Biomedical Research).
MaMTH experiments
MMD (BC026324) and MBOAT7 (BC003164) cDNAs were obtained from the Horizon Discovery Mammalian Gene Collection. memACSL4 cDNA was obtained from GenScript (OHu23492, transcript variant 2, mRNA). All cDNAs were PCR amplified to add attB sites for Gateway cloning (see Table S3). The resulting PCR products were separated on an agarose gel, purified, and combined with the Gateway pDONR221 vector (Invitrogen) in BP Clonase II reactions (Invitrogen) according to manufacturer instructions. The terminated BP clonase reactions were used to transform One Shot MAX Efficiency DH5α-T1R competent cells (Invitrogen) and transformants were plated on kanamycin agar plates. Single colonies from these plates were verified by Sanger sequencing to contain the desired inserts, and these plasmids were combined with the appropriate destination vectors (see Table S3) in LR Clonase II reactions (Invitrogen) according to manufacturer instructions. The terminated LR clonase reactions were used to transform One Shot MAX Efficiency DH5α-T1R competent cells (Invitrogen) and transformants were plated on ampicillin agar plates. Plasmids were purified from single colonies and verified to contain the desired inserts using primers in Table S3. The PEX7-N prey plasmid was generated by the Stagljar Lab.50
The generation of the HEK293 MaMTH reporter cell lines was described previously.49 The OVCAR-8 parental MaMTH reporter line (B0755) was generated using the same protocol, however all growth was carried out in OVCAR-8 media conditions as described in the cell culture conditions section. OVCAR-8 NT sg and MMD KO MaMTH reporter lines (B0781 and B0772, respectively) were generated in the same manner as the parental, however the reporter vector used for integration was modified (by Gibson Assembly) to contain a hygromycin resistance marker in place of puromycin and selection was performed using 50 μg/mL hygromycin.
For MaMTH assays, HEK293 or OVCAR-8 MaMTH reporter cells stably expressing Gaussia princeps luciferase (New England Biolabs) under the control of a 5xGAL4 UAS were seeded in 96-well tissue culture-treated plates (~15,000 cells/well) and grown at 37°C/5% CO2 overnight in DMEM/10% FBS/1% PS (HEK293) or 1:1 Medium 199:MCDB105/10% FBS/1% PS (OVCAR-8) to approximately 50% confluency. Cells were then transiently transfected with MaMTH bait and prey plasmids using X-tremeGENE9 DNA transfection reagent (Roche) following manufacturer’s instructions. Four hours post-transfection, tetracycline was added to wells to a final concentration of 0.5 μg/mL to induce bait and prey expression. After 48 h additional growth, luciferase activity was assayed from cell supernatants using 4 μM coelenterazine substrate (Nanolight) and measurement of chemiluminescence with a CLARIOstar plate reader (BMG).
Compound treatment and cell viability assays
For cell viability experiments, cells were treated with a range of concentrations of the indicated small molecule compounds, and relative viability was measured by the CellTiter-Glo Assay. Cells were seeded in 96-well black-wall tissue culture-treated plates at 2,000 to 3,000 cells per well (or 10,500 for primary human monocytes and macrophages), to reach 30% confluency the following day. Cells were treated with compounds 18–24 h after seeding using an HP D300e Digital Dispenser with three biological replicates per condition. For BSA-palmitate and BSA-control experiments, compounds were added manually to cells. 66–72 h after compound treatment, viability was measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) as per the manufacturer’s instructions. Unless otherwise specified, relative viability was normalized to the untreated condition. Regression fit curves were computed in Prism 8 or 9 (GraphPad) using four-parameter inhibition nonlinear regression. The mean and standard deviation for three biological replicates of each data point were calculated. The following compounds were used: RSL3 (Selleck Chem), ML210 (Sigma Aldrich), erastin (Selleck Chem), FIN56 (Selleck Chem), Ferrostatin-1 (Sigma Aldrich), Z-VAD-FMK (Selleck Chem), Necrostatin-1 (Selleck Chem), BSA-palmitate (Cayman Chem), BSA-control (Cayman Chem).
Lipid peroxidation measurement
The redox-sensitive dye, 581/591 C11-BODIPY (Invitrogen), was used to measure relative levels of lipid peroxidation. Cells were seeded to 30% confluency and treated with ML210 (1 μM for OVCAR-8, 0.25 μM for 786-O) for 3.5 h. 5 μM 581/591 C11-BODIPY was also added for the last 30 min of the incubation. Cells were trypsinized, washed with PBS 3 times, and strained through a 35 μm cell strainer for flow cytometry. Cells from each condition were analyzed on a LSRFortessa cytometer using the PE channel for reduced C11-BODIPY and FITC channel for oxidized C11-BODIPY.
RNA purification and RT-qPCR
Cells were pelleted and washed once with PBS. Total RNA was isolated using the RNeasy Mini Kit (QIAGEN) and reverse-transcribed into cDNA using the QuantiTect Reverse Transcription Kit (QIAGEN). qPCR was performed on cDNA using SYBR Green Master Mix (Thermo) according to manufacturer instructions. Relative mRNA expression was calculated by the ΔΔCT method with ACTB as reference. The primer sequences are as follows, listed from 5′ to 3’: MMD qPCR Custom F: CCGCTACAAGCCAACTTGCTAT, MMD qPCR Custom R: AGTCCCATTCCATAAATCCATG.
Immunoblotting
For immunoblots labeled “membrane,” lysates were prepared using the Mem-PER Plus Membrane Protein Extraction Kit (Thermo) using manufacturer buffers and instructions—this kit helped us ensure that integral membrane proteins such as MMD were included in the lysate. We later optimized our lysate preparation protocol for membrane proteins such that we did not need to use the kit: To prepare lysates for other immunoblots, cell pellets were washed in PBS and resuspended in lysis buffer containing 50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 1% digitonin, protease & phosphatase inhibitors (Thermo), and EDTA (Thermo). Alternatively, a commercial 10X Cell Lysis Buffer (Cell Signaling Technology, Cat#9803S) with PMSF (Cell Signaling Technology, Cat#8553S) was used. Lysates were incubated at 4°C for 30 min with rotation and centrifuged at 4°C for 20 min at 15,000 rpm in a microcentrifuge. Supernatants were collected and protein concentration was determined by the DC assay (Bio-Rad) using BSA to generate a standard curve.
