

Abstract
Lysergic Acid Diethylamide (LSD), a semi-synthetic ergoline alkaloid analogue and hallucinogen, is a potent psychoplastogen with promising therapeutic potential. While a variety of synthetic strategies for accessing ergoline alkaloids have emerged, the complexity of the tetracyclic ring system results in distinct challenges in preparing analogues with novel substitution patterns. Methods of modulating the hallucinogenic activity of LSD by functionalization at previously inaccessible positions are of continued interest, and efficient syntheses of the ergoline scaffold are integral towards this purpose. Here, we report novel C–C bond forming strategies for preparing the ergoline tetracyclic core, focused on the relatively unexplored strategy of bridging the B- and D-ring systems last. Following cross-coupling to first join the A- and D-rings, we explored a variety of methods for establishing the C-ring including intramolecular α-arylation, borrowing hydrogen alkylation, and rhodium-catalyzed C–H insertion. Our results led to a 7-step formal synthesis of LSD and the first methods for readily introducing substitution on the C-ring. These strategies are efficient for forming ergoline-like tetracyclic compounds and analogues, though they each face unique challenges associated with elaboration to ergoline natural products. Taken together, these studies provide important insights that will guide future synthetic strategies towards ergolines.
GRAPHICAL ABSTRACT

INTRODUCTION
Lysergic acid diethylamide (LSD) (Figure 1A) is a semi-synthetic ergoline alkaloid that has shown promise for the treatment of a variety of neuropsychiatric disorders such as addiction and anxiety.,1,2 Neuronal atrophy in the cortex is a key pathological feature of a variety of neuropsychiatric disorders,3,4,5 and LSD is a potent psychoplastogen,6 possessing the remarkable ability to rapidly promote cortical neuron growth and increase dendritic spine density.7,8 However, LSD is also among the most potent hallucinogenic compounds discovered to date,9 and this characteristic has hindered its development as a therapeutic. The advent of nonhallucinogenic psychoplastogens has established that in principle, it might be possible to reduce the hallucinogenic properties of LSD while maintaining its beneficial effects on cortical neuron growth.10,11,12
Figure 1. Bridging the B- and D-rings is an uncommon strategy for the synthesis of ergolines.

(A) Ergoline numbering and retrosynthetic disconnections for the de novo construction of lysergic acid and LSD. The methylene unit bridging the B- and D-rings B is highlighted in blue. The bond bridging the A- and D-rings is highlighted in orange. *Hendrickson’s synthesis of lysergic acid has been disputed. (B) Reported synthesis of lysergic acid by Hendrickson (top). Subsequently, Nichols attempted to replicate this synthesis and instead observed a rearrangement of 1 to 3. Nichols was unable to convert 3 into lysergic acid using a number of conditions, including those reported by Hendrickson (bottom). (C) Vollhardt’s short synthesis of (±)-LSD via cobalt-mediated cyclization and late-stage reduction.
As a class, ergolines have demonstrated a wide-range of exceptional biological properties, and their structural complexity has inspired a variety of synthetic strategies, including many methods for the preparation of lysergic acid, the direct precursor to LSD.13,14,15 While certain structural modifications are possible via semisynthesis from naturally sourced lysergic acid, the efficient preparation of unnatural LSD analogues that may modulate hallucinogenic potential and psychoplastogenicity remains a significant challenge using existing total synthesis strategies.
Lysergic acid diethylamide (LSD) is typically prepared via semisynthesis from lysergic acid, which itself is derived from ergotamine and other lysergamides present in ergot. However, due to biosynthesis of lysergic acid from tryptophan, analogues of LSD substituted at ring A are not readily available.16 The biological activities of simpler tryptamine-based compounds can be dramatically altered by A-ring substitution,17 however such analogues of LSD have not materialized. Since the first total synthesis of lysergic acid over 60 years ago, dozens of ergoline alkaloid synthetic strategies have emerged,14 yet only two examples of A-ring substituted analogues have been reported—a C13-fluorinated analogue of lysergol, and a recently reported C12-chlorinated lysergic acid derivative.18,19 Moreover, no methods published to date have enabled the reliable introduction of substituents on the C-ring. The limited study of unnatural ergoline analogues, including those with A- and C-ring substitution, is unsurprising given the synthetic complexity of the ergoline scaffold. To facilitate structure-activity relationship (SAR) studies, novel approaches for constructing the tetracyclic ergoline core will be necessary.
