

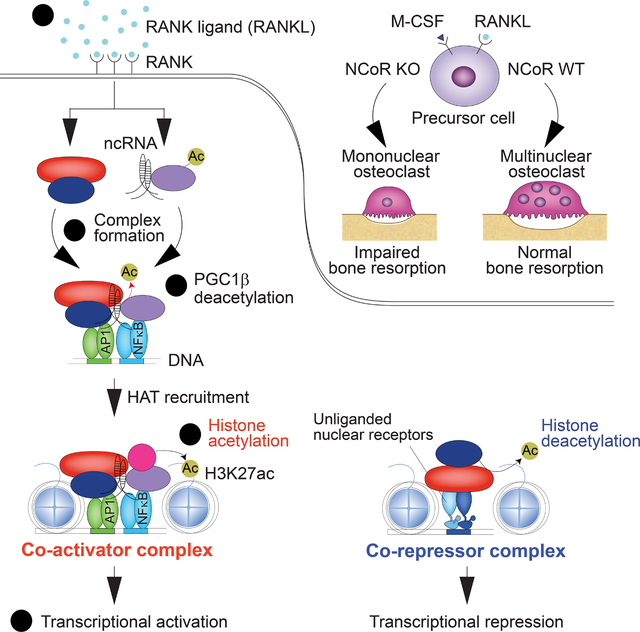
SUMMARY
The nuclear receptor co-repressor (NCoR) complex mediates transcriptional repression dependent on histone deacetylation by histone deacetylase 3 (HDAC3) as a component of the complex. Unexpectedly, we found that signaling by receptor activator of nuclear factor kappa-Β (RANK) converts the NCoR/HDAC3 co-repressor complex to a co-activator of AP-1 and NFκB target genes that are required for mouse osteoclast differentiation. Accordingly, the dominant function of NCoR/HDAC3 complexes in response to RANK signaling is to activate, rather than repress, gene expression. Mechanistically, RANK signaling promotes RNA-dependent interaction of the transcriptional co-activator PGC1β with the NCoR/HDAC3 complex, resulting in activation of PGC1β and inhibition of HDAC3 activity for acetylated histone H3. Non-coding RNAs Dancr and Rnu12 that are associated with altered human bone homeostasis promote NCoR/HDAC3 complex assembly and are necessary for RANKL-induced osteoclast differentiation in vitro. These findings may be prototypic for signal-dependent functions of NCoR in other biological contexts.
In brief
The nuclear receptor co-repressor NCoR is found to be necessary for normal bone density by co-activating RANKL-dependent gene transcription in osteoclasts. Abe et al. resolve this paradox by demonstrating that RANKL induces RNA-dependent assembly of an NCoR/HDAC3/PGC1β complex that co-activates NFκB and AP-1 target genes required for osteoclast differentiation.
Graphical Abstract
INTRODUCTION
Nuclear receptor co-repressor (NCoR) and silencing mediator of retinoid and thyroid hormone receptor (SMRT, also known as NCoR2) are critical transcriptional co-repressors of nuclear receptors and other signal-dependent transcription factors,1–3 underlying their diverse and essential roles in development, homeostasis and immunity. The co-repressor functions of NCoR and SMRT are dependent on histone deacetylase 3 (HDAC3), which mainly deacetylates K27 on the histone H3 N-terminal tail (H3K27).4–6 NCoR/HDAC3 and SMRT/HDAC3 co-repressor complexes interact with a subset of unliganded nuclear receptors bound to their cognate recognition motifs in enhancers and promoters, including thyroid hormone receptors, liver X receptors (LXRs) and retinoic acid receptors, resulting in histone deacetylation and transcriptional repression. The binding of activating ligands to each of these receptors causes NCoR/HDAC3 and SMRT/HDAC3 co-repressor complexes to dissociate in exchange for co-activator complexes with histone acetyltransferase activity that promote transcriptional activation.3 This paradigm of co-repressor/co-activator exchange for signal-dependent gene activation was subsequently extended to other classes of transcription factors, including recombination signal binding protein for immunoglobulin Kappa J region (Rbpj)7 and members of the activator protein 1 (AP-1) family.8
Unexpectedly, deletion of NCoR from macrophages was associated with an attenuated response to toll-like receptor 4 (TLR4) signaling and an anti-inflammatory/insulin sensitive phenotype under high fat diet.9 This result could be partially explained by de-repression of LXR target genes encoding enzymes that catalyze the synthesis of anti-inflammatory omega 3 fatty acids. However, omega 3 fatty acids only suppressed a small fraction of the genes that were hypo-responsive to TLR4 signaling in NCoR-deficient macrophages,9 leaving the majority of the attenuated response unexplained. To gain further insights into functions of NCoR in regulating macrophage phenotypes, we examined the consequences of NCoR deletion on the development and function of osteoclasts. These studies revealed the requirement of NCoR for activation of receptor activator of nuclear factor kappa-Β ligand (RANKL)-dependent genes and osteoclast differentiation. Consequently, mice lacking NCoR in the myeloid lineage exhibit a high bone mass phenotype. Genome-wide location analysis of NCoR and HDAC3 in RANKL-treated bone marrow cells indicated a paradoxical gain of local histone acetylation and gene activation, rather than the expected loss of histone acetylation and gene repression.
We present evidence that the RANKL-induced co-activator function of NCoR involves HDAC3-dependent deacetylation of peroxisome proliferator-activated receptor gamma (PPARγ) co-activator 1 beta (PGC1β), analogous to the prior finding that NCoR/HDAC3 complexes deacetylate PGC1α as a prerequisite for coactivation of estrogen-related receptor α (ERRα) and induction of thermogenesis in brown adipose tissue.10 We extend this concept to demonstrate that RANK signaling promotes the assembly of a ~2 MDa NCoR/HDAC3/PGC1β complex that is dependent on non-coding RNAs and results in the conversion of NCoR from a constitutive co-repressor to a signal-dependent co-activator of gene expression. These findings raise the possibility that NCoR complexes are broadly used as signal-dependent co-activators and therefore challenge the current view that they primarily function as co-repressors.
RESULTS
NCoR is required for osteoclast differentiation and normal bone development
To explore potential roles of NCoR in osteoclast function, we crossed LysM-Cre mice to Ncorf/f mice to establish myeloid-specific NCoR-deficient (NKO) mice9 (Figure S1A). Bone microstructure imaging by high-resolution micro-computed tomography (μCT) of femurs revealed that NKO male mice exhibited a high bone mass phenotype accompanied by significantly increased trabecular bone volume (top panels, Figures 1A and 1B) and trabecular bone number (top panel, Figure 1B), compared to WT mice. These findings were further supported by markedly decreased trabecular bone separation and increased bone mineral density in femurs of NKO male mice (top panels, Figure 1B). To investigate the extent to which SMRT might compensate for loss of NCoR, we also evaluated bone homeostasis in mice which both NCoR and SMRT were deleted (DKO) in the myeloid lineage by crossing LysM-Cre mice to Ncorf/f Smrtf/f mice (Figure S1B). DKO male mice exhibited a slightly more severe phenotype than NKO male mice as shown by the significantly increased trabecular bone thickness in femurs (Figure 1B) and cortical thickness of diaphysis (Figure S1C) by NCoR/SMRT deficiency not observed in NKO male mice (Figure 1B). These results suggest that SMRT partially compensates for the loss of NCoR in the regulation of bone homeostasis. NKO or DKO female mice also exhibited a high bone mass phenotype compared to WT female mice (Figure S1D). Notably, the expression of tartrate-resistant acid phosphatase 5 (ACP5) protein as a mature osteoclast marker was decreased in whole tibia and femur bone lysates of NKO or DKO mice (Figure 1C). Additionally, the serum level of deoxypyridinoline, a bone resorption marker was lower in both NKO and DKO mice, but there was no difference in the serum level of osteocalcin, a bone formation maker (Figure 1D), which suggests that the high bone mass phenotype in NCoR deficiency results from altered bone resorption, not formation. Osteoclasts were visualized in femoral longitudinal sections by cell staining for tartrate-resistant acid phosphatase (TRAP). Quantitative analysis of osteoclast number and surface area indicated significant reductions in NKO mice (Figures 1E and S1E), consistent with a defect in osteoclast differentiation and the increase in bone mass.
Figure 1. NCoR is required for osteoclast differentiation and normal bone development.
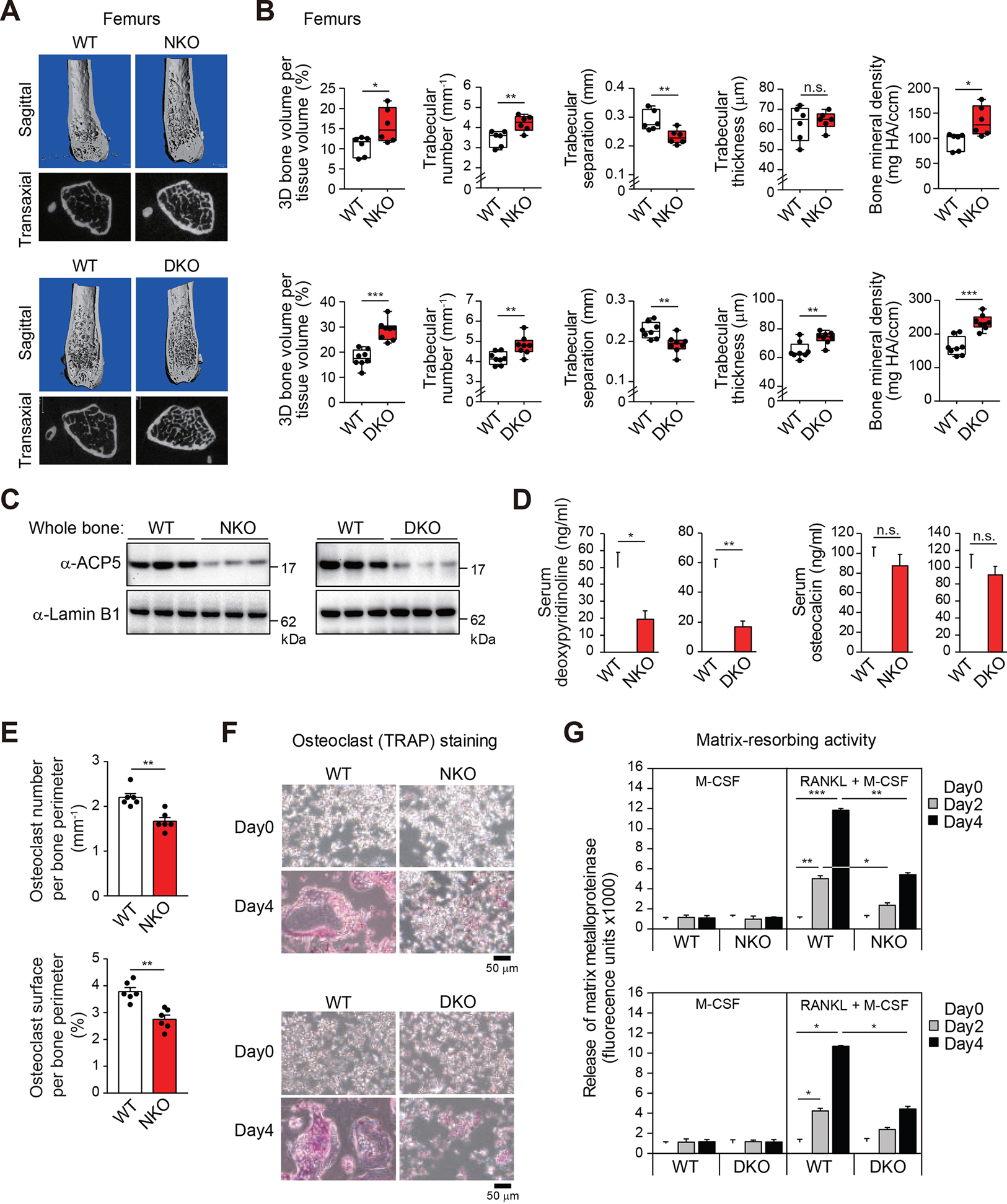
(A, B) Representative microcomputed tomography (μCT) images of the femurs (A) in 5-month-old male WT and NKO or DKO mice. Trabecular bone volume, trabecular number, trabecular separation, trabecular thickness and bone mineral density in the femurs (B) determined by μCT analysis. All box plots show the interquartile range. Data are mean ± s.d. (n=6–8 each). Student’s t-test was performed for comparisons.
(C) Immunoblot analysis for ACP5 protein in whole bone (tibia and femur) from 5-month-old male WT and NKO or DKO mice (n=3 each).
(D) Serum deoxypyridinoline and osteocalcin concentrations in 5-month-old male WT and NKO or DKO mice. Data are mean ± s.e.m. (n=4 biological replicates). Student’s t-test was performed for comparisons.
(E) Quantitative analysis of osteoclast number (upper panel) and surface area (lower panel) in the femurs from 5-month-old male WT and NKO mice. Data are mean ± s.e.m. (n=6 each). Student’s t-test was performed for comparisons.
(F) Representative TRAP-stained cell images showing the effect of NKO or DKO on bone marrow-derived osteoclast formation at Day0 and Day4 after RANKL treatment (12-week-old male mice).
(G) Matrix-resorbing activity on bone marrow derived-osteoclasts from WT and NKO or DKO mice (12-week-old male) in the presence of only M-CSF or M-CSF plus RANKL. Data are mean ± s.d. (n=3 biological replicates). Analysis of variance was performed followed by Tukey’s post hoc comparison.
For panels B, D, E and G; *p < 0.05, **p < 0.01 and ***p < 0.001.
See also Figure S1.
As RANKL is required for the development of osteoclasts from myeloid progenitor cells,11–13 we examined the impact of NCoR or NCoR/SMRT deficiency on RANKL-induced osteoclast differentiation in vitro. Bone marrow cells were cultured with macrophage colony stimulating factor (M-CSF) alone for 3 days and then stimulated with M-CSF and RANKL for 4 days (Figure S1F). Osteoclasts were stained with TRAP. RANKL promoted the formation of multinucleated osteoclasts in bone marrow cells from WT mice, but this effect was suppressed in cells from NKO or DKO mice (Figure 1F). Next, to examine osteoclast function, bone marrow cells were seeded on a collagen-coated plate and the resorption activity of osteoclasts, as reflected by matrix metalloproteases released in culture media, was measured. RANKL-induced matrix resorption activity in NKO or DKO cells was lower compared to WT cells (Figure 1G). Collectively, these findings establish signal-dependent roles of NCoR downstream of RANK, and a previously unknown requirement of NCoR for osteoclast differentiation and function.
Signal-specific NCoR-dependent gene expression
NCoR deficiency had a substantial impact on RANKL-dependent gene expression (Figure 2A). Of the 1686 mRNAs induced > 1.5-fold by RANKL (FDR < 0.05) following the full differentiation process in WT cells, 757 exhibited significantly attenuated responses in NKO cells (hypo-responsive genes) (Figure 2A), exemplified by transcription factor genes Jdp2 (encoding Jun dimerization protein 2), Fosl2 (encoding Fos like 2) and Nfatc1 (encoding nuclear factor of activated T-cells 1), required for osteoclast differentiation,14–16 and osteoclast specific genes Acp5 (encoding tartrate-resistant acid phosphatase 5, also known as TRAP) and Ocstamp (encoding osteoclast stimulatory transmembrane protein) (Figure 2B). The 757 genes exhibiting reduced responsiveness to RANKL in NKO cells were also significantly enriched for functional annotations related to mitochondrial organization and mitochondrial gene expression (Figure S2A), suggesting NCoR regulates mitochondrial biogenesis and activity, which has been linked to osteoclast differentiation and function.17 In contrast, the LXR target genes Abca1 (encoding ATP-binding cassette subfamily A member 1) and Abcg1 (encoding ATP-binding cassette subfamily G member 1) were de-repressed (Figure 2B), consistent with conventional co-repressor activity of NCoR in the same cells. In contrast to the comparison of wild type and NKO cells following four days of treatment with RANKL, the comparison between WT Day0 and NKO Day0 resulted in only 7 upregulated genes, including Abca1 as shown in Figure 2B, and only 3 downregulated genes (Figure S2B). These findings indicate that in this context, the major transcriptional function of NCoR is acquired in a signal dependent manner.
Figure 2. NCoR/HDAC3 complexes bind to RANKL-induced enhancers and promoters.
