

Abstract
Peptides that bind to inorganic materials can be used to functionalize surfaces, control crystallization, or assist in interfacial self-assembly. In the past, inorganic-binding peptides have been found predominantly through peptide library screening. While this method has successfully identified peptides that bind to a variety of materials, an alternative design approach that can intelligently search for peptides and provide physical insight for peptide affinity would be desirable. In this work, we develop a computational, physics-based approach to design inorganic-binding peptides, focusing on peptides that bind to the common plastics polyethylene, polypropylene, polystyrene, and PET. The PepBD algorithm, a Monte Carlo method that samples peptide sequence and conformational space, was modified to include simulated annealing, relaxed hydration constraints, and an ensemble of starting conformations. These modifications led to the discovery of peptides with significantly better scores compared to those obtained using original PepBD. PepBD scores were found to improve with increasing van der Waals interactions, although strengthening the intermolecular van der Waals interactions comes at the cost of introducing unfavorable electrostatic interactions. The best designs are enriched in amino acids with bulky sidechains and possess hydrophobic and hydrophilic patches whose location depends on the adsorbed conformation. Future work will evaluate the top peptide designs in experiment, enabling their application in microplastic pollution remediation and plastic-based biosensors.
Keywords: Computational peptide design, solid-binding peptides, Monte Carlo, microplastic remediation
Introduction
Discovering peptides that bind to inorganic materials is useful for developing new biotechnological tools like biosensors1, biological solar cells2, or bio-based mineralization processes3. Peptides that bind to a variety of inorganic materials including gold4, silica5, and graphene6 have been found in previous work by others. The peptides were discovered primarily by using library screening, an experimental method that randomly samples a large number of peptides, often 109 or 1011 unique sequences, to search for those with affinity for a target material7. While library screening has had success in finding inorganic-binding peptides, it has some downsides, including the need for substantial human labor, potential bias in the identified peptides due to experimental design8, and the need for downstream experiments to quantitatively measure peptide affinity9. An alternative method of discovering inorganic-binding peptides that circumvents these issues is desirable.
Computational design is a promising approach for discovering peptides that bind to inorganic materials. The advantages of computational design include a systematic exploration of peptide sequences via design rules and intelligent sampling schemes, minimal human labor, and lower cost due to the reduction in experimental testing. Computational design can also provide insight into the physical basis for a peptide’s affinity to an inorganic material. Such insight opens the door for iterative design as the computational model is improved to match experimental results and/or to incorporate insights from previous designs. Understanding the physical basis for peptide affinity can also improve our ability to predict how peptide binding is affected by environmental conditions or surface properties such as pH or surface oxidation. This would open the door to designing peptides that bind specifically to certain materials or whose affinity can be modulated by changing environmental conditions.
A few computational methods exist for designing inorganic-binding peptides or for predicting peptide affinity to an inorganic material, which we now review. Rosetta Surface Design by Masica et al.10 searches for inorganic-binding peptides via Monte Carlo sampling of sequence and conformational space, and scores peptides with the Rosetta energy function. The method was used to design hydroxyapatite-binding peptides, which are essential in bone formation. Another approach was developed by Schwaminger et al.11 to design peptides that bind to ionic magnetic nanoparticles. Their procedure used experimental measurements of peptide adsorption free energy via fluorescence spectroscopy and computational measurements via Monte Carlo simulations to generate the next round of peptides for testing. Such an iterative procedure can also form the basis for Bayesian optimization, as was done by Hughes et al. who used library screening data to tune peptide selectivity for gold or silver12. Other inorganic-binding peptides have been designed on an ad hoc basis by choosing a secondary structural element, e.g. an alpha helix, that matches the periodicity of a feature on an inorganic surface, and then crafting a polypeptide sequence that folds into the chosen secondary structure. This approach was used by DeOliveira et al. to design a peptide that binds to calcite surfaces13 and by Pyles et al. to design a protein that self-assembles on mica surfaces14. While the simplicity of this approach is appealing, it only applies to inorganic surfaces with a targetable pattern, such as crystal faces. Finally, machine learning models have been developed to predict if a peptide will bind to an inorganic material. Especially relevant to this work is PS-Binder15,16, a support vector machine that predicts if a peptide will bind to polystyrene. While PS-Binder provides rapid predictions of whether or not a peptide will bind to polystyrene, it does not provide quantitative predictions of peptide affinity.
Our aim is to develop an improved method of computationally designing inorganic-binding peptides, and we chose plastics as the target inorganic material. Plastics were selected because plastic-binding peptides could be a useful tool for addressing micro- and nano-plastic (MNP) pollution. MNPs are a concern as millions of tons contaminate the oceans17, they are challenging to capture and detect, and they pose environmental and health concerns18. Plastic-binding peptides could mitigate the risks of MNPs by helping capture, detect, and degrade microplastic waste, as suggested by previous studies19,20. The large specific surface area of MNPs makes peptide-based remediation apt as MNPs expose a large area for interaction with peptides. Plastic-binding peptides also have many other possible applications outside of MNP remediation, such as antimicrobial surfaces and plastic-based biosensors.
This paper describes and evaluates a new computational method that designs solid-binding peptides. The Peptide Binder Design (PepBD) algorithm, previously developed in our group to design peptides that bind to proteins21,22,23,24, was modified to design peptides that bind to four of the most commonly used plastics: polyethylene, polystyrene, polypropylene, and PET. Three major modifications were made to PepBD to achieve this design goal. First, we used an ensemble of different adsorbed conformations of a peptide on a plastic surface as starting points for design, which improved sampling of the conformational space available to peptides. Second, we added simulated annealing to increase sampling of the conformational and sequence space local to each starting peptide conformation. Third, we relaxed the hydration constraints that govern the allowed peptide sequences, further increasing sampling of sequence space.
Highlights of our results include the following. We show that adding simulated annealing combined with multiple starting conformations and relaxed hydration constraints to the PepBD algorithm leads to the discovery of peptides with significantly better scores than the designs using unmodified PepBD. Analysis of the top peptides reveals that good scores are accompanied by van der Waals interactions that strongly promote adsorption and electrostatic interactions that weakly disfavor adsorption. The best peptide designs for the four plastics are enriched in amino acids with bulky sidechains like tryptophan and arginine, and the frequency of each amino acid in the top designs is positively correlated with the amino acid mass. We also find patches of hydrophobic and hydrophilic amino acids whose location depend on the peptide. Overall, our work is a promising step in the underexplored area of computationally designing solid-binding peptides.
Methods
Overview of Original PepBD Method
The original Peptide Binder Design (PepBD) method is a Monte Carlo algorithm that searches the sequence and conformational space of peptides over thousands of steps to find candidates that bind strongly to a receptor. The best peptide designs are identified by the score function shown in Equation 1 below, where a more negative score corresponds to stronger binding affinity. A simplified flowchart of the design algorithm is shown in Figure S1. A detailed description of the method can be found in previous papers21,22,23,24.
The PepBD algorithm can be broken into five steps. Step one is to obtain a starting structure in which the peptide is bound to the receptor. This step uses a reference peptide known to have affinity for the receptor and is performed outside of PepBD using methods such as docking. The structure of the receptor is fixed throughout the entire design process. Step two is to propose a random change to either the peptide’s sequence or conformation. A sequence change consists either of a point mutation or swapping two amino acids in the peptide. Point mutations must satisfy hydration constraints specified by the user; the hydration properties of amino acids are defined in Table 1. A conformational change consists of displacing a contiguous section of the peptide backbone using the concerted rotation algorithm25. Step three is to repack the sidechains using the Lovell rotamer library26 and the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm27 to minimize the energy of the peptide-receptor complex. Step four is to score peptides using the peptide-receptor binding energy calculated via molecular mechanics with generalized Born and surface area solvation (MM/GBSA)28
| (1) |
where is the change in internal energy from bond, bond angle, and dihedral angle terms, is the change in Coulombic energy, is the change in the van der Waals energy, and is the polar solvation free energy calculated by the generalized Born (GB) model. The deltas are the energies for the peptide-receptor complex minus the peptide and receptor energies in the same conformation but not interacting with each other. The peptide parameters used to calculate these energies were taken from the Amber ff14SB forcefield29. The parameters used to describe plastic interactions will be discussed in the section “Parameterization of the Plastics” below. We also note that the non-polar solvation free energy, , that is typically included in the MM/GBSA method is excluded in the score function as past designs show that this energy changes negligibly between peptide mutations (a discussion on this point is provided in the conclusion). Finally, step five is to accept or reject the peptide modification using the Metropolis criterion,
| (2) |
where is the Boltzmann constant, is a reference temperature that controls the likelihood of accepting a modification, and the scores are those for the peptide before and after the modification. The reference temperatures for sequence changes, , and conformation changes, , can be different. Steps two through five are repeated thousands of times to sample many peptide conformations and sequences.