Structurally, LSD and related ergolines are comprised of an indole (ring AB) and a tetrahydropyridine (ring D) bridged by a central C-ring (Figure 1A). Many strategies have emerged for synthesizing this tetracyclic ring system, with the majority of modern syntheses joining rings A and D via sp2–sp2 coupling in the final stage.20,21,22,23 Initial efforts employing this strategy were relatively inefficient, though several recent useful improvements have streamlined this approach and should be considered for future efforts towards LSD analogues.24,19 In sharp contrast, strategies that join rings A and D prior to the closure of ring C have received relatively little attention. Only a single example has been reported using this strategy, and subsequent evidence questioned whether lysergic acid could be produced using this synthetic route (Figure 1B). Hendrickson first reported the synthesis of key intermediate 1 by Suzuki coupling. Base-mediated cyclization was purported to form tetracyclic alcohol 2 (Figure 1B), which led to the completed synthesis of lysergic acid in a few short steps thereafter.25 However, later attempts to replicate this methodology were unsuccessful. A full study by Nichols suggested that these results may have been misinterpreted, and that the treatment of 1 with NaOMe actually led to the formation of 3 (Figure 1B).26 This intermediate could not be further converted to the final ergoline scaffold under the conditions originally reported by Hendrickson.
Regardless of these issues, the formation of an ergoline-like tetracycle retaining the indole and pyridine prior to a late-stage reduction appeared promising as a highly efficient synthetic strategy towards LSD. Moreover, Vollhardt had reported that tetracycle 4 could be formed following a challenging cobalt-mediated cyclization, which could be further converted to LSD by pyridine N-methylation and reduction (Figure 1C).27 Given this result, we hypothesized that the source of instability in 2 was related to the additional oxidation of the C ring and that structures lacking a leaving group on the C ring, such as 4 or a related derivative, would be viable for further conversion to LSD. Accessing such an intermediate could potentially provide a new route to the ergoline scaffold with increased efficiency and versatility.
RESULTS AND DISCUSSION
We envisioned intercepting 4 en route to LSD following the decarboxylation of an ester substituted tetracycle (Figure 2A). We anticipated that the requisite tetracyclic compound could be formed by Suzuki coupling to join rings A and D, followed by intramolecular α-arylation of a pyridylacetate with a halogenated indole. As indole halogenation and decarboxylation of pyridylacetates28 are typically facile, we suspected that the key to this retrosynthetic approach would be accessing a uniquely substituted pyridylacetate
Figure 2. Intramolecular α-arylation for the construction of ergoline-like tetracycles.

(A) Retrosynthetic analysis for preparing LSD via an α-arylation strategy. (B) Synthesis of 12, an unstable ergoline-like tetracycle. (C) Synthesis of tetracyclic compound 15. (D) 15 and its deprotected counterpart 17 are indefinitely stable to a variety of indole reduction conditions.
Our synthesis commenced with the conversion of commercially available carboxylic acid 5 to its tert-butyl ester 6 using Boc2O and catalytic DMAP.29 Subsequently, we found that 6 underwent SNAr displacement of the chloride with diethyl malonate in near quantitative yields. Prior to this discovery, we explored similar SNAr reactivity with a diethyl amide derivative of 5. However, minimal product was observed when the amide was employed, presumably due to the subtle decrease in electron withdrawing strength of the amide relative to the ester. After forming the pyridylmalonate, the tert-butyl group was readily deprotected using methanesulfonic acid in MeCN to produce 7, which was used without further purification. The resulting carboxylic acid was converted to the diethylamide 8 following treatment with carbonyldiimidazole (CDI) and Et2NH. The 3-step procedure transforming 6 to 8 proceeded in excellent yield (79%). The malonate substituent was readily hydrolyzed by aqueous NaOH in methanol to form the monocarboxylate salt, which was isolated as its respective acid following pH adjustment during extraction. This unpurified material was subjected to esterification with TMSCl in MeOH to afford pyridylacetate intermediate 9 (Figure 2B).30
