

Abstract
Lateral Meningocele Syndrome (LMS) is a monogenic disorder associated with NOTCH3 pathogenic variants that result in the stabilization of NOTCH3 and a gain-of-function. A mouse model (Notch3em1Ecan) harboring a 6691TAATGA mutation in the Notch3 locus that results in a functional outcome analogous to LMS exhibits cancellous and cortical bone osteopenia. We tested Notch3 antisense oligonucleotides (ASOs) specific to the Notch36691- TAATGA mutation for their effects on Notch3 downregulation and on the osteopenia of Notch3em1Ecan mice. Twenty-four mouse Notch3 mutant ASOs were designed and tested for toxic effects in vivo, and 12 safe ASOs were tested for their impact on the downregulation of Notch36691-TTATGA and Notch3 mRNA in osteoblast cultures from Notch3em1Ecan mice. Three ASOs downregulated Notch3 mutant transcripts specifically and were tested in vivo for their effects on the bone microarchitecture of Notch3em1Ecan mice. All three ASOs were well tolerated. One of these ASOs had more consistent effects in vivo and was studied in detail. The Notch3 mutant ASO downregulated Notch3 mutant transcripts in osteoblasts and bone marrow stromal cells and had no effect on other Notch receptors. The subcutaneous administration of Notch3 mutant ASO at 50 mg/Kg decreased Notch6691-TTATGA mRNA in bone without apparent toxicity; microcomputed tomography demonstrated that the ASO ameliorated the cortical osteopenia of Notch3em1Ecan mice but not the cancellous bone osteopenia. In conclusion, a Notch3 ASO that downregulates Notch3 mutant expression specifically ameliorates the cortical osteopenia in Notch3em1Ecan mice. ASOs may become useful strategies in the management of monogenic disorders affecting the skeleton.
Keywords: Notch receptors, NOTCH3, gene mutations, antisense oligonucleotides, cortical bone, osteopenia
1. INTRODUCTION
Notch receptors (Notch1 through 4) are determinants of cell fate and function in multiple cellular organizations including the skeleton [1, 2]. Notch1, 2 and 3 and low levels of Notch4 transcripts are detected in bone cells, where each receptor plays a distinct role influencing the fate of cells of the osteoblast and osteoclast lineages [3]. Interactions of Notch receptors with ligands of the Jagged and Delta-like families result in the cleavage of NOTCH and activation of signaling following the release of the NOTCH intracellular domain (NICD), and its translocation into the nucleus to induce the transcription of target genes [4–6]. These include genes encoding Hairy Enhancer of Split (HES)1, 5 and 7 and HES-related with YRPW motif (HEY)1, 2 and L [7].
Each Notch receptor has distinct patterns of cellular expression and activity and pathogenic variants associated with each receptor result in distinct clinical syndromes. NOTCH3 is expressed in mural vascular cells where it plays a critical function in vascular homeostasis [8]. Pathogenic variants of NOTCH3 associated with mutations in its extracellular domain cause cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) [8–11]. In bone, Notch3 is expressed in osteocytes and osteoblasts and its activation in these cells increases intracortical bone remodeling [12].
Lateral meningocele syndrome (LMS), also known as Lehman Syndrome, (Online Mendelian Inheritance in Man 130720) is a devastating disorder associated with pathogenic variants of NOTCH3 and presenting with meningoceles, distinct facial features, developmental delay, decreased muscle mass, cardiac valve defects, short stature, scoliosis and craniofacial abnormalities [13–16]. LMS is associated with mutations or deletions in exon 33 of NOTCH3, that result in the creation of a STOP codon upstream of sequences coding for the proline (P), glutamic acid (E), serine (S) and threonine (T) (PEST) domain. As a result, the PEST domain is not translated and NOTCH3 is presumably stable, and a gain-of-function occurs [14]. Autosomal dominant inheritance and de novo heterozygous mutations are reported. Treatment of LMS is not available. To understand mechanisms operating in the pathogenesis of LMS and develop potential therapeutic agents to influence the outcome of LMS, we created a mouse model, Notch3em1Ecan (syn Notch3tm1.1Ecan), that recapitulates the genetic variant from LMS. Notch3em1Ecan mice harbor a 6691-TAATGA mutation in exon 33 that results in a STOP codon upstream of the PEST domain [17]. The mice exhibit a NOTCH3 gain-of-function, cortical and cancellous bone osteopenia [17].
A number of approaches have been devised to downregulate Notch signaling and could serve to temper conditions where a gain-of-function exits. Often Notch inhibitors are not specific for Notch signaling or specific to a given Notch receptor [18–23]. Antibodies to the negative regulatory region (NRR) of Notch have been effective at preventing the activation of individual Notch receptors, including NOTCH3, but they do not target the Notch pathogenic variants and as a consequence they downregulate wild type as well as the mutant Notch receptor [24–26].
The use of antisense oligonucleotides (ASO) is a novel therapeutic approach of RNA modulation, and ASOs have been effective in a variety of tissues including the central and peripheral nervous system, retina, liver, muscle and bone [27–36]. ASOs are single-stranded synthetic nucleic acids that bind target mRNA by Watson-Crick pairing resulting in mRNA degradation by RNase H [37, 38]. In previous studies, we employed the systemic administration of ASOs targeting either wild type Notch2 or Notch3 to downregulate these genes in mice harboring Notch2 or Notch3 mutations resulting in a gain-of-function [36, 39]. Although the approach was successful in ameliorating the osteopenia of both mouse models, the ASOs employed were not allele selective and they did not target the mutant gene specifically.
The purpose of the present work was to evaluate if by targeting the mutant allele in heterozygous Notch3em1Ecan mice, we could preserve the wild type allele creating a more normal genetic outcome. Because Notch3em1Ecan mice present with osteopenia, we asked whether a more favorable genetic outcome would ameliorate this aspect of the Notch3em1Ecan phenotype. Ionis pharmaceuticals designed and synthesized ASOs that would target specifically the Notch36691-TAATGA mutation harbored by the Notch3em1Ecan mouse. Notch3 mutant specific ASOs were tested in vitro and in vivo for their tolerability and for effects on the downregulation of wild type and mutant Notch3 transcripts and on the skeletal microarchitecture of heterozygous Notch3em1Ecan mice.
2. MATERIALS AND METHODS
2.1. Notch3em1Ecan Mutant Mice
Heterozygous Notch3em1Ecan (syn Notch3tm1.1Ecan) mice in a C57BL/6 genetic background were used to test the efficacy of ASOs in vitro and in vivo. Notch3em1Ecan mice harbor a tandem termination at bases 6691–6696 (ACCAAG→TAATGA) in exon 33 of Notch3 that results in the same functional outcome as the one found in LMS [17]. Genotypes were determined by PCR analysis of tail DNA using forward primer 5’-GTGCTCAGCTTTGGTCTGCTC-3’ and either reverse primer 5’-CGCAGGAAGCGCGCTCATTA-3’ for Notch3em1Ecan mutant or 5’-CGCAGGAAGCGGGCCT TGG-3’ for the wild type allele (Integrated DNA Technologies, Coralville, IA). To characterize and study the effect of Notch3 ASOs in Notch3em1Ecan mice, heterozygous Notch3em1Ecan mutants were crossed with wild type mice to generate ~50% heterozygous Notch3em1Ecan mice and 50% control littermates. Studies were approved by the Institutional Animal Care and Use Committee of UConn Health.
2.2. Notch3 Antisense Oligonucleotides
ASOs used in the current study consist of a central stretch of 10 DNA nucleotides flanked on either side by 3 nucleotides containing constrained ethyl (cEt) modifications. Additionally, the phosphodiester internucleotide linkages were replaced with phosphorothioate (PS). Twenty-four ASOs targeting the Notch36691-TAATGA mutant pre-mRNA were designed in silico by Ionis Pharmaceuticals (Carlsbad, CA). ASOs covering at least one of the nucleotides that are mutated in the Notch3em1Ecan mouse were designed (Figure 1). A control ASO that does not hybridize to any specific mRNA sequence was included in all experiments. Oligonucleotides were synthesized at Ionis Pharmaceuticals using an AKTA Oligopilot, as described previously [40]. Initially, 24 Notch3 mutant ASOs were tested for toxicity at Ionis Pharmaceuticals (Carlsbad, CA) in 8-week-old BALB/c mice. To this end, male mice were administered ASOs at a dose of 50 mg/Kg subcutaneously once a week for a total of 4.5 weeks (5 doses) and euthanized 72 h after the last dose of ASO. Body weights were determined weekly, and liver, kidney, and spleen were weighed after euthanasia and normalized to body weight and compared with organs from control mice. Blood was obtained by cardiac puncture, and plasma was collected for the measurement of alanine aminotransferase, aspartate aminotransferase, total bilirubin, albumin, and blood urea nitrogen. These procedures were performed at, and approved by, the Animal Care and Use Committee of Ionis Pharmaceuticals. Twelve well tolerated mouse ASOs targeting the mutant Notch3 sequence, and a control ASO that does not hybridize to any specific mRNA sequence, were selected for further study.
Figure 1.