Samples were prepared by diluting lysates in lysis buffer, NuPAGE LDS Sample Buffer (Thermo), and NuPAGE Sample Reducing Agent (Thermo), and heating at 70°C for 10 min. Samples were resolved by gel electrophoresis using NuPAGE 4–12% Bis-Tris gels (Thermo) and proteins were transferred onto nitrocellulose membranes. Membranes were blocked in 5% milk/TBST, washed 3 times with 1X TBST, and incubated with primary antibodies in 5% BSA/TBST overnight. The following day, membranes were washed with 1X TBST 3 times, incubated with HRP-linked secondary antibodies in 5% milk/TBST for 1 h, and washed with 1X TBST 3 times. Membranes were then incubated with SuperSignal West Dura Extended Duration Substrate (Thermo) or SuperSignal West Femto Extended Duration Substrate (Thermo) and developed using X-ray film. The following antibodies were used at indicated concentrations: HA-Tag (rabbit, CST #3724, 1:500 to 1:1000), MMD polyclonal (rabbit, CST, not commercially available, 1:500), MMD monoclonal (rabbit, CST #20226, 1:500), ACSL4 (rabbit, Invitrogen PA527137, 1:10,000 to 1:20,000), MBOAT7 (rat, gift from Dr. Nozomu Kono, 1:500 to 1:1000), FSP1 (CST #24972S, 1:1000), GPX4 (abcam ab125066, 1:10,000 to 1:20,000), Calnexin (mouse, Invitrogen, MA3–027, 1:1000), COXIV (rabbit, CST #11967, 1:20,000), GAPDH (rabbit, CST #2118, 1:20,000), HRP-linked anti-rabbit IgG (CST #7074, 1:5000), HRP-linked anti-mouse IgG (CST #7076, 1:5000), and HRP-linked anti-rat IgG (CST #7077, 1:5000).
Immunofluorescence microscopy
Cells plated on uncoated (786-O) or poly-L-lysine-coated (OVCAR-8) glass coverslips were washed briefly with 1X PBS and then fixed with 4% paraformaldehyde in PBS at room temperature for 10 min. After two PBS washes, cells were permeabilized with 0.1% Triton X-100 in PBS at room temperature for 5 min. After three more PBS washes, coverslips were blocked in 3% normal donkey serum (NDS) in PBS at room temperature for 1 h and incubated with primary antibody in 3% NDS/PBS at 4°C overnight. For experiments where a fluorophore-conjugated primary antibody was used, the overnight incubation and all following steps were performed in the dark. The next day, coverslips were washed with PBS at room temperature 3 times for 15 min each, and then incubated with fluorophore-conjugated secondary antibodies in 3% NDS/PBS at room temperature for 1 h in the dark. After three more 15-min washes with PBS at room temperature, coverslips were incubated for 5 min at room temperature with DAPI and phalloidin-iFluor 488 in PBS, washed twice with PBS and once with deionized water, and mounted onto slides using Prolong Gold Antifade Mountant. Coverslips were cured at room temperature in the dark for 16–24 h before imaging. For immunofluorescence experiments staining FLAG-MBOAT7, this protocol was modified to permeabilize cells with 0.1% saponin in PBS instead of Triton X-100, and 0.1% saponin was included in solutions in all subsequent steps until DAPI addition. The following antibodies and fluorescent dyes were used at the indicated concentrations: HA-Tag Alexa Fluor 647 conjugate (mouse, CST #3444, 1:50), HA-Tag (rabbit, CST #3724, 1:250), ACSL4 (rabbit, Invitrogen PA5–27137, 1:250), Calnexin (mouse, MA3–027, 1:250), GM130 (rabbit, CST #12480, 1:3200), DYKDDDDK (FLAG) Tag (rabbit, CST #14793, 1:500), DAPI (0.1ug/ml), phalloidin-iFluor488 (Abcam ab176753, 1:1000), CF 555 donkey anti-rabbit IgG (H + L) 555 secondary antibody (Biotium #20038, 1:500), CF633 donkey anti-mouse IgG (H + L) secondary antibody (Biotium #20124, 1:500).
Coverslips were imaged on a DeltaVision Elite Widefield Imaging System using the 60× objective. Images were deconvolved during post-acquisition processing. Quantification of colocalization was performed in Imaris using deconvolved imaging files. Within Imaris, phalloidin staining was used to mask images so that colocalization was measured only in regions containing cells. Pixel intensity thresholds for colocalized channels were set using negative controls, such that less than or equal to 2 percent of pixels were above each threshold in coverslips stained with secondary antibody only. Pearson’s thresholded coefficients in the entire colocalized volume were presented in figures. Representative images shown in figures represent one z stack and are taken from Imaris.
Tracing of radiolabeled [3H]arachidonic acid
Measurement of the rate of arachidonic acid incorporation into cellular phospholipid pools was accomplished by kinetic radiolabeling using [3H]arachidonic acid substrate. Briefly, one million cells of each cell line were seeded onto 6-well plates, and grown until they reached 80% confluence. The next day serum free medium was added and the cells were incubated with 0.5 μCi of [3H]arachidonic acid (PerkinElmer) briefly for either five, fifteen, or 30 min. Following pulse labeling, the cells were rinsed with phosphate buffer saline (PBS) twice, and then total lipid extracts were generated as previously described.68 Total lipid extracts were separated via two dimensional thin-layer chromatography (TLC) using chloroform/methanol/ammonium hydroxide/water (60:33.33:2.66:4, v/v) as the first dimension followed by chloroform/methanol/acetic acid/water (64:8:10:2, v/v) as the second-dimension solvent systems. Individual phospholipid species were identified by migration with respect to mass standards, spots resolving with the PI, PC, PE, PS, PG, or PA standard were scraped from the TLC plate, and the radioactivity was quantified with a liquid scintillation counter. [3H]-containing phospholipids were normalized to the amount of cellular protein, as quantified by the BCA assay (Pierce).
Untargeted lipidomics (datasets 1 and 3)
Lipids were isolated using the following chloroform-methanol extraction method. Cells were seeded in 6 well plates, 24 h prior, to reach 70% confluency at the time of extraction. Cells were washed with 0.9% cold NaCl on wet ice and then transferred to dry ice. 600 μL LC/MS grade methanol containing 500 nM internal standards (Amino Acid Metabolomics Mix; Cambridge Isotope Laboratories, Inc.) was added to each well. Adherent cells were scraped, collected and samples were transferred to wet ice. 300 μL of LC/MS grade water was added, followed by 400 μL of chloroform containing Splash LipidoMix (as an internal standard for the lipid fraction; Avanti, Cat. #330707), and samples were vortexed for 10 min at 4°C. Samples were centrifuged at 4°C for 10 min at 15,000 rpm in a microcentrifuge and the lower lipid-containing layer was collected and dried using a Speedvac. Samples were stored at −80°C until analysis.
Lipids were separated on an Ascentis Express C18 2.1 × 150 mm 2.7 μm column (Sigma-Aldrich) connected to a Vanquish Horizon UPLC system and an ID-X tribrid mass spectrometer (Thermo Fisher Scientific) equipped with a heated electrospray ionization (HESI) probe. External mass calibration was performed using the standard calibration mixture every seven days. Dried lipid extracts were reconstituted in 50 μl 65:30:5 acetonitrile: isopropanol: water (v/v/v). Typically, 2 μL of sample were injected onto the column, with separate injections for positive and negative ionization modes. Mobile phase A in the chromatographic method consisted of 60:40 water: acetonitrile with 10 mM ammonium formate and 0.1% formic acid, and mobile phase B consisted of 90:10 isopropanol: acetonitrile, with 10 mM ammonium formate and 0.1% formic acid. The chromatographic gradient was adapted from previous work.77,78 Briefly, the elution was performed with a gradient of 40 min; during 0–1.5 min isocratic elution with 32% B; from 1.5 to 4 min increase to 45% B, from 4 to 5 min increase to 52% B, from 5 to 8 min to 58% B, from 8 to 11 min to 66% B, from 11 to 14 min to 70% B, from 14 to 18 min to 75% B, from 18 to 21 min to 97% B, during 21–35 min 97% B is maintained; from 35 to 35.1 min solvent B was decreased to 32% and then maintained for another 4.9 min for column re-equilibration. The flow rate was set to 0.260 mL/min. The column oven and autosampler were held at 55°°C and 15°°C, respectively. The mass spectrometer parameters were as follows: The spray voltage was set to 3.25 kV in positive mode and 3.0 kV in negative mode, and the heated capillary and the HESI were held at 300°°C and 375°°C, respectively. The S-lens RF level was set to 45, and the sheath and auxillary gas were set to 40 and 10 units, respectively. These conditions were held constant for both positive and negative ionization mode acquisitions.