After extraction, 9 was used directly in a Suzuki coupling with indole-4-Bpin to form 10 in 90% yield over 3-steps. Iodination of the indole with N-iodosuccinimide proceeded rapidly, and subsequent indole N-Boc protection produced 11 in 84% yield over both steps. To achieve the key α-arylation cyclization, we first employed standard Pd(0)-catalyzed conditions for enolate arylation, but these reaction conditions did not yield any product. Fortunately, we discovered that conditions utilizing Cu(I) catalysis, originally developed for the coupling of malonates with aryl halides,31 proved effective for forging the key C–C bond. Formation of a 6-membered bidentate copper enolate complex has been proposed to be instrumental for the coupling of malonates with aryl halides,32,33 and we suspect that an analogous bidentate copper enolate complex is responsible for the high reactivity of 11 in this reaction. While C–C bond formation proceeded smoothly with clean conversion by LC-MS analysis, we unfortunately discovered that 12 was highly unstable and could not be isolated. Upon exposure to air, LC-MS analysis revealed that the mass of an oxygen atom had been added to compound 12. Given that C4 was benzylic and adjacent to two carbonyl derivatives, we hypothesize that initial α-oxidation may precede further decomposition. Similar α-hydroxylations in air are known to occur with malonates.34
While it was challenging to isolate the decomposition products of 12, we reasoned that we could test this hypothesis by eliminating the proposed decomposition pathway entirely by establishing a quaternary center at C4 between rings B and C (Figure 2C). Returning to malonate intermediate 8, we performed an analogous Suzuki coupling, indole C3-iodination, and indole protection to afford the pre-cyclization intermediate 14 in excellent yield (87% over 3 steps). The same Cu(I)-catalyzed α-arylation conditions yielded tetracycle 15 in 92% yield, which was readily isolable with no observed stability issues, supporting our hypothesis about the decomposition of 12. However, in this case, hydrolysis and decarboxylation with aqueous LiOH in MeOH followed by acidification only resulted in monodecarboxylation. Concerningly, N-Boc groups are typically cleaved from aromatic systems such as indole under these conditions, but we observed no deprotection either. A series of NMR and LC-MS experiments confirmed that the product obtained was 16, not unlike the aromatic tricyclic structure 3 previously observed by Nichols.26
Among ideas to solve this rearrangement issue, we felt the most viable was to reduce indole 15 to the indoline prior to decarboxylation, leaving the system unable to rearrange (Figure 2D). However, no reactivity was observed under various reduction conditions that typically reduce N-Boc indoles including Pd/C in alcoholic solvents or polymethylhydrosiloxane and Pd(OH)2,35 even at pressures up to 100 bar or extensive heating. Next, we deprotected the indole to form 17 in the hopes that we could reduce a more electron-rich indole using standard conditions such as NaCNBH3 with AcOH or TFA with Et3SiH.36,37 Unfortunately, 17 was unreactive under these conditions. While the presence of the quaternary center in 15 prevented unfavorable rearrangements, it also hindered our attempts to convert 15 to LSD or related analogues. Thus, we ultimately concluded that our α-arylation strategy was not a viable option for accessing ergolines, despite the ease of tetracycle formation.
Next, we revised our strategy to focus on directly accessing 4 without proceeding through an intermediate possessing C-ring substitution. An attractive option was to prepare a 2-(hydroxymethyl)pyridyl substituted indole via Suzuki coupling as before, and perform the final cyclization using borrowing hydrogen indole alkylation methodology38 (Figure 3A). We hoped that by avoiding C-ring ester substituents we would also circumvent the need for decarboxylation, thus mitigating the potential for undesired rearrangements like we had observed previously. To access this intermediate, we performed an SNAr reaction with commercially available methyl 5-bromo-6-chloronicotinate (18) and diethyl malonate. Global hydrolysis and decarboxylation afforded 19 in 85% yield over two steps, with purification being easily accomplished via direct precipitation and filtration from the reaction mixture. Activation of the carboxylic acid with CDI and displacement of the acyl imidazole with Et2NH yielded the unpurified diethylamide, which was oxidized with mCPBA to form N-oxide 20 in 91% over two steps. Boekelheide rearrangement mediated by trifluoroacetic anhydride (TFAA) and subsequent hydrolysis of the resulting trifluoroacetate during workup provided the key 2-(hydroxymethyl)pyridine 21 in 86% yield.39 Suzuki coupling to indole-4-Bpin afforded 22 without issue (Figure 3B).
Figure 3. Intramolecular borrowing hydrogen indole alkylation for the construction of ergoline-like tetracycles.