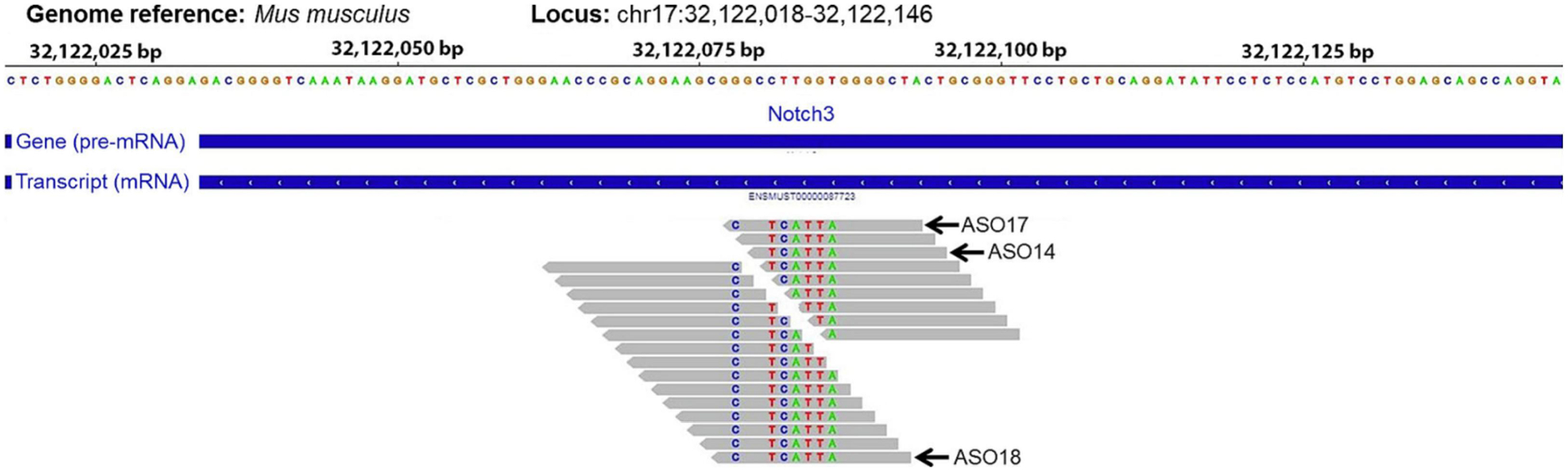
Twenty-four ASOs were designed to target the mouse mutant Notch3 pre-mRNA specifically and induce degradation of the LMS-causing transcript. Grey bars represent the site targeted by each ASO. The colored letters inside the grey bars represent the residues that are mutated in the LMS compared to the wild-type mouse Notch3 transcript. The wild-type mouse Notch3 pre-mRNA sequence is reported at top of the figure. Arrows indicate the three ASOs studied in detail (ASO 14, 17 and 18).
2.3. Osteoblast-enriched Cell Cultures
Osteoblasts were isolated following the digestion of parietal bones from 3 to 5 day old control and Notch3em1Ecan mice with liberase TL 1.2 units/ml (Sigma-Aldrich St. Louis, MO) for 20 min at 37°C for 5 consecutive reactions [41]. The cells isolated from the last 3 digestions were pooled and seeded at a density of 10 × 104 cells/cm2 [42]. Osteoblast-enriched cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with non-essential amino acids (both from Thermo Fisher Scientific, Waltham, MA) and 10% heat-inactivated fetal bovine serum (FBS; Atlanta Biologicals, Norcross, GA) in a humidified 5% CO2 atmosphere at 37°C. Confluent cells were exposed to DMEM supplemented with 10% heat-inactivated FBS, 100 μg/ml ascorbic acid and 5 mM β-glycerophosphate (both from Sigma-Aldrich) in the absence or presence of Notch3 mutant or control ASOs at various doses and periods of time as indicated in text and legends.
2.4. Bone Marrow Stromal Cell Cultures
Femurs and tibiae from 4 to 8 week old Notch3em1Ecan mice and littermate controls were dissected, the epiphysis removed and bone marrow stromal cells recovered by centrifugation [43]. Cells were pooled and seeded at a density of 1.25 × 106 cells/cm2 in α-minimum essential medium (α-MEM; Thermo Fisher Scientific) containing heat-inactivated 15% FBS and cultured at 37°C in a humidified 5% CO2 incubator. At confluence, cells were exposed to α-MEM supplemented with 10% FBS, 100 μg/ml ascorbic acid and 5 mM β-glycerophosphate and cultured in the absence or presence of Notch3 mutant or control ASOs at distinct doses and periods of time as indicated in text and legends.
2.5. In vivo Administration of Notch3 ASOs
One month old male Notch3em1Ecan heterozygous mutant and sex-matched wild type littermates were administered Notch3 mutant specific ASOs or control ASO suspended in PBS subcutaneously a dose of 50 mg/Kg once a week for 4 consecutive weeks to 1 month old Notch3em1Ecan and control mice, and mice were euthanized at 2 months of age.
2.6. Microcomputed Tomography (μCT)
Bone microarchitecture of femurs from experimental and control mice was determined using a μCT 40 microcomputed tomography instrument (Scanco Medical AG, Bassersdorf, Switzerland), which was calibrated periodically using a phantom provided by the manufacturer [44, 45]. Femurs were placed in 70% ethanol and scanned at high resolution, energy level of 55 kVp, intensity of 145 μA and integration time of 200 ms. One hundred and sixty slices at the distal femoral metaphysis were acquired at an isotropic voxel size of 216 μm3 and a slice thickness of 6 μm and chosen for analysis of cancellous bone microarchitecture. Trabecular bone volume fraction and microarchitecture were evaluated starting ~1.0 mm proximal from the femoral condyles. Contours were manually drawn a few voxels away from the endocortical boundary every 10 slices to define the region of analysis. The remaining slice contours were iterated automatically. Trabecular regions were assessed for total volume, bone volume, bone volume fraction (bone volume/total volume), trabecular thickness, trabecular number, trabecular separation, connectivity density and structure model index, using a Gaussian filter (σ = 0.8), and a threshold of 240 permil equivalent to 355.5 mg/cm3 hydroxyapatite [44, 45]. Femoral cortical bone architecture was performed along the cortex of the femoral midshaft, excluding the marrow cavity. Contours were iterated across 100 slices and analysis of bone volume/total volume, porosity, cortical thickness, total cross-sectional and cortical bone area, and polar moment inertia were performed using a Gaussian filter (σ = 0.8, support = 1), and a threshold of 400 permil equivalent to 704.7 mg/cm3 hydroxyapatite.
2.7. Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from cells, homogenized femurs or tibiae, following the removal of the bone marrow by centrifugation with the RNeasy kit or microRNeasy Kit (Qiagen, Valencia, CA), in accordance with manufacturer’s instructions [46–49]. The integrity of the RNA was assessed in random samples by microfluidic electrophoresis on an Experion instrument (BioRad, Hercules, CA), and only RNA with a quality indicator number equal to or higher than 7.0 was used for subsequent analysis. Equal amounts of RNA were reverse-transcribed using the iScript RT-PCR kit (BioRad) and amplified in the presence of specific primers (IDT) (Table 1A) with the iQ SYBR Green Supermix or SsoAdvanced Universal SYBR Green Supermix (BioRad) at 60°C for 35 cycles. Transcript copy number was estimated by comparison with a serial dilution of cDNA for Notch1 (from J.S. Nye, Cambridge, MA), Notch2 (from Thermo Fisher Scientific) and Notch4 (from Y. Shirayoshi, Tottori, Japan) [50, 51]. Notch3 wild type copy number was estimated by comparison to a serial dilution of a 100 to 200 base pair synthetic DNA template (IDT) cloned into pcDNA3.1 (Thermo Fischer Scientific) by isothermal single reaction assembly using commercially available reagents (New England BioLabs, Ipswich, MA) [52].
Table 1.
Primers used for qRT-PCR determinations. GenBank accession numbers identify transcript recognized by primer pairs.
| Gene | Strand | Sequence | GenBank Accession Number |
|---|---|---|---|
| Notch1 | Forward Reverse |
5'-GTCCCACCCATGACCACTACCCAGTTC-3' 5'-GGGTGTTGTCCACAGGGGA-3' |
NM_008714 |
| Notch2 | Forward Reverse |
5'-TGACGTTGATGAGTGTATCTCCAAGCC-3' 5'-GTAGCTGCCCTGAGTGTTGTGG-3' |
NM_010928 |
| Notch3* | Forward Reverse |
5'-CCGATTCTCCTGTCGTTGTCTCC-3' 5'-TGAACACAGGGCCTGCTGAC-3' |
NM_008716 |
| Notch4 | Forward Reverse |
5'-CCAGCAGACAGACTACGGTGGAC-3' 5'-GCAGCCAGCATCAAAGGTGT-3' |
NM_010929 |
| Rpl38 | Forward Reverse |
5'-AGAACAAGGATAATGTGAAGTTCAAGGTTC-3' 5'-CTGCTTCAGCTTCTCTGCCTTT-3' |
NM_001048057; NM_001048058; NM_023372 |
| Gene | Strand/Flurophore | Sequence | GenBank Accession Number |
| Notch3 | Forward Reverse HEX Fluorophore |
5'-AGGACATGGAGAGGAATA-3' 5'-GGTCAAATAAGGATGCTC-3' 5'-ACCAAGGCCCGCTTCC-3' |
Not Applicable |
| Notch3 6691-TAATGA | Forward Reverse FAM Fluorophore |
5'-AGGACATGGAGAGGAATA-3' 5'-GGTCAAATAAGGATGCTC-3' 5'-AGTAGCCCCTAATGAGCGCG-3' |
Not Applicable |
Used for Notch3 wild type mRNA determination in Figure 4, Panel B.
