

SUMMARY
MLL/KMT2A amplifications and translocations are prevalent in infant, adult and therapy-induced leukemia. However, the molecular contributor(s) to these alterations are unclear. Here we demonstrate that histone H3 lysine 9 mono- and di-methylation (H3K9me1/2) balance at the MLL/KMT2A locus regulates these amplifications and rearrangements. This balance is controlled by the cross-talk between lysine demethylase KDM3B and methyltransferase G9a/EHMT2. KDM3B depletion increases H3K9me1/2 levels and reduces CTCF occupancy at the MLL/KMT2A locus, and in turn, promotes amplification and rearrangements. Depleting CTCF is also sufficient to generate these focal alterations. Furthermore, the chemotherapy Doxorubicin (Dox), which associates with therapy-induced leukemia and promotes MLL/KMT2A amplifications and rearrangements, suppresses KDM3B and CTCF protein levels. KDM3B and CTCF overexpression rescues Dox-induced MLL/KMT2A alterations. G9a inhibition in human cells or mice also suppresses MLL/KMT2A events accompanying Dox treatment. Therefore, MLL/KMT2A amplifications and rearrangements are controlled by epigenetic regulators that are tractable drug targets, which has clinical implications.
Graphical Abstract
In Brief
An imbalance in histone modifications contributes to both transient amplifications and integrated rearrangements and amplifications observed in leukemia, which can be induced by a commonly used chemotherapy but prevented by chemical intervention.
Introduction
Genomic instability is a hallmark of cancer.1,2 Cancer is often associated with copy number changes (e.g., gains/losses of chromosome arms and/or whole chromosomes, amplification/deletion of genomic regions) and structural rearrangements.3 These events can be genetically stable; however, focal DNA copy gains can also be extrachromosomal, transiently appearing and disappearing based on their environment.3-5 A key question remains as to whether the appearance of low- or high-copy extrachromosomal DNA (ecDNA) gains are associated with or precede the integration events that result in genomic rearrangements, and subsequently genetic heterogeneity.
Recent discoveries demonstrated that epigenetic regulators control transient site-specific extrachromosomal copy gains (TSSGs) of regions impacting therapeutic response and drug resistance.3,6-10 For example, the histone 3 lysine 9/36 (H3K9/36) tri-demethylase KDM4A enzyme was shown to promote selective extrachromosomal TSSGs.7 Subsequently, a collection of methyl-lysine modifying enzymes were shown to regulate these TSSGs.9,10 These studies suggest additional chromatin modulators could be involved in fine-tuning local and global chromatin states and regulating unknown TSSGs.
Acute myeloid leukemia (AML) and myelodysplasia (MDS) are characterized by genomic amplifications and translocations of the 11q23 region, including MLL/KMT2A and other target genes.11-18 MLL/KMT2A rearrangements are observed in greater than 70 percent of infant leukemias,19,20 as well as adult primary and therapy-related leukemias.21-26 KMT2A rearrangements result in the fusion of the gene to more than 100 partner genes leading to protein chimeras26 and a number of noncoding regions throughout the genome.26 Therapy-related acute myeloid leukemia (t-AML) is a clinical syndrome occurring long after chemotherapy treatment with agents such topoisomerase II (topo II) inhibitors.27-29 Approximately 10% of all AML cases arise after a patient's exposure to therapy for a primary malignancy,27 and t-AML patients have a significantly worse outcome than those who develop AML de novo.27,29,30 To date, there is a clinically unmet need regarding the mechanistic understanding of how chemotherapy promotes DNA rearrangements.
The H3K9me1/2 lysine demethylase KDM3B, originally named 5qNCA, resides in the frequently deleted region of 5q31 associated with loss of heterozygosity (LOH).31-33 MLL/KMT2A copy gains often occur with 5q LOH.34,35 KDM3B has been implicated as a myeloid leukemia tumor suppressor through oncogene regulation and contributes to genome stability; however, a full appreciation for the role KDM3B plays in genome regulation is understudied.31,36-39 We previously reported that loss of a region on chromosome 19, containing microRNA mir-23 promoted TSSGs through KDM4A stabilization.8 These observations prompted us to assess whether reduced KDM3B directly promotes the MLL/KMT2A copy gains and associated genomic insertions.
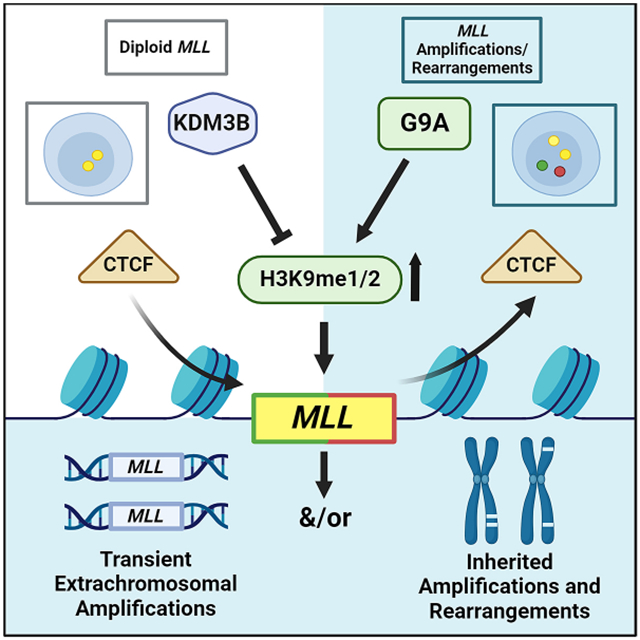
Consistent with these observations,34,35 we demonstrate with DNA Fluorescent In Situ Hybridization (FISH) that KDM3B depletion or chemical inhibition promotes transient and integrated site-specific MLL/KMT2A copy gains and rearrangements. These events are directly antagonized by depletion/inhibition of a H3K9me1/2 lysine methyltransferase (KMT) G9a/EHMT2. This axis controls H3K9me1/2 at KMT2A, especially in the region most frequently associated with genomic break aparts and rearrangements. We further demonstrate that a KDM3B-G9a balance controls CTCF occupancy in the H3K9 methylation enriched region, and in turn, the ability of the KMT2A/MLL locus to undergo site-specific copy gains and genomic rearrangement. We then establish that the chemotherapeutic agent Doxorubicin (Dox) reduces KDM3B and CTCF protein levels, and as a consequence, promotes KMT2A copy gains and rearrangements. KDM3B overexpression rescues Dox-induced KMT2A changes. Furthermore, knockdown or chemical inhibition of G9a rescues MLL/KMT2A alterations in Dox-treated cells and mice. Collectively, these data highlight a critical role for H3K9me1/2 balance through KDM3B/G9a in regulating selective amplification and rearrangement of KMT2A. This discovery has major clinical implications in understanding the genesis of extrachromosomal amplifications and associated chromosomal rearrangements, and sheds light on how to therapeutically control the emergence of treatment-induced KMT2A amplifications and rearrangements in cancer.
Results
Loss of KDM3B causes site-specific copy gains of MLL/KMT2A locus.
KDM3B is a H3K9me1/2 demethylase located in the 5q31.1 region, associated with KMT2A amplification and rearrangements.31,35,37,40 To confirm this relationship, TCGA Acute Myeloid Leukemia (LAML) samples containing >50% KDM3B loss were assessed for KMT2A copy number. Most samples with >50% KDM3B loss have KMT2A gains, with some showing >50% copy gain (p = 6.45e-07; Figure 1A, green dots). Furthermore, DNA FISH for KDM3B and KMT2A on leukemic cell lines with KDM3B LOH (KG1a and HL60) had cells within the population with an increased KMT2A baseline copy number (Figure 1B-C).
Figure 1. KDM3B depletion induces KMT2A DNA copy gains and break aparts.

(A) Analysis of TCGA LAML samples showing most LAML samples with KDM3B loss also have KMT2A copy gain with a p-value of 6.45e-07. Statistical significance was computed by Wilcoxon rank-sum test, which provides a non-parametric hypothesis test on two independent samples.
(B) Representative DNA FISH with the 5qDel probe (5q probe covers KDM3B locus) demonstrating 5q LOH status (red) of KG1a (upper) and HL60 (lower) (left panels). Examples with KMT2A DNA FISH probe (panel C) demonstrate a baseline increase in KMT2A copies in KG1a (upper) and HL60 (lower) (right panels). Arrowheads highlight the FISH signal.
(C) A schematic of DNA Fluorescent In Situ Hybridization (FISH) probe genomic locations that are used for KMT2A locus.
(D) Representative siCTRL (upper) and siKDM3B (lower) DNA FISH images with the KMT2A-1 probe (orange probe in panel C and centromere 11 (11C; control probe)).
(E) KDM3 family siRNA screen demonstrates that only KDM3B depletion generates KMT2A copy gains (KMT2A-1; orange) but not copy gains of centromere 11 (11C; grey).
(F) Representative images with the clinical KMT2A DNA FISH break apart probe (red and green probes in panel C) that show no copy gain in siCTRL (top panel), DNA copy gains (middle 3 panels) and break apart events (bottom panel) upon KDM3B siRNA depletion. Arrowheads highlight the FISH signal.
(G and H) DNA FISH showing KDM3B siRNA knockdown results in KMT2A copy gains (black) and break aparts (purple) but not CD3 (grey).
(I) Representative images with the clinical KMT2A DNA FISH break apart probe (red and green probes in schematic C) that show KMT2A copy gains with siRNA-mediated KDM3B knockdown in U937 leukemia cells. Arrowheads highlight the FISH signal.
(J and K) DNA FISH showing KDM3B siRNA knockdown results in KMT2A copy gains and break aparts but not CD3 alterations in U937 cells.
(L) Input-normalized H3K9me1-3 ChIP-seq tracks of the region containing KMT2A. Publicly available ChIP-seq shows KDM3B binding within the BCR in HCT-116 cells which is lost upon shKDM3 treatment (green tracks) (49).
(M) Bar graphs of ChIP-seq data in panel L demonstrating increased fold enrichments of H3K9me1/2 within the KMT2A locus.
Error bars represent the SEM. *p < 0.05 by two-tailed Student’s t-test. Scale bar represents 5μm.
We then tested whether depletion of KDM3B and/or other KDM3 family members generate KMT2A copy gains and genomic structural changes. Specifically, immortalized retinal pigment epithelial cells (RPEs) were siRNA depleted for each KDM3 family member.7,10,41 These cells are ideal for assessing DNA amplification and rearrangement mechanisms because they have a stable genome, do not harbor cancer mutations and are near diploid.7,10,41-45 Each independent set of siRNAs was validated and assessed for major cell cycle defects by flow cytometry analysis before being assayed by DNA FISH (Figure S1A-C). A DNA FISH probe against the KMT2A gene (Figure 1C; noted in orange) and a centromeric region at chromosome 11 (11C) were used to evaluate site-specific DNA copy gains. Copy number gain evaluation for each FISH probe was measured in percentages as previously described.7-10,46
While KDM3 family members have comparable H3K9me1/2 activity in vitro (Figure S1D-F), only KDM3B siRNA depletion caused a significant increase in KMT2A copy gains with no significant changes to the 11C control region (Figure 1D-E). We then assessed the site-specific impact of KDM3B depletion on KMT2A by leveraging a clinically relevant two color DNA FISH probe that covers KMT2A (green: 5’-end of KMT2A and red: 3’-end of KMT2A) and an adjacent and partially overlapping FISH probe (called CD3) (Figure 1C). The dual KMT2A FISH probes allow both locus rearrangement (referred to as break apart, BA) and DNA copy gains to be identified. KDM3B knockdown caused a significant increase in KMT2A gains (black bars) and break apart (BA; purple bars) events (Figure 1F-G). The amplifications did not have N-terminal (green) or C-terminal (red) bias for the KMT2A gene, including the whole gene (both probes- pseudo-colored yellow)(Figure 1F). The adjacent FISH probe CD3 (Figure 1C; grey) did not change upon KDM3B depletion (Figure 1H), emphasizing the site-specific control of KDM3B depletion.
We then explored the specificity of KDM3B siRNA-mediated depletion in generating leukemia-associated amplifications and/or rearrangements by conducting FISH for a panel of leukemia-associated amplified and/or rearranged genes (e.g., ENL/MLLT1, AF9/MLLT3; Figure S1G-O). Most regions did not change in their basal DNA copy number, with the exception of TCF3/E2A and AFF3/LAF4 (Figure S1M-O). We also observed KMT2A site-specific copy gains and rearrangements in the U937 leukemia cell line47,48 with KDM3B depletion, but not TCF3/E2A or AFF3/LAF4 (Figure 1I-K and S1P-R). These data suggest that unlike the KMT2A locus, these other regions are not consistently regulated by KDM3B across cell lines.
KDM3B depletion alters H3K9me1/2 across the KMT2A genomic locus
KDM3B is an H3K9me1/2 demethylase (Figure S1D-F).38,49 Therefore, we performed chromatin immunoprecipitation (ChIP) sequencing for H3K9me1/2/3 methylation marks in control and KDM3B siRNA transfected RPE cells (Figure S1S). KDM3B depletion produced genome-wide H3K9me1 changes that were preferentially skewed towards an increase of these marks (points above the upper red line: >1.5-fold increase)(Figure S1T). The magnitude of H3K9me1 increase across the middle of the KMT2A gene (red points) was one of the strongest events across the genome (Figure S1T). The list of all 10 Kb genomic bins with increased H3K9me1 and their nearest associated genes can be found in Table S1. A similar genome-wide increase of H3K9me2 was observed (Figure S1U).
Both H3K9me1/2 increased upon KDM3B depletion across the KMT2A gene body, particularly H3K9me1 within the 8.3kb breakpoint cluster region (BCR) spanning exons 8-14, which is enriched for KMT2A rearrangements (Figure 1L-M and S1S-U).50 In control cells, H3K9me1 at BCR was lower than the adjacent regions (Figure 1L-M). KDM3B knockdown led to a strong increase of H3K9me1 uniformly across the BCR (Figure 1L). H3K9me2 increased on the flank to the BCR (Figure 1L-M). We also observed altered H3K9me1/2 at other amplified and rearranged targets TCF3 and AFF3 regulated by KDM3B (Figure S1T-W). Consistent with a direct effect of KDM3B, analysis of published KDM3B ChIP-seq data51 demonstrated that KDM3B binds across KMT2A, with a strong peak within the BCR that was lost upon shRNA-mediated KDM3B depletion (Figure 1L). KDM3B also binds across TCF3 and AFF3 (Figure S1V-W). Collectively, our data establish that KDM3B depletion alters H3K9me1/2 methylation landscape of KMT2A, especially H3K9me1 at the BCR region, and contributes to KMT2A copy gains and break apart events.
Inhibition or depletion of KDM3B causes inherited KMT2A copy gains and genomic alterations
Using a KDM3 family inhibitor (JDI-12, referred to as KDM3i),52 we suppressed KDM3B enzymatic activity in vitro (Figure S2A) and noted a modest suppression of growth at 1μM with no impact at 25nM in RPE cells (Figure S2B). These doses were sufficient to promote significant KMT2A DNA copy gains and genomic rearrangements without altering KDM3B protein levels (Figure 2A and S2C-D). Furthermore, KDM3B inhibition significantly increased KMT2A copy gains and genomic rearrangements in a panel of primary and cancer cell lines (KG1a cells, a primary AML derived cell line, a primary AML organoid model, and primary Hematopoietic Stem and Progenitor Cells (HSPCs); Figure 2B-E).
Figure 2. KDM3B chemical inhibition (KDM3i) promotes transient KMT2A copy gains and break aparts.

(A) Schematic (top) and quantification of DNA FISH (bottom) demonstrating that KMT2A amplification and break apart events occur with KDM3B inhibitor treatment but no change in copy number at the adjacent CD3 locus in RPE cells.
(B-E) DNA FISH showing that KDM3B inhibition results in KMT2A copy gains and break aparts in KG1a, AML organoids, primary AML cells, and Hematopoietic Stem and Progenitor Cells with no change in copy number at the CD3 locus.
(F) Schematic (top) and quantification of DNA FISH (bottom) showing that KDM3 inhibition (KDM3i) results in KMT2A copy gains and break aparts. Upon KDM3i washout (12hrs Washout), copy gains and break aparts no longer occur. No significant change occurred with the CD3 probe.
Error bars represent the SEM. *p < 0.05 by two-tailed Student’s t-test.
KMT2A amplifications occur as both extrachromosomal and integrated events.35,53 By adding and removing KDM3i, the transient or permanent behavior of the KMT2A copy gains and genomic alterations was assessed. KMT2A DNA copy gains and break apart events occur 12 hours after KDM3i treatment (Figure 2F, KDM3i 12hrs), but are not observed upon KDM3i removal (Figure 2F, KDM3i 12hrs washout). The total cell number was not reduced under these conditions (Figure S2E). These data suggest that KDM3B inhibition promotes transient KMT2A amplifications (TSSGs) and altered genomic rearrangement in a short timeframe, which raises the question if longer KDM3i treatment could result in inherited DNA amplifications through insertion/rearrangement. Therefore, we treated cells for 72 hours (approximately 3 cell divisions), then passaged into fresh media (wash-off) for additional passages, which ensured that no active drug was present before assessing KMT2A alterations (Figure 3A). The longer suppression resulted in both KMT2A copy gains and genomic rearrangements being inherited (Figure 3B), which was confirmed with DNA FISH on mitotic chromosomes (Figure 3C-D).
Figure 3. KDM3B suppression leads to integration and inheritance of KMT2A copy gains and break aparts.

(A) A KDM3i treatment schematic and associated passaging of RPE cells. Cells were treated with 25nM of KDM3i. Cells were passaged in media without KDM3i every 3 days for sequential passages.
(B) KMT2A and CD3 DNA FISH at passage 0 and passage 10 after KDM3i treatment, which demonstrates KMT2A copy gains and break aparts are inherited in RPE cells after 10 passages (P10). No significant change occurred with the CD3 probe.
(C) Example metaphase spreads for KMT2A FISH for Vehicle and KDM3i treated cells at passage 10. Arrowheads highlight the FISH signal.
(D) Quantification of the metaphase spreads with KMT2A FISH in KDM3i treated and passage 10 cells demonstrating increased copies of KMT2A are retained.
(E) A KDM3B siRNA schematic and associated passaging of RPE cells (left). Western blots for KDM3B at cell passages used for DNA FISH demonstrates KDM3B protein levels return to baseline by passage 3 (P3; right).
(F) KMT2A and CD3 FISH of KDM3B siRNA passaged cells demonstrates inheritance at passage 3, 5 and 15. No significant change occurred with the CD3 probe at any passage.
