

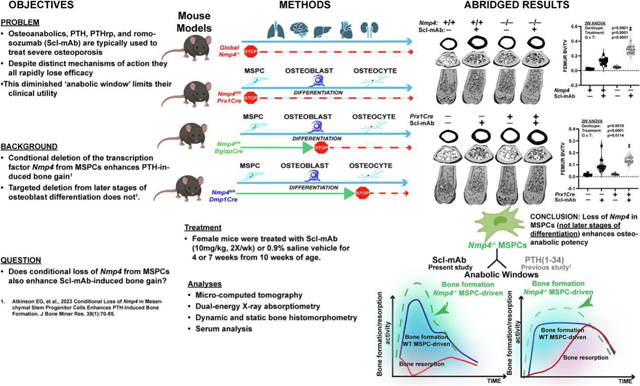
Abstract
Severe osteoporosis is often treated with one of three Food and Drug Administration (FDA)-approved osteoanabolics. These drugs act by (1) parathyroid hormone (PTH) receptor stimulation using analogues to PTH (teriparatide) or PTH-related peptide (abaloparatide) or by (2) monoclonal antibody neutralization of sclerostin, an innate Wnt inhibitor (Scl-mAb, romosozumab-aqqg). The efficacies of both strategies wane over time. The transcription factor Nmp4 (Nuclear Matrix Protein 4) is expressed in all tissues yet mice lacking this gene are healthy and exhibit enhanced PTH-induced bone formation. Conditional deletion of Nmp4 in mesenchymal stem progenitor cells (MSPCs) phenocopies the elevated response to PTH in global Nmp4−/− mice. However, targeted deletion in later osteoblast stages does not replicate this response. In this study we queried whether loss of Nmp4 improves Scl-mAb potency. Experimental cohorts included global Nmp4−/− and Nmp4+/+ littermates and three conditional knockout models. Nmp4-floxed (Nmp4fl/fl) mice were crossed with mice harboring one of three Cre-drivers (i) Prx1Cre+ targeting MSPCs, (ii) BglapCre+ (mature osteocalcin-expressing osteoblasts), and (iii) Dmp1Cre+ (osteocytes). Female mice were treated with Scl-mAb or 0.9% saline vehicle for 4 or 7 weeks from 10 weeks of age. Skeletal response was assessed using micro-computed tomography, dual-energy X-ray absorptiometry, bone histomorphometry, and serum analysis. Global Nmp4−/− mice exhibited enhanced Scl-mAb-induced increases in trabecular bone in the femur and spine and a heightened increase in whole body areal bone mineral density compared to global Nmp4+/+ controls. This improved Scl-mAb potency was primarily driven by enhanced increases in bone formation. Nmp4fl/fl;PrxCre+ mice showed an exaggerated Scl-mAb-induced increase in femoral bone but not in the spine since Prrx1 is not expressed in vertebra. The Nmp4fl/fl;BglapCre+ and Nmp4fl/fl;Dmp1Cre+ mice did not exhibit an improved Scl-mAb response. We conclude that Nmp4 expression in MSPCs interferes with the bone anabolic response to anti-sclerostin therapy.
Keywords: bone anabolism, osteoanabolics, osteoporosis, romosozumab-aqqg
Graphical Abstract
INTRODUCTION:
There are three osteoanabolic drugs used for treating severe osteoporosis and they exploit two distinct therapeutic strategies for adding new bone to the osteoporotic skeleton1,2. These Food and Drug Administration (FDA)-approved medications include teriparatide and abaloparatide, which are analogues to parathyroid hormone (PTH 1–34) and PTH related peptide (PTHrp 1–34), respectively3. Chiefly, both act by binding the PTH receptor (PTH1R), which elevates the second messenger cAMP activating protein kinase A4–6. This, in turn, mobilizes several transcription factors that drive changes in gene expression underlying the anabolic response7–9. The most recent FDA-approved osteoanabolic, romosozumab-aqqg, is a monoclonal antibody that blocks the action of sclerostin, a natural inhibitor of the Wnt pathway, a major determinant of bone mass and strength10–12. Canonical Wnt signaling is triggered when Wnt ligands bind to the receptor complex of the low-density lipoprotein receptor-related protein 5 or 6 (LRP5/6) and frizzled (Fzd)13,14. This interaction prevents the formation of the β-catenin destruction complex13,14. As a result, β-catenin is free to move into the nucleus and associate with T-cell factor (TCF)/lymphoid-enhancer binding factor (LEF) DNA binding partners, activating the Wnt anabolic transcriptomic program13–15. Wnt also suppresses bone resorption by enhancing the expression of osteoprotegrin (OPG), a decoy receptor for activator of nuclear- β ligand (RANKL), an obligate cytokine required for osteoclast recruitment and differentiation16. Sclerostin binds to LRP5/6-Fzd, which results in the stabilization of the β-catenin destruction complex, diminishing Wnt anabolic potency17,18. Additionally, sclerostin enhances RANKL release by osteocytes, driving osteoclast maturation and bone resorption19. Romosozumab-aqqg obstructs the sclerostin-LRP5/6 interaction thus inhibiting this inhibitor of osteoanabolism20. Multiple pathways exist that couple the canonical Wnt and PTH-induced cAMP/PKA pathways, but whether these links underlie osteoanabolic potency remains to be determined21.
These osteoanabolics also differ in their action on skeletal dynamics leading to bone gain. The PTH/PTHrp analogues primarily elevate bone remodeling increasing both bone formation and bone resorption, but with a net positive balance, i.e., formation outpaces resorption22,23. Romosozumab-aqqg is a dual-acting drug and chiefly increases model-based bone formation while temporarily decreasing bone resorption23,24.
The PTH/PTHrp analogues and romosozumab-aqqg do share two attributes: (i) their osteoanabolic activity largely derives from an induced increase in the number of osteoblasts and heightened output21,25 and (ii) their clinical potency is relatively short-lived2. These therapies augment osteoblast number by favoring osteogenic over adipogenic commitment of mesenchymal stem/progenitor cells (MSPC)26–28, suppressing osteoblast apoptosis26,27,29, and stimulation of bone lining cells28,30. Romosozumab-aqqg significantly boosts matrix secretion by osteoblasts in experimental rats31. All these osteoanabolics are relatively costly32,33 and exhibit a limited period of potency, sometimes referred to as a treatment plateau or anabolic window1,2, and addressed by concomitant anti-resorptive therapies. This may present an obstacle for treating osteoporosis, a chronic debilitating disease and has stimulated investigations to improve osteoanabolic potency.
We have identified a transcription factor, Nuclear Matrix Protein 4 (Nmp4, aka Zfp384, ZNF384, Ciz) that is expressed in all tissues34, yet when globally deleted in experimental mice results in no measurable baseline effect but dramatically enhances bone gain induced by PTH treatment35–40. This exaggerated boost in bone gain is mediated by an increase in bone formation. Naive Nmp4−/− mice harbor more osteoprogenitors, suggesting a bias toward the osteo-lineage before drug challenge37, and isolated Nmp4−/− MSPCs induced to differentiate exhibit precocious mineralization, enhanced protein synthesis and collagen secretion38,40,41. Nmp4 influences the expression of thousands of genes in the MSPC/osteoblast transcriptome many of which are involved in protein production and secretion38,40,41. This includes the activation of the physiological unfolded protein stress response (UPR)40,41, an adaptive stress program for handling the anticipated high endoplasmic reticulum (ER) client protein loads42,43. Additionally, UPR proteins play significant roles in lineage commitment and differentiation, including osteoblasts44,45. Nmp4 has the target gene profile of a scaling factor, single transcription factors that influence the expression of large sets of genes for establishing the machinery of the nascent secretory cell46–53. Indeed, this unique transcription factor also regulates the activity of other secretory cells54–56. Moreover, we recently demonstrated that conditionally removing Nmp4 from MSPCs in experimental mice virtually phenocopies the PTH response observed in global Nmp4−/− animals57. Surprisingly (and unexpectedly), conditional deletion of Nmp4 from mature osteoblasts or osteocytes failed to alter the skeletal response to hormone57. The enhanced PTH-induced increases in bone formation rate were driven primarily by increases in active osteoblast coverage (MS/BS) and these data suggested that loss of Nmp4 in osteoprogenitors accelerated osteoprogenitor differentiation, consistent with previous in vitro data38,40,57.
Therefore, we hypothesize that Nmp4−/− MSPCs are pre-programmed to drive an enhanced response to any osteoanabolic drug. To test this premise, we evaluated the skeletal response to Scl-mAb as a function of Nmp4 status. We employed global Nmp4−/− mice35 and three conditional knockout models, in which Nmp4 was selectively deleted from MSPCs, mature osteoblasts, and osteocytes as previously described57. These mice were treated with sclerostin monoclonal antibody (Scl-mAb) or vehicle control. The global Nmp4−/− mice and mice in which Nmp4 had been conditionally removed from MSPCs exhibited a significantly enhanced bone formation response to Scl-mAb. However, the mice in which Nmp4 had been removed from mature osteoblasts or osteocytes showed little to no change in bone phenotype or response to drug. These results are consistent with our novel concept that Nmp4 principally sets osteoanabolic peak efficacy early in bone cell development before drug delivery.
MATERIALS AND METHODS:
Mice:
Global Nmp4−/− mice and their global Nmp4+/+ littermates were generated as previously described35. Nmp4-floxed (Nmp4fl/fl) conditional knockout mice were produced by targeted homologous recombination as reported57. Exons 4–7 were removed from our global Nmp4−/− mice35, whereas the loxP sites flank exons 3–6 in the Nmp4fl/fl mice57. Briefly, Nmp4 was conditionally removed from (i) MSPCs, (ii) osteocalcin-expressing mature osteoblasts, or (iii) osteocytes by crossing Nmp4fl/fl mice with mice harboring either Prx1Cre (B6.Cg-Tg(Prxx1-cre)1Cjt/J, stock no: 005584; The Jackson Laboratory, Bar Harbor, ME), BglapCre (B6N.FVB-Tg(BGLAP-cre)Clem/J, stock no: 019509) or the 8kb Dmp1Cre transgene, respectively57,58. Targeted removal of Nmp4 from specific cell types was confirmed using PCR and Western analyses57.
Treatment:
The Indiana University Institutional Animal Care and Use Committee approved the experimental procedures described in this study. Virgin female mice were grouped into four cohorts by genotype and weight at 10 weeks of age. Typically, two mice were housed per cage under a 12:12-h light-dark regimen. Mice were fed Labdiet Rodent 5001 and provided water ad libitum. A ratized version of a sclerostin mouse monoclonal antibody (Scl-mAb) was administered subQ at 10mg/kg, 200μl/injection, twice/week beginning at 10 weeks of age. Vehicle control treatment was 0.9% saline59.
Dual-energy X-ray absorptiometry (DXA):
An X-ray PIXImus mouse densitometer (Piximus II; GE-Lunar Corp., Madison, WI, USA) was used to evaluate areal bone mineral density (aBMD, mg/cm2) of the post-cranial skeleton, excluding the tail (whole body [WB]). Additionally, femur, spine (L3 to L5), and tibia aBMD were assessed. These measurements were performed at the end of treatment35,36,57.