In experiments where cells or tissues were obtained from Notch3em1Ecan mice with the intent to determine an ASO effect on Notch36691-TAATGA mutant transcripts, fluorescent tagged products were used to perform RT-PCR. Total RNA was reverse transcribed with Moloney murine leukemia virus reverse transcriptase in the presence of reverse primers of Notch3 (5’-ATAAGGATGCTCGC TGGGAACC-3’) and Rpl38 (Table 1A). Notch3 cDNA was amplified by qPCR in the presence of SsoAdvanced Universal Probes Supermix (BioRad) gene expression assay mix, Notch3 and Notch3 mutant primers and HEX labeled Notch3 and FAM labeled Notch36691-TAATGA probes (Table 1B) (BioRad) at 95°C for 10 secs then 60°C for 30 secs and repeated for 45 cycles [53]. Notch3 or Notch36691-TAATGA mutant transcript copy number was estimated by comparison to a serial dilution of a 100 to 200 base pair (bp) synthetic DNA fragment (IDT) with or without the 6691TAATGA mutation in the Notch3 locus cloned into pcDNA3.1(−) and copy number was corrected for Rpl38 expression [52].
2.8. Statistics
Data are expressed as individual sample values, and as means ± SD. Data represent biological replicates except for osteoblast-enriched and stromal cell cultures, which represent technical replicates. qRT-PCR values were derived from two technical replicates of technical or biological replicates as indicated in the text and legends. Statistical differences were determined by unpaired t test for pairwise comparisons or two-way analysis of variance for multiple comparisons with Holm Šidák post-hoc analysis using GraphPad Prism version 9.3.1 for Mac OS, GraphPad Software (San Diego, CA).
3. RESULTS
3.1. Effect of Notch3 ASOs on Notch3 Expression in Cells of the Osteoblast Lineage
Twenty-four ASOs to target the Notch36691-TTATGA mutation were synthesized and tested initially for toxicity in vivo following their administration to BALB/c mice at 50 mg/Kg/week for 4.5 weeks. Most of the ASOs tested were well tolerated in mice, with only a few showing mild elevation in liver transaminase levels (Figure 2). We selected 12 safe ASOs and examined them further for activity and specificity in vitro (Table 2). The effect of Notch3 mutant ASOs was tested in cells of the osteoblast lineage from Notch3em1Ecan mice since previous work demonstrated that Notch3 is preferentially expressed in cells of this lineage including osteocytes [12, 17, 54]. Five of the 12 ASOs tested decreased Notch36691-TAATGA mRNA by ~60% to 100% 72 h after the addition of the ASO at 20 μM to the culture medium of osteoblast- enriched cells from Notch3em1Ecan mice (Table 2). The effect of three ASOs (ASO14; ASO17; ASO18) was specific to the mutant Notch3 mRNA since these ASOs did not downregulate Notch3 wild type mRNA (Figure 3). These three ASOs were studied further, and ASO18 was tested at multiple doses to confirm the downregulation of Notch36691-TAATGA mutant transcripts. Notch3 mutant ASO18 at 1 to 20 μM for 72 h downregulated Notch36691-TAATGA mRNA 80% to 100% in Notch3em1Ecan osteoblasts and did not alter the expression of Notch1, Notch2, wild type Notch3 or Notch 4 mRNA (Figure 4, Panels A and B). In addition, this Notch3 mutant ASO downregulated Notch36691TAATGA transcripts by 65% without affecting Notch3 mRNA in cultures of bone marrow stromal cells (Figure 4, Panel C) confirming the effect observed in calvarial osteoblasts. As expected, the Notch36691TAATGA transcript was not detected in control wild type cells.
Figure 2.
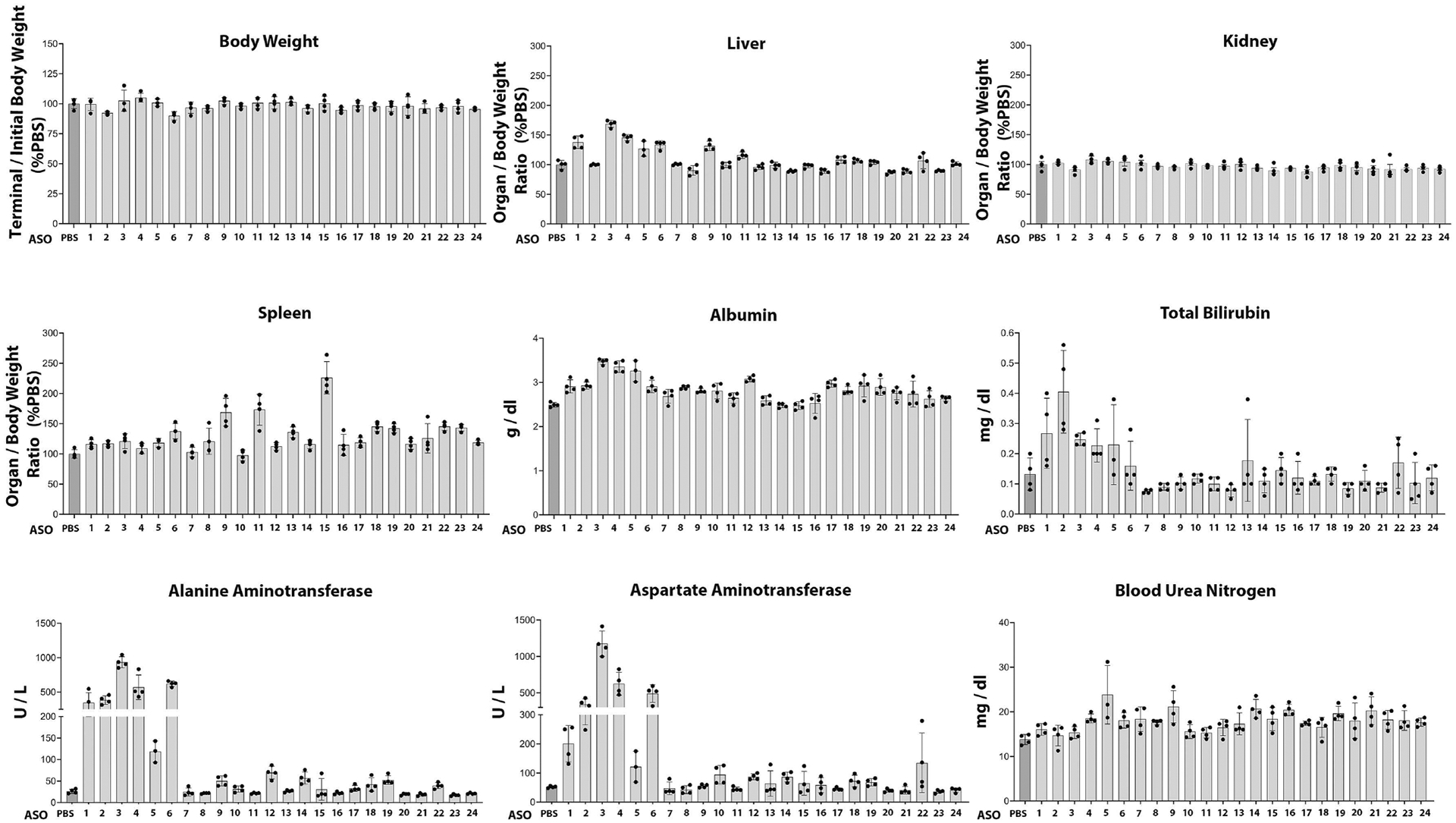
The safety profile of 24 mutant Notch3 ASOs was evaluated in mice following subcutaneous administration once a week for 4.5 weeks at 50 mg/Kg. At the end of the study, body, liver, kidney, and spleen weights were assessed, and plasma levels of alanine aminotransferase, aspartate aminotransferase, total bilirubin, albumin, and blood urea nitrogen were measured. Individual values are shown, and bars and ranges represent means ± SD; n = 4 mice for each ASO tested. Twelve ASOs considered safe were selected for subsequent activity studies.
Table 2.
Screening of Notch3 mutant ASOs for activity in vitro.
| ASO | Notch3 Downregulation | Notch36691TAATGA Downregulation |
|---|---|---|
| 07 | 0% | 0% |
| 08 | 0% | 0% |
| 10 | 0% | 0% |
| 11 | 0% | 0% |
| 13 | 0% | 0% |
| 14 | ~15% | 100% |
| 16 | ~40% | 100% |
| 17 | 0% | 70% |
| 18 | 0% | 60% |
| 19 | ~40% | 0% |
| 20 | ~25% | 0% |
| 21 | 100% | 100% |
Twelve ASOs considered safe were tested at 20 μM for their effect on Notch3 wild type or Notch36691TAAGTAmutant transcripts in osteoblast-enriched cells from Notch3em1Ecan mice. Notch3 mutant ASOs that were not safe or borderline safe in vivo were not tested in vitro.