(G) Example metaphase spreads for KMT2A FISH for siCTRL and siKDM3B cells at passage 3. Arrowheads highlight the FISH signal.
(H) Quantification of the metaphase spreads with KMT2A FISH in cells treated with siCTRL and siKDM3B from two independently propagated siCTRL and siKDM3B cells at passages 3 and 9 demonstrating increased copies of KMT2A are retained.
Error bars represent the SEM. *p < 0.05 by two-tailed Student’s t-test. Scale bar represents 5μm.
Upon transient siRNA depletion, KDM3B protein levels return to baseline levels by the third passage (P3) (Figure 3E). Inherited copy gains and genomic structure changes were still present in later passage interphase nuclei and mitotic chromosomes following siRNA-mediated KDM3B depletion, however, no change occurred to the adjacent region (Figure 3F-H). Increased inherited copies were confirmed with KMT2A Digital Droplet PCR, using the adjacent CD3E gene as a control (Figure S3A). While TCF3 copy gains were inherited, AFF3 copy gains were not present in later passages (Figure S3B-D). These data demonstrate that KDM3B inhibition and depletion promote transient amplification and integrated rearrangement events with extended suppression.
G9a and KDM3B cross-talk controls KMT2A copy gains and rearrangements
We previously demonstrated that co-depletion of specific H3K4 KMTs with the KDM5A enzyme prevents the KDM5A-driven TSSGs.9,10 Our results suggest that proper balance of H3K9me1/2 is critical in regulating site-specific copy gains and genomic rearrangements at KMT2A (Figure 1). Therefore, we pre-depleted either of the two H3K9me1/2 KMTs, G9a/EHMT2 and EHMT1,54 before KDM3B and assessed KMT2A DNA copy gains or rearrangements (Figure S4A-D). Pre-depletion of G9a, but not EHMT1, rescued/prevented the KMT2A amplification and genomic alterations caused by KDM3B depletion (Figure 4A). Rescue was also observed for TCF3 and AFF3 copy gains (Figure S4E-G). Furthermore, co-treatment with KDM3i and a dual inhibitor for G9a/EHMT2 and EHMT1 (EHMTi)55 completely rescued KMT2A amplification and rearrangements with no impact on cell growth (Figure 4B and Figure S4H). To strengthen the relationship between G9a and KMT2A amplification regulation, we transiently overexpressed G9a for 24 hours and assessed KMT2A genomic alterations (Figure 4C and Figure S4I-J). G9a overexpression was sufficient to promote KMT2A copy gains and genomic alterations (Figure 4C), emphasizing the role of H3K9me1/2 methylation balance in regulating focal amplification of KMT2A.
Figure 4. H3K9me1 balance controls KMT2A copy gains and break aparts.

(A) A schematic (upper) and DNA FISH (lower) for co-depletion of KDM3B with EHMT1 or EHMT2/G9a. siRNA depleted G9a but not EHMT1 prevents KMT2A copy gains and break aparts upon KDM3B siRNA depletion. No significant change occurred with the CD3 probe.
(B) A schematic (upper) and DNA FISH (lower) for KDM3i and EHMTi treatment. EHMT1/2 chemical inhibition prevents KMT2A copy gains and break aparts upon KDM3i treatment. No significant change occurred with the CD3 probe.
(C) A schematic (upper) and DNA FISH (lower) shows G9a overexpression promotes KMT2A copy gains and break aparts. Halo-EV- Halo empty vector. No significant change occurred with the 11C probe.
(D) A schematic (upper) and DNA FISH (lower) for depletion of G9a in HL60 cells (KDM3B LOH cell line). G9a depletion modestly but significantly suppresses KMT2A copy gains in HL60 cells. No significant change occurred with the CD3 probe.
(E) A schematic (upper) and DNA FISH (lower) for depletion of G9a in RPE-WT or RPE-inherited KMT2A cells. G9a depletion does not suppress KMT2A copy gains or break aparts in the RPE-inherited KMT2A cells. No significant change occurred with the CD3 probe.
(F) Input-normalized H3K9me1/2/3 tracks at the KMT2A gene upon siKDM3B or siG9a alone or in combination in RPE cells.
(G) Bar graphs representing H3K9 methylation ChIP-seq fold enrichment over input in three parts of KMT2A gene shown in (F).
(H) A model depicting interplay between KDM3B-G9a regulating H3K9me1/2 and modulating KMT2A amplifications/rearrangements.
Error bars represent the SEM. *p < 0.05 by two-tailed Student’s t-test. NS- not significant to control.
We then hypothesized that G9a depletion could rescue the extrachromosomal amplifications caused by constantly reduced KDM3B levels. Therefore, we siRNA depleted G9a in the KDM3B LOH leukemia cell line HL60, and evaluated whether the increased KMT2A copy number observed in these cells would be reduced or reset (Figure S4K). While KMT2A copy gains were significantly suppressed (Figure 4D), the levels were still higher than the baseline in RPE cells, suggesting that HL60 contain both transient extrachromosomal and inherited forms. To further explore this relationship, we siRNA depleted G9a in the KMT2A-inherited RPE cell lines after their level of KDM3B had returned to baseline (Figure 3E-G) and assessed whether depletion of G9a could rescue the inherited KMT2A gains (Figure S4L). G9a depletion did not impact the inherited KMT2A copy gains (Figure 4E), suggesting that inherited extra copies are stable and unable to be rescued. Collectively, these data demonstrate that KDM3B/G9a coordinate KMT2A amplification and rearrangements.
Since G9a depletion rescued KMT2A amplifications and genomic alterations caused by KDM3B suppression (Figure 4A-B), we hypothesized that co-depletion of G9a with KDM3B would reset the H3K9me1/2 patterns at KMT2A, strengthening the importance of H3K9me1/2 balance in regulating the KMT2A locus. In fact, the preferential genome-wide increase of H3K9me1 and H3K9me2 caused by siKDM3B was mostly rescued by double KDM3B and G9a knockdown. H3K9me1 increase was rescued across 81% of regions genome-wide (88 Mb out of total 109 Mb), whereas H3K9me2 increase was rescued across 75% of regions genome-wide (219 Mb out of total 292 Mb) (Figure S4M). These data suggest that maintaining a KDM3B-G9a balance is critical for controlling H3K9me1/2 levels genome-wide.
G9a depletion was able to completely reset the H3K9me1 patterns at the BCR (exon 8-14) in KMT2A (Figure 4F-G and Figure S4N). Consistent with KMT2A, we also observed similar rescue at TCF3 and AFF3 (Figure S4N). KDM3B and G9a knockdowns produced genome-wide H3K9me1 changes with opposite preferential patterns, whereas the double knockdown rescued these skews. For example, the KDM3B knockdown (Figure S4N, left plot) resulted in a preferential increase of H3K9me1. By contrast, G9a knockdown (Figure S4N, middle plot) resulted in a decrease of H3K9me1. In the double knockdown (Figure S4N, right plot), H3K9me1 changes were strongly reduced compared to siKDM3B alone, with smaller extent of differences from control in either direction. Similar results for G9a rescue of H3K9me2 across the genome, including KMT2A, TCF3, and AFF3 are shown in Figure S4O. The list of all 10 Kb genomic bins that had changes in H3K9me1/2 and their nearest associated genes can be found in Table S2. These data suggest that KDM3B-G9a balance controls H3K9me1/2 levels, and in turn, site-specific DNA copy gains and genomic rearrangements (Figure 4H).
Reduced CTCF occupancy promotes KMT2A copy gains and rearrangements
Prior studies suggest that CTCF binding could impact genome integrity, rearrangement, or duplication, especially at the KMT2A locus;56,57 however, the direct role of CTCF in controlling amplification and rearrangement has not been resolved. Upon evaluating multiple cell lines and tissues from ENCODE, we observed a highly conserved occupancy for CTCF at exon 11 within the BCR of KMT2A, directly overlapping with KDM3B binding (Figure 5A). We hypothesized that KDM3B depletion could disrupt CTCF binding and promote KMT2A genomic alterations. Therefore, KDM3B and CTCF were depleted individually or in combination before assessing KMT2A by DNA FISH (Figure S5A-B). CTCF depletion alone promoted significant KMT2A site-specific copy gains and genomic rearrangements that were not enhanced by KDM3B depletion (Figure 5B). Since the CTCF peak within exon 11 of KMT2A directly overlapped with the KDM3B peak (Figure 5A), we hypothesized that KDM3B depletion may be disrupting CTCF binding. We did not observe a global change in CTCF protein levels upon KDM3B depletion but did observe a significant reduction in CTCF binding at KMT2A exon 11 within the BCR by ChIP-Seq and ChIP-qPCR (Figure 5C-E and Figure S5C).
Figure 5. Reduced CTCF occupancy leads to KMT2A copy gains and break aparts.

(A) Publicly available ENCODE input-normalized ChIP-seq tracks densities of CTCF in multiple ENCODE cell lines or tissues at the KMT2A locus. CTCF binding at exon 11 of KMT2A is conserved in multiple cell lines and directly overlaps with KDM3B binding in HCT116 cells 51.
(B) DNA FISH demonstrating single and co-siRNA depletion of KDM3B and CTCF promotes KMT2A copy gains and break aparts. No significant change occurred with the CD3 probe.
(C) Quantification of western blots for CTCF in KDM3B siRNA depleted RPE cells. No significant change in steady state total CTCF protein levels were observed.
(D) Publicly available input-normalized ChIP-seq tracks of KDM3B 51 in control and shKDM3 cells. KDM3B binds at exon 11 and is lost upon shKDM3 (green tracks). Lower tracks: input-normalized ChIP-seq tracks of CTCF showing that siKDM3B reduced CTCF binding at exon 11 in RPE cells.
(E) ChIP-qPCR demonstrating suppression of CTCF occupancy at KMT2A exon 11 (KMT2A ex 11; black) or a negative control for CTCF binding (CTCF negative site; yellow) following KDM3B siRNA depletion.
(F) Venn diagram of the overlap between KDM3B ChIP-seq peaks from a public dataset and CTCF ChIP-seq peaks in this study. 6,386 of all KDM3B binding sites (41.5%) co-localize with a CTCF binding site (P-value=1.0e-07).
(G) A total of 17,077 CTCF sites out of 46,340 (36.9%) had reduced occupancy with KDM3B depletion. Among all 6,386 KDM3B binding sites coinciding with CTCF binding, 1,005 sites show a significant decrease in CTCF binding upon KDM3B knockdown. Z-score=143.38 corresponding to a P-value close to 0.
(H) Double KDM3B and G9a knockdown rescued the increase of H3K9me1 at the majority of CTCF peaks reduced by siKDM3B. Barplot showing genome-wide number of CTCF proximal regions (+/− 5Kb from a CTCF peak) that decreased CTCF and increased H3K9me1 level upon KDM3B knockdown (points above upper red line in I, left scatterplot). Red, the fraction of regions where this increase was rescued by double knockdown (points moved below upper red line in I, right scatterplot).
(I) Genome-wide effects of siKDM3B, siG9a, and double knockdown on H3K9me1 levels at the subset of CTCF binding sites where CTCF binding was decreased by siKDM3B (17,077 sites). KDM3B and G9a knockdowns have opposite skews, whereas the double knockdown strongly reduces these H3K9me1 changes. Left, scatterplot comparing input-normalized H3K9me1 ChIP-seq densities in +/− 5Kb proximity of all these individual CTCF peaks across the genome in control vs siKDM3B; H3K9me1 changes are skewed towards increase (points above upper red line corresponding to > 1.5 fold increase in siKM3B cells). Middle, scatterplot for control vs siG9a cells; H3K9me1 changes are skewed towards decrease (points below lower red line corresponding to > 1.5 fold decrease in siG9a cells). Right, scatterplot for control vs siKDM3B + siG9a cells, with much fewer H3K9me1 changes in either direction. Red point, +/−5-Kb vicinity of CTCF binding site within KMT2A gene.
(J) DNA FISH demonstrating siRNA depletion of G9a prevents KMT2A copy gains and break aparts upon CTCF siRNA depletion. No significant change occurred with the CD3 probe.
(K) DNA FISH demonstrating EHMT1/2 chemical inhibition prevents KMT2A copy gains and break aparts upon CTCF siRNA depletion. No significant change occurred with the CD3 probe.
(L) A model depicting interplay between KDM3B-G9a-CTCF upon H3K9me1/2 modulation.
Error bars represent the SEM. *p < 0.05 by two-tailed Student’s t-test.
Upon analyzing KDM3B and CTCF occupancy patterns genome-wide from public data51 and our RPE ChIP-seq respectively, we observed that 6,386 KDM3B peaks in the public data (41.5% of all strong KDM3B peaks) directly overlapped with the 46,340 CTCF peaks in RPE cells (P-value=1.0e-07; Figure 5F). Upon KDM3B depletion, 17,077 CTCF peaks reduced their intensity. As much as 16% of these CTCF binding sites with reduced occupancy overlapped with KDM3B binding (1,005 peaks out of 6,386 total co-occupied sites; P-value < 1.0e-216; Z-Score=143.38) (Figure 5G). Despite the public KDM3B binding data being from a different cell line, the association between KDM3B and CTCF binding suggest a functional interplay between KDM3B and CTCF genome-wide.
To understand the genome-wide behavior of H3K9me1 at the 17,077 CTCF binding sites with reduced occupancy upon KDM3B depletion, we analyzed the impact of siKDM3B, siG9a, and double knockdown on the levels of H3K9me1 in the vicinity of all CTCF peaks (±5kb flanks from the peak center). KDM3B and G9a knockdowns produced opposite changes in H3K9me1 at the CTCF proximal regions, whereas the double knockdown rescued these changes. Upon KDM3B knockdown, H3K9me1 levels increased at least 1.5-fold in the 5 Kb proximity of approximately 1,000 CTCF peaks genome-wide (Figure 5H). Upon the double knockdown of KDM3B and G9a, this increase was rescued at the majority (~80%) of these peaks (Figure 5H). For example, KDM3B knockdown (Figure 5I, left plot) resulted in a preferential increase of H3K9me1 (above upper red line: > 1.5-fold increase). The H3K9me1 increase at the CTCF binding site within BCR of the KMT2A gene (red point) was among the strongest changes of all CTCF sites genome-wide. G9a knockdown (Figure 5I, middle plot) resulted in decreased H3K9me1 (below lower red line: > 1.5-fold decrease). However, in the double KDM3B/G9a knockdown (Figure 5I, right plot), H3K9me1 changes were strongly reduced, with smaller extent of differences from control in either direction. The level of H3K9me1 at CTCF binding site within BCR of the KMT2A gene (red point) was close to control in the double depletion. These data indicate that KDM3B and G9a control H3K9me1/2 at CTCF peaks genome-wide, but the methylation control surrounding the KMT2A BCR CTCF is a strong outlier among all sites.
Since CTCF is known to regulate gene expression,58 we assessed whether CTCF depletion regulated KDM3B expression levels. While depletion of CTCF modestly suppressed KDM3B transcript levels (Figure S5D), both KDM3B and G9a protein levels were not significantly reduced (Figure S5E-G), suggesting that CTCF is a downstream effector of KDM3B loss and H3K9me1/2 disruption. We tested this by depleting or chemically inhibiting G9a in combination with CTCF depletion. G9a depletion/inhibition prevented CTCF-induced alterations (Figure 5J-K and S5H-K). Consistent with this data, the CTCF site in the KMT2A BCR had increased K9me1 upon KDM3B depletion that was completely rescued upon G9a co-depletion (Figure 4F-G). Taken together, these data suggest that KDM3B and G9a coordinate H3K9me1/2 levels at and around the CTCF site, and in turn, impact CTCF occupancy and the predilection of KMT2A to undergo amplification and genomic rearrangement (Figure 5L).
Doxorubicin promotes KMT2A amplification and rearrangement as well as reduces KDM3B and CTCF protein levels
KMT2A rearrangements are observed in pediatric and therapy-induced leukemia when conventional chemotherapy is used to treat several cancer types.24,59,60 For example, KMT2A amplified and rearranged MDS and AML are generated after topoisomerase II (topo II) inhibitor treatment (e.g., Doxorubicin, Dox).29 Consistent with these clinical observations, Dox treatment promoted KMT2A, AFF3 and TCF3 copy gains and genomic alterations with no significant impact on control regions (Figure 6A and S6A-B). To reduce pleiotropic defects in RPE cells, we used lower doses of Dox (1 pg/μl and 5pg/μl). Dox also promoted KMT2A copy gains and rearrangements in primary HSPCs (Figure 6B). These observations are consistent with prior reports showing that the topo II inhibitor etoposide induces heterogeneous rearrangements of KMT2A in a variety of primary and non-primary human cells.57,61 We then treated mixed C57BL/6-129/Sv mice with Dox before isolating their spleen and assessing Kmt2a copy number by DNA FISH. Consistent with the human primary HSPCs (Figure 6B), cells isolated from the spleen of mice treated with Dox had increased Kmt2a copy gains, with no change in the adjacent Control 9 probe compared to control mice (Figure 6C). These data demonstrate the conserved impact of Dox treatment on site-specific Kmt2a copy gain events in the mice.
Figure 6. Doxorubicin promotes KMT2A amplification and rearrangement as well as reduces KDM3B and CTCF protein levels.

(A and B) Schematic of human KMT2A and adjacent CD3 DNA FISH probes (top). Graph of the DNA FISH for RPE and HSPCs treated with Dox for 72hrs (bottom). Dox treatment causes significant copy gains and break aparts at the KMT2A locus; while the control region (CD3) changes were not significant.
(C) A schematic of DNA FISH probe genomic locations used for mouse Kmt2a/Con9 locus (top). Graph of DNA FISH quantification demonstrating that cells isolated from the Spleen of mice treated with Dox have increased copy gains of Kmt2a but not the adjacent Control 9 region (bottom).
(D) RT-qPCR demonstrating that Dox significantly reduced KDM3B transcript levels after 72 hours of exposure in RPE cells.
(E) Representative western blot illustrating Dox reducing KDM3B protein levels after 72 hours of exposure in RPE cells.
(F) Quantification of western blots (n=4) showing a significant reduction in KDM3B protein levels following Dox treatment after 72 hours of exposure in RPE cells.
(G) RT-qPCR demonstrating that Dox significantly reduced CTCF transcript levels after 72 hours of exposure in RPE cells.
(H) Representative western blot illustrating Dox reducing CTCF protein levels after 72 hours of exposure in RPE cells.
(I) Quantification of western blots (n=4) showing a significant reduction in CTCF protein levels following Dox treatment after 72 hours of exposure in RPE cells.
(J) Graph of the quantification of western blots in Figure S6F showing a significant reduction in KDM3B (black) and CTCF (blue) protein levels following Dox treatment in KG1a cells.