Micro computed tomography (μCT):
We have previously described the analysis of cortical and trabecular skeletal architectures35,36,39,40,57. Following euthanasia of experimental mice, femurs and L5 vertebra were removed, and cleaned of connective tissue and muscle. The bones were placed in 10% buffered formalin at 4°C for 48–72 hours and then transferred to 70% ethanol for storage at 4°C. A Scanco μCT-35 (Scanco Medical AG, Brüttisellen, Switzerland) was used to obtain the architectural parameters from these bones. We scanned a 9.4 mm section of the distal femur and used 2.6 mm of the metaphysis for trabecular and total bone analysis. The entire L5 vertebra was scanned and analyzed. Femurs and L5 vertebra were scanned at 10 μm resolution, 55 kV peak tube potential, and 151 ms integration time. The Scanco software provided three dimensional reconstructions of the following skeletal elements: trabecular bone volume/total volume (BV/TV), trabecular thickness (Tb.Th; mm), trabecular number (Tb.N; mm−1 ), trabecular spacing (Tb.Sp; mm), bone area (mm2), total area (mm2), cortical thickness (mm), marrow area (mm2), polar moment of inertia (pMOI; mm4), maximum moment of inertia (Imax; mm4), and minimum moment of inertia (Imin; mm4).
Serum Biochemistry:
Activated serum osteocalcin (Gla-osteocalcin) and serum C-terminal telopeptides (CTX) were measured to examine bone formation and bone resorption, respectively. Serum osteocalcin was evaluated using the Mouse Gla-Osteocalcin High Sensitive EIA Kit (TaKaRa, Shiga, Japan)57,60. Serum CTX was measured using the RatLaps enzyme-linked immunosorbent assay (Immunodiagnostic System, Scottsdale, AZ)39,57.
Bone histomorphometry:
Dynamic and static histomormphometric data were obtained from the four cohorts (i) global Nmp4+/+ vehicle, (ii) global Nmp4+/+ Scl-mAb, (iii) global Nmp4−/− vehicle, and (iv) global Nmp4−/− Scl-mAb mice. To obtain dynamic histomorphometric data, mice under a 4-week regimen were administered calcein green (20 mg/kg; Sigma-Aldrich) and alizarin complexone (25mg/kg; Sigma-Aldrich) 11 and 4 days before euthanasia, respectively. Mice under the 7-week therapy were administered oxytetracycline (60 mg/kg; Sigma-Aldrich) and alizarin complexone (25 mg/kg; Sigma-Aldrich) 11 and 4 days before euthanasia, respectively. The Histology Core of the Indiana Center for Musculoskeletal Health prepared the tissues harvested from our experimental mice. Femurs were acquired and processed as described above. The anterior face of the epiphyseal plate was cut to expose the marrow cavity. Bones were dehydrated with graded alcohols, embedded in methyl-methacrylate (Sigma-Aldrich), and sectioned (4μm) using a Leica RM2255 microtome (Leica Microsystems, Buffalo Grove, IL). Unstained tissue sections were then mounted on microscope slides, coverslipped, and scored using the OsteoMeasure High Resolution Digital Video System (OsteoMetrics, Decatur, GA) microscope system. Bone formation rate (BFR/BS, μm3/μm2/day), mineralizing surface/bone surface (MS/BS, %), and mineral apposition rate (MAR, μm/day) were obtained from a 0.75- to 1-mm2 metaphyseal region of interest approximately 200–400μm from the distal femur growth plate57. For static histomorphometric analyses, osteoclasts were quantified in TRAPase/Toluidine blue-stained bone sections from mice under the 4-week therapy61. From these slides we evaluated osteoclast number over bone perimeter (N Oc/B Pm, N mm−1), osteoclast surface over bone surface (Oc S/BS %), eroded surface over bone surface (ES/BS %), number of osteoclasts over the total area (N Oc/T Ar, N mm−2). Finally, we evaluated osteoid thickness (O Th μm).
Statistics:
The DXA, μCT, and histomorphometry, and serum data were analyzed using a two-way ANOVA to evaluate whether treatment or genotype affected the parameter under investigation and to determine whether an interaction took place between these main effects. A post-hoc Student’s t-test was employed when genotype and/or treatment were identified to influence the parameter. If an interaction between genotype and treatment was indicated, all pairwise post hoc comparisons were carried out to distinguish intergroup differences with p values ≤0.05 using a Tukey’s honestly significant difference (HSD) test. A significant genotype × treatment interaction signified that global Nmp4−/− and global Nmp4+/+ mice or the Nmp4fl/flCre+ and Nmp4fl/flCre− mice responded differently to Scl-mAb under our treatment regimen when comparing the four cohorts (i) vehicle-treated global Nmp4+/+ or Nmp4fl/flCre-mice, (ii) Scl-mAb-treated global Nmp4+/+ or Nmp4fl/flCre− mice (iii) vehicle-treated global Nmp4−/− or Nmp4fl/flCre+ mice, (iv) Scl-mAb-treated global Nmp4−/− or Nmp4fl/flCre+ mice. Data are displayed using violin plots with staggered data points, showing the quartiles and median. JMP Pro version 16 (SAS Institute, Cary, NC) was used for all statistical analyses.
RESULTS:
Global loss of Nmp4 augments the skeleton’s response to Scl-mAb:
To determine whether Nmp4 suppression can enhance the bone-building effects of Scl-mAb (as we have observed for other skeletal anabolics35–40,57), we randomly recruited global Nmp4−/− and wild type (Nmp4+/+) mice at 10 weeks of age, then treated them with vehicle or Scl-mAb for 7 weeks and measured changes in skeletal properties. Consistent with previous reports, μCT analysis indicated that global deletion of Nmp4 had either no effect (femoral BV/TV and Tb.Th, vertebral Tb.Th) or very mild effects (femoral Tb.Sp and Tb.N, vertebral BV/TV, Tb,Sp, and Tb.N) on bone mass and architecture under vehicle treatment—i.e., the effect of the knockout alone was marginal, if present at all (Fig. 1 and Fig. 2, Supplemental Table S1). Also consistent with our previous work, we found robust increases in all bone properties among Scl-mAb-treated mice, regardless of genotype, highlighting the potent effects of sclerostin inhibition (Fig. 1 and Fig. 2, Supplemental Table S1). However, the Scl-mAb-induced increases in bone properties were significantly greater in global Nmp4−/− mice compared to Nmp4+/+ mice, as revealed by a significant interaction term for most of the cancellous-derived parameters (Fig. 1 and Fig. 2, Supplemental Table S1).
Figure 1:

Global loss of Nmp4 enhances Scl-mAb-induced osteoanabolism. (A) μCT reconstructions of the distal femur from (i) global Nmp4+/+ vehicle, (ii) global Nmp4+/+ Scl-mAb, (iii) global Nmp4−/− vehicle, and (iv) global Nmp4−/− Scl-mAb mice at 7 weeks therapy. The images were from mice exhibiting the median values of their respective cohorts [in (C) as well]. (B) Statistical analysis and raw data distribution are shown for the distal femoral bone volume/tissue volume (BV/TV) after 7 weeks of therapy for the four treatment groups. (C) The μCT reconstructions of the lumbar L5 of the four cohorts after 7 weeks therapy. (D) Raw data distribution and statistical evaluation are shown for L5 BV/TV for the four cohorts. Violin plots represent the data distribution of the individual mice (n=15–16 mice/cohort) using dashed lines to show the median and the quartiles. We employed 2-way ANOVAs setting genotype and treatment as the independent variables. The p values for individual main effects and for the interaction term are shown in both panels. ‡p ≤ 0.05 for comparison between Scl-mAb-treated cohorts (testing for genotype difference among Scl-mAb-treated groups). *p ≤ 0.05 for comparison between vehicle-treated cohorts (testing for genotype difference among vehicle-treated groups). Post hoc analyses including Tukey’s HSD comparisons between all cohorts and Student’s t test are presented in Supplemental Table S1.
Figure 2:

Global loss of Nmp4 enhances Scl-mAb-induced femoral and L5 trabecular thickness. Statistical analyses and raw data distribution profiles for trabecular number (Tb.N), trabecular thickness (Tb.Th), and trabecular spacing (Tb.Sp) are shown for distal femur (A–C) and L5 vertebra (D–F) after 7-week treatment. Violin plots are used to show data distribution for the individual mice (n = 15–16 mice/cohort) from the four treatment groups (i) global Nmp4+/+ vehicle, (ii) global Nmp4+/+ Scl-mAb, (iii) global Nmp4−/− vehicle, and (iv) global Nmp4−/− Scl-mAb. The dashed lines represent the median and the quartiles for each cohort. The Scl-mAb-induced increases in femoral and L5 Tb.Th were improved in the global Nmp4−/− mice as compared to the global Nmp4+/+ mice (Fig. 2B and Fig. 2E). The statistical approach employed 2-way ANOVAs using genotype and treatment as the independent variables. The p values for the parameter main effects and for the interaction effect are presented in each panel. ‡p ≤ 0.05 for comparison between Scl-mAb-treated groups (testing for genotype difference among Scl-mAb-treated groups). Post hoc analyses including Student’s t test and Tukey’s HSD comparisons between groups are presented in Supplemental Table S1.
We have previously determined that global loss of Nmp4 augments the PTH-induced increase in trabecular bone in addition to amplifying improvements in trabecular architecture without enhancing or diminishing these kinds of changes in cortical parameters35–40,57. In other words, the booster effect provided by Nmp4 deletion is compartment specific. To determine whether these mice respond in a similar compartment-specific manner to 7 weeks of Scl-mAb therapy, we used DXA to assess postcranial whole body aBMD. Although DXA cannot distinguish between trabecular and cortical bone, whole body aBMD is chiefly driven by cortical bone mass62,63. Scl-mAb increased whole body aBMD as expected, however, the global Nmp4−/− mice exhibited a significantly enhanced gain in both whole body and femoral aBMD as indicated by significant genotype × interactions (Fig. 3, Table 1, Supplemental Table S1). However, μCT analysis showed that several cortical architectural parameters of the midshaft femur did not exhibit heightened response to Scl-mAb therapy upon loss of Nmp4 including total area, bone area, cortical thickness, marrow area, pMOI, Imax, and Imin (Table 1). Therefore, Nmp4 status impacts antibody response of some but not all parameters in the cortical bone compartment.
Figure 3:

Global loss of Nmp4 enhances Scl-mAb-induced increases in whole-body (WB) areal bone mineral density (aBMD). Violin plots are used to show data distribution of the postcranial WB aBMD for the individual mice (n = 12–16 mice/cohort) from the four treatment groups (i) global Nmp4+/+ vehicle, (ii) global Nmp4+/+ Scl-mAb, (iii) global Nmp4−/− vehicle, and (iv) global Nmp4−/− Scl-mAb after 7 weeks of treatment. The median and the quartiles are shown as dashed lines. Statistical analyses employed 2-way ANOVAs using genotype and treatment as the independent variables. The p values for individual main effects and for the interaction term are presented in the panel. ‡p ≤ 0.05 for comparison between Scl-mAb-treated groups (testing for genotype difference among Scl-mAb-treated groups). Post hoc analyses are presented in Supplemental Table S1.