Figure 3.
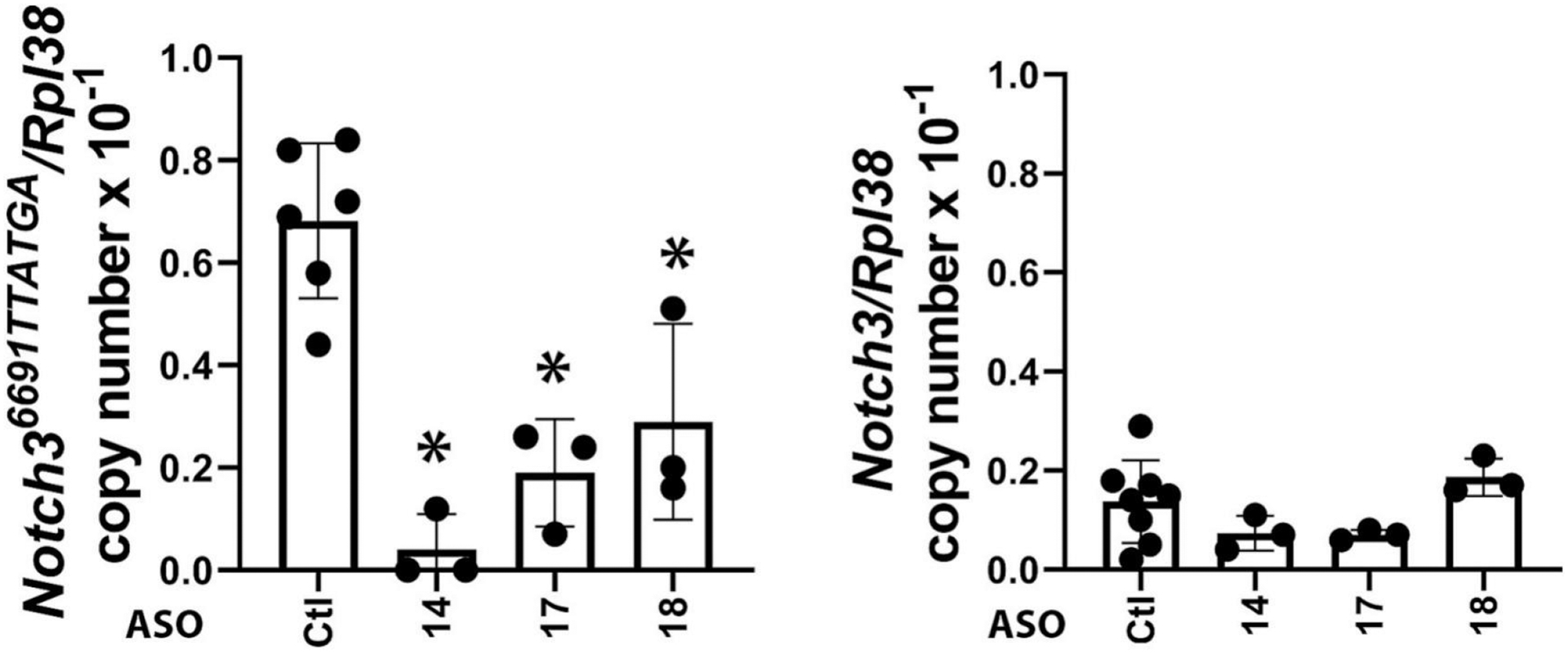
Effect of control or Notch3 mutant ASOs (14, 17 and 18) on Notch3 and Notch36691-TAATGA mRNA expression in calvarial osteoblast-enriched cells from Notch3em1Ecan mice. Notch3 and Notch36691-TAATGA mRNA levels were obtained 72 h after the addition of Notch3 mutant or control ASO at 20 μM to the culture medium. Transcript levels are expressed as copy number following correction for Rpl38. Individual values are shown, and bars and ranges represent means ± SD; n = 3 technical replicates. *Significantly different between Notch3 mutant ASO and control ASO by ANOVA with post-hoc analysis by Holm Šidák, p <0.05.
Figure 4.
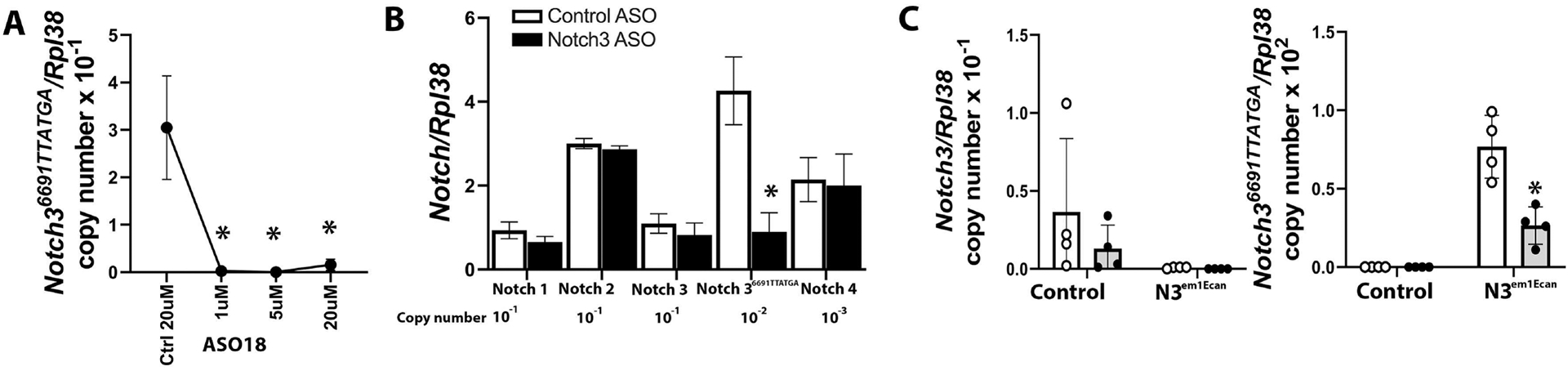
Effect of control or Notch3 mutant ASO 18 on Notch36691-TAATGA mRNA expression in calvarial osteoblast-enriched (A and B) and bone marrow stromal (C) cells from Notch3em1Ecan mice. In Panel A, Notch36691-TAATGA mRNA levels were obtained 72 h after the addition of Notch3 mutant or control ASO at the indicated doses. In Panel B, Notch1, Notch2, Notch3, Notch36691-TAATGA and Notch4 mRNA were detected 72 h after the addition of Notch3 mutant or control ASO at 20μM. In Panel C, Notch3 and Notch36691TAATGA mRNA levels were obtained after the addition of Notch 3 mutant (closed circles grey bars,) or control ASO (open circles, white bars,) at 20μM for 72 h. Transcript levels are expressed as copy number following correction for Rpl38. In Panels A and B, values are means ± SD. In Panel C, individual values are shown, and bars and ranges represent mean ± SD; n = 3 or 4 technical replicates. *Significantly different between Notch3 mutant ASO and control ASO by ANOVA with post-hoc analysis by Holm Šidák (Panels A and C) or unpaired t-test (Panel B), p <0.05.
3.2. Effect of Notch3 Mutant ASO on General Characteristics and Femoral Microarchitecture of Notch3em1Ecan Mice
Heterozygous Notch3em1Ecan mutant male mice were compared to wild type sex-matched littermate controls in a C57BL/6 genetic background. Male mice were studied since they have an osteopenic phenotype that is sustained for up to 3 to 4 months of age whereas female Notch3em1Ecan mutants tend to have a more transient osteopenia [17]. Homozygous Notch3em1Ecan mice were not studied since the homozygous mutation is developmentally lethal [17]. Three of the Notch3 mutant ASOs that suppressed Notch36691-TAATGA transcripts in vitro specifically (Figure 3) were tested for their effects in vivo and were tolerated well without overt signs of toxicity. Two of 3 Notch3 mutant ASOs had no effect or inconsistent effects on the cancellous or cortical bone osteopenia of Notch3em1Ecan mice and were not studied further (data not shown).