The mass spectrometer was operated in full-scan-ddMS/MS mode with an orbitrap resolution of 120,000 (MS1) and 30,000 (MS/MS). Internal calibration using Easy IC was enabled. Quadrupole isolation was enabled, the AGC target was 1 × 105, the maximum injection time was 50 ms, and the scan range was m/z = 200–2000. For data-dependent MS/MS, the cycle time as 1.5 s, the isolation window was 1, and an intensity threshold of 1 × 103 was used. HCD fragmentation was achieved using a stepwise collision energy of 15, 25, and 35 units, and detected in the orbitrap with an AGC target of 5 × 104 and a maximum injection time of 54 ms. Isotopic exclusion was on, a dynamic exclusion window of 2.5 s was used, and an exclusion list was generated using a solvent bank.
High-throughput annotation and relative quantification of lipids was performed using LipidSearch v4.2.27 (ThermoFisher Scientific/Mitsui Knowledge Industries)75,76 using the HCD database. LipidSearch matches MS/MS data in the experimental data with spectral data in the HCD database. Precursor ion tolerance was set to 5 ppm, product ion tolerance was set to 10 ppm. LipidSearch nomenclature uses underscores to separate the fatty acyl chains to indicate the lack of sn positional information (e.g., PC(16:0_18:1) and not (PC(16:0/18:1)). In cases where there is insufficient MS/MS data to identify all acyl chains, only the sum of the chains is displayed (i.e., PC(34:1)). Following the peak search, positive and negative mode data were aligned together where possible (where positive and negative mode data were collected on separate occasions data were aligned separately) and raw peak areas for all annotated lipids were exported to Microsoft Excel and filtered according to the following predetermined quality control criteria: Rej (“Reject” parameter calculated by LipidSearch) equal to 0; PQ (“Peak Quality” parameter calculated by LipidSearch software) greater than 0.75; CV (standard deviation/mean peak area across triplicate injections of a represented (pooled) biological sample) below 0.4; R (linear correlation across a three-point dilution series of the representative (pooled) biological sample) greater than 0.9. Typically, ~70% of annotated lipids passed all four quality control criteria. Redundant lipid ions (those with identical retention times and multiple adducts) were removed such that only one lipid ion per species/per unique retention time is reported in merged alignments. For data where positive and negative mode data were aligned separately some redundancies may still exist. Raw peak areas of the filtered lipids were normalized to total lipid signal (positive or negative ionization mode) in each sample to control for sample loading.
Untargeted lipidomics (datasets 2 and 4)
Lipids were isolated as follows. Cells were seeded in 6 well plates the previous day to reach 70% confluency at the time of extraction. Cells were washed with cold (4°C) PBS without magnesium or calcium. Immediately after that, cells were scraped into 800 μL of cold (4°C) isopropanol and extracts were incubated at 4°C on ice, protected from light, for 1 h. Extracts were vortexed and then centrifuged at 9000xg for 10 min at 4°C. The supernatant was taken, kept on ice, and stored at −80°C until analysis.
Reversed-phase C8 chromatography/positive ion mode MS detection was used to measure lipids. Analyses of polar and non-polar plasma lipids were conducted using an LC-MS system comprised of a Shimadzu Nexera X2 U-HPLC (Shimadzu Corp.) coupled to an Exactive Plus orbitrap mass spectrometer (Thermo Fisher Scientific). 2 μL of cells in IPA were injected directly onto a 100 × 2.1 mm, 1.7 μm ACQUITY BEH C8 column (Waters). The column was eluted isocratically with 80% mobile phase A (95:5:0.1 v/v/vol 10 mM ammonium acetate/methanol/formic acid) for 1 min followed by a linear gradient to 80% mobile-phase B (99.9:0.1 v/v methanol/formic acid) over 2 min, a linear gradient to 100% mobile phase B over 7 min, then 3 min at 100% mobile-phase B. MS analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis over 200–1100 m/z at 70,000 resolution and 3 Hz data acquisition rate. Other MS settings were: sheath gas 50, in source CID 5 eV, sweep gas 5, spray voltage 3 kV, capillary temperature 300°C, S-lens RF 60, heater temperature 300°C, microscans 1, automatic gain control target 1e6, and maximum ion time 100 ms. Raw data were processed using TraceFinder software (Thermo Fisher Scientific) for targeted peak integration and manual review of a subset of identified lipids and using Progenesis QI (Nonlinear Dynamics) for peak detection and integration of both lipids of known identify and unknowns. Lipid identities were determined based on comparison to reference plasma extracts and are denoted by total number of carbons in the lipid acyl chain(s) and total number of double bonds in the lipid acyl chain(s). Human metabolome database (HMDB) IDs were used to further identify the acyl chain composition of lipid species.
Downstream analysis of lipidomics data
Triacylglycerol and cholesteryl ester species were excluded from all downstream analyses. The measured value of each species in each sample was normalized to the summed values of all lipids in that sample to compare relative abundance of a given lipid species between samples. Original code used to analyze all lipidomics datasets is deposited at Zenodo; DOIs are listed in the key resources table.
Polar metabolomics
Polar metabolites were isolated by the same following chloroform-methanol extraction method described above (“untargeted lipidomics (datasets 1 and 3)”). After the final centrifugation step, the polar metabolites were collected as the top layer and dried using the Speedvac. Samples were stored at −80°C until analysis.
All solvents, including water, were purchased from Fisher and were Optima LC/MS grade. Metabolite profiling was conducted on a QExactive bench top orbitrap mass spectrometer equipped with an Ion Max source and a HESI II probe, which was coupled to a Dionex UltiMate 3000 HPLC system (Thermo Fisher Scientific, San Jose, CA). External mass calibration was performed using the standard calibration mixture every 7 days and an additional custom mass calibration was performed weekly alongside standard mass calibrations to calibrate the lower end of the spectrum (m/z 70–1050 positive mode and m/z 60–900 negative mode) using the standard calibration mixtures spiked with glycine (positive mode) and aspartate (negative mode). Typically, samples were reconstituted in 50 μL water and 2 μL were injected onto a SeQuant ZIC-pHILIC 150 × 2.1 mm analytical column equipped with a 2.1 × 20 mm guard column (both 5 mm particle size; Millipore-Sigma). Buffer A was 20 mM ammonium carbonate, 0.1% ammonium hydroxide; Buffer B was acetonitrile. The column oven and autosampler tray were held at 25°° and 4°°C, respectively. The chromatographic gradient was run at a flow rate of 0.150 mL/min as follows: 0–20 min: linear gradient from 80 to 20% B; 20–20.5 min: linear gradient form 20–80% B; 20.5–28 min: hold at 80% B. The mass spectrometer was operated in full-scan, polarity-switching mode, with the spray voltage set to 3.0 kV, the heated capillary held at 275°°C, and the HESI probe held at 350°°C. The sheath gas flow was set to 40 units, the auxiliary gas flow was set to 15 units, and the sweep gas flow was set to 1 unit. MS data acquisition was performed in a range of m/z = 70–1000, with the resolution set at 70,000, the AGC target at 1 × 106, and the maximum injection time at 20 ms. Relative quantitation of polar metabolites was performed with TraceFinder 4.1 (Thermo Fisher Scientific) using a 5 ppm mass tolerance and referencing an in-house library of chemical standards. Data were filtered according to predetermined QC metrics: CV of pools <25%; R of linear dilution series <0.975.