TABLE 1:
Global Nmp4−/− and Nmp4+/+ mice skeletal parameters: Dual energy X-ray absorptiometry (DEXA) for areal bone mineral density and micro computed tomography (μCT) for femoral cortical parameters
| DEXA aBMD g/cm2 (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
|---|---|---|---|---|---|---|---|
| FEMUR | 0.072±0.002 | 0.093±0.003 | 0.073±0.002 | 0.090±0.003 | p=0.2752 | p<0.0001 | p=0.0022 |
| TIBIA | 0.052±0.001 | 0.062±0.003 | 0.053±0.002 | 0.062±0.003 | p=0.9138 | p<0.0001 | p=0.6533 |
| SPINE (L3–L5) | 0.065±0.003 | 0.084±0.003 | 0.064±0.004 | 0.083±0.004 | p=0.3949 | p<0.0001 | p=0.8672 |
| μCT Femoral cortical architecture (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
| Total area mm2 | 1.741±0.073 | 1.847±0.076 | 1.740±0.149 | 1.843±0.083 | p=0.9118 | p=0.0001 | p=0.9674 |
| Bone area mm2 | 0.865±0.024 | 1.134±0.044 | 0.888±0.082 | 1.140±0.051 | p=0.2757 | p<0.0001 | p=0.5281 |
| Cortical Th mm | 0.209±0.007 | 0.274±0.008 | 0.212±0.022 | 0.278±0.009 | p=0.2685 | p<0.0001 | p=0.9974 |
| Marrow area mm2 | 0.877±0.070 | 0.713±0.059 | 0.851±0.082 | 0.703±0.048 | p=0.2881 | p<0.0001 | p=0.0674 |
| pMOI mm4 | 0.370±0.024 | 0.476±0.041 | 0.389±0.042 | 0.485±0.045 | p=0.1623 | p<0.0001 | p=0.6444 |
| Imax mm4 | 0.227±0.017 | 0.297±0.032 | 0.247±0.029 | 0.316±0.034 | p=0.0099 | p<0.0001 | p=0.9502 |
| Imin mm4 | 0.143±0.010 | 0.179±0.011 | 0.142±0.016 | 0.169±0.013 | p=0.0873 | p<0.0001 | p=0.2003 |
VEH=vehicle; Scl-mAb=sclerostin monoclonal antibody; aBMD=areal bone mineral density; Cortical Th=cortical thickness; Imax=maximum moment of inertia; Imin=minimum moment of inertia; L3–L5=lumbar vertebra 3 to 5; pMOI=polar moment of inertia. Global Nmp4−/− and global Nmp4+/+ mice were treated with Scl-mAb (10mg/kg, twice/week) or vehicle control from 10 weeks to 17 weeks of age. DXA at 7 weeks of treatment showed that Scl-mAb significantly increased femur, tibial, and spine (L3–L5) aBMD (treatment p < 0.0001) for both Nmp4−/− and Nmp4+/+ mice. Furthermore, the Nmp4−/− mice exhibited significantly greater WB aBMD and a significant genotype x treatment interaction (see Fig. 3). Data represent average ± SD; n = 12–16 mice/cohort. Femoral cortical architecture at 7 weeks of treatment showed there was a significant treatment effect for all parameters (p<0.0001) and a genotype effect for Imax, which was significantly higher in the wild-type mice. Data represent average ± SD; n = 15–16 mice/cohort. All statistical analyses were performed using 2-way ANOVA tests setting genotype and treatment as the independent variables. Statistical significance was set at p ≤ 0.05.
Next, we measured fluorochrome-based bone formation parameters of the trabecular bone of the distal femoral metaphysis, but no differences in Scl-mAb efficacy related to Nmp4 status were detected (data not shown). The lack of effect from such a sensitive readout was puzzling since we saw such large and significant increases in cancellous bone gain by μCT; those results suggested that we might need to look earlier in the treatment period to capture the dynamic changes to bone formation. Indeed, this had been the case for determining the impact of Nmp4 on these parameters during PTH therapy36,57. Therefore, we repeated the experiment but sacrificed the mice after 4 weeks of treatment instead of 7 weeks. Even as early as the 4-week time point in therapy, the Nmp4−/− mice showed a strikingly improved Scl-mAb-induced increase in distal femur cancellous bone formation as evidenced by the strong genotype × treatment interaction (Fig. 4A, Supplemental Table S1). The Nmp4−/− mice also exhibited a significantly heightened Scl-mAb-induced increase in mineralizing surface/bone surface (MS/BS), which is a measure of the active bone formation surface and reflects the number of active osteoblasts (Fig. 4B, Supplemental Table S1). However, the bone formation rate (BFR/BS) showed only a strong treatment effect meaning Scl-mAb equally increased this parameter in both Nmp4−/− and wild type mice at this point in therapy (Fig. 4C, Supplemental Table S1). Unexpectedly, the wild type mice exhibited the anticipated Scl-mAb-induced increase in mineral apposition rate (MAR), but the Nmp4−/− mice did not show a change in this parameter (genotype × treatment interaction, Fig. 4D, Supplemental Table S1). Osteoid thickness (O.Th) showed no genotype × treatment interaction (Fig. 4E, Supplemental Table S1). Nevertheless, there were significant genotype and treatment effects. This indicates that these two independent factors have individual effects on O.Th. Specifically, the Nmp4−/− mice had increased O.Th compared to wild type mice regardless of treatment. Similarly mineralization lag time (MLT, O.Th/MAR), the time interval between the deposition and mineralization of bone matrix, was longer in the Nmp4−/− mice independent of therapy, i.e., there was only a genotype effect (Fig. 4F, Supplemental Table S1). Collectively, the μCT and histomorphometry data (Figs. 4A – 4G, Supplemental Table S1) suggest that Scl-mAb-induced bone formation occurs earlier in therapy in the Nmp4−/− mice. This may be largely driven by an increased number of mineralizing osteoblasts. The greater MAR in the wild type mice may reflect this shift in the timing of the anabolic window between the genotypes, i.e., the bone formation in the knockout mice has already peaked. Independent of treatment, the larger O.Th and longer MLT in the Nmp4−/− mice, may represent the generally higher rate of matrix deposition initially outpacing mineralization.
Figure 4:

Global loss of Nmp4 enhances Scl-mAb-induced bone formation at 4 weeks of treatment primarily driven by increases in osteoblast coverage (MS/BS). (A) Statistical analysis and raw data distribution are shown for the distal femoral bone volume/tissue volume (BV/TV) after 4 weeks of therapy for the four treatment groups, which include (i) global Nmp4+/+ vehicle, (ii) global Nmp4+/+ Scl-mAb, (iii) global Nmp4−/− vehicle, and (iv) global Nmp4−/−Scl-mAb. (n= 9–11/cohort). Raw data distribution and statistical analysis are shown for (B) mineralizing surface/bone surface (MS/BS), (C) bone formation rate/bone surface (BFR/BS), (D) matrix apposition rate (MAR) (n= 9–11/cohort), and (E) Osteoid thickness (O.Th.) (F) mineralization lag time (MLT), (G) Representative longitudinal sections of fluorochrome-labeled (calcein green and alizarin red) femurs from the four treatment cohorts. Global Nmp4−/− mice showed a significantly enhanced Scl-mAb-induced increase in BV/TV at 4 weeks of treatment compared to their wild type littermates. These mice also showed a significant increase in MS/BS. O.Th showed no genotype × treatment interaction. However, there were significant genotype and treatment effects. MLT only exhibited a genotype effect. Violin plots represent the data distribution of the individual mice using dashed lines to show the median and the quartiles. We employed 2-way ANOVAs setting genotype and treatment as the independent variables. The p values for individual main effects and for the interaction term are shown in the panels. ‡p ≤ 0.05 for comparison between Scl-mAb-treated cohorts (testing for genotype difference among Scl-mAb-treated groups). *p ≤ 0.05 for comparison between vehicle-treated cohorts (testing for genotype difference among vehicle-treated groups). Post hoc analyses including Tukey’s HSD comparisons between all cohorts and Student’s t test are presented in Supplemental Table S1. Additionally, dynamic histomorphometry at 7 weeks treatment are shown in Supplemental Table S1
Nmp4 status had only a modest effect on the response of osteoclasts to Scl-mAb at 4 weeks therapy. Antibody weakly but significantly decreased the normalized osteoclast parameters of N Oc/B Pm, Oc.S/BS, and ES/BS for the Nmp4−/− mice compared to their vehicle-treated cohorts, but the wild type mice were unresponsive i.e., there were genotype × treatment interactions (Fig. 5A – Fig. 5D, Supplemental Table S1). This suggests that global loss of Nmp4 may modestly sensitize the response of the resorption arm to Scl-mAb therapy, playing at most a limited role in the observed enhanced bone gain. Resorption plays even less of role during the enhanced PTH-induced bone formation in these mice38,39.