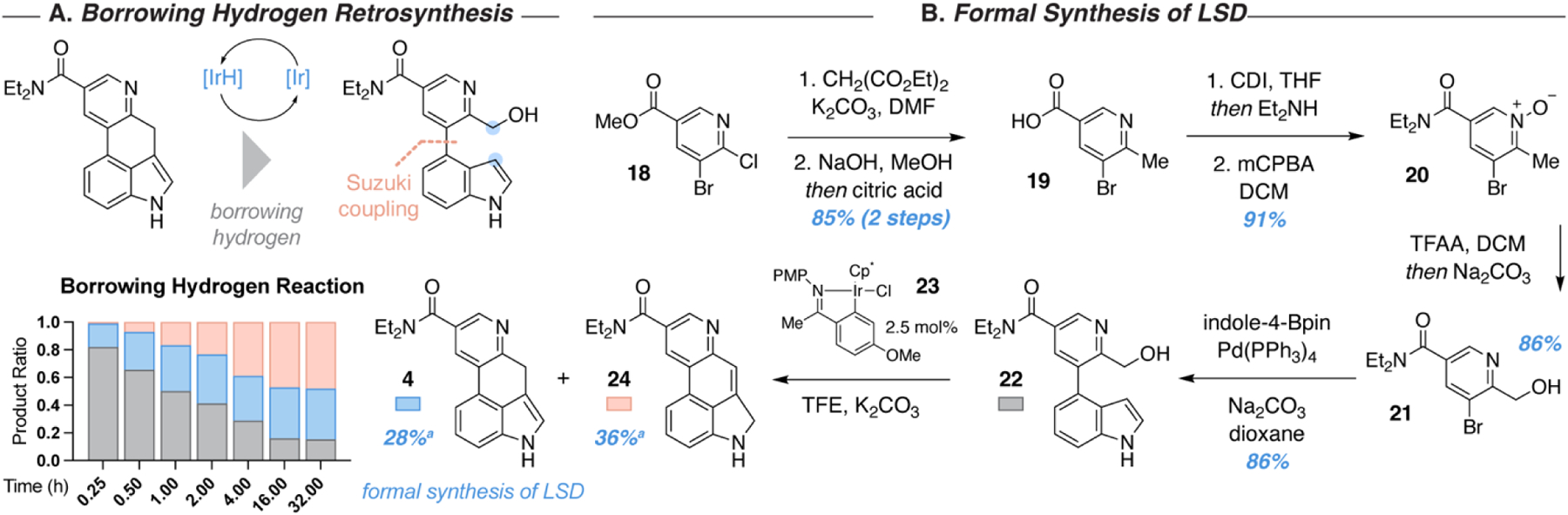
(A) Retrosynthetic analysis for the preparation of 4. (B) Formal synthesis of LSD by preparation of 4 by a Suzuki coupling and borrowing hydrogen indole alkylation strategy. aNMR yield determined using an internal standard.
Next, we subjected 22 to borrowing hydrogen indole alkylation conditions. Typically, these types of reactions require a large excess of one component, but an intramolecular process necessitates an equal ratio of alcohol and indole components. Thus, we elected to utilize the highly effective cyclometalated iridium(III) complex 23, which has been reported to catalyze these reactions without requiring an excess of either component.40 Reactivity was high under these conditions, although we found that two isomeric products of the same mass were formed in approximately equal amounts. These products were identified as 4 and 24 on the basis of NMR experiments and were formed in moderate yields (64% combined yield). It appears that 4 is the product of kinetic reduction, as it is the only product formed initially. However, 24 is calculated to be ~0.02 kcal/mol more stable,41 and over the course of the reaction, the concentration of 24 increases over time until the ratios of 4 and 24 are stable, suggesting that the initially formed 4 may be rapidly converted into a thermodynamic ratio of 4 and 24 (Figure 3B).
The spectral data of 4 matched that reported by Vollhardt, completing a 7-step formal synthesis of LSD.27 Despite the efficiency of this overall route, we found that the reported intermediate 4 is unstable, albeit less so than intermediate 12. Fortunately, 4 and 24 are separable by chromatography on either silica or alumina, but after isolation and concentration, impurities form quickly. Many related compounds in the initial report by Vollhardt and co-workers were reported to be unstable. However, in depth information about the stability of 4 was not disclosed. Additionally, attempts to re-purify the intermediates resulted in partial decomposition and loss of material, and regardless of conditions, traces of 24 and other impurities appear in the spectra of 4. Due to the late-stage Suzuki coupling, it is plausible that this strategy would allow for versatile differentiation of ring A by using substituted indole-4-Bpin compounds, but the practical challenges associated with compound stability and lack of selectivity following C-ring closure dissuaded us from pursuing this path further. Given that 24 is slightly more stable than 4, we assume that any tetracyclic structure of this class containing both an indole and pyridine would likely suffer from isomerization to the benzoquinoline tautomer.
Despite the ease and efficiency of preparing various advanced intermediates using the intramolecular α-arylation and borrowing hydrogen indole alkylation strategies, we ultimately determined that stability issues associated with pyridine- and indole-containing tetracycles would limit the usefulness of these strategies. To circumvent this issue, we elected to reduce the pyridine prior to the C–C bond-forming reaction that would bridge the B- and D-rings. We envisioned that Suzuki coupling of an N-protected tetrahydropyridine would join the A- and D-rings as before, but subsequent direct allylic C–H insertion of an indole-3-carbaldehyde derived carbene would complete the ergoline core scaffold (Figure 4A). The insertion of donor/acceptor carbenes into C–H bonds adjacent to nitrogen centers is well precedented with pioneering work by Davies and co-workers.42,43 However, we hoped that a donor carbene would exhibit similar reactivity.