Table 1.
Amino Acid Categorizations used by PepBD.
| Category | Amino Acids |
|---|---|
| Hydrophobic | Leu, Ile, Met, Phe, Trp, Tyr, Val |
| Hydrophilic | Asn, Gln, His, Ser, Thr |
| Anionic | Asp, Glu |
| Cationic | Arg, Lys |
| Other | Ala, Cys, Pro |
| Glycine | Gly |
Classifications are for pH of 7. Cysteine and proline were excluded from all designs in this paper.
We now describe the specific parameters used for peptide design using the original PepBD algorithm. The hydration and reference temperature parameters will be changed in PepBD modifications described in the sections “Incorporating Simulated Annealing into Peptide Design” and “Relaxing the Hydration Constraints”. All designed peptides are 12 residues long to match the length of peptides often used in library screening. Sequence changes were performed 67% of the time, while conformation changes were performed 33% of the time. When performing a sequence change, point mutations were performed 75% of the time and amino acid swaps were performed 25% of the time. Proline and cysteine were excluded from all designs to avoid potential issues with solid-phase peptide synthesis30,31,32, proline because its secondary amine has lower reactivity than the primary amine in all other canonical amino acids and cysteine because it is prone to oxidation. Designs using original PepBD were constrained to have exactly 5 hydrophobic amino acids, 5 hydrophilic amino acids, 1 cationic amino acid, and 1 anionic amino acid. This set of values, termed hydration constraints, was selected because they are a reasonable guess for plastic-binding peptides. The reference temperatures were chosen so and . It should also be noted that the score function in past designs included a term which was removed in this study. Briefly, the term is intended to penalize peptides that have a high energy when they adopt the bound state conformation in solution. The penalty is relevant when the peptide must adopt the bound conformation before binding, such as when binding to proteins with binding pockets that hinder peptide conformational changes after binding. The flat plastic surfaces used for designs in this paper do not restrict peptide conformation changes after adsorption, and thus the term was removed.
Reference Plastic-Binding Peptides
We searched for reference peptides known to have affinity for plastics. These peptides can produce starting conformations for PepBD and can serve as useful benchmarks for evaluating the efficacy of designs in future computational and experimental tests. We focus on four plastics in this study: polyethylene, polypropylene, polystyrene, and polyethylene terephthalate (PET). Suitable 12-residue peptides were discovered in published library screening studies for polyethylene32, polypropylene33, and polystyrene34. No such peptide could be found for PET, so a 25 residue protein domain termed Dermaseptin 1 (DS1) with affinity for PET20 was used as a starting point. To keep all the reference peptides the same length, we searched for a 12-residue subsequence of DS1 with high affinity for PET. This was accomplished by conducting molecular dynamics simulations on three different 12 residue subsequences of the 25-residue DS1 sequence in the vicinity of the PET surface: LWSTIKQKGKEA, AAKAAGQAALGA, and KGKEAAIAAAKA. The subsequence with the lowest intermolecular potential energy of interaction with the PET surface was selected as the reference peptide. Combining this peptide with the reference peptides for the other plastics gives the final list of reference sequences shown in Table 2.
Table 2.
Reference Sequences for Plastic-Binding Peptides.
| Plastic | Polyethylene | Polypropylene |
| Sequence | HNKSSPLTAALP | TSDIKSRSPHHR |
| Plastic | Polystyrene | Polyethylene Terephthalate |
| Sequence | KRNHWQRMHLSA | KGKEAAIAAAKA |
Parameterization of the Plastics
Atomistic force field parameters for polyethylene, polystyrene, polypropylene, and PET were obtained to model the peptide-plastic interactions. Partial charges for the plastic monomers were calculated in three steps: 1) obtain an electrostatic surface potential using Gaussian16 with the HF/6–31G(d) basis set35,36 to optimize the geometry of the plastic monomer, 2) calculate the electrostatic surface potential using the B3LYP/6–31G(d) basis set37, and 3) find atomic partial charges that best fit the electrostatic surface potential using the two-stage restrained electrostatic surface potential (RESP) charge fitting tool of AMBER38. In conducting two-stage RESP, the first stage allowed all partial charges to vary, while the second stage required all atoms that are equivalent by symmetry to have equal partial charges. The capping groups used for the plastic monomers are provided in the supplemental information. All other force field parameters for the plastic monomers, i.e. the bond lengths, bond angles, dihedral angles, Lennard-Jones parameters and Generalized Born parameters, were taken from Amber’s Generalized Force Field 239. To determine if the parameterization could reproduce a plastic’s basic physical properties, the radii of gyration of the polymer chains as well as the density and heat capacity for polymer melts were calculated for all four plastics, and then compared to experimental or theoretical values. Good agreement was found in all cases. The values of the plastics parameters and details of the calculations and comparisons to experimental data can be found in the supplementary information.
Generation of Atomistic Plastic Surfaces
Two types of atomistic models for plastic surfaces were generated, as shown in Figure 1. The first atomistic surface model was crystalline and was generated only for polyethylene. The crystal was created by replicating the unit cell of polyethylene40 to produce crystals of size 7.4 Å by 44.6 Å by 45.6 Å for the peptide design algorithm, and 22.2 Å by 59.5 Å by 58.3 Å for molecular dynamics (MD) simulations. MD simulations required a larger crystal than those used in PepBD to prevent the peptide from interacting with itself across the periodic boundaries. Note that PepBD does not use periodic boundaries. In both cases, the crystal was rotated so that the shortest dimension was parallel to the z-axis. The surface of the plastic, which is perpendicular to the smallest dimension of the rotated crystal, was exposed for peptide adsorption.
Figure 1.

Atomistic models of plastic. (A) Crystalline polyethylene, and (B) amorphous polypropylene. C) Procedure for generating amorphous plastic surfaces. Two walls of carbon atoms, shown in red, were placed above and below the plastic, and then the top wall was incrementally moved down to compress the plastic until the experimental density was reached. Polyethylene is shown in cyan and white, polypropylene is shown in blue and white, and polystyrene is shown in black and white.
The second atomistic model of a plastic surface was amorphous. This type of surface was generated for polypropylene, polystyrene, and polyethylene terephthalate (PET) using a method adopted from Hirvi et al41. First, multiple polymer chains were placed in a periodic box. The number and length of the polymer chains were chosen such that the plastic surface at the target density would be by by . Next, two rigid walls of carbon atoms were placed above and below the polymer chains, as shown in Figure 1. Harmonic position restraints of 5000 kJ/mol/nm2 were applied to each carbon to maintain the wall’s shape. Third, the top wall was moved down towards the bottom wall by slowly changing the location of the harmonic restraint minimum in 0.1 nm increments, resulting in compression of the plastic. After each movement of the wall, the system was equilibrated for 100 ps in the NVT ensemble at 500K. The plastic was compressed until its average density within 5 Å of the central plane parallel to the carbon walls was in the known density range for the plastic. The plastic surface was then equilibrated in a 5 ns NVT simulation at 500K before quenching to 300K and running a second NVT simulation for 5 ns. The two walls of carbon atoms were then removed to expose the final amorphous surface. For PepBD designs, a by by portion of the amorphous surface was extracted. The shape of the final polymer surface was maintained in PepBD designs by keeping all atoms static, and maintained in molecular dynamics simulations by using position restraints.