Public dataset queries
The Cancer Therapuetics Response Portal was accessed at https://portals.broadinstitute.org/ctrp.v2.1/?page=#ctd2BodyHome on February 23, 2020. Data from the Cancer Dependency Map (DepMap) was accessed at https://depmap.org/portal/on October 23, 2022. Original code used to calculate ranked correlations between all genes and MMD, MBOAT7, or ACSL4 is deposited at Zenodo; DOIs are listed in the key resources table.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed in GraphPad Prism and R. Except for Figures 5C and S5D, and S5E, a two-tailed, unpaired student’s two-sample t test was used to compute p values. In Figure 5C, an ordinary one-way ANOVA with Dunnett’s multiple comparisons test was used to compute p values. In Figures S5D and S5E, an ordinary one-way ANOVA with Tukey’s multiple comparisons test was used to compute p values. An alpha value of 0.05 was used. Other statistical details of experiments are included in the figure legends. SnapGene and ApE were used for molecular biology analyses. Synthego’s ICE software was used to infer CRISPR editing in bulk populations. Imaris was used for microscopy image analyses. LipidSearch was used for annotation of lipidomics datasets 1 and 3; TraceFinder and Progenesis QI were used for annotation of lipidomics datasets 2 and 4; and TraceFinder was used for annotation of the polar metabolomics dataset.
Supplementary Material
Highlights.
The Golgi scaffold protein MMD heightens cancer cell sensitivity to ferroptosis
MMD interacts with lipid metabolic enzymes ACSL4 and MBOAT7
MMD increases flux through the ACSL4-MBOAT7 pathway, leading to higher AA-PI levels
ACSL4 and MBOAT7 mediate the pro-ferroptotic effects of MMD
ACKNOWLEDGMENTS
We thank E.C. Holland at Fred Hutchinson Cancer Center, Seattle, WA, for providing laboratory space and support to conduct this research from July 2020 to February 2021 due to circumstances of the COVID-19 pandemic. We are grateful to T. Shibue (Broad Institute) for providing the pDONR221 plasmid and N. Kono (University of Tokyo) for providing the MBOAT7 antibody. We acknowledge the following Core Facilities at the Whitehead Institute for their technical assistance: K. Crowder from the Metabolomics Core, K. Daniels and P. Autissier from the Flow Cytometry Core, C. Rogers and B. Braswell from the Keck Microscopy Core, and G. Bell and T. Whitfield from the Bioinformatics core. We thank N. Boehnke, E.C. Lien, F. Szulzewsky, T. Oni, M.G. Vander Heiden, and all members of the Weinberg lab for helpful discussions.
This work is partly funded by the NIH (R35 CA220487), Virginia and D.K. Ludwig Fund for Cancer Research, Nile Albright Research Foundation, and the Brendan Bradley Gift. Research contributions from the Stagljar lab were supported by funding from the Canadian Cancer Society Research Institute (#703889), Genome Canada via Ontario Genomics (#9427 and #9428), Ontario Research fund (ORF/DIG-501411 and RE08-009), CQDM (Quantum Leap) and Brain Canada (Quantum Leap), Cancer Research Society (#23235), Genentech Inc. US (CLL-018879), Canadian Institutes of Health Research (CIHR #420989), and Toronto Innovation Acceleration Partners (TIAP L150-066 #72058826). Research contributions from the Brown lab were supported in part by funding from the NIH (P01 HL147823, P50 AA024333, R01 DK130227, and R01 DK120679) and the United States Department of Defense (DOD KC2210163).
V.V.P. was supported by the MIT Undergraduate Research Opportunities Program, Peter J. Eloranta Summer Research Fellowship and award number T32GM144273 from the National Institute of General Medical Sciences. K.W. was supported by the Valhalla Foundation, NIH T32 CA09172, A Breath of Hope Lung Foundation, ASCO Conquer Cancer Foundation Young Investigator Award, and AACR-AstraZeneca Career Development Award for Physician-Scientists in Honor of José Baselga. W.S.H. was supported by postdoctoral fellowships from the Jane Coffin Childs Memorial Fund and the Ludwig Center at MIT’s Koch Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113023.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
DECLARATION OF INTERESTS
K.W. declares relationships pertaining to macrophage-directed therapies, including patents and royalties (Stanford University, Whitehead Institute, Gilead Sciences), is a co-founder, SAB member, and equity holder (ALX Oncology, DEM Biopharma), and is a scientific advisor (Carisma Therapeutics).