Figure 4. Intramolecular C–H insertion for the construction of ergoline-like tetracycles.

(A) Retrosynthetic analysis of LSD synthesis via C–H insertion. (B) While C-H insertion from an aldehyde-derived carbene was unsuccessful, it was possible to achieve this transformation using a donor-acceptor stabilized carbene.
Starting from readily available 5-bromonicotinic acid (25), we synthesized the diethylamide by treating the acid with oxalyl chloride under DMF catalysis followed by quenching with Et2NH. The unpurified mixture was methylated using MeI in EtOAc, from which the resulting pyridinium salt 26 readily precipitated over the course of the reaction. This compound was then reduced with NaCNBH3 in acidic methanol to afford tetrahydropyridine 27. The N-methyl group was exchanged for an N-carbamoyl group to attenuate the nucleophilicity of the nitrogen atom. Derivative 28 was obtained following treatment of 27 with ethyl chloroformate and loss of chloromethane.44 Suzuki coupling with indole-4-Bpin generated 29 in excellent yield (84%). From here, we prepared indole-3-carbaldehyde derivative 30 by Vilsmeier-Haack formylation, which was then protected as the tert-butyl carbamate 31. Condensation with hydrazine readily afforded 32 (Figure 4B).
We initially attempted to use MnO2 as the oxidant and Rh2OAc4 as the catalyst for achieving a one-pot oxidation and C–H insertion transformation as has been reported previously.45 Many examples exist for performing this reaction using donor/acceptor or donor/donor carbenes, but there are a limited number of examples that employ electron-rich donor carbenes like our substrate, presumably due to the fact that electron-rich donor carbenes exhibit diminished stability relative to carbenes derived from donor/acceptor diazo compounds.46,47 We confirmed that MnO2 was capable of oxidizing hydrazone 32 to the corresponding diazo compound, as subsequent treatment with benzoic acid yielded the corresponding benzoate ester in 46% yield (see SI for details). Similarly, we found that Swern oxidation also efficiently converted 32 to its corresponding diazo compound.48 However, the addition of a Rh(II) catalyst (e.g., Rh2(OAc)4, Rh2(esp)2, Rh2(R-PTAD)4) never produced the desired C–H insertion product 33, and instead yielded only aldehyde 31.
Given that prior work demonstrated facile insertion of donor/acceptor rhodium carbenoids into related tetrahydropyridine systems, we opted to assess the reactivity of a donor/acceptor carbenoid as well. We used 29 to prepare methyl indole-3-glyoxylate derivative 34 by acylation with oxalyl chloride and subsequent addition of methanol. This material was protected, as before, to afford 35 in 89% yield over two steps. Condensation with hydrazine49 proceeded cleanly, and subsequent C–H insertion using MnO2 as the oxidant in the presence of Rh2OAc4 generated the desired ergoline compound 36 in 60% yield over both steps as a single diastereomer. Based on these results, it appears that the success of this intramolecular C–H insertion reaction is highly dependent on the metal-carbenoid electrophilicity, which is increased with donor-acceptor substitution. Relative configuration of 36 was tentatively assigned based on a combination of COSY and NOESY NMR experiments (Figure S1, see Supporting Information for details).
While this C–H insertion methodology was ultimately successful in forming the core ergoline scaffold, several challenges must be overcome to prepare LSD or related derivatives. First, due to the required presence of the ester group for insertion to proceed, a challenging decarboxylation still must be developed, followed by further deprotection and methylation. Additionally, the C–H insertion route is lengthy, which is problematic for planned derivatives, particularly because the indole is installed in the early stages. Despite these disadvantages, the prospect of imparting enantioselectivity via chiral Rh(II) catalysis is promising.43 However, the fact that we only observed a single diastereomer suggests that a chiral catalyst would have to overcome the inherent diastereoselectivity that is dictated by the relative stereochemistry of the amide substituent and associated conformation of the tetrahydropyridine, otherwise only diastereomer kinetic resolution is viable. Alternatively, asymmetric reduction of 26 to produce an enantiopure tetrahydropyridine would allow for diastereoselective C–H insertion without a chiral catalyst. However, this may also prove challenging as C8 will likely be epimerized by base during the Suzuki coupling or other intermediary steps prior to C–H insertion.50 If epimerization proves problematic, then replacement of the diethyl amide with a chiral auxiliary to induce thermodynamic preference of one epimer at C8 could similarly solve this issue, and these factors should be considered in subsequent C–H insertion methods for forming ergolines.