Figure 5:
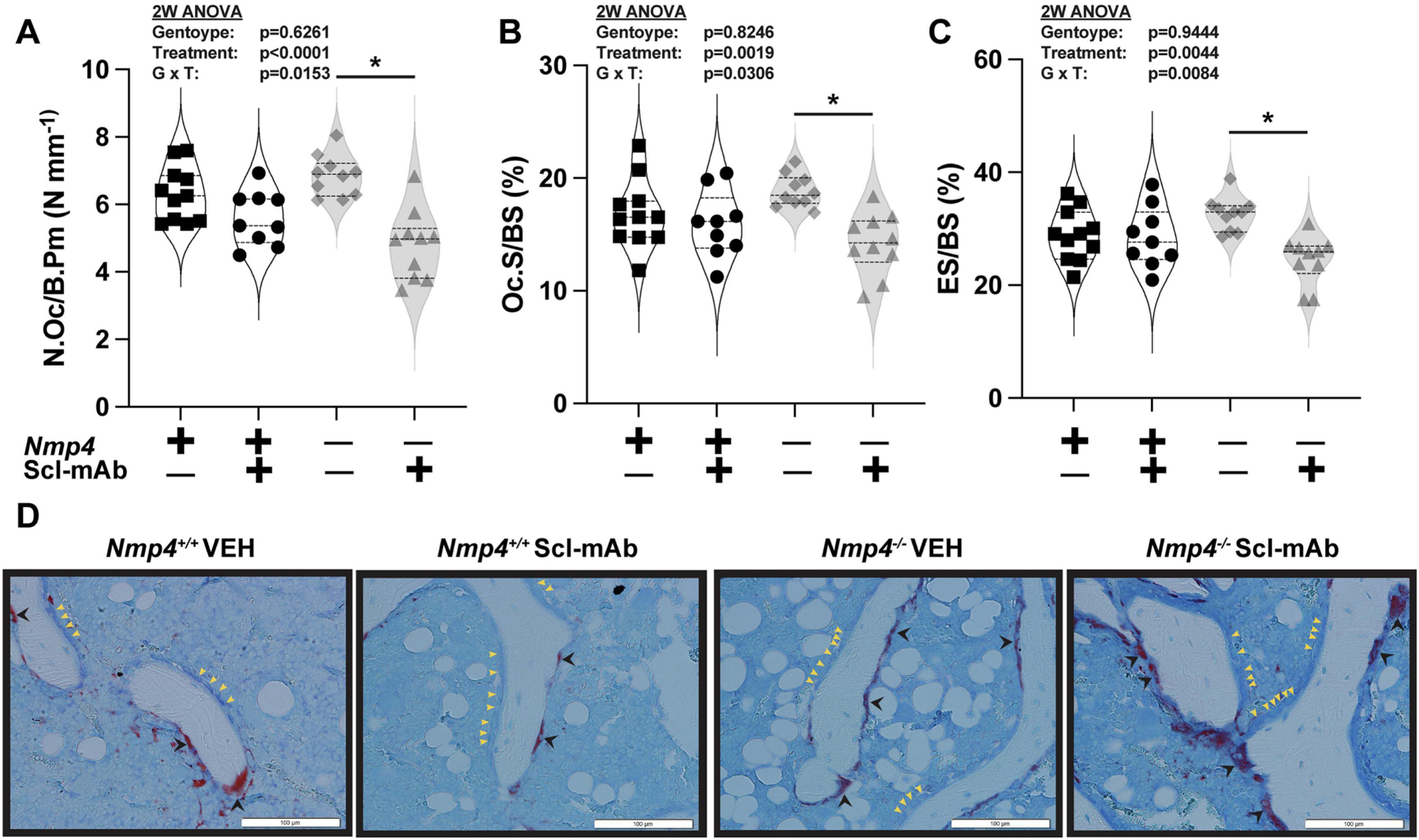
Nmp4 status had little effect on osteoclast density and activity at 4 weeks therapy. Statistical analysis and raw data distribution are shown for (A) the number of osteoclasts/bone perimeter (N.Oc/B.Pm) (B) osteoclast surface/bone surface (Oc.S/BS), (C) eroded surface/bone surface (ES/BS). after 4 weeks of therapy for the four treatment groups, which include (i) global Nmp4+/+ vehicle, (ii) global Nmp4+/+ Scl-mAb, (iii) global Nmp4−/− vehicle, and (iv) global Nmp4−/− Scl-mAb. (D) Representative longitudinal sections of TRAP-stained femurs from the four treatment cohorts. There were no statistical differences between the wild type and Nmp4−/− mice under Scl-mAb therapy for the normalized osteoclast parameters of N Oc/B Pm, Oc.S/BS, and ES/BS. The post hoc Tukey’s HSD tests showed Scl-mAb therapy weakly but significantly decreased these parameter values for the Nmp4−/− mice compared to their vehicle-treated cohorts (as indicated with *), whereas the wild-type mice were unresponsive. Violin plots represent the data distribution of the individual mice using dashed lines to show the median and the quartiles. We employed 2-way ANOVAs setting genotype and treatment as the independent variables. The p values for individual main effects and for the interaction term are shown in the panels (n= 9–11/cohort) Post hoc analyses including Tukey’s HSD comparisons between all cohorts and Student’s t test are presented in Supplemental Table S1.
Conditional loss of Nmp4 from MSPCs amplifies the skeleton response to Scl-mAb:
To begin identifying the cell type of action for boosted Scl-mAb effects, identical 7-week treatments to those using the global Nmp4−/− mice were conducted with the Nmp4fl/fl;Prx1Cre mouse model. The mice in which Nmp4 had been conditionally removed from MSPCs of the long bone (Nmp4fl/fl;Prx1Cre+) exhibited a similar exaggerated response to Scl-mAb in the femur as observed with the global Nmp4−/− mice (Fig. 6A and Fig. 6B; Fig. 7A – Fig. 7C, Supplemental Table S1). Additionally, Nmp4 status had little to no influence on femoral BV/TV or other architectural parameters in the vehicle-treated cohorts. Distinct from the global Nmp4−/− mice, the Nmp4fl/fl;Prx1Cre+ cohorts showed no enhanced response to Scl-mAb for L5 BV/TV, Tb.N, Tb.Th. and Tb.Sp (Fig. 6C and Fig. 6D; Fig. 7D – Fig. 7F, Supplemental Table S1). This was expected since Prrx1 is not expressed in the vertebral MSPCs64, thus acting as an internal control. It is also consistent with the response of these conditional knockout mice to intermittent PTH57.
Figure 6:

Conditional loss of Nmp4 from mesenchymal stem/progenitor cells (MSPCs) enhances Scl-mAb-induced osteoanabolism. (A) μCT reconstructions of the distal femur from (i) Nmp4fl/fl;Prx1Cre− vehicle, (ii) Nmp4fl/fl;Prx1Cre− Scl-mAb, (iii) Nmp4fl/fl;Prx1Cre+ vehicle, and (iv) Nmp4fl/fl;Prx1Cre+ Scl-mAb mice at 7 weeks therapy. The images were from mice exhibiting the median values of their respective cohorts. (B) Statistical analysis and raw data distribution are shown for the distal femoral bone volume/tissue volume (BV/TV) after 7 weeks of therapy for the four treatment groups. (C) The μCT reconstructions of the lumbar L5 of the four cohorts after 7 weeks therapy. (D) Raw data distribution and statistical evaluation are shown for L5 BV/TV for the four cohorts. Violin plots represent the data distribution of the individual mice (n=14–15 mice/cohort) using dashed lines to show the median and the quartiles. We employed 2-way ANOVAs setting genotype and treatment as the independent variables. The p values for individual main effects and for the interaction term are shown in both panels. ‡p ≤ 0.05 for comparison between Scl-mAb-treated cohorts (testing for genotype difference among Scl-mAb-treated groups). Post hoc analyses including Tukey’s HSD comparisons between all cohorts and Student’s t test are presented in Supplemental Table S1.
Figure 7:

Conditional loss of Nmp4 from MSPCs enhances Scl-mAb-induced femoral but not L5 trabecular thickness. Statistical analyses and raw data distribution profiles for trabecular number (Tb.N), trabecular thickness (Tb.Th), and trabecular spacing (Tb.Sp) are shown for distal femur (A–C) and L5 vertebra (D–F) after 7-week treatment. Violin plots are used to show data distribution for the individual mice (n = 14–15 mice/cohort) from the four treatment groups (i) Nmp4fl/fl;Prx1Cre− vehicle, (ii) Nmp4fl/fl;Prx1Cre− Scl-mAb, (iii) Nmp4fl/fl;Prx1Cre+ vehicle, and (iv) Nmp4fl/fl;Prx1Cre+ Scl-mAb mice at 7 weeks therapy. The dashed lines represent the median and the quartiles for each cohort. The Scl-mAb-induced increases in femoral Tb.Th was improved in the Nmp4fl/fl;Prx1Cre+ mice as compared to the Nmp4fl/fl;Prx1Cre− mice (Fig. 7B). However, this exaggerated response to Scl-mAb was not observed in L5 Tb.Th (Fig. 7E) since Prrx1 is not expressed in vertebral MSPCs. The statistical approach employed 2-way ANOVAs using genotype and treatment as the independent variables. The p values for the parameter main effects and for the interaction effect are presented in each panel. ‡p ≤ 0.05 for comparison between Scl-mAb-treated groups (testing for genotype difference among Scl-mAb-treated groups). Post hoc analyses including Student’s t test and Tukey’s HSD comparisons between groups are presented in Supplemental Table S1.
Serum markers of bone metabolism in global knockout (Nmp4−/−) and MSPC-selective (Nmp4fl/fl;Prx1Cre+) mutant models:
Given that both the global Nmp4−/− mice and the Nmp4fl/fl;Prx1Cre+ mice exhibited an enhanced Scl-mAb-induced increase in bone, we analyzed their serum bone turnover markers including osteocalcin and CTX. Serum was obtained from each mouse during treatment at 0 weeks (10 weeks of age, baseline), 3 weeks (13 weeks of age), and 7 weeks (17 weeks of age, termination). Loss of Nmp4 had no significant impact on baseline serum osteocalcin and CTX for the global or conditional mouse models (data not shown).
At the end of therapy (7 weeks) wild type mice (global Nmp4+/+ and Nmp4fl/fl;Prx1Cre−) treated with Scl-mAb exhibited higher levels of both serum osteocalcin and CTX compared to their vehicle-treated cohorts (Fig. 8A – Fig. 8H, Supplemental Table S1). Global loss of Nmp4 abolished Scl-mAb-induced increases in both serum osteocalcin and serum CTX compared to their vehicle-treated cohorts, resulting in genotype × treatment interactions for both (Fig. 8C, Fig. 8G, Supplemental Table S1). However, like the osteoclast histomorphometry data, there was no statistical difference for serum CTX between Scl-mAb-treated wild type and Nmp4−/− mice (Supplemental Table S1). Wild type mice treated with antibody did exhibit higher serum osteocalcin than the Nmp4−/− mice under this therapy (Supplemental Table S1). Conditional loss of Nmp4 from MSPCs did not significantly impact the Scl-mAb-induced increases in serum osteocalcin or CTX (Fig. 8D, Fig. 8H, Supplemental Table 1). Comparison of the serum profiles between the global, and conditional Nmp4 knockout models, suggests that loss of Nmp4 in non-osteoblast lineage cells, e.g., osteoclasts, attenuates Scl-mAb-mediated bone turnover and the release of osteocalcin and CTX from the matrix. This is consistent with the static histomorphometry data showing a modest but significant Scl-mAb-mediated decrease in osteoclast density in the global Nmp4−/− mice but not the wild type cohorts (Figs. 5A –5C, Supplemental Table S1).