Mouse Notch3 mutant ASO18 or control ASO were administered once a week at 50 mg/Kg for 4 weeks. Notch3 mutant ASO18 was administered to 26 Notch3em1Ecan mice and to 32 wild type littermates, and Notch3 control ASO was administered to 23 Notch3em1Ecan mice and 34 control littermates. The weight of Notch3em1Ecan heterozygous male mice was not different from those of control wild type mice and the femoral length was slightly decreased in Notch3em1Ecan mice. These parameters were not altered by the administration of the Notch3 mutant ASO compared to control ASO. The administration of the Notch3 mutant ASO for a 4-week period decreased Notch36691-TAATGA mRNA expression by ~50% in tibiae from Notch3em1Ecan mutant mice without affecting Notch3 mRNA (Figure 5). In accordance with previous observations, μCT of the femoral mid-diaphysis revealed that 2 month old Notch3em1Ecan male mice had decreased cortical bone volume/total volume (BV/TV) associated with a 26% increase in porosity, a 18% decrease in cortical thickness and a 9% decrease in polar moment of inertia (Figure 6) [17]. The administration of the Notch3 mutant ASO increased cortical BV/TV and resulted in a 8% increase in cortical thickness and a 6% decrease in cortical porosity in Notch3em1Ecan mice. In addition, when compared to control ASO, the Notch3 mutant ASO increased polar moment of inertia in Notch3em1Ecan mice by 16% (p 0.07 by ANOVA; p < 0.05 by unpaired t-test), indicating an amelioration of skeletal fragility. No effect was noted in wild type mice by the administration of Notch3 ASO since the ASO was designed to target the Notch3 mutation. Cancellous bone microarchitecture demonstrated that Notch3em1Ecan mice had a 37% decrease in cancellous BV/TV and in trabecular number when compared to wild type littermates (Figure 6). Administration of Notch3 mutant ASO did not increase femoral BV/TV significantly in Notch3em1Ecan (16%) mice and had no effect on control littermates (Figure 7). Therefore, the effect of the Notch3 mutant ASO was restricted to the cortical bone osteopenia of Notch3em1Ecan mice.
Figure 5.
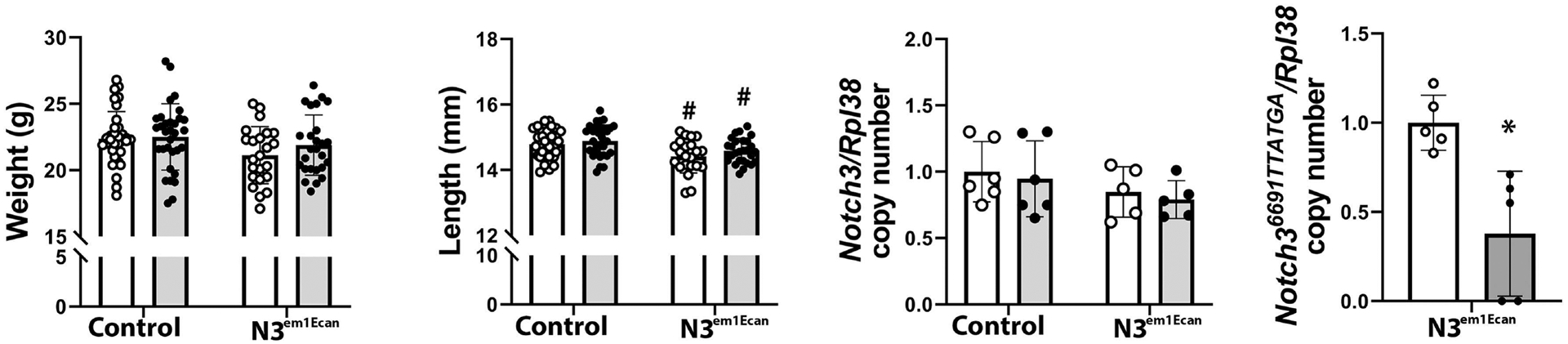
Body weight, femoral length and Notch3 and Notch36691-TAATGA transcripts in tibiae of 2-month-old male Notch3em1Ecan mutant mice and littermate controls treated with Notch3 mutant ASO 18 (closed circles, grey bars) or control ASO (open circles, white bars) at 50 mg/Kg subcutaneously, once a week for 4 weeks. Individual values are shown, and bars and ranges represent means ± SD; n = 34 wild type mice treated with control ASO, n = 32 wild type mice treated with Notch3 ASO and n = 23 Notch3em1Ecan mice treated with control ASO and n = 26 Notch3em1Ecan mice treated with Notch3 mutant ASO, except for Notch36691-TAATGA mRNA values n = 5 biological replicates. *Significantly different between Notch3 ASO and control, p < 0.05. #Significantly different between Notch3em1Ecan mutant and control mice, p < 0.05, by ANOVA with post-hoc analysis by Holm Šidák or unpaired t-test for Notch36691-TAATGA mRNA.
Figure 6.
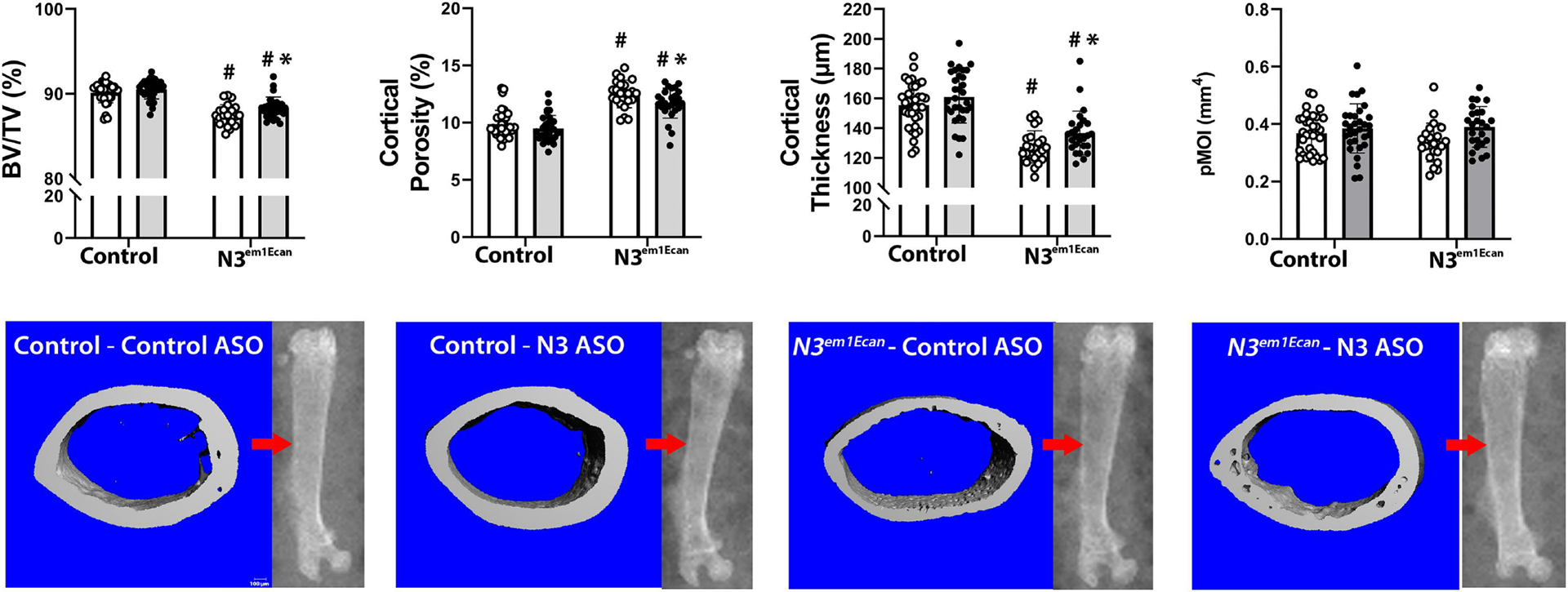
Cortical bone microarchitecture assessed by μCT of the mid-diaphyseal femur from 2 month old Notch3em1Ecan mutant male mice and sex-matched littermate controls treated with Notch3 mutant ASO 18 (closed circles, grey bars; n = 32 for wild type mice, n = 26 for Notch3em1Ecan) or control ASO (open circles, white bars; n = 34 for wild type mice, n = 23 for Notch3em1Ecan) both at 50 mg/Kg subcutaneously, once a week for 4 weeks prior to sacrifice. Parameters shown are cortical bone volume/total volume (BV/TV, %), cortical porosity (%), cortical thickness (mm) and polar moment of inertia (pMOI). Individual values are shown, and bars and ranges represent means ± SD of biological replicates. *Significantly different between Notch3 mutant and control ASO, p < 0.05. #Significantly different between Notch3em1Ecan and control, p < 0.05, by ANOVA with post-hoc analysis by Holm Šidák. Representative images show cortical bone osteopenia in Notch3em1Ecan mutant mice and its amelioration by Notch3 mutant ASOs. Scale bars in the right corner represent 100 μm. Arrow on scout μCT images point to the area analyzed.
Figure 7.