For future synthetic efforts towards LSD and related ergolines, we caution others from pursuing routes in which the aromaticity of the pyridine and indole rings both remain intact prior to formation of the tetracyclic core. Regardless of substitution or lack thereof, our work and previous reports in the literature suggest that these types of structures are prone to oxidation, rearrangement, and decomposition. The further development of C–H insertion methodology for ergoline synthesis could be advantageous and streamline efforts to develop indole-substituted ergoline analogues, especially if enantioselective methods could be developed utilizing donor carbenes. Despite the many challenges encountered in this work, we report several efficient strategies for constructing the tetracyclic core common to ergoline natural products and related analogues like LSD. As the ergoline core is capable of displaying a wide-range of important biological activities, we hope that our work will prove useful for the development of novel ergoline analogues with medicinal value.
Supplementary Material
Acknowledgments
We would like to thank Dean Tantillo for assisting with stability calculations. This work was supported by funds from the National Institutes of Health (NIH R01GM128997 and NIH R35GM148182 to DEO). Funding for NMR spectrometers was provided by the National Science Foundation (NSF 9808183) and National Institutes of Health (NIH NIEHS ES005707–13). Analysis for this project was performed in the UC Davis Campus Mass Spectrometry Facilities, with instrument funding provided by the NIH (NIH 1S10OD025271–01A1).
Footnotes
Supporting Information
The Supporting Information is available free of charge at (article link).
Detailed synthetic procedures, experimental data for all compounds, and 1H and 13C NMR Spectra (PDF)
Disclosure
DEO is a co-founder of Delix Therapeutics, Inc., serves as the chief innovation officer of head of the scientific advisory board, and has sponsored research agreements with Delix Therapeutics. Delix has licensed technology from UC Davis related to analogues of ergoline alkaloids.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
References
- 1.Fuentes JJ, Fonseca F; Elices M; Farré M; Torrens M Therapeutic use of LSD in psychiatry: a systematic review of randomized-controlled clinical trials. Front Psychiatry 2020, 10:943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dos Santos RG; Osório FL; Crippa JAS; Riba J; Zuardi AW; Hallak JEC Antidepressive, anxiolytic, and antiaddictive effects of ayahuasca, psilocybin and lysergic acid diethylamide (LSD): a systematic review of clinical trials published in the last 25 years. Ther Adv Psychopharmacol 2016, 6, 3, 193–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pittnenger C; Duman RS; Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacol 2008, 33, 88–109. [DOI] [PubMed] [Google Scholar]
- 4.Duman RS; Aghajanian GK; Sanacora G; Krystal JH Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat. Med 2016, 22, 238–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vargas MV; Meyer R; Avanes AA; Rus M; Olson DE Psychedelics and other psychoplastogens for treating mental illness. Front Psychiatry 2021, 12, 727117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson DE Psychoplastogens: A Promising Class of Plasticity-Promoting Neurotherapeutics. J. of Exp. Neurosci 2018, 12, 1–4. 10.1177/1179069518800508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ly C; Greb A; Cameron P; Wong J; Barragan E; Wilson P; Burbach K; Zarandi S; Sood A; Paddy M; Duim W; Dennis M; McAllister A; Ori-McKinney K; Gray J; Olson D Psychedelics promote structural and functional neural plasticity. Cell Rep 2018, 23, 3170–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ly C; Greb CA; Vargas MV; Duim WC; Grodzki ACG; Lein PJ; Olson DE Transient Stimulation with Psychoplastogens is Sufficient to Initiate Neuronal Growth. ACS Pharmacol. Transl. Sci, 2020, 4, 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Passie T; Halpern JH; Stichtenoth DO; Emrich HM; Hintzen A The pharmacology