Figure 8:
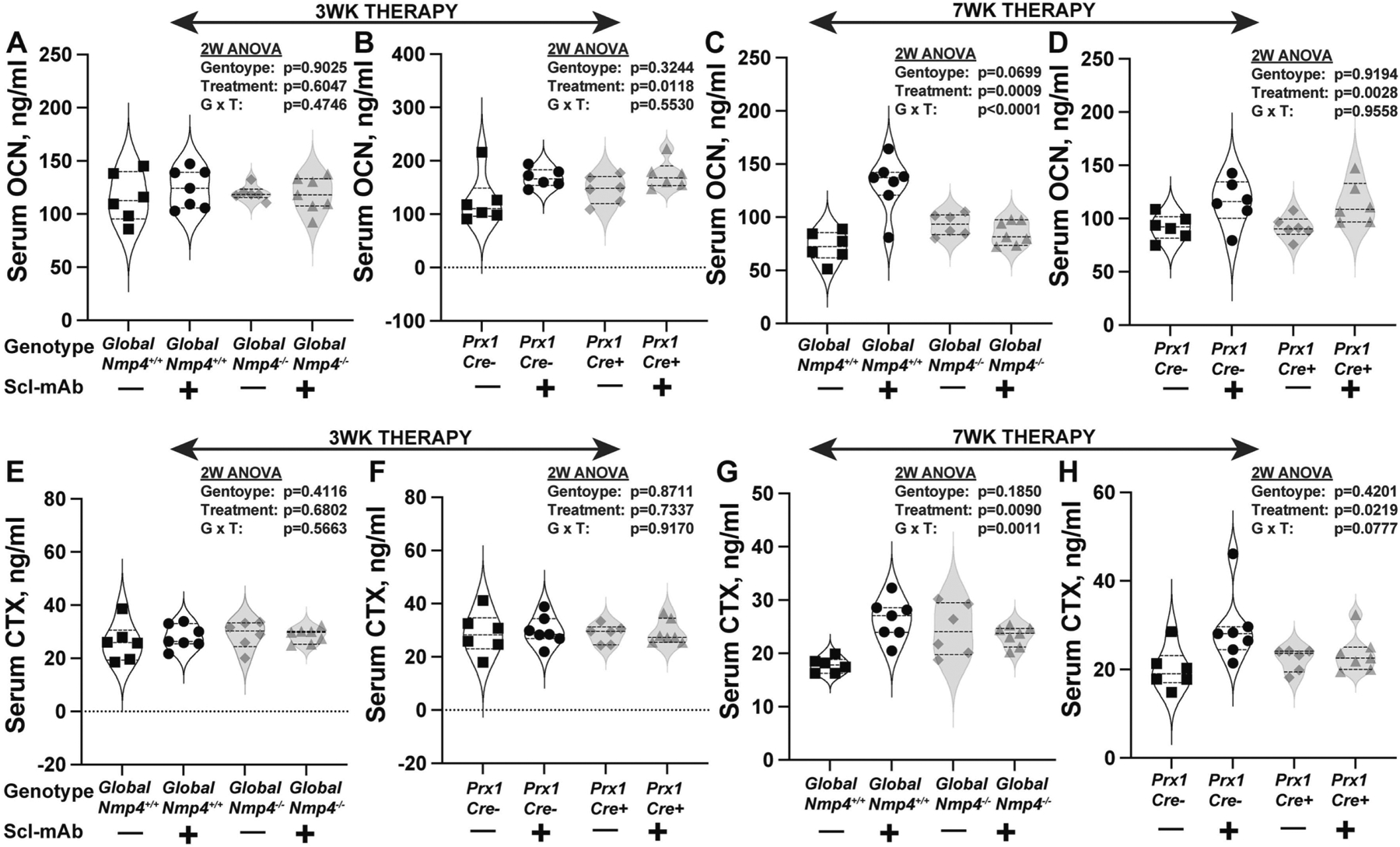
Scl-mAb-induced changes in the bone turnover markers (A-D) serum osteocalcin, OCN and (E-H) C-terminal telopeptides, CTX. Global loss of Nmp4 abrogates Scl-mAb-induced increases in serum OCN (A, C) and CTX (E, H) at both 3 weeks and 7 weeks of therapy. Conditional loss of Nmp4 from MSPCs, does not influence Scl-mAb-stimulated increases in serum OCN and may weaken rises in serum CTX. Violin plots with staggered data points show the values for individual mice at each serum collection and these data were evaluated using 2-way ANOVAs. The dashed lines represent the median and the quartiles for each cohort. Statistical significance was set at p ≤ 0.05 (n = 6–7 mice/cohort). Student’s t test and Tukey’s HSD comparisons between groups are presented in Supplemental Table S1.
Conditional loss of Nmp4 in MSPCs had little to no effect on cortical bone or its response to Scl-mAb (Table 2), in contrast to the augmented improvement in whole body and femoral aBMD in the global Nmp4−/− mice (Fig. 3, Table 1, Supplemental Table 1). This argues that a cell(s) other than or in addition to Nmp4−/− MSPCs and their progeny may contribute to the heightened whole body and femur aBMD response in the global Nmp4−/− mice.
TABLE 2:
Nmp4fl/fl;Prx1Cre mice skeletal parameters: Dual energy X-ray absorptiometry (DEXA) for areal bone mineral density and micro computed tomography (μCT) for femoral cortical parameters
| DEXA aBMD g/cm2 (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
|---|---|---|---|---|---|---|---|
| WB | 0.054±0.002 | 0.065±0.003 | 0.054±0.002 | 0.065±0.003 | p=0.7266 | p<0.0001 | p=0.5204 |
| FEMUR | 0.071±0.002 | 0.087±0.005 | 0.070±0.002 | 0.086±0.004 | p=0.1322 | p<0.0001 | p=0.9428 |
| TIBIA | 0.052±0.002 | 0.059±0.002 | 0.051±0.002 | 0.058±0.003 | p=0.0811 | p<0.0001 | p=0.8186 |
| SPINE (L3–L5) | 0.058±0.002 | 0.079±0.005 | 0.060±0.004 | 0.081±0.007 | p=0.1151 | p<0.0001 | p=0.6410 |
| μCT Femoral cortical architecture (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
| Total area mm2 | 1.623±0.101 | 1.708±0.094 | 1.593±0.092 | 1.657±0.102 | p=0.1153 | p=0.0048 | p=0.6738 |
| Bone area mm2 | 0.848±0.043 | 1.080±0.071 | 0.845±0.040 | 1.044±0.064 | p=0.1874 | p<0.0001 | p=0.2512 |
| Cortical Th mm | 0.215±0.009 | 0.278±0.017 | 0.217±0.008 | 0.271±0.014 | p=0.3902 | p<0.0001 | p=0.2152 |
| Marrow area mm2 | 0.775±0.079 | 0.628±0.068 | 0.748±0.065 | 0.613±0.075 | p=0.2610 | p<0.0001 | p=0.7467 |
| pMOI mm4 | 0.332±0.038 | 0.415±0.047 | 0.327±0.036 | 0.390±0.046 | p=0.1916 | p=0.0001 | p=0.3789 |
| Imax mm4 | 0.203±0.027 | 0.261±0.033 | 0.208±0.027 | 0.247±0.031 | p=0.5741 | p<0.0001 | p=0.2230 |
| Imin mm4 | 0.129±0.012 | 0.154±0.016 | 0.119±0.011 | 0.143±0.016 | p=0.0071 | p<0.0001 | p=0.9549 |
VEH=vehicle; Scl-mAb=sclerostin monoclonal antibody; aBMD=areal bone mineral density; Cortical Th=cortical thickness; Imax=maximum moment of inertia; Imin=minimum moment of inertia; L3–L5=lumbar vertebra 3 to 5; pMOI=polar moment of inertia, WB=whole body. Nmp4fl/fl;Prx1Cre+ and Nmp4fl/fl;Prx1Cre− mice were treated with Scl-mAb (10mg/kg, twice/week) or vehicle control from 10 weeks to 17 weeks of age. DXA at 7 weeks of treatment showed that Scl-mAb significantly increased WB, femur, tibial, and spine (L3–L5) aBMD (treatment p < 0.0001) for both Nmp4fl/fl;Prx1Cre+ and Nmp4fl/fl;Prx1Cre− mice. Data represent average ± SD; n = 13–14 mice/cohort. Femoral cortical architecture at 7 weeks of treatment showed there was a significant treatment effect for all parameters and a genotype effect for Imin, which was significantly higher in the wild-type mice. Consistent with these results, the Imin, an index of mechanical resistance to bending, was significantly higher in the Nmp4fl/fl;Prx1Cre+ mice compared with the Nmp4fl/fl;Prx1Cre− controls (p = 0.0071). Data represent average ± SD; n = 14–15 mice/cohort. All statistical analyses were performed using 2-way ANOVA tests setting genotype and treatment as the independent variables. Statistical significance was set at p ≤ 0.05.
Scl-mAb is a stronger osteoanabolic than PTH but the humanized version of the former is associated with some adverse events31,65,66. Therefore, we asked whether loss of Nmp4 in MSPCs elevates PTH potency to that of Scl-mAb observed in wild type mice? Juxtaposing PTH57- and Scl-mAb-induced increases in femoral BV/TV in the Nmp4fl/fl;Prx1Cre mice showed that Scl-mAb is a significantly stronger anabolic (Supplemental Figure 1), consistent with previous rodent and clinical studies31,65,66. However, PTH-induced increases in femoral BV/TV in Nmp4fl/fl;Prx1Cre+ mice were equivalent to Scl-mAb gains in Nmp4fl/fl;Prx1Cre− cohorts, i.e., conditional loss of Nmp4 in MSPCs endowed PTH with the efficacy of Scl-mAb observed in wild type mice (Supplemental Figure 1).
Conditional loss of Nmp4 from osteocalcin-expressing osteoblasts (Nmp4fl/fl;BglapCre+) or from osteocytes (Nmp4fl/fl;Dmp1Cre+) did not enhance bone response to Scl-mAb:
Given the observation that MSPC-selective deletion of Nmp4 enhances Scl-mAb-induced bone gain, we continued targeting down the osteoblast differentiation pathway to determine how late in the lineage we could go, regarding Nmp4 deletion, and still find Scl-mAb enhancing effects. Our mouse models in which Nmp4 was conditionally removed from later osteoblast lineage stages showed strong and significant response to Scl-mAb throughout the skeleton, but the loss of Nmp4 in the osteocalcin-expressing cells did not boost Scl-mAb efficacy. At the end of 7 weeks treatment, femoral BV/TV was equivalent in Nmp4fl/fl;BglapCre+ and Nmp4fl/fl;BglapCre− mice, as was L5 BV/TV, (Fig. 9A, Fig. 9B, Supplemental Table S1). Similar femoral and L5 BV/TV profiles were obtained with the Nmp4fl/fl;Dmp1Cre mice (Fig. 9C, Fig. 9D, Supplemental Table S1). Incidentally, our earlier work showed these models also failed to show improved response to PTH, i.e., genotype x treatment interactions for femur and L5 BV/TV57. Lastly, most of several skeletal parameters exhibited only strong Scl-mAb treatment effects in these conditional knockout models (Table 3 and Table 4).