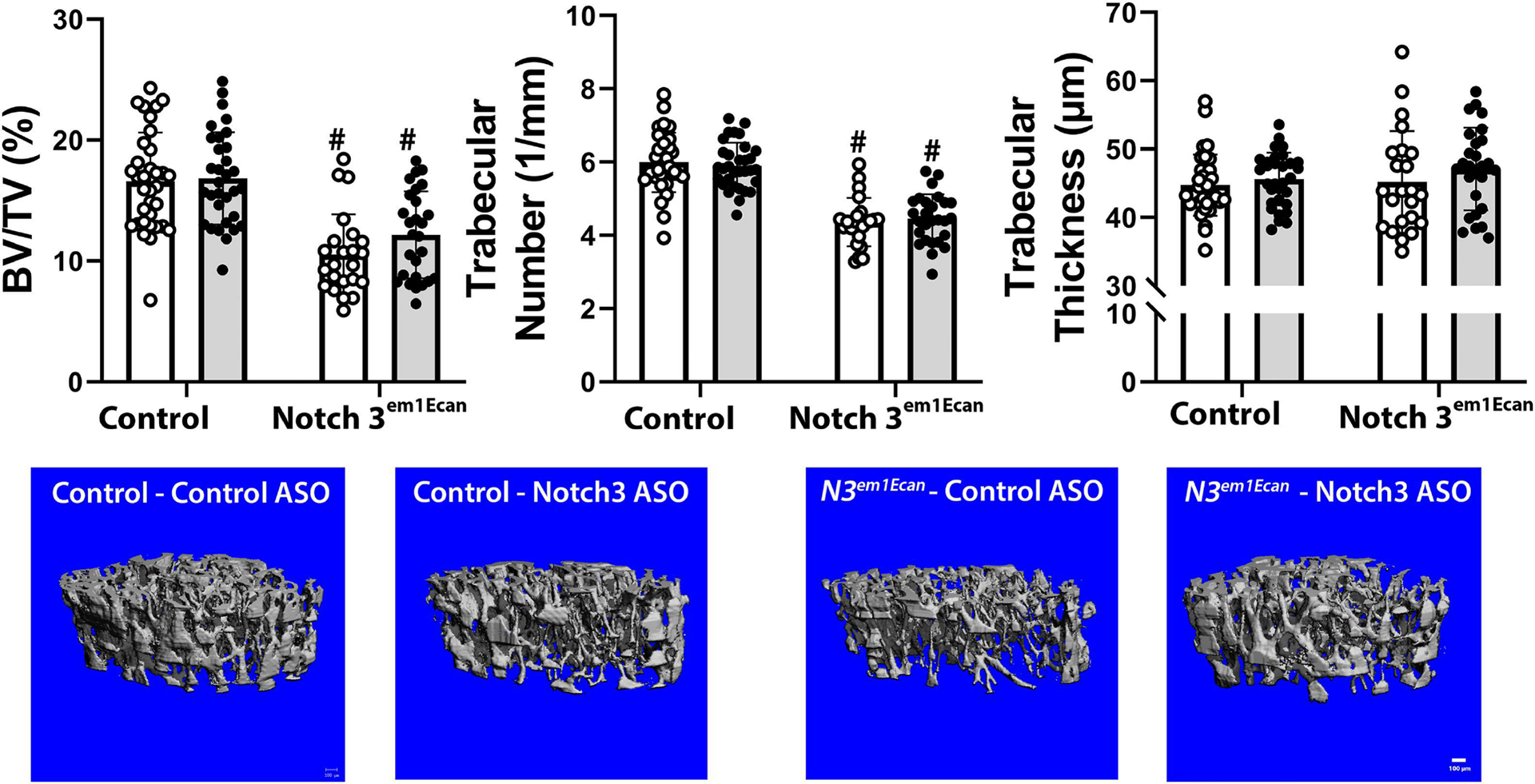
Cancellous bone microarchitecture assessed by μCT of the distal femur from 2 month old Notch3em1Ecan mutant male mice and sex-matched littermate controls treated with Notch3 mutant ASO 18 (closed circles, grey bars; n = 32 for wild type mice, n = 26 for Notch3em1Ecan) or control ASO (open circles, white bars; n = 34 for wild type mice, n = 23 for Notch3em1Ecan) both at 50 mg/Kg subcutaneously, once a week for 4 weeks prior to sacrifice. Parameters shown are trabecular bone volume/total volume (BV/TV, %); trabecular number (1/mm) and thickness (um). Individual values are shown, and bars and ranges represent means ± SD of biological replicates. #Significantly different between Notch3em1Ecan and control, p < 0.05, by ANOVA with post-hoc analysis by Holm Šidák. Representative images show cancellous bone osteopenia in Notch3em1Ecan mutant mice and no effect by Notch3 mutant ASO. Scale bars in the right corner represent 100 μm.
4. DISCUSSION
The current work was undertaken to determine whether ASOs could be developed to target and downregulate a Notch3 mutation in a mouse model (Notch3em1Ecan) replicating the genetic variant found in LMS. Because Notch3em1Ecan mice present with osteopenia, as a therapeutic end point we tested whether an improvement in genetic outcome would translate in amelioration of this aspect of the Notch3em1Ecan phenotype. About 50% of the Notch3 mutant specific ASOs designed were found not to be toxic in vivo and were studied further for their activity in skeletal cells. Three of the 12 ASOs considered safe downregulated the Notch36691-TAATGA mutation specifically without an effect on wild type Notch3 transcripts in osteoblast cultures. One of the ASOs was pursued in detail and found to inhibit Notch3 mutant transcripts in vivo and ameliorate phenotypic manifestations of Notch3em1Ecan mice.
Although a variety of approaches to downregulate Notch signaling have been reported, they often are not specific to Notch activity or to a specific Notch receptor. Anti-Notch antibodies targeting the NRR are an exception and we have demonstrated that anti-NOTCH2 NRR and anti-NOTCH3 NRR antibodies are effective in reversing the skeletal phenotype of Notch2tm1.1Ecan and of Notch3em1Ecan mice, models of Hajdu Cheney Syndrome and LMS, respectively [25, 26]. Although anti-Notch NRR antibodies are effective in their ability to downregulate a specific Notch receptor, they do not discriminate between the wild type and mutant forms of the Notch receptor. They induce substantial downregulation of Notch signaling with a potential of gastrointestinal toxicity. Because of these reasons, we search for other alternatives to reverse/ameliorate skeletal phenotypes associated with Notch2 or Notch3 mutations.
We demonstrated that Notch2 ASOs downregulate Notch2 expression in vitro and in vivo and ameliorate the osteopenia of mice harboring a Notch2 mutation replicating the one found in HCS [36]. Similarly, we found that Notch3 ASOs targeting Notch3 downregulate Notch3 wild type and mutant transcripts and ameliorate the cortical osteopenia of Notch3em1Ecan mice [39].
The current approach was designed to target the Notch36691-TAATGA mutation specifically in a heterozygous mouse model that replicates the genetic variant of LMS with the intent to preserve the activity of the wild type allele and create a state of functional normality. This approach would seem ideal for the targeting of mutations found in monogenic disorders with dominant inheritance, such as LMS. The downregulation of the Notch3 mutant transcript by the Notch3 ASO was effective in in vitro models and in vivo it ameliorated the cortical osteopenia of Notch3em1Ecan mice. However, Notch3 mutant ASOs did not modify significantly the cancellous bone osteopenia of Notch3em1Ecan mice. The present study limited the administration of the Notch3 mutant specific ASO to 50 mg/Kg for a 4-week period, and it is possible that a longer period of administration could have a greater beneficial effect on the bone microarchitecture of Notch3em1Ecan mice. The effect of Notch3 mutant ASOs in vivo was similar to the one that we reported recently on the effect of Notch3 ASOs. It is possible that Notch3 ASOs are more effective in the cortical compartment which is rich in osteocytes because Notch3 is preferentially expressed in this cellular environment [12, 16, 54, 55]. It is also possible that ASOs are transported more efficiently to cortical than to trabecular bone since the blood vessel network differs between these two skeletal compartments. A central nutrient artery and periosteal arteries irrigate cortical long bones, whereas epiphyseal arteries irrigate metaphyseal, cancellous-rich bone [56, 57]. However, perfusion efficiency studies do not reveal apparent differences between epiphyseal and diaphyseal bone vascularization [58].
Although the subcutaneous administration of Notch3 ASOs is practical from a possible therapeutic point of view, the present and prior studies suggest that ASOs are partially effective in the downregulation of the Notch3 mutant transcripts in skeletal tissue and the reversal of skeletal phenotypes. Greater efficacy might be attained by enhancing the transport of the ASO to the target cell [59]. This has been achieved for skeletal muscle by conjugating ASOs to antigen binding fragments to the transferrin receptor 1 [60]. Although our study demonstrates amelioration of the cortical osteopenia in a mouse model that harbors a genetic variant found in LMS, there was not complete reversal of the phenotype. Future work developing strategies to target ASOs to specific skeletal cells may serve to improve their efficacy and potential therapeutic outcomes.
The present findings confirm that a mouse model replicating a mutation found in LMS displays femoral cancellous and cortical bone osteopenia [17]. There are some limitations of this study. The phenotype of the Notch3em1Ecan mutant mouse recapitulates aspects of LMS including osteopenia, but not the neurological manifestations of the disease [16, 17]. Because only male mice were reported in the present study, it is important not to extrapolate the results observed to female mice. Phenotypic alterations of experimental and control mice were assessed by μCT, and analyses required the ex vivo exam of bone following the sacrifice of mice. Therefore, the same animal could not be analyzed before and after the administration of Notch3 ASOs. Cells were obtained from mice of both sexes for in vitro experiments since sex cell specific culture is at times impractical. The effects of Notch3 ASOs were assessed in cells of the osteoblast lineage and not in the myeloid/osteoclast lineage because Notch3 is not expressed in this lineage and NOTCH3 does not have direct effects on osteoclastogenesis [3, 16, 17].
5. CONCLUSIONS
In conclusion, a Notch3 mutant ASO downregulated Notch3 mutant expression specifically and ameliorated the cortical osteopenia of a mouse model that replicates the genetic variant found in LMS.
Highlights.