Obtaining Adsorbed Peptide Conformations for Peptide Design
The starting conformations for the peptides for PepBD were obtained by conducting molecular dynamics (MD) simulations on each reference peptide in Table 2 when placed roughly 10 Å above the corresponding plastic surface. Two types of simulations were conducted (see below for MD simulation details). The first simulation type was equilibrium MD; the final conformation at the end of the simulation was used as the starting conformation for design. The second simulation type was well-tempered metadynamics (WTMetaD), which generates multiple unique adsorbed conformations by applying a bias potential to the distance between the peptide’s center of mass and the top of the plastic surface. The bias potential induced the peptide to adsorb and desorb multiple times during the simulation. Each distinct adsorption event was selected as a starting conformation, with adsorption events considered distinct if the peptide’s center of mass was at least 10 Å from the top of the plastic surface for at least 1 ns after the previous adsorption event. WTMetaD simulations were found to generate 20 unique conformations for polyethylene and 11 unique conformations each for polystyrene, polypropylene, and PET. All the conformations were used for design.
Molecular Dynamics Simulations: Generating Starting Conformations and Evaluating Designed Peptides
Molecular dynamics simulations were performed to obtain adsorbed peptide conformations on plastic surfaces. tLEaP42 was used to generate input files in Amber format, then parmEd43 was used to convert to Gromacs format. The conversion from Amber to Gromacs format was necessary as the Amber MD engine cannot simulate infinite crystals such as in our polyethylene model. In simulations of crystalline polyethylene, the terminal carbons on each polymer chain in the crystal were bonded to each other across the periodic boundaries to mimic an infinite plane. TIP3P water molecules44 were added to the simulation box and extended at least 30 Å in the z-direction above both faces of the plastic surface. Simulations were run using Gromacs version 2019.645. The first stage of the simulations consisted of energy minimization using the method of steepest descent for a maximum of 1000 steps. The second stage was an NVT ensemble simulation at 300K for 100 ps using the velocity rescale temperature control algorithm. Separate thermostats were used for water and non-water molecules. Both thermostats used a time constant of 0.1 ps. For the NVT simulations and all subsequent simulations, periodic boundary conditions were used, hydrogens were constrained using the LINCS algorithm46, position restraints of 5000 kJ/mol/nm2 were applied to the non-hydrogen atoms in the plastic surface, and a time step of 2 fs was used. The third simulation stage was an NPT simulation at 300K and 1 bar for 200 ps. The semi-isotropic Berendsen barostat47 was used so that changes to the simulation box size in the direction normal to the plastic surface were independent of changes in the other two directions. An isothermal compressibility of was used in all directions, and the barostat time constant was 5 ps. The final stage was either equilibrium MD or Well-tempered Metadynamics (WTMetaD)48. Equilibrium MD was used to obtain a single adsorbed conformation for the peptide and was run for 100 ns. WTMetaD was used to obtain an ensemble of starting conformations and was performed for least 200 ns using Plumed Version 2.6.1 ,51. The collective variable in WTMetaD simulations was the distance from the center of mass of the peptide to the top of the plastic surface; the corresponding bias induced the peptide to adsorb and desorb from the plastic surface multiple times. The initial strength of the bias potential was 4.0 kJ/mol, biases were added every 0.5 ps, the bias factor was 20, and the Gaussian bias width was 0.1 Å.
Results and Discussion
Improving Conformational Sampling by using Multiple Starting Conformations
The first designs of plastic-binding peptides revealed that PepBD ineffectively sampled peptide conformational space. Crystalline polyethylene was selected as the inorganic material for these initial efforts. The starting conformation was obtained from a molecular dynamics simulation of the reference polyethylene-binding peptide shown in Table 2. Figure 2A shows the score profile, a plot of the peptide score at every step during peptide design. The score rapidly decreases in the first 50 steps then oscillates around -30 kcal/mol for the remaining 1500 steps. This oscillation led us to hypothesize that PepBD was trapped in a local minimum in the design space. Subsequent analysis revealed that the peptide backbone changed negligibly during the design procedure (Figure 2B), indicating that PepBD was stuck in the initial conformation. This is an issue as a peptide can adopt a vast number of conformations in the adsorbed state.
Figure 2.
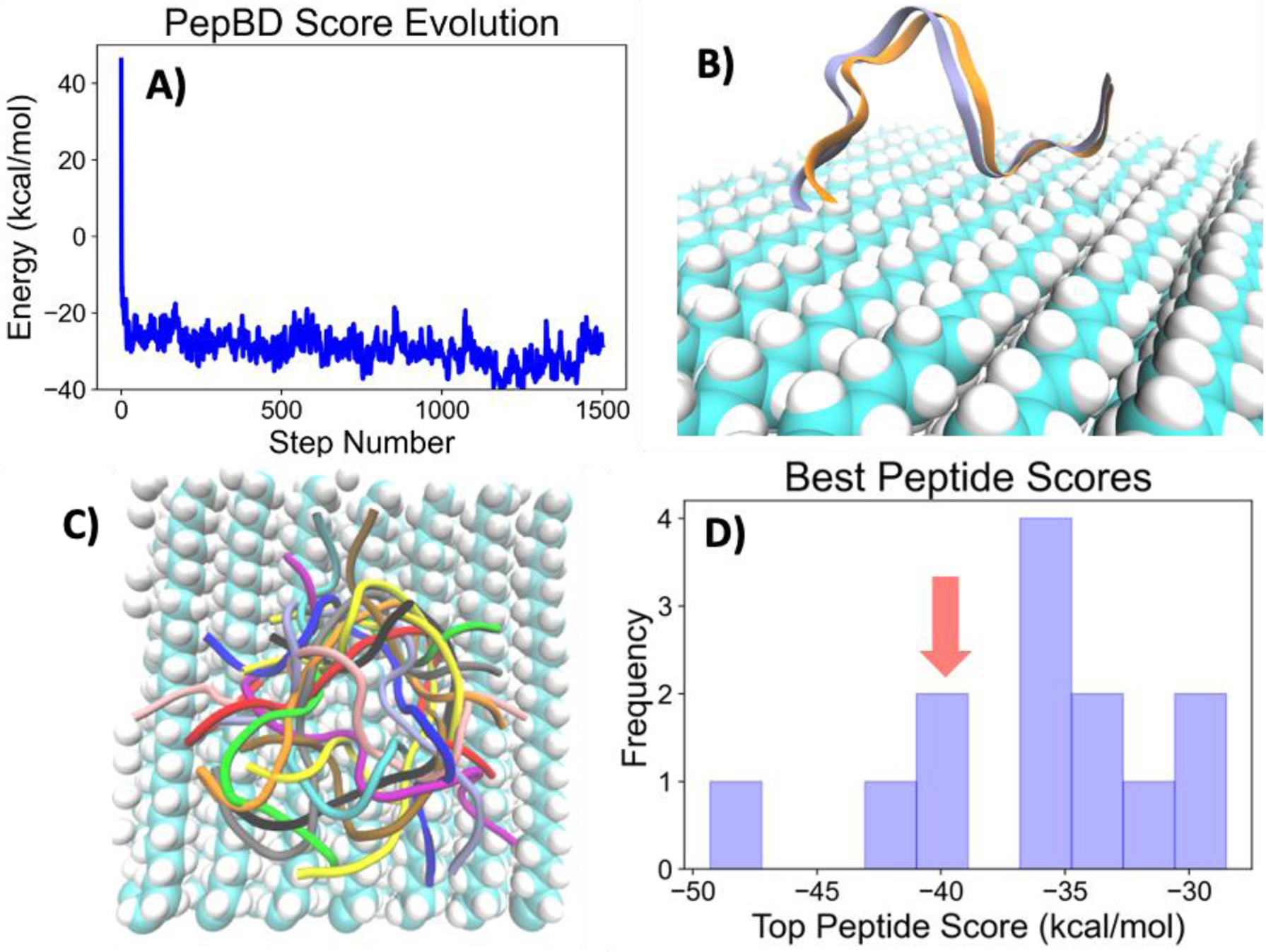
Evolution of Peptide Score During Design with Different PepBD Variations. A) Evolution in score using unmodified version of PepBD. B) Comparison of the peptide backbone structure at end (blue) and beginning (orange) of unmodified PepBD design. C) The twenty different starting conformations used for designing polyethylene-binding peptides. D) Distribution of the best scoring polyethylene-binding peptides resulting from 13 unique starting conformations. Red arrow points to the score of the best peptide found using the initial starting conformation shown in a) and b).