REFERENCES
- 1.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM,Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 149, 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang X, Stockwell BR, and Conrad M (2021). Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282. 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 116, 2672–2680. 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan Y, Liang X, Ma Z, Hu H, Liu H, Miao Z, Linkermann A, Hell-wege JN, Voight BF, and Susztak K (2021). A single genetic locus controls both expression of DPEP1/CHMP1A and kidney disease development via ferroptosis. Nat. Commun. 12, 5078. 10.1038/s41467-021-25377-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J,Wang Z, Jiang C, et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2, e90777. 10.1172/jci.insight.90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu Y, Jiang L, Wang H, Shen Z, Cheng Q, Zhang P, Wang J, Wu Q, Fang X, Duan L, et al. (2020). Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 136, 726–739. 10.1182/blood.2019002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, Sell A, Wei S,Grove S, Johnson JK, et al. (2022). CD8+ T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 40, 365–378.e6. 10.1016/j.ccell.2022.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang W, Green M, Choi JE, Gijó n M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. (2019). CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274. 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH,Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, Paradkar S, Boehnke N, Deik AA, Reinhardt F, et al. (2020). Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608. 10.1038/s41586-020-2732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I,Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng H, and Stockwell BR (2018). Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 16, e2006203. 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, and Stockwell BR (2014). Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523. 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I,Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 18.Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zand-karimi F, Merl-Pham J, Bao X, Anastasov N, Kö ssl, J., et al. (2020). GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 6, 41–53. 10.1021/acscentsci.9b01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 156, 317–331. 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng Y-Y,Deasy R, Kost-Alimova M, Dančík V, et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 10, 1617. 10.1038/s41467-019-09277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin T, Ding Q, Huang H, Xu D, Jiang Y, Zhou B, Li Z, Jiang X, He J, Liu W, et al. (2012). PAQR10 and PAQR11 mediate Ras signaling in the Golgi apparatus. Cell Res. 22, 661–676. 10.1038/cr.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rehli M, Krause SW, Schwarzfischer L, Kreutz M, and Andreesen R(1995). Molecular Cloning of a Novel Macrophage Maturation-Associated Transcript Encoding a Protein with Several Potential Transmembrane Domains. Biochem. Biophys. Res. Commun. 217, 661–667. 10.1006/bbrc.1995.2825. [DOI] [PubMed] [Google Scholar]
- 23.Tan X, Banerjee P, Guo H-F, Ireland S, Pankova D, Ahn YH, Niko-laidis IM, Liu X, Zhao Y, Xue Y, et al. (2017). Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J. Clin. Invest. 127, 117–131. 10.1172/JCI88736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang YT, Hu T, Arterburn M, Boyle B, Bright JM, Emtage PC, andFunk WD (2005). PAQR Proteins: A Novel Membrane Receptor Family Defined by an Ancient7-Transmembrane Pass Motif. J. Mol. Evol. 61, 372–380. 10.1007/s00239-004-0375-2. [DOI] [PubMed] [Google Scholar]
- 25.Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, Zimmermann K, Cai LL, Niehues M, Badock V, et al. (2020). Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 16, 497–506. 10.1038/s41589-020-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ,Brown LM, Valenzuela CA, Wolpaw AJ, and Stockwell BR (2016). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503. 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, et al. (2013). An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161. 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV,Gill S, Javaid S, Coletti ME, Jones VL, Bodycombe NE, et al. (2016). Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 12, 109–116. 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J, et al. (2015). Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 5, 1210–1223. 10.1158/2159-8290.CD-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piccolis M, Bond LM, Kampmann M, Pulimeno P, Chitraju C, Jayson CBK, Vaites LP, Boland S, Lai ZW, Gabriel KR, et al. (2019). Probing the Global Cellular Responses to Lipotoxicity Caused by Saturated Fatty Acids. Mol. Cell 74, 32–44.e8. 10.1016/j.molcel.2019.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu XG, Nicholson Puthenveedu S, Shen Y, La K, Ozlu C, Wang T,Klompstra D, Gultekin Y, Chi J, Fidelin J, et al. (2019). CHP1 Regulates Compartmentalized Glycerolipid Synthesis by Activating GPAT4.Mol. Cell 74, 45–58.e7. 10.1016/j.molcel.2019.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan X, Banerjee P, Shi L, Xiao G-Y, Rodriguez BL, Grzeskowiak CL, Liu X, Yu J, Gibbons DL, Russell WK, et al. (2021). p53 loss activates prometastatic secretory vesicle biogenesis in the Golgi. Sci. Adv. 7, eabf4885. 10.1126/sciadv.abf4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Behan FM, Iorio F, Picco G, Gonç alves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. (2019). Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568, 511–516. 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- 34.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784. 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan J, Meyers RM, Michel BC, Mashtalir N, Sizemore AE, Wells JN, Cassel SH, Vazquez F, Weir BA, Hahn WC, et al. (2018). Interrogation of Mammalian Protein Complex Structure, Function, and Membership Using Genome-Scale Fitness Screens. Cell Syst. 6, 555–568.e7. 10.1016/j.cels.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Küch EM, Vellaramkalayil R, Zhang I, Lehnen D, Brügger B, Sreemmel W, Ehehalt R, Poppelreuther M, and Füllekrug J (2014). Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta 1841, 227–239. 10.1016/j.bbalip.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 37.Caddeo A, Jamialahmadi O, Solinas G, Pujia A, Mancina RM, Pingitore P, and Romeo S (2019). MBOAT7 is anchored to endomembranes by six transmembrane domains. J. Struct. Biol. 206, 349–360. 10.1016/j.jsb.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Cao Y, Traer E, Zimmerman GA, McIntyre TM, and Prescott SM(1998). Cloning, Expression, and Chromosomal Localization of Human Long-Chain Fatty Acid-CoA Ligase 4 (FACL4). Genomics 49, 327–330. 10.1006/geno.1998.5268. [DOI] [PubMed] [Google Scholar]