(K) Western blot illustrating etoposide dose-dependently reduces KDM3B (upper) and CTCF (lower) protein levels after 72 hours of exposure in RPE cells.
(L and M) Western blot illustrating MG132 partially rescues KDM3B and CTCF protein levels in the presence of Dox treatment in RPE cells. Average quantification of 3 experiments in Figure S6G are below. Protein levels were quantified using ImageJ and normalized to α-Actinin.
Error bars represent the SEM. *p < 0.05 by two-tailed Student’s t-test.
Since reduction of KDM3B or CTCF levels promote KMT2A copy gains and rearrangement (Figures 1-4), we assessed whether KDM3B or CTCF expression is altered upon Dox treatment. Multiple doses resulted in a significant reduction in KDM3B and CTCF transcript and protein levels in RPE cells (Figure 6D-I); however, no change was observed with G9a transcripts (Figure S6C). We detected the same trend in KG1a cells, where Dox significantly reduced both KDM3B and CTCF transcript and protein levels (Figure 6J and S6D-F). Our observations are consistent with a prior report noting a loss of CTCF protein in Dox-treated patient-derived mammary epithelial cells.62 To assess whether this was specific to Dox, we treated RPEs with another topo II inhibitor, etoposide. Consistent with Dox, etoposide significantly reduced the protein levels of KDM3B and CTCF (Figure 6K), suggesting that these effects are a result of topo II inhibition.
Previous studies have shown that Dox treatment can activate the ubiquitin-proteasome system (UPS), leading to increased protein degradation.63 Therefore, we hypothesized that Dox could suppress KDM3B and CTCF levels through activation of the UPS. Consequently, cells were treated with Dox followed by the proteasome inhibitor MG132 before assessing KDM3B and CTCF protein levels (Figure 6L-M and S6G). Treatment with Dox and MG132 partially rescued the levels of both KDM3B (Figure 6L and S6G) and CTCF (Figure 6M and S6G) compared to Dox alone. Taken together, these data emphasize that Dox regulates KDM3B and CTCF levels through both transcriptional and post-transcriptional mechanisms.
KDM3B and CTCF regulation controls Doxorubicin-induced KMT2A amplification and rearrangement
Since KDM3B and CTCF are reduced upon Dox treatment, the imbalance in H3K9me1/2 and CTCF occupancy at KMT2A could be a key driver in promoting Dox-induced KMT2A amplification and genomic alterations. Consistent with this relationship, Dox treatment resulted in a similar increase in H3K9me1 at the KMT2A CTCF site (Figure 7A; upper graph comparing siKDM3B to Dox and S7A) and H3K9me2 at the flanking region when compared to KDM3B depletion (Figure S7A-B). Increased H3K9me1/2 was accompanied by reduced CTCF occupancy upon Dox treatment (Figure 7A, lower bar graph). Furthermore, CTCF overexpression was sufficient to prevent KMT2A copy gains upon Dox treatment (Figure 7B and S7C). CTCF overexpression appears to alter local chromosomal organization at the locus, with increased separation observed between red and green probe. Therefore we could not assess the impact on Dox-induced rearrangements as determined by the break apart events.
Figure 7. KDM3B and CTCF regulation controls Doxorubicin-induced KMT2A amplification and rearrangement.

(A) ChIP-qPCR demonstrating increase of H3K9me1 at KMT2A exon 11 (KMT2A CTCF site) following KDM3B siRNA depletion (left; upper) and Dox treatment at 1pg/μL for 24hr (right; upper). ChIP-qPCR demonstrating suppression of CTCF occupancy at KMT2A exon 11 (KMT2A ex 11; black) or a negative control for CTCF binding (CTCF negative site; yellow) following Dox treatment at 1pg/μL for 24hr (lower).
(B) Treatment schematic (upper) and DNA FISH (lower) demonstrating that Dox treatment causes KMT2A amplification and rearrangements. CTCF overexpression significantly rescues KMT2A amplifications.
(C) Treatment schematic (upper) and DNA FISH (lower) demonstrating that Dox treatment causes KMT2A amplification and rearrangement. G9a depletion significantly rescues KMT2A amplifications and rearrangements.
(D) Treatment schematic (upper) and DNA FISH (lower) demonstrating that Dox treatment causes KMT2A amplification and rearrangements that are significantly rescued with EHMT1/2 inhibition (EHMTi).
(E) Treatment schematic (upper) and DNA FISH (lower) demonstrating that Dox treatment causes Kmt2a copy gains in mouse cells isolated from the spleen, however, pretreatment with EHMT1/2 inhibitor (EHMTi) blocked the Dox-induced Kmt2a copy gains. The control region on chr 9 had no significant changes with any condition (Control 9).
(F) Treatment schematic (upper) and DNA FISH (lower) demonstrating that Dox treatment causes KMT2A amplification and rearrangements that are significantly rescued with KDM3B overexpression.
(G) Model summarizing the data from Figures 1-7. The model illustrates that KDM3B and CTCF are suppressed with Dox treatment, leading to increased H3K9 mono- and di-methylation and reducing CTCF occupancy, which in turn promotes KMT2A amplification and rearrangements (BA). G9a is critical in promoting the KMT2A copy gains and rearrangements.
Error bars represent the SEM. *p < 0.05 by two-tailed Student’s t-test.
Since Dox reduced KDM3B levels (Figure 6) and increased H3K9me1/2 in KMT2A (Figure 7A), we tested whether G9a depletion would prevent Dox-induced KMT2A changes. KMT2A alterations caused by Dox were completely rescued upon G9a depletion (Figure 7C and S7D-E). Furthermore, G9a inhibition significantly reduced Dox-induced KMT2A copy gains and prevented genomic alterations in human cells (EHMTi; Figure 7D and S7F). In addition, mice treated with EHMTi prior to Dox treatment did not generate Kmt2a DNA copy gains (EHMTi; Figure 7E), emphasizing the conserved importance of H3K9 methylation balance in promoting Dox-induced KMT2A alterations. In fact, transient KDM3B overexpression blocked KMT2A copy gains and rearrangements induced by Dox treatment (Figure 7F and S7G-H). Collectively, these results suggest that Dox suppresses KDM3B and CTCF protein levels, which drives the copy gains and rearrangements through KDM3B/G9a imbalance and CTCF displacement, establishing a potential mechanism to therapeutically target chemotherapy induced KMT2A rearrangements (Figure 7G).
Discussion
This study uncovered a molecular basis for therapy-induced amplification and rearrangement of KMT2A. Similar observations were noted for TCF3/E2A, another rearranged loci in leukemia.64-66 The findings reported in this study have broad implications because they: 1) establish that epigenetic regulation controls amplification and rearrangements; 2) set the stage to discover the secondary hit(s) required for the generation of oncogenic KMT2A fusions; and 3) identify potential biomarkers and therapeutic targets to consider during treatments with chemotherapy and to monitor in patients post treatment.
KDM3B depletion in relationship to KMT2A rearrangements and fusion partners
Currently, more than 100 known KMT2A rearrangement partners are documented.26 However, not all rearrangement events generate functional fusion proteins.67 These data suggest that the molecular mechanism(s) leading to the generation of KMT2A rearrangements, including those that do not generate translatable products, could be key to understanding tumors containing amplifications and rearrangements.
Based on our data, it is unlikely that KDM3B loss alone provides a major cellular fitness advantage for the inherited rearranged over the non-rearranged cells. This observation is not surprising since therapy-related AML has a latency period of up to 15 years after initial treatment with chemotherapy.29 These data suggest that KDM3B suppression or loss alone is likely just the first step necessary to promote or allow selection of the rearrangement events resulting in functional fusion proteins providing a cellular growth advantage. Studies show that non-homologous end-joining is required for topo II inhibitor driven leukemia-associated KMT2A rearrangements,57,68 suggesting that mis-regulated DNA damage response is another possible factor involved in generating/selecting for the leukemia-associated fusion events. However, additional influences could also potentiate the driver fusion events to emerge: cellular ageing, stress exposures, and/or acquired mutations.
KDM3B, 5q and KMT2A amplification and rearrangements
Not all del(5q) regions contain KDM3B.69 However, patients with del(5q) alone have a better prognosis compared to those presenting with del(5q) as well as other mutations or abnormalities.70 We suspect that additional gene mutations and/or the dysregulation of additional epigenetic regulators are likely required to promote copy gains and rearrangements of the oncogenic fusion partners, providing the secondary hit(s) necessary. Furthermore, a number of other candidate tumor suppressor genes have been identified within the del(5q) region who may also play an oncogenic role that is independent of generating KMT2A amplifications and rearrangements.33,71,72
Epigenetics, amplification, and in turn, rearrangements
When KDM3B was inhibited for short time intervals, the expected amplifications and break aparts at KMT2A locus were observed but resolved quickly with drug removal, highlighting their transient extrachromosomal nature (Figure S7I). However, upon longer treatment, these genomic events become inherited and are observed on the same chromosome or other chromosomes (Figure 3 and S7I). These data illustrate that the aberrant regulation of the epigenome promotes transient DNA amplifications that can be inherited when the stimuli is maintained through multiple cell divisions (Figure S7I). Therefore, we speculate that sustained amplification and break aparts are likely being incorporated into the genome through DNA damage repair pathways. Our data is consistent with prior proposed mechanisms.73 Future studies need to determine the exact integration sites and build complete sequence maps to identify the molecular features and pathways affiliated with the inherited amplifications. This study has now generated the roadmap to investigate these inherited genomic events.
Longer inhibition of KDM3B did not further increase the percent of cells within the population containing KMT2A alterations compared to short treatment (Figures 2-3). Yet when inherited-KMT2A cell lines were exposed to KDM3i for a short time (3h, 6h), a significant increase in KMT2A copy gains compared to the inherited baseline was observed (Figure S7J). Upon longer treatment (12h), KMT2A copy gains returned to baseline, suggesting that those cells containing genomic aberrations of KMT2A are being negatively selected for, while a new population of cells with these aberrations emerge. This model is supported by increased Annexin V in the inherited-KMT2A cell lines treated with KDM3i at the later time points (6h and 12h; Figure S7K). Consistent with a prior study,52 these data suggest that KMT2A-rearranged cells have increased susceptibility to KDM3B inhibition. This sensitivity provides a promising therapeutic window in KMT2A-rearranged cancers.
KDM3B and CTCF as a bridge to Dox-induced KMT2A amplification and rearrangement
Upon topo II inhibitor treatment (e.g., Doxorubicin), MDS and AML occur and are accompanied by amplification and rearrangement of the KMT2A locus.29 Topo II inhibitors promote non-leukemia and leukemia associated KMT2A rearrangements in various cell types,57,61 suggesting a universal regulatory mechanism controlling KMT2A alterations. A population-based study demonstrated that younger individuals developing secondary leukemia have a significantly worse prognosis compared to de novo.74 Therefore, preventing the emergence of secondary cancer caused by chemotherapy would have a profound clinical impact. This study demonstrates that epigenetic therapies could provide a much-needed tool to combat these cancers. Furthermore, we establish the possibility for controlling chemo-induced KMT2A amplification and rearrangements by pretreating or co-treating patients receiving these therapies with a G9a inhibitor or CTCF/KDM3B agonist. Collectively, these observations provide a molecular basis to develop treatment protocols to prevent therapy-associated KMT2A rearrangements by targeting epigenetic regulators.
Limitations of the study
Our findings establish that epigenetic mechanisms control the amplification and genomic rearrangements of KMT2A. We demonstrate that these events are directly promoted by Dox through suppression of KDM3B and CTCF protein levels, and can be blocked by co-depletion or inhibition of G9a/EHMT2. We also show that Dox suppresses KDM3B and CTCF protein levels through transcriptional and post-transcriptional mechanisms. However, our study does not (1) demonstrate the exact mechanism by which oncogenic KMT2A rearrangements are generated leading to leukemia development or (2) demonstrate exactly how Dox suppresses KDM3B and CTCF protein levels. While we have discovered the first hit required to generate leukemia-associated KMT2A rearrangements, the additional hit(s) required could be additional epigenetics perturbations, cellular ageing, stress exposures, acquired mutations, or alterations to DNA damage repair pathways. Future studies are needed to (1) systematically test the ability to promote oncogenic KMT2A fusion events driving leukemia, and (2) discover the exact mechanisms by which Dox suppresses KDM3B and CTCF protein levels.
STAR METHODS
Resource Availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Johnathan Whetstine (Johnathan.Whetstine@fccc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data
Original ChIP-sequencing data has been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-KDM3B, clone C69G2 | Cell Signaling | Cat# 3314; RRID: AB_1264294 |
| Anti-JMJD1B | Invitrogen | Cat# PA5-53459 |
| Anti-G9a | Sigma | Cat# G6919; RRID: AB_262007 |
| Anti-beta Actin | Millipore | Cat# MAB1501; RRID: AB626633 |
| Anti-Actinin | Santa Cruz | Cat# sc-17829; RRID: AB_626633 |
| Goat anti-mouse HRP | Biorad | Cat# 170-6516; RRID: AB11125547 |
| Goat anti-rabbit HRP | GenScript | Cat# A00167 |
| Anti-H3K9me1 | Abcam | Cat# ab8896 |
| Anti-H3K9me2 | Abcam | Cat# ab1220 |
| Anti-H3K9me3 | Abcam | Cat# ab8898 |
| Anti-CTCF, clone D31H2 | Cell Signaling | Cat# 3418 |
| Biological samples | ||
| Primary AML | Cihangir Duy 75 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Pierce Protease Inhibitor Tablets | Thermo Scientific | Cat# A32953 |
| Pierce Phosphatase Inhibitor Tablets | Thermo Scientific | Cat# A32957 |
| Propidium Iodide Solution | Sigma Aldrich | Cat# P4864 |
| Doxorubicin | Sigma Aldrich | ab142052 |
| Doxorubicin (Mouse study) | Selleckchem | Cat# E2516 |
| Dulbecco’s Modified Eagles Medium – High Glucose | Sigma Aldrich | Cat# D5648 |
| Roswell Park Memorial Institute Medium (RPMI)1640 | Sigma Aldrich | Cat# R6504 |
| Opti-Mem | Life Technologies | Cat# 31985070 |
| Trypsin (0.25%) EDTA | Life Technologies | Cat# 2520056 |
| L-Glutamine | Life Technologies | Cat# 25030-081 |
| Penicillin and Streptomycin | Life Technologies | Cat# 15140122 |
| Fetal Bovine Serum (FBS) | Gibco | Cat# 26140-079 |
| Lipofectamine 3000 | Life Technologies | Cat# L30000015 |
| EHMTi | Jian Jin | UNC0642 |
| KDM3i | Xu et al.52 | JDI-12 |
| Critical commercial assays | ||
| miRNeasy Mini Kit | Qiagen | Cat# 217004 |
| Superscript IV 1st Strand System | Life Technologies | Cat# 18091050 |
| CL-XPosure™ Film | Thermo Scientific | Cat# 34091 |
| Lumi-Light Western Blotting Substrate | Roche | Cat# 12015200001 |
| Pierce BCA Protein Assay | Thermo Scientific | Cat# 23223 and 23224 |
| Protein A Dynabeads | Thermo Scientific | Cat# 10002D |
| Protein G Dynabeads | Thermo Scientific | Cat# 10004D |
| TruSeq ChIP Sample Prep Kit Set A (48 samples) | Illumina | Cat# 10748010 |
| NextSeq® 500/550 High Output Kit v2 (75 cycles) | Illumina | Cat# FC-404-2005 |
| Bac-to-Bac™ Baculovirus Expression System | Thermo Scientific | Cat# 10359016 |
| Dead Cell Apoptosis Kits with Annexin V | Life Technologies | Cat# V13241 |
| Deposited data | ||
| Raw ChIP-sequencing | This paper | GEO: GSE210480 |
| KDM3B ChIP-seq | Li et al.51 | GEO: GSE71885 |
| RPE CTCF ChIP-seq | ENCODE | GEO: GSM749673 |
| GM12878 CTCF ChIP-seq | ENCODE | GEO: GSM733752 |
| H1 CTCF ChIP-seq | ENCODE | GEO: GSM733672 |
| bronchial_epithelial CTCF ChIP-seq | ENCODE | GEO: GSM749779 |
| Bcell CTCF ChIP-seq | ENCODE | GEO: GSM1003474 |
| HL-60 CTCF ChIP-seq | ENCODE | GEO: GSM749688 |
| HCT116 CTCF ChIP-seq | ENCODE | GEO: GSM1022652 |
| Experimental models: Cell lines | ||
| RPE-hTERT1 | Nick Dyson | N/A |
| U937 | FCCC Cell Culture Facility | CRL-1593.2 |
| HL60 | ATCC | CCL-240 |
| KG1a | ATCC | CCL-246.1 |
| HSPC | Cihangir Duy; 75 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 / 129/Sv | Jackson Labs | Strain# 101043 |
| Oligonucleotides | ||
| See Table S3 for Oligonucleotides used | This paper | N/A |
| Recombinant DNA | ||
| Halo-CTCF | Promega | Cat# FHC01807 |
| Halo-KDM3B | Promega | Cat# FHC05559 |
| Halo-Tag Alone | Promega | Cat# G6591 |
| MLL-1 Probe | Oxford Gene Technology | Cat# LPH 506-A |
| MLL breakapart | Oxford Gene Technology | Cat# LPH 013-A |
| AML1 breakapart | Oxford Gene Technology | Cat# LPH 027-SA |
| BCL6 breakapart | Oxford Gene Technology | Cat# LPH 035-SA |
| EVL1 breakapart | Oxford Gene Technology | Cat# LPH 036-SA |
| TCRB breakapart | Oxford Gene Technology | Cat# LPH 048-SA |
| MLLT1 | Oxford Gene Technology | Cat# LPH 508-A |
| MLLT3 | Oxford Gene Technology | Cat# LPH 509-A |
| Chromosome 11 Alpha Satellite Red | Oxford Gene Technology | Cat# LPE 011R-A |
| E2A breakapart | Oxford Gene Technology | Cat# LPH 019-SA |
| 5q del probe | Oxford Gene Technology | Cat# LPH 024 |
| 19p probe | Oxford Gene Technology | Cat# LPT19 PR-A |
| NMYC/LAF4 probe | Oxford Gene Technology | Cat# LPS-009A |
| MLL adjacent probe (CD3) | Empire Genomics | RPCI-11 215H18 |
| Kmt2a/Chr9 Mouse | Empire Genomics | Mouse KMT2A-Chr09 |
| Software and algorithms | ||
| Scaffold 6.0 | 3i-intelligent imaging Innovations | https://www.intelligent-imaging.com/slidebook |
| BWA 0.7.13 | Li et al.78 | https://biobwa.sourceforge.net/bwa.shtml |
| DeepTools 2.4.3 | Ramirez et al.79 | https://deeptools.readthedocs.io/en/develop/content/installation.html |
| HOMER v4.10.3 | Heinz et al.80 | http://homer.ucsd.edu/homer/ |
| DiffBind | Ross-Innes et al.81 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
Code
No original code was used in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell Culture
Retinal pigment epithelial (RPE) cells were cultured in DMEM-high glucose (Sigma) media supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100U/ml penicillin, 100μg/ml streptomycin, and 2mM L-glutamine. U937 cells were cultured in RPMI 1640 media supplemented with 10% heat-inactivated FBS, 100U/ml penicillin, 100μg/ml streptomycin and 2mM L-glutamine. HL60 and KG1a cells were cultured in RPMI 1640 media supplemented with 20% heat-inactivated FBS, 100U/ml penicillin, 100μg/ml streptomycin and 2mM L-glutamine. Cell line identities were authenticated by short tandem repeat analysis and Mycoplasma tested using the MycoAlert Detection Kit (Lonza, LT07-218). We are appreciative to the Cell Culture Facility at Fox Chase Cancer Center for their support.