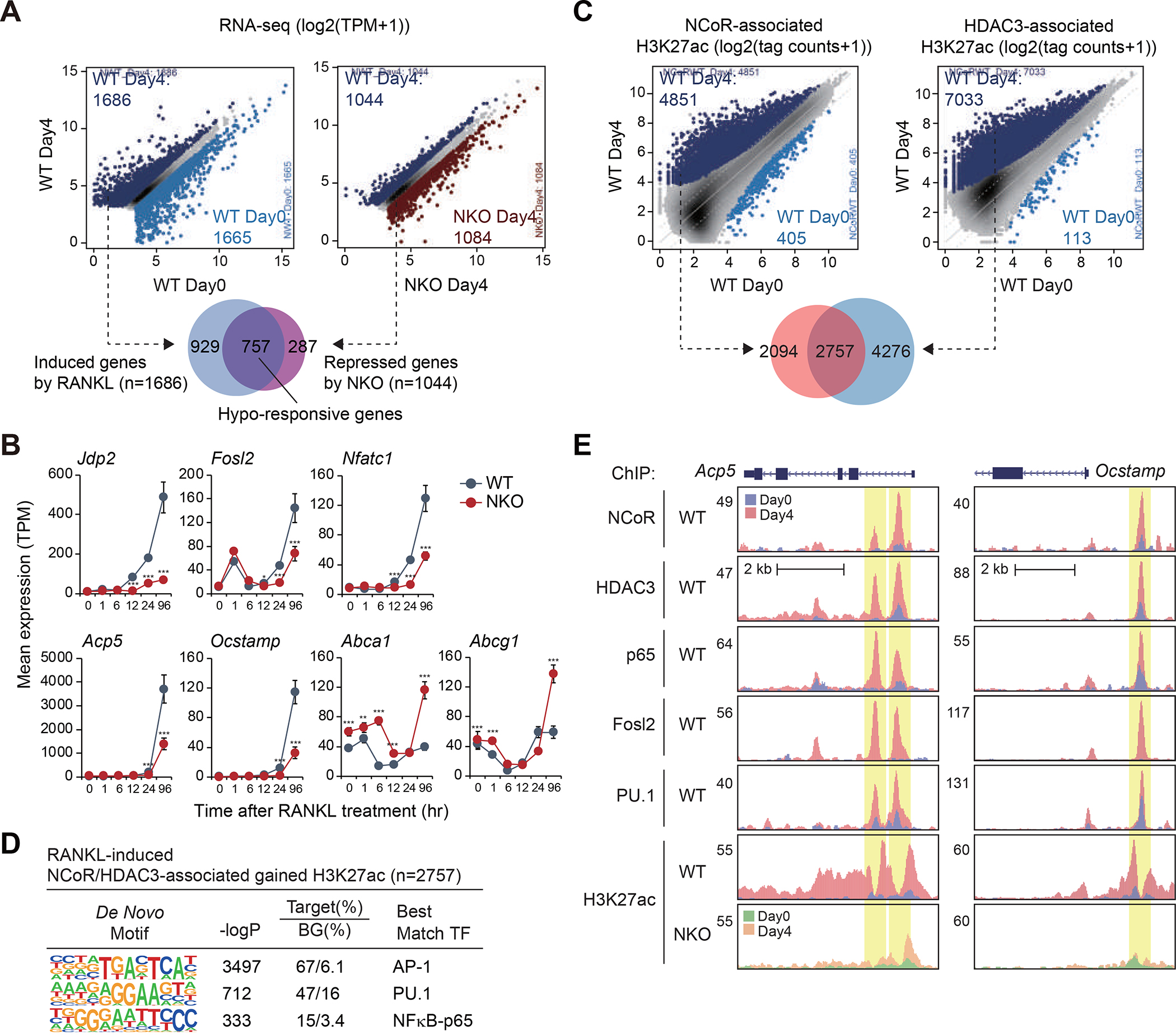
(A) Scatter plots of RNA-seq data showing RANKL-regulated gene expression in WT cells (left panel) and NKO-regulated gene expression in the presence of RANKL (right panel) (light blue dots in left panel: significantly RANKL-suppressed genes, dark blue dots in left panel: significantly RANKL-induced genes, dark red dots in right panel: significant NKO-induced genes, dark blue dots in right panel: significant NKO-suppressed genes, FDR < 0.05, FC > 1.5). The overlap between RANKL-induced genes in WT cells (n=1686) and NKO-suppressed genes in the presence of RANKL (n=1044) is shown by Venn diagram.
(B) Expression of Jdp2, Fosl2, Nfatc1, Acp5, Ocstamp, Abca1 and Abcg1 in WT and NKO cells as a function of time following RANKL treatment. The significance symbols indicate statistical significance comparing NKO to WT, *p-adj < 0.05, **p-adj < 0.01 and ***p-adj < 0.001.
(C) Scatter plot of distal NCoR (41058 peaks in Figure S2C)- (left panel) or HDAC3 (31832 peaks in Figure S2D)- (right panel) associated H3K27ac in WT at Day0 vs. WT at Day4. RANKL-induced NCoR- or HDAC3-associated H3K27ac peaks (FDR < 0.05, FC > 2) are color-coded (light blue dots: significantly NCoR- or HDAC3-associated lost H3K27ac in WT at Day4, dark blue dots: significantly NCoR- or HDAC3-associated gained H3K27ac in WT at Day4). The overlap between NCoR-associated gained H3K27ac (n=4851) and HDAC3-associated gained H3K27ac (n=7033) is shown by Venn diagram.
(D) De novo motif enrichment analysis of RANKL-induced NCoR and HDAC3-associated gained H3K27ac peaks (n=2757 in Figure 2C) using a GC-matched genomic background.
(E) Genome browser tracks of NCoR, HDAC3, p65, Fosl2, PU.1 and H3K27ac ChIP-seq peaks in the vicinity of the Acp5 and Ocstamp loci at Day0 and Day4 after RANKL treatment. Yellow shading: RANKL-induced peaks.
See also Figure S2.
NCoR/HDAC3 complexes bind to RANKL-induced enhancers and promoters
To investigate mechanisms underlying the requirement of the NCoR complex for RANKL-induced gene expression, we used chromatin immunoprecipitation sequencing (ChIP-seq) to define the genome wide locations of NCoR and HDAC3 in WT cells under control conditions and in response to RANKL. RANKL resulted in substantial increases in the number of confident genomic binding sites of NCoR (Figure S2C) and HDAC3 (Figure S2D). In parallel, we performed ChIP-seq for H3K27ac under control and RANKL treatment conditions as a surrogate for promoter and enhancer activity.18 RANKL treatment resulted in significant increases in H3K27ac at 1525 locations (Figure S2E). To define the relationship between NCoR/HDAC3 binding and gain or loss promoter/enhancer activity, we annotated NCoR and HDAC3 peaks for local H3K27ac (+/− 500 bp) under control and RANKL treatment conditions and determined their overlaps. In the case of RANKL treatment, > 2700 of the genomic locations exhibiting constitutive or gained presumptive NCoR/HDAC3 complexes localized to promoters and putative enhancers that exhibited a paradoxical > 2-fold increase in H3K27ac, whereas less than 120 of such regions were associated with the expected loss of H3K27ac (Figure 2C). NCoR/HDAC3-associated peaks gaining H3K27ac were most highly enriched for motifs recognized by AP-1 and NFκB-p65 family members, along with the motif for the macrophage lineage determining transcription factor PU.1 (Figure 2D). Signal-dependent recruitment of NCoR/HDAC3 to sites of gained H3K27ac is exemplified at the promoters and enhancers associated with Acp5 and Ocstamp (Figure 2E). ChIP-sequencing experiments confirmed RANKL-dependent increases in the binding of NFκB-p65, the AP-1 factor Fosl2, and PU.1 at sites of NCoR/HDAC3 binding (Figure 2E).
We previously demonstrated that IL1β causes nuclear export of NCoR, resulting in de-repression of a specific subset of NFκB-p50 (but not p65)-regulated genes in 293T and CV-1 cells.19 In addition, we also reported that IL1β signaling leads NCoR/TAB2 complexes to dissociate from nuclear receptors via TAB2-phosphorylation followed by inflammatory gene activation in LNCaP cells.20 In the present studies, the effect of RANK signaling to significantly increase, rather than decrease, the binding of NCoR to osteoclast-related genes is clearly established by ChIP assays (Figures 2C and 2E). Consistent with the ChIP-seq experiments, RANKL induced the amount of NCoR in nuclear fraction (Figure S2F). Importantly, NCoR did not interact with TAB2 or NFκB-p50 either under basal or RANKL treatment conditions (Figure S2F). In contrast, NCoR was associated with NFκB-p65 in response to RANKL treatment (Figure S2F), which is consistent with the enriched motifs of transcription factor for RANKL-induced NCoR/HDAC3-associated gained H3K27ac (Figure 2D). Collectively, these findings indicate that cellular trafficking of NCoR complexes is regulated in a cell and signal-dependent manner.
NCoR and HDAC3 activity are required for RANKL-induced H3K27 acetylation
To investigate whether there was a causal relationship between NCoR/HDAC3 binding and increased H3K27ac, we performed ChIP-seq for H3K27ac in NCoR-deficient bone marrow cells. Remarkably, NCoR deficiency resulted in a marked reduction in H3K27ac at genomic locations in which NCoR/HDAC3 binding was associated with RANKL-induced H3K27ac (Figures 2C and 3A). In parallel, we investigated the role of HDAC3 deacetylase activity by examining the consequences of the HDAC3-specific inhibitor RGFP96621 on RANKL-dependent gene expression and histone acetylation. Treatment of bone marrow cells with the combination of RANKL and RGFP966 resulted in an attenuated activation of a large fraction of genes (Figure 3B) including Acp5 and Ocstamp (Figure 3D) that were hypo-responsive in the context of the NCoR knockout (Figure S3A) and significant loss of H3K27ac peaks (Figures 3C and 3E). At a functional level, RANKL-induced bone resorption activity was attenuated by HDAC3 inhibition (Figure 3F). Collectively, these results provide evidence that the NCoR/HDAC3 complex is paradoxically required for histone acetylation and gene activation in response to RANK signaling.
Figure 3. NCoR and HDAC3 activity are required for RANKL-induced H3K27 acetylation.
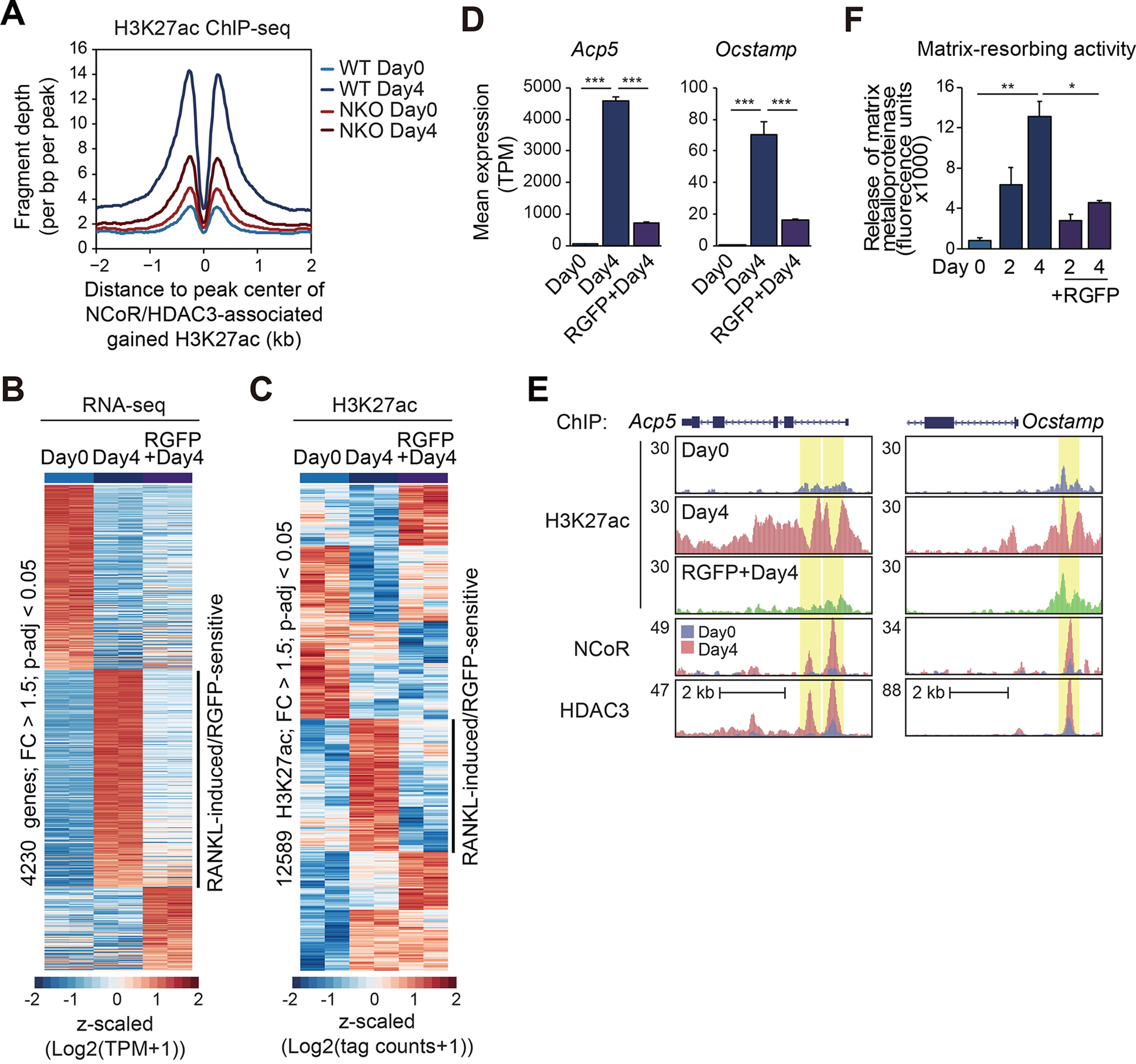
(A) Normalized distribution of H3K27ac tag density in WT and NKO at the vicinity of NCoR and HDAC3-associated gained H3K27ac peaks in WT at Day4 after RANKL treatment (n=2757 in Figure 2C).
(B) Heatmap of differential gene expression (FC > 1.5, p-adj < 0.05) in WT cells treated with the combination of RGFP966 with RANKL.
(C) Heatmap of differential H3K27ac ChIP-seq IDR peaks associated with ATAC-seq IDR peaks (FC > 1.5, p-adj < 0.05) at Day0 in a 1000 bp window.
(D) Bar plots for expression of Acp5 and Ocstamp. ***p-adj < 0.001.
(E) Genome browser tracks of H3K27ac ChIP-seq peaks in WT at Day0 and Day4 with or without RGFP966, and NCoR and HDAC3 ChIP-seq peaks in WT at Day0 and Day4 in the vicinity of the Acp5 and Ocstamp loci. Yellow shading: RANKL-induced RGFP966-sensitive peaks.
(F) RANKL-induced matrix-resorbing activity on bone marrow cells from WT mice (12-week-old male) in the presence or absence of RGFP966. Data are mean ± s.e.m. (n=3 biological replicates). Analysis of variance was performed followed by Tukey’s post hoc comparison. *p < 0.05 and **p < 0.01.
See also Figure S3.
RANK signaling induces NCoR/HDAC3/PGC1β interaction required for H3K27 acetylation
Recent studies linked HDAC3 to gene activation in brown adipose tissue by a mechanism involving deacetylation of PGC1α, which was found to be required for its ability to function as a co-activator for ERRα and enable a transcriptional program supporting thermogenesis.10 Although PGC1α mRNA (Ppargc1a) is not expressed in bone marrow cells, PGC1β mRNA (Ppargc1b) is expressed (Figure 4A) and has been previously suggested to contribute to alternative macrophage activation in response to IL4 signaling by serving as a PPARγ co-activator22 but to activation of osteoclastogenesis linking to bone destruction in rheumatoid arthritis.23 Intriguingly, Ppargc1b is strongly induced by RANKL (Figure 4A). To investigate the possibility that PGC1β might have a previously unrecognized function required for NCoR/HDAC3-dependent gene activation, we evaluated its genome wide locations under control and RANKL treatment conditions. ChIP-sequencing experiments revealed ~9000 high confidence PGC1β binding sites in bone marrow cells under control conditions (Day0) (Figure 4B). RANKL treatment (Day4) led to increased binding at > 35000 sites (Figure 4B). Intersection of PGC1β binding sites under RANKL treatment conditions with the 1525 regions of open chromatin exhibiting > 2-fold increases in H3K27ac indicated that 577 (38%) were occupied by the combination of NCoR and HDAC3 (Figure 4C), which were enriched for AP-1, PU.1 and NFκB-p65 recognition motifs (Figure S4A). In contrast, this combination was only observed at 190 (17%) of the 1112 regions of open chromatin exhibiting > 2-fold decreases in H3K27ac (Figures S2E and S4B), which were enriched for binding sites for PU.1, but not AP-1 and NFκB-p65 (Figure S4C).
Figure 4. RANK signaling induces NCoR/HDAC3/PGC1β interaction required for H3K27 acetylation.
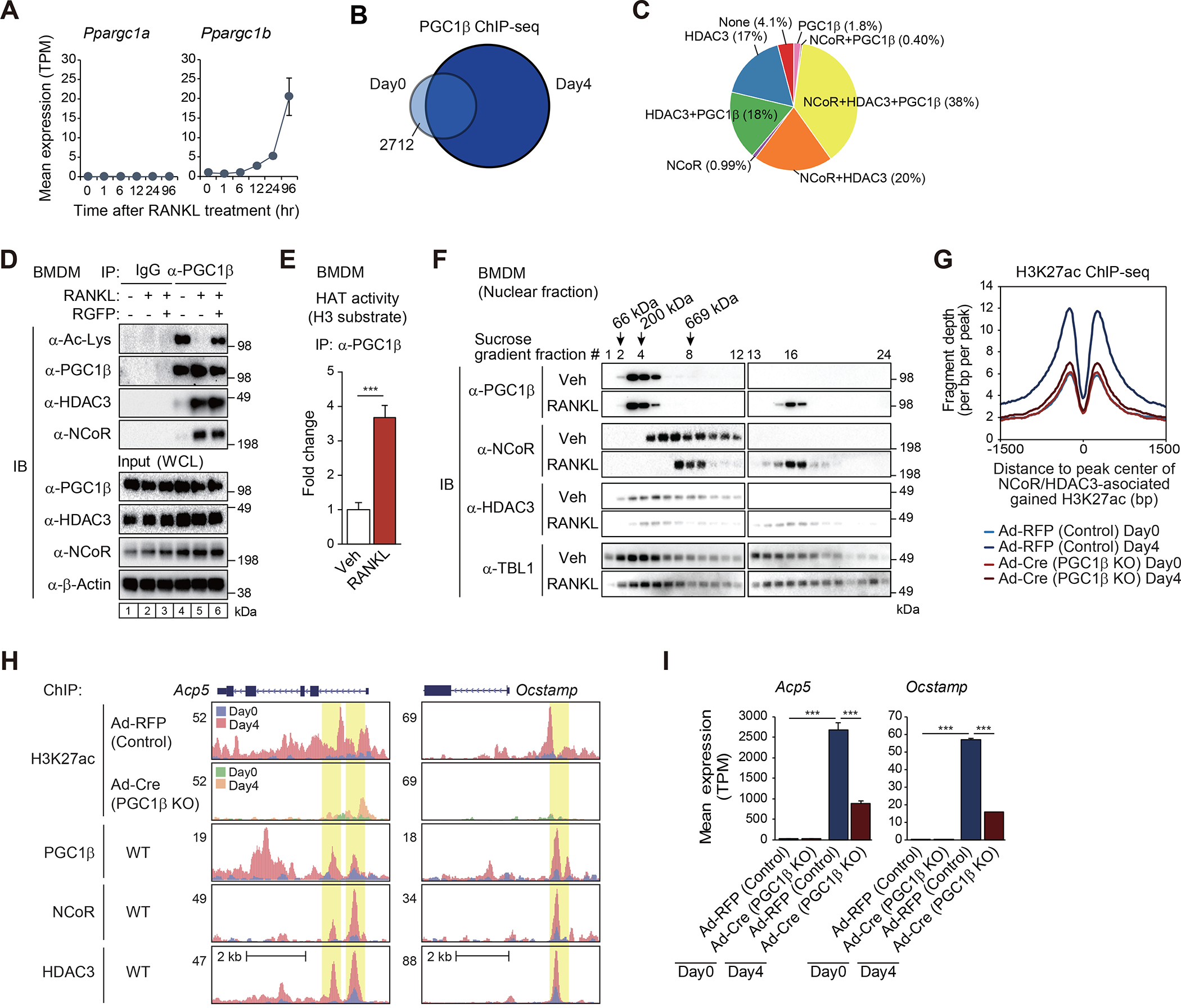
(A) Expression of Ppargc1a and Ppargc1b in WT cells as a function of time following RANKL treatment.