Figure 9:
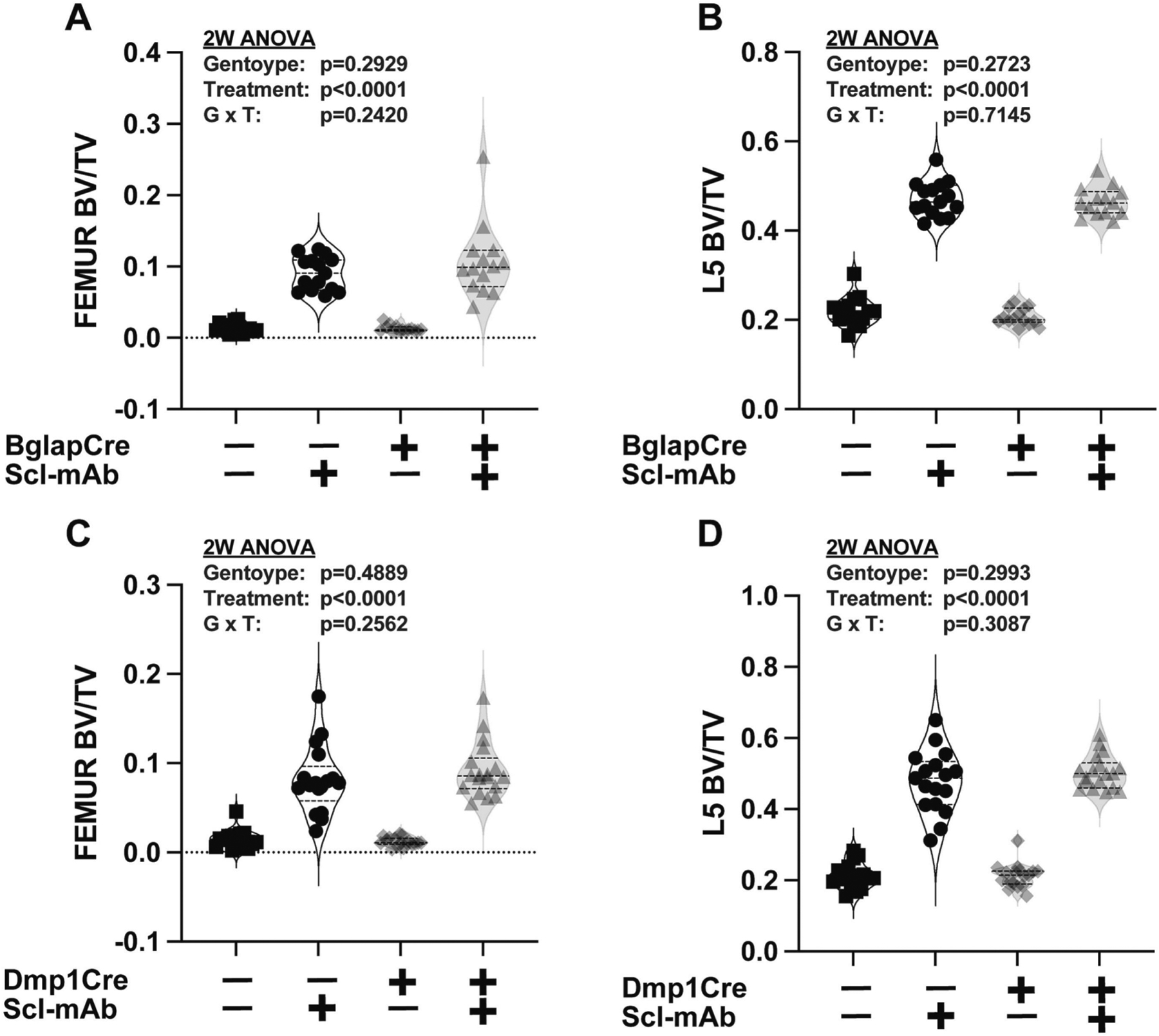
Conditional loss of Nmp4 in either (A, B) mature osteocalcin-expressing osteoblasts or (C, D) osteocytes has no effect on femoral or L5 vertebra bone volume/tissue volume (BV/TV) or Scl-mAb-induced increases in these parameters. Raw data distribution and statistical analysis are shown for the distal femoral BV/TV and the L5 vertebra BV/TV from the four cohorts (i) Nmp4fl/fl;BglapCre− or Nmp4fl/fl;Dmp1Cre− vehicle, (ii) Nmp4fl/fl;BglapCre− or Nmp4fl/fl;Dmp1Cre− Scl-mAb, (iii) Nmp4fl/fl;BglapCre+ or Nmp4fl/fl;Dmp1Cre+ vehicle, (iv) Nmp4fl/fl;BglapCre+ or Nmp4fl/fl;Dmp1Cre+ Scl-mAb, n = 14–18 at 7 weeks of treatment. Data distribution is represented using violin plots showing the median and the quartiles as dashed lines. Statistical analyses employed 2-way ANOVAs using treatment and genotype as the independent variables. Statistical significance was set at p ≤ 0.05. The p values for individual main effects and for the interaction term are presented in each panel. Post hoc analyses are presented in Supplemental Table S1
TABLE 3:
Nmp4fl/fl;BglapCre mice skeletal parameters: Micro computed tomography (μCT) for femoral trabecular and cortical parameters; Dual energy X-ray absorptiometry (DEXA) for areal bone mineral density
| μCT Femoral trabecular parameters (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
|---|---|---|---|---|---|---|---|
| TbN (mm− 1) | 2.125±0.322 | 2.760±0.517 | 2.004±0.329 | 2.705±0.351 | p=0.3906 | p<0.0001 | p=0.7496 |
| TbTh (mm) | 0.039±0.004 | 0.068±0.007 | 0.042±0.008 | 0.063±0.006 | p=0.5826 | p<0.0001 | p=0.0407 |
| TbSp (mm) | 0.481±0.075 | 0.365±0.080 | 0.510±0.085 | 0.367±0.052 | p=0.4242 | p<0.0001 | p=0.4764 |
| μCT Femoral cortical parameters (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
| Total area mm2 | 1.616±0.058 | 1.699±0.078 | 1.566±0.071 | 1.702±0.107 | p=0.2751 | p<0.0001 | p=0.2234 |
| Bone area mm2 | 0.814±0.029 | 1.047±0.067 | 0.815±0.053 | 1.060±0.056 | p=0.6201 | p<0.0001 | p=0.6638 |
| Cortical Th mm | 0.205±0.008 | 0.268±0.014 | 0.208±0.013 | 0.270±0.011 | p=0.3681 | p<0.0001 | p=0.8593 |
| Marrow area mm2 | 0.802±0.051 | 0.652±0.039 | 0.752±0.057 | 0.642±0.069 | p=0.0416 | p<0.0001 | p=0.1732 |
| pMOI | 0.324±0.0 | 0.406±0.0 | 0.313±0.0 | 0.412±0.0 | p=0.771 | p<0.000 | p=0.347 |
| mm4 | 21 | 40 | 29 | 48 | 4 | 1 | 4 |
| Imax mm4 | 0.206±0.016 | 0.257±0.027 | 0.200±0.018 | 0.264±0.033 | p=0.9474 | p<0.0001 | p=0.2830 |
| Imin mm4 | 0.118±0.007 | 0.148±0.015 | 0.113±0.012 | 0.147±0.017 | p=0.3602 | p<0.0001 | p=0.5427 |
| DEXA aBMD g/cm2 (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
| WB | 0.052±0.002 | 0.063±0.003 | 0.052±0.002 | 0.063±0.002 | p=0.7003 | p<0.0001 | p=0.7399 |
| FEMUR | 0.069±0.003 | 0.086±0.003 | 0.068±0.003 | 0.086±0.003 | p=0.8525 | p<0.0001 | p=0.9993 |
| TIBIA | 0.046±0.002 | 0.054±0.004 | 0.046±0.002 | 0.054±0.003 | p=0.9459 | p<0.0001 | p=0.6791 |
| SPINE(L3–L5) | 0.058±0.006 | 0.076±0.005 | 0.057±0.004 | 0.077±0.004 | p=0.8688 | p<0.0001 | p=0.3632 |
VEH=vehicle; Scl-mAb=sclerostin monoclonal antibody; aBMD=areal bone mineral density; Cortical Th=cortical thickness; Imax=maximum moment of inertia; Imin=minimum moment of inertia; L3–L5=lumbar vertebra 3 to 5; pMOI=polar moment of inertia; Tb.N=trabecular number; Tb.Th=trabecular thickness; Tb.Sp=trabecular space. Nmp4fl/fl;BglapCre+ and Nmp4fl/fl;BglapCre− mice were treated with Scl-mAb (10mg/kg, twice/week) or vehicle control from 10 weeks to 17 weeks of age. Femoral trabecular bone architecture at 7 weeks of treatment showed that loss of Nmp4 in mature osteoblasts had little impact on trabecular architecture. There was a modest genotype x treatment effect for Tb.Th Data represent average ± SD; n = 14–15 mice/group. Femoral cortical architecture at 7 weeks of treatment showed that Scl-mAb induced significant changes in all the cortical parameters in both genotypes. Data represent average ± SD; n = 14–15 mice/group. DXA at 7 weeks of treatment of the post-cranial skeleton (WB, whole body), femur, tibia, and spine (L3–L5) showed that Scl-mAb treatment significantly increased aBMD, but there was no difference in hormone response between the two genotypes. Data represent average ± SD; n = 11–15 mice/group. All analyses were performed using 2-way ANOVA setting genotype and treatment as the independent variables. Statistical significance was set at p ≤ 0.05.
TABLE 4:
Nmp4fl/fl;Dmp1Cre mice skeletal parameters: Micro computed tomography (μCT) for femoral trabecular and cortical parameters; Dual energy X-ray absorptiometry (DEXA) for areal bone mineral density
| μCT Femoraltrabecular parameters (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
|---|---|---|---|---|---|---|---|
| TbN (mm− 1) | 2.080±0.365 | 2.817±0.322 | 2.210±0.495 | 2.707±0.529 | p=0.9240 | p<0.0001 | p=0.2549 |
| TbTh (mm) | 0.038±0.006 | 0.060±0.007 | 0.037±0.005 | 0.059±0.005 | p=0.5486 | p<0.0001 | p=0.6675 |
| TbSp (mm) | 0.498±0.114 | 0.346±0.042 | 0.473±0.107 | 0.374±0.086 | p=0.9197 | p<0.0001 | p=0.2291 |
| μCT Femoral cortical parameters (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
| Total area mm2 | 1.579±0.062 | 1.684±0.082 | 1.562±0.092 | 1.697±0.104 | p=0.9340 | p<0.0001 | p=0.4773 |
| Bone area mm2 | 0.809±0.045 | 1.042±0.039 | 0.819±0.041 | 1.066±0.043 | p=0.0902 | p<0.0001 | p=0.4883 |
| Cortical Th mm | 0.209±0.015 | 0.266±0.009 | 0.212±0.011 | 0.274±0.011 | p=0.0396 | p<0.0001 | p=0.4120 |
| Marrow area mm2 | 0.770±0.077 | 0.643±0.071 | 0.743±0.077 | 0.631±0.086 | p=0.3101 | p<0.0001 | p=0.6775 |
| pMOI | 0.312±0.0 | 0.398±0.0 | 0.310±0.0 | 0.408±0.0 | p=0.558 | p<0.000 | p=0.465 |
| mm4 | 21 | 35 | 31 | 44 | 4 | 1 | 8 |
| Imax mm4 | 0.196±0.015 | 0.251±0.026 | 0.195±0.020 | 0.258±0.030 | p=0.5138 | p<0.0001 | p=0.4723 |
| Imin mm4 | 0.116±0.008 | 0.147±0.010 | 0.115±0.013 | 0.150±0.015 | p=0.7097 | p<0.0001 | p=0.5138 |
| DEXA aBMD g/cm2 (7wks treatment) | VEH | Scl-mAb | VEH | Scl-mAb | Genotype | Treatment | Genotype x Treatment |
| WB | 0.054±0.002 | 0.066±0.002 | 0.053±0.002 | 0.066±0.002 | p=0.8317 | p<0.0001 | p=0.3796 |
| FEMUR | 0.068±0.003 | 0.084±0.003 | 0.068±0.004 | 0.085±0.003 | p=0.7482 | p<0.0001 | p=0.5845 |
| TIBIA | 0.049±0.002 | 0.057±0.003 | 0.049±0.003 | 0.055±0.002 | p=0.1885 | p<0.0001 | p=0.2298 |
| SPINE (L3–L5) | 0.059±0.004 | 0.082±0.003 | 0.059±0.004 | 0.083±0.004 | p=0.7952 | p<0.0001 | p=0.4438 |
VEH=vehicle; Scl-mAb=sclerostin monoclonal antibody; aBMD=areal bone mineral density; Cortical Th=cortical thickness; Imax=maximum moment of inertia; Imin=minimum moment of inertia; L3–L5=lumbar vertebra 3 to 5; pMOI=polar moment of inertia, Tb.N=trabecular number; Tb.Th=trabecular thickness; Tb.Sp=trabecular space. Nmp4fl/fl;Dmp1Cre+ and Nmp4fl/fl;Dmp1Cre− mice were treated with Scl-mAb (10mg/kg, twice/week) or vehicle control from 10 weeks to 17 weeks of age. Femoral trabecular bone architecture at 7 weeks of treatment showed that loss of Nmp4 in osteocytes had no impact on trabecular architecture. Data represent average ± SD; n = 17–18 mice/group. Femoral cortical architecture at 7 weeks of treatment shows that Scl-mAb significantly equally altered several parameters in both genotypes. Cortical Th was lower in the Nmp4fl/fl;Dmp1Cre+ mice. Data represent average ± SD; n = 17–18 mice/group. DXA at 7 weeks of treatment of the post-cranial skeleton (whole body), femur, tibia, and spine (L3–L5) showed that Scl-mAb treatment significantly increased aBMD, but there was no difference in hormone response between the two genotypes. Data represent average ± SD; n = 14–15 mice/group. Statistical analyses were performed using 2-way ANOVA tests setting genotype and treatment as the independent variables. Statistical significance was set at p ≤ 0.05.