Human primary patient-derived AML cases were obtained from Dr. Cihangir Duy’s laboratory75 and maintained in Iscove’s modified Dulbecco’s medium (IMDM; Thermo Fisher Scientific, Waltham, MA) containing 20% fetal bovine serum (Corning Premium FBS), 100 IU ml–1 penicillin, 100 μg ml–1 streptomycin, and 50 μM 2-mercaptoethanol. Cytokines, purchased from StemCell Technologies (Vancouver, BC, Canada), were added twice a week with SCF (50 ng ml–1), IL-3 (20 ng ml–1), IL-6 (20 ng ml–1), GM-CSF (20 ng ml–1), G-CSF (20 ng ml–1), and FLT-3 ligand (50 ng ml–1). For the AML organoid model, Human primary patient-derived AML cases were expanded using OP9 stroma feeder layers. OP9 feeder layers were generated using irradiation with 30 Gy before seeding the stroma cells on 0.01% poly-L-lysine-coated cell culture dishes to a confluency of 80-90%. The next day, patient-derived AML cells were seeded and maintained on the OP9 dishes in Iscove’s modified Dulbecco’s medium (IMDM; Thermo Fisher Scientific, Waltham, MA) containing 20% fetal bovine serum (Corning Premium FBS), 100 IU ml–1 penicillin, 100 μg ml–1 streptomycin, and 50 μM 2-mercaptoethanol. Cytokines were added twice a week with SCF (50 ng ml–1), IL-3 (20 ng ml–1), IL-6 (20 ng ml–1), GM-CSF (20 ng ml–1), G-CSF (20 ng ml–1), and FLT-3 ligand (50 ng ml–1). Expanding AML cells were transferred every 1–2 weeks after reaching a cell density of more than 1 million ml–1 onto fresh OP9 dishes supplemented with cytokines. For drug treatments, KDM3i (1μM) was supplemented directly to the media for 72 hrs.
Isolation of HSPCs
Mononuclear cells (MNC) were isolated from fresh human umbilical cord blood samples (New York Blood Center) using Ficoll (Atlanta Biologicals) density gradient centrifugation. These cells were obtained from Dr. Cihangir Duy’s laboratory. After lysis of red blood cells, HSPCs were selected via immunomagnetic enrichment of CD34+ MNCs using CD34 MicroBead Kit and Automacs from Miltenyi Biotech. HSPCs were maintained in Iscove’s modified Dulbecco’s medium (IMDM; Thermo Fisher Scientific, Waltham, MA) containing 20% fetal bovine serum (Corning Premium FBS), 100 IU ml–1 penicillin, 100 μg ml–1 streptomycin, and 50 μM 2-mercaptoethanol. OP9 feeder layers were generated using irradiation with 30 Gy before seeding the stroma cells on 0.01% poly-L-lysine-coated cell culture dishes to a confluency of 80-90%. The next day, HSPCs were seeded and maintained on the OP9 dishes in Iscove’s modified Dulbecco’s medium (IMDM; Thermo Fisher Scientific, Waltham, MA) containing 20% fetal bovine serum (Corning Premium FBS), 100 IU ml–1 penicillin, 100 μg ml–1 streptomycin, and 50 μM 2-mercaptoethanol. Cytokines were added twice a week with SCF (50 ng ml–1), IL-3 (20 ng ml–1), IL-6 (20 ng ml–1), GM-CSF (20 ng ml–1), G-CSF (20 ng ml–1), and FLT-3 ligand (50 ng ml–1). Cells were regularly selected for CD34+ to maintain a pool of HSPCs. For drug treatments, KDM3i (1μM) was supplemented directly to the media for 72 hrs.
Mouse Model Details
The Institutional Animal Care and Use Committees review board at Temple University (approval number 5025) approved all mouse experiments. Mice used in this study were obtained from the Jackson Laboratory and maintained at Temple University in conventional housing with LabDiet 5053 irradiated food and Hydropacs filter sterilized water available ad libitum, on a 12L:12D light cycle, in 30-70% humidity with 10-15 air exchanges per hour. For the in vivo Doxorubicin treatment in Figure 6C, 3-5 months old mice of B6129SF1/J strain (The Jackson Laboratory 101043) were used. For the in vivo combination treatment in Figure 7E, 8-10 weeks old mice of B6129SF1/J strain (The Jackson Laboratory 101043) were used. Both male and female mice were tested in all conditions, and sex did not influence the results of the study.
Method details
Transfection Procedure for RPE cells
Cells were plated in 10 cm cell culture dishes and allowed to adhere for 16-20 hours. Cell culture medium was removed, cells were rinsed with phosphate buffered saline (PBS) and then replaced with OPTI-MEM medium (Life Technologies) prior to siRNA transfections (5nM-10nM/transfection). Transfections were changed to complete cell culture media after 4 hrs of transfection, and cells were collected 72 hrs post transfection. Transient overexpression transfections with 1-2μg of plasmid were performed using Lipofectamine 3000 transfection reagent and P3000 reagent (Life Technologies) in OPTI-MEM medium for 4 hrs, followed by changing to complete DMEM media for 24 hrs before collection. Silencer select negative controls and siRNAs were purchased from Life Technologies. Their sequences and unique identification numbers are tabulated in Key Resources Table. For Figures 4A and S4A-G, cells were first transfected with siEHMT1/G9a siRNAs for 24 hrs followed by KDM3B siRNA transfection for 48hr. For co-transfection experiments (Figures 5B,J, Figures S5A-B, H-l), both the siRNAs were transfected at the same time and collected at 72 hrs from transfection.
Transfection procedure for U937, HL60 and KG1a cells
U937, KG1a and HL60 were transfected using Neon System (Invitrogen) following manufacturer’s instructions. 500,000 cells were mixed with 10nM siRNA constructs in 10μl of supplied buffer. Cell mixture was loaded in Neon syringe and submerged in electrode buffer. For U937 cells, 3 pulses of 1400mV at 10ms was applied. For HL60 cells, 1 pulse of 1350 mV at 35ms was applied. For KG1a cells, 1 pulse of 1650mV at 20ms was applied. Cells were immediately transferred into fresh media in 6 well plates and allowed to grow for 72 hours.
Long-term passage of siRNA transfected cells
Control siRNA transfected and KDM3B siRNA transfected cells were considered as passage 0 (P=0) 72 hrs post transfection. After 72 hours post transfection, the 2.5X105 cells were plated and cultured for 72 hrs as passage P=1. The cells were subsequently plated, passaged and harvested at indicated passage numbers. For example, passage 3 is ~9 days in culture and ~9 doublings for RPEs and ~6 doublings for U937 cells, respectively.
RNA extraction and quantitative real-time PCR
Cells were washed and collected by trypsinization after two PBS washes. Cell pellet was resuspended in Qiazol reagent (QIAGEN) for lysis and stored at −80°C before further processing. Total RNA was extracted using miRNAeasy Mini Kit (QIAGEN) with an on-column DNase digestion according to the manufacturer’s instructions. RNA was quantified using NanoDrop 2000 or One (Thermo Scientific). Single strand cDNA was prepared using Super Script IV first strand synthesis kit (Invitrogen) using random hexamers. Expression levels were analyzed using FastStart Universal SYBR Green Master (ROX) (Roche) according to the manufacturer’s instructions on a LightCycler 480 PCR machine (Roche) or QuantStudio 5 Real-time PCR machine (Applied Biosystems). Samples were normalized to β-actin. Primer sequences are provided in Key Resources Table.
Immunoblotting
Cells were trypsinized and washed two times with PBS before resuspending in RIPA lysis buffer [50mM Tris pH 7.4, 150mM NaCl, 0.25% Sodium Deoxycholate, 1% NP40, 1mM EDTA, 10% Glycerol] freshly supplemented with Pierce Protease and Phosphatase inhibitor cocktails (ThermoFisher). Cells were lysed on ice for 15 min and stored at 80°C until further processing. Lysates were sonicated for 15 min (30sec ON and 30sec OFF cycle) at 70% amplitude in QSonica Q700 sonicator (Qsonica) followed by centrifugation at 12,000rpm for 15min. Cell lysate was transferred to a fresh tube and protein quantification was performed with Pierce BCA reagent (Thermo Scientific). Equal amounts of proteins were separated by SDS gel electrophoresis and transferred on nitrocellulose membrane (BioTrace NT, Pall Life Sciences) for at least 3 hrs at a constant current. The membranes were blocked for at least 1 hr in 5% BSA-PBST (1X PBS with 0.5% Tween-20) or 5% milk-PBST and probed over night with specific antibodies as follows at the following dilutions: anti-KDM3B (Cell Signaling) (1:1000); anti-β-Actin (Millipore) (1:10,000); anti-G9A (Sigma) (1:5000); anti-Actinin (Santacruz) (1:2000). Catalog numbers for all antibodies used in this study can be found in the Key Resources Table. Membranes were washed three times in PBST the next day, incubated with goat anti-mouse IgG peroxidase conjugated secondary antibody (170-6516, Biorad) or goat anti-rabbit peroxidase conjugated secondary antibody (A00167, GenScript) at 1:2500 in 5% milk-PBST for at least 1hr at room temperature, washed 3 times with PBST and incubated in Lumi-Light western blotting substrate (12015200001, Roche) 1min. Membranes were developed with CL-XPosure Films (34091, Thermo). The western blot images displayed in the figures have been cropped and auto-contrasted. Protein levels were quantified using ImageJ and normalized to the corresponding control protein levels.
Cell Cycle Analysis
Samples were washed with PBS, centrifuged at 1400rpm for 5 min, and permeabilized with 500mL PBS containing 0.5% Triton X-100 for 30 min. After this incubation, cells were washed with PBS and centrifuged at 1400rpm for 5 min. Samples were then stained with 1:100 dilutions of 1mg/mL PI solution and 0.5M EDTA with 100 mg RNase A, overnight at 4°C. Cell cycle distribution was analyzed by flow cytometry using the LSRII flow cytometry system (BD Biosciences). We are grateful to the Cell Sorting Facility at Fox Chase Cancer Center for assistance with flow cytometry.
DNA Fluorescent In Situ Hybridization (FISH)
The FISH protocol was performed as described previously in7. Briefly, cell suspensions were fixed in ice-cold methanol:glacial acetic acid (3:1) solution for a minimum of four hours, before being centrifuged onto 8 Chamber Polystyrene vessel tissue culture treated glass slides (Falcon, Fisher Scientific) at 900rpm. The slides were air-dried and incubated in 2X SSC buffer for 2 min, followed by serial ethanol dilution (70%, 85% and 100%) incubations for 2 min each, for a total of 6 min. Air-dried slides were hybridized with probes that were diluted in appropriate buffer overnight at 37°C. The slides were washed the next day for 3 to 4 mins in appropriate wash buffers at 69°C with 0.4X SSC for Cytocell probes, Agilent Buffer1 for Agilent probes, or 0.4X SSC + 0.3% NP-40 for Empire Genomic probes followed by washing in 2X SSC with 0.05% Tween-20 (Cytocell probes), Agilent Buffer 2 (Agilent) or 2X SSC+0.1% NP-40 (Empire). The slides were incubated in 1mg/mL DAPI solution made in 1% BSA-PBS, followed by a final 1X PBS wash. After the wash, the slides were mounted with ProLong Gold antifade reagent (Invitrogen).
FISH images were acquired using an Olympus IX81 or Olympus IX83 spinning disk microscope at 40X magnification and analyzed using Slidebook 6.0 software. A minimum of 20 z-planes with 0.5μm step size was acquired for each field. Copy number gains for MLL1,11C, NMYC/LAF4 were scored in RPE cells as three or more foci. For MLL break apart probe, copy gains were scored as 3 or more foci for the N terminus flanking probe (green) and C terminus flanking probe (red). Complete separation of red and green probe with no overlap was called break apart for the MLL locus, TCF3 locus and any other locus FISHed with dual break apart probe. A minimum of 200 nuclei are scored for each independent experiment unless otherwise specified. Extended list of probes used are provided in the Key Resources Table.
Metaphase Spreads
RPE cells were transfected with siRNAs and passed 3 times. Cells were seeded for 48 hours. The cells were treated with KaryoMAX colcemid solution (Gibco) at a final concentration of 2μg/mL for 3 hrs and were collected by mitotic shake off, washed with 1X PBS followed by 0.59% KCl (w/v) hypotonic solution for 40 mins for expansion and swelling. The reaction was terminated by addition of 3:1 solution of cold methanol:acetic acid, followed by 4 washes. The cells were then resuspended in fixative solution. The cells were pipetted and dropped on a glass slide from a height of 12-15 inches to make the metaphase spread. FISH was performed for the indicated probes post drying of the slides. The images were taken with 30 z-planes with 0.5 μm step size using the Olympus IX83 microscope. The images were analyzed using Slidebook 6.0 software.
Protein purification
Human full length of KDM3A, KDM3B and KDM5A were cloned into pFastbac1 with flag tag at N-terminal, then Bacmid were made to produce baculovirus in insect cells (sf9) using the Bac-to-Bac™ Baculovirus Expression System (Thermo Scientific). Cells were harvested 42 hours after baculovirus infection by centrifugation (2000rpm, 4 degree), then the pellet was lysed in lysis buffer (Tris-HCL(PH7.5) 25mM, NaCl 300mM, Triton 0.1%, PMSF 1mM, DTT 1mM) by sonication and centrifuged at 14000rpm for 30 minutes. The supernatant was incubated with Flag-M2 agarose beads for 5 hours, unbound proteins were removed by washing the beads with lysis buffer 4 times, and the proteins enriched were eluted with 3Xflag peptide (0.15mg/ml) diluted in elution buffer (Tris-HCL(PH7.5) 25mM, NaCl 150mM, DTT 1mM). Proteins purified were used for SDS-PAGE and biochemical reaction analysis. Coomassie staining of KDM3B and KDM5A in Figure S1D was spliced to place the KDM3B and KDM5A lanes next to the ladder due to other purified proteins being placed between them.
Histone demethylase reactions
400ng KDM3A, KDM3B and KDM5A were incubated with 1μg bulk histones (Histone from calf thymus) or 3.3μM H3K9me2 peptide in 30μl reaction system at 27 C for 5 hours. Reaction buffer: Hepes(PH7.5) 50mM, 2-OG 50μM, Fe(NH4)2(SO4)2 50μM, Sodium L-Acorbate 400μM, TCEP 1mM.
Recombinant human KDM3B/JMJD1B protein (abcam ab271569) was incubated with 1μM KDM3i, 1M Tris, 5M NaCl, 10mM Asorbic Acid, 10mM α-Ketoglutarate, 10mM Fe(NH4)2(SO4)2(H2O)6 at 27 C, 30 minutes. 1μl_ Histone from Calf Thymus (1mg/mL) in H2O was added to make reaction 100μL then incubated at 27 C for 5 hrs. 4X Laemmli Loading buffer with 5% β-Mercaptoethanol was added to reaction and then heated 95 C for 10 mins. Samples were snap frozen and then used for western blots.
Digital Droplet PCR
The Digital Droplet PCR (ddPCR) was performed using 10 μL of 2 × ddPCR Supermix for Probes (no dUTP) (Bio-Rad), 900 nM of each primer, 250 nM probe, 50 ng of digested DNA template using HindIII restriction enzyme (NEB) and r2.1 Buffer (NEB), and nuclease free water to a total volume of 20 μL. The QX200 droplet generator (Bio-Rad) was used to generate the droplet mixture. The droplet mixture was then transferred to a PCR reaction plate and amplified with the following conditions: denaturation of 95 °C for 10 min, followed by 40 cycles of a two-step thermal profile consisting of 95 °C for 15 s and 60 °C for 60 s, then incubated at 98 °C for 10 min and cooled to 8°C until the droplets were read. Once complete, the plate was transferred to the QX200 droplet reader (Bio-Rad) and analyzed for copy number variation (CNV). The number of positive (high level of fluorescence) and negative (low and constant level of fluorescence) droplets obtained were analyzed using QuantaSoft software (Bio-Rad, Pleasanton, CA, USA). Ratios of KMT2A to CD3E gene were used to determine copy number. Primer and probe sequences are provided in Key Resources Table.
Drug Treatment Conditions
For Doxorubicin treatment, RPE cells were plated in 10 cm tissue culture plates at a density of 2.5x105. Cells were allowed to adhere for approximately 16 hrs before Doxorubicin (Sigma) (dissolved in DMSO) was supplemented to media in different concentrations. Final concentrations used were 5, 2.5 and 1pg/μl. Cells were cultured in Doxorubicin for a total of 72 hrs before harvesting. For Figures 7B,F and S7C,G cells were transfected with KDM3B or CTCF plasmid for 4 hrs. After removal of transfection mixture, cells were supplemented with 1 pg/μl Doxorubicin supplemented media and cultured for 20 hours before harvesting. For Figures 7C and Figure S7D-E, Doxorubicin was supplemented to the media 48hrs post G9a transfection and 24 hrs before harvesting. For Figure 6J and Figure S6D-F, KG1a cells were cultured at a density of 2.5x105/ml before Doxorubicin was added at a concentration of 60pg/μl for a total of 72 hrs before harvesting.