Sampling of the peptide conformational space was improved by using multiple starting conformations to initiate design. While one solution is to add new conformational moves to PepBD, a simpler solution is to use different adsorbed peptide conformations to begin design. In this scheme, the ensemble of starting conformations samples the global conformational space, while each design samples conformational space local to the starting structure. Different starting structures were obtained by running WTMetaD simulations on the reference polyethylene-binding peptide. The bias potential in WTMetaD induces the peptide to repeatedly adsorb and desorb from the plastic, providing the 20 unique adsorbed conformations shown in Figure 2C. Each starting conformation was used for a single peptide design run, but 7 of the 20 conformations could not be used during design (this is due to the hydration constraints, which we revisit in the Section “Relaxing the Hydration Constraints”). The distribution of the best scoring peptide from each run is shown in Figure 2D. The top peptide scores vary significantly between different starting structures; three of the thirteen designs using unmodified PepBD gave better scores than the first design (indicated by the red arrow in Figure 2D). These results indicate the importance of sampling different conformations in the design of plastic-binding peptides.
Incorporating Simulated Annealing into Peptide Design
Simulated annealing (SA) was added to PepBD to further improve sampling of conformation and sequence space. While global sampling of conformation space was improved by using multiple starting conformations, each design poorly sampled the local conformational space as shown in Figure 2B. As mentioned previously, we believed that this can be attributed to PepBD being trapped in a local minimum, so SA52 was added to aid escape from local minima. Briefly, the SA design process begins with a high temperature in Equation 2, and slowly reduces the temperature with design conducted at each temperature until a minimum value is reached. The high temperature at the start of design increases the likelihood of accepting conformational and sequence changes that worsen the score, allowing the peptide conformation to change rapidly. Reducing the temperature decreases the likelihood of accepting unfavorable conformational changes. PepBD converges to a final conformation as the temperature reaches the minimum value. In general, the final state reached by SA is guaranteed to be the most stable conformation only if cooling is performed infinitely slowly53. Since cooling in peptide design must be done at a finite rate, the final peptide conformation and sequence are not guaranteed to be the global minimum. We adopt a common solution to this issue and perform multiple designs with each starting conformation to increase the odds of finding a deep local minimum.
Details of our SA implementation are the following. The temperature begins at and is reduced by a fixed ratio after steps. The design terminates when the temperature reaches . The same value is used for both sequence and conformation changes. When the temperature is decreased, the peptide sequence and structure with the best score over the previous steps is used as the starting point for the following design steps. We tested a variety of parameter combinations to search for an effective SA cooling schedule (Table S5), and selected , , , and . We note that different values can be selected prior to peptide design, if desired.
We found that SA improves conformational sampling and leads PepBD to sample new peptide structures and sequences. We repeated the design of polyethylene-binding peptides using the 20 starting conformations in Figure 2C, with four design runs for each starting conformation. As previously mentioned, multiple runs are performed with the same starting conformation because different SA designs may converge to different minima. Figure 3A shows an example score plot from a design using SA. We see that the peptide score does not change significantly in the final 500 steps, indicating that the design has converged. Figure 3B compares one starting peptide conformation to the final conformations of the four associated designs. In contrast to designs without SA (Figure 2B), the final peptide conformation changes significantly over the course of design. This was observed for all 20 starting structures, showing that SA improves sampling of the local conformational space. Another interesting feature of Figure 3B is that each final peptide conformation is distinct from all other final conformations. The corresponding final sequences are also distinct between designs (Figure 3C). It thus appears that different designs do indeed converge to different local minima.
Figure 3.

Impact of Incorporating Simulated Annealing (SA) into PepBD. A) Evolution of peptide score and system temperature using SA. Blue curve is the peptide score, red curve is the RT value in Equation 2. B) Comparison of starting peptide conformation (in orange) and the final conformation in different designs using SA (red, black, green, and blue). The starting conformation is the same as that shown in Figure 2C. C) Peptide sequence with highest score obtained from four designs using same starting conformation.
Relaxing the Hydration Constraints
A final modification made to PepBD was to address the restriction of peptide sequence sampling due to the hydration constraints. Previously, PepBD required that the number of amino acids of each type (Table 1) remain fixed during design. The hydration constraints are intended to endow peptides with desired properties like solubility in water or positive charge. However, fixing the numbers of each type of amino acid prevents many peptide sequences from being sampled, possibly precluding the discovery of high-affinity peptides. Previous design efforts addressed this issue by running separate designs with different hydration constraints, but the vast number hydration of constraint permutations meant that many peptide sequences still were never sampled. The hydration constraints also prevented the use of seven of the starting conformations in Figure 2C, as an amino acid sequence satisfying the hydration constraints could not be overlaid on the peptide structure without steric clashes. We note that the designs using SA could use all 20 starting conformations as the large starting temperature tolerated the high energy associated with steric clashes until a conformational change removed the steric clash.
We addressed these issues by relaxing the hydration constraints. Rather than fixing the number of each amino acid type at a single value, the relaxed constraints only require that the number of amino acids of each type stay between an upper and lower limit. This greatly expands the sequences that can be sampled while still offering control over peptide properties. The hydration constraints can be specified prior to design, and the ranges used in this study were 0 to 6 hydrophobic, 3 to 10 hydrophilic, 0 to 2 anionic, 0 to 2 cationic, 0 to 2 glycine, and 0 to 2 other amino acids. The maximum of 6 hydrophobic amino acids and minimum of 3 hydrophilic amino acids were chosen to maintain peptide solubility in water. The other ranges were selected as preliminary tests without hydration constraints showed that high-scoring peptides were not found outside these ranges.
Simulated Annealing and Relaxed Hydration Constraints Improve Peptide Design
Combining SA with the relaxed hydration constraints led PepBD to discover better scoring peptides. Crystalline polyethylene was again used as the inorganic material. The designs were performed using PepBD, PepBD with SA, and PepBD with SA and relaxed hydration constraints. Eighty total designs were performed with each PepBD variant (20 unique starting conformations 4 designs per starting conformation). As previously noted, designs with original PepBD only used 13 of the 20 conformations. Figure 4A shows a histogram of the top-scoring peptide from all the designs using the three PepBD versions. Comparing the average scores shows that SA alone reduces the score by about 9 kcal/mol, and that adding in the relaxed hydration constraints gives an additional modest improvement of 2 kcal/mol. The best peptide score using both modifications is nearly 15 kcal/mol lower than the best score with original PepBD. Thus, SA and relaxed hydration constraints significantly improve peptide design.
Figure 4.

Comparison of PepBD Versions and Analysis of Variability for Repeated Designs with the Same Starting Conformation. A) Score distributions of three PepBD variants: 1) original PepBD (blue), 2) PepBD with simulated annealing (SA) (yellow) and 3) PepBD with SA and relaxed hydration constraints (green). For each PepBD version, 80 designs were performed (20 unique starting conformations 4 designs per starting conformation). The legend provides the average and standard deviation of the best peptide’s score using each PepBD variant. B) Distribution of the top scores from 50 designs using the same starting conformation. C) Distribution of score differences between best and worst scores using the same starting conformation, with five designs performed per starting conformation. Values correspond to the range of scores of the best peptide from the five designs.
Features of the Design Space: Local Minima and Number of Designs to Perform
We next characterized the number and depth of local minima in the design space when simulated annealing and the relaxed hydration constraints are used. It was previously shown in Figure 2D that different starting structures, i.e. different locations in the global conformational space, result in significant variability in the associated best peptide scores. The question investigated now is whether such variability is also present in the local conformational space, specifically how much the best PepBD score varies between designs that have the same starting conformation. Borrowing terminology from the protein folding literature, we will term such variability as roughness in the sequence/conformational space. Evidence of this variability is present in Figure 3B, where different designs with the same starting conformation converged to different final structures. To further investigate this question, 50 designs of polyethylene-binding peptide with SA and relaxed hydration constraints were conducted with one single starting conformation. The distribution of the top peptide’s scores is shown in Figure 4B and spans a range of 20 kcal/mol. The best-scoring sequence in each design is unique, like in Figure 3C. It thus appears that the local space is significantly rough.