of lysergic acid diethylamide: a review. CNS Neurosci. Ther 2008, 14, 4, 263–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunlap LE; Azinfar A; Ly C; Cameron LP; Viswanathan J; Tombari RJ; Myers-Turnbull D; Taylor JC; Grodzki AC; Lein PJ; Kokel D; Olson DE Identification of Psychoplastogenic N,N-Dimethylaminoisotryptamine (isoDMT) Analogs Through Structure-Activity Relationship Studies. J. Med. Chem, 2020, 63, 1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cameron LP; Tombari RJ; Lu J; Pell AJ; Hurley ZQ; Ehinger Y; Vargas MV; McCarroll MN; Taylor JC; Myers-Turnbull D; Liu T; Yaghoobi B; Laskowski LJ; Anderson EI; Zhang G; Viswanathan J; Brown BM; Tjia M; Dunlap LE; Rabow ZT; Fiehn O; Wulff H; McCorvy JD; Lein PJ; Kokel D; Ron D; Peters J; Zuo Y; Olson DE A Non-Hallucinogenic Psychedelic Analogue with Therapeutic Potential. Nature, 2021, 589, 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis V; Bonniwell EM; Lanham JK; Ghaffari A; Sheshbaradaran H; Cao AB; Calkins MM; Bautista-Carro MA; Arsenault E; Telfer A; Taghavi-Abkuh F; Malcolm NJ; El Sayegh F; Abizaid A; Schmid Y; Morton K; Halberstadt AL; Aguilar-Valles A; McCorvy JD A non-hallucinogenic LSD analog with therapeutic potential for mood disorders. Cell Rep, 2023, 42(3):112203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schiff PL Jr. Ergot and its alkaloids. Am J Pharm Educ 2006, 70, 5, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H; Jia Y Ergot alkaloids: synthetic approaches to lysergic acid and clavine alkaloids. Nat. Prod. Rep 2017, 34, 4, 411–432. [DOI] [PubMed] [Google Scholar]
- 15.Chen J; Han M; Gong T; Yang J; Zhu P Recent progress in ergot alkaloid research. RSC Adv 2017, 7, 27384. [Google Scholar]
- 16.Tasker NR; Wipf P Biosynthesis, total synthesis, and biological profiles of ergot alkaloids. Alkaloids. Chem. Biol 2021,85, 1–112. [DOI] [PubMed] [Google Scholar]
- 17.Glennon RA; Young R; Jacyno JM; Slusher M; Rosecrans JA DOM-stimulus generalization to LSD and other hallucinogenic indolealkylamines. Eur. J. Pharmacol 1983, 86, 453–59. [DOI] [PubMed] [Google Scholar]
- 18.Yuan H; Guo Z; Luo T Synthesis of (+)-lysergol and its analogues to assess serotonin receptor activity. Org. Lett 2017, 19, 3, 624–627. [DOI] [PubMed] [Google Scholar]
- 19.Knight BJ; Harbit RC; Smith JM Six-step synthesis of (±)-lysergic acid. J. Org. Chem 2023, 88, 4, 2158–2165. [DOI] [PubMed] [Google Scholar]
- 20.Kurokawa T; Isomura M; Tokuyama H; Fukuyama T Synthesis of lysergic acid methyl ester via the double cyclization strategy. Synlett 2009, 5, 0775–0778. [Google Scholar]
- 21.Umezaki S; Yokoshima S; Fukuyama T Total synthesis of lysergic acid. Org. Lett 2013, 15, 16, 4230–4233. [DOI] [PubMed] [Google Scholar]
- 22.Liu Q; Jia Y Total synthesis of (+)-lysergic acid. Org. Lett 2011, 13, 18, 4810–4813. [DOI] [PubMed] [Google Scholar]
- 23.Liu Q; Zhang Y; Xu P; Jia Y Total synthesis of (+)-lysergic acid. J. Org. Chem 2013, 78, 10885–10893. [DOI] [PubMed] [Google Scholar]
- 24.Tasker NR; Wipf P A short synthesis of ergot alkaloids and evaluation of the 5-HT1/2 receptor selectivity of lysergols and isolysergols. Org. Lett 2022, 24, 40, 7255–7259. [DOI] [PubMed] [Google Scholar]
- 25.Hendrickson JB; Wang J A new synthesis of lysergic acid. Org. Lett 2004, 6, 1, 3–5. [DOI] [PubMed] [Google Scholar]
- 26.Bekkam M; Mo H; Nichols DE A reported “new synthesis of lysergic acid” yields only the derailment product: methyl 5-methoxy-4,5-dihydroindolo[4,3-f,g]quinoline-9-carboxylate. Org. Lett 2012, 14, 1, 296–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saá C; Crotts DD; Hsu G; Vollhardt KPC A cobalt-catalyzed entry into the ergot alkaloids: total syntheses of (±)-lysergene and (±)-LSD. Synlett 1994, 7, 487–489. [Google Scholar]
- 28.Donald C; Boyd S An efficient synthesis of 2-alkylpyridines using an alkylation/double decarboxylation strategy. Tetrahedron Lett 2012, 53, 30, 3853–3856. [Google Scholar]
- 29.Burgey CS; Ginetti A; Ashley N; Paone DV Fused heterocyclic azaindane carboxamide CGRP receptor antagonists. WO 2013066360, May, 10, 2013. [Google Scholar]