For JDI12 (KDM3i) treatment, KDM3i52 was synthesized for these studies. 3x105 RPE cells were plated in 10cm tissue culture plates. Cells were allowed to adhere to the plate for a minimum of 24 hrs before KDM3i (dissolved in DMSO) was supplemented to media at 25nM unless specified differently. For cancer and primary cell lines in Figures 2B-E, cells were treated with 1 μM KDM3i. Cells were cultured for a total of 72 hrs before harvesting. For washout experiment (Figure 2F), cells were allowed to adhere 24 hrs before treatment with JDI12/KDM3i. Cells were treated for 12 hrs before media was removed, plates were washed with 1X PBS, and cells were either harvested or fresh complete media was added back to the plate without KDM3i. For passage experiments, RPE cells were plated at 1.5x105 cells. Cells were allowed to adhere to the plate for 24 hrs before KDM3i was supplemented to media at 25nM. Cells were cultured in KDM3i for 72 hrs further before being harvested and passaged at 3x105 per 10cm plate. Cells were passaged every 3 days and seeded at the same amount each passage.
For KG1a, Primary AML, AML Organoid, and HSPC KDM3i treatments in Figure 2, media was supplemented with 1μM KDM3i for 72 hrs before harvesting as described in the DNA FISH methods section.
For KDM3i + EHMTi (UNC0642) experiments, cells were seeded at 3x105 in 10cm tissue culture plates and allowed to adhere for 60 hrs before media was supplemented with KDM3i (25nM) and EHMTi (2.5μM). Cells were harvested 12 hrs post treatment. For siCTCF + EHMTi experiments, cells were seeded in 10cm tissue culture plates at 2.1x105 and allowed to adhere for 24 hrs before following the transfection procedure described above. 24 hrs post-transfection, media was supplemented with 1.5 μM EHMTi. Cells were cultured for a further 48 hrs (72 hrs total transfection) before being harvested. For Dox+EHMTi experiments, cells were plated at 1.5x105 and allowed to adhere for 24 hrs. EHMTi was supplemented to media at a final concentration of 1.5μM. 48 hrs later Dox was supplemented to the media at 1pg/μl. 24 hrs after Dox supplement (72 hrs total EHMTi, 24 hrs total Dox), cells were harvested.
For MG132 treatment, cells were seeded at 1.5x105 in 10cm tissue culture plates and allowed to adhere for 24hrs before media was supplemented with 5pg/μL of Dox. 48hrs later, 10μM MG132 was supplemented to the media. 24hrs after MG132 addition, plates were placed on ice, media was collected and cells were scraped into media before being washed twice in PBS and resuspended in RIPA lysis buffer.
Chromatin Immunoprecipitation
Chromatin was prepared and ChIP were performed as described in76. Specifically, sonication of chromatin was done with the Qsonica Q800R2 system (Qsonica). RPE cells were seeded in 10cm plates. At ~80% confluence, crosslinking of the cells was done by adding 1% formaldehyde to the media for 13 min at 37°C and stopped with 0.125M glycine, pH2.5. Plates were washed with ice cold PBS and scraped off, followed by centrifugation at 800 rpm for 2 min at 4°C. The pellet was resuspended in cellular lysis buffer (5mM PIPES pH8.00, 85mM KCl, 0.5% NP40) supplemented with protease inhibitors, incubated 5 min on ice and centrifuged at 800 rpm, 2 min at 4°C. The pellet was resuspended in nuclear lysis buffer supplemented with protease inhibitors (NLB, 50mM Tris, pH 8.0, 1.0% SDS, or 0.2% SDS for CTCF ChIP).
Chromatin was sonicated at 70% amplitude 15sec on 45sec off setting for 35 min or 45 min for CTCF ChIP. 5 μL of chromatin was RNase treated, and reverse cross-linked at least 4 hrs at 65°C in presence of proteinase K. DNA was isolated by phenol:chloroform extraction and checked on 1.3% agarose gel for a smear below 300bp. Chromatin was precleared by centrifugation at 14,000rpm for 10min at 4°C. Chromatin concentration was then quantified on a NanoDrop One. For each IP, 1-10μg of chromatin was immunoprecipitated with 0.2-2μg of antibody in dilution IP buffer (16.7mM Tris pH 8.0, 1.2mM EDTA pH 8.0, 167mM NaCl, 0.2% or 0.1% SDS, 0.24% Triton-X-100 or 1.84% for CTCF ChIP) at 4°C overnight. % SDS for dilution IP depended on % SDS used in Nuclear Lysis Buffer. Final concentration for IP was always 0.2% SDS. Chromatin was precleared for 2 hrs each with protein A agarose and magnetic protein A or protein G beads (Invitrogen; to match antibody isotype) rotating at 4°C before immunoprecipitation. The immunoprecipitated material was washed 2 times in dilution IP buffer, 1 time in TSE buffer (20mM Tris pH 8.0, 2mM EDTA pH8.0, 500mM NaCl, 1% Triton X-100, 0.1% SDS), 1 time in LiCI buffer (100mM Tris pH 8.0, 500mM LiCI, 1% deoxycholic acid, 1% NP40) and 2 times in TE (10mM Tris pH 8.0, 1mM EDTA pH 8.0) before elution in elution buffer (50mM NaHCO3, 140mM NaCl, 1% SDS) with RNase treatment, followed by 10μg proteinase K at 1 hr 55°C 1000 rpm. The samples were removed from beads and reverse cross-linked at 65°C for 4 hrs. Immunoprecipitated DNA was purified using either PCR purification columns (Promega) or AMPureXP beads. All the ChIPs were performed with at least two independent chromatin preparations from two independent siRNAs or two independent RPE cell lines. Antibodies used for ChIP are as follows: H3K9me1 Abcam ab8896-100, H3K9me2 Abcam ab1220, H3K9me3 Abcam ab8898, CTCF (D31H2) Cell Signaling #3418. ChIP sequencing libraries were prepped using the TruSeq ChIP Sample Preparation kit (Illumina). Libraries were single-end sequenced (75 cycles) using a NextSeq500 (Illumina). ChIP-qPCR in Figures 5E and 7A was performed with 1μl of ChIP DNA with the following primers: (KMT2A Ex11) Forward - 5’-TCTGTCACGTTTGTGGAAG-3’, Reverse - 5’-GCCCAGCTGTAGTTCTATTAC-3’. (CTCF negative site) Forward - 5’-GAATCAGACTGAGACCCTAAAC-3’, Reverse 5’-GCCAATCCAGTCTTCTCATAC-3’. ChIP-qPCR in Figure S7B was performed with 1μl of ChIP DNA with the following primers: (KMT2A CTCF flanking site) Forward - 5’-CAGCCAGAATCCCAGTAGA-3’, Reverse 5’-CTTTCAGAGGAGGCTACAGA-3’.
In vivo Doxorubicin
For the in vivo Doxorubicin treatment in Figure 6C, 3-5 months old mice of the mixed background (C57BL/6 and 129/Sv) 4 males and 4 females were used. The mice were randomly assigned to two groups – Dox (treated with 1.5mg/kg doxorubicin (Selleckchem) at the total volume of 100μl administered via i.v.) and control (treated with 100μl saline i.v. for 3 days). 24hrs after the last dose was administered the mice were euthanized. The spleen was isolated, any connective tissue was trimmed, and the organ was passed through 70μm cell strainer to receive suspension of single cells. Red blood cells were lysed using ACK buffer (Gibco) for 5 min on ice, then washed twice with PBS. Cells were then fixed for DNA FISH as described above.
For the in vivo combination treatment in Figure 7E, Doxorubicin (Selleckchem) was solubilized in saline and EHMTi was first solubilized in DMSO to 100 mg/ml and then a stock solution was prepared in saline. 7 male and 8 female mice of B6129SF1/J strain (The Jackson Laboratory 101043) were assigned into 4 groups treated with: i) vehicle (n=4, two males, two females); ii) 4 doses of EHMTi (daily, i.p. 5mg/kg) (n=4, two males, two females); iii) 3 doses of Doxorubicin (daily i.v. 1.5 mg/kg) (n=4, two males, two females); iv) 4 doses of EHMTi with 3 doses of Doxorubicin implemented into the treatment starting day 2 (n=3, one male, two females). The day after the final treatment, mice were euthanized, and the cells were isolated from spleen for double blinded examination by Kmt2a and control FISH (Control 9).
Annexin V Staining
3x105 control or KMT2A inherited cells were seeded in a 10 cm plate and grown asynchronously for 72 hours. Cells were treated with 25nM of KDM3i for 12, 6, or 3 hours before prior to harvesting. Collected cells were processed using ALEXA FLUOR 488 conjugated Annexin V and PI staining following the manufacturer’s instructions (Life Technologies). Briefly, 1X annexin-binding buffer and 100 μg/mL PI solution were prepared. Harvested cells were washed once with 1X PBS before being resuspended in 1X annexin-binding buffer at a density of 1x10 6 cells/mL in 100 μL. 5 μL of FITC Annexin V and 1 μL of the 100 μg/mL PI solution were added to each 100 μL cell suspension. Cells were incubated at room temperature for 15 minutes and then 400 μL of 1X annexin-binding buffer was added to each sample. Stained cells were analyzed by flow cytometry, measuring fluorescence at 530 nm and 600 nm. Data was collected on a BD Biosciences Symphony A5 flow cytometer and analyzed using FACSDiva software.
Quantification and Statistical Analysis
DNA FISH quantification
All pairwise comparisons were done using two-tailed Student’s t-test unless otherwise stated. Significance was determined if the p value was ≤ 0.05. All FISH experiments were carried out with at least two independent siRNAs and at least 200 nuclei per replicate were counted for all the FISH studies conducted unless otherwise stated. All FISH studies had a minimum of 2 replicates, and therefore at least 400 nuclei were scored for each panel unless otherwise stated. All error bars represent the SEM.
ChIP-seq analysis
ChIP-seq analysis was performed as previously described9,76,77. Sequencing reads were aligned against the human hg19 reference genome using BWA78. Alignments were filtered for uniquely mapped reads and duplicates were removed. Input-normalized ratio coverage tracks were generated using Deeptools79. Peaks were called using HOMER80 with default parameters.
For genome-wide analyses of H3K9me1 and H3K9me2 densities, we calculated input-normalized densities at all 10 Kb bins across the genome using bamcompare function of DeepTools. For genome-wide analysis of CTCF peaks, we mapped CTCF ChIP-seq reads using BWA and normalized ChIP read density by input using bamcompare function of DeepTools. Peaks were called using the Homer package80. Differential peaks between control and KDM3B knockdown were identified using DiffBind package81, with cutoffs of >2-fold change in KDM3B knockdown compared to control. We used the resulting ~17000 decreasing CTCF peaks to compare the ChIP-seq density of H3K9me1 and H3K9me2 between control and KDM3B knockdown cells at two types of regions, the vicinity of each CTCF peak (10 Kb region of ± 5 Kb from the peak center) and two flanking 10 Kb regions to the left and right of this central region.
Genome-Wide analysis of CTCF peaks
CTCF ChIP-seq reads were mapped using BWA and normalized ChIP read density by input using bamcompare function of DeepTools. Peaks were called using the Homer package80. Differential peaks between control and KDM3B knockdown were identified using DiffBind package81, with additional cutoffs of >2-fold change and P-value < 0.05. The resulting set of differential peaks was used to compare the ChIP-seq density of H3K9me1 and H3K9me2 between control and KDM3B knockdown cells at the vicinity of each CTCF peak (10 Kb region of ± 5 Kb from the peak center).
Supplementary Material
Supplemental Figure 1. KDM3B depletion induces KMT2A DNA copy gains and break aparts. Related to Figure 1
(A) qRT-PCR analysis was performed to validate specific gene siRNA knockdowns for the KDM3 family. Samples were normalized to β-actin.
(B) Western blot demonstrates that KDM3B protein levels are reduced when compared to controls using two different siRNAs in RPE cells.
(C) FACS analysis was performed after KDM3 family member siRNA depletion after 72 hrs.
(D) Coomassie gel of purified KDM enzymes used in the demethylase assay.
(E) Western blot of histones after being incubated with purified KDM3A,B and KDM5A. CTH alone- calf thymus histones alone
(F) MALDI-TOF analyses of H3K9me2 peptides incubated with KDM3A or KDM3B.
(G-N) DNA FISH for various genomic regions tested upon KDM3B siRNA depletion. Only TCF3/E2A (M) and AFF3/LAF4 (N) has significant copy gains and break aparts, or copy gains, respectively, upon KDM3B depletion in RPE cells when compared to the other loci tested.
(O) DNA FISH quantification for Subtel19 (appropriate control for M) that does not change upon KDM3B siRNA depletion
(P) Western blot shows decreased KDM3B protein levels in U937 cells upon siKDM3B.
(Q) DNA FISH quantification for TCF3/E2A demonstrates no significant copy gains or break aparts in KDM3B siRNA depleted U937 cells.
(R) DNA FISH quantification for AFF3/LAF4 demonstrates no significant copy gains in KDM3B siRNA depleted U937 cells.
(S) qPCR validation shows decreased KDM3B transcript levels compared to control in KDM3B siRNA depleted cells. These cells were used for experiments represented in Figure 1L-M and S1T-V.
(T) Genome-wide effects of siKDM3B on H3K9me1 levels. Scatterplots shows all 10 Kb bins across the genome, with input-normalized H3K9me1 ChIP-seq densities in two conditions at each bin represented as a point. Red points, 10 Kb bins overlapping the central region of KMT2A gene (containing exons 8-14: chr11 bins 118.35-118.36 and 118.36-118.37 Mbp). Green points, 10 Kb bins overlapping the 5’ region of KMT2A (exons 1-8: chr11 bins 118.30-118.31, 118.31-118.32, 118.32-118.33, 118.33-118.34 and 118.34-118.35 Mbp). Blue points, 10 Kb bins overlapping the 3’ region of KMT2A (exons 14-36: chr11 bins 118.37-118.38, 118.38-118.39, and 118.39-118.40 Mbp). Purple points, 10 Kb bins overlapping with TCF3 (chr11 bins 1.61-1.62, 1.62-1.63 Mbp). Orange point, 10 Kb bin overlapping with AFF3 (chr2 bin 100.17-100.18 Mbp). Genomic positions are based on GRCh37/hg19.
(U) Genome-wide effects of siKDM3B on H3K9me2 levels. Scatterplots shows all 10 Kb bins across the genome, with input-normalized H3K9me2 ChIP-seq densities in two conditions at each bin represented as a point. Colored points correspond to the bins listed in (S1T). Red points, 10 Kb bins overlapping the central region of KMT2A gene (containing exons 8-14). Green points, 10 Kb bins overlapping the 5’ region of KMT2A (exons 1-8). Blue points, 10 Kb bins overlapping the 3’ region of KMT2A (exons 14-36). Purple points, 10 Kb bins overlapping with TCF3. Orange point, 10 Kb bin overlapping with AFF3. Genomic positions are based on GRCh37/hg19.
(V) Genomic tracks of input-normalized ChIP-seq densities of H3K9me1-3 and the public input-normalized KDM3B ChIP-seq density 51 for the region flanking TCF3/E2A upon depletion of KDM3B.
(W) Genomic tracks of input-normalized ChIP-seq densities of H3K9me1-3 and the public input-normalized KDM3B ChIP-seq density 51 for the AFF3 region upon depletion of KDM3B.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 100 nuclei were scored per replicate per experiment. BA- Break aparts.
Supplemental Figure 2. KDM3B chemical inhibition (KDM3i) promotes transient KMT2A copy gains and break aparts. Related to Figure 2
(A) In vitro KDM assay with commercial full length purified KDM3B protein incubated with histones and western blotted for KDM3B, H3K9me1 and H3K9me3. Blot demonstrates reduction in H3K9me1 levels upon incubation with KDM3B is rescued with KDM3i addition.
(B) Cell growth assay for KDM3i treated RPE cells. Treatment with KDM3i at 25nM does not impact cell growth, whereas 250nM and 1μM modestly but significantly suppresses growth.
(C) Western blot demonstrating KDM3B protein levels are not reduced with KDM3i treatment in RPE cells. A representative example from quantification in panel D.
(D) Quantification of western blots for KDM3B in KDM3i treated RPE cells (n=3). KDM3B levels are not reduced upon treatment with KDM3i.
(E) Cell growth assay for KDM3i treated RPE cells corresponding to Figure 2F. Cell growth is not impacted after 12hrs KDM3i treatment, or after 12hr KDM3i washout.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted.
Supplemental Figure 3. KDM3B suppression leads to integration and inheritance of KMT2A copy gains and break aparts. Related to Figure 3.
(A) Copy Number Variation analysis using Digital Droplet PCR (ddPCR) shows KMT2A copy number increase in later passage of the inherited KMT2A cell lines used in Figures 3G-H. Copy number is quantified by computing ratio of KMT2A to CD3E reference gene (4 biological replicates with 2 technical replicates).
(B) Quantification of DNA FISH for TCF3/E2A after passaging KDM3B siRNA depleted RPE cells. TCF3/E2A copy gain and break aparts are retained after 15 passages in KDM3B siRNA depleted cells.
(C) DNA FISH for xSubTel19 after passaging KDM3B siRNA depleted RPE cells. Subtel19 copy gain does not change in KDM3B siRNA depleted cells.
(D) DNA FISH for AFF3/LAF4 after passage KDM3B siRNA depleted RPE cells. AFF3 copy gains occur at PO, but are not retained after 3 passages in KDM3B siRNA depleted cells. NMYC copy gain does not change.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 200 nuclei were scored per replicate per experiment. BA- Break aparts.
Supplemental Figure 4. H3K9me1 balance controls KMT2A copy gains and break aparts. Related to Figure 4.
(A) qRT-PCR analysis was performed to validate gene specific siRNA knockdowns. Samples were normalized to β-actin.
(B) Western blot images demonstrating successful siRNA-mediated depletion of KDM3B and G9a protein in RPE cells.
(C) FACS analysis evaluating cell cycle dynamics was performed for the siRNA mediated co-depletion experiments.
(D) Cell growth assays for siRNA co-depletion of KDM3B, EHMT1, and/or G9a.
(E) DNA FISH quantification for siRNA co-depletion of KDM3B, EHMT1, and/or G9a. siRNA depleted G9a but not EHMT1 prevents TCF3/E2A copy gains and break aparts upon KDM3B depletion in RPE cells.
(F) DNA FISH quantification of SubTel19 (control for TCF3 region) for siRNA co-depletion of KDM3B, EHMT1, and/or G9a.
(G) DNA FISH quantification for siRNA co-depletion of KDM3B, EHMT1, and/or G9a. siRNA depleted G9a but not EHMT1 prevents AFF3/LAF4 copy gains upon KDM3B depletion.
(H) Cell growth assays for KDM3/EHMT inhibitor versus control treatments corresponding to Figure 4B.
(I) FACS analysis evaluating cell cycle dynamics was performed for cells with KDM3B and/or G9a overexpression.
(J) qRT-PCR analysis for KDM3B and G9a overexpression in RPE cells. Samples were normalized to β-actin.