DISCUSSION:
The mechanisms of action for teriparatide, abaloparatide, and romosozumab-aqqg differ by varying degrees but all three exhibit a rapid loss of potency during osteoporosis therapy1,2, raising the question as to whether there is a single, common barrier limiting their bone anabolic output. We have reported that the global loss of Nmp4 significantly enhanced PTH-induced bone formation in experimental mice, and conditional loss of this gene from MSPCs phenocopied this improved osteoanabolic response35–40,57. However, this increased sensitivity to PTH was lost in animals in which Nmp4 had been conditionally disabled in the later stages of lineage development including osteoblasts and osteocytes57.
For the present study we hypothesized that Nmp4−/− MSPCs are pre-programmed to drive an enhanced response to any osteoanabolic drug. That being so, we reasoned that our Nmp4 mouse models should exhibit similar patterns of skeletal response to Scl-mAb as observed for PTH therapies, despite their mechanistic differences for increasing bone formation. Indeed, this was generally the case. Global loss of Nmp4 strikingly heightened Scl-mAb-induced increases in cancellous bone of the femur and spine, just as we previously reported for PTH35–40. Additionally, as observed with PTH therapy57, conditional deletion of Nmp4 from MSPCs largely phenocopied these improved Scl-mAb-induced increases in the trabecular compartment. Finally, targeting Nmp4 in mature osteoblasts or osteocytes failed to improve bone response to Scl-mAb.
Osteoanabolic induced bone formation is more rapid and robust in both global Nmp4−/− and conditional knockout (Nmp4fl/fl;Prx1Cre+) mice compared to wild type littermates but the dynamics of bone formation between Scl-mAb and PTH differ somewhat (this study and refs37,38,57). The enhanced Scl-mAb- and PTH-induced femoral bone growth at 4 weeks of treatment were accompanied by increases in Nmp4−/− active osteoblast coverage (MS/BS) (this study and in ref57). The increased number of bone marrow Nmp4−/− osteoprogenitors in naïve mice37 may contribute to these altered dynamics of bone formation. Curiously, mineral apposition rates (MAR) were different between the two therapies. Under PTH treatment Nmp4 status had no impact on this histomorphometric parameter57. But, in the present study the Nmp4−/− mice treated with Scl-mAb exhibited a lower MAR than the wild type mice. Overall, this may reflect differences in the anabolic window profiles between our knockout and wild type mice combined with the contrast between the actions of PTH and Scl-mAb. A more detailed bone histomorphometric time course might clarify the basis of these differences.
The primary effect of Nmp4 status on both Scl-mAb and PTH therapies is the magnitude of trabecular bone gain that appears pre-programmed in bone marrow MSPCs; but we documented some differences in outcomes between Scl-mAb and PTH therapies. These distinctions may be related to Scl-mAb functioning as a dual action drug and differences in how Nmp4 regulates drug-induced bone remodeling (primary mode of PTH action) vs. modeling (primary mode of Scl-mAb action)22–24. Global Nmp4−/− mice showed a significant boost in the Scl-mAb-mediated increase in whole body and femur aBMD, which are primarily driven by cortical bone. However, this augmented gain was lost in the conditional knockout mice. Perhaps Nmp4−/− osteoclasts are more susceptible to the anti-resorptive action of Scl-mAb. Indeed, the global Nmp4−/− mice showed a Scl-mAb-orchestrated decrease in osteoclast density not observed in wild type mice. The serum profile is consistent with a decrease in Scl-mAb-induced bone turnover by Nmp4−/− osteoclasts. Specifically, antibody treatment increased serum osteocalcin and CTX in the wild type mice under our treatment regimen. Global, but not conditional loss of Nmp4 in MSPCs abrogated this Scl-mAb-stimulated increase in these markers. The change in osteoclast density and putative shift in bone turnover are small and it is unclear how functionally significant they are under our Scl-mAb regimen. Similarly, an earlier study showed that Nmp4−/− osteoblasts protected global knockout mice from unloading-induced bone loss but osteoclastic bone resorption parameters were unaffected by Nmp4 status67. Additionally, loss of Nmp4 failed to enhance Scl-mAb-induced increases in several other femoral cortical parameters, suggesting that the impact of Nmp4 status on the cortical compartment is site-specific. Conversely, global, or conditional loss of Nmp4 in MSPCs increased the PTH-induced rise in serum osteocalcin, had no effect on serum CTX, and did not influence increases in cortical bone gain37–39,57. Further investigations into the common and distinguishing mechanisms underlying Nmp4 control of Scl-mAb and PTH therapies are required.
Juxtaposing the PTH57 and Scl-mAb femoral trabecular BV/TV responses of the Nmp4fl/fl;Prx1Cre mice demonstrates that Nmp4−/− MSPCs endowed PTH with the equivalent potency of Scl-mAb observed in wild-type mice. This has potential clinical significance. Romosozumab-aqqg is a stronger osteoanabolic than teriparatide31,65,66 but its long term safety continues to be investigated because the FDA has indicated that it can increase the risks of serious cardiovascular events68–70. Therefore, targeting the Nmp4 pathway in osteoprogenitors may provide a novel adjuvant strategy for bestowing PTH therapy with the efficacy of romosozumab-aqqg treatment with appropriate safety.
Our data support the hypothesis that the potency of any osteoanabolic drug is largely pre-programmed by the expression of Nmp4 in a single cell type at a specific point during differentiation. This markedly reframes our understanding about the scope and agency regarding Nmp4 control of pharmacologically induced bone gain. However, these findings are tempered by the limitations of the present data, which provide the basis for the next experiments. Whether Nmp4 deletion combined with these treatment modalities sustains continued enhanced efficacy for longer time periods of therapy, such as 15 or 30 weeks after initiating treatment needs to be determined. Indeed, our histomorphometric data suggest that this exaggerated response to osteoanbolics may wane after 7 weeks. The mechanisms that convert the Nmp4−/− MSPC into an agent of heightened osteoanabolic response must be identified. These cells appear to harbor a unique physiological UPR warranting interrogation40,41,71. This altered pathway may bias the cells toward the osteo-lineage while at the same time proactively expanding their capacity for high matrix production without taxing delivery and triggering apoptosis. Indeed, Nmp4−/− MSPC transcriptomic data predicted that protein-folding activity is elevated, and UPR-induced apoptosis is attenuated in these cells40. Therefore, investigation on whether loss of Nmp4 enhances PTH and Scl-mAb anti-apoptotic activity is of interest. The ultrastructural analysis of the Nmp4−/− MSPC endoplasmic reticulum and Golgi apparatus will address whether loss of this gene pre-emptively expands the key organelles regulating secretory capacity. The use of scRNA-seq will provide an in vivo measure of how Nmp4−/− MSPCs alter the cellular taxonomy of the bone marrow stroma thus priming it for the exaggerated response to osteoanabolics. Investigations to determine whether Nmp4−/− osteoclasts play a minor but significant role during Scl-mAb response in cortical bone are required. Finally, to extend earlier experiments interrogating the impact of Nmp4 on skeletal unloading67, we are investigating whether Nmp4 suppresses the bone anabolic response to mechanical loading with or without osteoanabolic drug therapy.
In summary, to determine the influence of Nmp4 on Scl-mAb osteoanabolic therapy, we introduced a loss-of-function mutation, either globally or conditionally in osteogenic lineage cells at distinct stages of differentiation. Global, or conditional loss of Nmp4 in MSPCs enhanced Scl-mAb-induced increases in bone formation, whereas conditional deletion in later stages of osteoblast differentiation did not. These results are very similar to the previously reported effect of Nmp4 on bone response to PTH therapy, with some evidence that Nmp4−/− osteoclasts may make a minor contribution to the Scl-mAb improved response. We conclude that the expression of Nmp4 in MSPCs largely determines the magnitude of the skeletal response to any osteoanabolic.