These results raise the question of how many designs should be performed for a given starting conformation to improve the likelihood of sampling a deep local minimum. Realizing that there is no simple universal answer to this question, we aimed to develop a simple design criterion. The target metric we used was to have a 95% chance of being within 5 kcal/mol of the best score. While the global best score is unknown, the best score in Figure 4B is believed to be a good approximation as 50 designs were performed. A bootstrap analysis of the data in Figure 4B (Supplemental Information) showed that when 5 designs are performed, there is a 95% chance that at least one of the five designs will have a score within 5 kcal/mol of the best score (see Figure S2 and Supplemental Text for details). We thus adopted five designs per starting conformation as our design criterion.
While Figure 4B shows that the local space around that starting conformation is rough, a lingering question is whether such roughness is present around other starting structures. Investigation of this question began by first performing an additional design for each of the 20 different starting conformations in Figure 2C to satisfy our design criterion of five designs per starting conformation. We next calculated the score range for each starting conformation, by finding the top score for each of the five designs and then calculating the difference between the best and worst scores. The score ranges are shown in Figure 4C and span from 5 kcal/mol to 17 kcal/mol. Roughness thus appears to be a general feature of the design space, where the magnitude of the roughness varies with the starting peptide conformation.
Designing Peptide Binders for the Four Plastics
Encouraged by the improvement in peptide design using SA, relaxed hydration constraints, and multiple starting conformations, all three modifications were applied to design peptides that bind to polystyrene, polypropylene, and PET. Eleven starting peptide conformations were obtained for each plastic via WTMetaD simulations of the reference peptides in Table 2. Five designs were performed per starting conformation; Table 3 shows the best-scoring peptides for each plastic. It is notable that the best score for PET is nearly 20 kcal/mol lower than the best score for polystyrene, which we suspect is due to the greater roughness of the polystyrene surface compared to the PET surface. Table 3 also shows that more negative van der Waals energies are paired with more positive electrostatic energies, suggesting that strengthening the van der Waals interactions comes at the price of introducing unfavorable electrostatic interactions. The electrostatic energies were found to consist primarily of the polar solvation energy, which is the electrostatic interaction associated with the continuous dielectric representation of water in the implicit solvent Generalized Born model.
Table 3.
Top Peptide Binder Designs for Different Plastics.
| Plastic | Sequence | PepBD Score | ||
|---|---|---|---|---|
| Polyethylene | HWMWAMKWHMRH | −59.6 | −90.7 | 31.1 |
| Polypropylene | WWQRHMFHFRTW | −52.3 | −80.5 | 28.1 |
| Polystyrene | WHWQRIIWQQMR | −44.5 | −69.7 | 25.2 |
| PET | WEWWHVFHHRLR | −63.8 | −105.0 | 41.2 |
: van der Waals; sum of electrostatics and polar solvation energy.
Properties of Top Peptide Designs: Amino Acid Frequencies
Analysis of the PepBD designs reveals that high scoring peptides are enriched in amino acids with bulky sidechains. The sequences in Table 3 show a high frequency of certain amino acids, like tryptophan (W), arginine (R), and histidine (H), which led us to calculate the frequency of each amino acid in the best peptide designs. The analysis considered the top 50 peptide sequences for all designs using PepBD with SA and relaxed hydration constraints, corresponding to 5,000 peptides for polyethylene (20 starting conformations designs per starting conformation 50 peptides per design) and 2,750 peptides each for polystyrene, polypropylene, and PET (11 starting conformations 5 designs per state 50 peptides per design). The results in Figure 6A show that tryptophan (W), arginine (R), histidine (H), and glutamine (Q) are common, while residues such as alanine (A), glycine (G) and lysine (K) do not occur much. Figure 6B shows that the amino acid frequency correlates positively with amino acid mass, likely because PepBD scores are linearly correlated with van der Waals interactions (Figure 6C, Figure S4) and van der Waals interactions strengthen with increasing mass. Increasing van der Waals interactions is associated with a decrease in the electrostatic interactions (Figure 6D), largely due to the change in the dielectric environment between the plastic surface and bulk water. Interestingly, several of the most frequently appearing amino acids are either charged (e.g. R) or hydrophilic (e.g. H or Q). This seems counterintuitive because placing a hydrophilic residue near a hydrophobic plastic surface could reduce or eliminate hydrogen bonding or electrostatic interactions between the residue and water. However, these residues all have relatively large sidechains compared to the other amino acids, so they can desolvate a larger area of the plastic surface and have stronger van der Waals interactions with the plastic than other amino acids. These pros apparently outweigh the cons in our PepBD model, given the high frequency of these residues. It is also noteworthy that the amino acid frequencies for all four plastics shown in Figure 6A are similar, possibly indicating that peptides with affinity for different plastics will have similar compositions.
Figure 6.

Properties of top polyethylene designs. A) Frequency of amino acids found in the top peptide designs for all four plastics. Cysteine and proline have 0 frequency as they were excluded from design. Data taken from top 50 peptides from all designs for each plastic. B) Correlation of amino acid mass with the frequency with which the amino acid is found in the top polyethylene designs. Data taken from top 50 peptides from all designs for polyethylene. C) Correlation of PepBD score with der Waals binding energy. D) Correlation of van der Waals binding energy with sum of electrostatic and polar solvation binding energy. Data for figures C and D taken from all peptides generated during the 100 designs for polyethylene (PE). The results in B, C, and D are qualitatively similar for the other plastics (Figure S3–S5).
Properties of Top Peptides Designs: Hydration Properties
We found that the top peptide designs were not purely hydrophobic, even in the absence of hydration constraints. Because the four plastics investigated in this study are hydrophobic, it was reasonable to expect that the best plastic-binding peptides would consist entirely of hydrophobic amino acids. Purely hydrophobic peptides are not suitable, however, for biotechnology or MNP remediation as they would be difficult to synthesize, poorly soluble in water, and likely to aggregate. The design of purely hydrophobic peptides can be avoided via the hydration constraints, but it still is worth investigating if PepBD produces purely hydrophobic peptides if the hydration constraints are removed. To answer this question, we first analyzed the hydration properties of our 100 polyethylene-binding peptide designs (20 starting conformations 5 designs per starting conformation) using SA and relaxed hydration constraints. Recall that these designs used the hydration ranges specified in Section 3.3. Figure 7A shows the number of amino acids of each type at each step averaged over all 100 designs. Because the designs were performed with SA, the values at the end of design capture what PepBD believes to be the optimal peptide hydration properties. Figure 7A shows convergence to six hydrophobic amino acids at the end of design for polyethylene-binding peptides, which is the maximum allowable value. Plots for the other plastics (Figure S6) show the same trend. We suspected that PepBD would insert more hydrophobic amino acids if the hydration constraints were completely removed, so an additional round of peptide design was performed for all four plastics without any hydration constraints. 20 designs were performed for each plastic (10 starting conformations 2 designs per starting conformation). The corresponding hydration profiles (Figure 7B) shows that the designs now converge to eight hydrophobic amino acids. The trend was similar for the other plastics (Figure S6). Thus, PepBD does not converge to purely hydrophobic peptides, even in the absence of hydration constraints. Investigation of the sequences revealed that the hydrophilic amino acids were primarily histidine, arginine, and glutamine. Revisiting Figure 6B, it appears that the bulky sidechains of these amino acids make them favorable to include in the peptide.
Figure 7.

Evolution of Hydration Properties with Variable Hydration. A) Evolution of peptide hydration properties for design of polyethylene-binding peptides using hydration constraints of 0 to 6 hydrophobic amino acids, 3 to 10 hydrophilic amino acids, 0 to 2 positive amino acids, 0 to 2 negative amino acids, 0 to 2 other amino acids, and 0 to 2 glycine. B) Evolution of peptide hydration properties for design of polyethylene-binding peptides using no hydration constraints. Amino acids are classified as per Table 1.