(K) qRT-PCR analysis was performed to validate gene specific siRNA knockdowns in HL60 cells. Samples were normalized to β-actin.
(L) qRT-PCR analysis was performed to validate gene specific siRNA knockdowns in WT versus Inherited KMT2A cells. Samples were normalized to β-actin. Two independent stable cell lines were used per condition with two independent siRNAs.
(M) Across the genome, the increase of H3K9me1 and H3K9me2 caused by siKDM3B was mostly rescued by double KDM3B and G9a knockdown. Barplots showing the fractions of the genome (based on 10 Kb genomic bins) that increased H3K9me1 (left) and H3K9me2 (right) upon KDM3B knockdown. Red, genomic fraction where this increase was rescued by double knockdown.
(N) Genome-wide effects of siKDM3B, siG9a, and double knockdown on H3K9me1 levels. Scatterplots shows all 10 Kb bins across the genome, with input-normalized H3K9me1 ChIP-seq densities in two conditions at each bin represented as a point. KDM3B and G9a knockdowns have opposite skews, whereas the double knockdown strongly reduces these H3K9me1 changes. Left, scatterplot comparing H3K9me1 densities in control vs siKDM3B; H3K9me1 changes are skewed towards increase (points above upper red line corresponding to > 1.5 fold increase in siKM3B cells). Middle, scatterplot for control vs siG9a cells; H3K9me1 changes are skewed towards decrease (points below lower red line corresponding to > 1.5 fold decrease in siG9a cells). Right, scatterplot for control vs siKDM3B + siG9a cells, with much fewer H3K9me1 changes in either direction. Red points, 10 Kb bins overlapping the central region of KMT2A gene (containing exons 8-14: chr11 bins 118.35-118.36 and 118.36-118.37 Mbp). Green points, 10 Kb bins overlapping the 5’ region of KMT2A (exons 1-8: chr11 bins 118.30-118.31, 118.31-118.32, 118.32-118.33, 118.33-118.34, and 118.34-118.35 Mbp). Blue points, 10 Kb bins overlapping the 3’ region of KMT2A (exons 14-36: chr11 bins 118.37-118.38, 118.38-118.39, and 118.39-118.40 Mbp). Purple points, 10 Kb bins overlapping with TCF3 (chr19 bins 1.61-1.62, 1.62-1.63 Mbp). Orange point, 10 Kb bins overlapping with AFF3 (chr2 bin 100.17-100.18 Mbp). Genomic positions are based on GRCh37/hg19.
(O) Genome-wide effects of siKDM3B, siG9a, and double knockdown on H3K9me2 levels. Scatterplots shows all 10 Kb bins across the genome, with input-normalized H3K9me2 ChIP-seq densities in two conditions at each bin represented as a point. Left, scatterplot comparing H3K9me2 densities in control vs siKDM3B. Middle, scatterplot for control vs siG9a cells. Right, scatterplot for control vs siKDM3B + siG9a cells. Red lines correspond to > 1.5 fold increase or decrease compared to control. Colored points correspond to the bins listed in (N). Red points, 10 Kb bins overlapping the central region of KMT2A gene (containing exons 8-14). Green points, 10 Kb bins overlapping the 5’ region of KMT2A (exons 1-8). Blue points, 10 Kb bins overlapping the 3’ region of KMT2A (exons 14-36). Purple points, 10 Kb bins overlapping with TCF3. Orange point, 10 Kb bin overlapping with AFF3. Genomic positions are based on GRCh37/hg19.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 200 nuclei were scored per replicate per experiment. BA- Break aparts. NS-not significant to control.
Supplemental Figure 5. Reduced CTCF occupancy leads to KMT2A copy gains and break aparts. Related to Figure 5.
(A) Western blot validating siRNA depletion of KDM3B and CTCF in RPE cells.
(B) Cell growth assays for siRNA KDM3B and/or CTCF co-depletion in RPE cells.
(C) qRT-PCR analysis of KDM3B transcript levels relative to β-actin upon KDM3B siRNA depletion in RPE cells. Corresponding to Figure 5D-E.
(D) qRT-PCR analysis of KDM3B transcript levels relative to β-actin upon CTCF siRNA depletion in RPE cells from three independent experiments.
(E) Western blot for three independent experiments evaluating CTCF, KDM3B and G9a protein levels in cells siRNA depleted for CTCF.
(F) Western blot quantification of KDM3B protein in panel E that was normalized to α-Actinin.
(G) Western blot quantification of G9a protein in panel E that was normalized to α-Actinin.
(H) Western blot demonstrating siRNA depletion of CTCF and G9a.
(I) Cell growth assays for siRNA CTCF and/or G9a/EHMT2 co-depletion.
(J) Western blot demonstrating siRNA depletion of CTCF and relative protein level expression of G9A after EHMTi treatment.
(K) Cell growth for siRNA CTCF depletion and/or EHMTi treated cells.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted.
Supplemental Figure 6. Doxorubicin promotes KMT2A amplification and rearrangement as well as reduces KDM3B and CTCF protein levels. Related to Figure 6.
(A) Graph of DNA FISH quantification for RPE cells treated with 1pg/μL Dox for 72hrs. Dox treatment causes significant copy gains and break aparts at the TCF3/E2A locus; while the control region (Subtel19) did not have significant change.
(B) Graph of the DNA FISH quantification for RPE cells treated with 1pg/μL Dox for 72hrs. Dox treatment causes significant copy gains AFF3/LAF4 locus; while the control region (NMYC) was not significant.
(C) qRT-PCR of G9a transcript levels in RPE cells after 72hrs of Dox. Samples were normalized to β-actin.
(D) RT-qPCR analysis demonstrating that Dox modestly but significantly reduced KDM3B transcript levels relative to β-actin in KG1a cells.
(E) RT-qPCR analysis demonstrating that Dox modestly but significantly reduced CTCF transcript levels relative to β-actin in KG1a cells.
(F) Western blots of two independent experiments illustrating Dox reducing KDM3B (top) and CTCF (bottom) protein levels in KG1a cells.
(G) Western blots illustrating MG132 partial rescue of KDM3B (top) and CTCF (bottom) protein levels of individual experiments. Quantification of protein levels of KDM3B (top) or CTCF (bottom) normalized to α-Actinin are below each blot. These quantifications were used to generate the avg. ratio shown in Figure 6L and 6M. Representative images used in Figure 6L and 6M are on the far right.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 200 nuclei were scored per replicate per experiment. BA- Break aparts. NS- not significant to control.
Supplemental Figure 7. KDM3B and CTCF regulation controls Doxorubicin-induced KMT2A amplification and rearrangement. Related to Figure 7.
(A) Graph of the qRT-PCR validation for siKDM3B-mediated depletion RPE cells. Samples used in Figure 7A and S7B. Samples were normalized to β-actin.
(B) ChIP-qPCR demonstrating increase of H3K9me2 at KMT2A (KMT2A CTCF flanking site) following KDM3B siRNA depletion (left; blue) and Dox treatment at 1pg/μL for 24hr (right; red).
(C) Graph of the qRT-PCR validation for Halo-CTCF overexpression and 24hrs Doxorubicin treatment in RPE cells. Samples were normalized to β-actin.
(D) Graph of the cell growth assays for siRNA-mediated G9a depletion with or without Dox treatment for 24 hrs.
(E) Western blot demonstrating siRNA-mediated depletion of G9a and Dox treatment. Dox treatment alone does not impact G9a protein levels.
(F) Graph of the cell growth assays for Dox and/or EHMTi treated RPE cells for 24 hrs.
(G) Graph of the cell growth assays with or without Dox treatment for 24 hrs and/or KDM3B overexpression in RPE cells.
(H) Western blot illustrating overexpression of HALO-KDM3B (left) and HALO-CTCF (right) in RPE cells. Dox reduced both exogenous Halo-tagged (upper band) and endogenous (lower band) KDM3B and CTCF protein levels.
(I) A schematic depicting the regulation of KMT2A DNA copy gains and rearrangements. KDM3B and/or CTCF control KMT2A copy gains and break aparts, while G9a promotes the events (I; Figures 1, 2, 4, and 5). Upon removal of transient suppression of KDM3B (<1 cell cycle), KMT2A extrachromosomal DNA copy gains and break aparts recover and return to baseline (II; Figure 2F). With longer suppression of KDM3B (multiple cell cycles), KMT2A copy gains and rearrangement events become inherited in the genome (Bottom; Figure 3). Continuous depletion of KDM3B (e.g., KDM3B LOH) generates transient extrachromosomal and inherited KMT2A amplifications (III; Figure 4D). Recovery from KDM3B depletion or suppression after multiple cell cycles results in inherited KMT2A DNA copy gains as rearrangements, which cannot be removed or rescued (IV; Figure 4E).
(J) DNA FISH quantification for Inherited KMT2A RPE cells generated in Figure 3F, treated with KDM3i for 3-12hr. Inherited KMT2A cells have a significant increase in KMT2A copy number after 3 and 6hr KDM3i exposure. No increase in KMT2A copy number is observed at 12hr.
(K) Annexin V staining for Inherited KMT2A RPE cells generated in Figure 3F, treated with KDM3i for 3-12hr. Longer exposure to KDM3i results in increased apoptosis for the Inherited KMT2A cells, but not the Control cells.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 200 nuclei were scored per replicate per experiment. BA- Break aparts. NS- not significant to control.
Table S1. List of 10k genomic bins and nearest associated genes with increased H3K9me1 upon siKDM3B in RPE cells, related to Figure 1.
Table S2.List of 10k genomic bins and nearest associated genes with reduced or increased H3K9me1/2 upon siKDM3B, siG9a, or both in RPE cells, related to Figure 4.
Table S3. Oligonucleotides used, related to STAR Methods.
HIGHLIGHTS.
KDM3B and G9a regulate MLL/KMT2A copy gains and rearrangements through H3K9me1,2
CTCF depletion and reduced binding associate with KMT2A copy gains and break aparts
Doxorubicin reduces KDM3B and CTCF levels, promoting MLL/KMT2A alterations
G9a inhibition suppresses Dox-induced MLL/KMT2A alterations in mice and human cells
ACKNOWLEDGEMENTS
We would like to thank the glass washing, cell culture facility, genomics resource and cell sorting facilities at Fox Chase Cancer Center for support. We also thank Dr. Thomas L. Clarke for technical assistance. Drs. Capucine Van Rechem, Alfonso Bellacosa, Jon Chernoff, and Jade Wilson and Ethan Sumner for comments on the manuscript. Work related to this study is supported by R01GM097360 and R35GM144131 (J.R.W.), NIH/NCI Cancer Center Support grant P30 CA006927 (J.R.W.), and the American Lung Association Lung Cancer Discovery Award (J.R.W.). O.G. is supported by NIH (R35 GM139569). D.C. is a recipient of Ovarian Cancer Research Fund Alliance (Ann and Sol Schreiber Mentored Investigator Award-543667). B.I.F. is supported by NIH (T32 GM142606). T.S. is supported by NIH (R01 CA244044, R01 CA237386). C.D. is supported by V Foundation (V2021-017) and the W.W. Smith Charitable Trust (C2101). R.I.S. is supported by NIH (P30 DK040561).
Footnotes
Declaration of Interests
J.R.W. has served or is serving as a consultant or advisor for Qsonica, Salarius Pharmaceuticals, Daiichi Sankyo, Inc., Vyne Therapeutics and Lily Asia Ventures. J.R.W also receives funding for research from Salarius Pharmaceuticals and Oryzon Genomics. O.G. is a scientific co-founder and shareholder of EpiCypher, Inc, K36 Therapeutics, Inc., and Alternative Bio, Inc. J.J. received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc. and Cullinan Oncology, Inc. J.J. is a cofounder and equity shareholder in Cullgen, Inc., a scientific cofounder and scientific advisory board member of Onsero Therapeutics, Inc., and a consultant for Cullgen, Inc., EpiCypher, Inc., and Accent Therapeutics, Inc. C.D. receives research funds from Janssen outside the submitted work. The other authors do not declare any conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D. (2022). Hallmarks of Cancer: New Dimensions. Cancer Discov 12, 31–46. 10.1158/2159-8290.Cd-21-1059. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Mishra S, and Whetstine JR (2016). Different Facets of Copy Number Changes: Permanent, Transient, and Adaptive. Mol Cell Biol. 10.1128/MCB.00652-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey C, Shoura MJ, Mischel PS, and Swanton C (2020). Extrachromosomal DNA-relieving heredity constraints, accelerating tumour evolution. Ann Oncol 31, 884–893. 10.1016/j.annonc.2020.03.303. [DOI] [PubMed] [Google Scholar]
- 5.Song K, Minami JK, Huang A, Dehkordi SR, Lomeli SH, Luebeck J, Goodman MH, Moriceau G, Krijgsman O, Dharanipragada P, et al. (2022). Plasticity of Extrachromosomal and Intrachromosomal BRAF Amplifications in Overcoming Targeted Therapy Dosage Challenges. Cancer Discov 12, 1046–1069. 10.1158/2159-8290.CD-20-0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black JC, Atabakhsh E, Kim J, Biette KM, Van Rechem C, Ladd B, Burrowes PD, Donado C, Mattoo H, Kleinstiver BP, et al. (2015). Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes & development 29, 1018–1031. 10.1101/gad.259796.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Black JC, Manning AL, Van Rechem C, Kim J, Ladd B, Cho J, Pineda CM, Murphy N, Daniels DL, Montagna C, et al. (2013). KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 154, 541–555. 10.1016/j.cell.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Black JC, Zhang H, Kim J, Getz G, and Whetstine JR (2016). Regulation of Transient Site-specific Copy Gain by MicroRNA. J Biol Chem 291, 4862–4871. 10.1074/jbc.M115.711648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarke TL, Tang R, Chakraborty D, Van Rechem C, Ji F, Mishra S, Ma A, Kaniskan HU, Jin J, Lawrence MS, et al. (2020). Histone Lysine Methylation Dynamics Control EGFR DNA Copy-Number Amplification. Cancer Discov 10, 306–325. 10.1158/2159-8290.CD-19-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mishra S, Van Rechem C, Pal S, Clarke TL, Chakraborty D, Mahan SD, Black JC, Murphy SE, Lawrence MS, Daniels DL, and Whetstine JR (2018). Cross-talk between Lysine-Modifying Enzymes Controls Site-Specific DNA Amplifications. Cell 174, 803–817 e816. 10.1016/j.cell.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schichman SA, Caligiuri MA, Gu Y, Strout MP, Canaani E, Bloomfield CD, and Croce CM (1994). ALL-1 partial duplication in acute leukemia. Proc Natl Acad Sci U S A 91, 6236–6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poppe B, Vandesompele J, Schoch C, Lindvall C, Mrozek K, Bloomfield CD, Beverloo HB, Michaux L, Dastugue N, Herens C, et al. (2004). Expression analyses identify MLL as a prominent target of 11q23 amplification and support an etiologic role for MLL gain of function in myeloid malignancies. Blood 103, 229–235. 10.1182/blood-2003-06-2163. [DOI] [PubMed] [Google Scholar]
- 13.Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H, Zhao Y, Baty J, Heath S, Westervelt P, Watson MA, et al. (2009). Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci U S A 106, 12950–12955. 10.1073/pnas.0903091106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang G, DiNardo C, Zhang L, Ravandi F, Khoury JD, Huh YO, Muzzafar T, Medeiros LJ, Wang SA, and Bueso-Ramos CE (2015). MLL gene amplification in acute myeloid leukemia and myelodysplastic syndromes is associated with characteristic clinicopathological findings and TP53 gene mutation. Hum Pathol 46, 65–73. 10.1016/j.humpath.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Dolan M, McGlennen RC, and Hirsch B (2002). MLL amplification in myeloid malignancies: clinical, molecular, and cytogenetic findings. Cancer Genet Cytogenet 134, 93–101. [DOI] [PubMed] [Google Scholar]
- 16.Maitta RW, Cannizzaro LA, and Ramesh KH (2009). Association of MLL amplification with poor outcome in acute myeloid leukemia. Cancer Genet Cytogenet 192, 40–43. 10.1016/j.cancergencyto.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 17.Cox MC, Panetta P, Venditti A, Del Poeta G, Maurillo L, Tamburini A, Del Principe MI, and Amadori S (2003). Fluorescence in situ hybridization and conventional cytogenetics for the diagnosis of 11q23+/MLL+ translocation in leukaemia. Br J Haematol 121, 953–955. [DOI] [PubMed] [Google Scholar]
- 18.Sperling AS, Gibson CJ, and Ebert BL (2017). The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 17, 5–19. 10.1038/nrc.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woo JS, Alberti MO, and Tirado CA (2014). Childhood B-acute lymphoblastic leukemia: a genetic update. Exp Hematol Oncol 3, 16. 10.1186/2162-3619-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rice S, and Roy A (2020). MLL-rearranged infant leukaemia: A 'thorn in the side' of a remarkable success story. Biochim Biophys Acta Gene Regul Mech 1863, 194564. 10.1016/j.bbagrm.2020.194564. [DOI] [PubMed] [Google Scholar]
- 21.Winters AC, and Bernt KM (2017). MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front Pediatr 5, 4. 10.3389/fped.2017.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Super HJ, McCabe NR, Thirman MJ, Larson RA, Le Beau MM, Pedersen-Bjergaard J, Philip P, Diaz MO, and Rowley JD (1993). Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 82, 3705–3711. [PubMed] [Google Scholar]
- 23.Andersen MK, Christiansen DH, Jensen BA, Ernst P, Hauge G, and Pedersen-Bjergaard J (2001). Therapy-related acute lymphoblastic leukaemia with MLL rearrangements following DNA topoisomerase II inhibitors, an increasing problem: report on two new cases and review of the literature since 1992. Br J Haematol 114, 539–543. [DOI] [PubMed] [Google Scholar]
- 24.Pedersen-Bjergaard J, Andersen MK, and Johansson B (1998). Balanced chromosome aberrations in leukemias following chemotherapy with DNA-topoisomerase II inhibitors. J Clin Oncol 16, 1897–1898. 10.1200/JCO.1998.16.5.1897. [DOI] [PubMed] [Google Scholar]
- 25.Dulak AM, Schumacher SE, van Lieshout J, Imamura Y, Fox C, Shim B, Ramos AH, Saksena G, Baca SC, Baselga J, et al. (2012). Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res 72, 4383–4393. 10.1158/0008-5472.Can-11-3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer C, Larghero P, Almeida Lopes B, Burmeister T, Gröger D, Sutton R, Venn NC, Cazzaniga G, Corral Abascal L, Tsaur G, et al. (2023). The KMT2A recombinome of acute leukemias in 2023. Leukemia. 10.1038/s41375-023-01877-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leone G, Mele L, Pulsoni A, Equitani F, and Pagano L (1999). The incidence of secondary leukemias. Haematologica 84, 937–945. [PubMed] [Google Scholar]