Lateral Meningocele Syndrome (LMS) is associated with NOTCH3 mutations
A mouse model of LMS (Notch3em1Ecan) harbors a Notch3 mutation and exhibits cortical and trabecular osteopenia
Antisense oligonucleotides targeting the Notch3 mutation in the mouse environment ameliorate the cortical osteopenia of Notch3em1Ecan mice
ACKNOWLEDGMENTS
The authors thank Mary Yurczak for secretarial support, and Emily Denker for technical assistance and creating figures for the manuscript.
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) [grant numbers AR076747 and AR072987]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- α-MEM
α-minimum essential medium
- ASO
antisense oligonucleotide
- bp
base pair
- BV/TV
bone volume/total volume
- CADASIL
cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- cEt
constrained ethyl
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- HES
Hairy Enhancer of Split
- HEY
HES-related with YRPM
- LMS
Lehman Syndrome or Lateral Meningocele Syndrome
- μCT
microcomputed tomography
- NRR
negative regulatory region
- NICD
NOTCH intracellular domain
- PS
phosphorothiate
- pMOI
polar moment of inertia
- PEST
proline (P), glutamic acid (E), serine (S) and threonine (T)
- qRT-PCR
quantitative reverse transcript-polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
Ernesto Canalis: Conceptualization, Methodology, Validation, Writing – Original Draft, Writing – Review & Editing, Supervision, Project Administration, Funding Acquisition. Lauren Schilling: Investigation, Visualization. Magda Mocarska: Investigation. Paymaan Jafar-nejad: Resources, Writing – Review and Editing, Visualiztion. Michele Carrer: Resource, Writing – Review and Editing, Visualization.
Conflict of Interest
The authors declare no conflicts of interest with the contents of this article.
REFERENCES
- [1].Siebel C, Lendahl U, Notch Signaling in Development, Tissue Homeostasis, and Disease, Physiol. Rev 97(4) (2017) 1235–1294. 10.1152/physrev.00005.2017 [DOI] [PubMed] [Google Scholar]
- [2].Zanotti S, Canalis E, Notch Signaling and the Skeleton, Endocr. Rev 37(3) (2016) 223–253. 10.1210/er.2016-1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Canalis E, Notch in skeletal physiology and disease, Osteoporos.Int 29(12) (2018) 2611–2621. 10.1007/s00198-018-4694-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kovall RA, More complicated than it looks: assembly of Notch pathway transcription complexes, Oncogene 27(38) (2008) 5099–5109. [DOI] [PubMed] [Google Scholar]
- [5].Nam Y, Sliz P, Song L, Aster JC, Blacklow SC, Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes, Cell 124(5) (2006) 973–983. [DOI] [PubMed] [Google Scholar]
- [6].Wilson JJ, Kovall RA, Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA, Cell 124(5) (2006) 985–996. [DOI] [PubMed] [Google Scholar]
- [7].Iso T, Kedes L, Hamamori Y, HES and HERP families: multiple effectors of the Notch signaling pathway, J. Cell. Physiol 194(3) (2003) 237–255. 10.1002/jcp.10208 [DOI] [PubMed] [Google Scholar]
- [8].Liu H, Zhang W, Kennard S, Caldwell RB, Lilly B, Notch3 is critical for proper angiogenesis and mural cell investment, Circ Res 107(7) (2010) 860–870. 10.1161/CIRCRESAHA.110.218271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mizuno T, Mizuta I, Watanabe-Hosomi A, Mukai M, Koizumi T, Clinical and Genetic Aspects of CADASIL, Front Aging Neurosci 12 (2020) 91. 10.3389/fnagi.2020.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Prakash N, Hansson E, Betsholtz C, Mitsiadis T, Lendahl U, Mouse Notch 3 expression in the pre- and postnatal brain: relationship to the stroke and dementia syndrome CADASIL, Exp. Cell Res 278(1) (2002) 31–44. 10.1006/excr.2002.5544 [DOI] [PubMed] [Google Scholar]
- [11].Henshall TL, Keller A, He L, Johansson BR, Wallgard E, Raschperger E, Mae MA, Jin S, Betsholtz C, Lendahl U, Notch3 is necessary for blood vessel integrity in the central nervous system, Arterioscler Thromb Vasc Biol 35(2) (2015) 409–420. 10.1161/ATVBAHA.114.304849 [DOI] [PubMed] [Google Scholar]
- [12].Canalis E, Zanotti S, Schilling L, Eller T, Yu J, Activation of Notch3 in osteoblasts/osteocytes causes compartment-specific changes in bone remodeling, J. Biol. Chem 296 (2021) 100583. 10.1016/j.jbc.2021.100583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gripp KW, Scott CI Jr., Hughes HE, Wallerstein R, Nicholson L, States L, Bason LD, Kaplan P, Zderic SA, Duhaime AC, Miller F, Magnusson MR, Zackai EH, Lateral meningocele syndrome: three new patients and review of the literature, Am. J. Med. Genet 70(3) (1997) 229–239. [PubMed] [Google Scholar]
- [14].Gripp KW, Robbins KM, Sobreira NL, Witmer PD, Bird LM, Avela K, Makitie O, Alves D, Hogue JS, Zackai EH, Doheny KF, Stabley DL, Sol-Church K, Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome, Am. J. Med. Genet. A 167A(2) (2015) 271–281. 10.1002/ajmg.a.36863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lehman RA, Stears JC, Wesenberg RL, Nusbaum ED, Familial osteosclerosis with abnormalities of the nervous system and meninges, J. Pediatr 90(1) (1977) 49–54. [DOI] [PubMed] [Google Scholar]
- [16].Canalis E, The Skeleton of Lateral Meningocele Syndrome, Front Genet 11 (2020) 620334. 10.3389/fgene.2020.620334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Canalis E, Yu J, Schilling L, Yee SP, Zanotti S, The lateral meningocele syndrome mutation causes marked osteopenia in mice, J. Biol. Chem 293(36) (2018) 14165–14177. 10.1074/jbc.RA118.004242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ryeom SW, The cautionary tale of side effects of chronic Notch1 inhibition, J. Clin. Invest 121(2) (2011) 508–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R, A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain, Nature 398(6727) (1999) 518–522. 10.1038/19083 [DOI] [PubMed] [Google Scholar]
- [20].Duggan SP, McCarthy JV, Beyond gamma-secretase activity: The multifunctional nature of presenilins in cell signalling pathways, Cell. Signal 28(1) (2016) 1–11. 10.1016/j.cellsig.2015.10.006 [DOI] [PubMed] [Google Scholar]
- [21].Ilagan MX, Kopan R, Selective blockade of transport via SERCA inhibition: the answer for oncogenic forms of Notch?, Cancer Cell 23(3) (2013) 267–269. 10.1016/j.ccr.2013.02.020 [DOI] [PubMed] [Google Scholar]
- [22].Moellering RE, Cornejo M, Davis TN, Del BC, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE, Direct inhibition of the NOTCH transcription factor complex, Nature 462(7270) (2009) 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lehal R, Zaric J, Vigolo M, Urech C, Frismantas V, Zangger N, Cao L, Berger A, Chicote I, Loubery S, Choi SH, Koch U, Blacklow SC, Palmer HG, Bornhauser B, Gonzalez-Gaitan M, Arsenijevic Y, Zoete V, Aster JC, Bourquin JP, Radtke F, Pharmacological disruption of the Notch transcription factor complex, Proc. Natl. Acad. Sci. U. S. A 117(28) (2020) 16292–16301. 10.1073/pnas.1922606117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, Choy R, Howard P, Kadyk L, Yan M, Zha J, Callahan CA, Hymowitz SG, Siebel CW, Therapeutic antibody targeting of individual Notch receptors, Nature 464(7291) (2010) 1052–1057. 10.1038/nature08878 [DOI] [PubMed] [Google Scholar]
- [25].Canalis E, Sanjay A, Yu J, Zanotti S, An Antibody to Notch2 Reverses the Osteopenic Phenotype of Hajdu-Cheney Mutant Male Mice, Endocrinology 158(4) (2017) 730–742. 10.1210/en.2016-1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yu J, Siebel CW, Schilling L, Canalis E, An antibody to Notch3 reverses the skeletal phenotype of lateral meningocele syndrome in male mice, J. Cell. Physiol 235(1) (2020) 210–220. 10.1002/jcp.28960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Murray SF, Jazayeri A, Matthes MT, Yasumura D, Yang H, Peralta R, Watt A, Freier S, Hung G, Adamson PS, Guo S, Monia BP, LaVail MM, McCaleb ML, Allele-Specific Inhibition of Rhodopsin With an Antisense Oligonucleotide Slows Photoreceptor Cell Degeneration, Invest Ophthalmol Vis Sci 56(11) (2015) 6362–6375. 10.1167/iovs.15-16400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shy ME, Antisense oligonucleotides offer hope to patients with Charcot-Marie-Tooth disease type 1A, J. Clin. Invest 128(1) (2018) 110–112. 10.1172/JCI98617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hung G, Bennett CF, Freier SM, Hayden MR, Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin, Mol. Ther 19(12) (2011) 2178–2185. 10.1038/mt.2011.