(B) The overlap between IDR-defined PGC1β ChIP-seq peaks at Day0 and at Day4 is shown by Venn diagram.
(C) The overlaps of ATAC-defined gained H3K27ac peaks in the presence of RANKL (n=1525 in Figure S2E) with NCoR, HDAC3 and/or PGC1β ChIP-seq peaks at Day4 are shown by pie chart.
(D) BMDMs were treated with or without RANKL for 6 hours in the presence or absence of RGFP966, and then the whole-cell lysates (WCL) were subjected to immunoprecipitation (IP) using anti-PGC1β antibody and immunoblot (IB) analysis with anti-acetylated lysine, PGC1β, HDAC3 or NCoR antibody.
(E) Histone acetyltransferase (HAT) activity of immunoprecipitated PGC1β protein in whole-cell lysates from BMDMs treated with or without RANKL for 6 hours was measured in the presence of acetyl-CoA and histone H3 substrate. Data are mean ± s.d. (n=3 biological replicates). Student’s t-test was performed for comparisons. ***p < 0.001.
(F) 10–30% sucrose density gradient centrifugation was performed on nuclear fractions from BMDMs treated with or without RANKL for 6 hours. All fractions (1–24, top to bottom) were subjected to IB analysis with anti-PGC1β, NCoR, HDAC3 or TBL1 antibody. The molecular weight standards are indicated at the top of the panel; 66 kDa, bovine serum albumin; 200 kDa, β-amylase; 669 kDa, thyroglobulin.
(G) Normalized distribution of H3K27ac ChIP-seq tag density in control (Ad-RFP) and PGC1β KO (Ad-Cre) at the vicinity of NCoR and HDAC3-associated gained H3K27ac peaks in WT at Day4 after RANKL treatment (n=2757 in Figure 2C).
(H) Genome browser tracks of H3K27ac, PGC1β, NCoR and HDAC3 ChIP-seq peaks in the vicinity of Acp5 and Ocstamp loci. Yellow shading: lost H3K27ac by PGC1β KO at RANKL-induced NCoR, HDAC3 and PGC1β binding regions.
(I) Bar plots for expression of Acp5 and Ocstamp in control (Ad-RFP) and PGC1β KO (Ad-Cre) at Day0 and Day4 after RANKL treatment. ***p-adj < 0.001.
See also Figure S4.
These findings suggest the signal-dependent assembly of PGC1β/NCoR/HDAC3 complexes with co-activator function at specific genomic loci. As shown in Figure 4A, PGC1β is expressed at low levels in undifferentiated bone marrow cells before RANKL treatment (Day0). In contrast, bone marrow-derived macrophages (BMDMs) express PGC1β at appreciable levels in the absence of RANKL (Figure 4D), providing a relevant system to monitor acute changes of the complex formation and the state of PGC1β acetylation in response to RANKL signaling by co-immunoprecipitation assays. Treatment of BMDMs with RANKL for 6 hours resulted in co-immunoprecipitation of PGC1β with NCoR/HDAC3 and its deacetylation (Figure 4D, compare lanes 4 and 5). The HDAC3-specific inhibitor RGFP966 prevented signal-dependent deacetylation of PGC1β, but not its interaction with NCoR/HDAC3 (Figure 4D, compare lanes 5 and 6). In addition, immunoprecipitated NCoR complexes acquired histone acetyltransferase (HAT) activity following RANKL treatment (Figure 4E), consistent with NCoR-dependent H3K27 acetylation in response to RANKL (Figure 3A).
To examine the properties of the protein complexes in the absence or presence of RANKL, we performed sucrose density gradient centrifugation and analyzed gradient fractions by immunoblotting (Figure 4F). In vehicle-treated BMDMs, PGC1β was primarily found in fractions 3–5, consistent with its monomeric molecular weight of 113 kDa. In contrast, NCoR and HDAC3 were both observed in fractions 5–12, consistent with previously described NCoR/HDAC3 co-repressor complexes. Following treatment with RANKL for 6 hours, substantial portions of NCoR, HDAC3 and PGC1β shifted to high molecular weight fractions, with co-sedimentation of all three proteins in fractions 15–17 (Figure 4F), consistent with the signal-dependent induction of a distinct stable NCoR/HDAC3/PGC1β complex. Based on the sedimentation of a thyroglobulin reference in a parallel gradient tube,24 the protein size of the PGC1β/NCoR/HDAC3 complex is estimated to be approximately 2 MDa (Figure S4D). On the other hand, TBL1 (transducin β-like protein 1), which is a well-known component of NCoR/HDAC3 complex in other cell types,25,26 was distributed throughout the gradient regardless of RANKL treatment, suggesting participation in complexes in addition to NCoR/HDAC3 complexes that are not affected by RANKL signaling (Figure 4F).
To confirm the functional importance of PGC1β in osteoclasts, we utilized an adenovirus to express Cre recombinase (Ad-Cre) in bone marrow cells from homozygous floxed PGC1β (Pgc1bf/f) mice.27 An adenovirus directing expression of red fluorescent protein (Ad-RFP) was used as a control. Loss of function of PGC1β resulted in a global loss of RANKL-induced H3K27ac at genomic locations bound by NCoR and HDAC3 (Figures 2C and 4G), exemplified at the Acp5 and Ocstamp genes (Figure 4H). In accordance with these findings, expression of NCoR/HDAC3-dependent RANKL-induced genes was reduced in Pgc1bf/f bone marrow cells expressing adenovirus-directed Cre recombinase (Figure 4I). As expected, RANKL-induced bone resorption activity in the PGC1β-deficient cells was lower compared to control cells (Figure S4E). On enhancer/promoter regions of classical NCoR LXR target genes (Abcg1, Abga1, Scd2), H3K27ac signals were higher in NKO cells, while there was no change of H3K27ac signals between PGC1β KO and the control cells (yellow shading in Figure S4F). These findings are consistent with NCoR/HDAC3 functioning as classical co-repressors at LXR target genes in the same cells in which NCoR/HDAC3/PGC1β complexes function as co-activators of AP-1/NFκB target genes.
RANK and TLR4-induced interaction of PGC1β with NCoR/HDAC3 prevents histone H3K27 deacetylation by HDAC3
Although HDAC3-dependent deacetylation of PGC1β provides an explanation for acquisition of co-activator activity, a remaining paradox is the lack of histone deacetylation at sites of NCoR/HDAC3/PGC1β binding. To investigate whether the association of PGC1β with the NCoR/HDAC3 complex alters HDAC3 substrate specificity, we established estrogen receptor (ER)-Hoxb8-immortalized hematopoietic progenitor cells from Cas9 knock-in mice as described28 and PGC1β guide RNAs (gRNAs) were transduced to establish a clonal knockout cell line (Figure 5A, compare lanes 1–3 and 4–6 of input). After differentiation into macrophages by M-CSF, the Hoxb8 cell-derived macrophages were used for the assays of immunoprecipitation for NCoR. The immunoprecipitated NCoR complex in control cells fully deacetylated nucleosomal H3K27ac under basal condition, while RANKL treatment attenuated deacetylation of nucleosomal H3K27ac accompanied by interaction of NCoR with PGC1β (Figure 5A, compare lanes 1 and 2). In contrast, the immunoprecipitated NCoR in PGC1β-deficient macrophages completely deacetylated nucleosomal H3K27ac in the presence of RANKL (Figure 5A, compare lanes 2 and 5). To confirm that the interaction of PGC1β with the NCoR/HDAC3 complex inhibits HDAC3-dependent H3K27 deacetylation, pCMV-mouse PGC1β plasmid which has silent mutations at gRNA-targeted sequences was transfected into PGC1β gRNA-induced knockout cells (Figure 5A, compare lanes 4–6 and 7–9 of input). The re-expression of PGC1β rescued the knockdown phenotype by decreasing the deacetylation of H3K27 by immunoprecipitated NCoR (Figure 5A, compare lanes 5 and 8).
Figure 5. RANK and TLR4-induced interaction of PGC1β with NCoR/HDAC3 prevents histone deacetylation by HDAC3.
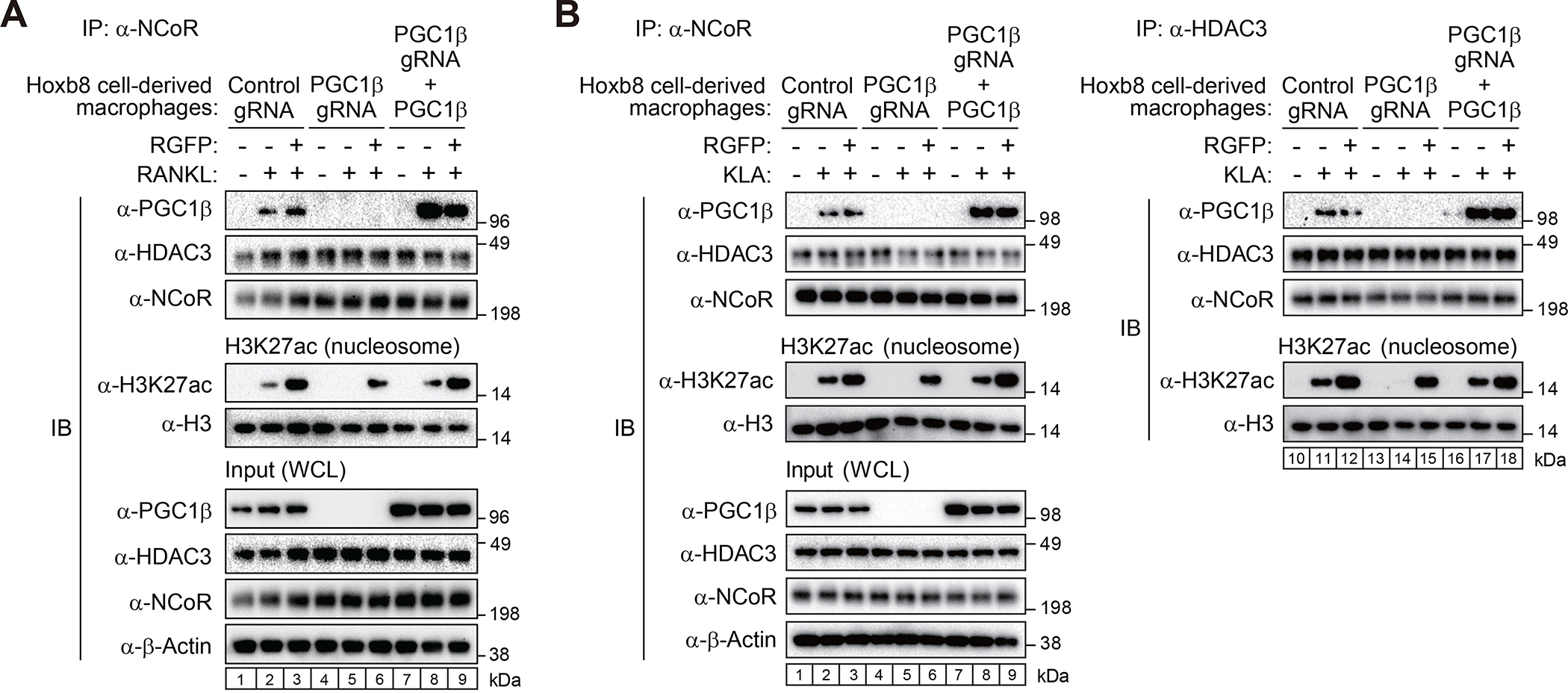
(A, B) Exogenous PGC1β was transiently expressed in PGC1β gRNA-introduced Cas9-Hoxb8 macrophages. Immunoprecipitants with anti-NCoR or HDAC3 antibody in the cells treated with or without RANKL (A) or KLA (B) for 6 hours in the presence or absence of RGFP966 were incubated with nucleosomal H3K27ac followed by immunoblotting with anti-H3K27ac or H3 antibody.
Based on our prior finding that deletion of NCoR from macrophages was associated with an attenuated response to toll-like receptor 4 (TLR4) signaling and an anti-inflammatory/insulin sensitive phenotype under high fat diet,9 we investigated whether TLR4 signaling also induced interaction of NCoR/HDAC3 complexes with PGC1β and altered the substrate specificity of HDAC3. The control and PGC1β KO Hoxb8 cell-derived macrophages used for RANKL studies were treated with the specific TLR4 agonist KLA29 in the presence and absence of RGFP966. As in the case of RANKL, treatment of Hoxb8 cell-derived macrophages with KLA induced interaction of NCoR with PGC1β in a manner that was not affected by RGFP966 (left panel, Figure 5B, compare lanes 1, 2 and 3. NCoR complexes completely deacetylated nucleosomal H3K27ac in the absence of KLA treatment, but this activity was significantly reduced following KLA treatment (left panel, Figure 5B, compare lanes 1 and 2). Complete H3K27 deacetylation in the presence of KLA was observed when NCoR complexes were immunoprecipitated from PGC1β KO cells, while inhibition of acetylation was restored by rescue of PGC1β expression (Figure 5B, left panel, compare lanes 2, 5 and 8). Similar results were obtained by directly evaluating HDAC3 immunoprecipitates following treatment of Hoxb8 cell-derived macrophages with KLA (right panel, Figure 5B). These findings support a mechanism in which the signal-dependent interaction of NCoR/HDAC3 complexes with PGC1β switches HDAC3 substrate specificity from H3K27ac to acetylated PGC1β.
The PGC1β RRM mediates RNA-dependent interaction with NCoR/HDAC3 complexes
All full-length isoforms of the PGC1 protein family (PGC1α, PGC1β and PRC) share a conserved C-terminal RNA-recognition motif (RRM), pointing to potential RNA-binding activity.30 While PGC1α has been shown to bind RNAs with metabolic functions,31 the functional significance of these interactions have not been established and the potential RNA-binding activity of PGC1β has not been probed. To evaluate whether RNA species might facilitate the interaction between PGC1β and NCoR/HDAC3 complex, PGC1β immunoprecipitants from BMDMs treated with or without RANKL were incubated with RNase or DNase. Immunoblotting after immunoprecipitation showed RNase treatment repressed the RANKL-induced interaction of PGC1β with NCoR/HDAC3 complex but DNase did not (Figure 6A, compare lanes 6 and 7/8). These results suggest that RNAs are necessary for the assembly of the NCoR/HDAC3/PGC1β complex. To confirm whether this RNA-dependent complex formation is dependent specifically on an interaction between RNAs and the PGC1β RRM region, we transiently introduced FLAG-tagged PGC1β WT or the RRM deletion mutant (ΔRRM) in RAW 264.7 cells and pulled down the FLAG-tagged PGC1β followed by immunoblotting. The RANKL-induced interaction between FLAG-tagged PGC1β WT and NCoR/HDAC3 (Figure 6B, compare lanes 2 and 5) was nearly abolished by the deletion of RRM in FLAG-tagged PGC1β (Figure 6B, compare lanes 5 and 6). These findings prompted us to identify RNAs that interact with PGC1β.
Figure 6. The PGC1β RRM mediates RNA-dependent interaction with NCoR/HDAC3 complexes.
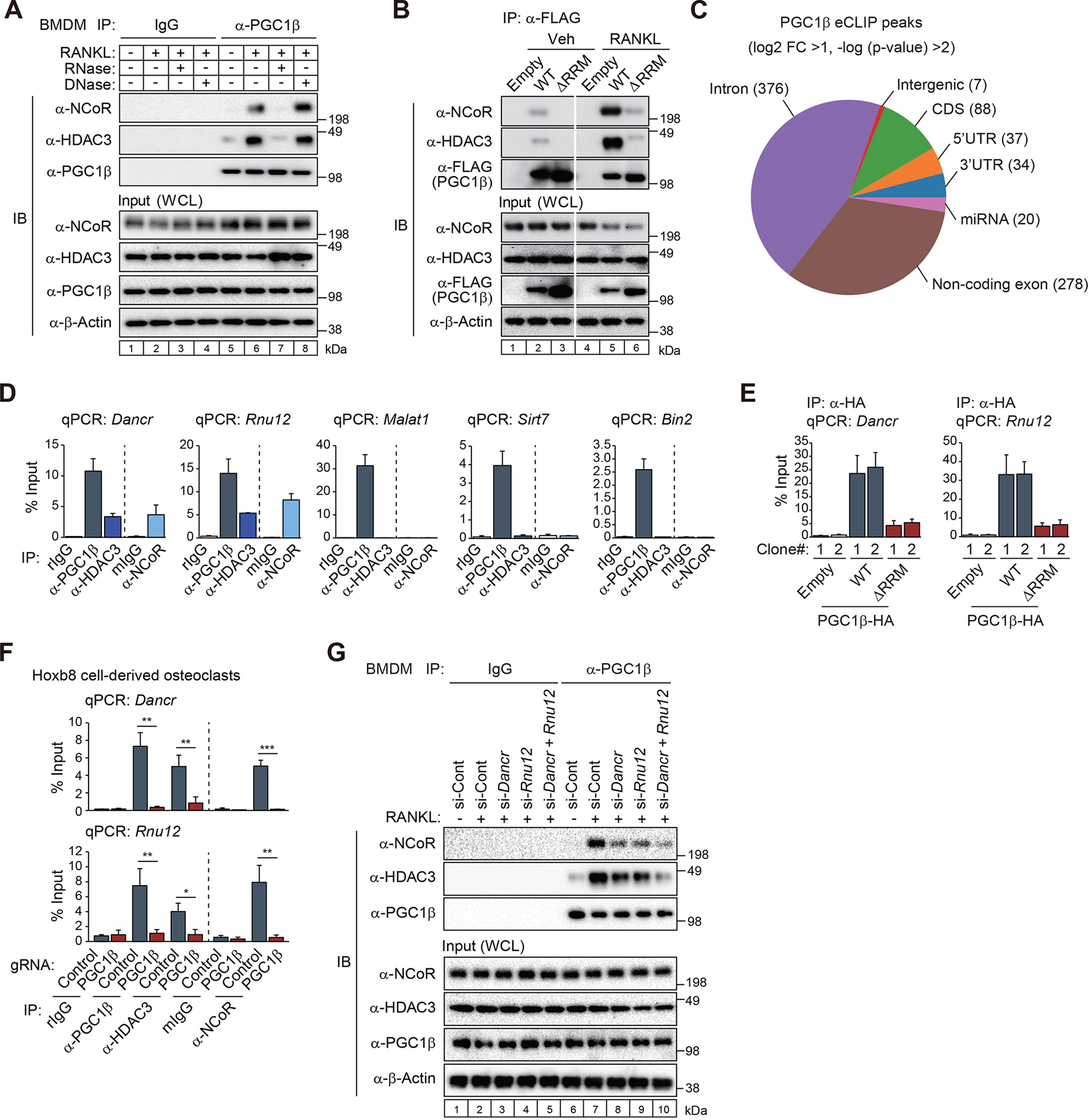
(A) Bone marrow-derived macrophages (BMDMs) were treated with or without RANKL for 6 hours, and then the whole-cell lysates (WCL) were subjected to immunoprecipitation (IP) using anti-PGC1β antibody. The immunoprecipitants were incubated with RNase or DNase followed by immunoblot (IB) analysis with anti-NCoR, HDAC3 or PGC1β antibody.