- 28.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, and Jaffe ES (2011). The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 117, 5019–5032. 10.1182/blood-2011-01-293050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Godley LA, and Larson RA (2008). Therapy-related myeloid leukemia. Semin Oncol 35, 418–429. 10.1053/j.seminoncol.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borthakur G, and Estey AE (2007). Therapy-related acute myelogenous leukemia and myelodysplastic syndrome. Curr Oncol Rep 9, 373–377. 10.1007/s11912-007-0050-z. [DOI] [PubMed] [Google Scholar]
- 31.Hu Z, Gomes I, Horrigan SK, Kravarusic J, Mar B, Arbieva Z, Chyna B, Fulton N, Edassery S, Raza A, and Westbrook CA (2001). A novel nuclear protein, 5qNCA (LOC51780) is a candidate for the myeloid leukemia tumor suppressor gene on chromosome 5 band q31. Oncogene 20, 6946–6954. 10.1038/sj.onc.1204850. [DOI] [PubMed] [Google Scholar]
- 32.Yoo J, Jeon YH, Cho HY, Lee SW, Kim GW, Lee DH, and Kwon SH (2020). Advances in Histone Demethylase KDM3A as a Cancer Therapeutic Target. Cancers (Basel) 12. 10.3390/cancers12051098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebert BL (2010). Genetic deletions in AML and MDS. Best Pract Res Clin Haematol 23, 457–461. 10.1016/j.beha.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schoch C, Kern W, Kohlmann A, Hiddemann W, Schnittger S, and Haferlach T (2005). Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer 43, 227–238. 10.1002/gcc.20193. [DOI] [PubMed] [Google Scholar]
- 35.Herry A, Douet-Guilbert N, Guéganic N, Morel F, Le Bris MJ, Berthou C, and De Braekeleer M (2006). Del(5q) and MLL amplification in homogeneously staining region in acute myeloblastic leukemia: a recurrent cytogenetic association. Ann Hematol 85, 244–249. 10.1007/s00277-005-0059-z. [DOI] [PubMed] [Google Scholar]
- 36.MacKinnon RN, Kannourakis G, Wall M, and Campbell LJ (2011). A cryptic deletion in 5q31.2 provides further evidence for a minimally deleted region in myelodysplastic syndromes. Cancer Genet 204, 187–194. 10.1016/j.cancergen.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Xu X, Nagel S, Quentmeier H, Wang Z, Pommerenke C, Dirks WG, Macleod RAF, Drexler HG, and Hu Z (2018). KDM3B shows tumor-suppressive activity and transcriptionally regulates HOXA1 through retinoic acid response elements in acute myeloid leukemia. Leuk Lymphoma 59, 204–213. 10.1080/10428194.2017.1324156. [DOI] [PubMed] [Google Scholar]
- 38.Kim JY, Kim KB, Eom GH, Choe N, Kee HJ, Son HJ, Oh ST, Kim DW, Pak JH, Baek HJ, et al. (2012). KDM3B is the H3K9 demethylase involved in transcriptional activation of lmo2 in leukemia. Mol Cell Biol 32, 2917–2933. 10.1128/mcb.00133-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saavedra F, Gurard-Levin ZA, Rojas-Villalobos C, Vassias I, Quatrini R, Almouzni G, and Loyola A (2020). JMJD1B, a novel player in histone H3 and H4 processing to ensure genome stability. Epigenetics Chromatin 13, 6. 10.1186/s13072-020-00331-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zatkova A, Merk S, Wendehack M, Bilban M, Muzik EM, Muradyan A, Haferlach C, Haferlach T, Wimmer K, Fonatsch C, and Ullmann R (2009). AML/MDS with 11q/MLL amplification show characteristic gene expression signature and interplay of DNA copy number changes. Genes Chromosomes Cancer 48, 510–520. 10.1002/gcc.20658. [DOI] [PubMed] [Google Scholar]
- 41.Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, and Chiu CP (1999). Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet 21, 111–114. 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- 42.Janssen A, van der Burg M, Szuhai K, Kops GJ, and Medema RH (2011). Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333, 1895–1898. 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- 43.Maciejowski J, Li Y, Bosco N, Campbell PJ, and de Lange T (2015). Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–1654. 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mardin BR, Drainas AP, Waszak SM, Weischenfeldt J, Isokane M, Stutz AM, Raeder B, Efthymiopoulos T, Buccitelli C, Segura-Wang M, et al. (2015). A cell-based model system links chromothripsis with hyperploidy. Mol Syst Biol 11, 828. 10.15252/msb.20156505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, and Pellman D (2015). Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184. 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Black JC, Atabakhsh E, Kim J, Biette KM, Van Rechem C, Ladd B, Burrowes PD, Donado C, Mattoo H, Kleinstiver BP, et al. (2015). Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes Dev 29, 1018–1031. 10.1101/gad.259796.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Necochea-Campion R, Diaz Osterman CJ, Hsu HW, Fan J, Mirshahidi S, Wall NR, and Chen CS (2015). AML sensitivity to YM155 is modulated through AKT and Mcl-1. Cancer Lett 366, 44–51. 10.1016/j.canlet.2015.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sánchez-Reyes K, Pedraza-Brindis EJ, Hernández-Flores G, Bravo-Cuellar A, López-López BA, Rosas-González VC, and Ortiz-Lazareno PC (2019). The supernatant of cervical carcinoma cells lines induces a decrease in phosphorylation of STAT-1 and NF-κB transcription factors associated with changes in profiles of cytokines and growth factors in macrophages derived from U937 cells. Innate Immun 25, 344–355. 10.1177/1753425919848841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, Fan H, Xu C, Jiang G, Wang H, and Zhang J (2019). KDM3B suppresses APL progression by restricting chromatin accessibility and facilitating the ATRA-mediated degradation of PML/RARalpha. Cancer Cell Int 19, 256. 10.1186/s12935-019-0979-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Broeker PL, Super HG, Thirman MJ, Pomykala H, Yonebayashi Y, Tanabe S, Zeleznik-Le N, and Rowley JD (1996). Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood 87, 1912–1922. [PubMed] [Google Scholar]
- 51.Li J, Yu B, Deng P, Cheng Y, Yu Y, Kevork K, Ramadoss S, Ding X, Li X, and Wang C-Y (2017). KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/β-catenin signalling. Nature Communications 8, 15146. 10.1038/ncomms15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu X, Wang L, Hu L, Dirks WG, Zhao Y, Wei Z, Chen D, Li Z, Wang Z, Han Y, et al. (2020). Small molecular modulators of JMJD1C preferentially inhibit growth of leukemia cells. Int J Cancer 146, 400–412. 10.1002/ijc.32552. [DOI] [PubMed] [Google Scholar]
- 53.Streubel B, Valent P, Jäger U, Edelhäuser M, Wandt H, Wagner T, Büchner T, Lechner K, and Fonatsch C (2000). Amplification of the MLL gene on double minutes, a homogeneously staining region, and ring chromosomes in five patients with acute myeloid leukemia or myelodysplastic syndrome. Genes Chromosomes Cancer 27, 380–386. [PubMed] [Google Scholar]
- 54.Black JC, Van Rechem C, and Whetstine JR (2012). Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 48, 491–507. 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh V, Huang XP, Allali-Hassani A, Janzen WP, Roth BL, Frye SV, et al. (2013). Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem 56, 8931–8942. 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atkin ND, Raimer HM, Wang Z, Zang C, and Wang YH (2021). Assessing acute myeloid leukemia susceptibility in rearrangement-driven patients by DNA breakage at topoisomerase II and CCCTC-binding factor/cohesin binding sites. Genes Chromosomes Cancer 60, 808–821. 10.1002/gcc.22993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gothe HJ, Bouwman BAM, Gusmao EG, Piccinno R, Petrosino G, Sayols S, Drechsel O, Minneker V, Josipovic N, Mizi A, et al. (2019). Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Mol Cell 75, 267–283 e212. 10.1016/j.molcel.2019.05.015. [DOI] [PubMed] [Google Scholar]
- 58.Phillips JE, and Corces VG (2009). CTCF: master weaver of the genome. Cell 137, 1194–1211. 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Felix CA (1998). Secondary leukemias induced by topoisomerase-targeted drugs. Biochim Biophys Acta 1400, 233–255. 10.1016/s0167-4781(98)00139-0. [DOI] [PubMed] [Google Scholar]
- 60.Sanjuan-Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, and Menendez P (2015). Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 126, 2676–2685. 10.1182/blood-2015-09-667378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Libura J, Slater DJ, Felix CA, and Richardson C (2005). Therapy-related acute myeloid leukemia-like MLL rearrangements are induced by etoposide in primary human CD34+ cells and remain stable after clonal expansion. Blood 105, 2124–2131. 10.1182/blood-2004-07-2683. [DOI] [PubMed] [Google Scholar]
- 62.Lehman BJ, Lopez-Diaz FJ, Santisakultarm TP, Fang L, Shokhirev MN, Diffenderfer KE, Manor U, and Emerson BM (2021). Dynamic regulation of CTCF stability and sub-nuclear localization in response to stress. PLoS Genet 17, e1009277. 10.1371/journal.pgen.1009277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, Zheng H, and Wang X (2005). A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. Faseb j 19, 2051–2053. 10.1096/fj.05-3973fje. [DOI] [PubMed] [Google Scholar]
- 64.Mullighan CG (2012). Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest 122, 3407–3415. 10.1172/JCI61203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andersen MK, Autio K, Barbany G, Borgstrom G, Cavelier L, Golovleva I, Heim S, Heinonen K, Hovland R, Johannsson JH, et al. (2011). Paediatric B-cell precursor acute lymphoblastic leukaemia with t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br J Haematol 155, 235–243. 10.1111/j.1365-2141.2011.08824.x. [DOI] [PubMed] [Google Scholar]
- 66.Kager L, Lion T, Attarbaschi A, Koenig M, Strehl S, Haas OA, Dworzak MN, Schrappe M, Gadner H, Mann G, and Austrian, B.F.M.S.G. (2007). Incidence and outcome of TCF3-PBX1-positive acute lymphoblastic leukemia in Austrian children. Haematologica 92, 1561–1564. 10.3324/haematol.11239. [DOI] [PubMed] [Google Scholar]
- 67.Aplan PD (2006). Chromosomal translocations involving the MLL gene: molecular mechanisms. DNA Repair (Amst) 5, 1265–1272. 10.1016/j.dnarep.2006.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gómez-Herreros F, Zagnoli-Vieira G, Ntai I, Martínez-Macías MI, Anderson RM, Herrero-Ruíz A, and Caldecott KW (2017). TDP2 suppresses chromosomal translocations induced by DNA topoisomerase II during gene transcription. Nat Commun 8, 233. 10.1038/s41467-017-00307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eisenmann KM, Dykema KJ, Matheson SF, Kent NF, DeWard AD, West RA, Tibes R, Furge KA, and Alberts AS (2009). 5q- myelodysplastic syndromes: chromosome 5q genes direct a tumor-suppression network sensing actin dynamics. Oncogene 28, 3429–3441. 10.1038/onc.2009.207. [DOI] [PubMed] [Google Scholar]
- 70.Giagounidis AA, Germing U, Wainscoat JS, Boultwood J, and Aul C (2004). The 5q- syndrome. Hematology 9, 271–277. 10.1080/10245330410001723824. [DOI] [PubMed] [Google Scholar]
- 71.Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, and Golub TR (2008). Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 451, 335–339. 10.1038/nature06494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, Hirst M, Hogge D, Marra M, Wells RA, et al. (2010). Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat Med 16, 49–58. 10.1038/nm.2054. [DOI] [PubMed] [Google Scholar]
- 73.Aplan PD (2006). Causes of oncogenic chromosomal translocation. Trends Genet 22, 46–55. 10.1016/j.tig.2005.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hulegardh E, Nilsson C, Lazarevic V, Garelius H, Antunovic P, Rangert Derolf A, Mollgard L, Uggla B, Wennstrom L, Wahlin A, et al. (2015). Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: a report from the Swedish Acute Leukemia Registry. Am J Hematol 90, 208–214. 10.1002/ajh.23908. [DOI] [PubMed] [Google Scholar]
- 75.Duy C, Teater M, Garrett-Bakelman FE, Lee TC, Meydan C, Glass JL, Li M, Hellmuth JC, Mohammad HP, Smitheman KN, et al. (2019). Rational Targeting of Cooperating Layers of the Epigenome Yields Enhanced Therapeutic Efficacy against AML. Cancer discovery 9, 872–889. 10.1158/2159-8290.CD-19-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Rechem C, Ji F, Chakraborty D, Black JC, Sadreyev RI, and Whetstine JR (2021). Collective regulation of chromatin modifications predicts replication timing during cell cycle. Cell Rep 37, 109799. 10.1016/j.celrep.2021.109799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Rechem C, Ji F, Mishra S, Chakraborty D, Murphy SE, Dillingham ME, Sadreyev RI, and Whetstine JR (2020). The lysine demethylase KDM4A controls the cell-cycle expression of replicative canonical histone genes. Biochim Biophys Acta Gene Regul Mech 1863, 194624. 10.1016/j.bbagrm.2020.194624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. KDM3B depletion induces KMT2A DNA copy gains and break aparts. Related to Figure 1
(A) qRT-PCR analysis was performed to validate specific gene siRNA knockdowns for the KDM3 family. Samples were normalized to β-actin.
(B) Western blot demonstrates that KDM3B protein levels are reduced when compared to controls using two different siRNAs in RPE cells.
(C) FACS analysis was performed after KDM3 family member siRNA depletion after 72 hrs.
(D) Coomassie gel of purified KDM enzymes used in the demethylase assay.
(E) Western blot of histones after being incubated with purified KDM3A,B and KDM5A. CTH alone- calf thymus histones alone
(F) MALDI-TOF analyses of H3K9me2 peptides incubated with KDM3A or KDM3B.
(G-N) DNA FISH for various genomic regions tested upon KDM3B siRNA depletion. Only TCF3/E2A (M) and AFF3/LAF4 (N) has significant copy gains and break aparts, or copy gains, respectively, upon KDM3B depletion in RPE cells when compared to the other loci tested.
(O) DNA FISH quantification for Subtel19 (appropriate control for M) that does not change upon KDM3B siRNA depletion
(P) Western blot shows decreased KDM3B protein levels in U937 cells upon siKDM3B.
(Q) DNA FISH quantification for TCF3/E2A demonstrates no significant copy gains or break aparts in KDM3B siRNA depleted U937 cells.
(R) DNA FISH quantification for AFF3/LAF4 demonstrates no significant copy gains in KDM3B siRNA depleted U937 cells.
(S) qPCR validation shows decreased KDM3B transcript levels compared to control in KDM3B siRNA depleted cells. These cells were used for experiments represented in Figure 1L-M and S1T-V.
(T) Genome-wide effects of siKDM3B on H3K9me1 levels. Scatterplots shows all 10 Kb bins across the genome, with input-normalized H3K9me1 ChIP-seq densities in two conditions at each bin represented as a point. Red points, 10 Kb bins overlapping the central region of KMT2A gene (containing exons 8-14: chr11 bins 118.35-118.36 and 118.36-118.37 Mbp). Green points, 10 Kb bins overlapping the 5’ region of KMT2A (exons 1-8: chr11 bins 118.30-118.31, 118.31-118.32, 118.32-118.33, 118.33-118.34 and 118.34-118.35 Mbp). Blue points, 10 Kb bins overlapping the 3’ region of KMT2A (exons 14-36: chr11 bins 118.37-118.38, 118.38-118.39, and 118.39-118.40 Mbp). Purple points, 10 Kb bins overlapping with TCF3 (chr11 bins 1.61-1.62, 1.62-1.63 Mbp). Orange point, 10 Kb bin overlapping with AFF3 (chr2 bin 100.17-100.18 Mbp). Genomic positions are based on GRCh37/hg19.
(U) Genome-wide effects of siKDM3B on H3K9me2 levels. Scatterplots shows all 10 Kb bins across the genome, with input-normalized H3K9me2 ChIP-seq densities in two conditions at each bin represented as a point. Colored points correspond to the bins listed in (S1T). Red points, 10 Kb bins overlapping the central region of KMT2A gene (containing exons 8-14). Green points, 10 Kb bins overlapping the 5’ region of KMT2A (exons 1-8). Blue points, 10 Kb bins overlapping the 3’ region of KMT2A (exons 14-36). Purple points, 10 Kb bins overlapping with TCF3. Orange point, 10 Kb bin overlapping with AFF3. Genomic positions are based on GRCh37/hg19.
(V) Genomic tracks of input-normalized ChIP-seq densities of H3K9me1-3 and the public input-normalized KDM3B ChIP-seq density 51 for the region flanking TCF3/E2A upon depletion of KDM3B.
(W) Genomic tracks of input-normalized ChIP-seq densities of H3K9me1-3 and the public input-normalized KDM3B ChIP-seq density 51 for the AFF3 region upon depletion of KDM3B.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 100 nuclei were scored per replicate per experiment. BA- Break aparts.
Supplemental Figure 2. KDM3B chemical inhibition (KDM3i) promotes transient KMT2A copy gains and break aparts. Related to Figure 2
(A) In vitro KDM assay with commercial full length purified KDM3B protein incubated with histones and western blotted for KDM3B, H3K9me1 and H3K9me3. Blot demonstrates reduction in H3K9me1 levels upon incubation with KDM3B is rescued with KDM3i addition.
(B) Cell growth assay for KDM3i treated RPE cells. Treatment with KDM3i at 25nM does not impact cell growth, whereas 250nM and 1μM modestly but significantly suppresses growth.
(C) Western blot demonstrating KDM3B protein levels are not reduced with KDM3i treatment in RPE cells. A representative example from quantification in panel D.
(D) Quantification of western blots for KDM3B in KDM3i treated RPE cells (n=3). KDM3B levels are not reduced upon treatment with KDM3i.
(E) Cell growth assay for KDM3i treated RPE cells corresponding to Figure 2F. Cell growth is not impacted after 12hrs KDM3i treatment, or after 12hr KDM3i washout.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted.
Supplemental Figure 3. KDM3B suppression leads to integration and inheritance of KMT2A copy gains and break aparts. Related to Figure 3.
(A) Copy Number Variation analysis using Digital Droplet PCR (ddPCR) shows KMT2A copy number increase in later passage of the inherited KMT2A cell lines used in Figures 3G-H. Copy number is quantified by computing ratio of KMT2A to CD3E reference gene (4 biological replicates with 2 technical replicates).