Supplementary Material
HIGHLIGHTS:
FDA-approved osteoanabolics include PTH, PTHrp and sclerostin antibody (Scl-mAb)
These drugs have distinct mechanisms of action, but all rapidly lose efficacy
Earlier we showed conditional loss of Nmp4 in MSPCs improves PTH-induced bone gain
Deletion of Nmp4 in later osteoblast stages did not replicate this response
In this study Nmp4−/− MSPCs similarly improved Scl-mAb-induced bone gain
We conclude Nmp4 sets osteoanabolic potency early in osteoblast differentiation
ACKNOWLEDGEMENTS:
This work was supported by National Institutes of Health Grants (NIH) 1R01 AR073739 to J.P.B. and R01 AR053237 to A.G.R.; VA grants I01 BX005861 and IK6 BX 003783 to A.G.R.; and support from T32 AR065971 to E.G.A and C.K, and NIH R21 AG078861, VA 1I0 1BX005154 to L.I.P. We thank Alexander Jackson for his technical assistance with some aspects of this project. Sclerostin antibody was provided by Amgen Inc., (Thousand Oaks, CA) and UCB (Brussels, Belgium).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES: L.I.P. is a member of the FD/MAS Alliance Scientific Advisory Council
DATA AVAILABILITY STATEMENT:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES:
- 1.Sims NA Overcoming natural Wnt inhibition to optimize therapy. Nature Reviews Rheumatology 15, 67–68 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Tabacco G & Bilezikian JP Osteoanabolic and dual action drugs. Br J Clin Pharmacol 85, 1084–1094, doi: 10.1111/bcp.13766 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tay D, Cremers S & Bilezikian JP Optimal dosing and delivery of parathyroid hormone and its analogues for osteoporosis and hypoparathyroidism - translating the pharmacology. Br J Clin Pharmacol 84, 252–267, doi: 10.1111/bcp.13455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jüppner H et al. AG protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science 254, 1024–1026 (1991). [DOI] [PubMed] [Google Scholar]
- 5.Abou-Samra A-B et al. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proceedings of the National Academy of Sciences 89, 2732–2736 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang D et al. Contributions of parathyroid hormone (PTH)/PTH-related peptide receptor signaling pathways to the anabolic effect of PTH on bone. Bone 40, 1453–1461, doi: 10.1016/j.bone.2007.02.001 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu S et al. Parathyroid hormone increases activating transcription factor 4 expression and activity in osteoblasts: requirement for osteocalcin gene expression. Endocrinology 149, 1960–1968 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu F, Lee S-K, Adams DJ, Gronowicz GA & Kream BE CREM deficiency in mice alters the response of bone to intermittent parathyroid hormone treatment. Bone 40, 1135–1143 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang R et al. Transcriptional regulation of BMP2 expression by the PTH-CREB signaling pathway in osteoblasts. PloS one 6, e20780 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui Y et al. Lrp5 functions in bone to regulate bone mass. Nature medicine 17, 684–691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Padhi D, Jang G, Stouch B, Fang L & Posvar E Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 26, 19–26, doi: 10.1002/jbmr.173 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Padhi D et al. Multiple doses of sclerostin antibody romosozumab in healthy men and postmenopausal women with low bone mass: a randomized, double-blind, placebo-controlled study. The Journal of Clinical Pharmacology 54, 168–178 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Angers S & Moon RT Proximal events in Wnt signal transduction. Nature reviews Molecular cell biology 10, 468–477 (2009). [DOI] [PubMed] [Google Scholar]
- 14.MacDonald BT & He X Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harbor perspectives in biology 4, a007880 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J & Long F β-catenin promotes bone formation and suppresses bone resorption in postnatal growing mice. Journal of Bone and Mineral Research 28, 1160–1169 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glass DA et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Developmental cell 8, 751–764 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Li X et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. Journal of Biological Chemistry 280, 19883–19887 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Semënov M, Tamai K & He X SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. Journal of Biological Chemistry 280, 26770–26775 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Wijenayaka AR et al. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PloS one 6, e25900 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krupa K, Parmar M & Delo LF in StatPearls [Internet] (StatPearls Publishing, 2022). [Google Scholar]
- 21.Wein MN & Kronenberg HM Regulation of Bone Remodeling by Parathyroid Hormone. Cold Spring Harbor perspectives in medicine 8, doi: 10.1101/cshperspect.a031237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin TJ, Sims NA & Seeman E Physiological and pharmacological roles of PTH and PTHrP in bone using their shared receptor, PTH1R. Endocrine reviews 42, 383–406 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Ramchand SK & Seeman E Reduced Bone Modeling and Unbalanced Bone Remodeling: Targets for Antiresorptive and Anabolic Therapy. Handb Exp Pharmacol 262, 423–450, doi: 10.1007/164_2020_354 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Ominsky MS, Niu QT, Li C, Li X & Ke HZ Tissue-level mechanisms responsible for the increase in bone formation and bone volume by sclerostin antibody. Journal of Bone and Mineral Research 29, 1424–1430 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Ke HZ, Roberts SJ & Holdsworth G in Principles of Bone Biology 1711–1731 (Elsevier, 2020). [Google Scholar]
- 26.Balani DH, Trinh S, Xu M & Kronenberg HM Sclerostin Antibody Administration Increases the Numbers of Sox9creER+ Skeletal Precursors and Their Progeny. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 36, 757–767, doi: 10.1002/jbmr.4238 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miao D et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1–34. The Journal of clinical investigation 115, 2402–2411, doi: 10.1172/jci24918 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan Y et al. Parathyroid Hormone Directs Bone Marrow Mesenchymal Cell Fate. Cell metabolism 25, 661–672, doi: 10.1016/j.cmet.2017.01.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jilka RL et al. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. The Journal of clinical investigation 104, 439–446, doi: 10.1172/jci6610 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim SW et al. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 27, 2075–2084, doi: 10.1002/jbmr.1665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ominsky MS et al. Differential temporal effects of sclerostin antibody and parathyroid hormone on cancellous and cortical bone and quantitative differences in effects on the osteoblast lineage in young intact rats. Bone 81, 380–391, doi: 10.1016/j.bone.2015.08.007 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Strom O et al. Osteoporosis: burden, health care provision and opportunities in the EU: a report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch Osteoporos 6, 59–155, doi: 10.1007/s11657-011-0060-1 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Conley RB et al. Secondary Fracture Prevention: Consensus Clinical Recommendations from a Multistakeholder Coalition. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 35, 36–52, doi: 10.1002/jbmr.3877 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Thunyakitpisal P et al. Cloning and functional analysis of a family of nuclear matrix transcription factors (NP/NMP4) that regulate type I collagen expression in osteoblasts. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 16, 10–23, doi: 10.1359/jbmr.2001.16.1.10 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Robling AG et al. Nmp4/CIZ suppresses parathyroid hormone-induced increases in trabecular bone. Journal of cellular physiology 219, 734–743, doi: 10.1002/jcp.21717 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Childress P et al. Nmp4/CIZ suppresses the response of bone to anabolic parathyroid hormone by regulating both osteoblasts and osteoclasts. Calcified tissue international 89, 74–89, doi: 10.1007/s00223-011-9496-y (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He Y et al. Nmp4/CIZ suppresses the parathyroid hormone anabolic window by restricting mesenchymal stem cell and osteoprogenitor frequency. Stem cells and development 22, 492–500, doi: 10.1089/scd.2012.0308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Childress P et al. Genome-Wide Mapping and Interrogation of the Nmp4 Antianabolic Bone Axis. Molecular endocrinology 29, 1269–1285, doi: 10.1210/me.2014-1406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shao Y et al. Improving Combination Osteoporosis Therapy In a Preclinical Model of Heightened Osteoanabolism. Endocrinology 158, 2722–2740, doi: 10.1210/en.2017-00355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shao Y et al. Loss of Nmp4 optimizes osteogenic metabolism and secretion to enhance bone quality. American journal of physiology. Endocrinology and metabolism 316, E749–e772, doi: 10.1152/ajpendo.00343.2018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Young SK, Shao Y, Bidwell JP & Wek RC Nuclear Matrix Protein 4 is a Novel Regulator of Ribosome Biogenesis and Controls the Unfolded Protein Response Via Repression of Gadd34 Expression. The Journal of biological chemistry, doi: 10.1074/jbc.M116.729830 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gass JN, Gifford NM & Brewer JW Activation of an unfolded protein response during differentiation of antibody-secreting B cells. Journal of Biological Chemistry 277, 49047–49054 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Van Anken E et al. Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity 18, 243–253 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Turishcheva E, Vildanova M, Onishchenko G & Smirnova E The Role of Endoplasmic Reticulum Stress in Differentiation of Cells of Mesenchymal Origin. Biochemistry (Mosc) 87, 916–931, doi: 10.1134/s000629792209005x (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwakoshi NN et al. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nature immunology 4, 321–329 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Mills JC & Taghert PH Scaling factors: transcription factors regulating subcellular domains. Bioessays 34, 10–16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Al-Maskari M et al. Site-1 protease function is essential for the generation of antibody secreting cells and reprogramming for secretory activity. Scientific reports 8, 14338, doi: 10.1038/s41598-018-32705-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dekaney CM, King S, Sheahan B & Cortes JE Mist1 expression is required for Paneth cell maturation. Cellular and Molecular Gastroenterology and Hepatology 8, 549–560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lo H-YG et al. A single transcription factor is sufficient to induce and maintain secretory cell architecture. Genes & development 31, 154–171 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khetchoumian K et al. Pituitary cell translation and secretory capacities are enhanced cell autonomously by the transcription factor Creb3l2. Nature communications 10, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fox RM & Andrew DJ Transcriptional regulation of secretory capacity by bZip transcription factors. Frontiers in biology 10, 28–51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hess DA et al. MIST1 links secretion and stress as both target and regulator of the unfolded protein response. Molecular and cellular biology 36, 2931–2944 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang M et al. MIST1 and PTF1 Collaborate in Feed-Forward Regulatory Loops That Maintain the Pancreatic Acinar Phenotype in Adult Mice. Mol Cell Biol 36, 2945–2955, doi: 10.1128/mcb.00370-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang S et al. NMP4 regulates the innate immune response to influenza A virus infection. Mucosal Immunol 14, 209–218, doi: 10.1038/s41385-020-0280-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bidwell J et al. Nmp4, a Regulator of Induced Osteoanabolism, Also Influences Insulin Secretion and Sensitivity. Calcified tissue international 110, 244–259, doi: 10.1007/s00223-021-00903-7 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakamoto T et al. Mice Deficient in CIZ/NMP4 Develop an Attenuated Form of K/BxN-Serum Induced Arthritis. Journal of cellular biochemistry 117, 970–977, doi: 10.1002/jcb.25382 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Atkinson EG et al. Conditional Loss of Nmp4 in Mesenchymal Stem Progenitor Cells Enhances PTH-Induced Bone Formation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, doi: 10.1002/jbmr.4732 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bivi N et al. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 27, 374–389, doi: 10.1002/jbmr.548 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lim K-E et al. Co-deletion of Lrp5 and Lrp6 in the skeleton severely diminishes bone gain from sclerostin antibody administration. Bone 143, 115708 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iwamoto R et al. Chemokine ligand 28 (CCL28) negatively regulates trabecular bone mass by suppressing osteoblast and osteoclast activities. Journal of bone and mineral metabolism 39, 558–571, doi: 10.1007/s00774-021-01210-9 (2021). [DOI] [PubMed] [Google Scholar]
- 61.Deosthale P et al. Sex-specific differences in direct osteoclastic versus indirect osteoblastic effects underlay the low bone mass of Pannexin1 deletion in TRAP-expressing cells in mice. Bone reports 16, 101164 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robling AG et al. Anabolic and catabolic regimens of human parathyroid hormone 1–34 elicit bone- and envelope-specific attenuation of skeletal effects in Sost-deficient mice. Endocrinology 152, 2963–2975, doi: 10.1210/en.2011-0049 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Migotsky N et al. Multi-Scale Cortical Bone Traits Vary in Two Mouse Models of Genetic Diversity. bioRxiv, 2023.2006. 2002.543484 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Logan M et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80, doi: 10.1002/gene.10092 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Poole KE et al. Romosozumab enhances vertebral bone structure in women with low bone density. Journal of Bone and Mineral Research 37, 256–264 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keaveny TM et al. Greater gains in spine and hip strength for romosozumab compared with teriparatide in postmenopausal women with low bone mass. Journal of Bone and Mineral Research 32, 1956–1962 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Hino K et al. Deficiency of CIZ, a nucleocytoplasmic shuttling protein, prevents unloading-induced bone loss through the enhancement of osteoblastic bone formation in vivo. Bone 40, 852–860, doi: 10.1016/j.bone.2006.03.019 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Saag KG et al. Romosozumab or Alendronate for Fracture Prevention in Women with Osteoporosis. The New England journal of medicine 377, 1417–1427, doi: 10.1056/NEJMoa1708322 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Lv F et al. Denosumab or romosozumab therapy and risk of cardiovascular events in patients with primary osteoporosis: Systematic review and meta- analysis. Bone 130, 115121, doi: 10.1016/j.bone.2019.115121 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Huang W et al. Evaluation of the efficacy and safety of romosozumab (evenity) for the treatment of osteoporotic vertebral compression fracture in postmenopausal women: A systematic review and meta-analysis of randomized controlled trials (CDM-J). Pharmacoepidemiology and Drug Safety (2023). [DOI] [PubMed] [Google Scholar]
- 71.Korff C et al. NMP4, an Arbiter of Bone Cell Secretory Capacity and Regulator of Skeletal Response to PTH Therapy. Calcified tissue international, 1–16 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.