(B) FLAG-tagged PGC1β WT or RNA-recognition motif deletion mutant (ΔRRM) expressed RAW 264.7 cells were treated with or without RANKL for 6 hours, and then the whole-cell lysates (WCL) were subjected to IP using anti-FLAG antibody followed by IB analysis with anti-NCoR, HDAC3 or FLAG antibody.
(C) High confidence PGC1β eCLIP peaks in RAW 264.7 cells treated with RANKL for 4 days (n=2 biological replicates) were evenly split between protein-coding and non-coding transcripts.
(D) RNA immunoprecipitation (RIP)-qPCR for Dancr, Rnu12, Malat1, Sirt7 or Bin2 using anti-PGC1β, HDAC3 or NCoR antibody in RAW 264.7 cells treated with RANKL for 4 days. Control rabbit IgG (rIgG) for anti-PGC1β and HDAC3 antibodies and control mouse IgG (mIgG) for anti-NCoR antibody were used. Data are mean ± s.d. (n=2 biological replicates).
(E) RIP-qPCR for Dancr or Rnu12 using anti-HA tag antibody in HA-tagged PGC1β (WT or ΔRRM) or the empty (Emp) vector-introduced RAW 264.7 cells in the presence of RANKL and doxycycline (Dox) for 4 days. Data are mean ± s.e.m. (n=4 biological replicate).
(F) RIP-qPCR for Dancr or Rnu12 using anti-PGC1β, HDAC3 or NCoR antibody in PGC1β gRNA or control gRNA-introduced Cas9-Hoxb8 cells treated with RANKL for 4 days. Data are mean ± s.e.m. (n=3 biological replicates). Student’s t-test was performed for comparisons. *p < 0.05, **p < 0.01 and ***p < 0.001.
(G) IB analysis showing interaction of PGC1β with NCoR/HDAC3 in Dancr and Rnu12 siRNA knockdown BMDMs. Whole-cell lysates (WCL) were subjected to IP using anti-PGC1β antibody followed by IB analysis with anti-NCoR, HDAC3 or PGC1β antibody.
See also Figure S5.
We performed enhanced crosslinking and immunoprecipitation (eCLIP)32 in RAW 264.7 cells treated with RANKL for 4 days. Among the 609 high-confidence peaks called across both replicates, log2-fold change compared to size-matched input was highly correlated (R2=0.83) (Figure S5A). Although coding and non-coding transcripts were similarly represented among PGC1β binding targets (Figure S5B), there were more unique PGC1β binding sites on non-coding exons than on protein-coding regions, indicating that the non-coding transcripts contain multiple PGC1β binding sites more often than the coding transcripts (Figure 6C). Motif analysis of eCLIP peaks on non-coding versus coding targets revealed divergent sequences, but most enriched motifs were GC-rich (Figure S5C), a feature shared with the enriched motifs found in PGC1α eCLIP peaks.31 The results of the eCLIP assay were validated for noncoding RNAs Dancr, Rnu12 and Malat1 and mRNAs Sirt7 and Bin2 by RNA immunoprecipitation (RIP)-qPCR with anti-PGC1β antibody using RANKL-treated RAW 264.7 cells (Figure 6D).
Of the many non-coding PGC1β interacting RNAs, two in particular - Dancr (differentiation antagonizing non-protein coding RNA) and Rnu12 (U12 Small Nuclear) - were of interest because they have previously determined associations with bone homeostasis and notably robust eCLIP peak signals (Figure S5D). The expression of Dancr is elevated in circulating monocytes from patients with low bone density, pointing to Dancr as a potential biomarker associated with osteoporosis.33 Meanwhile, Rnu12 is highly expressed in peripheral blood mononuclear cells from psoriatic arthritis patients.34 It is of note that the 5-mer UGCCC, which is partially represented in the 2nd and 3rd most significantly enriched motifs when looking at all non-coding regions (Figure S5C), was found within both the Dancr and Rnu12 peaks. We therefore investigated whether PGC1β interactions with NCoR/HDAC3 complexes were associated with specific RNAs. To do this, we independently immunoprecipitated PGC1β, NCoR or HDAC3 from RANKL-treated cells and performed RIP-qPCR for Dancr, Rnu12, Malat1, Sirt7 and Bin2. Strikingly, while these assays confirmed interaction of all 5 RNAs with PGC1β, only Dancr and Rnu12 were associated with NCoR and HDAC3 (Figure 6D), despite levels of expression of Malat1, Sirt7 and Bin2 that were similar or greater than Dancr or Rnu12.
To confirm the contribution of RRM in PGC1β to these binding events, we generated HA-tagged PGC1β WT or RRM deletion mutant (ΔRRM)-induced RAW 264.7 cell lines using lentiviral integration. The HA-tagged and mRuby-fused PGC1β can be overexpressed via doxycycline (Dox) induction. Upon doxycycline induction and mRuby-based FACS, two single clones of each cell line expressing high levels of the transgene (Figure S5E) were used for further RIP-qPCR assays. Immunoprecipitation with anti-HA tag antibody was performed following 4 days of RANKL treatment, followed by qPCR for Dancr and Rnu12. While WT-PGC1β interacted with Dancr and Rnu12, the deletion of RRM nearly abolished these interactions (Figure 6E). In addition, to confirm whether the binding of Dancr or Rnu12 to NCoR/HDAC3 complex would be dependent on PGC1β, we performed RIP-qPCR assay using anti-NCoR, HDAC3 or PGC1β antibody in PGC1β KO Hoxb8 cells treated with RANKL for 4 days. PGC1β transcript (Ppargc1b) was suppressed in the PGC1β KO cells at Day0 and Day4 after RANKL treatment, but the loss of PGC1β did not affect Ncor1, Hdac3, Dancr and Rnu12 expressions (Figure S5F). In the control cells, PGC1β, NCoR or HDAC3 immunoprecipitants interacted with Dancr or Rnu12 in the presence of RANKL (Figure 6F). In contrast, PGC1β KO completely abolished the interaction of NCoR or HDAC3 with Dancr or Rnu12 (Figure 6F), suggesting PGC1β is necessary for NCoR/HDAC3 complex to be associated with Dancr or Rnu12. We then investigated whether Dancr or Rnu12 were required for RANKL-induced complex formation. Individual knockdowns each modestly reduced the interaction of PGC1β with NCoR/HDAC3, while combined knockdown substantially reduced interaction (Figure 6G, compare lanes 5 and 6). Collectively, these findings indicate that Dancr and Rnu12 are preferentially incorporated into PGC1β/NCoR/HDAC3 complexes in response to RANKL and play quantitatively important roles in their assembly.
Dancr and Rnu12 are required for RANKL-induced osteoclast differentiation
To investigate possible roles of Dancr or Rnu12 in osteoclastogenesis, we performed siRNA-mediated knockdown studies in bone marrow-derived osteoclastogenesis (Figures S6A and S6B), using two independent siRNAs for each target. 1466 mRNAs were induced > 1.5-fold by RANKL (FDR < 0.05) following the full differentiation process in control cells (Figure 7A). Notably, 190 of these genes exhibited significantly attenuated responses for both Dancr siRNAs and 245 of these genes exhibited significantly attenuated responses for both Rnu12 siRNAs (Figure 7A), containing 139 common genes exhibiting osteoclast differentiation as the top term in gene ontology enrichment analysis (Figure 7B). Genes affected by knockdown of both RNAs not only included osteoclast-specific genes such as Acp5 and Ocstamp, but also the RANKL-induction of PGC1β (Ppargc1b) itself (Figure 7C). In addition, knockdown of Dancr and Rnu12 severely impaired RANKL-induced bone resorption activity (Figure 7D).
Figure 7. Dancr and Rnu12 are required for RANKL-induced osteoclast differentiation.
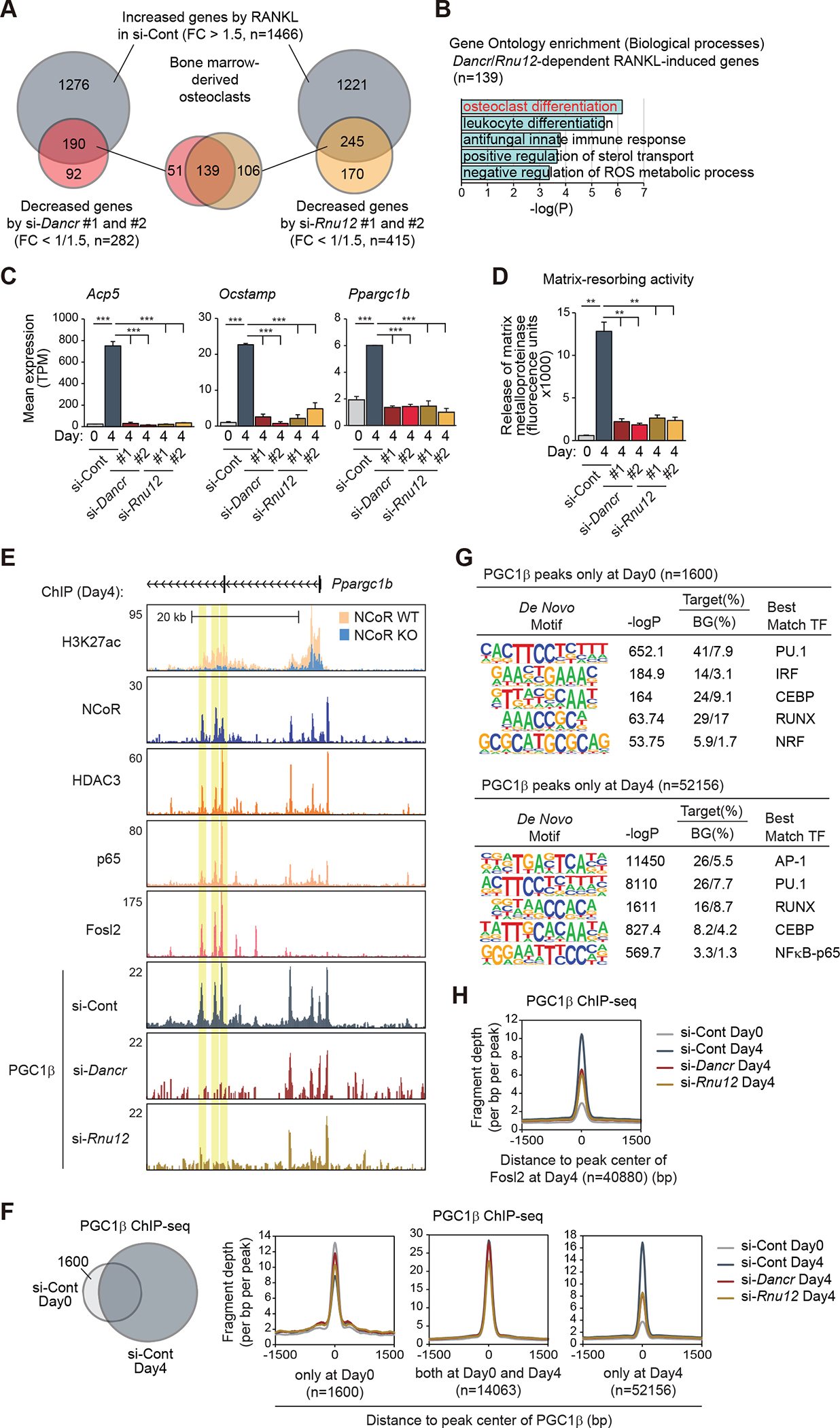
(A) The overlaps between RANKL-induced genes in si-Cont (FC > 1.5, FDR < 0.05) and suppressed genes by si-Dancr (left, #1 and #2) or si-Rnu12 (right, #1 and #2) (FC < 1/1.5, FDR < 0.05) in bone marrow-derived osteoclasts are shown by Venn diagram.
(B) Significant gene ontology terms associated with Dancr/Rnu12-dependent RANKL-induced genes (n=139 in Figure 7A).
(C) Bar plots for expression of Acp5, Ocstamp and Ppargc1b in siRNA (si-Cont, si-Dancr or si-Rnu12)-introduced bone marrow cells at Day0 and Day4 after RANKL treatment. ***p-adj < 0.001.
(D) Matrix-resorbing activity on siRNA (si-Cont, si-Dancr or si-Rnu12)-introduced bone marrow cells at Day0 and Day4 after RANKL treatment. Data are mean ± s.e.m. (n=3 biological replicates). Analysis of variance was performed followed by Tukey’s post hoc comparison. **p < 0.01.
(E) Genome browser tracks of H3K27ac, NCoR, HDAC3, p65, Fosl2 and PGC1β ChIP-seq peaks in bone marrow cells at Day4 after RANKL treatment in the vicinity of Ppargc1b locus. Yellow shading: lost PGC1β peak by knockdown of Dancr/Rnu12.
(F) The overlap between IDR-defined PGC1β ChIP-seq peaks in control siRNA-introduced bone marrow cells at Day0 and Day4 after RANKL treatment is shown by Venn diagram. Normalized distribution of PGC1β ChIP-seq tag density in siRNA (si-Cont, si-Dancr or si-Rnu12)-introduced bone marrow cells at the vicinity of PGC1β binding regions at Day0 and/or Day4.
(G) De novo motif enrichment analysis of PGC1β peaks at Day0 (n=1600 in Figure 7F) and at Day4 (n=52156 in Figure 7F) using a GC-matched genomic background.
(H) Normalized distribution of PGC1β ChIP-seq tag density in siRNA (si-Cont, si-Dancr or si-Rnu12)-introduced bone marrow cells at the vicinity of Fosl2 binding regions at Day4.
See also Figure S6.
The observation that siRNA-mediated knockdown of Dancr and Rnu12 resulted in reduction of RANKL-induced expression of PGC1β but not its basal expression (Figure 7C) raised the question of whether PGC1β is a direct target of the NCoR/HDAC3/PGC1β complex in response to RANK signaling. Consistent with this possibility, RANKL-induced expression of PGC1β was attenuated in NCoR KO mice (Figure S6C). To investigate this further, we performed ChIP-seq for PGC1β in si-Dancr or si-Rnu12-transfected bone marrow cells in comparison to si-control-transfected cells treated with RANKL for 4 days. As expected, due to the lack of induction of PGC1β expression, these experiments demonstrated significant reduction in the global PGC1β signal at NCoR/HDAC3 binding sites exhibiting gain of H3K27ac in response to RANKL in comparison to control cells (Figure S6D). Overall, more than 6000 PGC1β peaks exhibited a >1.5-fold reduction in normalized tag counts in si-Dancr-transfected cells, and more than 9000 peaks exhibited a similar reduction in si-Rnu12-transfected cells (Figure S6E). Consistent with the result of H3K27ac ChIP-seq in PGC1β KO cells (Figure 4G), Dancr/Rnu12 knockdown suppressed H3K27ac in NCoR/HDAC3 binding sites exhibiting gain of H3K27ac in response to RANKL (right panel, Figure S6F). This is shown at a composite level for all of the NCoR/HDAC3 peaks that gain H3K27ac in response to RANKL. In contrast, Dancr/Rnu12 knockdown had much less effect on the binding of NCoR or HDAC3 at these locations (middle and right panels, Figure S6F). Thus, Dancr and Rnu12 are required to form a NCoR/HDAC3/PGC1β co-activator complex, but not for localization of NCoR/HDAC3 to the genomic loci at which histone acetylation occurs in response to RANKL signaling.
Notably, the Ppargc1b gene exhibits numerous enhancer-like regions that gain H3K27ac in response to RANKL in an NCoR-dependent manner in concert with the recruitment of NCoR/HDAC3/PGC1β complexes (Figure 7E). Of particular interest, siRNA knockdown of Dancr or Rnu12 did not alter binding of PGC1β at promoter proximal locations, but strongly reduced binding at multiple downstream enhancer-like regions (denoted by vertical yellow stripes in Figure 7E). These regions are distinguished from stable PGC1β binding sites in the vicinity of the promoter by strong induction of the occupancy of p65 and Fosl2 in response to RANKL. These findings provide evidence for a feed forward loop in which RANK signaling directly upregulates PGC1β expression by a mechanism involving Dancr/Rnu12-dependent assembly of NCoR/HDAC3/PGC1β complexes at enhancers that are activated by AP-1 and NFκB transcription factors.
To investigate whether this relationship was observed at a genome-wide level, we compared the effects of siRNA knockdown of Dancr or Rnu12 on the RANKL-induced binding of PGC1β at genomic locations characterized by PGC1β binding in the absence or presence of RANKL. This analysis indicated RANKL-induced PGC1β binding was strongly suppressed by the knockdown of Dancr or Rnu12 at the locations of RANKL-induced PGC1β peaks (n=52156), while pre-existing PGC1β peaks regardless of RANK signaling (n=1600 and 14063) were much less affected (Figure 7F). De novo motif enrichment analysis identified recognition elements for AP-1 and NFκB transcription factors among the top motifs on RANKL-induced PGC1β binding regions (bottom panel, Figure 7G), but not at pre-existing PGC1β binding regions (top panel, Figure 7G). Consistent with these findings, knockdown of Dancr or Rnu12 significantly reduced PGC1β binding at sites occupied by Fosl2 (Figure 7H). These findings are consistent with Dancr and Rnu12 regulating osteoclast differentiation by mechanisms that include roles in RANKL-dependent assembly of NCoR/HDAC3/PGC1β complexes on AP-1/NFκB-dependent enhancers.