- 39.Kuwata H, and Hara S (2019). Role of acyl-CoA synthetase ACSL4 inarachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 144, 106363. 10.1016/j.prostaglandins.2019.106363. [DOI] [PubMed] [Google Scholar]
- 40.O’Donnell VB (2022). New appreciation for an old pathway: the LandsCycle moves into new arenas in health and disease. Biochem. Soc. Trans. 50, 1–11. 10.1042/BST20210579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee H-C, Inoue T, Imae R, Kono N, Shirae S, Matsuda S, Gengyo-Ando K, Mitani S, and Arai H (2008). Caenorhabditis elegans mboa-7, a Member of the MBOAT Family, Is Required for Selective Incorporation of Polyunsaturated Fatty Acids into Phosphatidylinositol. Mol. Biol. Cell 19, 1174–1184. 10.1091/mbc.e07-09-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caddeo A, Hedfalk K, Romeo S, and Pingitore P (2021). LPIAT1/MBOAT7 contains a catalytic dyad transferring polyunsaturated fatty acids to lysophosphatidylinositol. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1866, 158891. 10.1016/j.bbalip.2021.158891. [DOI] [PubMed] [Google Scholar]
- 43.Wang K, Lee C-W, Sui X, Kim S, Wang S, Higgs AB, Baublis AJ, Voth GA, Liao M, Walther TC, and Farese RV Jr. (2023). The structure of phosphatidylinositol remodeling MBOAT7 reveals its catalytic mechanism and enables inhibitor identification. Nat. Commun. 14, 3533. 10.1038/s41467-023-38932-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petschnigg J, Groisman B, Kotlyar M, Taipale M, Zheng Y, Kurat CF, Sayad A, Sierra JR, Mattiazzi Usaj M, Snider J, et al. (2014). The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat. Methods 11, 585–592. 10.1038/nmeth.2895. [DOI] [PubMed] [Google Scholar]
- 45.Pathmanathan S, Yao Z, Coelho P, Valla R, Drecun L, Benz C, Snider J, Saraon P, Grozavu I, Kotlyar M, et al. (2022). B cell linker protein (BLNK) is a regulator of Met receptor signaling and trafficking in non-small cell lung cancer. iScience 25, 105419. 10.1016/j.isci.2022.105419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lim SH, Snider J, Birimberg-Schwartz L, Ip W, Serralha JC, Botelho HM, Lopes-Pacheco M, Pinto MC, Moutaoufik MT, Zilocchi M, et al. (2022). CFTR interactome mapping using the mammalian membrane two-hybrid high-throughput screening system. Mol. Syst. Biol. 18, e10629. 10.15252/msb.202110629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aboualizadeh F, Yao Z, Guan J, Drecun L, Pathmanathan S, Snider J, Umapathy G, Kotlyar M, Jurisica I, Palmer R, and Stagljar I (2021). Mapping the Phospho-dependent ALK Interactome to Identify Novel Components in ALK Signaling. J. Mol. Biol. 433, 167283. 10.1016/j.jmb.2021.167283. [DOI] [PubMed] [Google Scholar]
- 48.Grozavu I, Stuart S, Lyakisheva A, Yao Z, Pathmanathan S, Ohh M, and Stagljar I (2022). D154Q Mutation does not Alter KRAS Dimerization. J. Mol. Biol. 434, 167392. 10.1016/j.jmb.2021.167392. [DOI] [PubMed] [Google Scholar]
- 49.Saraon P, Snider J, Kalaidzidis Y, Wybenga-Groot LE, Weiss K, Rai A, Radulovich N, Drecun L, Vučković N, Vučetić A, et al. (2020). A drug discovery platform to identify compounds that inhibit EGFR triple mutants. Nat. Chem. Biol. 16, 577–586. 10.1038/s41589-020-0484-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saraon P, Grozavu I, Lim SH, Snider J, Yao Z, and Stagljar I (2017). Detecting Membrane Protein-protein Interactions Using the Mammalian Membrane Two-hybrid (MaMTH) Assay. Curr. Protoc. Chem. Biol. 9, 38–54. 10.1002/cpch.15. [DOI] [PubMed] [Google Scholar]
- 51.Hashidate-Yoshida T, Harayama T, Hishikawa D, Morimoto R, Hamano F, Tokuoka SM, Eto M, Tamura-Nakano M, Yanobu-Takanashi R, Mukumoto Y, et al. (2015). Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. Elife 4, e06328. 10.7554/eLife.06328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao Y, Chen Y-Q, Bonacci TM, Bredt DS, Li S, Bensch WR, Moller DE, Kowala M, Konrad RJ, and Cao G (2008). Identification and characterization of a major liver lysophosphatidylcholine acyltransferase. J. Biol. Chem. 283, 8258–8265. 10.1074/jbc.M710422200. [DOI] [PubMed] [Google Scholar]
- 53.Blunsom NJ, and Cockcroft S (2020). Phosphatidylinositol synthesis at the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1865, 158471. 10.1016/j.bbalip.2019.05.015. [DOI] [PubMed] [Google Scholar]
- 54.Neumann CKA, Silver DJ, Venkateshwari V, Zhang R, Traughber CA, Przybycin C, Bayik D, Smith JD, Lathia JD, Rini BI, and Brown JM (2020). MBOAT7-driven phosphatidylinositol remodeling promotes the progression of clear cell renal carcinoma. Mol. Metab. 34, 136–145. 10.1016/j.molmet.2020.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kadamur G, and Ross EM (2013). Mammalian Phospholipase C. Annu. Rev. Physiol. 75, 127–154. 10.1146/annurev-physiol-030212-183750. [DOI] [PubMed] [Google Scholar]
- 56.Hishikawa D, Hashidate T, Shimizu T, and Shindou H (2014). Diversityand function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J. Lipid Res. 55, 799–807. 10.1194/jlr.R046094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saliakoura M, Reynoso-Moreno I, Pozzato C, Rossi Sebastiano M,Galié M, Gertsch J, and Konstantinidou G (2020). The ACSL3-LPIAT1 signaling drives prostaglandin synthesis in non-small cell lung cancer. Oncogene 39, 2948–2960. 10.1038/s41388-020-1196-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pedley AM, Pareek V, and Benkovic SJ (2022). The Purinosome: ACase Study for a Mammalian Metabolon. Annu. Rev. Biochem. 91, 89–106. 10.1146/annurev-biochem-032620-105728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang C, Zhao Y, Wang L, Guo Z, Ma L, Yang R, Wu Y, Li X, Niu J, Chu Q, et al. (2023). De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat. Cell Biol. 25, 836–847. 10.1038/s41556-023-01146-4. [DOI] [PubMed] [Google Scholar]
- 60.Liang D, Feng Y, Zandkarimi F, Wang H, Zhang Z, Kim J, Cai Y,Gu W, Stockwell BR, and Jiang X (2023). Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 186, 2748–2764.e22. 10.1016/j.cell.2023.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reed A, Ware T, Li H, Fernando Bazan J, and Cravatt BF (2023). TMEM164 is an acyltransferase that forms ferroptotic C20:4 ether phospholipids. Nat. Chem. Biol. 19, 378–388. 10.1038/s41589-022-01253-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY,Bayır H, Abhari BA, Angeli JPF, Choi SM, et al. (2018). Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest. 128, 3341–3355. 10.1172/JCI99032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, Vandenabeele P, Wullaert A, and Vanden Berghe T (2020). Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 11, 922–1011. 10.1038/s41419-020-03118-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pope LE, and Dixon SJ (2023). Regulation of ferroptosis by lipid metabolism. Trends Cell Biol. 10.1016/j.tcb.2023.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang H-L, Hu B-X, Li Z-L, Du T, Shan J-L, Ye Z-P, Peng X-D,Li X, Huang Y, Zhu X-Y, et al. (2022). PKCbII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 24, 88–98. 10.1038/s41556-021-00818-3. [DOI] [PubMed] [Google Scholar]