(B) Quantification of DNA FISH for TCF3/E2A after passaging KDM3B siRNA depleted RPE cells. TCF3/E2A copy gain and break aparts are retained after 15 passages in KDM3B siRNA depleted cells.
(C) DNA FISH for xSubTel19 after passaging KDM3B siRNA depleted RPE cells. Subtel19 copy gain does not change in KDM3B siRNA depleted cells.
(D) DNA FISH for AFF3/LAF4 after passage KDM3B siRNA depleted RPE cells. AFF3 copy gains occur at PO, but are not retained after 3 passages in KDM3B siRNA depleted cells. NMYC copy gain does not change.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 200 nuclei were scored per replicate per experiment. BA- Break aparts.
Supplemental Figure 4. H3K9me1 balance controls KMT2A copy gains and break aparts. Related to Figure 4.
(A) qRT-PCR analysis was performed to validate gene specific siRNA knockdowns. Samples were normalized to β-actin.
(B) Western blot images demonstrating successful siRNA-mediated depletion of KDM3B and G9a protein in RPE cells.
(C) FACS analysis evaluating cell cycle dynamics was performed for the siRNA mediated co-depletion experiments.
(D) Cell growth assays for siRNA co-depletion of KDM3B, EHMT1, and/or G9a.
(E) DNA FISH quantification for siRNA co-depletion of KDM3B, EHMT1, and/or G9a. siRNA depleted G9a but not EHMT1 prevents TCF3/E2A copy gains and break aparts upon KDM3B depletion in RPE cells.
(F) DNA FISH quantification of SubTel19 (control for TCF3 region) for siRNA co-depletion of KDM3B, EHMT1, and/or G9a.
(G) DNA FISH quantification for siRNA co-depletion of KDM3B, EHMT1, and/or G9a. siRNA depleted G9a but not EHMT1 prevents AFF3/LAF4 copy gains upon KDM3B depletion.
(H) Cell growth assays for KDM3/EHMT inhibitor versus control treatments corresponding to Figure 4B.
(I) FACS analysis evaluating cell cycle dynamics was performed for cells with KDM3B and/or G9a overexpression.
(J) qRT-PCR analysis for KDM3B and G9a overexpression in RPE cells. Samples were normalized to β-actin.
(K) qRT-PCR analysis was performed to validate gene specific siRNA knockdowns in HL60 cells. Samples were normalized to β-actin.
(L) qRT-PCR analysis was performed to validate gene specific siRNA knockdowns in WT versus Inherited KMT2A cells. Samples were normalized to β-actin. Two independent stable cell lines were used per condition with two independent siRNAs.
(M) Across the genome, the increase of H3K9me1 and H3K9me2 caused by siKDM3B was mostly rescued by double KDM3B and G9a knockdown. Barplots showing the fractions of the genome (based on 10 Kb genomic bins) that increased H3K9me1 (left) and H3K9me2 (right) upon KDM3B knockdown. Red, genomic fraction where this increase was rescued by double knockdown.
(N) Genome-wide effects of siKDM3B, siG9a, and double knockdown on H3K9me1 levels. Scatterplots shows all 10 Kb bins across the genome, with input-normalized H3K9me1 ChIP-seq densities in two conditions at each bin represented as a point. KDM3B and G9a knockdowns have opposite skews, whereas the double knockdown strongly reduces these H3K9me1 changes. Left, scatterplot comparing H3K9me1 densities in control vs siKDM3B; H3K9me1 changes are skewed towards increase (points above upper red line corresponding to > 1.5 fold increase in siKM3B cells). Middle, scatterplot for control vs siG9a cells; H3K9me1 changes are skewed towards decrease (points below lower red line corresponding to > 1.5 fold decrease in siG9a cells). Right, scatterplot for control vs siKDM3B + siG9a cells, with much fewer H3K9me1 changes in either direction. Red points, 10 Kb bins overlapping the central region of KMT2A gene (containing exons 8-14: chr11 bins 118.35-118.36 and 118.36-118.37 Mbp). Green points, 10 Kb bins overlapping the 5’ region of KMT2A (exons 1-8: chr11 bins 118.30-118.31, 118.31-118.32, 118.32-118.33, 118.33-118.34, and 118.34-118.35 Mbp). Blue points, 10 Kb bins overlapping the 3’ region of KMT2A (exons 14-36: chr11 bins 118.37-118.38, 118.38-118.39, and 118.39-118.40 Mbp). Purple points, 10 Kb bins overlapping with TCF3 (chr19 bins 1.61-1.62, 1.62-1.63 Mbp). Orange point, 10 Kb bins overlapping with AFF3 (chr2 bin 100.17-100.18 Mbp). Genomic positions are based on GRCh37/hg19.
(O) Genome-wide effects of siKDM3B, siG9a, and double knockdown on H3K9me2 levels. Scatterplots shows all 10 Kb bins across the genome, with input-normalized H3K9me2 ChIP-seq densities in two conditions at each bin represented as a point. Left, scatterplot comparing H3K9me2 densities in control vs siKDM3B. Middle, scatterplot for control vs siG9a cells. Right, scatterplot for control vs siKDM3B + siG9a cells. Red lines correspond to > 1.5 fold increase or decrease compared to control. Colored points correspond to the bins listed in (N). Red points, 10 Kb bins overlapping the central region of KMT2A gene (containing exons 8-14). Green points, 10 Kb bins overlapping the 5’ region of KMT2A (exons 1-8). Blue points, 10 Kb bins overlapping the 3’ region of KMT2A (exons 14-36). Purple points, 10 Kb bins overlapping with TCF3. Orange point, 10 Kb bin overlapping with AFF3. Genomic positions are based on GRCh37/hg19.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 200 nuclei were scored per replicate per experiment. BA- Break aparts. NS-not significant to control.
Supplemental Figure 5. Reduced CTCF occupancy leads to KMT2A copy gains and break aparts. Related to Figure 5.
(A) Western blot validating siRNA depletion of KDM3B and CTCF in RPE cells.
(B) Cell growth assays for siRNA KDM3B and/or CTCF co-depletion in RPE cells.
(C) qRT-PCR analysis of KDM3B transcript levels relative to β-actin upon KDM3B siRNA depletion in RPE cells. Corresponding to Figure 5D-E.
(D) qRT-PCR analysis of KDM3B transcript levels relative to β-actin upon CTCF siRNA depletion in RPE cells from three independent experiments.
(E) Western blot for three independent experiments evaluating CTCF, KDM3B and G9a protein levels in cells siRNA depleted for CTCF.
(F) Western blot quantification of KDM3B protein in panel E that was normalized to α-Actinin.
(G) Western blot quantification of G9a protein in panel E that was normalized to α-Actinin.
(H) Western blot demonstrating siRNA depletion of CTCF and G9a.
(I) Cell growth assays for siRNA CTCF and/or G9a/EHMT2 co-depletion.
(J) Western blot demonstrating siRNA depletion of CTCF and relative protein level expression of G9A after EHMTi treatment.
(K) Cell growth for siRNA CTCF depletion and/or EHMTi treated cells.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted.
Supplemental Figure 6. Doxorubicin promotes KMT2A amplification and rearrangement as well as reduces KDM3B and CTCF protein levels. Related to Figure 6.
(A) Graph of DNA FISH quantification for RPE cells treated with 1pg/μL Dox for 72hrs. Dox treatment causes significant copy gains and break aparts at the TCF3/E2A locus; while the control region (Subtel19) did not have significant change.
(B) Graph of the DNA FISH quantification for RPE cells treated with 1pg/μL Dox for 72hrs. Dox treatment causes significant copy gains AFF3/LAF4 locus; while the control region (NMYC) was not significant.
(C) qRT-PCR of G9a transcript levels in RPE cells after 72hrs of Dox. Samples were normalized to β-actin.
(D) RT-qPCR analysis demonstrating that Dox modestly but significantly reduced KDM3B transcript levels relative to β-actin in KG1a cells.
(E) RT-qPCR analysis demonstrating that Dox modestly but significantly reduced CTCF transcript levels relative to β-actin in KG1a cells.
(F) Western blots of two independent experiments illustrating Dox reducing KDM3B (top) and CTCF (bottom) protein levels in KG1a cells.
(G) Western blots illustrating MG132 partial rescue of KDM3B (top) and CTCF (bottom) protein levels of individual experiments. Quantification of protein levels of KDM3B (top) or CTCF (bottom) normalized to α-Actinin are below each blot. These quantifications were used to generate the avg. ratio shown in Figure 6L and 6M. Representative images used in Figure 6L and 6M are on the far right.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 200 nuclei were scored per replicate per experiment. BA- Break aparts. NS- not significant to control.
Supplemental Figure 7. KDM3B and CTCF regulation controls Doxorubicin-induced KMT2A amplification and rearrangement. Related to Figure 7.
(A) Graph of the qRT-PCR validation for siKDM3B-mediated depletion RPE cells. Samples used in Figure 7A and S7B. Samples were normalized to β-actin.
(B) ChIP-qPCR demonstrating increase of H3K9me2 at KMT2A (KMT2A CTCF flanking site) following KDM3B siRNA depletion (left; blue) and Dox treatment at 1pg/μL for 24hr (right; red).
(C) Graph of the qRT-PCR validation for Halo-CTCF overexpression and 24hrs Doxorubicin treatment in RPE cells. Samples were normalized to β-actin.
(D) Graph of the cell growth assays for siRNA-mediated G9a depletion with or without Dox treatment for 24 hrs.
(E) Western blot demonstrating siRNA-mediated depletion of G9a and Dox treatment. Dox treatment alone does not impact G9a protein levels.
(F) Graph of the cell growth assays for Dox and/or EHMTi treated RPE cells for 24 hrs.
(G) Graph of the cell growth assays with or without Dox treatment for 24 hrs and/or KDM3B overexpression in RPE cells.
(H) Western blot illustrating overexpression of HALO-KDM3B (left) and HALO-CTCF (right) in RPE cells. Dox reduced both exogenous Halo-tagged (upper band) and endogenous (lower band) KDM3B and CTCF protein levels.
(I) A schematic depicting the regulation of KMT2A DNA copy gains and rearrangements. KDM3B and/or CTCF control KMT2A copy gains and break aparts, while G9a promotes the events (I; Figures 1, 2, 4, and 5). Upon removal of transient suppression of KDM3B (<1 cell cycle), KMT2A extrachromosomal DNA copy gains and break aparts recover and return to baseline (II; Figure 2F). With longer suppression of KDM3B (multiple cell cycles), KMT2A copy gains and rearrangement events become inherited in the genome (Bottom; Figure 3). Continuous depletion of KDM3B (e.g., KDM3B LOH) generates transient extrachromosomal and inherited KMT2A amplifications (III; Figure 4D). Recovery from KDM3B depletion or suppression after multiple cell cycles results in inherited KMT2A DNA copy gains as rearrangements, which cannot be removed or rescued (IV; Figure 4E).
(J) DNA FISH quantification for Inherited KMT2A RPE cells generated in Figure 3F, treated with KDM3i for 3-12hr. Inherited KMT2A cells have a significant increase in KMT2A copy number after 3 and 6hr KDM3i exposure. No increase in KMT2A copy number is observed at 12hr.
(K) Annexin V staining for Inherited KMT2A RPE cells generated in Figure 3F, treated with KDM3i for 3-12hr. Longer exposure to KDM3i results in increased apoptosis for the Inherited KMT2A cells, but not the Control cells.
Error bars represent the SEM. Asterisk indicates significant difference from indicated (p < 0.05) by two-tailed Student’s t-test. A minimum of 2 replicates per experiment were conducted. For FISH, a minimum of 200 nuclei were scored per replicate per experiment. BA- Break aparts. NS- not significant to control.
Table S1. List of 10k genomic bins and nearest associated genes with increased H3K9me1 upon siKDM3B in RPE cells, related to Figure 1.
Table S2.List of 10k genomic bins and nearest associated genes with reduced or increased H3K9me1/2 upon siKDM3B, siG9a, or both in RPE cells, related to Figure 4.
Table S3. Oligonucleotides used, related to STAR Methods.
Data Availability Statement
Data
Original ChIP-sequencing data has been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-KDM3B, clone C69G2 | Cell Signaling | Cat# 3314; RRID: AB_1264294 |
| Anti-JMJD1B | Invitrogen | Cat# PA5-53459 |
| Anti-G9a | Sigma | Cat# G6919; RRID: AB_262007 |
| Anti-beta Actin | Millipore | Cat# MAB1501; RRID: AB626633 |
| Anti-Actinin | Santa Cruz | Cat# sc-17829; RRID: AB_626633 |
| Goat anti-mouse HRP | Biorad | Cat# 170-6516; RRID: AB11125547 |
| Goat anti-rabbit HRP | GenScript | Cat# A00167 |
| Anti-H3K9me1 | Abcam | Cat# ab8896 |
| Anti-H3K9me2 | Abcam | Cat# ab1220 |
| Anti-H3K9me3 | Abcam | Cat# ab8898 |
| Anti-CTCF, clone D31H2 | Cell Signaling | Cat# 3418 |
| Biological samples | ||
| Primary AML | Cihangir Duy 75 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Pierce Protease Inhibitor Tablets | Thermo Scientific | Cat# A32953 |
| Pierce Phosphatase Inhibitor Tablets | Thermo Scientific | Cat# A32957 |
| Propidium Iodide Solution | Sigma Aldrich | Cat# P4864 |
| Doxorubicin | Sigma Aldrich | ab142052 |
| Doxorubicin (Mouse study) | Selleckchem | Cat# E2516 |
| Dulbecco’s Modified Eagles Medium – High Glucose | Sigma Aldrich | Cat# D5648 |
| Roswell Park Memorial Institute Medium (RPMI)1640 | Sigma Aldrich | Cat# R6504 |
| Opti-Mem | Life Technologies | Cat# 31985070 |
| Trypsin (0.25%) EDTA | Life Technologies | Cat# 2520056 |
| L-Glutamine | Life Technologies | Cat# 25030-081 |
| Penicillin and Streptomycin | Life Technologies | Cat# 15140122 |
| Fetal Bovine Serum (FBS) | Gibco | Cat# 26140-079 |
| Lipofectamine 3000 | Life Technologies | Cat# L30000015 |
| EHMTi | Jian Jin | UNC0642 |
| KDM3i | Xu et al.52 | JDI-12 |
| Critical commercial assays | ||
| miRNeasy Mini Kit | Qiagen | Cat# 217004 |
| Superscript IV 1st Strand System | Life Technologies | Cat# 18091050 |
| CL-XPosure™ Film | Thermo Scientific | Cat# 34091 |
| Lumi-Light Western Blotting Substrate | Roche | Cat# 12015200001 |
| Pierce BCA Protein Assay | Thermo Scientific | Cat# 23223 and 23224 |
| Protein A Dynabeads | Thermo Scientific | Cat# 10002D |
| Protein G Dynabeads | Thermo Scientific | Cat# 10004D |
| TruSeq ChIP Sample Prep Kit Set A (48 samples) | Illumina | Cat# 10748010 |
| NextSeq® 500/550 High Output Kit v2 (75 cycles) | Illumina | Cat# FC-404-2005 |
| Bac-to-Bac™ Baculovirus Expression System | Thermo Scientific | Cat# 10359016 |
| Dead Cell Apoptosis Kits with Annexin V | Life Technologies | Cat# V13241 |
| Deposited data | ||
| Raw ChIP-sequencing | This paper | GEO: GSE210480 |
| KDM3B ChIP-seq | Li et al.51 | GEO: GSE71885 |
| RPE CTCF ChIP-seq | ENCODE | GEO: GSM749673 |
| GM12878 CTCF ChIP-seq | ENCODE | GEO: GSM733752 |
| H1 CTCF ChIP-seq | ENCODE | GEO: GSM733672 |
| bronchial_epithelial CTCF ChIP-seq | ENCODE | GEO: GSM749779 |
| Bcell CTCF ChIP-seq | ENCODE | GEO: GSM1003474 |
| HL-60 CTCF ChIP-seq | ENCODE | GEO: GSM749688 |
| HCT116 CTCF ChIP-seq | ENCODE | GEO: GSM1022652 |
| Experimental models: Cell lines | ||
| RPE-hTERT1 | Nick Dyson | N/A |
| U937 | FCCC Cell Culture Facility | CRL-1593.2 |
| HL60 | ATCC | CCL-240 |
| KG1a | ATCC | CCL-246.1 |
| HSPC | Cihangir Duy; 75 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 / 129/Sv | Jackson Labs | Strain# 101043 |
| Oligonucleotides | ||
| See Table S3 for Oligonucleotides used | This paper | N/A |
| Recombinant DNA | ||
| Halo-CTCF | Promega | Cat# FHC01807 |
| Halo-KDM3B | Promega | Cat# FHC05559 |
| Halo-Tag Alone | Promega | Cat# G6591 |
| MLL-1 Probe | Oxford Gene Technology | Cat# LPH 506-A |
| MLL breakapart | Oxford Gene Technology | Cat# LPH 013-A |
| AML1 breakapart | Oxford Gene Technology | Cat# LPH 027-SA |
| BCL6 breakapart | Oxford Gene Technology | Cat# LPH 035-SA |
| EVL1 breakapart | Oxford Gene Technology | Cat# LPH 036-SA |
| TCRB breakapart | Oxford Gene Technology | Cat# LPH 048-SA |
| MLLT1 | Oxford Gene Technology | Cat# LPH 508-A |
| MLLT3 | Oxford Gene Technology | Cat# LPH 509-A |
| Chromosome 11 Alpha Satellite Red | Oxford Gene Technology | Cat# LPE 011R-A |
| E2A breakapart | Oxford Gene Technology | Cat# LPH 019-SA |
| 5q del probe | Oxford Gene Technology | Cat# LPH 024 |
| 19p probe | Oxford Gene Technology | Cat# LPT19 PR-A |
| NMYC/LAF4 probe | Oxford Gene Technology | Cat# LPS-009A |
| MLL adjacent probe (CD3) | Empire Genomics | RPCI-11 215H18 |
| Kmt2a/Chr9 Mouse | Empire Genomics | Mouse KMT2A-Chr09 |
| Software and algorithms | ||
| Scaffold 6.0 | 3i-intelligent imaging Innovations | https://www.intelligent-imaging.com/slidebook |
| BWA 0.7.13 | Li et al.78 | https://biobwa.sourceforge.net/bwa.shtml |
| DeepTools 2.4.3 | Ramirez et al.79 | https://deeptools.readthedocs.io/en/develop/content/installation.html |
| HOMER v4.10.3 | Heinz et al.80 | http://homer.ucsd.edu/homer/ |
| DiffBind | Ross-Innes et al.81 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
Code
No original code was used in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.