DISCUSSION
In concert, these studies provide evidence that RANK signaling converts NCoR/HDAC3 co-repressor complexes to PGC1β-dependent co-activator complexes that are required for normal osteoclast differentiation. Although an RRM has long been recognized as a conserved feature of the PGC1 family of co-activators, its function has been enigmatic. Here, we provide evidence that the RRM is required for RNA-dependent interaction of PGC1β with the NCoR/HDAC3 complex that mediates its deacetylation and activation. The identification of Dancr and Rnu12 as two of the many RNAs that associate with PGC1β and the consequences of their loss function suggest that they play functionally important roles in osteoclasts that may underlie associations with human bone diseases. The present finding that knockdown of Dancr inhibits osteoclast differentiation and bone resorption in vitro is consistent with the prior report that high levels of Dancr in monocytes are associated with osteoporosis.33 Similarly, the finding that knockdown of Rnu12 inhibits osteoclast differentiation is consistent with the possibility that elevated Rnu12 expression reported in individuals with psoriatic arthritis34 could drive osteoclast activity and contribute to bone erosion.
The observation that many RNAs interact with PGC1β but Dancr and Rnu12 were strongly preferred among the comparisons tested implies that there is specificity in the mechanism by which RNAs promote PGC1β interactions with the NCoR/HDAC3 complex. The finding that knockdown of Dancr and Rnu12 was not enough to completely abolish the interaction of PGC1β with NCoR/HDAC3 complexes could be due to incomplete loss of their expression. However, it is likely that additional RNAs make quantitative contributions to the complex formation. It will also be of interest to determine whether these observations extend to PGC1α and whether alternative RNAs are utilized to mediate NCoR/HDAC3 interactions in other signaling contexts, such as signals driving thermogenesis. It is also important to note that Dancr and Rnu12 have previously identified functions that may contribute to the observed consequences of their loss of function. In the case of Rnu12, prior studies have shown that it is a component of the minor spliceosome, which contains four stem-loops.35 Based on PGC1β eCLIP peak footprints, PGC1β interacts with the stem-loop III element in Rnu12, which also binds to the RRM of U11/U12–65K protein, which is essential for splicing activity.36 These findings raise the possibility that PGC1β also plays a role in shaping the alternative splicing landscape in osteoclastogenesis. Intriguingly, Dancr has previously been reported as a central node to enhance multiple kinase cascades, leading to gene activation in cancer cells.37–40 From the perspective that Dancr activates the NFκB signaling pathway,38 the localization of PGC1β/Dancr to sites of NFκB-p65 binding raises the possibility of an additional function of Dancr to facilitate local activation of NFκB.
In concert, the present studies provide support for a model in which RANK signaling induces the interaction of PGC1β with NCoR/HDAC3 complexes on NFκB/AP-1-containing enhancers or promotors in a manner that is dependent on the PGC1β RRM and RNAs. In this context, the dominant function of NCoR is as a co-activator, with only minimal evidence for co-repressor activity at nuclear receptor target genes. In view of the large size of the RANKL-induced complex, it will be of interest to determine the other components that contribute to its co-activator function. Notably, signaling through TLR4 induces an analogous complex formation that may contribute to the reduced magnitude of LPS responses observed in NCoR-deficient macrophages.9 These findings therefore raise the broader question of whether NCoR/HDAC3/PGC1β complexes also function as co-activators downstream of other signaling pathways that regulate development and homeostasis.
Limitations of the study
The biochemical studies demonstrating that the association of PGC1β with NCoR/HDAC3 alters the substrate specificity of HDAC3 does not exclude the possibility that the specific HAT that acetylates and deactivates PGC1β under basal conditions might also have its substrate switched to histones as a consequence of RANKL-induced NCoR/HDAC3 interaction. Although Dancr/Rnu12 knockdown suppressed H3K27ac in NCoR/HDAC3 binding sites exhibiting gain of H3K27ac in response to RANKL, the knockdown had much less effect on the binding of NCoR or HDAC3. Thus, Dancr and Rnu12 are required to form a NCoR/HDAC3/PGC1β co-activator complex, but not for localization of NCoR/HDAC3 to the genomic loci at which histone acetylation occurs in response to RANK signaling, suggesting that NCoR/HDAC3-PGC1β interaction mediated by Dancr/Rnu12 as an intermolecular glue might change the complex structure which would facilitate HDAC3-dependent PGC1β deacetylation and gene activation. While the present findings establish the principle that RANKL-dependent assembly of NCoR/HDAC3/PGC1β complexes is dependent on RNA in an in vitro model of osteoclastogenesis, the relative contributions of Dancr and Rnu12 to osteoclast differentiation and function in vivo will require further study.
STAR METHODS
RESOUCE AVAILABLITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christopher K. Glass (ckg@ucsd.edu).
Material availability
All materials used in this study are either commercially available or obtained through collaboration, are available from the corresponding author upon request.
Data and code availability
All sequencing data including RNA-seq, ChIP-seq, ATAC-seq, and eCLIP assay were deposited in the Gene Expression Omnibus (GEO) under the accession number GSE211676.
This paper does not report any original code.
Any information required to reanalyze the data reported in this paper is available from the Lead contact, Christopher K. Glass (ckg@ucsd.edu) upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animal studies
All animal procedures were in accordance with University of California, San Diego research guidelines for the care and use of laboratory animals. Mice were maintained under a 12 hr light/12 hr dark cycle at constant temperature (20–23°C) with free access to food and water. Animals were fed a normal chow diet (T8604, Envigo). Ncorf/f and Ncorf/f LysM-Cre mice were described previously.9 Smrtf/f mice described previously 41 were crossbred with Ncorf/f mice9 to generate Ncorf/f Smrtf/f mice. Ncorf/f and Ncorf/f Smrtf/f mice were used as WT mice for Ncorf/f LysM-Cre and Ncorf/f Smrtf/f LysM-Cre mice, respectively. Pgc1bf/f mice were described previously.27 5-month-old male and female mice were used for in in vivo experiments. Bone marrow cells were isolated from 8- to 12-week-old male mice and differentiated to macrophages or osteoclasts for each in vitro experiment.
Bone marrow-derived osteoclast culture
Bone marrow cells were obtained by flushing the tibia and femur from 8- to 12-week-old C57BL/6 mice with alpha MEM (Sigma-Aldrich) containing 10% FBS (Omega Biosciences), 1% penicillin/streptomycin+L-glutamine (Thermo Fisher Scientific) and lysed using red blood cell lysis buffer (Invitrogen). 0.1 million cells per wells of 96-well plates (Sigma-Aldrich) for TRAP staining and osteoclast bone resorption assay, 3 million cells per wells of 6-well plates for ATAC-seq and RNA-seq, 18 million cells per 10 cm tissue culture plates (Thermo Fisher Scientific) for immunoblotting and immunoprecipitation or 48 million cells per 15 cm tissue culture plates (Thermo Fisher Scientific) for ChIP-seq were cultured in alpha MEM containing 10% FBS, 1% penicillin/streptomycin+L-glutamine and 10 ng/ml M-CSF (Shenandoah Biotechnology) for 3 days to generate osteoclast precursor cells. After non-adherent cells were washed off with alpha MEM, adherent osteoclast precursor cells were cultured in alpha MEM containing 10% FBS, 1% penicillin/streptomycin+L-glutamine, 10 ng/ml M-CSF and 50 ng/ml RANKL (PeproTech) for 4 days to differentiate to osteoclasts. The M-CSF plus RANKL containing culture media was replaced every 2 days. 5 μM RGFP966 (Selleckchem) was treated alone with M-CSF and RANKL for 4 days.
RAW 264.7 cell-derived osteoclast culture
RAW 264.7 cells were maintained in RPMI 1640 (Corning) containing 10% FBS, 1% penicillin/streptomycin+L-glutamine. 3.5 million cells per 10 cm tissue culture plates for RIP-qPCR or 20 million cells per 15 cm tissue culture plates for eCLIP were seeded. To differentiate osteoclasts, the cells were cultured in RPMI 1640 containing 10% FBS, 1% penicillin/streptomycin+L-glutamine and 50 ng/ml RANKL for 4 days. The RANKL containing culture media was replaced every 2 days.
Bone marrow-derived macrophage (BMDM) culture
Bone marrow cells were obtained by flashing the tibia and femur from 8- to 12-week-old C57BL/6 mice with RPMI 1640 containing 10% FBS, 1% penicillin/streptomycin+L-glutamine and lysed using red blood cell lysis buffer. After counting, 20 million cells were seeded per 15 cm non-tissue culture plates in RPMI 1640 containing 10% FBS, 30% L929 cell conditioned laboratory-made media (as source of M-CSF) and 1% penicillin/streptomycin+L-glutamine. After 3 days, 16.7 ng/ml M-CSF was added into the media. After an additional 4 days, non-adherent cells were washed off and adherent cells were harvested. 10 million cells per 15 cm tissue culture plates for immunoblotting and immunoprecipitation were cultured in RPMI 1640 containing 10% FBS, 1% penicillin/streptomycin+L-glutamine and 16.7 ng/ml M-CSF overnight. After the serum starvation in RPMI 1640 containing 1% FBS and 1% penicillin/streptomycin+L-glutamine for 12 hrs, cells were treated with 50 ng/ml RANKL for 6 hrs. The treatment of 5 μM RGFP966 was performed at the same time as the serum starvation.
METHOD DETAILS
Serum parameters
5-month-old male mice were humanely euthanized by exposure to CO2 and whole blood was collected in tubes. The blood was leaved to clot at room temperature for 30 min, and then at 4°C for 16 hrs. To remove the clot, the blood was centrifuged at 1200 g for 10 min at 4°C. The resulting supernatant was used as serum in the experiments. Serum deoxypyridinoline and osteocalcin concentrations were measured by Mouse Deoxypyridinoline ELISA Kit (LSBio) and Mouse Osteocalcin EIA Kit (Alfa Aesar) respectively, according to the manufacturer’s instructions.
Bone imaging
Micro-CT analysis (μCT40, Scanco Medical, Bruttisellen, Switzerland) was performed on femora dissected and cleaned from the muscle. Bones were fixed in 10% Millonig’s formalin, dehydrated with ethanol and kept in 100% ethanol until analysis. Femora were scanned at 12 μm nominal isotropic voxel size, 500 projection (medium resolution, E=55 kVp, I=72 μA, 4W, integration time 150 ms and threshold 200 mg/cm3), and integrated into 3-D voxel images (1024 × 1024 pixel matrices for each individual planar stack). Femora were scanned from the distal epiphysis to the mid-diaphysis to obtain a number of slices variable between 650 and 690. Two-dimensional evaluation of cancellous bone was performed on contours of the cross sectional acquired images; primary spongiosa and cortex were excluded. Contours were drawn from the distal metaphysis to the diaphysis of the femur to obtain 151 slices (12 μm/slice). For cancellous bone measurements, contours were drawn every 10 to 20 slices. Voxel counting was used for bone volume per tissue volume measurements and sphere filling distance transformation indices were used for cancellous microarchitecture with a threshold value of 200, without pre-assumptions about the bone shape as a rod or plate. Micro-CT measurements were expressed in 3-D nomenclature as recommended by the American Society for Bone and Mineral Research.42
Histology
Femurs fixed in Millonig’s 10% formalin were decalcified in 14% EDTA pH 7.4 for 7 days, dehydrated, and paraffin-embedded. 5 μm longitudinal sections were cut and stained for tartrate-resistant acid phosphatase (TRAP) activity and counterstained with toluidine blue after removal of paraffin and rehydration. The number and the surface of TRAP-positive cells on the trabecular perimeter were measured using the OsteoMeasure Analysis System (OsteoMetrics, Inc. Atlanta, GA) interfaced to an Axio image M2 (Carl Zeiss, NY). To exclude the primary spongiosum, bone within 440 μm of the growth plate was not included in the analysis. Histomorphometric measurements were restricted to the secondary spongiosa in an area extending for approximately 1100 μm. One section per sample was analyzed in a blinded fashion. The results are reported using the terminology recommended by the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research.43
Antibodies
Mouse monoclonal antibody immunoglobulin G-Y8129 (IgG-Y8129) against mouse NCoR (amino acids 1817–1879) was produced by immunizing mice with gp64 fusion protein expressed by baculoviral system as described previously.44 A list of other antibodies used in this article is shown in Table S1.
CRISPR knockout in Hoxb8 cells
Bone marrow cells were isolated from femurs and tibias of Cas9-expressiong transgenic mice (Jackson Laboratory, No.028555). Cas9-expressiong ER-Hoxb8 conditionally immortalized myoid progenitor cells were generated following established protocols.28 In brief, bone marrow cells were purified with a Ficoll gradient (Ficoll-Paque-Plus, Sigma-Aldrich) and resuspended in RPMI 1640 containing 10% FBS, 1% penicillin/streptomycin and 10 ng/ml each of SCF, IL3 and IL6 (PeproTech). After 48 hrs culture, 250000 cells in 1 ml were transduced with 2 ml of ER-Hoxb8 retrovirus (in DMEM with 30% FBS) containing 0.5 μl/ml lentiblast A (OZ Biosciences), 2.5 μl/ml lentiblast B (OZ Biosciences) and 8 μg/ml polybrene (Sigma-Aldrich) in a well of fibronectin (Sigma-Aldrich)-coated 6-well culture plates and centrifuged at 1000 g for 90 min at 22°C. Murine stem cell virus-based expression vector for ER-Hoxb8 was gifted from Dr. David Sykes (Massachusetts General Hospital, Boston, MA). After transduction, 6 ml of ER-Hoxb8 cell media (RPMI 1640 supplemented with 10% FBS, 1% penicillin/streptomycin, 0.5 μM β-estradiol (Sigma-Aldrich), and 20 ng/ml GM-CSF (PeproTech)) were added and an additional half-media exchange with ER-Hoxb8 media performed the next day. Transduced cells were selected with G418 (Thermo Fisher) at 1 mg/ml for 48 hrs. Thereafter, cells were maintained in ER-Hoxb8 media. Cells were single-cell sorted with a Sony MA900 and cells from a single clone used for the following experiments. gRNA lentiviruses were prepared as previously described45 with modifications as follows. LentiGuide-mCherry was generated by modifying lentiGuide-puro (Addgene) to remove a puromycin-resistant gene and replace it with mCherry. gRNA sequences directed against exons of the murine PGC1β gene were designed with CHOPCHOP web tool for genome engineering.46 2 CRISPR gRNA oligonucleotides were inserted for each target via PCR amplification with the H1 promoter (BsmBI site/guide 1/scaffold/H1 promoter/guide 2/BsmBI site) for 2 guides per virus (U6 and H1 driven). A list of gRNA targets used in this article is shown in KEY RESOUCES TABLE. Lenti-X 293T cells (Clontech) were seeded in poly-D-lysin (Sigma-Aldrich) coated 10 cm tissue culture plates at a density of 3.5 million cells per plate in 10 ml of DMEM containing 10% FBS and 1% penicillin/streptomycin, and then incubated overnight at 37°C. After replacement of the media to 6 ml of DMEM containing 30% FBS, plasmid DNAs (5 μg of lentiviral vector, 3.75 μg of psPAX2 (Addgene) and 1.25 μg of pVSVG (Addgene)) were transfected into LentiX-293T cells using 20 μl of X-tremeGENE™ HP DNA Transfection Reagent (Roche) at 37°C overnight. The media was replaced with 8 ml of DMEM containing 30% FBS and 1% penicillin/streptomycin, and then cultured at 37°C overnight. The supernatants were filtrated with 0.45 μm syringe filter and used as lentivirus media. Cell culture media was replaced and virus collected again after 24 hrs. 1 million Cas9-expressing ER-Hoxb8 cells were transduced with virus in 2 ml of lentivirus media and 1 ml of ER-Hoxb8 cell media containing 0.5 μl/ml lentiblast A, 2.5 μl/ml lentiblast B and 8 μg/ml polybrene in a well of fibronectin-coated 6-well culture plates and centrifuged at 1000 g for 90 min at 22°C. After the transduction, 6 ml of ER-Hoxb8 cell media was added to each well. Half of the media was exchanged the following day and in the following days, cells were passaged into flasks. After 5 days, cells were single-cell sorted into 96-well culture plates by FACS using a Sony MA900. Successful transduction was assessed by fluorescence of mCherry. After sorting, clones were expanded in ER-Hoxb8 cell media. Genomic DNA was isolated from clones using NucleoSpin Tissue kits (Macherey-Nagel), and PCR reactions were optimized with 100 ng of genomic DNA and 1 μl of KOD Xtreme™ Hot Start DNA Polymerase (Millipore) to amplify PGC1β gRNA target regions. PCR amplicons were evaluated using agarose gel electrophoresis and purified using NucleoSpin Gel and PCR Clean-Up kits (Macherey-Nagel). Successful knockout in clones with frameshift mutations at both alleles were confirmed by immunoblot for PGC1β. Cells transduced with control gRNA were grown in parallel and treated equally throughout as cells with PGC1β knockout. Presence of PGC1β in control gRNA clones was confirmed by immunoblot in the same experiment as the confirmation of the knockout. The Hoxb8 cells were differentiated into macrophages and osteoclasts as well as Bone marrow-derived macrophage (BMDM) culture and Bone marrow-derived osteoclast culture, respectively. Hoxb8-derived macrophages were treated with 100 ng/ml KLA (Avanti Polar Lipids) or 50 ng/ml RANKL for 6 hrs.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K27ac | Active Motif | Cat# 39133; RRID: AB_2561016 |