- 66.Leonard TA, Rózycki B, Saidi LF, Hummer G, and Hurley JH (2011). Crystal Structure and Allosteric Activation of Protein Kinase C bII. Cell 144, 55–66. 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang C, Wang Y, Wang F, Wang Z, Lu Y, Xu Y, Wang K, Shen H, Yang P, Li S, et al. (2017). Quantitative profiling of glycerophospholipids during mouse and human macrophage differentiation using targeted mass spectrometry. Sci. Rep. 7, 412. 10.1038/s41598-017-00341-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Helsley RN, Varadharajan V, Brown AL, Gromovsky AD, Schugar RC, Ramachandiran I, Fung K, Kabbany MN, Banerjee R, Neumann CK, et al. (2019). Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. Elife 8, e49882. 10.7554/eLife.49882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors andgenome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Golden RJ, Chen B, Li T, Braun J, Manjunath H, Chen X, Wu J, Schmid V, Chang T-C, Kopp F, et al. (2017). An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 542, 197–202. 10.1038/nature21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hayer A, Shao L, Chung M, Joubert L-M, Yang HW, Tsai F-C, Bi-saria A, Betzig E, and Meyer T (2016). Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat. Cell Biol. 18, 1311–1323. 10.1038/ncb3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Davis MW, and Jorgensen EM (2022). ApE, A Plasmid Editor: A Freely Available DNA Manipulation and Visualization Program. Front. Bio-informa 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Team RStudio (2020). RStudio (Integrated Development Environment for R (RStudio, PBC.). [Google Scholar]
- 74.R Core Team (2022). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing). [Google Scholar]
- 75.Taguchi R, and Ishikawa M (2010). Precise and global identification of phospholipid molecular species by an Orbitrap mass spectrometer and automated search engine Lipid Search. J. Chromatogr. A 1217, 4229–4239. 10.1016/j.chroma.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 76.Yamada T, Uchikata T, Sakamoto S, Yokoi Y, Fukusaki E, and Bamba T (2013). Development of a lipid profiling system using reverse-phase liquid chromatography coupled to high-resolution mass spectrometry with rapid polarity switching and an automated lipid identification software. J. Chromatogr. A 1292, 211–218. 10.1016/j.chroma.2013.01.078. [DOI] [PubMed] [Google Scholar]
- 77.Bird SS, Marur VR, Sniatynski MJ, Greenberg HK, and Kristal BS(2011). Serum lipidomics profiling using LC-MS and high-energy collisional dissociation fragmentation: focus on triglyceride detection and characterization. Anal. Chem. 83, 6648–6657. 10.1021/ac201195d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu C, van Dommelen J, van der Heijden R, Spijksma G, Reijmers TH, Wang M, Slee E, Lu X, Xu G, van der Greef J, and Hankemeier T (2008). RPLC-ion-trap-FTMS method for lipid profiling of plasma: method validation and application to p53 mutant mouse model. J. Proteome Res. 7, 4982–4991. 10.1021/pr800373m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw metabolomics data have been deposited at Metabolights and are publicly available as of the date of publication. The accession number is listed in the key resources table. Spreadsheets related to the four lipidomics datasets and one polar metabolomics dataset reported in this study have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-HA | Cell Signaling Technology | Cat#3724; RRID: AB_1549585 |
| Rabbit polyclonal anti-MMD | Cell Signaling Technology | N/A |
| Rabbit monoclonal anti-MMD | Cell Signaling Technology | Cat#20226 |
| Rabbit polyclonal anti-FSP1 | Cell Signaling Technology | Cat#24972S |
| Rabbit polyclonal anti-ACSL4 | Invitrogen | Cat#PA527137; RRID: AB_2544613 |
| Rabbit monoclonal anti-GPX4 | abcam | Cat#ab125066; RRID: AB_10973901 |
| Rat monoclonal anti-MBOAT7 | Dr. Nozomu Kono (The University of Tokyo, Tokyo, Japan); Lee et al.41 | N/A |
| Mouse monoclonal anti-COXIV | Cell Signaling Technology | Cat#11967; RRID: AB_2797784 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat#2118; RRID: AB_561053 |
| Mouse monoclonal anti-CANX | Invitrogen | Cat#MA3–027; RRID: AB_2069043 |
| Mouse monoclonal anti-HA-Tag Alexa Fluor 647 Conjugate |
Cell Signaling Technology | Cat#3444S; RRID: AB_10693329 |
| Rabbit monoclonal anti-GM130 | Cell Signaling Technology | Cat#12480; RRID: AB_2797933 |
| Rabbit monoclonal Anti-DYKDDDDK (FLAG) | Cell Signaling Technology | Cat#14793S; RRID: AB_2572291 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| One Shot™ MAX Efficiency™ DH5α-T1R competent cells | Invitrogen | 12297016 |
|
| ||
| Biological samples | ||
|
| ||
| Discarded human blood products | Crimson Core Specimen Bank (Brigham & Women’s Hospital, Boston, MA, USA) | https://crimson-core.partners.org/samples/ |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RSL3 | SelleckChem | Cat#S8155 |
| ML210 | Sigma-Aldrich | Cat#SML0521 |
| Erastin | SelleckChem | Cat#S7242 |
| FIN56 | SelleckChem | Cat#S8254 |
| Ferrostatin-1 | Sigma-Aldrich | Cat#SML0583 |
| Z-VAD-FMK | SelleckChem | Cat#S7023 |
| Necrostatin-1 | SelleckChem | Cat#S8037 |
| BSA-control | Cayman Chemical | Cat#29556 |
| BSA-palmitate | Cayman Chemical | Cat#29558 |
| PMA | Sigma-Aldrich | Cat#P8139 |
| Recombinant human M-CSF | Peprotech | Cat#300–25 |
| 3H-Arachidonic acid | Perkin Elmer | Cat#NET298Z050UC |
|
| ||
| Critical commercial assays | ||
|
| ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat#G7571 |
| BODIPY™ 581/591 C11 (Lipid Peroxidation Sensor) | Invitrogen | Cat#D3861 |
|
| ||
| Deposited data | ||
|
| ||
| Lipidomics dataset #1 (OVCAR-8 and 786-O NT sg vs. MMD KO cells) and associated code | This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169857 |
| Lipidomics dataset #3 (OVCAR-8 NT sg, MMD KO, EV + NT sg, and EV + MBOAT7 sg cells) and associated code |
This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169908 |
| Lipidomics dataset #4 (OVCAR-8 NT sg, MMD2 sg bulk, MMD2 KO1, MMD2 KO2, NT sg + NT sg, NT sg + MBOAT7 sg, MMD KO + NT sg, and MMD KO + MBOAT7 sg cells) and associated code | This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169917 |
| Polar metabolomics dataset (OVCAR-8 and 786-O NT sg vs. MMD KO cells) | This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169924 |
| Code for calculating all ranked codependencies of MMD, ACSL4, and MBOAT7 in DepMap | This paper; Zenodo | Zenodo: https://doi.org/10.5281/zenodo.8169937 |
| All raw mass spectrometry (metabolomics) data | This paper; Metabolights | Metabolights: MTBLS8280 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| OVCAR-8 parental | Dr. Joan Brugge (Harvard Medical School, Boston, MA, USA) | RRID: CVCL_1629 |