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Limmroth V, Barkhof F, Desem N, Diamond MP, Tachas G, Group ATLS, CD49d antisense drug ATL1102 reduces disease activity in patients with relapsing-remitting MS, Neurology 83(20) (2014) 1780–1788. 10.1212/WNL.0000000000000926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McCampbell, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, Schoch KM, Hoye ML, Shabsovich M, Sun L, Luo Y, Zhang M, Comfort N, Wang B, Amacker J, Thankamony S, Salzman DW, Cudkowicz M, Graham DL, Bennett CF, Kordasiewicz HB, Swayze EE, Miller TM, Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models, J. Clin. Invest 128(8) (2018) 3558–3567. 10.1172/JCI99081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhao HT, Damle S, Ikeda-Lee K, Kuntz S, Li J, Mohan A, Kim A, Hung G, Scheideler MA, Scherer SS, Svaren J, Swayze EE, Kordasiewicz HB, PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models, J. Clin. Invest 128(1) (2018) 359–368. 10.1172/JCI96499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, Meroni M, Graham MJ, Yates KP, Diehl AM, Schwabe RF, Tabas I, Valenti L, Lavine JE, Pajvani UB, Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis, Sci Transl Med 10(468) (2018) 10.1126/scitranslmed.aat0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li M, Jancovski N, Jafar-Nejad P, Burbano LE, Rollo B, Richards K, Drew L, Sedo A, Heighway J, Pachernegg S, Soriano A, Jia L, Blackburn T, Roberts B, Nemiroff A, Dalby K, Maljevic S, Reid CA, Rigo F, Petrou S, Antisense oligonucleotide therapy reduces seizures and extends life span in an SCN2A gain-of-function epilepsy model, J. Clin. Invest 131(23) (2021) 10.1172/JCI152079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nguyen Q, Yokota T, Antisense oligonucleotides for the treatment of cardiomyopathy in Duchenne muscular dystrophy, Am J Transl Res 11(3) (2019) 1202–1218. [PMC free article] [PubMed] [Google Scholar]
- [36].Canalis E, Grossman TR, Carrer M, Schilling L, Yu J, Antisense oligonucleotides targeting Notch2 ameliorate the osteopenic phenotype in a mouse model of Hajdu-Cheney syndrome, J. Biol. Chem 295(12) (2020) 3952–3964. 10.1074/jbc.RA119.011440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bennett CF, Baker BF, Pham N, Swayze E, Geary RS, Pharmacology of Antisense Drugs, Annu. Rev. Pharmacol. Toxicol 57 (2017) 81–105. 10.1146/annurev-pharmtox-010716-104846 [DOI] [PubMed] [Google Scholar]
- [38].Cerritelli SM, Crouch RJ, Ribonuclease H: the enzymes in eukaryotes, FEBS J. 276(6) (2009) 1494–1505. 10.1111/j.1742-4658.2009.06908.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Canalis E, Carrer M, Eller T, Schilling L, Yu J, Use of antisense oligonucleotides to target Notch3 in skeletal cells, PLoS One 17(5) (2022) e0268225. 10.1371/journal.pone.0268225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vasquez G, Freestone GC, Wan WB, Low A, De Hoyos CL, Yu J, Prakash TP, Ostergaard ME, Liang XH, Crooke ST, Swayze EE, Migawa MT, Seth PP, Site-specific incorporation of 5’-methyl DNA enhances the therapeutic profile of gapmer ASOs, Nucleic Acids Res. 49(4) (2021) 1828–1839. 10.1093/nar/gkab047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yesil P, Michel M, Chwalek K, Pedack S, Jany C, Ludwig B, Bornstein SR, Lammert E, A new collagenase blend increases the number of islets isolated from mouse pancreas, Islets 1(3) (2009) 185–190. 10.4161/isl.1.3.9556 [DOI] [PubMed] [Google Scholar]
- [42].McCarthy TL, Centrella M, Canalis E, Further biochemical and molecular characterization of primary rat parietal bone cell cultures, J. Bone Miner. Res 3(4) (1988) 401–408. [DOI] [PubMed] [Google Scholar]
- [43].Zanotti S, Kalajzic I, Aguila HL, Canalis E, Sex and genetic factors determine osteoblastic differentiation potential of murine bone marrow stromal cells, PLoS One 9(1) (2014) e86757. 10.1371/journal.pone.0086757 [doi];PONE-D-13–31354 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R, Guidelines for assessment of bone microstructure in rodents using micro-computed tomography, J. Bone Miner. Res 25(7) (2010) 1468–1486. [DOI] [PubMed] [Google Scholar]
- [45].Glatt V, Canalis E, Stadmeyer L, Bouxsein ML, Age-Related Changes in Trabecular Architecture Differ in Female and Male C57BL/6J Mice, J. Bone Miner. Res 22(8) (2007) 1197–1207. [DOI] [PubMed] [Google Scholar]
- [46].Canalis E, Schilling L, Yee SP, Lee SK, Zanotti S, Hajdu Cheney Mouse Mutants Exhibit Osteopenia, Increased Osteoclastogenesis and Bone Resorption, J. Biol. Chem 291 (2016) 1538–1551. 10.1074/jbc.M115.685453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zanotti S, Yu J, Sanjay A, Schilling L, Schoenherr C, Economides AN, Canalis E, Sustained Notch2 signaling in osteoblasts, but not in osteoclasts, is linked to osteopenia in a mouse model of Hajdu-Cheney syndrome, J. Biol. Chem 292(29) (2017) 12232–12244. 10.1074/jbc.M117.786129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nazarenko, Lowe B, Darfler M, Ikonomi P, Schuster D, Rashtchian A, Multiplex quantitative PCR using self-quenched primers labeled with a single fluorophore, Nucleic Acids Res. 30(9) (2002) e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nazarenko, Pires R, Lowe B, Obaidy M, Rashtchian A, Effect of primary and secondary structure of oligodeoxyribonucleotides on the fluorescent properties of conjugated dyes, Nucleic Acids Res. 30(9) (2002) 2089–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nye JS, Kopan R, Axel R, An activated Notch suppresses neurogenesis and myogenesis but not gliogenesis in mammalian cells, Development 120(9) (1994) 2421–2430. [DOI] [PubMed] [Google Scholar]
- [51].Shirayoshi Y, Yuasa Y, Suzuki T, Sugaya K, Kawase E, Ikemura T, Nakatsuji N, Proto-oncogene of int-3, a mouse Notch homologue, is expressed in endothelial cells during early embryogenesis, Genes Cells 2(3) (1997) 213–224. [DOI] [PubMed] [Google Scholar]
- [52].Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO, Enzymatic assembly of DNA molecules up to several hundred kilobases, Nat. Methods 6(5) (2009) 343–345. 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- [53].Kutyavin IV, Afonina IA, Mills A, Gorn VV, Lukhtanov EA, Belousov ES, Singer MJ, Walburger DK, Lokhov SG, Gall AA, Dempcy R, Reed MW, Meyer RB, Hedgpeth J, 3’-minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures, Nucleic Acids Res. 28(2) (2000) 655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zanotti S, Canalis E, Parathyroid hormone inhibits Notch signaling in osteoblasts and osteocytes, Bone 103 (2017) 159–167. 10.1016/j.bone.2017.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, Yoneda T, Mohammad KS, Plotkin LI, Roodman GD, Bellido T, Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma, Cancer Res. 76(5) (2016) 1089–1100. 10.1158/0008-5472.CAN-15-1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Filipowska J, Tomaszewski KA, Niedzwiedzki L, Walocha JA, Niedzwiedzki T, The role of vasculature in bone development, regeneration and proper systemic functioning, Angiogenesis 20(3) (2017) 291–302. 10.1007/s10456-017-9541-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Watson EC, Adams RH, Biology of Bone: The Vasculature of the Skeletal System, Cold Spring Harb Perspect Med 8(7) (2018) a031559. 10.1101/cshperspect.a031559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lassailly F, Foster K, Lopez-Onieva L, Currie E, Bonnet D, Multimodal imaging reveals structural and functional heterogeneity in different bone marrow compartments: functional implications on hematopoietic stem cells, Blood 122(10) (2013) 1730–1740. 10.1182/blood-2012-11-467498 [DOI] [PubMed] [Google Scholar]
- [59].Zhang G, Guo B, Wu H, Tang T, Zhang BT, Zheng L, He Y, Yang Z, Pan X, Chow H, To K, Li Y, Li D, Wang X, Wang Y, Lee K, Hou Z, Dong N, Li G, Leung K, Hung L, He F, Zhang L, Qin L, A delivery system targeting bone formation surfaces to facilitate RNAi-based anabolic therapy, Nature medicine 18(2) (2012) 307–314. 10.1038/nm.2617 [DOI] [PubMed] [Google Scholar]
- [60].Desjardins CA, Yao M, Hall J, O’Donnell E, Venkatesan R, Spring S, Wen A, Hsia N, Shen P, Russo R, Lan B, Picariello T, Tang K, Weeden T, Zanotti S, Subramanian R, Ibraghimov-Beskrovnaya O, Enhanced exon skipping and prolonged dystrophin restoration achieved by TfR1-targeted delivery of antisense oligonucleotide using FORCE conjugation in mdx mice, Nucleic Acids Res. 50(20) (2022) 11401–11414. 10.1093/nar/gkac641 [DOI] [PMC free article] [PubMed] [Google Scholar]