| H3 | Active Motif | Cat# 39163; RRID: AB_2614978 |
| NCoR | Gifted from Dr. Hamakubo | Cat# IgG-Y8129; RRID: N/A |
| SMRT | Novus | Cat# NB100-58826; RRID: AB_877767 |
| HDAC3 | GeneTex | Cat# GTX113303; RRID: AB_10721050 |
| PU.1 | Santa Cruz | Cat# sc-352; RRID: AB_632289 |
| p65 | Santa Cruz | Cat# sc-372; RRID: AB_632037 |
| p50 | Santa Cruz | Cat# sc-1190; RRID: AB_632033 |
| Fosl2 | Santa Cruz | Cat# sc-166102; RRID: AB_2107079 |
| PGC1β | Abcam | Cat# ab176328; RRID: N/A |
| Acetylated-Lysine | Cell Signaling Technology | Cat# 9441; RRID: AB_331805 |
| ACP5 | Millipore | Cat# MABF96; RRID: AB_10845145 |
| TAB2 | Proteintech | Cat# 14410-1-AP; RRID: AB_2281638 |
| TBL1 | Santa Cruz | Cat# sc-137006; RRID: AB_ 2199796 |
| FLAG | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| HA | Abcam | Cat# ab9110; RRID: AB_307019 |
| Lamin B | Santa Cruz | Cat# sc-374015; RRID: AB_10947408 |
| Lamin A/C | Cell Signaling Technology | Cat# 2032; RRID: AB_2136278 |
| β-Actin | Sigma-Aldrich | Cat# A2228; RRID: AB_476697 |
| Polyclonal Goat Anti-Mouse Immunoglobulins-HRP | Dako | Cat# P0447; RRID: AB_2617137 |
| Polyclonal Goat Anti-Rabbit Immunoglobulins-HRP | Dako | Cat# P0448; RRID: AB_2617138 |
| Mouse monoclonal anti-rabbit IgG light chain-HRP | Abcam | Cat# ab99697; RRID: AB_10673897 |
| Bacterial and Virus Strains | ||
| Ad-RFP | Vector Biolabs | Cat# 1660 |
| Ad-CMV-iCre | Vector Biolabs | Cat# 1045 |
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| PMI 1640 | Corning | Cat# 10-014-CV |
| alpha MEM | Sigma-Aldrich | Cat# M8042 |
| Opti-MEM | Thermo Fisher Scientific | Cat# 11058021 |
| FBS | Omega Biosciences | Cat# FB-02 |
| Penicillin/streptomycin + L-glutamine | Thermo Fisher Scientific | Cat# 10378016 |
| eBioscience™ 1X RBC Lysis Buffer | Invitrogen | Cat# 00-4333-57 |
| Polybrene | Sigma-Aldrich | Cat# H9268 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Invitrogen | Cat# 13778075 |
| Lipofectamine™ LTX Reagent with PLUS™ Reagent | Invitrogen | Cat# 15338030 |
| X-tremeGENE™ HP DNA Transfection Reagent | Roche | Cat# 6366546001 |
| Mouse M-CSF | Shenandoah Biotechnology | Cat# 200-08 |
| β-estradiol | Sigma-Aldrich | Cat# E2758 |
| GM-CSF | PeproTech | Cat# 315-03 |
| RGFP966 | Selleckchem | Cat# S7229 |
| KLA | Avanti Polar Lipids | Cat# 699500P |
| RANKL | PeproTech | Cat# 315-11 |
| SCF | PeproTech | Cat# 250-03 |
| IL3 | PeproTech | Cat# 213-13 |
| IL6 | PeproTech | Cat# 216-16 |
| Ficoll-Paque-Plus | Sigma-Aldrich | Cat# GE17-1440-02 |
| LentiBlast™ Transduction Reagent | OZ Biosciences | Cat# LB00500 |
| Fibronectin | Sigma-Aldrich | Cat#F0895 |
| G418 | Thermo Fisher Scientific | Cat# 10131035 |
| Poly-D-lysin | Sigma-Aldrich | Cat# DLW354210 |
| KOD Xtreme™ Hot Start DNA Polymerase | Millipore | Cat# 71975 |
| NuPAGE™ LDS Sample Buffer | Thermo Fisher Scientific | Cat# NP0007 |
| NuPAGE™ Sample Reducing Agent | Thermo Fisher Scientific | Cat# NP0009 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat #34095 |
| Luminate™ Forte Western HRP Substrate | Merck Millipore | Cat# WBLUF0100 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat# 10002D |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10004D |
| SpeedBeads magnetic carboxylate modified particles | GE Healthcare | Cat# 65152105050250 |
| Dynabeads My One Silane | Thermo Fisher Scientific | Cat# 37002D |
| TRIzol Reagent | Thermo Fisher Scientific | Cat# 15596018 |
| Formaldehyde | Thermo Fisher Scientific | Cat# BP531-500 |
| Disuccinimidyl glutarate | ProteoChem | Cat# c1104-100mg |
| Proteinase K | NEB | Cat# P8107S |
| Oligo d(T)25 Magnetic Beads | NEB | Cat# S1419S |
| DTT | Thermo Fisher Scientific | Cat# P2325 |
| SUPERase-In | Ambion | Cat# AM2696 |
| RNase Inhibitor | NEB | Cat# M0314S |
| FastAP | Thermo Fisher Scientific | Cat# EF0651 |
| Oligo dT primer | Thermo Fisher Scientific | Cat# 18418020 |
| RNA Clean XP Beads | Beckman Coulter | Cat# A63987 |
| 10 X Blue Buffer | Enzymatics | Cat# P7050L |
| dNTP mix | Thermo Fisher Scientific | Cat# R0191 |
| RNase A | NEB | Cat# T3018L |
| RNase H | Enzymatics | Cat# Y9220L |
| RNase I | Thermo Fisher Scientific | Cat# AM2295 |
| DNase I | NEB | Cat# M0303S |
| Turbo DNase | Thermo Fisher Scientific | Cat# AM2238 |
| DNA polymerase I | Enzymatics | Cat# P7050L |
| T4 PNK | NEB | Cat# M0201S |
| T4 RNA ligase | NEB | Cat# M0204S |
| NEXTflex® DNA Barcodes | Bioo Scientific | Cat# NOVA-514104 |
| Random primers | Thermo Fisher Scientific | Cat# 48190011 |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18080044 |
| 5X SuperScript III first-strand buffer | Thermo Fisher Scientific | Cat# 18080044 |
| Actinomycin D | Sigma-Aldrich | Cat# A1410 |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat# M0541S |
| KAPA SYBR FAST qPCR Master Mix | Kapa Biosystems | Cat# KR0389 |
| Exo SAP-IT | Applied Biosystems | Cat# 78201.1.ML |
| PMSF | Sigma-Aldrich | Cat# P7626 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| Sodium butylate | Sigma-Aldrich | Cat# B5887 |
| Recombinant Mononucleosomes H3K27ac | Active Motif | Cat# 81077 |
| Phenol/chloroform/isoamyl alcohol (125:24:1) | Sigma-Aldrich | Cat# P1944 |
| Critical Commercial Assays | ||
| HAT Assay Kit | Active Motif | Cat# 56100 |
| Direct-zol RNA MicroPrep Kit | Zymo Research | Cat# R2062 |
| ChIP DNA Clean & Concentrator Kit | Zymo Research | Cat# D5205 |
| NEBNext Ultra II Library Preparation Kit | NEB | Cat# E7645L |
| Nextera DNA Library Prep Kit | Illumina | Cat# 15028212 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| KAPA HiFi HotStart PCR Kit | Kapa Biosystems | Cat# KK2501 |
| Mouse Deoxypyridinoline ELISA Kit | LSBio | Cat# LS-F25774-1 |
| Mouse Osteocalcin EIA Kit | Alfa Aesar | Cat# 15406279 |
| OsteoLyse™ Assay Kit | Lonza | Cat# PA-1500 |
| TRAP Staining Kit | Cosmo Bio | Cat# PMC-AK04F |
| Q5 Site-Directed Mutagenesis Kit | NEB | Cat# E0554S |
| DC™ Protein Assay Kit | Bio-Rad Laboratories | Cat# 5000112 |
| NucleoSpin Tissue kits | Macherey-Nagel | Cat# 740952 |
| NucleoSpin Gel and PCR Clean-Up kits | Macherey-Nagel | Cat# 740609 |
| Deposited Data | ||
| UCSC hub of all sequencing data | This paper | GEO: GSE211676 |
| Experimental Models: Cell Lines | ||
| Lenti-X 293T cell | Clontech | Cat# 632180 |
| RAW 264.7 cell | ATCC | Cat# TIB-71; RRID: CVCL_0493 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Ncorflox/flox | Li et al., 2013 | N/A |
| Mouse: Smrtflox/flox | Lee et al., 2022 | N/A |
| Mouse: Ncorflox/flox Smrtflox/flox | This paper | N/A |
| Mouse: Pgc1bflox/flox | Sonoda et al., 2007 | N/A |
| Mouse: B6.129P2-Lyz2tm1(cre)Ifo/J | The Jackson Laboratory | Cat# 004781 |
| Mouse: B6(C)-Gt(ROSA)26Sorem1.1(CAG-cas9*,-EGFP)Rsky/J | The Jackson Laboratory | Cat# 028555 |
| Oligonucleotides | ||
| Mouse Dancr siRNA (individual #1), See METHOD DETALS | Dharmacon | Customized |
| Mouse Dancr siRNA (individual #2), See METHOD DETALS | Dharmacon | Customized |
| Mouse Rnu12 siRNA (individual #1), See METHOD DETALS | Dharmacon | Customized |
| Mouse Rnu12 siRNA (individual #2), See METHOD DETALS | Dharmacon | Customized |
| siGENOME Non-Targeting siRNA #2 | Dharmacon | Cat# D-001210-02 |
| Mouse PGC1β_1 gRNA GGAAGAGCTCGGAGTCATCG |
This paper | N/A |
| Mouse PGC1β_2 gRNA AGCGTCTGACGTGGACGAGC |
This paper | N/A |
| Mouse Control gRNA GCACTACCAGAGCTAACTCA |
This paper | N/A |
| Recombinant DNA | ||
| lentiGuide-puro | Addgene | Cat# 52963 |
| psPAX2 | Addgene | Cat# 12260 |
| pVSVG | Addgene | Cat# 138479 |
| pLIX403-APOBEC-HA-P2A-mRuby | Brannan et al., 2021 | N/A |
| pcDNA3.1-FLAG-mouse PGC1β | NovoPro Bioscience | Cat# 759974-2 |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/ |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| Irreproducibility Discovery Rate (IDR) | Li et al., 2011 | https://www.encodeproject.org/software/idr/ |
| Metascape | Tripathi et al., 2015 | http://metascape.org/gp/index.html#/main/step1 |
| R package: DeSeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| UCSC Genome Browser | Kent et al., 2002 | https://genome.ucsc.edu/ |
| GraphPad Prism 8 software | Dotmatics | https://www.graphpad.com/ |
| Other | ||
| N/A | ||
Adenoviral transduction
Bone marrow cells were obtained by flushing the tibia and femur from 12-week-old Pgc1bf/f mice gifted from Dr. Ronald M. Evans with alpha MEM containing 10% FBS and 1% penicillin/streptomycin+L-glutamine, and then lysed using red blood cell lysis buffer. 18 million cells per 10 cm tissue culture plates were cultured in alpha MEM containing 10% FBS, 1% penicillin/streptomycin+L-glutamine and 10 ng/ml M-CSF overnight. 20 ml of RFP-tagged adenovirus (Ad-RFP) (Vector Biolabs) or RFP-tagged Cre recombinase adenovirus (Ad-Cre) (Vector Biolabs) were incubated with 8 μg/ml polybrene in 10 ml of Opti-MEM (Thermo Fisher Scientific) for 5 min at room temperature. After non-adherent cells were washed off with PBS, the adenovirus mixtures were added into adherent osteoclast precursor cells and incubated at 37°C for 2 hrs. The cells were washed off with PBS and cultured in alpha MEM containing 10% FBS, 1% penicillin/streptomycin+L-glutamine and 10 ng/ml M-CSF for 2 days. To differentiate osteoclasts, 50 ng/ml RANKL was treated with cells and cultured for 4 days. The M-CSF plus RANKL containing culture media was replaced every 2 days.
Osteoclast resorbing activity assay
Bone marrow cells or Hoxb8 cells were seeded onto the surface of the 96-well OsteoLyse™ plate (Lonza) and cultured in the presence or absence of RANKL for 4 days as described above. 10 μl of culture media was sampled from each well every 2 days and stored at −20°C until ready for assays. Osteoclast resorbing activity was measured by OsteoLyse™ Assay Kit (Lonza) according to the manufacturer’s instructions. Briefly, 200 μl of Fluorophore Releasing Reagent was incubated with 10 μl of culture media for 10 min at room temperature in each well of 96-well black assay plate (Lonza). The fluorescence was determined at 340 nm of excitation and 615 nm of emission.
TRAP staining
Cells were rinsed with PBS and fixed with 10% (vol/vol) formaldehyde for 5 min at room temperature. After two washes in water, cells were stained with chromogenic substrate (TRAP Staining Kit, Cosmo Bio) for 1 hr at 37°C. The substrate was then removed, and the cells were washed twice with water and photographed by Fluorescence Microscope (BZ-X710, Keyence).
RNA interference
Non-coding RNA Dancr or Rnu12 was depleted from RAW 264.7 cells or bone marrow cells with siRNAs targeting mouse Dancr (Dharmacon, Customized individual #1 (5’-GCAAAGAUUUCAUGCUAGAUU) or #2 (5’-ACAAGAAACCCGUGACUGAUU-3’)) or Rnu12 (Dharmacon, Customized individual #1 (5’-GCUUAUGUGAGAAGAAUUUUU-3’) or #2 (5’-GGGUAUAGGUUGCAAUCUGUU-3’)). siGENOME Non-Targeting siRNA #2 (Dharmacon, D-001210–02) was used for a negative control. RAW 264.7 cells were cultured overnight as described in RAW 264.7 cell-derived osteoclast culture, and then transfected with each 30 nM siRNA using Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer’s instructions with modifications as follows. Lipofectamine/siRNA complexes were formed in Opti-MEM for 15 min at room temperature, and then added into cells in culture media containing 50 ng/ml RANKL. Cells were incubated for 4 days to differentiate to osteoclasts. For bone marrow-derived osteoclasts, each 30 nM siRNA was introduced at day −2, 0 and 2 after RANKL treatment as described in Bone marrow-derived osteoclast culture. The RANKL and siRNA-containing culture media was replaced every 2 days. For bone marrow-derived macrophages, each 30 nM siRNA was introduced two days before the cells were harvested as described in Bone marrow-derived macrophage (BMDM) culture.
Plasmid construction and transfection
For the construction of lentiviral vector pLIX403-mouse PGC1β (WT (a.a.1–1022) or ΔRRM (a.a.1–901))-APOBEC-HA-P2A-mRuby, a synthetic gene block (Integrated DNA technology) containing mouse PGC1β (WT or ΔRRM) sequence was inserted into MluI digested backbone pLIX403-APOBEC-HA-P2A-mRuby47 in frame and immediately upstream of the APOBEC. The open-reading frames (ORFs) of mouse PGC1β were obtained from pCMV-mouse PGC1β plasmid. Lenti-X 293T cells were seeded in 10 cm tissue culture plates at a density of 3.5 million cells per plate in 8 ml of DMEM (Gibco) containing 10% FBS and 1% penicillin/streptomycin, and then incubated overnight at 37°C. The plasmid DNAs (5 μg of lentiviral vector, 3.75 μg of psPAX2 and 1.25 μg of pVSVG) were transfected into LentiX-293T cells using 20 μl of X-tremeGENE™ HP DNA Transfection Reagent at 37°C overnight. The media was replaced with 7 ml of DMEM containing 10% FBS and 1% penicillin/streptomycin, and then cultured at 37°C overnight. The supernatants were filtrated with 0.45 μm syringe filter and used as lentivirus media. 4 million RAW 264.7 cells were transduced with virus in 4 ml of lentivirus media containing 0.5 μl/ml lentiblast A, 2.5 μl/ml lentiblast B and 8 μg/ml polybrene in 10 cm tissue culture plates and centrifuged at 1400 g for 1 hr at 22°C. After the transduction, 4 ml of DMEM containing 10% FBS and 1% penicillin/streptomycin was added to each dish. After 2 days, the media was exchanged to DMEM containing 10% FBS, 1% penicillin/streptomycin and 2 μg/ml puromycin (Sigma-Aldrich). The puromycin containing culture media was replaced every 2 days until negative control (non-infected) cells completely died. Lentivirus infected RAW 264.7 cells were cultured with 1 μg/ml doxycycline in RPMI 1640 containing 10% FBS and 1% penicillin/streptomycin+L-glutamine for 72 hrs. The cells with fluorescence of mRuby were single-cell sorted with a Sony MA900, and then maintained in RPMI 1640 containing 10% FBS and 1% penicillin/streptomycin+L-glutamine. The cells were cultured with 1 μg/ml doxycycline in RPMI 1640 containing 10% FBS, 1% penicillin/streptomycin+L-glutamine and 50 ng/ml RANKL for 4 days, followed by experiments.
For the transient expression of PGC1β in RAW 264.7 cells or Hoxb8 cells, 2 μg of pcDNA3.1-FLAG-mouse PGC1β (NovoPro Bioscience) (WT or ΔRRM) plasmid was incubated with 0.625 μl of PLUS™ Reagent (Invitrogen) in 300 μl of Opti-MEM for 5 min at room temperature followed by another incubation with 1.6 μl of Lipofectamine LTX Reagent (Invitrogen) for 30 min at room temperature. The DNA-lipofectamine complex was added into cells cultured in 10 cm tissue culture plates 2 days before the cells were harvested.
Immunoblotting and immunoprecipitation
Whole cell lysates were prepared as previously described48 with modifications as follows. Cells were sonicated in cell lysis buffer (50 mM HEPES-KOH (pH 7.9), 150 mM NaCl, 1.5 mM MgCl2, 1% NP-40, 1 mM PMSF (Sigma-Aldrich), 1X protease inhibitor cocktail (Sigma-Aldrich)) by ultrasound homogenizer (Bioruptor, Diagenode) for 10 min at 4°C. After centrifugation at 15000 g for 10 min at 4°C, the supernatant was used as whole cell lysate. To isolate nuclear fraction from cells, cells were homogenized in STM buffer (250 mM Sucrose, 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 1 mM PMSF, 1X protease inhibitor cocktail) and incubated on ice for 30 min. After centrifugation at 800 g for 15 min at 4°C, the pellet was washed with STM buffer twice and resuspended in NET buffer (20 mM HEPES (pH7.9), 1.5 mM MgCl2, 0.5 M NaCl, 0.2 mM EDTA, 20% Glycerol, 1% Triton-X, 1 mM PMSF, 1X protease inhibitor cocktail). The suspension was passed through the needle 18 gage 15 times and sonicated by ultrasound homogenizer for 10 min at 4°C. After centrifugation at 9000 g for 30 min at 4°C, the supernatant was used as nuclear fraction.