Properties of Top Peptides Designs: Sequence Motifs and Hydrophobic Patches
Analysis of the top peptide designs showed that they did not share a common sequence pattern. We wanted to determine if there was an underlying pattern, or sequence motif, in the best PepBD designs, as this was previously reported for polystyrene-binding peptides found experimentally54. Logos55 were created for the top 100 peptides (20 starting conformations 5 designs per starting conformation) for polyethylene and for the top 55 peptides (11 starting conformations 5 designs per starting conformation) for polypropylene, polystyrene, and PET. Only one peptide was selected from each design as the top sequences in each design are sampled from the same local minimum at the end of design and so are often very similar. Figure 8A shows the logo for polyethylene. The logos for the other plastics may be found in Figure S7. To interpret the logo, a larger letter corresponds to a higher probability of finding the amino acid at that position, and a sequence motif corresponds to a string of amino acids that collectively appear with high frequency. Referring to Figure 8A, tryptophan (W) appears with high frequency at positions 7 and 11, indicating W commonly appears at this position in the top designs. However, the small letter sizes at all other positions indicates significant variability between peptides and the absence of a sequence motif. A sequence motif was not found for the other plastics as well (Figure S7).
Figure 8.

Hydration Profiles of Top Peptides A) Sequence logo of the top 100 polyethylene designs using PepBD with SA and relaxed hydration constraints. Sequence logo created using WebLogo55. B) Frequency of finding an amino acid type at each position for the top 50 polyethylene-binding peptides for a single design (left) and averaged over the top 50 polyethylene-binding peptides from all 100 designs (right). Per Table 1, non-polar residues are amino acid types hydrophobic and other, while polar amino acids are all other amino acid types C) Distribution of largest non-polar patches in top 50 peptides for all 100 polyethylene designs vs. distribution of hydrophobic patches in random 12-residue peptide constrained to have either 5 or 6 hydrophobic amino acids. D) Same as C), except for polar amino acids.
We next searched for hydrophobic and hydrophilic patches in our top peptide designs. Referencing the amino acid classifications in Table 1, amino acids were divided into two groups: non-polar (type N) made up of amino acid types hydrophobic and other (which is only alanine) and polar (type P) made up of all other amino acid types. A patch is defined as consecutive Ns or Ps in the peptide. We first visualized patches by calculating the frequency of finding each residue type at each residue position for the 50 peptides in all peptide designs. Figure 8B shows the results for one polyethylene design (left) and the average over all 100 polyethylene designs (right). The single design does show polar patches at residues 1–2 and 8–10, along with a small non-polar patch at position 11–12. To assess peptide patchiness more rigorously, we compared the distribution of the largest polar and non-polar patches for the top PepBD designs to those of a random copolymer. Random copolymer sequences were generated by selecting either an N or P residue at each position of the 12-residue peptide, with each residue selected independently of all others. To align with the PepBD hydration constraints and the hydration properties of the top peptide designs in Figure 7B, random copolymer sequences required either 5 or 6 N residues and 7 or 6 P residues. Figure 8C/D compares the distributions for the largest polar and non-polar patches for the two peptide populations. The PepBD designs are more likely to possess large non-polar patches relative to random peptides, while a significant difference is not observed for polar patches. Referring to Figure 8B, right, it does not appear that the non-polar patch always occurs in the same location. This could be attributable to a dependence of the patch location on the adsorbed conformation, i.e. which residues are solvent-exposed or in contact with the plastic surface. Interestingly, the degree of non-polar patchiness varied between the top designs for different plastics, and the top designs for polystyrene and polypropylene have non-polar patch size distributions that closely resemble those of random copolymers (Figure S9).
Conclusions
This work describes the first steps in developing a computational method for designing peptides that bind to common plastics. The PepBD algorithm was modified to include simulated annealing, to relax the hydration properties, and to use an ensemble of starting conformations. The modifications improved sampling of conformational and sequence space, leading to the discovery of peptides with better scores than the peptides discovered by original PepBD. The updated algorithm was used to design peptides that bind to polyethylene, polypropylene, polystyrene, and PET. The best peptides were found to be enriched in amino acids with bulky sidechains that increase van der Waals interactions with the plastic. While a common sequence motif for the best designs was not found, the top designs show patches of hydrophobic or hydrophilic amino acids whose location are specific to each peptide backbone. Experimental and molecular dynamics testing of the top peptide designs is underway and will be important for evaluating PepBD in the design of plastic-binding peptides.
An important parameter in peptide design is the peptide length. We fixed all peptides to 12 residues to enable comparison to previous plastic-binding peptides found in peptide library screening experiments and because the 12-residue sequence could be repeated multiple times to obtain a larger, multivalent plastic-binding peptide if higher affinity is desired. However, peptide length is an important parameter in design, and its impact is worth investigating. Based on the linear correlation between van der Waals interactions and peptide scores, we predict that peptide scores will linearly increase as the peptide length increases. This raises the question of whether a short sequence repeated several times will have higher affinity to a plastic than one long, unique sequence. This could be explored in future work.
It is crucial to note that PepBD predictions require verification. PepBD scores are not binding free energies since the score does not include entropic terms in peptide adsorption, such as solvent restructuring or changes in the peptide’s conformational space. Entropy is deliberately excluded from the PepBD score because entropy requires sampling peptide conformational space or the water structure, a computationally intractable task when peptide sequence space is also being sampled. Instead, PepBD aims to identify potential “hit” peptides that bind strongly to a target material. The hits can be further evaluated using either computational methods, like umbrella sampling56 or metadynamics57, or experimental methods, like surface plasmon resonance58 or quartz crystal microbalance59. We omit these evaluations in the current work as our aim is to describe the PepBD design algorithm and its outputs. Computational and experimental characterization of the best peptide designs will be described in a future work.
Moving forward, additional modifications are planned to further improve peptide design. An improved treatment of solvent effects may be important given their key role in adsorption processes60. Particularly important for peptide adsorption to plastics are hydrophobic forces, as a major driving force in peptide adsorption is displacing water from the interface. Hydrophobic energy, attributed to van der Waals interactions with water as well as entropic energy resulting from restructuring of water, was neglected in previous PepBD designs of protein-binding peptides as the hydrophobic energy was found to be roughly constant. The constant energy stems from calculating the hydrophobic energy in terms of the peptide surface area, which is roughly constant in a protein binding pocket due to steric constraints. In contrast, the peptide surface area can vary significantly from adsorbed conformation to adsorbed conformation, so it is worth revisiting if the hydrophobic energy is an important factor in peptide design. We also plan to automate the generation of starting conformations both to eliminate the need for molecular dynamics simulations to generate these conformations and to permit intelligent sampling of peptide conformational space. Lastly, we hope to apply PepBD to design peptides that bind to other inorganic materials. This requires generating accurate atomistic surfaces for the material, obtaining parameters that accurately model peptide-surface interactions, and revising the score function to include new interactions such as that between thiols and gold61. Overall, this is the first of many steps in our journey to computationally designing peptides that bind to inorganic materials.
Supplementary Material
Acknowledgements
PepBD designs and molecular dynamics simulations were performed on the Hazel supercomputing center at North Carolina State University and Expanse at the San Diego Supercomputing Center. M.B. was funded from NIH grant 1T32GM133366. The authors acknowledge the support from the US National Science Foundation EFRI E3P - 2029327.
Abbreviations
- PE
polyethylene
- PP
polypropylene
- PS
polystyrene
- PET
polyethylene terephthalate
- WTMetaD
well-tempered metadynamics
- SA
simulated annealing
- GB
generalized Born solvent model
Footnotes
Supporting Information:
Methods and results for calculating plastic densities and heat capacities, plastic parameterization, simplified PepBD flowchart, optimization of PepBD parameters, and characterization of top peptide designs for all four plastics (DOCX).