- 30.Li J; Sha Y A convenient synthesis of amino acid methyl esters. Molecules 2008, 13, 5, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yip SF; Cheung HY; Zhou Z; Kwong FY Room-tempearture copper-catalyzed α-arylation of malonates. Org. Lett 2007, 9, 17, 3469–3472. [DOI] [PubMed] [Google Scholar]
- 32.Hennessy EJ; Buchwald SL A general and mild copper-catalyzed arylation of diethyl malonate. Org. Lett 2002, 4, 2, 269–272. [DOI] [PubMed] [Google Scholar]
- 33.Huang Z; Hartwig JF Copper(I) enolate complexes in α-arylation reactions: synthesis, reactivity and mechanism. Angew. Chem. Int. Ed 2012, 51- 1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe T; Ishikawa T Mild air-oxidation of 1,3-dicarbonyl compounds with cesium salts: novel α-hydroxylation accompanied by partial hydrolysis of malonate derivatives. Tetrahedron Lett 1999, 40, 7795–7798. [Google Scholar]
- 35.Chandrasekhar S Palladium-catalyzed reduction of N-(tert-butoxycarbonyl)indoles by polymethylhydrosiloxane. Synthesis 2007, 10, 1509–1512. [Google Scholar]
- 36.Kumar Y; Florvall L Convenient synthesis of indolines by reduction of indoles with sodium cyanoborohydride in carboxylic acids. Synth. Commun 1983, 13, 6, 489–493. [Google Scholar]
- 37.Lanzilotti AE; Littell R; Fanshawe WJ; McKenzie TC; Lovell FM Stereoselective reduction of some indoles with triethylsilane-trifluoroacetic acid. J. Org. Chem 1979, 44, 26, 4809–4813. [Google Scholar]
- 38.Bartoccini F; Retini M; Piersanti G C3-alkylation of indoles and oxindoles by alcohols by means of borrowing hydrogen methodology. Tetrahedron Lett 2020, 61, 22, 151875. [Google Scholar]
- 39.Fontenas C; Bejan E; Haddou HA; Balavoine GGA The Boekelheide reaction: trifluoroacetic anhydride as a convenient acylating agent. Synth. Commun 1995, 25, 5, 629–633. [Google Scholar]
- 40.Jiang X; Tang W; Xue D; Xiao J; Wang C Divergent dehydrogenative coupling of indolines with alcohols. ACS Catal 2017, 7, 1831–1835. [Google Scholar]
- 41.Calculated using Gaussian 16, Revision C.01, SMD(TFE)-M06–2X/6–311+G(2d,p)//M06–2X/6–31+G(d,p),; Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian, Inc, Wallingford CT, 2016. [Google Scholar]
- 42.Davies HML; Venkataramani C; Hansen T; Hopper DW New strategic reactions for organic synthesis: catalystic asymmetric C-H activation α to nitrogen as a surrogate for the mannich reaction. J. Am. Chem. Soc 2003, 125, 6262–6468. [DOI] [PubMed] [Google Scholar]
- 43.Davies HML; Beckwith REJ Catalytic enantioselective C-H activation by means of metal-carbenoid-induced C-H insertion. Chem. Rev 2003, 103, 2861–2903. [DOI] [PubMed] [Google Scholar]
- 44.Časar Z; Mesar T A DMAP-catalyzed approach to the industrial-scale preparation of N-6-demethylated 9,10-dihyrolysergic acid methyl ester: a key cabergoline and pergolide precursor. Org. Process Res. Dev 2014, 19, 378–385. [Google Scholar]
- 45.Soldi C; Lamb KN; Squiteri RA; González-López M; Di Maso MJ; Shaw JT Enantioselective intramolecular C-H insertion reaction of donor-donor metal carbenoids. J. Am. Chem. Soc 2014, 136, 43, 15142–15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bergstrom BD; Nickerson LA; Shaw JT; Souza LW Transition metal catalyzed insertion reactions with donor/donor carbenes. Angew. Chem. Int. Ed 2021, 60, 13, 6864–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu D; Chen L; Fan H; Yao Q; Zhu S Recent progress on donor and donor-donor carbenes. Che. Soc. Rev 2020, 49, 908. [DOI] [PubMed] [Google Scholar]
- 48.Javed MI; Brewer M Diazo preparation via dehydrogenation of hydrazones with “activated” DMSO. Org. Lett 2007, 9, 9, 1789–1792. [DOI] [PubMed] [Google Scholar]
- 49.Nicolle SM; Moody CJ Potassium N-iodo p-toluenesulfonamide (TsNIK, iodamine-T): a new reagent for the oxidation of hydrazones to diazo compounds. Chem. Eur. J 2014, 20, 15, 4420–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salamone SJ; Li Z; McNally AJ; Vitone S; Wu RS Epimerization studies of LSD using 1H nuclear magnetic resonance (NMR) spectroscopy. J. Anal. Toxicol 21, 492–497 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.