For immunoblotting, aliquots of whole cell lysate or nuclear fraction were boiled at 95°C for 5 min in NuPAGE™ LDS Sample Buffer (Thermo Fisher Scientific) with NuPAGE™ Sample Reducing Agent (Thermo Fisher Scientific), subjected to SDS-PAGE, and transferred to immobilon-P transfer membranes (Merck Millipore). Immunodetection was carried out with the indicated antibodies (Table S1) and bound antibodies were visualized with peroxidase-conjugated affinity-purified donkey anti-mouse or anti-rabbit IgG (Dako), or mouse monoclonal anti-rabbit IgG light chain (Abcam) using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) or Luminate™ Forte Western HRP Substrate (Merck Millipore), and luminescence images were analyzed by ChemiDoc XRS+ System (Bio-Rad Laboratories). For immunoprecipitation, whole cell lysates or nuclear fractions were immunoprecipitated in cell lysis buffer (50 mM HEPES-KOH (pH 7.9), 150 mM NaCl, 1.5 mM MgCl2, 1% NP-40, 1mM PMSF, 1X protease inhibitor cocktail) by wheel rotating overnight at 4°C in the presence of Dynabeads protein G (Thermo Fisher Scientific) and each antibody described in Table S1. After immunoprecipitation, beads were washed three times with cell lysis buffer and three times with PBS, and then eluted with sample buffer. The elution was subjected to immunoblotting as described above.
H3K27ac deacetylase activity assay
The endogenous NCoR was immunoprecipitated with anti-NCoR antibody in 300 μg of whole cell lysate from Hoxb8 cell-derived macrophages as described in Immunoblotting and immunoprecipitation. After washing the NCoR antibody-Dynabeads Protein G complex three times each with cell lysis buffer and HDAC3 assay buffer from HDAC3 Activity Assay Kit (Sigma-Aldrich), HDAC3 assay buffer in presence or absence of 5 μM RGFP966 was added, and then incubated for 10 min at 37°C. 500 ng of recombinant mononucleosomal H3K27ac (Active Motif) was added and reacted overnight at 37°C. The supernatants and beads were separated and boiled with sample buffer for 5 min at 95°C, and then subject to immunoblotting.
Histone acetyltransferase (HAT) activity assay
HAT activity in BMDMs was measured using a HAT Assay Kit (Active Motif) according to the manufacturer’s protocol. The HAT activity of immunoprecipitated PGC1β with anti-PGC1β antibody (Abcam) in 300 μg of whole cell lysate as described in Immunoblotting and immunoprecipitation. Briefly, 50 μl of 1X assay buffer containing 50 μM acetyl-CoA and 50 μM Histone H3 was added to the PGC1β antibody-Dynabeads Protein G complex and incubated for 30 min at room temperature. Once the incubation was complete, 50 μl of stop solution was mixed. Then 100 μl of developing solution was added and incubated for 15 min in the dark at room temperature. The reaction solutions were transferred to a 96-well plate and the fluorescent signals were determined at 380 nm of excitation and 450 nm of emission.
RNase/DNase treatment
The antibody-bound Dynabeads after immunoprecipitation as described in Immunoblotting and immunoprecipitation were washed three times with cell lysis buffer and three times with PBS, and then incubated with 1 μg/μl RNase A (NEB) plus 1 U/ μl RNase H (Enzymatics) or 0.4 U/μl DNase I (NEB) in 50 μl of each reaction buffer at 37°C for 1 hr. After washed twice with PBS, the beads were used for immunoblotting or HAT activity assay.
Quantitative real time PCR (RT-qPCR)
Total RNA was isolated from cells by Direct-zol RNA MicroPrep Kit (Zymo Research). First strand cDNA was synthesized from the total RNA with the use of a Super-Script III First-Strand Synthesis System (Thermo Fisher Scientific), and then PCR was performed with KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems) and mouse Ppargc1b primers (Forward; 5’-CTCTGACACGCAGGGTGG-3’, Revers; 5’-AGTCAAAGTCACTGGCGTCC-3’), mouse Dancr primers (Forward; 5’-AGAAACCCGTGACTGAATGG-3’, Revers; 5’-ACTCACATGGCCCTCACTTC-3’) or mouse Rnu12 primers (Forward; 5’-TTTGAGCGGGTATAGGTTGCA-3’, Revers; 5’-CAGATCGCGTCACCCAGG-3’) on StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). Relative mRNA abundance was calculated by the standard curve method and was normalized by the corresponding amount of mouse Actb (Forward primer; 5’-CTTCTACAATGAGCTGCGTG-3’, Revers primer; 5’-TCATGAGGTAGTCTGTCAGG-3’) or Gapdh (Forward primer; <monospace>5</monospace>‘-AATGTGTCCGTCGTGGATCT-3’, Revers primer; 5’-CATCGAAGGTGGAAGAGTGG-3’).
RNA immunoprecipitation (RIP)
RAW 264.7 cells or Hoxb8 cells cultured in media with 50 ng/ml RANKL for 4 days were cross-linked with 1% (vol/vol) formaldehyde (Thermo Fisher Scientific) in PBS for 10 min at room temperature. The cells were incubated in hypotonic buffer (10 mM NaCl, 10 mM Tris-HCl (pH 7.4), 0.5% NP-40, 1 mM DTT (Thermo Fisher Scientific), 200 U/ml RNase OUT (Thermo Fisher Scientific), 1X protease inhibitor cocktail (Sigma)) and homogenized for 1 min at 4°C. The cells were centrifuged at 1000g for 10 min at 4°C, and then nuclear pellets were resuspended in nuclei resuspension buffer (50 mM HEPES-NaCl (pH 7.0), 10 mM MgCl2, 1 mM DTT, 200 U/ml RNase OUT). The cells were sonicated at 4°C for 45 min by ultrasound homogenizer, and then incubated with 250 U/ml DNase I for 30 min at 37°C. The DNase reaction was quenched by 20 mM EDTA (Invitrogen) and mixed with 1% Triton X-100, 0.1% sodium deoxycholate, 0.01% SDS and 140 mM NaCl. The lysate was centrifuged at 20000 g for 10 min at 4°C and the supernatant was used for immunoprecipitation. Before immunoprecipitation, 1% of the lysate was kept as the paired-input sample. The nuclear lysate was rotated with each antibody pre-bound to 25 μl of beads (12.5 μl of Dynabeads protein A (Thermo Fisher Scientific) + 12.5 μl of Dynabeads protein G) at 4°C overnight. After the immunoprecipitation, beads were collected on a magnet and washed three times each with wash buffer (150 mM NaCl, 10 mM Tris-HCl (pH7.4), 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.5% NP-40, 1 mM DDT, 200 U/ml RNase OUT), and then resuspended in 150 μl of Proteinase K buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40, 1% SDS, 1.2 mg/ml Proteinase K (NEB)), followed by incubation for 30 min at 55°C. The isolated protein-associated RNAs by phenol/chloroform/isoamyl alcohol (125:24:1) (Sigma-Aldrich) were subjected to first strand cDNA synthesis, and then PCR was performed with KAPA SYBR FAST qPCR Master Mix and mouse Dancr primers (Forward; 5’-AGAAACCCGTGACTGAATGG-3’, Revers; 5’-ACTCACATGGCCCTCACTTC-3’), mouse Rnu12 primers (Forward; 5’-TTTGAGCGGGTATAGGTTGCA-3’, Revers; 5’-CAGATCGCGTCACCCAGG-3’), mouse Malat1 primers (Forward; 5’-GAGGCTGACCAGAGCAGTTT-3’, Reverse; 5’-GCCGACCTTCAAACTAGAACC-3’), mouse Sirt7 primers (Forward; 5’-GTGTCACGCATCCTGAGGAA-3’, Reverse; 5’-GGTCAT CACACACCTCCTCC-3’) or mouse Bin2 primers (Forward; 5’-CCCAGGAGAAGG TTCTGCAG-3’, Reverse; 5’-TGGTAGAAGTTGCTGGCACT-3’) on StepOnePlus Real-Time PCR System.
Preparation of bone cell extracts
Protein extracts were prepared from tibia and femur of 5-month-old male mice. The bones were pulverized on dry ice and the bone powder was sonicated at 4°C for 10 min in lysis buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM PMSF, 1X protease inhibitor cocktail) by ultrasound homogenizer. The bone lysates were centrifuged at 15000 rpm for 10 min at 4°C and the protein concentration in supernatants was measured by DC™ Protein Assay Kit (Bio-Rad Laboratories) according to the manufacturer’s instructions. The supernatants were boiled at 95°C for 5 min in NuPAGE™ LDS Sample Buffer with NuPAGE™ Sample Reducing Agent for SDS-PAGE.
Sucrose density gradient centrifugation
20 million BMDMs were homogenized in STM buffer (250 mM Sucrose, 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 1 mM PMSF, 1X protease inhibitor cocktail) and incubated on ice for 30 min. After centrifugation at 800 g for 15 min at 4°C, the pellet was washed with STM buffer twice and resuspended in 150 μl of lysis buffer (50 mM HEPES-KOH (pH 7.9), 150 mM NaCl, 1.5 mM MgCl2, 1% NP-40, 1 mM PMSF, 1X protease inhibitor cocktail) by ultrasound homogenizer (Bioruptor, Diagenode) for 10 min at 4°C. The suspension was passed through an 18 gauge needle 15 times and sonicated by ultrasound homogenizer for 10 min at 4°C. After centrifugation at 9000 g for 30 min at 4°C, the supernatant was used as nuclear fraction. 150 μl of nuclear fraction at a concentration of 5.1 μg/μL were layered on top of a 12 ml 10–30% (w/v) continuous sucrose gradient (in buffer containing 50 mM HEPES-KOH (pH 7.9), 150 mM NaCl, 1.5 mM MgCl2, 1 mM PMSF and 1X protease inhibitor cocktail). Gradients were centrifuged at 32000 rpm for 12 hrs at 4°C in an SW41 Ti rotor (Beckman). Molecular weight standards (Sigma-Aldrich) in lysis buffer were run in parallel. 24 fractions (500 μl each) were collected from each gradient, and 125 μl of 100% (w/v) trichloroacetic acid (Sigma-Aldrich) containing 4 mg/ml sodium deoxycholate was added to each fraction. After incubation on ice for 15 min, the samples were spun for 15 min, and the supernatant was removed. 1 ml of acetone (Sigma-Aldrich) was added to each pellet. Each sample was incubated for 10 min at room temperature and spun for 15 min. The supernatant was discarded, and the pellet was dissolved in NuPAGE™ LDS Sample Buffer (Thermo Fisher Scientific) with NuPAGE™ Sample Reducing Agent (Thermo Fisher Scientific). After heating at 95°C for 5 min, the samples were subjected to SDS-PAGE.
eCLIP assay
20 million RAW 264.7 cells per 15 cm tissue culture plates were cultured in RPMI 1640 containing 10% FBS, 1% penicillin/streptomycin+L-glutamine with 50 ng/ml RANKL for 4 days. eCLIP assay was performed as previously described 32 with modifications as follows. The UV-crosslinked cells were lysed in CLIP lysis buffer (50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1X protease inhibitor cocktail) and sonicated by ultrasound homogenizer (Bioruptor, Diagenode) for 5 min at 4°C, followed by treatment with 0.01 U/μl Turbo DNase (Thermo Fisher Scientific) and 0.04 U/μl RNase I (Thermo Fisher Scientific) for 5 min at 37°C. After treatment with 0.4 U/μl RNase Inhibitor (NEB), the cell lysates were centrifuged at 15000 g for 3 min at 4°C and the supernatants were used for the following immunoprecipitation. Anti-PGC1β antibody and dynabeads were added to the cell lysates and incubated overnight at 4°C. Before beads wash, 2% of sample was kept as the paired-input sample. The remining beads were washed three times with high-salt wash buffer (50 mM Tris-HCl (pH 7.4), 1M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5 % sodium deoxycholate) and three times with low-salt wash buffer (20 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 0.2 % Tween 20, 5 mM NaCl). The beads were incubated with 0.6 U/μl FastAP (Thermo Fisher Scientific), 1.6 U/μl RNase Inhibitor and 0.08 U/μl Turbo DNase for 10 min at 37°C for RNA dephosphorylation, and with 0.2 U/μl T4 PNK (NEB) for 20 min at 37°C for RNA phosphorylation. After beads wash two times with high-salt wash buffer and three times with low-salt wash buffer, 3’RNA adaptor was ligated with 3 U/μl T4 RNA ligase (NEB) and 0.64 U/μl RNase Inhibitor. After beads wash three times with high-salt wash buffer and three times with low-salt wash buffer, the beads were denatured in 1X NuPAGE LDS Buffer with 0.1 M DTT for 10 min at 70°C. The supernatants (RNA-protein complexes) were run on 4–12% Bis-Tris protein gel and transferred to PVDF membrane. The membrane (the region 65–140 kDa) was excised off and treated with proteinase K (NEB). RNA was then reverse transcribed with Superscript III (Thermo Fisher Scientific) followed by treatment with Exo SAP-IT (Applied Biosystems) to remove excess oligonucleotides. Samples were cleaned up with Dynabeads My One Silane (Thermo Fisher Scientific) and subject to qPCR to determine the appropriate number of PCR cycles. Libraries were amplified with NEBNext High-Fidelity 2X PCR Master Mix (NEB) and sequenced on the HiSeq 4000.
ATAC-seq library preparation
70000 cultured cells were washed once with PBS and once with cold lysis buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40). The cells were suspended in 50 μl of 1X Reaction Buffer (25 μl of Tagment DNA Buffer, 2.5 μl of Tagment DNA enzyme I, and 22.5 μl of water) (Nextera DNA Library Preparation Kit, Illumina) as previously described 49. Transposase reactions were carried out at 37°C for 30 min, and then DNA was purified using ChIP DNA Clean & Concentrator Kit (Zymo Research). DNA was amplified using the Nextera primer Ad1 and a unique Ad2.n barcoding primer using NEBNext High-Fidelity 2X PCR Master Mix (NEB) for 7 cycles. The amplified libraries were purified with 2 μl of SpeedBeads (GE Healthcare) in 20% PEG 8000/2.5 M NaCl (Final 13% PEG 8000), eluted with 15 μl of EB (Zymo Research), size selected using PAGE/TBE gel (Invitrogen) for 175–225 bp fragments by gel extraction, and single-end sequenced on HiSeq 4000 (Illumina).
ChIP-seq library preparation
Chromatin immunoprecipitation (ChIP) was performed in biological replicates as previously described50 with modifications as follows. For H3K27ac ChIP assays, cells were cross-linked with 1% (vol/vol) formaldehyde in PBS for 10 min at room temperature. For NCoR, HDAC3, p65, Fosl2, PU.1 and PGC1β ChIP assays, cells were cross-linked with 2 mM disuccinimidyl glutarate (DSG) (ProteoChem) in PBS for 30 min at room temperature, and then directly a second crosslinking was performed by the addition of 1% (vol/vol) formaldehyde in PBS for 10 min. The crosslinking reaction was quenched by 0.125 M glycine (Sigma-Aldrich). Cells were washed once with ice-cold PBS and pelleted at 1000 g for 5 min at 4°C. Crosslinked cells were resuspended in ice-cold hypotonic buffer (10 mM HEPES-KOH (pH 7.9), 85 mM KCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF, 0.5 mM sodium butyrate (Sigma-Aldrich), 1X protease inhibitor cocktail), and centrifuged at 1000 g for 5 min at 4°C to obtain a nuclear fraction. Nuclear pellets were resuspended in 100 μl of either LB3 buffer (10 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.5% N-laurosylsarcosine, 1 mM PMSF, 0.5 mM sodium butyrate, 1X protease inhibitor cocktail, for H3K27ac ChIP) or PIPA-NR buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.4% sodium deoxycholate, 0.1% SDS, 1% NP-40, 0.5 mM DTT, 1 mM PMSF, 0.5 mM sodium butyrate, 1X protease inhibitor cocktail, for NCoR, HDAC3, p65, Fosl2, PU.1 and PGC1β ChIP). Chromatin DNA was sonicated in a 96 Place microTUBE Rack (Covaris) using a Covaris E220 for 12–16 cycles with the following setting: time, 60 seconds; duty, 5.0; PIP, 140; cycles, 200; amplitude, 0.0; velocity, 0.0; dwell, 0.0. Samples were centrifuged at 15000 rpm for 10 min at 4°C, and the supernatant was used for immunoprecipitation. LB3 lysate was diluted 1.1-fold with 10% Triton X-100. 1% of the lysate was kept as ChIP input. The lysates were rotated with each antibody pre-bound to 20 μl of beads (10 μl of Dynabeads protein A + 10 μl of Dynabeads protein G) overnight at 4°C. After the immunoprecipitation, beads were collected on a magnet and washed three times each with wash buffer I (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM EDTA, 1 mM PMSF, 0.5 mM sodium butyrate, 1X protease inhibitor cocktail), wash buffer III (10 mM Tris-HCl (pH 7.5), 250 mM LiCl, 1% Triton X-100, 0.7% sodium deoxycholate, 1 mM EDTA, 1 mM PMSF, 0.5 mM sodium butyrate, 1X protease inhibitor cocktail) and twice with TET (10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 0.2% Tween-20, 1 mM PMSF, 0.5 mM sodium butyrate, 1X protease inhibitor cocktail), and then resuspended in 25 μl of TT (10mM Tris-HCl (pH 7.5), 0.05% Tween-20). The immunoprecipitated chromatin samples were used for library preparation with NEBNext Ultra II Library kit (NEB) according to the manufacturer’s instructions. DNA in 46.5 μl of NEB reactions was revers crosslinked by adding 4 μl of 10% SDS, 4.5 μl of 5 M NaCl, 3 μl of 500 mM EDTA, 1 μl of 20 mg/ml proteinase K and 20 μl of water by incubation for 1 hr at 55°C, and then 30 min at 65°C. DNA was purified with 2 μl of SpeedBeads in 20% PEG 8000/1.5 M NaCl (Final 12% PEG 8000), and eluted with 25 μl of TT. The eluted DNA was amplified for 14 cycles in 50 μl of PCR reactions using NEBNext High-Fidelity 2X PCR Master Mix and 0.5 mM each of primers Solexa 1GA and Solexa 1GB. The amplified libraries were purified with 2 μl of SpeedBeads in 20% PEG 8000/2.5 M NaCl (Final 13% PEG 8000), eluted with 20 μl of TT, size selected using PAGE/TBE gel for 225–325 bp fragments by gel extraction, and single-end sequenced on HiSeq 4000 or NovaSeq 6000 (Illumina). ChIP input material (1% of sheared DNA) in 46.5 μl of TE was revers crosslinked by adding 4 μl of 10% SDS, 4.5 μl of 5 M NaCl, 3 μl of 500 mM EDTA, 1 μl of 20 mg/ml proteinase K and 20 μl of water by incubation for 1 hr at 55°C, and then 30 min at 65°C. The input DNA was purified with SpeedBeads as described above, and eluted with 25 μl of TT. The eluted input DNA was prepared for libraries and amplified as described for ChIP samples.