References
- (1).Care A; Bergquist PL; Sunna A Solid-Binding Peptides: Smart Tools for Nanobiotechnology. Trends Biotechnol 2015, 33 (5), 259–268. 10.1016/j.tibtech.2015.02.005. [DOI] [PubMed] [Google Scholar]
- (2).Ihssen J; Braun A; Faccio G; Gajda-Schrantz K; Thöny-Meyer L Light Harvesting Proteins for Solar Fuel Generation in Bioengineered Photoelectrochemical Cells. Curr. Protein Pept. Sci 2014, 15 (4), 374–384. 10.2174/1389203715666140327105530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gungormus M; Fong H; Kim IW; Evans JS; Tamerler C; Sarikaya M Regulation of in Vitro Calcium Phosphate Mineralization by Combinatorially Selected Hydroxyapatite-Binding Peptides. Biomacromolecules 2008, 9 (3), 966–973. 10.1021/bm701037x. [DOI] [PubMed] [Google Scholar]
- (4).Lee DJ; Park HS; Koo K; Lee JY; Nam YS; Lee W; Yang MY Gold Binding Peptide Identified from Microfluidic Biopanning: An Experimental and Molecular Dynamics Study. Langmuir ACS J. Surf. Colloids 2019, 35 (2), 522–528. 10.1021/acs.langmuir.8b02563. [DOI] [PubMed] [Google Scholar]
- (5).Zhang X; Chen J; Li E; Hu C; Luo S-Z; He C Ultrahigh Adhesion Force Between Silica-Binding Peptide SB7 and Glass Substrate Studied by Single-Molecule Force Spectroscopy and Molecular Dynamic Simulation. Front. Chem 2020, 8:600918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).E. Hughes Z; R. Walsh T What Makes a Good Graphene-Binding Peptide? Adsorption of Amino Acids and Peptides at Aqueous Graphene Interfaces. J. Mater. Chem. B 2015, 3 (16), 3211–3221. 10.1039/C5TB00004A. [DOI] [PubMed] [Google Scholar]
- (7).Liu R; Enstrom AM; Lam KS Combinatorial Peptide Library Methods for Immunobiology Research. Exp. Hematol 2003, 31 (1), 11–30. 10.1016/S0301-472X(02)01008-1. [DOI] [PubMed] [Google Scholar]
- (8).Puddu V; Perry CC Peptide Adsorption on Silica Nanoparticles: Evidence of Hydrophobic Interactions. ACS Nano 2012, 6 (7), 6356–6363. 10.1021/nn301866q. [DOI] [PubMed] [Google Scholar]
- (9).Seker UOS; Demir HV Material Binding Peptides for Nanotechnology. Molecules 2011, 16 (2), 1426–1451. 10.3390/molecules16021426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Masica DL; Schrier SB; Specht EA; Gray JJ De Novo Design of Peptide−Calcite Biomineralization Systems. J. Am. Chem. Soc 2010, 132 (35), 12252–12262. 10.1021/ja1001086. [DOI] [PubMed] [Google Scholar]
- (11).Schwaminger SP; Anand P; Borkowska-Panek M; Blank-Shim SA; Fraga-García P; Fink K; Berensmeier S; Wenzel W Rational Design of Iron Oxide Binding Peptide Tags. Langmuir 2019, acs.langmuir.9b00729. 10.1021/acs.langmuir.9b00729. [DOI] [PubMed] [Google Scholar]
- (12).Hughes ZE; Nguyen MA; Wang J; Liu Y; Swihart MT; Poloczek M; Frazier PI; Knecht MR; Walsh TR Tuning Materials-Binding Peptide Sequences toward Gold- and Silver-Binding Selectivity with Bayesian Optimization. ACS Nano 2021, 15 (11), 18260–18269. 10.1021/acsnano.1c07298. [DOI] [PubMed] [Google Scholar]
- (13).DeOliveira DB; Laursen RA Control of Calcite Crystal Morphology by a Peptide Designed To Bind to a Specific Surface. J. Am. Chem. Soc 1997, 119 (44), 10627–10631. 10.1021/ja972270w. [DOI] [Google Scholar]
- (14).Pyles H; Zhang S; De Yoreo JJ; Baker D Controlling Protein Assembly on Inorganic Crystals through Designed Protein Interfaces. Nature 2019, 571 (7764), 251–256. 10.1038/s41586-019-1361-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li N; Kang J; Jiang L; He B; Lin H; Huang J PSBinder: A Web Service for Predicting Polystyrene Surface-Binding Peptides. BioMed Res. Int 2017, 2017, e5761517. 10.1155/2017/5761517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Meng C; Hu Y; Zhang Y; Guo F PSBP-SVM: A Machine Learning-Based Computational Identifier for Predicting Polystyrene Binding Peptides. Front. Bioeng. Biotechnol 2020, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pabortsava K; Lampitt RS High Concentrations of Plastic Hidden beneath the Surface of the Atlantic Ocean. Nat. Commun 2020, 11 (1), 4073. 10.1038/s41467-020-17932-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vethaak AD; Legler J Microplastics and Human Health. Science 2021, 371 (6530), 672–674. 10.1126/science.abe5041. [DOI] [PubMed] [Google Scholar]
- (19).Oh S; Hur H; Kim Y; Shin S; Woo H; Choi J; Lee HH Peptide Specific Nanoplastic Detection Based on Sandwich Typed Localized Surface Plasmon Resonance. Nanomaterials 2021, 11 (11), 2887. 10.3390/nano11112887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liu Z; Zhang Y; Wu J Enhancement of PET Biodegradation by Anchor Peptide-Cutinase Fusion Protein. Enzyme Microb. Technol 2022, 156, 110004. 10.1016/j.enzmictec.2022.110004. [DOI] [PubMed] [Google Scholar]
- (21).Xiao X; Hall CK; Agris PF The Design of a Peptide Sequence to Inhibit HIV Replication: A Search Algorithm Combining Monte Carlo and Self-Consistent Mean Field Techniques. J. Biomol. Struct. Dyn 2014, 32 (10), 1523–1536. 10.1080/07391102.2013.825757. [DOI] [PubMed] [Google Scholar]
- (22).Xiao X; Agris PF; Hall CK Introducing Folding Stability into the Score Function for Computational Design of RNA-Binding Peptides Boosts the Probability of Success. Proteins 2016, 84 (5), 700–711. 10.1002/prot.25021. [DOI] [PubMed] [Google Scholar]
- (23).Xiao X; Agris PF; Hall CK Designing Peptide Sequences in Flexible Chain Conformations to Bind RNA: A Search Algorithm Combining Monte Carlo, Self-Consistent Mean Field and Concerted Rotation Techniques. J. Chem. Theory Comput 2015, 11 (2), 740–752. 10.1021/ct5008247. [DOI] [PubMed] [Google Scholar]
- (24).Xiao X; Hung ME; Leonard JN; Hall CK Adding Energy Minimization Strategy to Peptide-Design Algorithm Enables Better Search for RNA-Binding Peptides: Redesigned λ N Peptide Binds BoxB RNA. J. Comput. Chem 2016, 37 (27), 2423–2435. 10.1002/jcc.24466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dodd LR; Boone TD; Theodorou DN A Concerted Rotation Algorithm for Atomistic Monte Carlo Simulation of Polymer Melts and Glasses. Mol. Phys 1993, 78 (4), 961–996. 10.1080/00268979300100641. [DOI] [Google Scholar]
- (26).Lovell SC; Word JM; Richardson JS; Richardson DC The penultimate rotamer library. Proteins Struct. Funct. Bioinforma 2000, 40 (3), 389–408. . [DOI] [PubMed] [Google Scholar]
- (27).Quasi-Newton Methods. In Numerical Optimization; Nocedal J, Wright SJ, Eds.; Springer: New York, NY, 2006; pp 135–163. 10.1007/978-0-387-40065-5_6. [DOI] [Google Scholar]
- (28).Wang E; Sun H; Wang J; Wang Z; Liu H; Zhang JZH; Hou T End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev 2019, 119 (16), 9478–9508. 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- (29).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput 2015, 11 (8), 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Spears RJ; McMahon C; Chudasama V Cysteine Protecting Groups: Applications in Peptide and Protein Science. Chem. Soc. Rev 2021, 50 (19), 11098–11155. 10.1039/D1CS00271F. [DOI] [PubMed] [Google Scholar]
- (31).Chiva C; Vilaseca M; Giralt E; Albericio F An HPLC-ESMS Study on the Solid-Phase Assembly of C-Terminal Proline Peptides. J. Pept. Sci 1999, 5 (3), 131–140. . [DOI] [PubMed] [Google Scholar]
- (32).Xiao X; Kilgore R; Sarma S; Chu W; Menegatti S; Hall CK De Novo Discovery of Peptide-Based Affinity Ligands for the Fab Fragment of Human Immunoglobulin G. J. Chromatogr. A 2022, 1669, 462941. 10.1016/j.chroma.2022.462941. [DOI] [PubMed] [Google Scholar]
- (33).Cunningham SD; Lowe DJ; O’Brien JP; Wang H; Wilkins AE Polyethylene Binding Peptides and Methods of Use US7906617B2, March 15, 2011. https://patents.google.com/patent/US7906617B2/en (accessed 2021-05-10).