| OVCAR-8 NT sg (puro) | This paper | N/A |
| OVCAR-8 NT sg (blast) | This paper | N/A |
| OVCAR-8 MMD KO (puro) | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-EF1a-IRES-blast] EV | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-EF1a-IRES-blast] MMD (HA-MMD) |
This paper | N/A |
| OVCAR-8 [pLJM1] EV | This paper | N/A |
| OVCAR-8 [pLJM1] MMD OE (a.k.a. MMD, HA-MMD) | This paper | N/A |
| OVCAR-8 HA-MMD + [pRH115] FLAG-MBOAT7 | This paper | N/A |
| OVCAR-8 MMD2 sg1 (puro) | This paper | N/A |
| OVCAR-8 MMD2 KO1 (puro) | This paper | N/A |
| OVCAR-8 MMD2 KO2 (puro) | This paper | N/A |
| OVCAR-8 ACSL4 sg (blast) | This paper | N/A |
| OVCAR-8 [pLJM1] EV + NT sg (blast) | This paper | N/A |
| OVCAR-8 [pLJM1] EV + ACSL4 sg (blast) | This paper | N/A |
| OVCAR-8 MMD + NT sg (blast) | This paper | N/A |
| OVCAR-8 MMD + ACSL4 sg (blast) | This paper | N/A |
| OVCAR8 [pLJM1] EV + MBOAT7 sg (blast) | This paper | N/A |
| OVCAR-8 MMD + MBOAT7 sg (blast) | This paper | N/A |
| OVCAR-8 [pLJM1] EV + LPCAT3 sg (blast) | This paper | N/A |
| OVCAR-8 MMD + LPCAT3 sg (blast) | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-mCherry] ACSL4-HA | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-mCherry] FLAG-MBOAT7 | This paper | N/A |
| OVCAR-8 MMD KO + [pLV-mCherry] ACSL4-HA-P2A-FLAG-MBOAT7 | This paper | N/A |
| OVCAR-8 NT sg (puro) + NT sg (blast) | This paper | N/A |
| OVCAR-8 NT sg (puro) + MBOAT7 sg (blast) | This paper | N/A |
| OVCAR-8 MMD KO (puro) + NT sg (blast) | This paper | N/A |
| OVCAR-8 MMD KO (puro) + MBOAT7 sg (blast) | This paper | N/A |
| 786-O parental | Broad Institute Biological Samples Platform (Broad Institute, Cambridge, MA, USA) | RRID: CVCL_1051 |
| 786-O NT sg (blast) | This paper | N/A |
| 786-O MMD KO (blast) | This paper | N/A |
| 786-O [pLJM1] MMD OE (puro) | This paper | N/A |
| 786-O MBOAT7 sg (blast) | This paper | N/A |
| Huh7 WT | Dr. J. Mark Brown (Cleveland Clinic, Cleveland, OH, USA); Helsley et al.68 | N/A |
| Huh7 MBOAT7 KO | Dr. J. Mark Brown (Cleveland Clinic, Cleveland, OH, USA); Helsley et al.68 | N/A |
| HT-1080 parental | Dr. Robert A. Weinberg (Whitehead Institute for Biomedical Research, Cambridge, MA, USA) | RRID: CVCL_0317 |
| HT-1080 NT sg (blast) | This paper | N/A |
| HT-1080 MBOAT7 sg (blast) | This paper | N/A |
| THP-1 | Dr. Jaime Cheah (Koch Institute for Integrative Cancer Research, Cambridge, MA, USA) | RRID: CVCL_0006 |
| HEK293T | ATCC | Cat#CRL-3216; RRID: CVCL_0063 |
| OVCAR-8 parental MaMTH Gaussia princeps luciferase reporter line (B0755) | This paper | N/A |
| OVCAR-8 NT sg MaMTH Gaussia princeps luciferase reporter line (B0781) | This paper | N/A |
| OVCAR-8 MMD KO MaMTH Gaussia princeps luciferase reporter line (B0772) | This paper | N/A |
| HEK293 | Thermo-Fisher | Cat#R78007; RRID: CVCL_0045 |
| HEK293 MaMTH Gaussia princeps luciferase reporter line | Dr. Igor Stagljar (University of Toronto, Toronto, Canada); Saraon et al.49 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| MMD qPCR primer (Fwd, 5’→3′): CCGCTACAAGCCAACTTGCTAT | This paper | N/A |
| MMD qPCR primer (Rev, 5’→3′): AGTCCCATTCCATAAATCCATG | This paper | N/A |
| ACTB qPCR primer (Fwd, 5’→3′): CACCATTGGCAATGAGCGGTTC | This paper | N/A |
| ACTB qPCR primer (Rev, 5’→3′): AGGTCTTTGCGGATGTCCACGT | This paper | N/A |
| sgRNA target sequences, see Table S2 | This paper | N/A |
| CRISPR Sanger sequencing primers, see Table S2 | This paper | N/A |
| MaMTH plasmid cloning primers, see Table S3 | This paper | N/A |
| MaMTH plasmid sequencing primers, see Table S3 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Vector for CRISPR cloning: lentiCRISPRv2-puro | Dr. Feng Zhang (Broad Institute, Cambridge, MA, USA); Sanjana et al.69 | Addgene #52961; RRID: Addgene_52961 |
| Vector for CRISPR cloning: lentiCRISPRv2-blast | Dr. Mohan Babu (University of Regina, Regina, Canada) | Addgene #83480; RRID: Addgene_83480 |
| Vector for cDNA expression: pLJM1 | Dr. Joshua Mendell (UT Southwestern, Dallas, TX, USA); Golden et al.70 | Addgene #91980; RRID: Addgene_91980 |
| Vector for cDNA expression: pLV-EF1a-IRES-blast | Dr. Tobias Meyer (Stanford University School of Medicine, Stanford, CA, USA); Hayer et al.71 | Addgene #85133; RRID: Addgene_85133 |
| Vector for FLAG-MBOAT7 cloning: pRH115 | Dr. Tobiloba Oni (Whitehead Institute for Biomedical Research, Cambridge, MA, USA) | N/A |
| MMD cDNA | Horizon Discovery Mammalian Gene Collection | Cat#BC026324 |
| MBOAT7 cDNA | Horizon Discovery Mammalian Gene Collection | Cat#BC003164 |
| memACSL4 cDNA | GenScript | Cat#OHu23492, transcript variant 2, mRNA |
| pLV-mCherry-EF1a-memACSL4-HA | VectorBuilder, custom made for this paper | N/A |
| pLV-mCherry-EF1a-FLAG-MBOAT7 | VectorBuilder, custom made for this paper | N/A |
| pLV-mCherry-EF1a-memACSL4-HA:P2A:FLAG-MBOAT7 | VectorBuilder, custom made for this paper | N/A |
| lentiCRISPRv2-puro_NT sg | This paper | N/A |
| lentiCRISPRv2-blast_NT sg | This paper | N/A |
| lentiCRISPRv2-puro_MMDsg10 | This paper | N/A |
| lentiCRISPRv2-blast_MMDsg8 | This paper | N/A |
| lentiCRISPRv2-puro_MMD2sg1 | This paper | N/A |
| lentiCRISPRv2-blast_ACSL4sg | This paper | N/A |
| lentiCRISPRv2-blast_MBOAT7sg3 | This paper | N/A |
| lentiCRISPRv2-blast_LPCAT3sg3 | This paper | N/A |
| pLV-EF1a-IRES-blast_HA-MMD_sgResistant | This paper | N/A |
| pLJM1_HA-MMD | This paper | N/A |
| pRH115_FLAG-MBOAT7 | This paper | N/A |
| Gateway cloning entry vector: pDONR221 | Dr. Tsukasa Shibue (Broad Institute, Cambridge, MA, USA) | N/A |
| MaMTH N-terminally tagged MMD prey: A1245 MMD | This paper | N/A |
| MaMTH C-terminally tagged memACSL4 prey: A1379 memACSL4 | This paper | N/A |
| MaMTH C-terminally tagged MBOAT7 bait: A1160 MBOAT7 | This paper | N/A |
| MaMTH C-terminally tagged memACSL4 bait: A1160 memACSL4 | This paper | N/A |
| MaMTH N-terminally tagged PEX7 prey: A1245 PEX7 | Dr. Igor Stagljar (University of Toronto, Toronto, Canada); Saraon et al.50 | N/A |
| MaMTH 5xGAL4 UAS Gaussia princeps reporter plasmid containing hygromycin resistance cassette (A1687) | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ApE Version 3.1.3 | Davis et al.72 | https://jorgensen.biology.utah.edu/wayned/ape/ |
| SnapGene Version 6.2.1 | Dotmatics | www.snapgene.com |
| GraphPad Prism Version 9.5.1 | Dotmatics | www.graphpad.com |
| RStudio Version 2022.07.0 + 548 | RStudio Team73 | www.rstudio.com |
| R Version 4.2.1 | R Core Team74 | https://www.R-project.org |
| ICE Analysis Tool Version 3.0 | Synthego | https://ice.synthego.com/#/ |
| Imaris Version 10.0 | Oxford Instruments | https://imaris.oxinst.com/ |
| LipidSearch Version 4.2.27 | Thermo Fisher Scientific, Mitsui Knowledge Industries75,76 | N/A |
| TraceFinder | Thermo Fisher Scientific | N/A |
| Progenesis QI | Nonlinear Dynamics | https://www.nonlinear.com/progenesis/qi/ |