RNA-seq library preparation
Total RNA was isolated from culture cells and purified using a Direct-zol RNA MicroPrep Kit as described by the manufacture. mRNAs were enriched by incubation with Oligo d(T)25 Magnetic Beads (NEB). To fragment Poly A-enriched mRNA, mRNAs were incubated in 2X Superscript III first-strand buffer (Thermo Fisher Scientific) with 10 mM DTT at 94°C for 9 min. The 10 μl of fragmented mRNAs were incubated with 0.5 μl of Random primers (3 μg/μl) (Thermo Fisher Scientific), 0.5 μl of Oligo dT primer (50 μM) (Thermo Fisher Scientific), 0.5 μl of SUPERase-In (Ambion) and 1 μl of dNTPs (10 mM) (Thermo Fisher Scientific) at 50°C for 1 min. After the incubation, 5.8 μl of water, 1 μl of DTT (100 mM), 0.1 μl of Actinomycin D (2 μg/μl) (Sigma-Aldrich), 0.2 μl of 1% Tween-20 and 0.5 μl of Superscript III (Thermo Fisher Scientific) were added and incubated on the following conditions: 25°C for 10 min, 50°C for 50 min, and 4°C hold. The mixture was purified with RNAClean XP beads (Beckman Coulter) as described by the manufacture and eluted with 10 μl of water. For second-strand synthesis with dUTP, the RNA/cDNA double-stranded hybrid was then added to 1.5 μl of Blue Buffer (Enzymatics), 1.1 μl of dUTP mix (10 mM dATP, dCTP, dGTP and 20 mM dUTP) (Promega), 0.2 μl of Rnase H (5 U/μl), 1.05 μl of water, 1 μl of DNA polymerase I (Enzymatics) and 0.15 μl of 1% Tween-20. The mixture was incubated at 16°C for 2.5 hrs. The resulting dUTP-marked dsDNA was purified with 2 μl of SpeedBeads in 20% PEG 8000/2.5M NaCl (final 13% PEG 8000), and eluted with 40 μl of EB. The eluted dsDNA was carried out end repair by blunting, A-tailing and adapter ligation as previously described 51 using barcoded adapters (Bioo Scientific). The end repaired dsDNA was amplified for 14–16 cycles using 0.05 U/μl KAPA High Fidelity HotStart DNA polymerase (Kapa Biosystems) and 1 μM each of primers Solexa 1GA and Solexa 1GB. The amplified libraries were selected using PAGE/TBE gel for 225–325 bp fragments by gel extraction, and single-end sequenced on HiSeq 4000 or NovaSeq 6000.
eCLIP analysis
The eCLIP peak calling computational tool Clipper52 was used to call peaks for both replicates of the PGC1β eCLIP experiment, using size-matched inputs as backgrounds for peak-calling. Replicate peaks were overlapped, and only shared peaks where both the log2 fold change over respective inputs and −log10 p-value were greater than 2 in both replicates were retained.
ATAC-seq and ChIP-seq analysis
FASTQ files were mapped to the mm10 mouse reference genome with Bowtie2.53 Peaks were called with HOMER (findPeaks) using parameters “-style factor -minDist 200 -size 200”. After merging these peaks, correlations among replicates from the same cell subset/treatment were evaluated by correlation using tag counts. The two most highly correlated samples were used for identifying the most robust peaks using the irreproducible discovery rate (IDR) method.54 For this step, peaks were called with HOMER’s findPeaks, using parameters “-L 0 -C 0 -fdr 0.9 -minDist 200 -size 200”. IDR peaks from different conditions involved in a comparison merged with HOMER’s mergePeaks and annotated with HOMER’s annotatePeaks.pl. The raw tags of all samples which had reasonable correlation were quantified with HOMER (annotatePeaks.pl) using parameter “-noadj”. Peaks which contained at least 4 tags in at least 1 sample were used to identify differentially bounded peaks (DBP) by DESeq2. Peaks were categorized as distal peaks which are 2 kb away from known TSS and promotor peaks which are located within 2 kb region of known TSS sites. Histone marks, such as H3K27ac single, were quantified by either ATAC IDR peaks under the same conditions or nuclear protein ChIP IDR peaks. ATAC peak quantification was normalized to the total tags in peaks, while the ChIP peak single was normalized to the sequence depth.
RNA-seq analysis
FASTQ files were processed to assess quality by determining general sequencing bias, clonality and adapter sequence contamination. RNA sequencing reads were aligned to the mm10 mouse reference genome using STAR.55 Gene expression levels were calculated using HOMER 51 by counting all strand specific reads within exons. Only the most abundant transcripts, including multiple alternative variants, were selected for each gene, and the genes with a length smaller than 250 bp were removed. Transcripts per million (TPM) were used to evaluate the correlation among replicates. Differential gene expression was calculated using DESeq256 to assess both biological and technical variability between experiments. Unsupervised hierarchical clustering was used to cluster the gene expression in the heatmaps.
Motif analysis
To identify motifs enriched in peak regions over the background, HOMER’s motif analysis “findMotifsGenome.pl” including known default motifs and de novo motifs was used.51 The background peaks used either from random genome sequences or from peaks in comparing condition were indicated throughout the main text and in the figure legends.
Gene ontology enrichment analysis
Metascape was used for the analysis. All genes in the genome have been used as the enrichment background.
Data visualization
ChIP-seq data were visualized in the UCSC genome browser.57
QUANTIFICATION AND STATISTICAL ANALYSIS
The significance of differences in the experimental data were determined using GraphPad Prism 8.0 software. All data involving statistics are presented as mean ± s.d. or s.e.m. The number of replicates and the statistical test used are described in the figure legends.
Supplementary Material
Highlights.
NCoR is required for osteoclast differentiation and normal bone density
RANK signaling converts NCoR/HDAC3 co-repressor complexes to co-activator complexes
RANK signaling induces RNA-dependent interaction of NCoR/HDAC3 with PGC1β
The major function of NCoR in osteoclasts is RANKL-dependent gene activation
ACKNOWLEDGEMENTS
We thank Jana G. Collier and Martina P. Pasillas for technical assistance, Ty D. Troutman and Zeyang Shen for discussion, Leslie Van Ael and Katelyn Griffith for secretary assistance, the German Gene Trap Consortium for Smartf/f mice and the RIKEN for NCoR antibody. These studies were supported by NIH grants DK091183, HL088083, DK063491, AG068449, GM118060 and GM085764 and Foundation Leducq grant 16CVD01. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a NIH SIG grant #S10 OD026929 and the UAMS Bone Biomechanics, Histology and Imaging Core funded by NIH P20GM125503. Y.A. was supported by the Japan Society for the Promotion of Science Overseas Research Fellowship 201860150 and Uehara Memorial Foundation Fellowship. E.R.K. was supported by an NSF Graduate Research Fellowship DGE-2038238.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as a gender minority in their field of research.
Footnotes
DECLARATION OF INTERESTS
C.K.G. is a cofounder, equity holder and member of the Scientific Advisory Board of Asteroid Therapeutics. J.T.K. is on the Molecular Cell advisory board. The other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at OOO.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFARENCES
- 1.Emmett MJ, and Lazar MA (2019). Integrative regulation of physiology by histone deacetylase 3. Nat Rev Mol Cell Biol 20, 102–115. 10.1038/s41580-018-0076-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong MM, Guo C, and Zhang J (2014). Nuclear receptor corepressor complexes in cancer: mechanism, function and regulation. Am J Clin Exp Urol 2, 169–187. [PMC free article] [PubMed] [Google Scholar]
- 3.Perissi V, Jepsen K, Glass CK, and Rosenfeld MG (2010). Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet 11, 109–123. 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- 4.Guenther MG, Barak O, and Lazar MA (2001). The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol 21, 6091–6101. 10.1128/mcb.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrante F, Giaimo BD, Bartkuhn M, Zimmermann T, Close V, Mertens D, Nist A, Stiewe T, Meier-Soelch J, Kracht M, et al. (2020). HDAC3 functions as a positive regulator in Notch signal transduction. Nucleic Acids Res 48, 3496–3512. 10.1093/nar/gkaa088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mondello P, Tadros S, Teater M, Fontan L, Chang AY, Jain N, Yang H, Singh S, Ying HY, Chu CS, et al. (2020). Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov 10, 440–459. 10.1158/2159-8290.Cd-19-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borggrefe T, and Oswald F (2009). The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci 66, 1631–1646. 10.1007/s00018-009-8668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogawa S, Lozach J, Jepsen K, Sawka-Verhelle D, Perissi V, Sasik R, Rose DW, Johnson RS, Rosenfeld MG, and Glass CK (2004). A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc Natl Acad Sci U S A 101, 14461–14466. 10.1073/pnas.0405786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li P, Spann NJ, Kaikkonen MU, Lu M, Oh DY, Fox JN, Bandyopadhyay G, Talukdar S, Xu J, Lagakos WS, et al. (2013). NCoR repression of LXRs restricts macrophage biosynthesis of insulin-sensitizing omega 3 fatty acids. Cell 155, 200–214. 10.1016/j.cell.2013.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emmett MJ, Lim HW, Jager J, Richter HJ, Adlanmerini M, Peed LC, Briggs ER, Steger DJ, Ma T, Sims CA, et al. (2017). Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature 546, 544–548. 10.1038/nature22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. (1998). Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93, 165–176. 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 12.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, et al. (1998). Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A 95, 3597–3602. 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, et al. (1999). OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397, 315–323. 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 14.Kawaida R, Ohtsuka T, Okutsu J, Takahashi T, Kadono Y, Oda H, Hikita A, Nakamura K, Tanaka S, and Furukawa H (2003). Jun dimerization protein 2 (JDP2), a member of the AP-1 family of transcription factor, mediates osteoclast differentiation induced by RANKL. J Exp Med 197, 1029–1035. 10.1084/jem.20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bozec A, Bakiri L, Hoebertz A, Eferl R, Schilling AF, Komnenovic V, Scheuch H, Priemel M, Stewart CL, Amling M, and Wagner EF (2008). Osteoclast size is controlled by Fra-2 through LIF/LIF-receptor signalling and hypoxia. Nature 454, 221–225. 10.1038/nature07019. [DOI] [PubMed] [Google Scholar]
- 16.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al. (2002). Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3, 889–901. 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 17.Zeng R, Faccio R, and Novack DV (2015). Alternative NF-κB Regulates RANKL-Induced Osteoclast Differentiation and Mitochondrial Biogenesis via Independent Mechanisms. J Bone Miner Res 30, 2287–2299. 10.1002/jbmr.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, and Rosenfeld MG (2002). Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell 110, 55–67. 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 20.Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, and Rose DW (2006). Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell 124, 615–629. 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 21.Cao F, Zwinderman MRH, and Dekker FJ (2018). The Process and Strategy for Developing Selective Histone Deacetylase 3 Inhibitors. Molecules 23. 10.3390/molecules23030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, and Chawla A (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4, 13–24. 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma JD, Jing J, Wang JW, Mo YQ, Li QH, Lin JZ, Chen LF, Shao L, Miossec P, and Dai L (2019). Activation of the Peroxisome Proliferator-Activated Receptor γ Coactivator 1β/NFATc1 Pathway in Circulating Osteoclast Precursors Associated With Bone Destruction in Rheumatoid Arthritis. Arthritis Rheumatol 71, 1252–1264. 10.1002/art.40868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin RG, and Ames BN (1961). A method for determining the sedimentation behavior of enzymes: application to protein mixtures. J Biol Chem 236, 1372–1379. [PubMed] [Google Scholar]
- 25.Perissi V, Scafoglio C, Zhang J, Ohgi KA, Rose DW, Glass CK, and Rosenfeld MG (2008). TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Mol Cell 29, 755–766. 10.1016/j.molcel.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, and Wong J (2003). Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. Embo j 22, 1336–1346. 10.1093/emboj/cdg120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sonoda J, Mehl IR, Chong LW, Nofsinger RR, and Evans RM (2007). PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc Natl Acad Sci U S A 104, 5223–5228. 10.1073/pnas.0611623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Häcker H, and Kamps MP (2006). Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods 3, 287–293. 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 29.Raetz CR, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC Jr., Ribeiro AA, Murphy RC, Ulevitch RJ, Fearns C, et al. (2006). Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J Lipid Res 47, 1097–1111. 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Scarpulla RC (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813, 1269–1278. 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tavares CDJ, Aigner S, Sharabi K, Sathe S, Mutlu B, Yeo GW, and Puigserver P (2020). Transcriptome-wide analysis of PGC-1α-binding RNAs identifies genes linked to glucagon metabolic action. Proc Natl Acad Sci U S A 117, 22204–22213. 10.1073/pnas.2000643117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13, 508–514. 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tong X, Gu PC, Xu SZ, and Lin XJ (2015). Long non-coding RNA-DANCR in human circulating monocytes: a potential biomarker associated with postmenopausal osteoporosis. Biosci Biotechnol Biochem 79, 732–737. 10.1080/09168451.2014.998617. [DOI] [PubMed] [Google Scholar]
- 34.Yue T, Ji M, Qu H, Guo M, Bai F, Zhang Z, Wang W, Gong X, and Zhang Z (2019). Comprehensive analyses of long non-coding RNA expression profiles by RNA sequencing and exploration of their potency as biomarkers in psoriatic arthritis patients. BMC Immunol 20, 28. 10.1186/s12865-019-0297-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sikand K, and Shukla GC (2011). Functionally important structural elements of U12 snRNA. Nucleic Acids Res 39, 8531–8543. 10.1093/nar/gkr530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benecke H, Lührmann R, and Will CL (2005). The U11/U12 snRNP 65K protein acts as a molecular bridge, binding the U12 snRNA and U11–59K protein. Embo j 24, 3057–3069. 10.1038/sj.emboj.7600765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang N, Wang X, Xie X, Liao Y, Liu N, Liu J, Miao N, Shen J, and Peng T (2017). lncRNA DANCR promotes tumor progression and cancer stemness features in osteosarcoma by upregulating AXL via miR-33a-5p inhibition. Cancer Lett 405, 46–55. 10.1016/j.canlet.2017.06.009. [DOI] [PubMed] [Google Scholar]
- 38.Ma Y, Zhou G, Li M, Hu D, Zhang L, Liu P, and Lin K (2018). Long noncoding RNA DANCR mediates cisplatin resistance in glioma cells via activating AXL/PI3K/Akt/NF-κB signaling pathway. Neurochem Int 118, 233–241. 10.1016/j.neuint.2018.03.011. [DOI] [PubMed] [Google Scholar]
- 39.Tang J, Zhong G, Zhang H, Yu B, Wei F, Luo L, Kang Y, Wu J, Jiang J, Li Y, et al. (2018). LncRNA DANCR upregulates PI3K/AKT signaling through activating serine phosphorylation of RXRA. Cell Death Dis 9, 1167. 10.1038/s41419-018-1220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Zeng X, Wang N, Zhao W, Zhang X, Teng S, Zhang Y, and Lu Z (2018). Long noncoding RNA DANCR, working as a competitive endogenous RNA, promotes ROCK1-mediated proliferation and metastasis via decoying of miR-335–5p and miR-1972 in osteosarcoma. Mol Cancer 17, 89. 10.1186/s12943-018-0837-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee RD, Knutson TP, Munro SA, Miller JT, Heltemes-Harris LM, Mullighan CG, Jepsen K, and Farrar MA (2022). Nuclear corepressors NCOR1/NCOR2 regulate B cell development, maintain genomic integrity and prevent transformation. Nat Immunol 23, 1763–1776. 10.1038/s41590-022-01343-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, and Müller R (2010). Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 25, 1468–1486. 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 43.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, and Parfitt AM (2013). Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 28, 2–17. 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka T, Takeno T, Watanabe Y, Uchiyama Y, Murakami T, Yamashita H, Suzuki A, Aoi R, Iwanari H, Jiang SY, et al. (2002). The generation of monoclonal antibodies against human peroxisome proliferator-activated receptors (PPARs). J Atheroscler Thromb 9, 233–242. 10.5551/jat.9.233. [DOI] [PubMed] [Google Scholar]
- 45.Fonseca GJ, Tao J, Westin EM, Duttke SH, Spann NJ, Strid T, Shen Z, Stender JD, Sakai M, Link VM, et al. (2019). Diverse motif ensembles specify non-redundant DNA binding activities of AP-1 family members in macrophages. Nat Commun 10, 414. 10.1038/s41467-018-08236-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montague TG, Cruz JM, Gagnon JA, Church GM, and Valen E (2014). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res 42, W401–407. 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brannan KW, Chaim IA, Marina RJ, Yee BA, Kofman ER, Lorenz DA, Jagannatha P, Dong KD, Madrigal AA, Underwood JG, and Yeo GW (2021). Robust single-cell discovery of RNA targets of RNA-binding proteins and ribosomes. Nat Methods 18, 507–519. 10.1038/s41592-021-01128-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abe Y, Fujiwara Y, Takahashi H, Matsumura Y, Sawada T, Jiang S, Nakaki R, Uchida A, Nagao N, Naito M, et al. (2018). Histone demethylase JMJD1A coordinates acute and chronic adaptation to cold stress via thermogenic phospho-switch. Nat Commun 9, 1566. 10.1038/s41467-018-03868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, Rialdi A, White KM, Albrecht RA, Pache L, et al. (2018). Transcription Elongation Can Affect Genome 3D Structure. Cell 174, 1522–1536.e1522. 10.1016/j.cell.2018.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lovci MT, Ghanem D, Marr H, Arnold J, Gee S, Parra M, Liang TY, Stark TJ, Gehman LT, Hoon S, et al. (2013). Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol 20, 1434–1442. 10.1038/nsmb.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. The Annals of Applied Statistics 5, 1752–1779, 1728. [Google Scholar]
- 55.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res 12, 996–1006. 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data including RNA-seq, ChIP-seq, ATAC-seq, and eCLIP assay were deposited in the Gene Expression Omnibus (GEO) under the accession number GSE211676.
This paper does not report any original code.
Any information required to reanalyze the data reported in this paper is available from the Lead contact, Christopher K. Glass (ckg@ucsd.edu) upon request.