- (34).Cunningham SD; Lowe DJ; O’Brien JP; Wang H; Wilkins AE Polypropylene Binding Peptides and Methods of Use US7928076B2, April 19, 2011. https://patents.google.com/patent/US7928076B2/en (accessed 2021-05-10).
- (35).Cunningham SD; Lowe DJ; O’brien JP; Wang H; Wilkins AE Polystyrene Binding Peptides and Methods of Use 7632919, December 15, 2009. https://www.freepatentsonline.com/7632919.html (accessed 2021-05-10).
- (36).Petersson GA; Bennett A; Tensfeldt TG; Al‐Laham MA; Shirley WA; Mantzaris J A Complete Basis Set Model Chemistry. I. The Total Energies of Closed‐shell Atoms and Hydrides of the First‐row Elements. J. Chem. Phys 1988, 89 (4), 2193–2218. 10.1063/1.455064. [DOI] [Google Scholar]
- (37).Petersson GA; Al‐Laham MA A Complete Basis Set Model Chemistry. II. Open‐shell Systems and the Total Energies of the First‐row Atoms. J. Chem. Phys 1991, 94 (9), 6081–6090. 10.1063/1.460447. [DOI] [Google Scholar]
- (38).Stephens PJ; Devlin FJ; Chabalowski CF; Frisch MJ Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem 1994, 98 (45), 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- (39).Cieplak P; Cornell WD; Bayly C; Kollman PA Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem 1995, 16 (11), 1357–1377. 10.1002/jcc.540161106. [DOI] [Google Scholar]
- (40).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25 (9), 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- (41).Teare PW The Crystal Structure of Orthorhombic Hexatriacontane, C 36 H 74. Acta Crystallogr 1959, 12 (4), 294–300. 10.1107/S0365110X59000901. [DOI] [Google Scholar]
- (42).Hirvi JT; Pakkanen TA Molecular Dynamics Simulations of Water Droplets on Polymer Surfaces. J. Chem. Phys 2006, 125 (14), 144712. 10.1063/1.2356470. [DOI] [PubMed] [Google Scholar]
- (43).CASE DA; CHEATHAM TE; DARDEN T; GOHLKE H; LUO R; MERZ KM; ONUFRIEV A; SIMMERLING C; WANG B; WOODS RJ The Amber Biomolecular Simulation Programs. J. Comput. Chem 2005, 26 (16), 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shirts MR; Klein C; Swails JM; Yin J; Gilson MK; Mobley DL; Case DA; Zhong ED Lessons Learned from Comparing Molecular Dynamics Engines on the SAMPL5 Dataset. bioRxiv September 25, 2016, p 077248. 10.1101/077248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- (46).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- (47).Hess B; Bekker H; Berendsen HJC; Fraaije JGEM LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem 1997, 18 (12), 1463–1472. . [DOI] [Google Scholar]
- (48).Berendsen HJC; Postma JPM; van Gunsteren WF; DiNola A; Haak JR Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys 1984, 81 (8), 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- (49).Bussi G; Laio A Using Metadynamics to Explore Complex Free-Energy Landscapes. Nat. Rev. Phys 2020, 2 (4), 200–212. 10.1038/s42254-020-0153-0. [DOI] [Google Scholar]
- (50).Bonomi M; Bussi G; Camilloni C; Tribello GA; Banáš P; Barducci A; Bernetti M; Bolhuis PG; Bottaro S; Branduardi D; et al. Promoting Transparency and Reproducibility in Enhanced Molecular Simulations. Nat. Methods 2019, 16 (8), 670–673. 10.1038/s41592-019-0506-8. [DOI] [PubMed] [Google Scholar]
- (51).Tribello GA; Bonomi M; Branduardi D; Camilloni C; Bussi G PLUMED 2: New Feathers for an Old Bird. Comput. Phys. Commun 2014, 185 (2), 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- (52).Dowsland KA; Thompson J Simulated Annealing. In Handbook of Natural Computing; Rozenberg G, Back T, Kok JN, Eds.; Springer-Verlag, 2012; pp 1623–1655. 10.1007/978-3-540-92910-9_49. [DOI] [Google Scholar]
- (53).Bertsimas D; Tsitsiklis J Simulated Annealing. Stat. Sci 1993, 8 (1), 10–15. 10.1214/ss/1177011077. [DOI] [Google Scholar]
- (54).Gebhardt K; Lauvrak V; Babaie E; Eijsink V; Lindqvist BH Adhesive Peptides Selected by Phage Display: Characterization, Applications and Similarities with Fibrinogen. Pept. Res 1996, 9 (6), 269–278. [PubMed] [Google Scholar]
- (55).Crooks GE; Hon G; Chandonia J-M; Brenner SE WebLogo: A Sequence Logo Generator. Genome Res 2004, 14 (6), 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Degen GD; Cunha KC; Levine ZA; Waite JH; Shea J-E Molecular Context of Dopa Influences Adhesion of Mussel-Inspired Peptides. J. Phys. Chem. B 2021, 125 (35), 9999–10008. 10.1021/acs.jpcb.1c05218. [DOI] [PubMed] [Google Scholar]
- (57).Mao CM; Sampath J; Sprenger KG; Drobny G; Pfaendtner J Molecular Driving Forces in Peptide Adsorption to Metal Oxide Surfaces. Langmuir 2019, 35 (17), 5911–5920. 10.1021/acs.langmuir.8b01392. [DOI] [PubMed] [Google Scholar]
- (58).Tamerler C; Oren EE; Duman M; Venkatasubramanian E; Sarikaya M Adsorption Kinetics of an Engineered Gold Binding Peptide by Surface Plasmon Resonance Spectroscopy and a Quartz Crystal Microbalance. Langmuir 2006, 22 (18), 7712–7718. 10.1021/la0606897. [DOI] [PubMed] [Google Scholar]
- (59).Mermut O; Phillips DC; York RL; McCrea KR; Ward RS; Somorjai GA In Situ Adsorption Studies of a 14-Amino Acid Leucine-Lysine Peptide onto Hydrophobic Polystyrene and Hydrophilic Silica Surfaces Using Quartz Crystal Microbalance, Atomic Force Microscopy, and Sum Frequency Generation Vibrational Spectroscopy. J. Am. Chem. Soc 2006, 128 (11), 3598–3607. 10.1021/ja056031h. [DOI] [PubMed] [Google Scholar]
- (60).Schneider J; Colombi Ciacchi L Specific Material Recognition by Small Peptides Mediated by the Interfacial Solvent Structure. J. Am. Chem. Soc 2012, 134 (4), 2407–2413. 10.1021/ja210744g. [DOI] [PubMed] [Google Scholar]
- (61).Vericat C; E. Vela M; Benitez G; Carro P; C. Salvarezza R Self-Assembled Monolayers of Thiols and Dithiols on Gold : New Challenges for a Well-Known System. Chem. Soc. Rev 2010, 39 (5), 1805–1834. 10.1039/B907301A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.